 Qingyi Wang1,2
Qingyi Wang1,2 Bin Xie
Bin Xie Yongguang Tao
Yongguang Tao Desheng Xiao
Desheng Xiao- 1Department of Pathology, Xiangya Hospital, Central South University, Changsha, China
- 2Department of Pathology, School of Basic Medicine, Key Laboratory of Carcinogenesis and Cancer Invasion (Ministry of Education), Central South University, Changsha, China
- 3National Health Commission (NHC) Key Laboratory of Carcinogenesis (Central South University), Cancer Research Institute and School of Basic Medicine, Central South University, Changsha, China
- 4Hunan Key Laboratory of Early Diagnosis and Precision Therapy, Second Xiangya Hospital, Central South University, Changsha, China
- 5Department of the 2nd Department of Thoracic Surgery, Hunan Cancer Hospital and The Affiliated Cancer Hospital of Xiangya School of Medicine, Central South University, Changsha, China
The fruitful results of tumor immunotherapy establish its indispensable status in the regulation of the tumorous immune context. It seems that the treatment of programmed cell death receptor 1 (PD-1) blockade is one of the most promising approaches for cancer control. The significant efficacy of PD-1 inhibitor therapy has been made in several cancer types, such as breast cancer, lung cancer, and multiple myeloma. Even so, the mechanisms of how anti-PD-1 therapy takes effect by impacting the immune microenvironment and how partial patients acquire the resistance to PD-1 blockade have yet to be studied. In this review, we discuss the cross talk between immune cells and how they promote PD-1 blockade efficacy. In addition, we also depict factors that may underlie tumor resistance to PD-1 blockade and feasible solutions in combination with it.
Background
Immune surveillance functions of innate and adaptive immune cells can be suppressed by multiple mechanisms in the tumor microenvironment (TME); the most noted one is the programmed cell death receptor 1 (PD-1)/programmed cell death ligand 1 (PD-L1) pathway. For example, PD-L1, as the ligand of PD-1, could overexpress on tumor cells to evade the antitumor immune response by repressing the activation and function of CD8+T cells (1). Anti-PD-1 is one of the most promising attractive anticancer immune checkpoint blockers (ICB). Growing evidence shows that not only T cells but also other immune cells can be promoted by anti-PD-1 directly or indirectly, to suppress the progression of tumors (2–5). However, despite PD-1 blockade therapies having durable responses for a minority of patients in clinical trials, there is still an unmet clinical need for the majority of patients who do not respond to anti-PD-1 (6). Thus, we firstly summarize the cross talk between immune cells and their possible transformation in the TME after PD-1 blockade therapy. In the second part, we discuss the primary impact factors of resistance to PD-1 inhibitors, such as tumor immune recognition, oncogenic signal pathways, interferon (IFN), immune contexture, angiogenesis, immunometabolism, intestinal microbiota, and new immune checkpoints. We also highlight feasible combined therapy strategies to re-sensitize tumors to PD-1 blockade.
The Role of PD-1 and PD-1 Inhibitors in Immune Response
PD-1
PD-1, a member of the B7-CD28 receptor family, is a transmembrane protein and widely expressed in B cells, T cells, natural killer (NK) cells, and myeloid cells (7). As the ligand of PD-1, programmed cell death ligand 1 (PD-L1) can be expressed in dendritic cells (DCs), macrophages, T cells, NK cells (8, 9), and tumor cells (10). Generally, when PD-L1 binds to PD-1 in the presence of the T cell receptor (TCR) signaling complex, PD-1 delivers a co-inhibitory signal, leading to the termination of TCR signaling and inhibition of T cell proliferation (11). PD-1 often uses mono-tyrosine signaling motifs which present in its cytoplasmic tail, such as immunoreceptor tyrosine-based inhibitory motif (ITIM) and immunoreceptor tyrosine-based switch motif (ITSM) (12), to end the CD28/TCR signal by PD-1 phosphorylation and the recruitment of SHP-2 and SHP-1 (13–15). In the tumor immune context, antigen-presenting cells (APCs) and tumor cells highly express PD-L1, and they can interact with PD-1-overexpressed T cells, leading to T-cell anergy or exhaustion (16, 17). Programmed cell death ligand 2 (PD-L2) is the second ligand for the PD-1 molecule, which is expressed predominantly by DCs, macrophages, B cells, and cancer cell populations, depending on microenvironmental stimulation (18, 19). Similar to PD-L1, PD-L2 plays a crucial role in evading antitumor immunity. The engagement of PD-1 and PD-L2 can lead to the downregulation of T cell responses, which inhibits TCR-mediated proliferation and cytokine production by CD4+ T cells by blocking cell cycle progression (18). Although PD-1/PD-L2 blockade must be considered for optimal immunotherapy in antitumor immunity (20), since most of the research results are focused on the PD-1/PD-L1 pathway, we mainly discuss the PD-1/PD-L1 axis in this article.
PD-1 Inhibitors
As surface molecules, the activity of PD-1 and PD-L1 can be easily inhibited by blocking antibodies. Anti-PD-1 therapy is one of the most successful immune checkpoint blockade therapies that have been approved to treat a wide variety of cancer types (Table 1). PD-1 inhibitors competitively bind to PD-1 and block PD-1/PD-L1 interactions, which subsequently regulate negative signals on the T cell surface to enhance the functions of effector T cells and promote the proliferation of T cells (54). Nivolumab and pembrolizumab are the primary clinically approved PD-1 inhibitors. They are humanized IgG4 antibodies targeting PD-1 with high affinity (55). To ensure that they elicit their inhibitory effects of PD-1/PD-L1 interactions primarily by direct occupancy and steric blockade of the PD-L1-binding site of PD-1 (56), they minimize the function of effector cells engaging other antibodies.
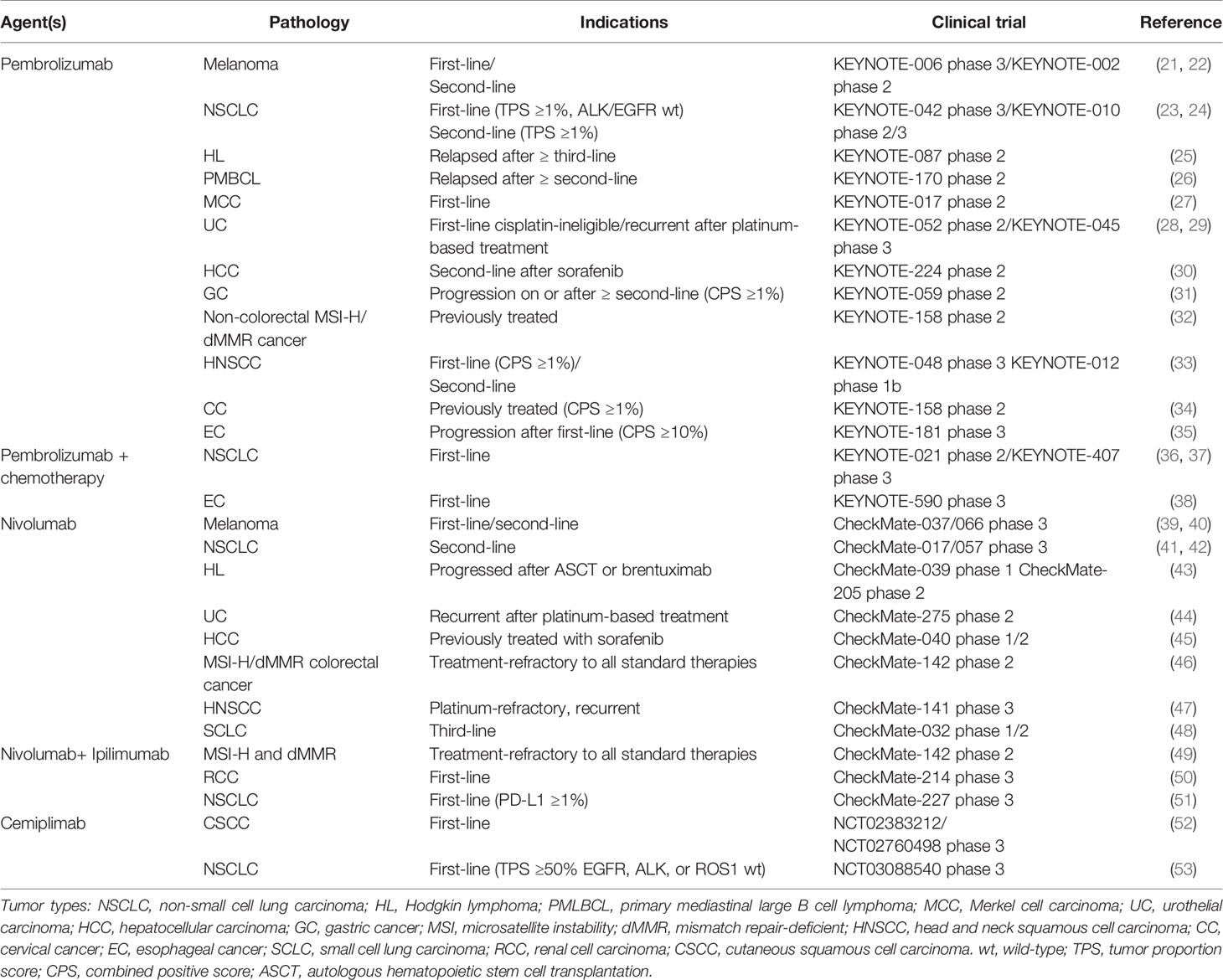
Table 1 Summary of FDA-approved PD-1 inhibitors in advanced/metastatic cancers.
Pembrolizumab was initially approved for refractory unresectable melanoma in 2014 (57), known as the first PD-1-targeted therapy to gain Food and Drug Administration (FDA) approval. Not long, in 2015, it becomes the first immune checkpoint inhibitor to be approved as a first-line treatment, also in melanoma therapy (21). Pembrolizumab is thus approved to treat a wide variety of cancer types. To date, pembrolizumab therapy has been licensed in many cancers (27, 30, 58, 59) and was often conducted primarily in patients with PD-L1-positive disease (31, 34). In general, a higher level of PD-L1 expression is associated with a more effective clinic outcome of pembrolizumab. However, in some cancer types, such as non-small cell lung cancer (NSCLC) (60), classical Hodgkin’s lymphoma (cHL) (25), and urothelial carcinoma (UC) (61), PD-L1 expression did not explicitly correlate with response to pembrolizumab.
Nivolumab also displays a good response and favorable safety profile, particularly in melanoma and NSCLC. Nivolumab was approved by FDA following its showing a clear advantage in response over chemotherapy in refractory unresectable melanoma (62). Soon after, the FDA approved nivolumab for the treatment of NSCLC after progression on a platinum-based chemotherapy regimen (41, 63). Also, nivolumab has been demonstrated durable effects in other cancers (47, 64, 65), and it appears that combination therapy may further improve them (50, 66). Nevertheless, research has demonstrated a low response rate in some hematological tumors, such as follicular lymphoma (FL) (67) and diffuse large B cell lymphoma (DLBCL) (68). It may appear to correlate positively with 9p24.1 translocation and increased PD-L1 expression (69). In addition to nivolumab and pembrolizumab, cemiplimab is also approved by FDA for the treatment of advanced cutaneous squamous cell carcinoma (70) and first-line NSCLC (53). Up till now, more than 1,500 clinical trials involving PD-1 inhibition are currently supported by the National Cancer Institute (NCI).
Immune Microenvironment
Immunotherapies based on PD-L1/PD-1 blockade have revolutionized the treatment paradigm for several cancer types. Their interaction regulates the activation of immune responses and specifically of T cell responses in physiological conditions. In the last years, increasing evidence has demonstrated that the elimination of tumor cells is mainly mediated by cytotoxic T lymphocytes (CTLs) (71). Several types of immune cells in the TME, such as tumor-associated macrophages (TAMs), DCs, NK cells, and immunosuppressive cells, can also interact with each other to promote or repress tumor progression in direct and indirect mechanisms by secreting cytokines and chemokines (71). Indeed, there is a complex picture of the relationship between checkpoint blockade and immune context. The precise molecular mechanisms of how PD-1 inhibitors function by stimulation/inhibition of immune-related cells remain to be fully understood. Here, we will attempt to discuss in detail the cross talk between immune cells and the critical role of some immune cells in the efficacy of PD-1 inhibitors therapy (Figure 1).
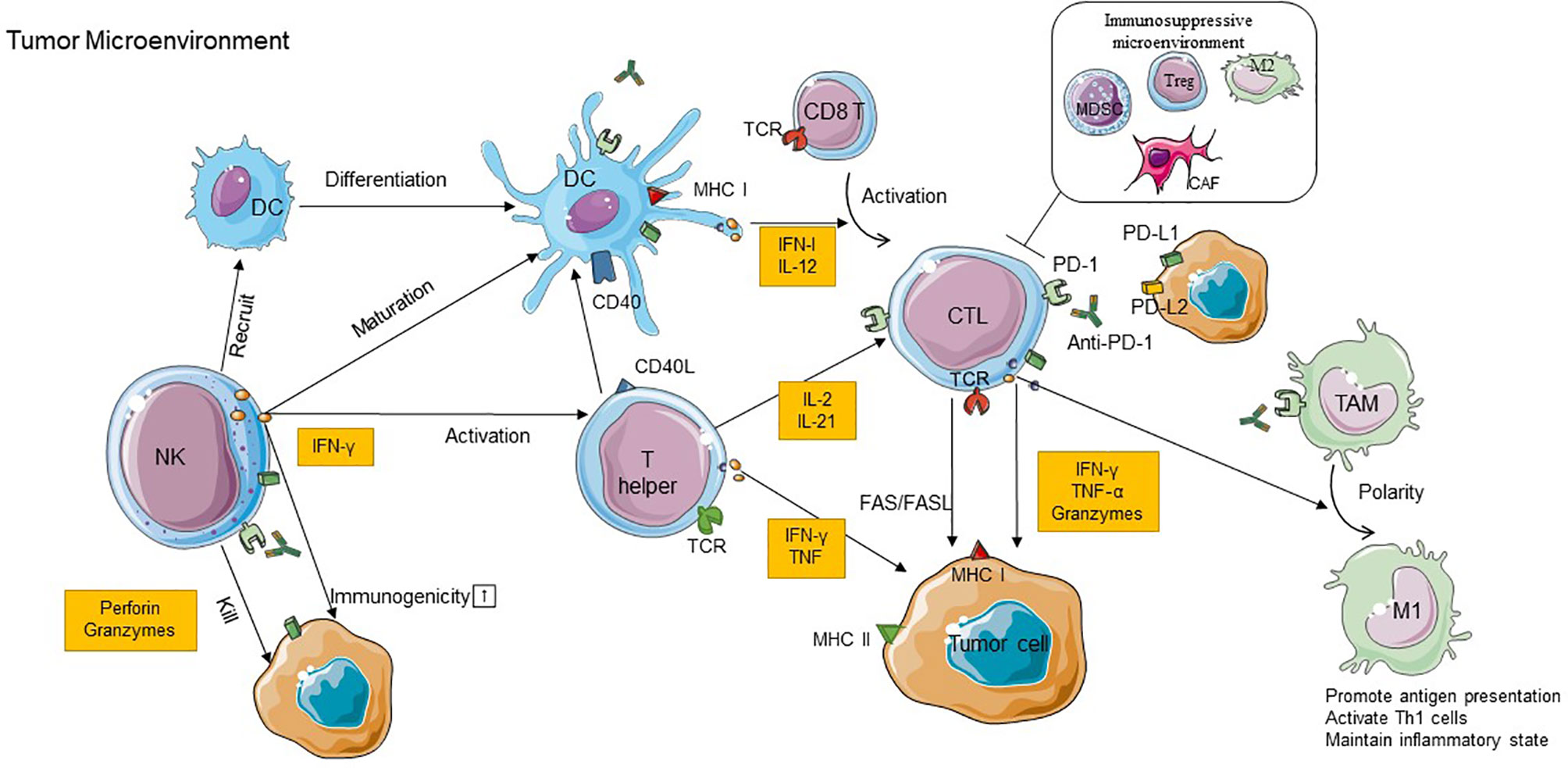
Figure 1 The cross talk between immune cells in TME and the role of immune cells in the efficacy of PD-1 inhibitor therapy. CD8+T cells recognize antigens requiring a corroboration work between CD4+T cells with NK cells and DCs, and M1 TAM can exert antitumoral effects due to the stimulation of IFN-γ produced by CD8+ T cells. In addition to these immunostimulatory cells, the immunoinhibitory cells including CAFs, Tregs, M2 TAMs, and MDSCs can construct an immunosuppressive microenvironment to restrict the antitumor effect. Anti-PD-1 binds to PD-1 on immune cells which can block PD-1/PD-L1 interactions and recover the antitumor function of those cells. PD-1, programmed cell death protein 1; PD-L1, programmed death-ligand 1; PD-L2, programmed death-ligand 2; IFN-γ, interferon gamma; TNF, tumor necrosis factor; CTL, cytotoxic T lymphocyte; DC, dendritic cell; NK, natural killer cell; MDSC, myeloid-derived suppressor cell; Treg, regulatory T cell; CAF, cancer-associated fibroblast; TAM, tumor-associated macrophage; M1, type 1 macrophage cell; M2, type 2 macrophage cell; MHC, major histocompatibility complex; TCR, T cell receptor; TME, tumor microenvironment.
T Cells
CD8+T Cells
CD8+T cells are a subset of lymphocytes developing in the thymus. They recognize antigen-presented cells expressing major histocompatibility complex (MHC) class I molecules and in turn exert antitumor function (3, 71). Initiation of a response from CD8+T cells against an antigen requires corroboration work between CD4+T cells with NK cells and DCs (3, 72). Activated, antigen-loaded DCs can launch the differentiation of CD8+T cells into CTLs by cross-presenting MHC class I molecules to cells (73). CD4+T cells can secrete cytokines following the interaction with antigens to simulate the optimal proliferation and activation of CD8+ T cells (74). On the other hand, NK cells and CD4+T cells can produce chemokines which indirectly induce the activation of CD8+ T cells by promoting the differentiation and maturation of DC cells (72, 75). Due to such cross talk, CTLs can initiate the antitumor effect through releasing IFN-γ and tumor necrosis factor α (TNF-α) to induce cytotoxicity in the cancer cells (76).
However, PD-1, as a coinhibitory receptor, could overexpress on activated CD8+ T cells (77). Once this happens, signals downstream of TCR may be attenuated and may cause the exhaustion of CD8+ T cells and ultimately contribute to the restriction of T cell activation and cytokine production (78). PD-1 blockade therapy seems to counteract tumor-induced T cell dysfunctionality by interfering with PD-1/PD-L1 signals; it releases the negative regulation of T cells and promotes T cells which produced higher levels of IFN-γ to activate antitumor immune response (79–81). Besides, PD-1 inhibitors reinvigorate preexisting CD8+T cells within the tumor and promote systemic T cell immunity priming. Nevertheless, the study revealed that preexisting tumor-specific T cells may have limited reinvigoration capacity and that the T cell response to checkpoint blockade derives from a distinct repertoire of T cell clones that may have just recently entered the tumor (82). The priming of antitumor T-cell immunity in lymphatic drainage might explain such consequence, which is further explained in another study. This study showed that tumor-draining lymph nodes (TDLNs) are enriched for tumor-specific PD-1+T cells which are closely associated with PD-L1+DCs (83). Suppression of DCs, accompanied by excess PD-L1 surface expression, may lead to restrained T cell priming and deviated CD8+ T cell differentiation in the TDLN. Therefore, it suggests that progenitor-exhausted T cells can be rescued by immune checkpoint blockade and then home to the tumor and populate the TME, to improve tumor control (83). However, the exact contribution of TDLN versus TME during PD-1/PD-L1 checkpoint blockade therapy remains to be elucidated.
On the other hand, the report found that PD-L1 can also be upregulated on T cells (84). PD-L1-expressing T cells can suppress immunity on neighboring T cells and polarize macrophages toward a tolerogenic phenotype via the PD-L1–PD-1 axis in the TME, which in turn both suppresses T cell activation and promotes tumor growth (84). It is still not clear whether PD-1 inhibitors also play a role based on this theory. Accordingly, the precise molecular mechanisms of T cell function stimulated by PD-1 inhibitors remain to be clarified.
CD4+T Cells (T Helper Cells)
CD4+T cells participate in the activation and expansion of CD8+T effectors; they induce an antitumor response by providing regulatory signals (85–87). In the tumor context, MHC class II molecules can present antigenic peptides recognized by CD4+T cells (88, 89). MHC-class II+ tumors can be directly killed by CD4+ CTLs. For the MHC-class II-negative tumor cells, CD4+ T cells can produce a vast range of cytokines that mediate inflammatory and effector immune responses (90, 91); TNF and IFN-γ are the most important cytokines that are mainly produced by T helper (Th) 1 cells. Additionally, CD4 Th1 cells also display antitumor responses by activating NK cells (90) and M1 TAM (92, 93), inhibition of angiogenesis (94), and/or induction of tumor senescence (95).
To date, the specific contribution of CD4 immunity to PD-1 blockade therapy efficacy is still unknown. In NSCLC, proliferation and low PD-1/LAG-3 co-expression of CD4 at baseline were responsive to PD-1 blockade ex vivo and in vivo (96). In cHL, PD-1 blockade therapy has strong antitumor effects on MHC-II-expressing tumors mediated by cytotoxic CD4+ T cells in murine models (97). These provide strong evidence that CD4 immunity might be an entry point to achieve efficacious clinical responses under PD-1 blockade therapies. Further research is needed to reveal the specific contribution of CD4+ T cells.
NK Cells
NK cells can spontaneously kill cells and thus are presumed to be key innate immune effectors in cancer immunosurveillance; it belongs to the family of innate lymphoid cells (ILCs) (98). IFN-γ produced by NK cells during early-phase immune responses can directly kill tumor cells and promote the differentiation of naive CD4+ T cells toward Th1 cells to facilitate cell-mediated immunity (99). Thus, NK cells are critical components both in humoral immunity and in cellular immunity.
As an inhibitory receptor, PD-1 can express on NK cells (100, 101) and prevent the activation of NK cell function when engaging with its ligand which is expressed on the surface of target tumor cells or APC (102). PD-1+ NK cells may be inhibited in killing tumor cells instead of being anergic in PD-L1+ tumors, which means that PD-1 is an important checkpoint for NK activation and PD-1 blockade might elicit an antitumor NK cell response (102). In high PD-L1 expression head and neck cancer (HNC) patients, the study observed that PD-1 blockade increased cetuximab-mediated NK cell activation and cytotoxicity (103). Besides, tumors might drive the development of PD-L1-expressing NK cells that acquire immunoregulatory functions; such cell population can directly inhibit CD8+ T cell proliferation in a PD-L1-dependent manner (104). These results show the importance of the PD-1/PD-L1 axis in inhibiting NK cell responses in vivo, and future research is needed to determine the specific mechanism of the PD-1 pathway in the antitumor response of NK cells.
DCs
DCs, known as specialized APC, transport tumor antigens to draining lymph nodes and cross-present antigens via MHC I and II to activate cytotoxic T lymphocytes (105). DC maturation is necessary to T cell proliferation and differentiation; the final antitumor immunity is also associated with co-stimulatory molecules and cytokines which are expressed as the mature markers on DCs, such as CD80/CD86 and IL-12 (106).
DCs are necessary for anti-PD-1 efficacy. Anti-PD-1-activated T cells secrete IFN-γ, which in turn primes a transcriptomic shift in DC phenotype; DCs produce IL-12 upon sensing IFN-γ to stimulate effector T cell responses (107–109). The activation of the non-canonical nuclear factor kappa-light-chain enhancer of the activated B cell (NF-κB) pathway is also required for checkpoint efficacy, for it can enrich IL-12-producing DCs (107). Additionally, evidence of direct regulation is still emerging. PD-1 expression has recently been identified on DCs in the specific tumor context (110, 111). The result of an ovarian study demonstrated that PD-1 expressed on the tumor-associated DC can suppress NF-κB activation and the release of immune regulatory cytokines and restrict the upregulation of co-stimulatory molecules (111), which mediate immune suppression. PD-1 inhibition seems to increase the co-stimulatory molecule expression of DCs (112). In addition, the specific ablation of PD-1 on intratumoral DCs resulted in enhanced priming of tumor-specific CD8+ T cells to secrete IL-2 and IFN-γ (110). While DCs are the major antigen-presenting cells for cross-presenting tumor antigens to T cells and promoting antitumor response, PD-L1 expression on DCs can be upregulated by inflammatory cytokines, especially IFNs. Such upregulation is likely to prevent the overexpansion of tumor-infiltrating lymphocytes and eventually dampen the antitumor responses (113, 114). These results might provide additional insights into the role PD-1/PD-L1 plays on DCs to facilitate antitumor response and the mechanisms of immune checkpoint blockade therapy efficacy.
TAMs
TAMs are major components of infiltrated leukocytes in tumors, which dominantly orchestrate cancer-related inflammation (115). They can be divided into two subtypes: M1 and M2. Anti-tumorigenic M1 macrophages express high levels of TNFα, inducible nitric oxide synthase (iNOS), and MHC class II molecules. They exert antitumoral effects due to the stimulation of IFN-γ produced by CD8+ T cells and CD4+T cells (71). Inversely, pro-tumorigenic M2 macrophages are marked with a high level of arginase 1 (ARG1) and CD206 expression (116). M2 cells can secrete STAT3 to the TME for impairing responses from CTLs when their number increases in the stroma (117). Besides, M2 cells can express inhibitory ligands PD-L1, which bind to inhibitory receptor PD-1 constitutively expressed in T cells to activate them, directly inhibiting TCR signals to restrain the antitumor function of T cells (118).
Primary macrophages transform into the M1 or M2 phenotype which can be induced by PD-1 signaling pathways (119). TAMs display detectable PD-1 levels in the tumor microenvironment; PD-1 blockade therapy contributed to both a direct and an indirect impact on TAMs. Indirectly, checkpoint blockade-activated T cells can accumulate TAMs by secrete factors (such as IFN-γ) to remodel the TME toward a tumor hostile environment rich in iNOS+ TAMs (119). In direct regulation, PD-1 deficiency in TAMs shifts their phenotype toward an antitumor profile, with higher levels of TNF-α, iNOS, and MHC II (120). Myeloid-specific PD-1 deletion was as effective at limiting tumor growth as global PD-1 deletion and more effective than selective ablation of PD-1 in T cells (121). TAM PD-1 expression negatively correlates with phagocytic potency against tumor cells; TAM infiltration is skewed toward high CD206 and ARG1 macrophages dampening antitumor immune responses (122, 123). Anti-PD-1 therapy can surprisingly reverse this trend, increasing the expression of iNOS, TNF-α, and IL-6, which may augment antitumor immunity (124). Accordingly, the inhibition of PD-1 expressed on TAMs can shift them to the M1 phenotype and form an antitumor TME.
Immunosuppressive Cells
Immunosuppressive cells, unlike immune cells, have a positive effect on antitumoral immunity. There are some immunoinhibitory cells that display negative cross talking in TME, including cancer-associated fibroblasts (CAFs), regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), and M2 TAMs (mentioned above). Tregs repress the proliferation of both CD8+ and CD4+T cells through releasing transforming growth factor β (TGF-β) (125). CAFs promote the rate of glycolytic metabolism and further constitute a glucose-deficient TME. CTLs tend to decrease their number when encountered with such conditions (126). It is not yet known whether these have a role in promoting the efficacy of PD-1, but studies have shown that they are crucial in immune resistance, which will be discussed in detail in a subsequent paragraph.
Drug Resistance and Combined Therapy
Anti-PD-1 therapy has shown significant efficacy in clinical trials and has been approved for treating several cancers in clinic therapy. However, the occurrence of primary or acquired drug resistance will cause the patient to be ineffective to PD-1 blockade therapy or eventually the recurrence of malignant tumors (127). There are internal and external causes of tumor resistance to PD-1 blockade. The internal causes focus on the inherent characteristics of tumor cells; these include defective tumor immunorecognition, epigenetic regulation, abnormal oncogenic signaling, and IFN-γ signal pathway, while the external causes are mainly emanated from the tumor microenvironment, such as exhaustion of T cells, immunosuppressive cells and cytokines, tumor metabolites, new immune checkpoints, and intestinal microflora (128). Here, we summarize the primary resistant mechanisms to anti-PD-1 (Figure 2). In addition, we highlight emerging combined treatment strategies that might prolong the efficacy of PD-1 blockade or enable immunotherapy to impinge on previously intractable cancer types.
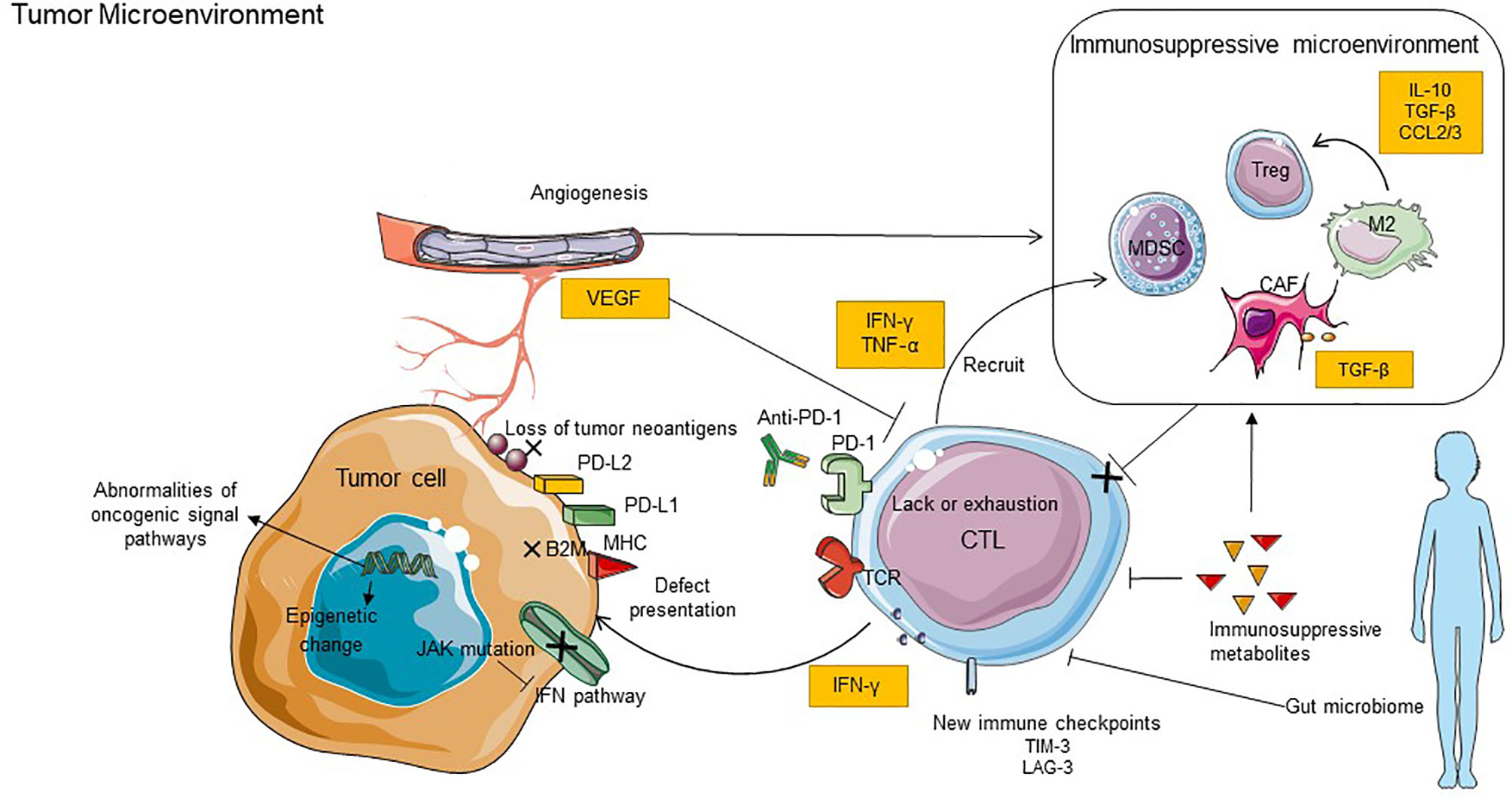
Figure 2 Key mechanisms of resistance to anti-PD-1 inhibitors. The mechanisms of resistance including internal and external causes. The internal causes focus on the inherent characteristics of tumor cells, it includes defective tumor immunorecognition, epigenetic regulation, abnormal oncogenic signaling, and IFN-γ signal pathway. The external causes are mainly emanated from the tumor microenvironment, such as exhaustion of T cells, immunosuppressive cells and cytokines, tumor metabolites, new immune checkpoints, and intestinal microflora. B2M, B2-microglobulin; LAG3, lymphocyte-activation gene 3; TIM3, T cell immunoglobulin and mucin domain-containing molecule 3; VEGF, vascular endothelial growth factor; JAK, Janus kinase.
Defective Tumor Immunorecognition
Some studies have shown that carcinomas with robust T cell immunosurveillance can evade recognition through diverse genetic and immune-related mechanisms, including loss of tumor neoantigens and defect in antigen presentation.
Loss of Tumor Neoantigens
Despite that cancer immunoediting can suppress tumor growth, it can establish favorable conditions within the tumor microenvironment to facilitate tumor outgrowth of the immune system which no longer recognizes the tumor (129). A neoantigen is an antigen encoded by the mutant gene of tumor cells. It is cross-presented via DCs and recognized by mature activated T cells. Emerging research supports the critical role of neoantigens in response to PD-1 blockade therapy. For instance, it highlights that neoantigen-specific CD8+T-cell responses were parallel to tumor regression in a responder of NSCLC patients treated with pembrolizumab (130), indicating that anti-PD-1 therapy enhances tumor neoantigen-specific T cell responses. In addition, in NSCLC patients who developed acquired drug resistance after single anti-PD-1 or anti-PD-1 combined with anti-CTLA-4 therapy, the loss of neoantigens has been found based on complete exome sequencing of tumor cells (131). It means that the PD-1-blocking therapy may be less effective if the tumor does not contain a mutation that can be a target. Despite the underlying mechanism being still unclear, evidence highlights that the combination of radiotherapy (RT) and anti-PD-1 is considered a promising strategy (132). Most likely, it is dependent on RT-induced cell damage that may express somatic mutations that generate neo-antigens, which have the potential to serve as targets for a more robust immune response (133). In preclinical triple-negative breast tumor models, data show that radiotherapy can enrich tumors of functionally active. Curative capacity has been enhanced when radiotherapy is combined with immunostimulatory and α-PD-1 monoclonal antibodies (mAbs) (134). Similarly, cancer cell death induced by chemotherapy is thought to promote tumor antigen release and antigen presentation and stimulate immune effectors. Combining checkpoint inhibitors with standard-of-care chemotherapy has been successful in non-small cell lung carcinoma (135, 136) and triple-negative breast cancer (137). Besides, individualized mutanome vaccines, an RNA-based poly-neo-epitope approach to mobilize immunity against a spectrum of cancer mutations, were applied to patients in melanoma and obtained a complete response to vaccination in combination with PD-1 blockade therapy (138). These results mean that the combination of PD-1 blockade with an agent that can facilitate tumor cells to generate neo-antigen may increase antitumor immunity.
Defective Antigen Presentation
Effective tumor antigen presentation to CD8+T cells relies on class I MHC (139, 140). Loss of heterozygosity and genetic deficiencies of β2-microglobulin (B2M) are both crucial ways that lead to the loss of MHC molecules (140–142), which promote resistance to PD-1 blockade due to the inability of CD8+T cells to recognize tumor antigens and specifically kill tumor cells (143). Thus, to recover the ability of antigen presentation may represent potential avenues that can be combined with immunotherapy.
The impairment of antigen presentation can be induced by epigenetic regulation. DNA methylation is thought to regulate the expression of tumor-associated antigens by downregulating the level of MHC class I. Studies have shown that the capability of DNA methyltransferase inhibitors (DNMTi) to upregulate MHC class I and MHC class II has appeared in many cancers (144, 145). Enhancer of zeste homolog 2 (EZH2), a catalytic component in the polycomb repressive complex 2 (PRC2), plays a crucial role in the mediation of histone h3 lysine 27 tri-methylation (H3K27me3) (146). Research revealed a negative correlation between the expression levels of EZH2 and MHC I antigen presentation molecules (147). The study also found that tumor progression of an anti-PD-1-resistant head and neck squamous cell carcinoma (HNSCC) model can be suppressed by combinatorial treatment of an EZH2 inhibitor and anti-PD-1. Paradoxically, in ovarian cancer models, EZH2 inhibition has nothing to do with the alteration of the class I antigen presentation of ovarian cancer cells (148), indicating that the regulation of EZH2 on antigen presentation may be cancer-type specific. Therefore, the impairment of antigen presentation may promote tumor immune escape while providing a potential strategy to overcome resistance to PD-1 inhibitor therapy.
Oncogenic Signal Pathways
Cancer is a genetic disease that can be induced by multiple genetic alterations, which are commonly caused by abnormalities of several key oncogenic pathways (149), like the phosphatase and tensin homolog (PTEN) signal pathway and mitogen-activated protein kinase (MAPK) signal pathway. Here, we mainly describe the two most common pathways, which have been proven to be closely related to PD-1 inhibitor resistance.
Research found that loss of PTEN in tumor cells in clinical patients of melanoma correlates with decreased T-cell infiltration, expansion, and inferior outcomes with PD-1 inhibitor therapy (150). PTEN loss-of-function mutations in tumors were significantly increased in non-responders who were treated with anti-PD-1 antibodies (151). Additionally, one of the most common pathways activated by loss of expression of the tumor suppressor PTEN is the phosphatidylinositol 3-kinase (PI3K) pathway, which plays a critical role in cancer by regulating several critical cellular processes. Thus, the PI3Kβ inhibitor, which is thought to regulate AKT activity in tumors with PTEN loss, has been applied to PTEN-deficient melanoma mouse models and demonstrated to enhance the efficacy of both PD-1 and CTLA-4 inhibitors (150). Accordingly, the regime that anti-PD-1 combined with PI3K-AKT pathway inhibitors may benefit cancer patients in the future.
The RAF/MEK/ERK pathway which is the classic routine in the MAPK pathway is also critical for human cancer; the pathway can be primed by activated RAS interacting with RAF kinase (152–154). Furthermore, RAS, RAF, and MEK are also frequently amplified or mutated in various cancers, accompanied by the activated MEK-ERK signaling pathway (155). KRAS, the component of RAS, is one of the most frequently mutated oncogenes in human cancers and participates in the mechanism of PD-1 inhibitor resistance (156). Similarly, BRAF, another mutated oncogene, has the vast majority in number harboring an activating point mutation (V600E) (157). This oncogenic mutation leads to constitutive activation of the MAPK signaling pathway and increased oncogenic potential through a variety of mechanisms, including reduced apoptosis, increased invasiveness, and increased metastatic behavior (158). Recent in vitro data suggest that BRAF V600E could also contribute to immune escape (157, 159). Based on these, selective inhibition of BRAF has been shown to induce an activated CD8+ T cell infiltrate, as well as increase melanoma MHC expression and melanoma antigen presentation early during treatment both in preclinical models and in human melanoma tissue samples (159–161). The study also suggested that combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma can increase the frequency of long-lasting antitumor responses (162). Thus, the inhibition of the RAF/MEK/ERK signaling pathway may be a promising therapeutic strategy for cancer dysregulated in this pathway.
IFNs
IFN-γ, effector cytokines of T cells, can directly exert an effective antitumor immune response by recognizing the corresponding receptors on tumor cells or indirectly promote the cross-activation of CD8+ T cells by upregulating antigen-presenting machinery to attack tumor cells (163). Classically, IFN-γ inhibits the proliferation of tumor cells and promotes their apoptosis, as it can activate signal transducer and activator of transcription 1 (STAT1) through using the Janus kinase (JAK) signal transducer and activator of the transcription pathway (127). Recent studies have implicated that defects in such pathways involved in IFN-receptor signaling and antigen presentation are associated with primary and acquired resistance to PD-1 blockades, such as inactivating mutations in JAK1 and JAK2 (143, 164). It may result in PD-L1 not being able to be reactively expressed and failing to attract T cell infiltration due to lack of chemokine production which is controlled by the IFN-γ pathway downstream of JAK1/2 (165). Considering that preexisting T cells in the tumor are a requisite for response to anti-PD-1 therapy (166), the absence of reactive PD-L1 expression may implicate a poor response to PD-1 blockade therapy, because of the impairment of tumor-infiltrating T cells (164).
IFN-β, belonging to type I IFN that is associated with innate immune responses (167), was proved to be suppressed by lysine-specific histone demethylase 1 (LSD1) (168). Ablation of LSD1 in cancer cells increases repetitive element expression; this leads to dsRNA stress and activation of type 1 IFN, which promotes antitumor T cell immunity and sensitizes refractory tumors to PD-1 blockade in a melanoma mouse model (168). The remarkable ability of LSD1 inhibition to convert a tumor resistant to PD-1 blockade to a tumor responsive to PD-1 blockade provides a means to increase the efficacy of anti-PD-1 cancer therapy and potentially turn “cold” tumors “hot” (169). It may suggest LSD1 inhibition combined with PD-1 blockade as a novel cancer treatment strategy. In addition, long-term IFN-β transcription can also promote the occurrence of resistance to anti-PD-1 therapy by inducing intratumoral augment of Tregs and myeloid cells, which cause T cell depletion and immunosuppression (170). Thus, IFNs display the consequence of resulting in T cell depletion and immunosuppression, although they can also promote the effect of tumor-specific CD8+T cells.
Immune Contexture
As noted, research of immune checkpoint blockade therapy was concentrated on reversing tumor-specific T cell dysfunction. CD8+T cells play an essential role in the scope of T cell-directed immunotherapy. Thus, the exhaustion of CD8+T cells induced by several factors can also be a crucial reason for PD-1 blockade resistance (143).
Epigenomic modifications might underlie CD8+T cell exhaustion. These long-lasting, exhaustion-associated epigenetic programs limit the rejuvenation of antigen-specific CD8 T cells during PD-1 blockade therapy. A study displayed that initial DNA-methylation programs could restrict T-cell expansion and clonal diversity during PD-1 blockade treatment (171). The administration of DNA-demethylating agents before ICB therapy reversed these programs and enhanced the reinvigoration of antitumor CD8 T cells. Moreover, the latest clinic trials concerning epigenetic therapies also suggest that histone deacetylase inhibitors may synergize with PD-1 blockade to overcome resistance (172, 173). What they found highlights epigenetic programs among exhausted T-cells as a potential mechanism to explain PD-1 blockade therapeutic failures. Besides, research found that co-stimulatory molecules like CD28 can also suppress the function of effector T cells and reduce the response to anti-PD-1 therapy by blocking the CD28-B7 co-stimulatory pathway (13). In addition to the regulation of epigenetic change and co-stimulatory pathway over CD8+T cells, other immune-suppressive cells also have more or less indirect effects on it, impacting drug resistance of anti-PD-1 therapy.
MDSCs are defined as immature myeloid cells, which can be induced to expand by tumor progression and play an immunosuppressive role in multiple cancers (174, 175). The recruitment of immunosuppressive MDSCs has shown complex protumorigenic outcomes following anti-PD-1 therapy (176). One mechanism of this recruitment may be driven by anti-PD-1-activated T cells, which partially trigger a tumor-intrinsic NLRP3 inflammasome signaling cascade (176, 177). This signaling cascade constitutes an adaptive resistance pathway, the genetic and pharmacological inhibition of which can enhance the efficacy of anti-PD-1 immunotherapy by inhibiting the tumor infiltration of MDSCs (176). On the other hand, checkpoint-activated CD8+ T cells can induce the differentiation and survival of protumorigenic TAMs and MDSCs by stimulating tumor production of CSF1 by secreting more TNF-α (178). These prompt us to hypothesize that neutralizing MDSCs and preserving T cell function may elicit robust immunotherapy responses by the combined actions of ICB agents together with targeted agents (179). Paradoxically, in HNSCC, it demonstrates reduced granulocytic MDSC infiltration post-PD-1 blockade (180). Thus, it is still unclear whether this model involves different mechanisms of MDSC recruitment or whether blockade of PD-1 inhibits MDSC proliferation directly.
TAM is another type of myeloid cells. It can impact the response to immunotherapy by activating triggering receptors expressed on myeloid cells 2 (TREM2) (181). TREM2 deficiency was associated with the transformation of macrophage subsets and an increase of intratumoral CD8+ T cells, some of which expressed PD-1. The observation found that tumor macrophage infiltrates enhanced T-cell-mediated control of tumor growth after the anti-TREM2 therapy; the anti-TREM2 mAb to tumor-bearing mice blunted tumor growth and strongly enhanced the efficacy of anti-PD-1 immunotherapy (181). Efforts are currently ongoing to complement checkpoint blockade with treatment targeting myeloid cells (115), including depletion of myeloid cells from tumors, blocking their pro-tumoral functions, or restoring their immunostimulatory properties (182, 183). These results may be applied as a theoretical basis to clinical trials.
Tregs can inhibit TCR-mediated activation and proliferation of CD4+/CD8+T cells to promote tumor immune evasion. Simultaneously, EZH2 has a critical role in maintaining the identity and function of Tregs; it has been proved that Ezh2 deficiency in Tregs stimulates antitumor immunity with enhanced T cell infiltration and elevated effector function (147). Mechanistically, Ezh2 functioned in regulating the stability of Foxp3 protein which is specifically expressed by Tregs. Based on these, the synergistic impact of the combination of EZH2 inhibition and anti-PD-1 has been found in an anti-PD-1-resistant model of HNSCC. It is explained that EZH2 inhibition can enhance tumor cell Class I MHC expression in vivo including in highly resistant models (147). Thus, it is promising that try to improve the efficacy of anti-PD-1 therapy by combining it with Ezh2 inhibitors.
CAFs are activated fibroblast cells during cancer development, contributing to the establishment of an immunosuppressive TME (184). Despite T cells being recovered from the capability against tumor cells following anti-PD-1 therapy, CAFs can act as a formidable barrier to T cells by secreting-related factors, resulting in T cell exclusion from tumor nests (185). TGF-β, a factor released by CAFs, promotes T-cell exclusion and blocks Th1 effector phenotype acquisition, which eventually results in resistance to PD-1 blockade therapy (186, 187). Inhibition of TGF-β unleashed a potent, enduring cytotoxic T-cell against tumor cells to prevent refractory. In mice with progressive liver metastatic disease, blockade of TGF-β signaling improves the susceptibility to anti-PD-1 therapy and suggests that TGF-β inhibition could prevent, but not reverse, CAF differentiation (186). NOX 4 is a specific downstream target of TGF-β. Inhibition of NOX 4 can “normalize” CAF to a quiescent phenotype and promote intratumoral CD8+ T-cell infiltration, overcoming the exclusion effect (185). These trials show that the regulation of CAFs through repressing the related downstream pathway or factors may have a synergistic effect on the anti-PD-1 therapy.
As mentioned above, one of the major obstacles that remain to be overcome is the restriction of T cells’ function in the immunosuppressive microenvironment formed by Tregs, MDSCs, and TAMs. The adoptive cell therapy (ACT) with chimeric antigen receptor (CAR)-redirected T cells is an attractive anticancer strategy. The breakthrough with CAR-T cell therapy was achieved, targeting B-cell hematologic tumors (188–191), while there is less efficacy in solid tumors. Research shows that TGF-β can be produced in most human tumors and markedly inhibits tumor antigen-specific cellular immunity. CAR-T lymphocytes have generated the resistance to TGF-β suppression, which expresses dominant-negative TGF-β receptors, to counteract these immunomodulatory activities (192). Such a result demonstrates their superior antitumor activity in animal models. Thus, combining engineered CAR-T cells with PD-1 antagonists makes a great deal of sense. There are promising results in both the pre-clinic model and case report (193, 194), presenting a large opportunity for the field of cellular engineering and immune checkpoint therapy.
Accordingly, the abovementioned studies indicate that the resistance to PD-1 inhibitors is directly related to the dysfunction of T cells caused by its epigenetic change, while other immune-related cells can also indirectly result in immune evasion via impacting the antitumor immunity progression of T cells.
Angiogenesis
The angiogenic tumor vasculature plays a vital role in regulating the response to cancer immunotherapy. Vascular abnormalities restrict T cell trafficking into the intratumor via upregulating vascular endothelial growth factor (VEGF) and gene-related to proangiogenic (195). Study has suggested that the VEGF signal induces the expression of the factor-related apoptosis antigen ligand (FasL)-mediated cell death on vascular endothelial cells, which in turn poses a formidable physical barrier to vascular material exchange (195). Additionally, the tumor neovasculature also decreases immature DCs and expands Treg cells and MDSC populations (195, 196). The modulation of tumor vasculature includes anti-angiogenesis and vascular normalization, which can induce the depletion of Tregs and regulatory B cells, enhancement of M1 TAMs, and activation of T cells, to reduce immunosuppression. The modulation can make favorable conditions for the infiltration of CD8+ cells and allow the effectiveness of immune checkpoint blockade (197). Immune checkpoint inhibitors have also shown promise in combination with anti-angiogenic in solid tumors (198), such as NSCLC and colorectal cancer (199). Thus, anti-angiogenesis and immunotherapy are documented to work synergistically together, showing promise for the resistance of PD-1 inhibitors.
Deregulation of Immunometabolism
Immune cells undergo complex shifts in metabolic states; immunosuppressive metabolites in TME can inhibit antitumor immunity by inhibiting immune cell infiltration (200–203).
Aerobic glycolysis is indispensable to CD8+T effector cells. It can be restricted by tumor cells that outcompete T cells for glucose uptake (81). In pretreatment of melanoma tumors, hypoxia-associated genes are highly expressed in the tumors that are subsequently resistant to PD-1 blockade compared with those from responding tumors (204). A high concentration of lactic acid can also blunt aerobic glycolysis of CD8+T cells and correlate with primary resistance on PD-1 blockade (205). A database analysis of patients with melanoma revealed strong negative associations between tumor lactate dehydrogenase expression and markers of CTL activation (201). Separately, indoleamine 2,3-dioxygenase (IDO), generated by tumors and immune cells, can enhance Treg and MDSC production and activity and inhibit the effect on T-cell immunity (206). IDO is the initial and rate-limiting enzyme in the degradation of tryptophan through the kynurenine pathway. A report found a significantly higher kynurenine/tryptophan ratio in NSCLC patients with early progression on nivolumab, suggesting that IDO might contribute to primary resistance to anti-PD-1 monoclonal antibodies (207). Despite that, the following clinical studies have shown that the efficacy of the IDO1 selective inhibitor plus PD-1 inhibitor is not as good as that of PD-1 blockade treatment alone (208). The combination therapy of IDO inhibitors and PD-1 antibodies may become a study direction for overcoming immunotherapy resistance. In addition, adenosine also is an immunosuppressive molecule that can suppress effector T cells and NK cells and increase Treg numbers (209, 210). Accordingly, metabolic disorders can encumber proper T cell activation and effector functions, which is a potential mechanism of resistance to PD-1 blockade. It is believed that the combined strategy based on this can bring gratifying results.
Disorder of Intestinal Microbiota
The gastrointestinal microbiome has been demonstrated to play an essential role in regulating the immune response function during cancer therapy (211–214). There is a group of active microorganisms that live in symbiosis with the host in the human intestinal tract and may cause tumor resistance to anti-PD-1 when it gets disordered (215, 216). Concordantly, a result has displayed that the responders to PD-1 blockade had a differential composition of gut bacteria (217). It has shown an “unfavorable” gut microbiome with low diversity and high relative abundance. Such a population may impair systemic and antitumor immune responses mediated by the limited intratumoral T cells, myeloid infiltration, and weakened antigen presentation capacity (211). Enhanced responses of anti-PD-1 therapy have been observed in mice that accepted fecal microbiome transplantation of the responder to PD-1 blockade. On the other hand, the efficacy of anti-PD-1 in mice receiving a non-responder could be restored by administration of specific genera enriched in responding patients in these mice. In addition, these specific genera were associated with increased intratumoral immune infiltrates mediated by the recruitment of CD4+T cells into the tumor bed and increased ratio of CD4+T cells to Tregs in response to PD-1 blockade (217). Besides, fecal microbiota transplant also overcomes resistance to anti-PD-1 therapy in melanoma patients (218). This suggests that regulating the gut microbiota may potentially enhance antitumor immune responses as well as response to immune checkpoint blockade.
New Immune Checkpoints
During checkpoint blockade with anti-PD-1 inhibitors, other inhibitory checkpoints might become coordinately upregulated and in turn lead to therapeutic failure (219). T-cell immunoglobulin mucin 3 (TIM-3), a member of the TIM family of immunomodulatory proteins, has been identified as a critical regulator of CTL exhaustion with co-expression of PD-1 (220). Such co-expression means that the most dysfunctional subgroup of T cells does not produce IL-2 and IFN-γ and eventually causes adaptive resistance. The mechanism has demonstrated that the increased Tim-3-mediated escape of exhausted TIL from PD-1 inhibition was mediated by PI3K/Akt complex downstream of TCR signaling in HNSCC (219). In vitro, the anti-Tim-3-blocking antibody reverses resistance to anti-PD-1 in PBMC from lung cancer patients (221). On the other hand, significant antitumor activity was observed after sequential addition of anti-Tim-3 mAb to overcome adaptive resistance to anti-PD-1 mAb in a murine HNSCC model (219). Thus, combination therapy targeting TIM-3 and PD-1 signaling pathways might be effective against the resistance of mono-immunotherapy.
Lymphocyte activation gene 3 (LAG-3) can selectively be expressed on activated T cells, NK cells, DCs and may get compensatory upregulation. The regulatory function of LAG-3 on T cells is similar to that of PD-1, which delivers suppressive signaling to hinder antitumor response (222). LAG-3 also competes for binding to MHC class II, which leads to decreased efficacy of MHC class II-mediated antigen presentation (223). The upregulation of LAG-3 in tumors of melanoma and lung cancer patients with acquired resistance to anti-PD-1 therapy has been demonstrated (223). There appeared to be a synergistic benefit of anti-LAG-3/anti-PD-1 combinatorial immunotherapy compared with anti-PD-1 monotherapy. In addition, a higher proportion of effector T cells were observed in mice treated with anti-LAG-3/anti-PD-1 than in PD-1 monotherapy groups. These suggest that anti-LAG-3/anti-PD-1 combinatorial immunotherapy may act synergistically (224). The roles of other checkpoints are still unconfirmed in anti-PD-1 resistance, such as TIGIT. Thus, a more particular knowledge of these new immune checkpoints may provide a rationale for designing combination treatments in the future.
Conclusions
In this review, we primarily describe a complex story of the relationship between anti-PD-1 and TME. The initiation of the antitumor effect depends on the cross talk between immune cells (Figure 1). Besides T cells, other immune-activating cells, like NK cells, DCs, and M1 TAMs, also contribute to anti-PD-1 efficacy through direct or indirect mechanisms. Furthermore, PD-1 blockade can target PD-1 expressed on these cells directly or reactivate CD8+ T cells to induce these immune-activating cell responses indirectly within the TME. Also, the review briefly displays the mechanisms that possibly contribute to primary or acquired resistance to PD-1 blockade, including the internal and external causes; the former focuses on the inherent characteristics of tumor cells while the other is mainly emanated from the tumor microenvironment (Figure 2). Due to the different reasons for drug resistance, the appropriate combination immunotherapy is also different, which is also discussed in detail in this article. It means that using a combination of such strategies is more suitable than using one approach alone for stimulating an antitumor immune response in some situations. A future challenge for researchers and clinicians is to achieve the satisfactory efficacy of immunotherapy. It means that the mechanisms of tumor immune evasion and immune drug resistance should be clarified as much as possible. It also plays a crucial role in the exploration of predictive markers, which are associated with the response rate of immunotherapy and improved clinical outcomes.
Author Contributions
QW wrote the manuscript; all authors were involved in the amendments and improvements in the text. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China [82072594 (YT), 82073097 (SL), 82073136 (DX), 81874139 (SL), and 81872285 (YS)], Shenzhen Science and Technology Program [KQTD20170810160226082 (YT)], Shenzhen Municipal Government of China [JCYJ20180507184647104 (YT)], and the Hunan Provincial Key Area R&D Program [2019SK2253 (YT)].
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors want to give the sincerest thanks to the other lab members of YT and DX for their helpful comments and suggestions.
Glossary
References
1. Baumeister SH, Freeman GJ, Dranoff G, Sharpe AH. Coinhibitory Pathways in Immunotherapy for Cancer. Annu Rev Immunol (2016) 34(February):539–73. doi: 10.1146/annurev-immunol-032414-112049
2. Liu X, Hogg GD, DeNardo DG. Rethinking Immune Checkpoint Blockade: “Beyond the T Cell”. J Immunother Cancer (2021) 9(1):e001460. doi: 10.1136/jitc-2020-001460
3. Borst J, Ahrends T, Bąbała N, Melief CJM, Kastenmüller W. CD4(+) T Cell Help in Cancer Immunology and Immunotherapy. Nat Rev Immunol (2018) 18(10):635–47. doi: 10.1038/s41577-018-0044-0
4. Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic Cells in Cancer Immunology and Immunotherapy. Nat Rev Immunol (2020) 20(1):7–24. doi: 10.1038/s41577-019-0210-z
5. Peterson EE, Barry KC. The Natural Killer–Dendritic Cell Immune Axis in Anti-Cancer Immunity and Immunotherapy. Front Immunol (2021) 11:621254. doi: 10.3389/fimmu.2020.621254
6. Yan Y, Zhang L, Zuo Y, Qian H, Liu C. Immune Checkpoint Blockade in Cancer Immunotherapy: Mechanisms, Clinical Outcomes, and Safety Profiles of PD-1/PD-L1 Inhibitors. Archivum Immunologiae Therapiae Experimentalis (2020) 68(6):1–15. doi: 10.1007/s00005-020-00601-6
7. Quezada SA, Peggs KS. Exploiting CTLA-4, PD-1 and PD-L1 to Reactivate the Host Immune Response Against Cancer. Br J Cancer (2013) 108(8):1560–5. doi: 10.1038/bjc.2013.117
8. Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive Correlates of Response to the Anti-PD-L1 Antibody MPDL3280A in Cancer Patients. Nature (2014) 515(7528):563–7. doi: 10.1038/nature14011
9. Alvarez M, Simonetta F, Baker J, Morrison AR, Wenokur AS, Pierini A, et al. Indirect Impact of PD-1/PD-L1 Blockade on a Murine Model of NK Cell Exhaustion. Front Immunol (2020) 11:7. doi: 10.3389/fimmu.2020.00007
10. Juneja VR, McGuire KA, Manguso RT, LaFleur MW, Collins N, Nicholas Haining W, et al. PD-L1 on Tumor Cells Is Sufficient for Immune Evasion in Immunogenic Tumors and Inhibits CD8 T Cell Cytotoxicity. J Exp Med (2017) 214(4):895–904. doi: 10.1084/jem.20160801
11. Okazaki T, Maeda A, Nishimura H, Kurosaki T, Honjo T. PD-1 Immunoreceptor Inhibits B Cell Receptor-Mediated Signaling by Recruiting Src Homology 2-Domain-Containing Tyrosine Phosphatase 2 to Phosphotyrosine. Proc Natl Acad Sci USA (2001) 98(24):13866–71. doi: 10.1073/pnas.231486598
12. He X, Xu C. Immune Checkpoint Signaling and Cancer Immunotherapy. Cell Res (2020) 30(8):660–9. doi: 10.1038/s41422-020-0343-4
13. Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T Cell Costimulatory Receptor CD28 Is a Primary Target for PD-1-Mediated Inhibition. Sci (New York NY) (2017) 355(6332):1428–33. doi: 10.1126/science.aaf1292
14. Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed Cell Death 1 Forms Negative Costimulatory Microclusters That Directly Inhibit T Cell Receptor Signaling by Recruiting Phosphatase SHP2. J Exp Med (2012) 209(6):1201–17. doi: 10.1084/jem.20112741
15. Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 Associate With Immunoreceptor Tyrosine-Based Switch Motif of Programmed Death 1 Upon Primary Human T Cell Stimulation, But Only Receptor Ligation Prevents T Cell Activation. J Immunol (2004) 173(2):945–54. doi: 10.4049/jimmunol.173.2.945
16. Zhao Y, Harrison DL, Song Y, Ji J, Huang J, Hui E. Antigen-Presenting Cell-Intrinsic PD-1 Neutralizes PD-L1 in Cis to Attenuate PD-1 Signaling in T Cells. Cell Rep (2018) 24(2):379–90.e6. doi: 10.1016/j.celrep.2018.06.054
17. Patsoukis N, Brown J, Petkova V, Liu F, Li L, Boussiotis VA. Selective Effects of PD-1 on Akt and Ras Pathways Regulate Molecular Components of The Cell Cycle and Inhibit T Cell Proliferation. Sci Signaling (2012) 5(230):ra46. doi: 10.1126/scisignal.2002796
18. Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 Is a Second Ligand for PD-1 and Inhibits T Cell Activation. Nat Immunol (2001) 2(3):261–8. doi: 10.1038/85330
19. Sharpe AH, Pauken KE. The Diverse Functions of the PD1 Inhibitory Pathway. Nat Rev Immunol (2018) 18(3):153–67. doi: 10.1038/nri.2017.108
20. Tanegashima T, Togashi Y, Azuma K, Kawahara A, Ideguchi K, Sugiyama D, et al. Immune Suppression by PD-L2 Against Spontaneous and Treatment-Related Antitumor Immunity. Clin Cancer Res (2019) 25(15):4808–19. doi: 10.1158/1078-0432.CCR-18-3991
21. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab Versus Ipilimumab in Advanced Melanoma. N Engl J Med (2015) 372(26):2521–32. doi: 10.1056/NEJMoa1503093
22. Ribas A, Puzanov I, Dummer R, Schadendorf D, Hamid O, Robert C, et al. Pembrolizumab Versus Investigator-Choice Chemotherapy for Ipilimumab-Refractory Melanoma (KEYNOTE-002): A Randomised, Controlled, Phase 2 Trial. Lancet Oncol (2015) 16(8):908–18. doi: 10.1016/S1470-2045(15)00083-2
23. Mok TSK, Wu YL, Kudaba I, Kowalski DM, Cho BC, Turna HZ, et al. Pembrolizumab Versus Chemotherapy for Previously Untreated, PD-L1-Expressing, Locally Advanced or Metastatic Non-Small-Cell Lung Cancer (KEYNOTE-042): A Randomised, Open-Label, Controlled, Phase 3 Trial. Lancet (London England) (2019) 393(10183):1819–30. doi: 10.1016/S0140-6736(18)32409-7
24. Herbst RS, Baas P, Kim DW, Felip E, Pérez-Gracia JL, Han J-Y, et al. Pembrolizumab Versus Docetaxel for Previously Treated, PD-L1-Positive, Advanced Non-Small-Cell Lung Cancer (KEYNOTE-010): A Randomised Controlled Trial. Lancet (London England) (2016) 387(10027):1540–50. doi: 10.1016/S0140-6736(15)01281-7
25. Chen R, Zinzani PL, Fanale MA, Armand P, Johnson NA, Brice P, et al. Phase II Study of the Efficacy and Safety of Pembrolizumab for Relapsed/Refractory Classic Hodgkin Lymphoma. J Clin Oncol: Off J Am Soc Clin Oncol (2017) 35(19):2125–32. doi: 10.1200/JCO.2016.72.1316
26. Armand P, Rodig S, Melnichenko V, Thieblemont C, Bouabdallah K, Tumyan G, et al. Pembrolizumab in Relapsed or Refractory Primary Mediastinal Large B-Cell Lymphoma. J Clin Oncol: Off J Am Soc Clin Oncol (2019) 37(34):3291–9. doi: 10.1200/JCO.19.01389
27. Nghiem P, Bhatia S, Lipson EJ, Sharfman WH, Kudchadkar RR, Brohl AS, et al. Durable Tumor Regression and Overall Survival in Patients With Advanced Merkel Cell Carcinoma Receiving Pembrolizumab as First-Line Therapy. J Clin Oncology: Off J Am Soc Clin Oncol (2019) 37(9):693–702. doi: 10.1200/JCO.18.01896
28. Balar AV, Castellano D, O’Donnell PH, Grivas P, Vuky J, Powles T, et al. First-Line Pembrolizumab in Cisplatin-Ineligible Patients With Locally Advanced and Unresectable or Metastatic Urothelial Cancer (KEYNOTE-052): A Multicentre, Single-Arm, Phase 2 Study. Lancet Oncol (2017) 18(11):1483–92. doi: 10.1016/S1470-2045(17)30616-2
29. Fradet Y, Bellmunt J, Vaughn DJ, Lee JL, Fong L, Vogelzang NJ, et al. Randomized Phase III KEYNOTE-045 Trial of Pembrolizumab Versus Paclitaxel, Docetaxel, or Vinflunine in Recurrent Advanced Urothelial Cancer: Results of >2 Years of Follow-Up. Ann Oncol: Off J Eur Soc Med Oncol (2019) 30(6):970–6. doi: 10.1093/annonc/mdz127
30. Zhu AX, Finn RS, Edeline J, Cattan S, Ogasawara S, Palmer D, et al. Pembrolizumab in Patients With Advanced Hepatocellular Carcinoma Previously Treated With Sorafenib (KEYNOTE-224): A Non-Randomised, Open-Label Phase 2 Trial. Lancet Oncol (2018) 19(7):940–52. doi: 10.1016/S1470-2045(18)30351-6
31. Fuchs CS, Doi T, Jang RW, Muro K, Satoh T, Machado M, et al. Safety and Efficacy of Pembrolizumab Monotherapy in Patients With Previously Treated Advanced Gastric and Gastroesophageal Junction Cancer: Phase 2 Clinical KEYNOTE-059 Trial. JAMA Oncol (2018) 4(5):e180013. doi: 10.1001/jamaoncol.2018.0013
32. Marabelle A, Le DT, Ascierto PA, Di Giacomo AM, De Jesus-Acosta A, Delord J-P, et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J Clin Oncol: Off J Am Soc Clin Oncol (2020) 38(1):1–10. doi: 10.1200/JCO.19.02105
33. Burtness B, Harrington KJ, Greil R, Souliéres D, Tahara M, de Castro GJ, et al. Pembrolizumab Alone or With Chemotherapy Versus Cetuximab With Chemotherapy for Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck (KEYNOTE-048): A Randomised, Open-Label, Phase 3 Study. Lancet (London England) (2019) 394(10212):1915–28. doi: 10.1016/S0140-6736(19)32591-7
34. Chung HC, Ros W, Delord JP, Perets R, Italiano A, Shapira-Frommer R, et al. Efficacy and Safety of Pembrolizumab in Previously Treated Advanced Cervical Cancer: Results From the Phase II KEYNOTE-158 Study. J Clin Oncology: Off J Am Soc Clin Oncol (2019) 37(17):1470–8. doi: 10.1200/JCO.18.01265
35. Kojima T, Shah MA, Muro K, Francois E, Adenis A, Hsu C-H, et al. Randomized Phase III KEYNOTE-181 Study of Pembrolizumab Versus Chemotherapy in Advanced Esophageal Cancer. J Clin Oncology: Off J Am Soc Clin Oncol (2020) 38(35):4138–48. doi: 10.1200/JCO.20.01888
36. Langer CJ, Gadgeel SM, Borghaei H, Papadimitrakopoulou VA, Patnaik A, Powell SF, et al. Carboplatin and Pemetrexed With or Without Pembrolizumab for Advanced, Non-Squamous Non-Small-Cell Lung Cancer: A Randomised, Phase 2 Cohort of the Open-Label KEYNOTE-021 Study. Lancet Oncol (2016) 17(11):1497–508. doi: 10.1016/S1470-2045(16)30498-3
37. Paz-Ares L, Luft A, Vicente D, Tafreshi A, Gümüş M, Mazières J, et al. Pembrolizumab Plus Chemotherapy for Squamous Non-Small-Cell Lung Cancer. N Engl J Med (2018) 379(21):2040–51. doi: 10.1056/NEJMoa1810865
38. Kato K, Shah MA, Enzinger P, Bennouna J, Shen L, Adenis A, et al. KEYNOTE-590: Phase III Study of First-Line Chemotherapy With or Without Pembrolizumab for Advanced Esophageal Cancer. Future Oncol (London England) (2019) 15(10):1057–66. doi: 10.2217/fon-2018-0609
39. Weber JS, D’Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, et al. Nivolumab Versus Chemotherapy in Patients With Advanced Melanoma Who Progressed After Anti-CTLA-4 Treatment (CheckMate 037): A Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet Oncol (2015) 16(4):375–84. doi: 10.1016/S1470-2045(15)70076-8
40. Larkin J, Minor D, D’Angelo S, Neyns B, Smylie M, Miller WHJ, et al. Overall Survival in Patients With Advanced Melanoma Who Received Nivolumab Versus Investigator’s Choice Chemotherapy in CheckMate 037: A Randomized, Controlled, Open-Label Phase III Trial. J Clin Oncol: Off J Am Soc Clin Oncol (2018) 36(4):383–90. doi: 10.1200/JCO.2016.71.8023
41. Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WEE, Poddubskaya E, et al. Nivolumab Versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N Engl J Med (2015) 373(2):123–35. doi: 10.1056/NEJMoa1504627
42. Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab Versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med (2015) 373(17):1627–39. doi: 10.1056/NEJMoa1507643
43. Younes A, Santoro A, Shipp M, Zinzani PL, Timmerman JM, Ansell S, et al. Nivolumab for Classical Hodgkin’s Lymphoma After Failure of Both Autologous Stem-Cell Transplantation and Brentuximab Vedotin: A Multicentre, Multicohort, Single-Arm Phase 2 Trial. Lancet Oncol (2016) 17(9):1283–94. doi: 10.1016/S1470-2045(16)30167-X
44. Sharma P, Retz M, Siefker-Radtke A, Baron A, Necchi A, Bedke J, et al. Nivolumab in Metastatic Urothelial Carcinoma After Platinum Therapy (CheckMate 275): A Multicentre, Single-Arm, Phase 2 Trial. Lancet Oncol (2017) 18(3):312–22. doi: 10.1016/S1470-2045(17)30065-7
45. El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in Patients With Advanced Hepatocellular Carcinoma (CheckMate 040): An Open-Label, Non-Comparative, Phase 1/2 Dose Escalation and Expansion Trial. Lancet (London England) (2017) 389(10088):2492–502. doi: 10.1016/S0140-6736(17)31046-2
46. Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz H-J, Morse MA, et al. Nivolumab in Patients With Metastatic DNA Mismatch Repair-Deficient or Microsatellite Instability-High Colorectal Cancer (CheckMate 142): An Open-Label, Multicentre, Phase 2 Study. Lancet Oncol (2017) 18(9):1182–91. doi: 10.1016/S1470-2045(17)30422-9
47. Ferris RL, Blumenschein GJ, Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med (2016) 375(19):1856–67. doi: 10.1056/NEJMoa1602252
48. Antonia SJ, López-Martin JA, Bendell J, Ott PA, Taylor M, Eder JP, et al. Nivolumab Alone and Nivolumab Plus Ipilimumab in Recurrent Small-Cell Lung Cancer (CheckMate 032): A Multicentre, Open-Label, Phase 1/2 Trial. Lancet Oncol (2016) 17(7):883–95. doi: 10.1016/S1470-2045(16)30098-5
49. Overman MJ, Lonardi S, Wong KYM, Lenz H-J, Gelsomino F, Aglietta M, et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J Clin Oncol: Off J Am Soc Clin Oncol (2018) 36(8):773–9. doi: 10.1200/JCO.2017.76.9901
50. Motzer RJ, Tannir NM, McDermott DF, Arén Frontera O, Melichar B, Choueiri TK, et al. Nivolumab Plus Ipilimumab Versus Sunitinib in Advanced Renal-Cell Carcinoma. N Engl J Med (2018) 378(14):1277–90. doi: 10.1056/NEJMoa1712126
51. Hellmann MD, Paz-Ares L, Bernabe Caro R, Zurawski B, Kim S-W, Carcereny Costa E, et al. Nivolumab Plus Ipilimumab in Advanced Non-Small-Cell Lung Cancer. N Engl J Med (2019) 381(21):2020–31. doi: 10.1056/NEJMoa1910231
52. Rischin D, Migden MR, Lim AM, Schmults CD, Khushalani NI, Hughes BGM, et al. Phase 2 Study of Cemiplimab in Patients With Metastatic Cutaneous Squamous Cell Carcinoma: Primary Analysis of Fixed-Dosing, Long-Term Outcome of Weight-Based Dosing. J Immunother cancer (2020) 8(1):e000775. doi: 10.1136/jitc-2020-000775
53. Sezer A, Kilickap S, Gümüş M, Bondarenko I, Özgüroğlu M, Gogishvili M, et al. Cemiplimab Monotherapy for First-Line Treatment of Advanced Non-Small-Cell Lung Cancer With PD-L1 of at Least 50%: A Multicentre, Open-Label, Global, Phase 3, Randomised, Controlled Trial. Lancet (London England) (2021) 397(10274):592–604. doi: 10.1016/S0140-6736(21)00228-2
54. Wong RM, Scotland RR, Lau RL, Wang C, Korman AJ, Kast WM, et al. Programmed Death-1 Blockade Enhances Expansion and Functional Capacity of Human Melanoma Antigen-Specific CTLs. Int Immunol (2007) 19(10):1223–34. doi: 10.1093/intimm/dxm091
55. Fessas P, Lee H, Ikemizu S, Janowitz T. A Molecular and Preclinical Comparison of the PD-1–Targeted T-Cell Checkpoint Inhibitors Nivolumab and Pembrolizumab. Semin Oncol (2017) 44(2):136–40. doi: 10.1053/j.seminoncol.2017.06.002
56. Lee JY, Lee HT, Shin W, Chae J, Choi J, Kim SH, et al. Structural Basis of Checkpoint Blockade by Monoclonal Antibodies in Cancer Immunotherapy. Nat Commun (2016) 7:1–10. doi: 10.1038/ncomms13354
57. Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R, et al. Anti-Programmed-Death-Receptor-1 Treatment With Pembrolizumab in Ipilimumab-Refractory Advanced Melanoma: A Randomised Dose-Comparison Cohort of a Phase 1 Trial. Lancet (London England) (2014) 384(9948):1109–17. doi: 10.1016/S0140-6736(14)60958-2
58. Robert C, Ribas A, Schachter J, Arance A, Grob J-J, Mortier L, et al. Pembrolizumab Versus Ipilimumab in Advanced Melanoma (KEYNOTE-006): Post-Hoc 5-Year Results From an Open-Label, Multicentre, Randomised, Controlled, Phase 3 Study. Lancet Oncol (2019) 20(9):1239–51. doi: 10.1016/S1470-2045(19)30388-2
59. Rini BI, Plimack ER, Stus V, Gafanov R, Hawkins R, Nosov D, et al. Pembrolizumab Plus Axitinib Versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med (2019) 380(12):1116–27. doi: 10.1056/NEJMoa1816714
60. Gandhi L, Rodríguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, et al. Pembrolizumab Plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N Engl J Med (2018) 378(22):2078–92. doi: 10.1056/NEJMoa1801005
61. Bellmunt J, de Wit R, Vaughn DJ, Fradet Y, Lee J-L, Fong L, et al. Pembrolizumab as Second-Line Therapy for Advanced Urothelial Carcinoma. N Engl J Med (2017) 376(11):1015–26. doi: 10.1056/NEJMoa1613683
62. Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob J-J, Cowey CL, et al. Overall Survival With Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med (2017) 377(14):1345–56. doi: 10.1056/NEJMoa1709684
63. Vokes EE, Ready N, Felip E, Horn L, Burgio MA, Antonia SJ, et al. Nivolumab Versus Docetaxel in Previously Treated Advanced Non-Small-Cell Lung Cancer (CheckMate 017 and CheckMate 057): 3-Year Update and Outcomes in Patients With Liver Metastases. Ann oncology: Off J Eur Soc Med Oncol (2018) 29(4):959–65. doi: 10.1093/annonc/mdy041
64. Antonia SJ, Lopez-Martin JA, Bendell JC, Ott PA, Taylor MH, Eder JP, et al. Checkmate 032: Nivolumab (N) Alone or in Combination With Ipilimumab (I) for the Treatment of Recurrent Small Cell Lung Cancer (SCLC). J Clin Oncol (2016) 34(15_suppl):100. doi: 10.1200/JCO.2016.34.15_suppl.100
65. Siefker-Radtke A, Baron A, Necchi A, Plimack E, Pal S, Bedke J, et al. Nivolumab Monotherapy in Patients With Advanced Platinum-Resistant Urothelial Carcinoma: Efficacy and Safety Update From CheckMate 275. J Clin Oncol (2019) 37:4524. doi: 10.1200/JCO.2019.37.15_suppl.4524
66. Overman MJ, Kopetz S, Lonardi S, McDermott R, Leone F, Leach J, et al. Nivolumab ± Ipilimumab Treatment (Tx) Efficacy, Safety, and Biomarkers in Patients (Pts) With Metastatic Colorectal Cancer (mCRC) With and Without High Microsatellite Instability (MSI-H): Results From the CheckMate-142 Study. Ann Oncol (2016) 27:vi158. doi: 10.1093/annonc/mdw370.27
67. Herrera AF, Goy A, Mehta A, Ramchandren R, Pagel JM, Svoboda J, et al. Safety and Activity of Ibrutinib in Combination With Durvalumab in Patients With Relapsed or Refractory Follicular Lymphoma or Diffuse Large B-Cell Lymphoma. Am J Hematology (2020) 95(1):18–27. doi: 10.1002/ajh.25659
68. Ansell SM, Minnema MC, Johnson P, Timmerman JM, Armand P, Shipp MA, et al. Nivolumab for Relapsed/Refractory Diffuse Large B-Cell Lymphoma in Patients Ineligible for or Having Failed Autologous Transplantation: A Single-Arm, Phase II Study. J Clin Oncol: Off J Am Soc Clin Oncol (2019) 37(6):481–9. doi: 10.1200/JCO.18.00766
69. Roemer MGM, Redd RA, Cader FZ, Pak CJ, Abdelrahman S, Ouyang J, et al. Major Histocompatibility Complex Class II and Programmed Death Ligand 1 Expression Predict Outcome After Programmed Death 1 Blockade in Classic Hodgkin Lymphoma. J Clin Oncol (2018) 36(10):942–50. doi: 10.1200/JCO.2017.77.3994
70. Migden MR, Rischin D, Schmults CD, Guminski A, Hauschild A, Lewis KD, et al. PD-1 Blockade With Cemiplimab in Advanced Cutaneous Squamous-Cell Carcinoma. N Engl J Med (2018) 379(4):341–51. doi: 10.1056/NEJMoa1805131
71. Farhood B, Najafi M, Mortezaee K. CD8+ Cytotoxic T Lymphocytes in Cancer Immunotherapy: A Review. J Cell Physiol (2019) 234(6):8509–21. doi: 10.1002/jcp.27782
72. Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. NK Cells Stimulate Recruitment of Cdc1 Into the Tumor Microenvironment Promoting Cancer Immune Control. Cell (2018) 172(5):1022–37.e14. doi: 10.1016/j.cell.2018.01.004
73. Palucka K, Banchereau J. Cancer Immunotherapy via Dendritic Cells. Nat Rev Cancer (2012) 12(4):265–77. doi: 10.1038/nrc3258
74. Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-Driven Biomarkers to Guide Immune Checkpoint Blockade in Cancer Therapy. Nat Rev Cancer (2016) 16(5):275–87. doi: 10.1038/nrc.2016.36
75. Peng X, He Y, Huang J, Tao Y, Liu S. Metabolism of Dendritic Cells in Tumor Microenvironment: For Immunotherapy. Front Immunol (2021) 12:613492. doi: 10.3389/fimmu.2021.613492
76. Thomas DA, Massagué J. TGF-Beta Directly Targets Cytotoxic T Cell Functions During Tumor Evasion of Immune Surveillance. Cancer Cell (2005) 8(5):369–80. doi: 10.1016/j.ccr.2005.10.012
77. Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, et al. Tumor Antigen-Specific CD8 T Cells Infiltrating the Tumor Express High Levels of PD-1 and Are Functionally Impaired. Blood (2009) 114(8):1537–44. doi: 10.1182/blood-2008-12-195792
78. Sfanos KS, Bruno TC, Meeker AK, De Marzo AM, Isaacs WB, Drake CG. Human Prostate-Infiltrating CD8+ T Lymphocytes Are Oligoclonal and PD-1+. Prostate (2009) 69(15):1694–703. doi: 10.1002/pros.21020
79. Pardoll DM. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi: 10.1038/nrc3239
80. Topalian SL, Drake CG, Pardoll DM. Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer Cell (2015) 27(4):450–61. doi: 10.1016/j.ccell.2015.03.001
81. Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell (2015) 162(6):1229–41. doi: 10.1016/j.cell.2015.08.016
82. Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, et al. Clonal Replacement of Tumor-Specific T Cells Following PD-1 Blockade. Nat Med (2019) 25(8):1251–9. doi: 10.1038/s41591-019-0522-3
83. Dammeijer F, van Gulijk M, Mulder EE, Lukkes M, Klaase L, van den Bosch T, et al. The PD-1/PD-L1-Checkpoint Restrains T Cell Immunity in Tumor-Draining Lymph Nodes. Cancer Cell (2020) 38(5):685–700.e8. doi: 10.1016/j.ccell.2020.09.001
84. Diskin B, Adam S, Cassini MF, Sanchez G, Liria M, Aykut B, et al. PD-L1 Engagement on T Cells Promotes Self-Tolerance and Suppression of Neighboring Macrophages and Effector T Cells in Cancer. Nat Immunol (2020) 21(4):442–54. doi: 10.1038/s41590-020-0620-x
85. Zuazo M, Arasanz H, Bocanegra A, Fernandez G, Chocarro L, Vera R, et al. Systemic CD4 Immunity as a Key Contributor to PD-L1/PD-1 Blockade Immunotherapy Efficacy. Front Immunol (2020) 11:586907. doi: 10.3389/fimmu.2020.586907
86. Marzo AL, Kinnear BF, Lake RA, Frelinger JJ, Collins EJ, Robinson BW, et al. Tumor-Specific CD4+ T Cells Have a Major “Post-Licensing” Role in CTL Mediated Anti-Tumor Immunity. J Immunol (Baltimore Md: 1950) (2000) 165(11):6047–55. doi: 10.4049/jimmunol.165.11.6047
87. Hung K, Hayashi R, Lafond-Walker A, Lowenstein C, Pardoll D, Levitsky H. The Central Role of CD4(+) T Cells in the Antitumor Immune Response. J Exp Med (1998) 188(12):2357–68. doi: 10.1084/jem.188.12.2357
88. Oh DY, Kwek SS, Raju SS, Li T, McCarthy E, Chow E, et al. Intratumoral CD4+ T Cells Mediate Anti-Tumor Cytotoxicity in Human Bladder Cancer. Cell (2020) 181(7):1612–25.e13. doi: 10.1016/j.cell.2020.05.017
89. Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, et al. Tumor-Reactive CD4(+) T Cells Develop Cytotoxic Activity and Eradicate Large Established Melanoma After Transfer Into Lymphopenic Hosts. J Exp Med (2010) 207(3):637–50. doi: 10.1084/jem.20091918
90. Perez-Diez A, Joncker NT, Choi K, Chan WFN, Anderson CC, Lantz O, et al. CD4 Cells can be More Efficient at Tumor Rejection Than CD8 Cells. Blood (2007) 109(12):5346–54. doi: 10.1182/blood-2006-10-051318
91. Mumberg D, Monach PA, Wanderling S, Philip M, Toledano AY, Schreiber RD, et al. CD4(+) T Cells Eliminate MHC Class II-Negative Cancer Cells In Vivo by Indirect Effects of IFN-Gamma. Proc Natl Acad Sci USA (1999) 96(15):8633–8. doi: 10.1073/pnas.96.15.8633
92. Eisel D, Das K, Dickes E, König R, Osen W, Eichmüller SB. Cognate Interaction With CD4(+) T Cells Instructs Tumor-Associated Macrophages to Acquire M1-Like Phenotype. Front Immunol (2019) 10:219. doi: 10.3389/fimmu.2019.00219
93. Fauskanger M, Haabeth OAW, Skjeldal FM, Bogen B, Tveita AA. Tumor Killing by CD4(+) T Cells Is Mediated via Induction of Inducible Nitric Oxide Synthase-Dependent Macrophage Cytotoxicity. Front Immunol (2018) 9:1684. doi: 10.3389/fimmu.2018.01684
94. Qin Z, Blankenstein T. CD4+ T Cell–Mediated Tumor Rejection Involves Inhibition of Angiogenesis That Is Dependent on IFN Gamma Receptor Expression by Nonhematopoietic Cells. Immunity (2000) 12(6):677–86. doi: 10.1016/s1074-7613(00)80218-6
95. Braumüller H, Wieder T, Brenner E, Aßmann S, Hahn M, Alkhaled M, et al. T-Helper-1-Cell Cytokines Drive Cancer Into Senescence. Nature (2013) 494(7437):361–5. doi: 10.1038/nature11824
96. Zuazo M, Arasanz H, Fernández-Hinojal G, García-Granda MJ, Gato M, Bocanegra A, et al. Functional Systemic CD4 Immunity Is Required for Clinical Responses to PD-L1/PD-1 Blockade Therapy. EMBO Mol Med (2019) 11(7):e10293. doi: 10.15252/emmm.201910293
97. Nagasaki J, Togashi Y, Sugawara T, Itami M, Yamauchi N, Yuda J, et al. The Critical Role of CD4+ T Cells in PD-1 Blockade Against MHC-II-Expressing Tumors Such as Classic Hodgkin Lymphoma. Blood advances (2020) 4(17):4069–82. doi: 10.1182/bloodadvances.2020002098
98. Diefenbach A, Colonna M, Romagnani C. The ILC World Revisited. In: Immunity. Cell Press (2017) 46:327–32. doi: 10.1016/j.immuni.2017.03.008
99. Castro F, Cardoso AP, Gonçalves RM, Serre K, Oliveira MJ. Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front Immunol (2018) 9:847. doi: 10.3389/fimmu.2018.00847
100. Pesce S, Greppi M, Tabellini G, Rampinelli F, Parolini S, Olive D, et al. Identification of a Subset of Human Natural Killer Cells Expressing High Levels of Programmed Death 1: A Phenotypic and Functional Characterization. J Allergy Clin Immunol (2017) 139(1):335–46.e3. doi: 10.1016/j.jaci.2016.04.025
101. Vari F, Arpon D, Keane C, Hertzberg MS, Talaulikar D, Jain S, et al. Immune Evasion via PD-1/PD-L1 on NK Cells and Monocyte/Macrophages Is More Prominent in Hodgkin Lymphoma Than DLBCL. Blood (2018) 131(16):1809–19. doi: 10.1182/blood-2017-07-796342
102. Hsu J, Hodgins JJ, Marathe M, Nicolai CJ, Bourgeois-Daigneault M-C, Trevino TN, et al. Contribution of NK Cells to Immunotherapy Mediated by PD-1/PD-L1 Blockade. J Clin Invest (2018) 128(10):4654–68. doi: 10.1172/JCI99317
103. Concha-Benavente F, Kansy B, Moskovitz J, Moy J, Chandran U, Ferris RL. PD-L1 Mediates Dysfunction in Activated PD-1 Þ NK Cells in Head and Neck Cancer Patients. Cancer Immunol Res (2018) 6(12):1548–60. doi: 10.1158/2326-6066.CIR-18-0062
104. Sierra JM, Secchiari F, Nuñez SY, Iraolagoitia XLR, Ziblat A, Friedrich AD, et al. Tumor-Experienced Human NK Cells Express High Levels of PD-L1 and Inhibit CD8+ T Cell Proliferation. Front Immunol (2021) 12:745939. doi: 10.3389/fimmu.2021.745939
105. Gardner A, Ruffell B. Dendritic Cells and Cancer Immunity. Trends Immunol (2016) 37(12):855–65. doi: 10.1016/j.it.2016.09.006
106. Mellman I, Steinman RM. Dendritic Cells: Specialized and Regulated Antigen Processing Machines. Cell (2001) 106(3):255–8. doi: 10.1016/S0092-8674(01)00449-4
107. Garris CS, Arlauckas SP, Kohler RH, Trefny MP, Garren S, Piot C, et al. Successful Anti-PD-1 Cancer Immunotherapy Requires T Cell-Dendritic Cell Crosstalk Involving the Cytokines IFN-γ and IL-12. Immunity (2018) 49(6):1148–61.e7. doi: 10.1016/j.immuni.2018.09.024
108. Frasca L, Nasso M, Spensieri F, Fedele G, Palazzo R, Malavasi F, et al. IFN-Gamma Arms Human Dendritic Cells to Perform Multiple Effector Functions. J Immunol (Baltimore Md: 1950) (2008) 180(3):1471–81. doi: 10.4049/jimmunol.180.3.1471
109. He T, Tang C, Xu S, Moyana T, Xiang J. Interferon Gamma Stimulates Cellular Maturation of Dendritic Cell Line DC2.4 Leading To Induction of Efficient Cytotoxic T Cell Responses and Antitumor Immunity. Cell Mol Immunol (2007) 4(2):105–11. doi: 10.1016/j.cellimm.2007.07.001
110. Lim TS, Chew V, Sieow JL, Goh S, Yeong JP-S, Soon AL, et al. PD-1 Expression on Dendritic Cells Suppresses CD8(+) T Cell Function and Antitumor Immunity. Oncoimmunology (2016) 5(3):e1085146. doi: 10.1080/2162402X.2015.1085146
111. Krempski J, Karyampudi L, Behrens MD, Erskine CL, Hartmann L, Dong H, et al. Tumor-Infiltrating Programmed Death Receptor-1+ Dendritic Cells Mediate Immune Suppression in Ovarian Cancer. J Immunol (Baltimore Md: 1950) (2011) 186(12):6905–13. doi: 10.4049/jimmunol.1100274
112. Karyampudi L, Lamichhane P, Krempski J, Kalli KR, Behrens MD, Vargas DM, et al. PD-1 Blunts the Function of Ovarian Tumor-Infiltrating Dendritic Cells by Inactivating NF-κb. Cancer Res (2016) 76(2):239–50. doi: 10.1158/0008-5472.CAN-15-0748
113. Chen L, Azuma T, Yu W, Zheng X, Luo L, Chen L. B7-H1 Maintains the Polyclonal T Cell Response by Protecting Dendritic Cells From Cytotoxic T Lymphocyte Destruction. Proc Natl Acad Sci USA (2018) 115(12):3126–31. doi: 10.1073/pnas.1722043115
114. Peng Q, Qiu X, Zhang Z, Zhang S, Zhang Y, Liang Y, et al. PD-L1 on Dendritic Cells Attenuates T Cell Activation and Regulates Response to Immune Checkpoint Blockade. Nat Commun (2020) 11(1):4835. doi: 10.1038/s41467-020-18570-x
115. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-Associated Macrophages as Treatment Targets in Oncology. Nat Rev Clin Oncol (2017) 14(7):399–416. doi: 10.1038/nrclinonc.2016.217
116. Waqas SFH, Ampem G, Röszer T. Analysis of IL-4/STAT6 Signaling in Macrophages. Methods Mol Biol (Clifton NJ) (2019) 1966:211–24. doi: 10.1007/978-1-4939-9195-2_17
117. Tan B, Shi X, Zhang J, Qin J, Zhang N, Ren H, et al. Inhibition of Rspo-Lgr4 Facilitates Checkpoint Blockade Therapy by Switching Macrophage Polarization. Cancer Res (2018) 78(17):4929–42. doi: 10.1158/0008-5472.CAN-18-0152
118. Bloch O, Crane CA, Kaur R, Safaee M, Rutkowski MJ, Parsa AT. Gliomas Promote Immunosuppression Through Induction of B7-H1 Expression in Tumor-Associated Macrophages. Clin Cancer Res (2013) 19(12):3165–75. doi: 10.1158/1078-0432.CCR-12-3314
119. Gubin MM, Esaulova E, Ward JP, Malkova ON, Runci D, Wong P, et al. High-Dimensional Analysis Delineates Myeloid and Lymphoid Compartment Remodeling During Successful Immune-Checkpoint Cancer Therapy. Cell (2018) 175(4):1014–30.e19. doi: 10.1016/j.cell.2018.09.030
120. Yao A, Liu F, Chen K, Tang L, Liu L, Zhang K, et al. Programmed Death 1 Deficiency Induces the Polarization of Macrophages/Microglia to The M1 Phenotype After Spinal Cord Injury in Mice. Neurotherapeutics: J Am Soc Exp NeuroTherapeutics (2014) 11(3):636–50. doi: 10.1007/s13311-013-0254-x
121. Strauss L, Mahmoud MAA, Weaver JD, Tijaro-Ovalle NM, Christofides A, Wang Q, et al. Targeted Deletion of PD-1 in Myeloid Cells Induces Antitumor Immunity. Sci Immunol (2020) 5(43):eaay1863. doi: 10.1126/sciimmunol.aay1863
122. Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 Expression by Tumour-Associated Macrophages Inhibits Phagocytosis and Tumour Immunity. Nature (2017) 545(7655):495–9. doi: 10.1038/nature22396
123. Rashidian M, LaFleur MW, Verschoor VL, Dongre A, Zhang Y, Nguyen TH, et al. Immuno-PET Identifies the Myeloid Compartment as a Key Contributor to the Outcome of The Antitumor Response Under PD-1 Blockade. Proc Natl Acad Sci USA (2019) 116(34):16971–80. doi: 10.1073/pnas.1905005116
124. Dhupkar P, Gordon N, Stewart J, Kleinerman ES. Anti-PD-1 Therapy Redirects Macrophages From an M2 to an M1 Phenotype Inducing Regression of OS Lung Metastases. Cancer Med (2018) 7(6):2654–64. doi: 10.1002/cam4.1518
125. Maj T, Wang W, Crespo J, Zhang H, Wang W, Wei S, et al. Oxidative Stress Controls Regulatory T Cell Apoptosis and Suppressor Activity and PD-L1-Blockade Resistance in Tumor. Nat Immunol (2017) 18(12):1332–41. doi: 10.1038/ni.3868
126. Kato T, Noma K, Ohara T, Kashima H, Katsura Y, Sato H, et al. Cancer-Associated Fibroblasts Affect Intratumoral CD8(+) and FoxP3(+) T Cells Via IL6 in the Tumor Microenvironment. Clin Cancer Research: An Off J Am Assoc Cancer Res (2018) 24(19):4820–33. doi: 10.1158/1078-0432.CCR-18-0205
127. Syn NL, Teng MWL, Mok TSK, Soo RA. De-Novo and Acquired Resistance to Immune Checkpoint Targeting. Lancet Oncol (2017) 18(12):e731–41. doi: 10.1016/S1470-2045(17)30607-1
128. Huang Q, Lei Y, Li X, Guo F, Liu M. A Highlight of the Mechanisms of Immune Checkpoint Blocker Resistance. Front Cell Dev Biol (2020) 8:580140. doi: 10.3389/fcell.2020.580140
129. Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, et al. Cancer Exome Analysis Reveals a T-Cell-Dependent Mechanism of Cancer Immunoediting. Nature (2012) 482(7385):400–4. doi: 10.1038/nature10755
130. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer Immunology. Mutational Landscape Determines Sensitivity to PD-1 Blockade in Non-Small Cell Lung Cancer. Sci (New York NY) (2015) 348(6230):124–8. doi: 10.1126/science.aaa1348
131. Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, et al. Evolution of Neoantigen Landscape During Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Discov (2017) 7(3):264–76. doi: 10.1158/2159-8290.CD-16-0828
132. Trommer M, Yeo SY, Persigehl T, Bunck A, Grüll H, Schlaak M, et al. Abscopal Effects in Radio-Immunotherapy-Response Analysis of Metastatic Cancer Patients With Progressive Disease Under Anti-PD-1 Immune Checkpoint Inhibition. Front Pharmacol (2019) 10:511. doi: 10.3389/fphar.2019.00511
133. Formenti SC, Demaria S. Combining Radiotherapy and Cancer Immunotherapy: A Paradigm Shift. J Natl Cancer Institute (2013) 105(4):256–65. doi: 10.1093/jnci/djs629
134. Verbrugge I, Hagekyriakou J, Sharp LL, Galli M, West A, McLaughlin NM, et al. Radiotherapy Increases the Permissiveness of Established Mammary Tumors to Rejection by Immunomodulatory Antibodies. Cancer Res (2012) 72(13):3163–74. doi: 10.1158/0008-5472.CAN-12-0210
135. Gadgeel SM, Garassino MC, Esteban E, Speranza G, Felip E, Hochmair MJ, et al. KEYNOTE-189: Updated OS and Progression After the Next Line of Therapy (PFS2) With Pembrolizumab (Pembro) Plus Chemo With Pemetrexed and Platinum vs Placebo Plus Chemo for Metastatic Nonsquamous NSCLC. J Clin Oncol (2019) 37(15_suppl):9013. doi: 10.1200/JCO.2019.37.15_suppl.9013
136. Paz-Ares L, Luft A, Vicente D, Tafreshi A, Gümüş M, Mazières J, et al. Pembrolizumab Plus Chemotherapy for Squamous Non–Small-Cell Lung Cancer. N Engl J Med (2018) 379(21):2040–51. doi: 10.1056/nejmoa1810865
137. Cortes J, Cescon DW, Rugo HS, Nowecki Z, Im SA, Yusof MM, et al. Pembrolizumab Plus Chemotherapy Versus Placebo Plus Chemotherapy for Previously Untreated Locally Recurrent Inoperable or Metastatic Triple-Negative Breast Cancer (KEYNOTE-355): A Randomised, Placebo-Controlled, Double-Blind, Phase 3 Clinical Trial. Lancet (2020) 396(10265):1817–28. doi: 10.1016/S0140-6736(20)32531-9
138. Sahin U, Derhovanessian E, Miller M, Kloke B-P, Simon P, Löwer M, et al. Personalized RNA Mutanome Vaccines Mobilize Poly-Specific Therapeutic Immunity Against Cancer. Nature (2017) 547(7662):222–6. doi: 10.1038/nature23003
139. Schumacher TN, Schreiber RD. Neoantigens in Cancer Immunotherapy. Science (2015) 348(6230):69–74. doi: 10.1126/science.aaa4971
140. Gettinger S, Choi J, Hastings K, Truini A, Datar I, Sowell R, et al. Impaired HLA Class I Antigen Processing and Presentation as a Mechanism of Acquired Resistance to Immune Checkpoint Inhibitors in Lung Cancer. Cancer Discov (2017) 7(12):1420–35. doi: 10.1158/2159-8290.CD-17-0593
141. McGranahan N, Rosenthal R, Hiley CT, Rowan AJ, Watkins TBK, Wilson GA, et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell (2017) 171(6):1259–71.e11. doi: 10.1016/j.cell.2017.10.001
142. Pereira C, Gimenez-Xavier P, Pros E, Pajares MJ, Moro M, Gomez A, et al. Genomic Profiling of Patient-Derived Xenografts for Lung Cancer Identifies B2M Inactivation Impairing Immunorecognition. Clin Cancer research: an Off J Am Assoc Cancer Res (2017) 23(12):3203–13. doi: 10.1158/1078-0432.CCR-16-1946
143. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations Associated With Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med (2016) 375(9):819–29. doi: 10.1056/nejmoa1604958
144. Dubovsky JA, McNeel DG, Powers JJ, Gordon J, Sotomayor EM, Pinilla-Ibarz JA. Treatment of Chronic Lymphocytic Leukemia With a Hypomethylating Agent Induces Expression of NXF2, an Immunogenic Cancer Testis Antigen. Clin Cancer Res: An Off J Am Assoc Cancer Res (2009) 15(10):3406–15. doi: 10.1158/1078-0432.CCR-08-2099
145. Fonsatti E, Nicolay HJM, Sigalotti L, Calabrò L, Pezzani L, Colizzi F, et al. Functional Up-Regulation of Human Leukocyte Antigen Class I Antigens Expression by 5-Aza-2’-Deoxycytidine in Cutaneous Melanoma: Immunotherapeutic Implications. Clin Cancer Res: An Off J Am Assoc Cancer Res (2007) 13(11):3333–8. doi: 10.1158/1078-0432.CCR-06-3091
146. Zhou L, Xu N, Shibata H, Saloura V, Uppaluri R. Epigenetic Modulation of Immunotherapy and Implications in Head and Neck Cancer. Cancer Metastasis Rev (2021) 40(1):141–52. doi: 10.1007/s10555-020-09944-0
147. Zhou L, Mudianto T, Ma X, Riley R, Uppaluri R. Targeting EZH2 Enhances Antigen Presentation, Antitumor Immunity, and Circumvents Anti-PD-1 Resistance in Head and Neck Cancer. Clin Cancer research: an Off J Am Assoc Cancer Res (2020) 26(1):290–300. doi: 10.1158/1078-0432.CCR-19-1351
148. Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, et al. Epigenetic Silencing of TH1-Type Chemokines Shapes Tumour Immunity and Immunotherapy. Nature (2015) 527(7577):249–53. doi: 10.1038/nature15520
149. Kobayashi Y, Lim SO, Yamaguchi H. Oncogenic Signaling Pathways Associated With Immune Evasion and Resistance to Immune Checkpoint Inhibitors in Cancer. Semin Cancer Biol (2020) 65:51–64. doi: 10.1016/j.semcancer.2019.11.011
150. Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov (2016) 6(2):202–16. doi: 10.1158/2159-8290.CD-15-0283
151. Zhao J, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T, et al. Immune and Genomic Correlates of Response to Anti-PD-1 Immunotherapy in Glioblastoma. Nat Med (2019) 25(3):462–9. doi: 10.1038/s41591-019-0349-y
152. Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK Mitogen-Activated Protein Kinase Cascade for the Treatment Of Cancer. Oncogene (2007) 26(22):3291–310. doi: 10.1038/sj.onc.1210422
153. Katz M, Amit I, Yarden Y. Regulation of MAPKs by Growth Factors and Receptor Tyrosine Kinases. Biochim Biophys Acta (2007) 1773(8):1161–76. doi: 10.1016/j.bbamcr.2007.01.002
154. Dhillon AS, Hagan S, Rath O, Kolch W. MAP Kinase Signalling Pathways in Cancer. Oncogene (2007) 26(22):3279–90. doi: 10.1038/sj.onc.1210421
155. Simanshu DK, Nissley DV, McCormick F. RAS Proteins and Their Regulators in Human Disease. Cell (2017) 170(1):17–33. doi: 10.1016/j.cell.2017.06.009
156. Liao W, Overman MJ, Boutin AT, Shang X, Zhao D, Dey P, et al. KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell (2019) 35(4):559–72.e7. doi: 10.1016/j.ccell.2019.02.008
157. Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, et al. Survival in BRAF V600–Mutant Advanced Melanoma Treated With Vemurafenib. N Engl J Med (2012) 366(8):707–14. doi: 10.1056/nejmoa1112302
158. Singh M, Lin J, Hocker TL, Tsao H. Genetics of Melanoma Tumorigenesis. Br J Dermatol (2008) 158(1):15–21. doi: 10.1111/j.1365-2133.2007.08316.x
159. Boni A, Cogdill AP, Dang P, Udayakumar D, Njauw CNJ, Sloss CM, et al. Selective BRAFV600E Inhibition Enhances T-Cell Recognition of Melanoma Without Affecting Lymphocyte Function. Cancer Res (2010) 70(13):5213–9. doi: 10.1158/0008-5472.CAN-10-0118
160. Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, et al. BRAF Inhibition Is Associated With Enhanced Melanoma Antigen Expression and a More Favorable Tumor Microenvironment in Patients With Metastatic Melanoma. Clin Cancer Res (2013) 19(5):1225–31. doi: 10.1158/1078-0432.CCR-12-1630
161. Hirata E, Girotti MR, Viros A, Hooper S, Spencer-Dene B, Matsuda M, et al. Intravital Imaging Reveals How BRAF Inhibition Generates Drug-Tolerant Microenvironments With High Integrin β1/FAK Signaling. Cancer Cell (2015) 27(4):574–88. doi: 10.1016/j.ccell.2015.03.008
162. Ribas A, Lawrence D, Atkinson V, Agarwal S, Miller WHJ, Carlino MS, et al. Combined BRAF and MEK Inhibition With PD-1 Blockade Immunotherapy in BRAF-Mutant Melanoma. Nat Med (2019) 25(6):936–40. doi: 10.1038/s41591-019-0476-5
163. Kammertoens T, Friese C, Arina A, Idel C, Briesemeister D, Rothe M, et al. Tumour Ischaemia by Interferon-γ Resembles Physiological Blood Vessel Regression. Nature (2017) 545(7652):98–102. doi: 10.1038/nature22311
164. Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov (2017) 7(2):188–201. doi: 10.1158/2159-8290.CD-16-1223
165. Fish EN, Platanias LC. Interferon Receptor Signaling in Malignancy: A Network of Cellular Pathways Defining Biological Outcomes. Mol Cancer Res (2014) 12(12):1691–703. doi: 10.1158/1541-7786.MCR-14-0450
166. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, et al. PD-1 Blockade Induces Responses by Inhibiting Adaptive Immune Resistance. Nature (2014) 515(7528):568–71. doi: 10.1038/nature13954
167. Parker BS, Rautela J, Hertzog PJ. Antitumour Actions of Interferons: Implications for Cancer Therapy. Nat Rev Cancer (2016) 16(3):131–44. doi: 10.1038/nrc.2016.14
168. Sheng W, LaFleur MW, Nguyen TH, Chen S, Chakravarthy A, Conway JR, et al. LSD1 Ablation Stimulates Anti-Tumor Immunity and Enables Checkpoint Blockade. Cell (2018) 174(3):549–63.e19. doi: 10.1016/j.cell.2018.05.052
169. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell (2017) 168(4):707–23. doi: 10.1016/j.cell.2017.01.017
170. Jacquelot N, Yamazaki T, Roberti MP, Duong CPM, Andrews MC, Verlingue L, et al. Sustained Type I Interferon Signaling as a Mechanism of Resistance to PD-1 Blockade. Cell Res (2019) 29(10):846–61. doi: 10.1038/s41422-019-0224-x
171. Ghoneim HE, Fan Y, Moustaki A, Abdelsamed HA, Dash P, Dogra P, et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell (2017) 170(1):142–57.e19. doi: 10.1016/j.cell.2017.06.007
172. Sheng W, Liu Y, Chakraborty D, Debo B, Shi Y. Simultaneous Inhibition of LSD1 and TGF-B Enables Eradication of Poorly Immunogenic Tumors With Anti-PD-1 Treatment. Cancer Discov (2021) 11(8):1970–81. doi: 10.1158/2159-8290.CD-20-0017
173. Hellmann MD, Jänne PA, Opyrchal M, Hafez N, Raez LE, Gabrilovich DI, et al. Entinostat Plus Pembrolizumab in Patients With Metastatic NSCLC Previously Treated With Anti-PD-(L)1 Therapy. Clin Cancer Res: An Off J Am Assoc Cancer Res (2021) 27(4):1019–28. doi: 10.1158/1078-0432.CCR-20-3305
174. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated Regulation of Myeloid Cells by Tumours. Nat Rev Immunol (2012) 12(4):253–68. doi: 10.1038/nri3175
175. Weber J, Gibney G, Kudchadkar R, Yu B, Cheng P, Martinez AJ, et al. Phase I/II Study of Metastatic Melanoma Patients Treated With Nivolumab Who Had Progressed After Ipilimumab. Cancer Immunol Res (2016) 4(4):345–53. doi: 10.1158/2326-6066.CIR-15-0193
176. Theivanthiran B, Evans KS, DeVito NC, Plebanek M, Sturdivant M, Wachsmuth LP, et al. A Tumor-Intrinsic PD-L1/NLRP3 Inflammasome Signaling Pathway Drives Resistance to Anti-PD-1 Immunotherapy. J Clin Invest (2020) 130(5):2570–86. doi: 10.1172/JCI133055
177. van Deventer HW, Burgents JE, Wu QP, Woodford RMT, Brickey WJ, Allen IC, et al. The Inflammasome Component Nlrp3 Impairs Antitumor Vaccine by Enhancing the Accumulation of Tumor-Associated Myeloid-Derived Suppressor Cells. Cancer Res (2010) 70(24):10161–9. doi: 10.1158/0008-5472.CAN-10-1921
178. Neubert NJ, Schmittnaegel M, Bordry N, Nassiri S, Wald N, Martignier C, et al. T Cell-Induced CSF1 Promotes Melanoma Resistance to PD1 Blockade. Sci Trans Med (2018) 10(436):eaan3311. doi: 10.1126/scitranslmed.aan3311
179. Lu X, Horner JW, Paul E, Shang X, Troncoso P, Deng P, et al. Effective Combinatorial Immunotherapy for Castration-Resistant Prostate Cancer. Nature (2017) 543(7647):728–32. doi: 10.1038/nature21676
180. Yu GT, Bu LL, Huang CF, Zhang W-F, Chen W-J, Gutkind JS, et al. PD-1 Blockade Attenuates Immunosuppressive Myeloid Cells Due to Inhibition of CD47/Sirpα Axis in HPV Negative Head and Neck Squamous Cell Carcinoma. Oncotarget (2015) 6(39):42067–80. doi: 10.18632/oncotarget.5955
181. Molgora M, Esaulova E, Vermi W, Hou J, Chen Y, Luo J, et al. TREM2 Modulation Remodels the Tumor Myeloid Landscape Enhancing Anti-PD-1 Immunotherapy. Cell (2020) 182(4):886–900.e17. doi: 10.1016/j.cell.2020.07.013
182. Hughes R, Qian BZ, Rowan C, Muthana M, Keklikoglou I, Olson OC, et al. Perivascular M2 Macrophages Stimulate Tumor Relapse After Chemotherapy. Cancer Res (2015) 75(17):3479–91. doi: 10.1158/0008-5472.CAN-14-3587
183. Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW, et al. CD24 Signalling Through Macrophage Siglec-10 Is a Target for Cancer Immunotherapy. Nature (2019) 572(7769):392–6. doi: 10.1038/s41586-019-1456-0
184. Ziani L, Chouaib S, Thiery J. Alteration of the Antitumor Immune Response by Cancer-Associated Fibroblasts. Front Immunol (2018) 9:414. doi: 10.3389/fimmu.2018.00414
185. Ford K, Hanley CJ, Mellone M, Szyndralewiez C, Heitz F, Wiesel P, et al. NOX4 Inhibition Potentiates Immunotherapy by Overcoming Cancer-Associated Fibroblast-Mediated CD8 T-Cell Exclusion From Tumors. Cancer Res (2020) 80(9):1846–60. doi: 10.1158/0008-5472.CAN-19-3158
186. Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. Tgfβ Drives Immune Evasion in Genetically Reconstituted Colon Cancer Metastasis. Nature (2018) 554(7693):538–43. doi: 10.1038/nature25492
187. Li MO, Flavell RA. TGF-β: A Master of All T Cell Trades. Cell (2008) 134(3):392–404. doi: 10.1016/j.cell.2008.07.025
188. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T Cells Expressing CD19 Chimeric Antigen Receptors for Acute Lymphoblastic Leukaemia In Children and Young Adults: A Phase 1 Dose-Escalation Trial. Lancet (London England) (2015) 385(9967):517–28. doi: 10.1016/S0140-6736(14)61403-3
189. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T Cells of Defined CD4+:CD8+ Composition in Adult B Cell ALL Patients. J Clin Invest (2016) 126(6):2123–38. doi: 10.1172/JCI85309
190. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N Engl J Med (2014) 371(16):1507–17. doi: 10.1056/NEJMoa1407222
191. Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and Toxicity Management of 19-28z CAR T Cell Therapy in B Cell Acute Lymphoblastic Leukemia. Sci Trans Med (2014) 6(224):224ra25. doi: 10.1126/scitranslmed.3008226
192. Foster AE, Dotti G, Lu A, Khalil M, Brenner MK, Heslop HE, et al. Antitumor Activity of EBV-Specific T Lymphocytes Transduced With a Dominant Negative TGF-β Receptor. J Immunother (2008) 31(5):500–5. doi: 10.1097/CJI.0b013e318177092b
193. John LB, Devaud C, Duong CPM, Yong CS, Beavis PA, Haynes NM, et al. Anti-PD-1 Antibody Therapy Potently Enhances the Eradication of Established Tumors by Gene-Modified T Cells. Clin Cancer Res: An Off J Am Assoc Cancer Res (2013) 19(20):5636–46. doi: 10.1158/1078-0432.CCR-13-0458
194. Chong EA, Melenhorst JJ, Lacey SF, Ambrose DE, Gonzalez V, Levine BL, et al. PD-1 Blockade Modulates Chimeric Antigen Receptor (CAR)-Modified T Cells: Refueling The CAR. Blood (2017) 129(8):1039–41. doi: 10.1182/blood-2016-09-738245
195. Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS, et al. Tumor Endothelium FasL Establishes a Selective Immune Barrier Promoting Tolerance in Tumors. Nat Med (2014) 20(6):607–15. doi: 10.1038/nm.3541
196. Gabrilovich D, Ishida T, Oyama T, Ran S, Kravtsov V, Nadaf S, et al. Vascular Endothelial Growth Factor Inhibits the Development of Dendritic Cells and Dramatically Affects the Differentiation of Multiple Hematopoietic Lineages. vivo Blood (1998) 92(11):4150–66. doi: 10.1016/S0950-3536(98)80044-9
197. Newport EL, Pedrosa AR, Njegic A, Hodivala-Dilke KM, Muñoz-Félix JM. Improved Immunotherapy Efficacy by Vascular Modulation. Cancers (2021) 13(20):5207. doi: 10.3390/cancers13205207
198. Fukumura D, Kloepper J, Amoozgar Z, Duda DG, Jain RK. Enhancing Cancer Immunotherapy Using Antiangiogenics: Opportunities and Challenges. Nat Rev Clin Oncol (2018) 15(5):325–40. doi: 10.1038/nrclinonc.2018.29
199. Ren S, Xiong X, You H, Shen J, Zhou P. The Combination of Immune Checkpoint Blockade and Angiogenesis Inhibitors in the Treatment of Advanced Non-Small Cell Lung Cancer. Front Immunol (2021) 12:689132. doi: 10.3389/fimmu.2021.689132
200. Delgoffe GM. Filling the Tank: Keeping Antitumor T Cells Metabolically Fit for the Long Haul. Cancer Immunol Res (2016) 4(12):1001–6. doi: 10.1158/2326-6066.CIR-16-0244
201. Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab (2016) 24(5):657–71. doi: 10.1016/j.cmet.2016.08.011
202. Young A, Mittal D, Stagg J, Smyth MJ. Targeting Cancer-Derived Adenosine: New Therapeutic Approaches. Cancer Discov (2014) 4(8):879–88. doi: 10.1158/2159-8290.CD-14-0341
203. Li T, Mao C, Wang X, Shi Y, Tao Y. Epigenetic Crosstalk Between Hypoxia and Tumor Driven by HIF Regulation. J Exp Clin Cancer Res: CR (2020) 39(1):224. doi: 10.1186/s13046-020-01733-5
204. Gide TN, Quek C, Menzies AM, Tasker AT, Shang P, Holst J, et al. Distinct Immune Cell Populations Define Response to Anti-PD-1 Monotherapy and Anti-PD-1/Anti-CTLA-4 Combined Therapy. Cancer Cell (2019) 35(2):238–55.e6. doi: 10.1016/j.ccell.2019.01.003
205. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory Effect of Tumor Cell-Derived Lactic Acid on Human T Cells. Blood (2007) 109(9):3812–9. doi: 10.1182/blood-2006-07-035972
206. Puccetti P, Grohmann U. IDO and Regulatory T Cells: A Role for Reverse Signalling and Non-Canonical NF-kappaB Activation. Nat Rev Immunol (2007) 7(10):817–23. doi: 10.1038/nri2163
207. Botticelli A, Cerbelli B, Lionetto L, Zizzari I, Salati M, Pisano A, et al. Can IDO Activity Predict Primary Resistance to Anti-PD-1 Treatment in NSCLC? J Trans Med (2018) 16(1):219. doi: 10.1186/s12967-018-1595-3
208. Long G v, Dummer R, Hamid O, Gajewski TF, Caglevic C, Dalle S, et al. Epacadostat Plus Pembrolizumab Versus Placebo Plus Pembrolizumab in Patients With Unresectable or Metastatic Melanoma (ECHO-301/KEYNOTE-252): A Phase 3, Randomised, Double-Blind Study. Lancet Oncol (2019) 20(8):1083–97. doi: 10.1016/S1470-2045(19)30274-8
209. Liu H, Kuang X, Zhang Y, Ye Y, Li J, Liang L, et al. ADORA1 Inhibition Promotes Tumor Immune Evasion by Regulating the ATF3-PD-L1 Axis. Cancer Cell (2020) 37(3):324–339.e8. doi: 10.1016/j.ccell.2020.02.006
210. Sun X, Wu Y, Gao W, Enjyoji K, Csizmadia E, Müller CE, et al. CD39/ENTPD1 Expression by CD4+Foxp3+ Regulatory T Cells Promotes Hepatic Metastatic Tumor Growth in Mice. Gastroenterology (2010) 139(3):1030–40. doi: 10.1053/j.gastro.2010.05.007
211. Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets T v, et al. Gut Microbiome Modulates Response to Anti-PD-1 Immunotherapy in Melanoma Patients. Science (2018) 359(6371):97–103. doi: 10.1126/science.aan4236
212. Gopalakrishnan V, Helmink BA, Spencer CN, Reuben A, Wargo JA. The Influence of the Gut Microbiome on Cancer, Immunity, and Cancer Immunotherapy. Cancer Cell (2018) 33(4):570–80. doi: 10.1016/j.ccell.2018.03.015
213. Si W, Liang H, Bugno J, Xu Q, Ding X, Yang K, et al. Lactobacillus Rhamnosus GG Induces cGAS / STING- Dependent Type I Interferon and Improves Response to Immune Checkpoint Blockade. Gut (2021) (2021):gutjnl-2020-323426. doi: 10.1136/gutjnl-2020-323426
214. Zhang Z, Tang H, Chen P, Xie H, Tao Y. Demystifying the Manipulation of Host Immunity, Metabolism, and Extraintestinal Tumors by the Gut Microbiome. Signal Transduction Targeted Ther (2019) 4:41. doi: 10.1038/s41392-019-0074-5
215. Derosa L, Routy B, Enot D, Baciarello G, Massard C, Loriot Y, et al. Impact of Antibiotics on Outcome in Patients With Metastatic Renal Cell Carcinoma Treated With Immune Checkpoint Inhibitors. J Clin Oncol (2017) 35:462. doi: 10.1200/JCO.2017.35.6_suppl.462
216. Gopalakrishnan V, Spencer CN, Reuben A, Karpinets T, Hutchinson D, Hoffman K, et al. Association of Diversity and Composition of the Gut Microbiome With Differential Responses to PD-1 Based Therapy in Patients With Metastatic Melanoma. J Clin Oncol (2017) 35:2. doi: 10.1200/JCO.2017.35.7_suppl.2
217. Routy B, le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, et al. Gut Microbiome Influences Efficacy of PD-1-Based Immunotherapy Against Epithelial Tumors. Science (2018) 359(6371):91–7. doi: 10.1126/science.aan3706
218. Davar D, Dzutsev AK, McCulloch JA, Rodrigues RR, Chauvin J-M, Morrison RM, et al. Fecal Microbiota Transplant Overcomes Resistance to Anti-PD-1 Therapy in Melanoma Patients. Sci (New York NY) (2021) 371(6529):595–602. doi: 10.1126/science.abf3363
219. Shayan G, Srivastava R, Li J, Schmitt N, Kane LP, Ferris RL. Adaptive Resistance to Anti-PD1 Therapy by Tim-3 Upregulation Is Mediated by the PI3K-Akt Pathway in Head and Neck Cancer. Oncoimmunology (2017) 6(1):e1261779. doi: 10.1080/2162402X.2016.1261779
220. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive Resistance to Therapeutic PD-1 Blockade Iis Associated With Upregulation of Alternative Immune Checkpoints. Nat Commun (2016) 7:10501. doi: 10.1038/ncomms10501
221. Limagne E, Richard C, Thibaudin M, Fumet JD, Truntzer C, Lagrange A, et al. Tim-3/Galectin-9 Pathway and mMDSC Control Primary and Secondary Resistances to PD-1 Blockade in Lung Cancer Patients. OncoImmunology (2019) 8(4):e1564505. doi: 10.1080/2162402X.2018.1564505
222. Lui Y, Davis SJ. LAG-3: A Very Singular Immune Checkpoint. Nat Immunol (2018) 19(12):1278–9. doi: 10.1038/s41590-018-0257-1
223. Johnson DB, Nixon MJ, Wang Y, Wang DY, Castellanos E, Estrada M v, et al. Tumor-Specific MHC-II Expression Drives a Unique Pattern of Resistance to Immunotherapy via LAG-3/FCRL6 Engagement. JCI Insight (2018) 3(24):e120360. doi: 10.1172/jci.insight.120360
Keywords: immunotherapy, PD-1 inhibitor, tumor microenvironment, cytotoxic T lymphocytes (CTLs), immunotherapy resistance, combined immunotherapy
Citation: Wang Q, Xie B, Liu S, Shi Y, Tao Y, Xiao D and Wang W (2021) What Happens to the Immune Microenvironment After PD-1 Inhibitor Therapy? Front. Immunol. 12:773168. doi: 10.3389/fimmu.2021.773168
Received: 09 September 2021; Accepted: 23 November 2021;
Published: 23 December 2021.
Edited by:
Nicolas Larmonier, Université de Bordeaux, FranceReviewed by:
Jaya Lakshmi Thangaraj, University of California, San Diego, United StatesMaite Alvarez, University of Navarra, Spain
Copyright © 2021 Wang, Xie, Liu, Shi, Tao, Xiao and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongguang Tao, taoyong@csu.edu.cn; Desheng Xiao, xdsh96@csu.edu.cn; Wenxiang Wang, wwx78@foxmail.com