 Zhijun Liu
Zhijun Liu Yifei Le
Yifei Le Hang Chen1
Hang Chen1 Ji Zhu
Ji Zhu Dezhao Lu
Dezhao Lu- 1School of Life Science, Zhejiang Chinese Medical University, Hangzhou, China
- 2The Third Affiliated Hospital of Zhejiang Chinese Medical University (Zhongshan Hospital of Zhejiang Province), Hangzhou, China
The cytokine storm is a marker of severity of various diseases and increased mortality. The altered metabolic profile and energy generation of immune cells affects their activation, exacerbating the cytokine storm. Currently, the emerging field of immunometabolism has highlighted the importance of specific metabolic pathways in immune regulation. The glycolytic enzyme pyruvate kinase M2 (PKM2) is a key regulator of immunometabolism and bridges metabolic and inflammatory dysfunction. This enzyme changes its conformation thus walks in different fields including metabolism and inflammation and associates with various transcription factors. This review summarizes the vital role of PKM2 in mediating immunometabolic reprogramming and its role in inducing cytokine storm, with a focus on providing references for further understanding of its pathological functions and for proposing new targets for the treatment of related diseases.
1 Introduction
The cytokine storm is defined as an overreaction of the immune system, which excessively activates macrophages and lymphocytes, and acutely secretes and releases high levels of proinflammatory cytokines resulting in a severe inflammatory response (1). The connections between metabolism and inflammatory response have been seen as the sally port that modulates the cytokine storm. Several novel studies have revealed the crucial role of cellular metabolism in controlling immune cell phenotype and functions. The term immunometabolism has been used to explain the intimate relationship between metabolic regulation and immune functionality. A switch of metabolic flux in immune cells from oxidative phosphorylation to aerobic glycolysis (Warburg effect) is a likely mechanism behind the sustained inflammation (2). The final rate limiting step of aerobic glycolysis is modulated by pyruvate kinase M2 (PKM2) enzyme, which been studied extensively for its important role in modulating metabolism (3). Studies report that PKM2 plays an important role in the development of some inflammatory diseases. The regulation of PKM2 can alleviate sepsis through numerous pathways (4, 5). A recent study documented that neutrophils in patients with COVID-19 exhibit immunometabolic reprogramming, with increased cytosolic PKM2, phosphorylated PKM2, hypoxia-inducible factor 1α (HIF-1α), and lactate (6). Previously, many studies demonstrated that PKM2-mediated immunometabolic reprogramming regulates the levels of cytokines through multiple pathways. The PKM2-mediated immunometabolic reprogramming promotes release of large amounts of proinflammatory cytokines, causing an excessive inflammatory response. Therefore, PKM2 is a potential therapeutic target for the treatment of cytokine storm-related inflammatory diseases. This study reviews the pathways implicated in PKM2-mediated immunometabolic reprogramming and their contribution to the cytokine storm. The findings of the study will provide a basis for the development of novel therapies for managing the cytokine storm and other inflammatory diseases.
2 PKM2: A Key Molecule In Modulating Inflammation in Metabolic Disorders
2.1 The PKM2 Metabolic Advantages Over Other Isoforms
Pyruvate kinase (PK) is a rate-limiting enzyme in glycolysis. It catalyses the transfer of a phosphate group from phosphoenolpyruvate (PEP) to adenosine diphosphate (ADP) generating pyruvate and adenosine triphosphate (ATP) (7). Pyruvate kinase exists in four isoforms including: PKL, PKR, PKM1, and PKM2. These isoforms are encoded by two PK genes, one of which encodes PKL (expressed in the liver) and PKR (expressed in erythrocytes), and the other gene encodes PKM1 and PKM2 isoforms. PKM1 and PKM2 isoforms are generated from the mutual exclusive alternative splicing of the PKM pre-mRNA, with PKM1 being formed through inclusion of exon 9, whereas PKM2 is formed through inclusion of exon 10 (8). PKM2 has low basal PK activity and is present at high levels in cancer cells, activated immune cells, and only few types of proliferating normal cells.
PKM2 occurs either as a tetramer or a dimer. The tetramer has a higher enzymatic activity and has a high level of affinity for PEP, thus can quickly catalyse PEP to form pyruvate. On the other hand, the PKM2 dimer has a lower catalytic activity and cannot produce pyruvate at a normal rate, resulting in accumulation of upstream glycolysis intermediates. These intermediates are then transferred to other pathways, such as the pentose phosphate pathway (PPP), to produce pyruvate to support biosynthesis in cells. Therefore, PKM2 is highly dimerized in cells or tissues with high nucleic acid synthesis. Studies report that PKM2 generally exists as a dimer in human monocytes and macrophages, whereas the major oligomeric status of PKM2 in murine macrophages is the monomer suggesting the existence of a species-specific regulatory mechanism (9, 10). Full activity of PKM2 requires allosteric activation by fructose 1, 6-bisphosphate (FBP) which is not the case for PKM1. The binding of FBP causes a the active site of PKM2 to combine with the ligand, therefore presenting a partially closed conformation (11). Thus, the binding of FBP remarkably increases the activity of PKM2 (12). Since the activity of PKM2 is much lower than PKM1 in the absence of FBP, proliferating cells still mainly express PKM2 but not PKM1. Thus, there exists a mechanism that can induce the transformation from PEP to pyruvate without PK. Studies have shown that PEP can be transformed into pyruvate through catalysis by phosphoglycerate mutase 1 (PGAM1), thus preventing ATP production, resulting in further inhibition of upstream steps of glycolysis (12). In addition, the PKM2 tetramer transforms into a dimer after processes, such as oxidation and acetylation, which inhibit the interaction between PKM2 and its activator FBP, thus reducing stability of the PKM2 tetramer (13). The low catalytic activity of PKM2 dimer blocks the glycolytic pathway at the last step, and the intermediates accumulate in cells. As a result, the composition of the intracellular environment becomes disordered, including accumulation of large amounts of acidic intermediates, leading to imbalance in acid-base equilibrium (14). The PKM2 dimer can enter the nucleus and bind to the transcription factors HIF-1α, Stat3 and Oct-4, thus promoting activation of the related signalling pathways and ultimately regulates biological processes including hypoxic injury and inflammation (10, 15).
Structural changes in the PKM2 tetramer deteriorate its enzymatic activity and thus mediate metabolic reprogramming, which is associated with various cellular stress responses. Upon activation, immune cells prefer glycolysis to oxidative phosphorylation to meet the urgent requirement for energy and micro-molecules. The role of PKM2 in disease pathology, such as tumour and inflammation has been extensively explored owing to the role it plays in glycometabolism and immunometabolism. Studies have shown that it plays essential roles in proliferation and transformation of tumour cells (16, 17).
2.2 Pharmacological Opportunities for PKM2 Modulation
This section describes activators and inhibitors under two groups including: those involved in the activation/inhibition of pyruvate kinase (PK) activity of PKM2, or those involved in isoenzyme-specific activation/inhibition of activity of PKM2. It is important to distinguish these two since they result in opposing effects (Table 1).
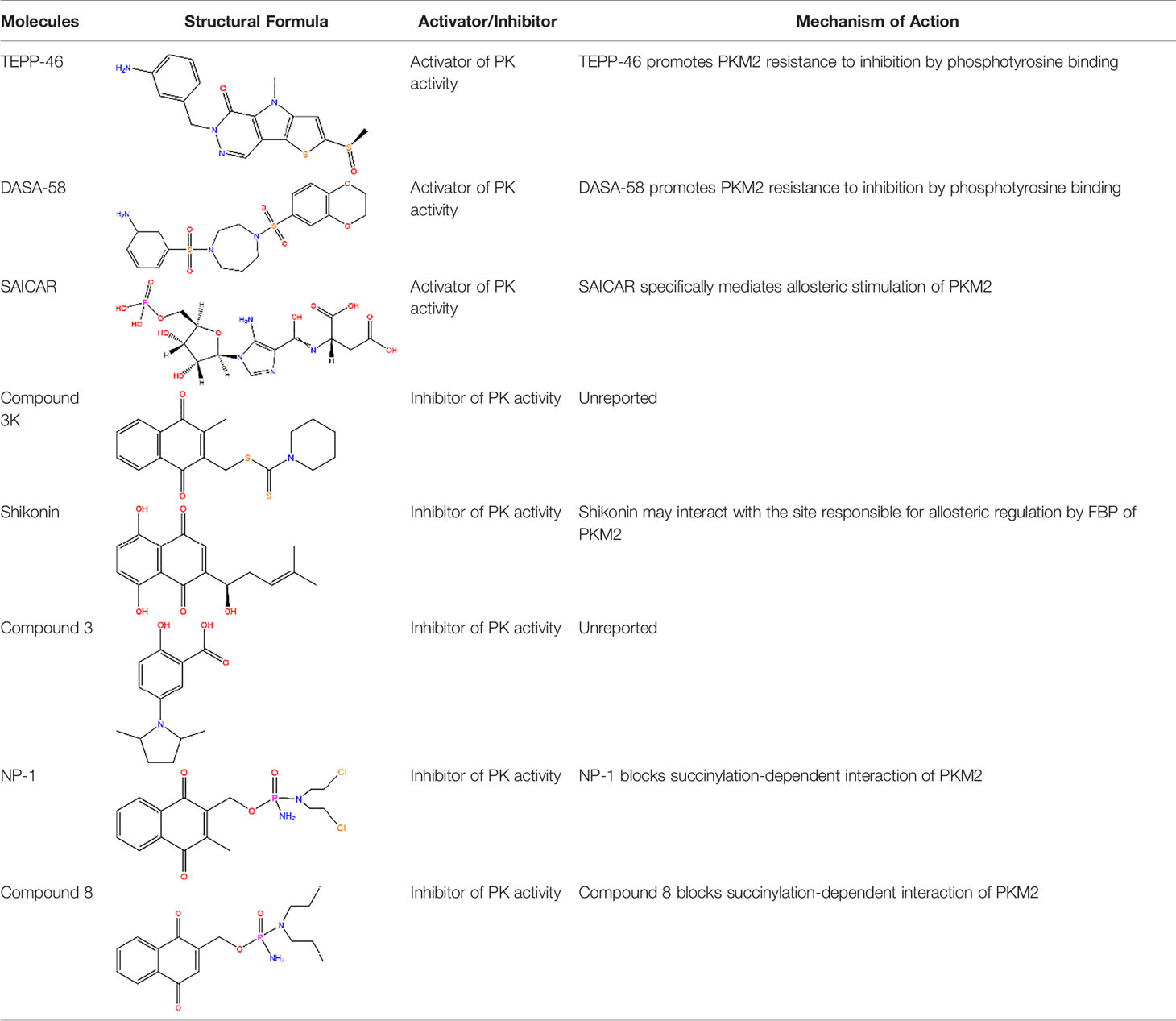
Table 1 Common activators and inhibitors of PKM2.
2.2.1 Activators of PKM2
Metabolic intermediates increase the PK activity of PKM2. PKM2 differs from PKM1 by a 56-amino-acid stretch encoded by the alternatively spliced region. This stretch of amino acids forms an allosteric pocket that allows binding of FBP (11). Therefore, FBP is one of the effector molecules that activates PKM2. Studies have reported that the PKM2 tetramer can adopt the inactive T-state or active R-state conformation. After the binding of FBP, the conformation of PKM2 tetramer is changed from the T-state to the R-state, which favours the binding of PEP in the active site and enhances its enzymatic activity (18). Notably, the binding of phosphotyrosine protein can catalyse the release of FBP from PKM2 allosteric site, hence inhibiting the PKM2 enzymatic activity (13). Studies have explored a small-molecule PKM2 activator that renders PKM2 resistant to inhibition through the binding of phosphotyrosine and hinders the release of FBP from PKM2, thus increasing the PK activity of PKM2 to levels that are comparable to those of PKM1 (19). TEPP-46 binds at the dimer-dimer interface between the two subunits of PKM2 to induce the PKM2 tetramer, which is a more active state of PKM2 (20). For instance, Thieno[3,2-b]pyrrole[3,2-d]pyridazinones (21), which is a member of the TEPP-46 class is commonly used in experiments (19), and a series of substituted N,N′-diarylsulfonamides have been reported as PKM2 activators[25]. In addition, DASA-58, a widely used PKM2 activator, specifically activates PKM2 through similar mechanisms as TEPP-46 (19). Moreover, succinylaminoimidazolecarboxamide ribose-5′-phosphate (SAICAR), an intermediate of the de novo purine nucleotide synthesis pathway, specifically mediates the allosteric stimulation of PKM2 (22). These molecules activate PKM2 and play a role in latent inhibition of proliferation of tumour and development of inflammation. However, activation of PKM2 in specific subtypes of cancer cells by this class of small molecule activators results in a metabolic adaption characterized by a strict dependency on the amino acid serine for continued cell proliferation (23). In addition, other regulators that promote the post-translational modification of PKM2 can also influence its activity. Among these kinds of modifications, acetylation of PKM2 is modulated by cellular sirtuins. Sirtuins are involved in sensing nutrient availability and directing metabolic activity to match energy needs with energy production and consumption. SIRT2 (sirtuin-2) and SIRT3 (sirtuin-3) are implicated in modulation of deacetylation of PKM2, thus increasing the level of PKM2 tetramer formation and suppressing the level of PKM2 dimer, which increase the level of PKM2 activity (24, 25). SIRT6 (sirtuin-6) mediates deacetylation by binding to the nuclear PKM2 resulting in the nuclear export of PKM2, which suppresses the oncogenic functions of PKM2 (26). Notably, the effect of acetylation on PKM2 is not limited to changes in its conformation. Studies have documented that acetylation of PKM2 reduces its enzymatic activity, mainly due to its decreased affinity toward the substrate PEP, and destabilizes PKM2 through HCS70-remediated chaperone-mediated autophagy (CMA) (14). Moreover, SIRT5 (sirtuin-5) desuccinylates and activates PKM2, which plays an anti-inflammation role (27). These sirtuins can also be seen as activators of PKM2.
There are other activators that isoenzyme-specific activates PKM2, leading to an inverse effect. For instance, serine, an activator of PKM2, stimulates the activity of PKM2 but not its PK activity. Studies have shown that serine can bind PKM2 and activate it. PKM2 activity in cells is reduced when they are deprived of serine (28). This reduction in PKM2 activity results in diversion of pyruvate to the mitochondria and more glucose derived carbon is directed to serine biosynthesis to support proliferation of cells (28). The death-associated protein kinase (DAPk) which is a multi-domain serine/threonine kinase functionally increases PK activity of PKM2, thus modulating cancer cell metabolism and inhibits cell proliferation (29).
2.2.2 Inhibitors of PKM2
Similarly, PKM2 has inhibitors which inhibit its activity. For example, the thyroid hormone 3,3,5-triiodo-L-thyronine (T3) and phenylalanine are the most effective enzyme inhibitors of PKM2 among more than 50 natural metabolites. T3 and phenylalanine stabilizes an inactive T-state tetrameric conformer, thus inhibiting the PK activity of PKM2 and increasing cell proliferation (30). L-cysteine amino acid induces dissociation of the PKM2 subunit, converting PKM2 from a tetramer to a dimer/monomer, thus suppressing PK activity of PKM2 (31). In addition, post-translational modifications such as phosphorylation, oxidation and acetylation can inhibit the PK activity of PKM2 (32–34).
Meanwhile, the isoenzyme-specific inhibitors that inhibit PKM2 but not its PK activity have not been reported. Isoenzyme-specific inhibitors can adjust glucose metabolism and reduce the harmful effects caused by PKM2. Compound 3K (35) and Shikonin (4, 36) are the most commonly used PKM2 inhibitors in experiments. Shikonin (SKN) is a naphthoquinone compound mainly isolated from Chinese herbal medicine Lithospermum erythrorhizon root and is a potent PKM2 inhibitor in cancer cells and macrophages (4, 36). Studies have reported that SKN inhibits cell proliferation and cell migration, and induces apoptosis partially through the PKM2 related pathways (37–40). However, the SKN can affect proliferation in a PKM2-independent manner, since its use may not be entirely related to PKM2 inhibition. The combination of SKN/paclitaxel overcome multidrug resistance in ovarian cancer through a PKM2-independent way of ROS generation (41). Also, whether SKN regulates other pathways through PKM2-independent ways remains unknown (37). In addition, three distinct structural classes of small molecules have been reported that exhibit selective inhibition of PKM2 over PKM1, and are the only current examples of isoenzyme-specific inhibitors (42). A small molecule referred as compound 8 can significantly reduce PKM2 activity by blocking the succinylation-dependent interaction of PKM2 and voltage-dependent anion channel proteins (VDAC)-3 thus inhibiting translocation of PKM2 into the mitochondria (43).
2.3 Unique Nuclear Functions of PKM2
PKM2 has unique nuclear functions which make it different from other metabolic enzymes. It contains a nuclear localization signal (NLS) encoded by exon 10. In cells activated with EGRF, PKM2 exon 10 also encodes an extracellular signal-regulated kinase (ERK) docking groove that can allow activated ERK1/2 mitogen-activated protein kinases to bind to PKM2 (44), leading to ERK1/2 phosphorylation of PKM2 at Ser37. Peptidyl-proline isomerase protein interacts with never in mitosis A (NIMA)-1 (PIN1), containing an NLS (45) and is recruited by phosphorylated PKM2 Ser37, binds to PKM2 and induces cis-trans isomerization of PKM2, which results in conversion of PKM2 from a tetramer to a dimer/monomer and then binds to importin α5. Importin α5, one of the six importin α family members (46), links NLS-containing proteins to importin β and then docks the ternary complex at the nuclear-pore complex (NPC), thus inducing translocation of PKM2 into the nucleus. In addition, a natural product micheliolide (MCL) selectively activates PKM2 through a covalent bond at residue cysteine 424 (C424) thus promoting tetramer formation, ultimately suppressing nuclear translocation of PKM2 (47). The transformation process of PKM2 is also regulated by post-translational modifications, such as acetylation and phosphorylation which shift PKM2 from tetrameric to dimeric form, leading to a higher level of nuclear transformation (48).
After localization in the nucleus, various mechanisms induce PKM2 to act as an active protein kinase. Epidermal growth factor receptor (EGFR) induces translocation of PKM2 into the nucleus through NF-κB-mediated PKM gene transcription (49, 50). Moreover, EGFR induces the transactivation of β-catenin through interaction with PKM2, which causes binding of PKM2 to Y333-phosphorylated β-catenin thus transactivating it and the protein complex subsequently binds to the cyclin D1 promoter (51). Studies have demonstrated that activation of EGFR promotes PKM2 directly by binding to histone H3 and induces phosphorylation of Thr11 on histone H3. This phosphorylation promotes dissociation of HDAC3 from cyclin D1 and MYC promoter regions, resulting in the subsequent acetylation of Lys9 on histone H3 (52). PKM2 acts as a histone kinase during PKM2-dependent histone H3 modifications and significantly upregulates the expression of c-Myc and cyclin D1. Notably, the oncogenic transcription factor c-Myc modulates different splicing of PKM1 and PKM2 by upregulating the transcription of heterogeneous nuclear ribonucleoprotein (hnRNP) proteins which bind to RNA sequences encoded by exon 9 and inhibit PKM1 mRNA splicing, leading to preferential PKM2 isoform expression (8).
HIF-1α plays an important role in metabolism reprogramming in inflammatory cells by promoting inflammatory gene expression (53). The interaction between HIF-1α and PKM2 results in a positive feedback loop. HIF-1α activates gene transcription of PKM2, whereas PKM2 interacts directly with HIF-1α subunit and promotes transactivation of HIF-1 target genes. Under hypoxic conditions, prolyl hydroxylation of HIF-1α leads to inhibition of HIF-1α proteasomal degradation, thus stabilizing and activating HIF-1α protein (54). HIF-1α dimerizes with HIF-1β in the nucleus and binds to hypoxia response element (HRE) of target genes. PKM2 contains HRE in the first intron, which allows binding of HIF-1 and directly activates its transcription (55). The PKM2 and HIF-1α complex can bind to the promoter of IL-1β (Interleukin-1β), which activates and regulates immune cells, thus playing an important role in inflammatory response (10). Moreover, hypoxia activates NF-κB, a critical component in the transcriptional pathway (56). PKM2 interacts with NF-κB subunit, p65 (57) and the interaction can trigger transcription of HIF-1α and its target genes, such as vascular endothelial growth factor (VEGF) (58). Hasan et al. reported cell-specific differences in hypoxic regulation of PKM2- HIF-1α axis. PKM2 expression is regulated by HIF-1α, however, HIF-1α expression is not regulated by PKM2 in LNCaP cells. On the contrary, PKM2 expression is not regulated by HIF-1α, but HIF-1α expression is regulated by PKM2 in PC3 cells (59). In addition, studies have reported molecules that regulate the PKM2- HIF-1α axis leading to cell metabolic reprogramming. For instance, Jumonji-C domain-containing protein 5 (JMJD5) can interact with PKM2, thus modulating nuclear translocation of PKM2 and promoting HIF-1α–mediated transactivation (60). Furthermore, the Six-Transmembrane Epithelial Antigen of the Prostate 4 (STEAP4) inhibits HIF1/PKM2 signalling (61). A combination therapy of simvastatin and sorafenib suppresses PKM2 expression and its nuclear translocation by inhibiting the interaction between PKM2 and HIF-1α/PPAR-γ (a factor that maintains energy homeostasis by modulation of glucose and lipid metabolism) (62).
Therefore, it is interesting to explore how the protein substrates bind to sites on PKM2. Studies demonstrated that the ADP binding site on PKM2 tetramer is a large hydrophobic cavity which is almost completely buried in the PKM2 tetramer structure but is accessible in the dimeric structure (11). Therefore, studies postulate that the ADP binding site can accommodate a protein substrate when PKM2 is converted to a dimer (63). Dimeric PKM2 localizes in the nucleus where it acts as a protein kinase and directly activates signal transducers and activators of transcription 3 (STAT3). However, studies have not fully elucidated whether STAT3 binds to the ADP binding site. STAT3 is a transcription activator that is activated in response to inflammatory cytokines (64). In addition, STAT3 is a unique target that can redirect inflammation to promote tumour or anti-tumour activities (65). PKM2 knockdown through intrathecal injection of siRNA has been shown to suppress chronic constriction injury (CCI)-induced STAT3 activation, thus attenuating neuropathic pain and inflammatory responses (66). Moreover, STAT3 participates in PKM2/HIF-1α feedback loop, since STAT3 activated by PKM2 can induce HIF-1α expression (67). After PKM2 transcription induced by HIF-1α, PKM2 activates STAT3 and trans-activates HIF-1α. The activated STAT3 then induces HIF-1a expression. Furthermore, cytokines, growth factors or oncogenes activate STAT3, leading to HIF-1α transcription. The STAT3/PKM2/HIF-1α feed forward loop then goes on. The function of STAT3 in promoting HIF-1α transcription has been reported. Serotonin (5-HT) phosphorylates Jak1, resulting in the phosphorylation of STAT3 which promotes ERK1/2 activation, Akt phosphorylation and HIF-1α expression (68). Although several studies report that PKM2 acts as a protein kinase, some studies oppose this theory. Hosios et al. reported that protein species labelled by [32P]-PEP dependent on presence of ADP but not PKM2, and PKM2-dependent transfer of phosphate from ATP directly to protein was not observed (69). This finding implies that further studies should be conducted to confirm the protein kinase function of PKM2.
Studies report that PKM2 is a critical regulator of expression and secretion of proinflammatory mediators thus controlling transcriptional activity of HIF-1α and STAT3 pathways during inflammation (70). Therefore, PKM2 is a potential target for controlling inflammatory response (Figure 1).
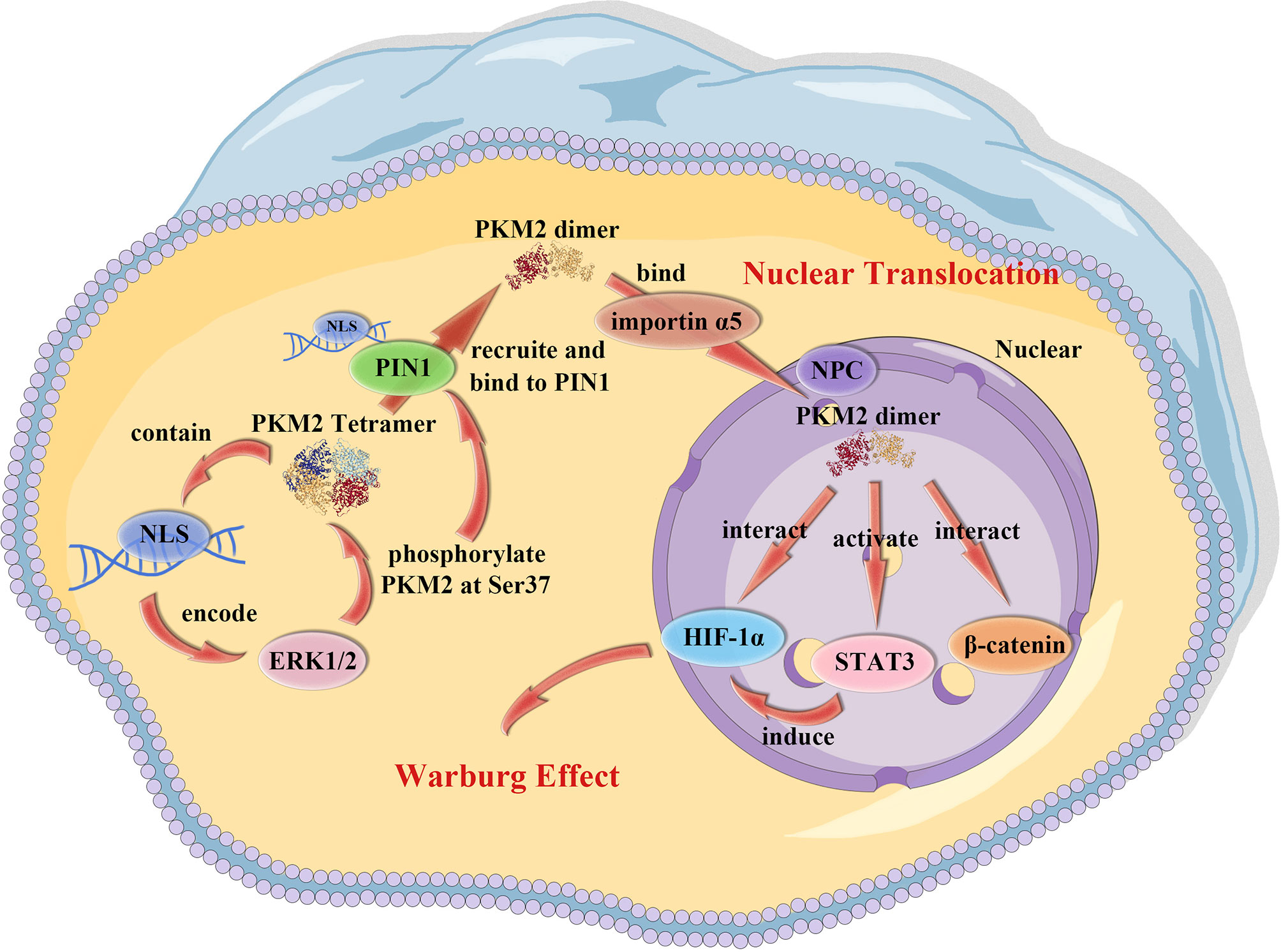
Figure 1 PKM2 nuclear translocation. PKM2 contains a NLS that allows ERK1/2 to bind to and phosphorylate PKM2 at Ser37. The PKM2 tetramer than recruits and binds to the NLS on PIN1, leading to the conversion of PKM2 tetramer into a dimer/monomer. The dimer/monomer PKM2 binds to importin α5 which links NLS-containing proteins to importin β and then docks the ternary complex at the NPC, inducing the translocation of PKM2 into the nucleus.
2.4 Warburg Effect: A Special Metabolic Reprogramming That Can Control Immune Cell Function
In the twentieth century, a German scientist known as Warburg reported a peculiar phenomenon of metabolic reprogramming, which is currently known as the Warburg effect (71). The Warburg effect, generally seen in tumour cells or activated immune cells (2), is characterized by cell metabolism shift from oxidative phosphorylation to aerobic glycolysis under aerobic conditions. The Warburg effect was initially observed in rapidly proliferating tumour cells, however, Dr Warburg reported the same phenomenon in activated leukocytes (72). Several studies on inflammatory response report that resting immune cells derive most of their ATP from oxidative phosphorylation, whereas activated immune cells derive most of their ATP from aerobic glycolysis. The activation of immune cells, such as T Lymphocytes occurs through a process that shifts from a relative quiescence state to rapid proliferative expansion, which is regulated by signals delivered through cytokine and antigen receptors (73–76). Therefore, controlling metabolic reprogramming can enhance or suppress specific immune cell functions.
Previous studies have shown that a phenomenon like Warburg effect reported in tumours occurs during inflammatory response of immune cells. In this case, glucose metabolism in an aerobic environment is highly dependent on glycolysis and not oxidative phosphorylation (this has also been reported in some cells undergoing an inflammatory response). Reprogramming of macrophage glucose metabolism under inflammation can lead to high dependence of cellular energy metabolism on glycolysis to meet the high demand of metabolites to fight infection in an inflammatory environment. LPS induces a metabolic shift towards aerobic glycolysis in macrophages, thus promoting transcriptional expression and release of early pro-inflammatory cytokines, such as IL-1β. This is manifested by accumulation of glycolytic intermediates, rapid low-level production of ATP and accumulation of lactic acid, as well as diversion of metabolites to the pentose phosphate pathway. Therefore, the citric acid cycle (TCA) is inhibited. In addition, the onset of aerobic glycolysis increases glucose uptake and promotes the glutamine-dependent back-supplementation effect to meet succinate requirement owing to inhibition of the TCA cycle. Succinate results in succinylation of malate dehydrogenase which blocks the breakdown of downstream products of succinate and ultimately promotes accumulation of succinate, an intermediate of the TCA cycle. Abnormal accumulation of succinate in the cytoplasm creates a state of cellular ‘pseudo-hypoxia’, thus stabilizing hypoxia-inducible factor-1α (HIF-1α) by inhibiting prolyl hydroxylase. HIF-1α then enters the nucleus and binds to various regulatory factors that inversely activate transcription of glycolytic enzyme-related genes and secretion of pro-inflammatory cytokines, thus maintaining high levels of cellular glycolysis and inflammation. PKM2 is an important molecule in mediating the Warburg effect (77). PKM2 catalyses conversion of PEP in the presence of ADP to form pyruvate and ATP, the final step of the glycolytic pathway reaction. The tetrameric form of PKM2 has a high affinity for its substrate phosphoenolpyruvate (PEP) and, therefore, PEP can be converted to pyruvate at a much faster rate. However, when the PKM2 protein undergoes acetylation, phosphorylation, succinylation and other post-translational modifications, it is converted from a tetramer to a dimer/monomer. The dimeric/monomeric form of pyruvate kinase has low activity; therefore, it does not produce pyruvate efficiently and rapidly, leading to accumulation of glycolytic intermediates. Therefore, metabolism shifts to the lactate pathway to meet the high demand for ATP, leading to disruption of the TCA cycle and activation of HIF-1α. PKM2 dimers/monomers translocate to the nucleus and activate inflammation-related target genes. In the nucleus, the monomeric/dimeric form of PKM2 interacts with HIF-1α and binds directly to the IL-1β promoter. Moreover, PKM2 and HIF-1α complex stimulates polarization of M1 macrophages. On the contrary, stabilization and regeneration of PKM2 tetramers prevents accumulation of glycolytic intermediates and succinate, thus HIF-1α is downregulated. In addition, PKM2 tetramers prevent PKM2 nuclear translocation, thus reducing activation of HIF-1α target genes.
Tyrosine phosphorylation of PKM2 inhibits the role of PKM2, thus promoting the Warburg effect and proliferation of tumour cells (32). It can inhibit the tetramerization of PKM2 by disrupting binding of the PKM2 cofactor FBP. Atg7 is an important molecule involved in autophagy, which inhibits the Warburg effect by preventing tyrosine phosphorylation of PKM2 (78). In addition, the nuclear translocation of PKM2 modulates the Warburg effect (44). Nuclear PKM2 functions as a histone kinase and upregulates c-Myc expression, promoting expression of glycolytic enzyme genes which induce the Warburg effect in a feedback loop (49, 52). Therefore, when PKM2 shifts from a dimer to a tetramer, it can suppress PK activity and nuclear translocation of PKM2, thus promoting the Warburg effect, which leads to the activation of immune cells. Naturally occurring flavonoids such as apigenin, proanthocyanidin B2 and shikonin are effective regulators of aerobic glycolysis that modulate the expression of PKM2 (79). Some flavonoids exhibit anti-inflammatory activities (80). Furthermore, PKM2 regulates cellular metabolism by reducing intracellular utilization of glucose, thus increasing the α-ketoglutarate/citrate ratio, and promote formation of the product of glutamine-derived acetylcoenzyme A through a reductive pathway (81). PKM2 effectively promotes reductive glutamine metabolism, which is important for lymphocyte proliferation and cytokine production (82), macrophage phagocytic and secretory activities, and neutrophil bacterial killing (83). These findings show that PKM2 modulates metabolic reprogramming of cells to activate immune cells and trigger inflammatory response, implying that PKM2 can be inhibited to control inflammation.
3 PKM2: A Key Regulator of Immune Cells Metabolism
3.1 PKM2 Triggers Macrophage Polarization
Macrophages are important components of the innate immunity, and they play a key role in inflammatory responses. Macrophages activated by lipopolysaccharides (LPS) shift their core metabolism from oxidative phosphorylation to glycolysis even under aerobic conditions similar to the Warburg effect (84). Moreover, macrophages possess high plasticity and can adapt their phenotype to the microenvironment (85). Various signals, such as Toll-like receptor (TLR) ligands and Interferon (IFN)-γ can stimulate macrophages to form a proinflammatory M1 phenotype, promoting the secretion of high levels of proinflammatory cytokines, such as tumour necrosis factor-α (TNF-α), IL-6, IL-1β, and reactive nitrogen and oxygen intermediates. On the contrary, IL-4 and IL-13 polarize macrophages towards an anti-inflammatory M2 phenotype which exhibits immunoregulatory functions. M2 macrophages exhibit phagocytic activity and release anti-inflammatory factors such as IL-10, Arg-1, and promote tissue remodelling (86–88). Studies have shown that macrophage M2 polarization is modulated by glucose metabolism and activated macrophage metabolism is highly dependent on glucose. In normal cellular metabolism, pyruvate produced by glycolysis mainly enters mitochondrial tricarboxylic acid (TCA) cycle and undergoes a series of oxidative reactions to produce high levels of ATP. However, in M1 macrophages, glucose metabolism relies on increased rate of glycolysis to produce energy, and pyruvate produced does not enter the TCA cycle to participate in oxidative phosphorylation, thus it is converted to lactate by the lactate dehydrogenase (LDH) enzyme (89).
As we have mentioned before, previous studies reported that PKM2 is an important modulator of IL-1β production, macrophage polarization, glycolytic reprogramming, and Warburg metabolism in LPS-activated macrophages (10). PKM2 is up-regulated and phosphorylated in an activated macrophage, therefore, PKM2 primarily forms an enzymatically inactive dimer or monomer in LPS activated macrophages, which is translocated into the nucleus and binds to HIF-1α forming a PKM2-HIF-1α complex (55). The complex binds to the IL-1β promoter, thus induces the secretion of proinflammatory cytokines and triggers an inflammatory response. Furthermore, studies have reported that the PKM2 tetramer promotes M1 macrophage polarization, whereas the PKM2 dimer promotes M2 polarization. Annexin A5 is an activator of PKM2 tetramer and can inhibit phosphorylation of PKM2, thus promoting shift of macrophage phenotype from M1 to M2 and alleviates inflammation (90). On the contrary, HSPA12A interacts with PKM2 and induces its nuclear translocation, thus promoting M1 polarization of macrophages and secretion of proinflammatory M1cytokines (91). Moreover, class IIa histone deacetylases (HDACs) interact with PKM2 to initiate TLR-inducible aerobic glycolysis and drive inflammatory responses in M1 macrophages, resulting in immunometabolism-associated inflammatory responses in macrophages (92).
3.2 PKM2 Modulates Inflammatory Activity and Differentiation of CD4+ T Cells
CD4+ T cells represent an immune cell population which play a key role in immune responses. CD4+ T cells induce B cells to make antibodies, induce enhanced microbicidal activity in macrophages, recruit neutrophils, eosinophils and basophils to sites of infection and produce cytokines and chemokines thus inducing immune responses (93). However, over-activation of CD4+T cell can lead to development of immune-related pathologies, such as inflammatory diseases. Therefore, several studies have explored the mechanisms of interfering with intracellular metabolic pathways in immune cells to treat inflammatory diseases (94, 95). Activation of innate and adaptive immune cells is characterized by the Warburg effect-dominated metabolic reprogramming (96). Furthermore, both the activation and differentiation of T cells are highly associated with metabolic reprogramming through ligation and co-stimulation of T cell receptor (TCR) (97). Previous studies have shown that CD4+ T cells preferentially express PKM2 over PKM1. Moreover, PKM2 is highly phosphorylated in activated T cells, a modification that results in high levels of dimerization and nuclear translocation of PKM2 (98). TEPP-46 is a commonly-used activator of PKM2 tetramer, which inhibits the nuclear translocation of PKM2 in CD4+ T cells (97). In addition, Myc, HIF-1a, and mTORC1 are transcription factors that regulate related metabolic functions. Myc-dependent global metabolic transcriptome drives metabolic reprogramming in activated T cells. It has been documented that the acute deletion of Myc markedly inhibits activation-induced glycolysis and glutaminolysis in T cells which compromise activation-induced T cell growth and proliferation (99). mTOR signalling have a profound effect on the fate of CD4+ T cells as they promote Th1 and Th17 cell differentiation and participate in effector-like Th cells differentiation (100, 101). The inflammatory CD4+ T cell subsets rely on mTOR signalling and glycolytic metabolism, which also require expression of glycolytic enzymes, such as glucose transporter 1 (Glut1) (97). However, HIF-1α, the downstream target of mTOR, may play a role in limiting the effective function in T cells. In an hypoxic environment, HIF-1α stabilization in inhibits Th1 function (102). Additionally, the HIF-1α–dependent glycolytic pathway orchestrates the differentiation of TH17 and Treg cells through a metabolic pathway (103). Studies have reported that TEPP-46 indirectly inhibits Myc, HIF-1a, and mTORC1 signalling, which are key factors in T cell activation and functionality (97). Therefore, PKM2 modulates the functions of HIF-1α, mTORC1, and Myc and engagement of aerobic glycolysis in TCR-activated CD4+ T cells, thus controlling activation and effector functions of CD4+ T cell (98). Moreover, the PKM2-dependent glycolytic-lipogenic axis, which is a novel mechanism of metabolic regulation, is important for hyperhomocysteinemia (HHcy)-induced CD4+ T cell activation (104). Homocysteine (Hcy)-activated CD4+ T cell increased the protein expression and activity of PKM2 through the phosphatidylinositol 3-kinase/AKT/mechanistic target of rapamycin signalling pathway. Shikonin, a PKM2 inhibitor, effectively suppresses glucose metabolism and reduces the accumulation of lipids in Hcy-activated CD4+ T cells through PKM2-dependent metabolic suppression, ultimately inhibiting CD4+T cell inflammatory activation (105).
Differentiation of CD4+ T cells modulated by cytokines elicited from pathogen-activated cells plays a vital role in adaptive immunity and innate immune response. Naive CD4+ T cells have significant plasticity which is modulated by signals from cells of the innate immune system (106). CD4+ T cells are characterized into several subsets including T helper 1 (Th1), Th2, Th17, and induced regulatory T (iTreg) cells (93). The Th cells play important roles in adaptive immune responses as they secret cytokines and chemokines that activate and further recruit target cells. Notably, Th17 cells is a special IL-17-producing subset and was reported recently. Th17 cells play key roles during host defence reactions and pathogenesis of inflammation in autoimmune disease. Th17 cells produce IL-17 which promotes local chemokine production thus recruiting monocytes and neutrophils to sites of infection and inflammation (107). In addition, Th17 is a proinflammatory T cell and its differentiation is inversely related with development of anti-inflammatory regulatory T (Treg) cell (108). PKM2 plays an indispensable role during Th17 differentiation. Although PKM2 is not a prerequisite for metabolic reprogramming and proliferative capacity of Th17 cells, it promotes Th17 cell differentiation thus it is a critical nonmetabolic regulator of Th17 cell differentiation (109). TCR activation of T cells significantly increases PKM2 expression to support their differentiation. The phosphorylated PKM2 at Y105 formed into dimer when translocated into nuclear of Th17 cells. It interacts with and phosphorylates STAT3 at Y705 resultant of Th17 differentiation (109). In addition, STAT3 combines with other transcription factors and retinoic acid orphan receptor gamma T synergize to regulate transcription of Th17 cell–signature genes, thus promoting Th17 differentiation (107). Moreover, PKM2-mediated metabolic reprogramming contributes to Th17 differentiation. A previous study reported that Th17-inducing cytokines promote increased levels of calcium/calmodulin-dependent protein kinase IV (CaMK4) in naive T cells, leading to Th17 cell differentiation (110). CaMK4 binds to PKM2 and promotes its PK activity, which is required for Th17 differentiation as Th17 cells depend on glycolysis as a source of energy (111). Interestingly, the fate of T cell differentiation is determined by glycolytic activity and expression of glycolytic enzymes. The inhibition of glycolytic pathway by treatment of glycolytic pathway inhibitor via blocking hexokinase in T cells promotes T cell differentiation to form Treg cells rather than Th17 cells (103). The high expression of HIF-1a that increases glycolytic activity was confirmed to mediate T cell differentiation. Reports indicated that PKM2 dimer can stabilize and activate HIF-1a (54), while its tetramer form counteracts excessive rate of glycolysis. Thus, modulation on PKM2-HIF-1α axis promote Th17 differentiation, whereas activation of PKM2 (PKM2 tetramer) implicated the increment of Treg development (112). Therefore, inhibiting PKM2 dimerization can block Th17 differentiation and induce naive T cells to differentiate into anti-inflammatory Tregs, thus abrogating excessive inflammation and its damage to multiple organs.
3.3 Other Immune Cell Responses Activated by PKM2
Natural killer (NK) cells are lymphocytes implicated in both innate and adaptive immunity. NK cells contribute to immune defence by killing unhealthy cells and regulating responses of antigen-presenting cells and adaptive T cells (113). NK cells function as an important source of cytokines and chemokines after recognizing aberrant cells, thus they are involved in the secretion of large amounts of cytokines, such as TNF-α, IFN-γ and IL family of proteins to amplify inflammatory responses (114). In addition, NK cells interact with dendritic cells, macrophages, T cells and endothelial cells, thus inhibiting or exacerbating immune responses (115). Glycolysis activation plays an important role in NK cell effector functions (116). Studies have reported that PKM2 expression is significantly induced in activated NK cells (117). Furthermore, PKM2 is the dominant pyruvate kinase isoform in NK cells and exists mainly as a monomer and tetramer (117). PKM2 is not required for the transcription of HIF-1α and STAT5α target genes in NK cells and does not significantly regulate gene expression in activated NK cells. However, PKM2-regulated glycolytic metabolism and redox status can promote an optimal response of NK cells. PKM2-controlled metabolism is required for NK cell responses, and administration of TEPP-46 inhibits the release of NK cell proinflammatory cytokines, including IFN-γ and TNF-α. These findings show that PKM2 contributes to activation of NK cells and the secretion of NK cell proinflammatory cytokines through the glycolytic metabolism pathway.
Dendritic cells (DC) are bone marrow-derived leukocytes that engage in innate and adaptive immunity and shape adaptive immune responses based on peripheral cues (118). DCs are the most potent antigen presenting cells, and are implicated in expressing lymphocyte co-stimulatory molecules, secreting cytokines in lymphoid organs and governing both T cell immunity and tolerance (119, 120). Therefore, DCs can be targeted to manipulate the immune system. Activation of DC is inextricably linked to metabolic reprogramming just like other immune cells. Metabolism in activated DC switches from oxidative phosphorylation to glycolysis, with increase in glucose consumption and lactate production (121). Furthermore, upregulation of glucose uptake during DC activation is often accompanied by fatty acid synthesis and other pathways that rewire metabolic reactions toward anabolism (122). Notably, early glycolytic activation of DC is a common phenomenon regardless whether the stimulation is strong or weak. The main difference is that weakly stimulated DCs lack long-term HIF-1α-dependent glycolytic reprogramming and retain mitochondrial oxidative phosphorylation (123). Early glycolysis activation supports the migratory ability of DCs regardless of mitochondrial bioenergetics. Studies have indicated that PKM2 modulates the activation of DC cells by integrating metabolic reprograming with essential gene expression (124). The JNK-p300 signalling axis induces the detetramerization of PKM2 and its nuclear translocation which are crucial for the activation of DC cells. After translocating into the nucleus, PKM2 interacts with transcription factor c-Rel thus enhances IL12p35 transcription. Therefore, PKM2 facilitates activation of DCs by upregulating expression of essential cytokines. PKM2 is thus a potential new target for controlling the activation of DC cells.
The cooperation between T cells and B cells is a central tenet of immune function (125). B cells and T cells work synergistically in the immune defence system and present several common characteristics during activation. However, metabolic reprogramming in B cells is different from that in activated T cells and most other immune cells, in that oxidative phosphorylation and glycolysis are increased in BCR or LPS stimulated B cells. In addition, glycolysis results in accumulation of intermediates which are channelled to the pentose phosphate pathway (126). Moreover, glucose metabolism plays a key role in B cell activation (127). PKM2 expression and enzyme activity are increased in HHcy-induced B cells, thus promoting metabolic reprogramming that induce oxidative phosphorylation and glycolysis (128). Of note, shikonin restores Hcy-induced metabolic reprogramming in B cells, and further inhibits B cell activation and proliferation. Therefore, PKM2 is required to support metabolic reprogramming for B cell activation and function, whereas inhibiting PKM2 expression can limit B cell activation and promote inflammatory responses.
These findings show that the expression of PKM2 promotes activation of multiple immune cells by modulating both metabolic and non- metabolic pathway. Therefore, it plays a crucial role in the activation and function of immune system. Studies have reported that pharmacologically inhibiting PKM2 can control activation and proliferation of immune cells and further suppress inflammatory responses. Regulating the expression of PKM2 can thus be targeted for the treatment of inflammatory diseases through immunometabolic reprogramming (Figure 2).
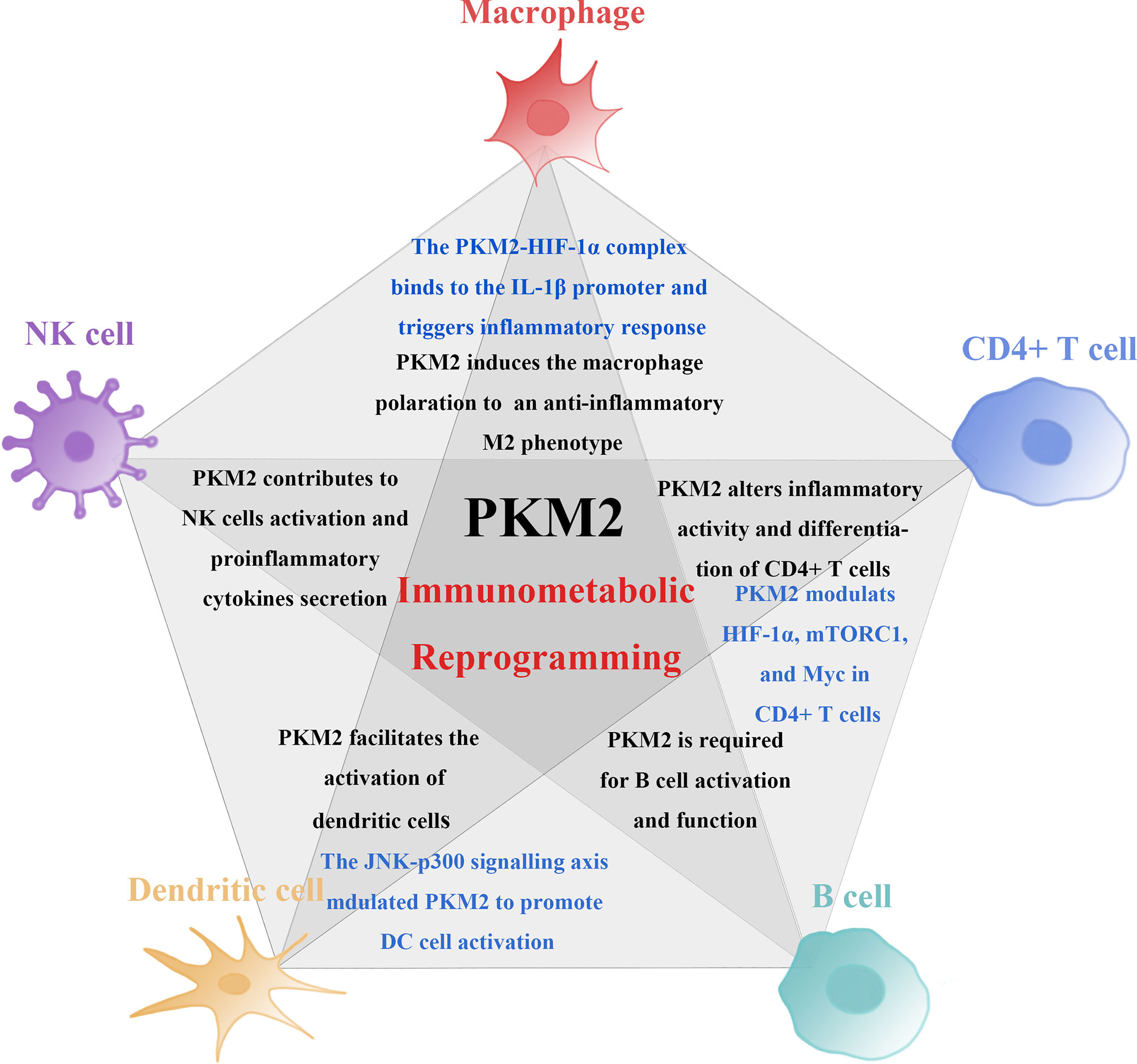
Figure 2 PKM2 mechanisms involved in the induction of immune cell activity and proinflammatory function. PKM2 is involved in the induction of immune cell activity and proinflammatory function. PKM2 medicates changes in the proliferation and function of immune cells through metabolic and non-metabolic pathways. PKM2-mediated immunometabolic reprogramming promotes abnormalization of immune cells. Among these immune cells, the non-metabolic pathway induced by PKM2 is in different ways (marked in blue). Firstly, PKM2 triggers macrophages to polarise from a proinflammatory M1 phenotype to an anti-inflammatory M2 phenotype. The interaction of PKM2 and HIF-1α forms a PKM2-HIF-1α complex, binding to the IL-1β promoter inducing the secretion of proinflammatory cytokines and triggers inflammatory response. Secondly, PKM2 controls the activation and effector functions of CD4+ T cell by modulating the functions of HIF-1α, mTORC1, and Myc and engagement of aerobic glycolysis in TCR-activated CD4+ T cells. Lastly, PKM2 detetramerization and nuclear translocation induced by JNK-p300 signalling axis can promote DC cell activation.
4 PKM2: A Potential Promotor of Proinflammatory Non-Apoptotic-Regulated Cell Death
4.1 PKM2-Dependent Glycolysis Promotes Activation of Inflammasome and Induces Pyroptosis
Pyroptosis is an inflammatory form of cell death triggered by certain inflammasomes, leading to the activation of inactive cytokines like IL-18 and IL-1β and thus inducing an inflammatory response. A previous study suggested that the development of pyroptosis is closely linked to the cytokine storm (129). Inflammasomes are large multimolecular complexes containing one or more Nod-like receptors (NLRs) that control activation of the proteolytic enzyme, caspase-1 (130, 131). Caspase-1 subsequently regulates the proteolytic maturation of IL-1β and IL-18, ultimately leading to pyroptosis (132–134). Inflammasomes are assembled and activated on recognition of exogenous and endogenous stress signals released from damaged or dying cells, thus promote the pathological inflammation in sterile inflammatory diseases, such as atherosclerosis, and inflammatory diseases of acute infection, such as sepsis (135, 136). Notably, autophagy accompanies inflammasome activation to modulate inflammation by eliminating active inflammasomes (137). The canonical activation of inflammasomes modulates immune and inflammatory responses by serving as platforms for the activation of canonical caspase-1 or non-canonical caspase-11 and secretion of both early (such as IL-1β and IL-18) and late (such as HMGB1) proinflammatory mediators. Eukaryotic translation initiation factor 2 alpha kinase 2 (EIF2AK2, also known as PKR) is a protein kinase activated by viral infection and is required for inflammasome-dependent IL-1β and HMGB1 release by macrophages (138). EIF2AK2 can physically interact with inflammasomes, including NLR family pyrin domain-containing 3 (NLRP3) and Absent in melanoma 2 (AIM2). In addition, phosphorylation of EIF2AK2 is required for the activation of various inflammasomes in macrophages.
Several studies have explored pyroptosis because of its association with innate immunity and disease (139). Pyroptosis is a novel form of pro-inflammatory programmed cell death and is known as Gasdermin-mediated programmed necrotic cell death (140, 141). The pyroptosis executioner gasdermin D (GSDMD) is a substrate of both caspase-1 and caspase-11/4/5 and represents a large gasdermin family with a novel membrane pore-forming activity. Pyroptosis is associated with the activity of caspase-1, thus it causes the secretion and maturation of proinflammatory cytokines, and it is implicated in autoimmune diseases and other diseases (142–145). Although PKM2 plays a role in activating inflammasomes and inflammasome activation contributes to pyroptosis, studies have not fully confirmed whether PKM2 can mediate pyroptosis through metabolic reprogramming. Elucidating the role of PKM2 in pyroptosis can provide a basis for the development of drugs targeting PKM2. Previous studies have shown that PKM2-dependent glycolysis promotes EIF2AK2 phosphorylation thus promoting NLRP3 and AIM2 inflammasome activation in macrophages (5). The pharmacological inhibition of the PKM2–EIF2AK2 pathway inhibits release of proinflammatory cytokines thus suppressing the cytokine storm. Moreover, studies have demonstrated that PKM2-mediated-upregulation of glycolysis activates NLRP3 inflammasome leading to pyroptosis in lesioned muscle cells (146). Patients with anti‐signal recognition particle autoantibody expression present with high levels of PKM2 and IL-1β. TEPP-46 plays a protective role in the development of thoracic aortic aneurysm and dissection mediated by PKM2-induced inflammasome activation. Thoracic aortic aneurysm and dissection increases secretion of pro-inflammatory cytokines, chemokines, and reactive oxygen species (ROS) (147). These findings show that PKM2 plays a key role in inflammasome activation, and indicate that PKM2 can be pharmacologically targeted for treatment of inflammatory diseases and to alleviate cytokine storm.
Studies have indicated that stress granules which are cytoplasmic compartments enable cells to overcome various stressors. The stress granule protein, DDX3X can interact with NLRP3 to promote inflammasome activation, whereas assembly of stress granules sequesters DDX3X and inhibits NLRP3 inflammasome activation (148). Competition for DDX3X between stress granules and NLRP3 subsequent cell fate under stress conditions, determines whether the cell will die or not. Therefore, activating stress granules through the PKM2 axis to prevent inflammatory cell death is a potential therapy for inflammatory diseases. Studies have shown that hyperglycaemia increases the activation of PKM2-mediated NLRP3 inflammasomes and stress granule signalling, thus increasing plaque vulnerability and is associated with poor prognosis (149).
4.2 Indirect Involvement of PKM2 in Cellular Ferroptosis
Ferroptosis is new type of cell death reported recently. Ferroptosis is an iron-dependent, oxidative form of non-apoptotic cell death characterized by lipid peroxidation. Ferroptosis-inducing factors affect glutathione peroxidase (GPX) through different pathways, leading to decreased antioxidant capacity and accumulation of ROS in cells, ultimately causing oxidative cell death (150). Therefore, ferroptosis can be triggered by small molecules or conditions that inhibit glutathione biosynthesis or the glutathione-dependent antioxidant enzyme GPX4 (151). Inactivation of GPX4 induces ferroptosis by inhibiting cystine/glutamate antiporter, resulting in accumulation of ROS in the form of lipid hydroperoxides (152). Furthermore, ferroptosis is morphologically characterized by decreased or diminished mitochondria cristae, a ruptured outer mitochondrial membrane, and a condensed mitochondrial membrane (153). These morphological changes result from loss of selective permeability of the plasma membrane due to significant membrane lipid peroxidation and occurrence of oxidative stress (154). Therefore, membrane lipid peroxidation caused by ferroptosis can damage cytomembranes and promote inflammatory reactions in cells. Dysregulation of ferroptosis has been reported in multiple physiological and pathological processes, such as cancer and metabolic diseases (155, 156). In addition, studies have reported that ferroptosis plays a positive role in inflammation through immunogenicity (157). Unlike the immunologically silent apoptosis, damage-associated molecular patterns (DAMPs) and alarmins are released in ferroptosis affected cells, thus triggering cell death and promoting a series of reactions related to inflammation (158–160). Interestingly, several ferroptosis inhibitors exhibit anti-inflammatory effects.
Studies have indicated that pyruvate metabolism is associated with ferroptosis factors. Mitochondrial pyruvate carrier (MPC)-1 is an inner membrane protein that transfers pyruvate to the mitochondria (161). MPC1 expression is regulated by histone lysine demethylase 5A (KDM5A), and the KDM5A-MPC1 axis promotes cancer cell progression (162). Recent studies have shown that the suppression of MPC1 increases ferroptosis vulnerability in vitro and in vivo by promoting mesenchymal traits and glutaminolysis (163). Regulation of the KDM5A-MPC1 axis promotes cancer ferroptosis susceptibility. In addition, pyruvate kinase deficiency (PKD) causes GPX4 deficiency, resulting in mitophagy perturbation and inhibits reticulocyte maturation (164). Cytosolic L-glutamine is preferentially used as α-ketoglutarate as a source of intermediates for the tricarboxylic acid (TCA) cycle at the expense of glutathione (GSH) production, leading to GSH depletion and GPX4 deficiency. In summary, dysregulated pyruvate metabolism contributes to GPX4 deficiency resulting in oxidative imbalance. Furthermore, previous studies established a potential association between PKM2 expression and GPX4 (165). RSL3 is a small molecular compound that induces ferroptosis by inactivating GPX4. ATP level, pyruvate content and protein levels of glycolytic enzymes including PKM2 were decreased in glioma cells treated by RSL3, indicating that RSL3 induces glycolysis dysfunction, resulting in autophagic cell death (165). Therefore, the inactivation of GPX4 promotes PKM2-related glycolysis dysfunction indirectly, indicating a potential association between PKM2 and GPX4. Targeting GPX4 can regulate ferroptosis, and affects other inflammatory-related cell death. GPX4 potentially coordinates lipid peroxidation, inflammasome activation, and pyroptosis during polymicrobial infection (166, 167). Upregulation of GPX4 counter-regulates GSDMD-N mediates pyroptotic cell death in response to infectious insults, preventing lethal systemic inflammation. Therefore, GPX4 is an essential negative regulator of pyroptosis that prevents a lethal inflammatory response.
PKM2-mediated Warburg effect can induce ferroptosis. Formaldehyde induces ferroptosis by upregulating the Warburg effect in hippocampal neuronal cells (168). Analysis of upregulation of PKM2 shows that formaldehyde upregulates the Warburg effect in hippocampal tissue, and increases the level of lipid ROS and iron content. In addition, inhibition of the Warburg effect by dichloroacetate (DCA) protects hippocampal neuronal cells against ferroptosis and cell death (168). Therefore, regulation of PKM2 to suppress the Warburg effect is a feasible method for modulating ferroptosis.
These findings show that PKM2 and PKM2-mediated metabolic reprogramming modulates proinflammatory non-apoptotic-regulated forms of cell death, such as pyroptosis and ferroptosis through multiple pathways. PKM2 can promote the secretion of proinflammatory cytokines and cause systemic inflammation. Therefore, studies should design molecules targeting PKM2 to regulate the inflammatory forms of cell death, thus suppressing inflammation and inhibiting cytokine storm (Figure 3).
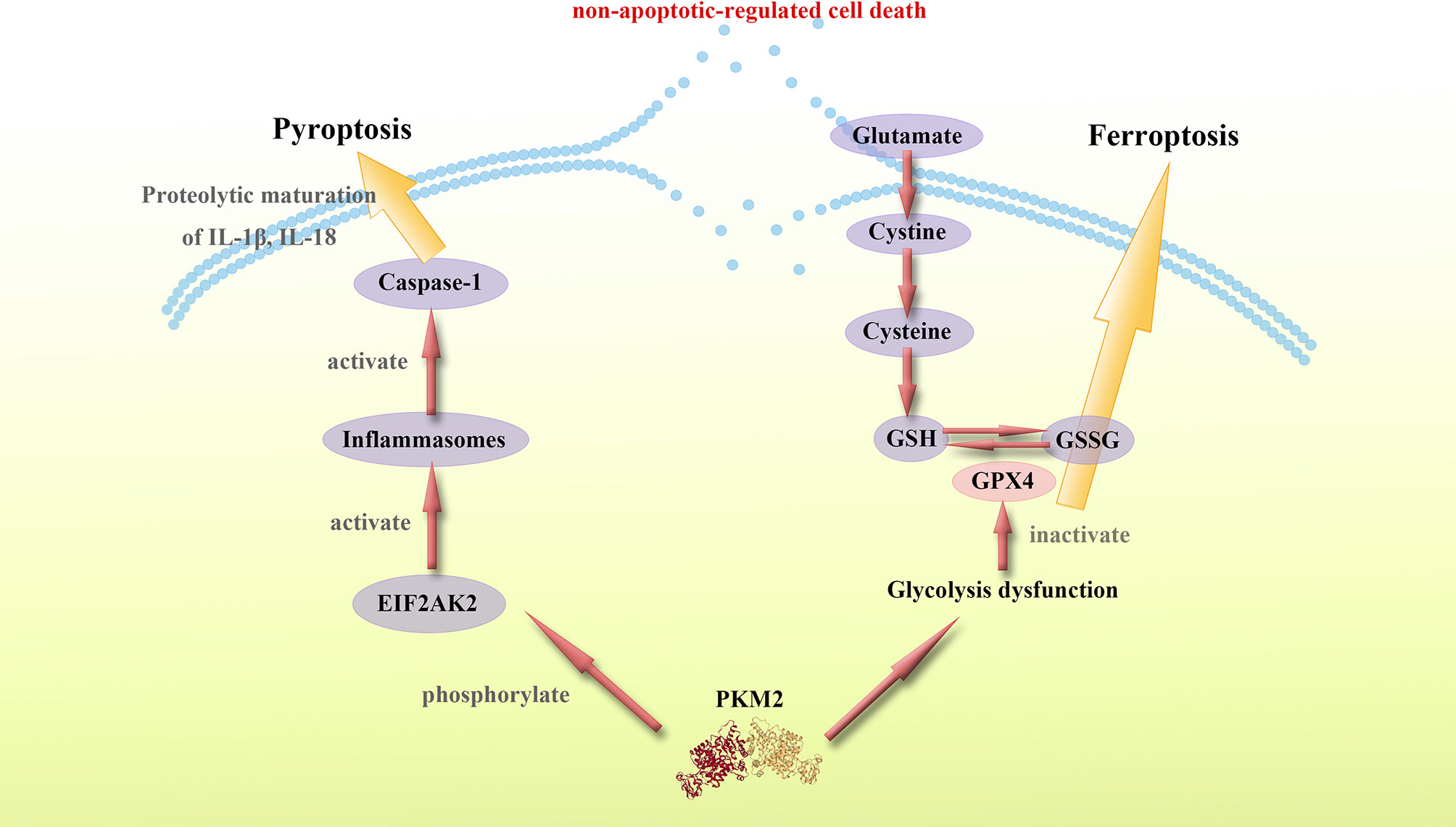
Figure 3 PKM2 mediates proinflammatory non-apoptotic-regulated cell death. PKM2 mediates proinflammatory non-apoptotic-regulated cell death such as pyroptosis and ferroptosis. The specific mechanisms by which PKM2 regulates pyroptosis and ferroptosis remain unknown, however, a considerable number of studies imply a potential connection between PKM2 and these non-apoptotic-regulated cell death. PKM2-dependent glycolysis promotes EIF2AK2 phosphorylation and inflammasome activation, while the activation of inflammasome also activates Caspase-1 and thus leading to pyroptosis. Moreover, PKM2-mediated Warburg effect can induce ferroptosis. The glucose metabolism dysfunction caused by PKM2 mediates the reducibility function of GSH, leading to ferroptosis. Thus, targeting PKM2 is a potential approach of regulating proinflammatory non-apoptotic-regulated cell death.
5 PKM2: A Potential Therapeutic Target of Oxidative Stress and Inflammatory Damage
Oxidative stress is defined as the imbalance between the production of ROS and the endogenous antioxidant defence system. Oxidative stress has been viewed as one of the potential common aetiologies of many inflammatory diseases (169, 170). Studies have reported that dysregulated fatty acid metabolism is related to oxidative stress from the mitochondria, which drives chronic inflammation (171). Therefore, the relationship between oxidative stress and immunometabolism should be explored.
5.1 PKM2 Directly Induces Release of Proinflammatory Cytokines
Cytokines are key modulators of immunity. Cytokines are involved in every facet of immunity and inflammation, including innate immunity, antigen presentation, bone marrow differentiation, cellular recruitment and activation, and expression of adhesion molecules (172). High levels of proinflammatory cytokines and hyperactivation of immune cells leads to a life-threatening systemic inflammatory syndrome, which is also known as cytokine storm (1). Several studies have shown that PKM2-mediated immunometabolic reprogramming promotes the secretion of proinflammatory cytokines. PKM2 forms a complex with HIF-1α in LPS-stimulated macrophages then the complex directly binds to the IL-1β promoter, and can be inhibited by PKM2 activators such as DASA-58 and TEPP-46 (10). The activation of PKM2 by both compounds inhibits glycolytic reprogramming and succinate production. In addition, activation of PKM2 by TEPP-46 in vivo blocks IL-1β production induced by LPS and Salmonella, whereas production of the anti-inflammatory IL-10 is increased. PKM2 mediates production of early proinflammatory mediators, and promotes secretion of late proinflammatory mediators. PKM2 interacts with HIF-1α and activates HIF-1α-dependent transcription of enzymes involved in the Warburg effect in macrophages. PKM2-mediated Warburg effect then regulates the release of HMGB1 (4). PKM2 knockdown and pharmacological inhibition reduces serum lactate and HMGB1 levels. Shikonin protects mice from lethal endotoxemia and sepsis and other inflammatory diseases by inhibiting PKM2 expression. In addition, studies have shown that the levels of proinflammatory cytokine in injured lungs induced by mechanical ventilation significantly increased whereas CXCL14 level decreased and PKM2 expression was increased (173). During this process, overexpression of CXCL14 can alleviate ventilator-induced lung injury and inhibition of pulmonary inflammation through downregulation of PKM2-mediated cytokine production. Previous studies have reported that shikonin, metformin, vitamin K (VK)3/5 exhibit significant inhibitory effects on PKM2 expression. Moreover, these agents exhibit an anti-inflammation effect that protects lungs against sepsis-associated lung injury (174–176). Therefore, novel agents targeting PKM2 can be explored to inhibit proinflammatory cytokine production and alleviate organ failure caused by the cytokine storm. Furthermore, PKM2 plays an important role in pathogenesis of allergic airways disease partly through mediating phosphorylation of STAT3, thus increasing IL-1β–induced proinflammatory signalling (177). In addition, the nuclear PKM2-STAT3 pathway is implicated in LPS-induced lung injury (178). These findings indicate that PKM2 exhibits a proinflammatory effect by phosphorylating STAT3. The PKM2 activator, TEPP-46 exerts an anti-inflammatory effect and low activation of STAT3 (177).
Therefore, PKM2 promotes the production of proinflammatory cytokines, and expression of PKM2 significantly promotes inflammatory responses. Shikonin and other PKM2 inhibitors exert an anti-inflammatory effect by preventing the production of proinflammatory cytokines. Hence, the design of drugs targeting PKM2 inhibition can reduce the release of pro-inflammatory cytokines and alleviate cytokines storms.
5.2 Bidirectional Role of PKM2 in Maintenance of Cellular Redox Homeostasis
Oxidative stress is an imbalance between production of free radicals and ROS, and their elimination by protective mechanisms, such as antioxidants. This imbalance can lead to the damage of important biomolecules and organs, with potential effects on the whole organism (179). Several studies have explored the mechanism through which continued oxidative stress can lead to chronic inflammation and further cause chronic diseases. Oxidative stress can activate various transcription factors such as NF-κB, HIF-1α, β-catenin and Nuclear factor E2-related factor (Nrf-2), resulting in the expression of over 500 different genes, including inflammatory cytokines and chemokines (180). Therefore, oxidative stress and inflammation are closely linked.
Some cells highly depend on PKM2 whereas some do not, therefore, their regulation of redox state through PKM2 modulation occurs through opposite mechanisms. Redox-regulated PKM2 directly binds to p53 and the redox status of cysteine-423 of PKM2 tetramer is important for differential regulation of p53 transcriptional activity. The PKM2 tetramer suppresses p53 transcriptional activity and apoptosis in a high oxidation state but enhances these two processes in a low oxidation state (181). TEPP-46 alleviates oxidative stress in cardiomyocytes and suppresses apoptosis of cardiomyocytes, thus preventing cardiac dysfunction, whereas it exacerbates oxidative stress and promotes apoptosis in lung cancer cells. These findings indicate the redox-dependent differences expressed in different tissues. In endothelial cells, cancer cells and neuronal cells, cellular metabolism is highly dependent on PKM2 expression and it’s mediated by aerobic glycolysis. Therefore, expression of PKM2 plays a protective role against oxidative stress in these cells. Studies have reported that PKM2 is a crucial neuroprotective target against oxidative stress (182). PKM2 drives GSH biosynthesis which is an antioxidant intermediate, thus protecting neurons from oxidative damage and conferring neuroprotective effects. In endothelial cells, active endothelial NO synthase (eNOS) interacts with PKM2 and S-nitrosates PKM2, thus reducing PKM2 activity. PKM2 inhibition increases substrate flux through the pentose phosphate pathway to generate reducing equivalents, including NADPH and GSH, thus protecting cells against oxidative stress (183). Moreover, PKM2 translocates to mitochondria under oxidative stress (184). In the mitochondria, PKM2 interacts with Bcl2 and phosphorylates it at threonine (T) 69, thus preventing Cul3-RBX1 ligase-mediated degradation of Bcl2, ultimately enhancing apoptosis resistance of tumour cells. ROS inhibits PKM2 activity thus contributes to cellular antioxidant responses (34). The inhibition of PKM2 activity diverts glucose flux into PPP to generate sufficient reducing potential for detoxification of ROS. PKM2 expression supports the survival of cancers under acute oxidative stress by promoting anti-oxidant responses, whereas PKM2 activators compromise both pro-anabolic and anti-oxidant functions of cancer cells by increasing PKM2 activity thus interfering with its metabolism. 3,3′,5-triiodothyroxine (T3), a hormone secreted from the thyroid gland protects cells by improving the redox state and regulates the expression of PKM (185). T3 can inhibit apoptosis and oxidative stress in human myocardial cells by upregulating the PKM2/PKM1 ratio. In addition, Nrf-2 regulates expression of several genes responsible for cellular detoxification, antioxidant function, anti-inflammatory effect, drug/xenobiotic transportation, and stress-related factors (186). Astrocytic DRD2 induces GSH synthesis through PKM2-mediated Nrf2 transactivation (187).
PKM2 expression plays different roles in oxidative stress in cells of different tissues. Selective protection against oxidative damage can be achieved by activating or inhibiting PKM2 activity. Although inhibition of PKM2 can significantly alleviate inflammation, complete knockdown of PKM2 is not feasible as it causes damage to cells that are highly dependent on PKM2, such as endothelial cells. Therefore, selective modulation of PKM2 is important to reduce the secretion of inflammatory cytokines and suppression of cytokine storm through alleviation of oxidative stress.
5.3 Alleviating Inflammatory Damage by Pharmacologically Targeting PKM2
PKM2 forms a bridge between immunometabolic reprogramming and inflammatory dysfunction, therefore, PKM2 is a potential therapeutic target for the treatment of inflammatory disease. Several agents have been reported that improve inflammatory damage by inhibiting PKM2 expression. Increased glucose uptake and glycolytic flux promotes generation of mitochondrial ROS in monocytes and macrophages of patients with atherosclerotic coronary artery disease (CAD), which in turn promotes PKM2 dimerization thus inducing its nuclear translocation (9). Nuclear PKM2 functions as a protein kinase and phosphorylates STAT3 and further promotes the production of IL-6 and IL-1β, resulting in systemic and tissue inflammation. PKM2 serves as a molecular integrator of metabolic dysfunction, oxidative stress, and tissue inflammation, representing a novel therapeutic target for treatment of cardiovascular disease and other inflammatory diseases. Notably, PKM2-mediated glycolysis is significantly increased in the complete Freund’s adjuvant-induced astrocyte activation, and platelet-rich plasma effectively inhibits inflammatory response and HMGB1 expression by regulating PKM2-mediated glycolysis and STAT3 signalling-mediated astrocyte activation (188). PKM2-mediated aerobic glycolysis and protein kinase interactions play important roles in cytokine production and inflammation and provide a basis for the development of PKM2-targeted anti-inflammatory drugs.
Activators of PKM2 activity and PKM2 dimer inhibitors can suppress cytokine production and abrogate inflammatory diseases by mediating immunometabolic reprogramming. In addition, several pharmacological active ingredients have been reported that target PKM2 to modulate inflammatory responses. Plumbagin (5-hydroxy-2-methyl-1, 4-naphthoquinone) is a quinone isolated from the roots of Plumbago zeylanica. Previous studies reported that plumbagin exerts an anti-inflammatory effect by regulating pro-inflammatory signalling (189–191). Plumbagin was shown to protect mice from lethal endotoxemia and polymicrobial sepsis induced by cecal ligation and puncture (CLP) by inhibiting the NADPH oxidase 4 (NOX4)/PKM2-dependent immunometabolism pathway, which inhibits LPS-induced PKM2 expression, lactate production and subsequent proinflammatory cytokine (IL-1β and HMGB1) release in macrophages (192). These findings indicate that plumbagin downregulates PKM2 expression to control proinflammatory cytokine production. The traditional Chinese medicine formula Xijiao Dihuang decoction (XJDHT) was first reported in the medical classic “Beiji Qianjin Yaofang” and TCM doctors utilize XJDHT to improve prognosis of patients with sepsis. The main components of XJDHT include Rehmannia, Peony, Cortex Moudan and Cornu Bubali. Studies have shown that XJDHT improves survival of rats with sepsis by modulating the HIF-1α signalling pathway and inhibiting the release of inflammatory cytokine, such as IL-6 (193). Recent studies reported that XJDHT alleviates sepsis by reducing the release of proinflammatory cytokines. XJDHT can improve sepsis by suppressing aerobic glycolysis through downregulation of the TLR4/HIF-1α/PKM2 signalling pathway (194). Therefore, the treatment effect of XJDHT on sepsis indicates that cytokine release can be inhibited by modulating PKM2. However, some agents reduce ROS production and alleviate oxidative stress by upregulating PKM2 in PKM2-dependent cells such as endothelial cells. Qiliqiangxin is a traditional Chinese medicine preparation, comprising 11 Chinese herbal medicines, including astragalus, ginseng and aconite. A multicentre, randomized, double-blind, parallel-group, placebo-controlled study reported effective treatment of 512 chronic heart failure patients using qiliqiangxin (195). The therapeutic effect of qiliqiangxin is through improving cardiomyocyte metabolism and inhibiting cardiomyocyte apoptosis (196–199). Qiliqiangxin improves glucose utilization and metabolism and increases ATP production by upregulating HIF-1α and several glycolysis-related enzymes, including PKM2, thus protecting cardiac microvascular endothelial cells against hypoxia injury (15). In addition, the effects of Chinese medicine in controlling macrophage polarization by modulating PKM2 have been reported. Lycium barbarum polysaccharide is the main bioactive component of Chinese wolfberry, and this compound inhibits PKM2 ubiquitination by upregulating the expression of ubiquitin ligases, thus suppressing LPS-induced inflammation by modulating glycolysis and M1 macrophage polarization (200). Therefore, targeting PKM2 to inhibit glycolysis in an overactive macrophage can provide a new therapeutic option for the treatment of inflammatory diseases.
Several drugs that inhibit cytokine release and improve inflammatory damage by regulating PKM2 expression have been reported. However, effective pharmacological interventions for effective treatment of cytokine storms have not been designed. Therefore, drugs that target and modulate PKM2 to inhibit inflammatory storms should be designed.
6 Protective Role of PKM2 in Specific Cells
PKM2 is highly expressed in rapidly dividing cells whereas its PK activity is significantly decreased. Notably, inhibition of PKM2 by cell growth signals can uncouple the ability of cells to assimilate nutrients into biosynthetic pathways from production of ATP (12). Therefore, PKM2 is an important regulator of cancer cell metabolism, and inhibition of PKM2 can block cancer cell proliferation and excessive immune cell responses (2). However, endothelial cells and a few other types of cells display similar rapid proliferation characteristics such as cancer cells. Therefore, metabolism is a key regulator of growth of endothelial cells, in particular angiogenesis (201, 202). Glycolysis is a driving force for endothelial cell proliferation, whereas the Warburg effect has been reported in endothelial cells (203). Studies have shown that endothelial cells express PKM2 almost exclusively over PKM1, which is significant for maintaining their tight junctions and barrier function (204). PKM2 is required for the suppression of p53 and for maintaining cell cycle progression in proliferating endothelial cells. On the contrary, PKM2 regulates vascular barrier function by suppressing NF-κB and its downstream target, the vascular permeability factor angiopoietin 2 in quiescent endothelial cells. Notably, the loss of PKM2 in endothelial cells results in TCA cycle dysfunction, changes in mitochondrial substrate utilization, thus impairing endothelial cell proliferation and migration (205). In addition, the loss of PKM2 can impair S-adenosylmethionine (SAM) synthesis, causing DNA hypomethylation, de-repression of endogenous retroviral elements and autoimmune activation. Moreover, infiltrated/activated neutrophils at wound site release PKM2 during early stages of wound repair, which facilitates early wound healing by promoting angiogenesis at the wound site (206). Furthermore, PKM2 activates the STAT3 pathway which in turn promotes tissue repair and functional recovery by enhancing neuroblast migration, neurogenesis, and angiogenesis after stroke (207). PKM2 has been proposed as a new myokine, which contributes to axonal extension and functional motor recovery in spinal cord injured mice (208). PKM2 targets valosin-containing protein (VCP) to increase the density of axons and motor function which are mediated by extracellular PKM2-VCP-driven ATPase activity (209). The expression of PKM2 dimer plays an important role in repairing injury by promoting endothelial cell proliferation.
The potential effects of systemic pharmaceutical PKM2 inhibition on normal cells have not been fully elucidated, thus potential on-target side effects of PKM2 modulation should be explored. Targeting PKM2 as an adjuvant antineoplastic therapy can damage the vascular barrier and increase permeability of the vascular endothelium, thus leading to sepsis in immuno-compromised patients. Therefore, it is necessary to be cautious when developing therapies against cancer or inflammation that target PKM2.
PKM2 exerts an antioxidant effect thus protecting PKM2-dependent cells, such as endothelial cells, from oxidative stress. This protective effect of PKM2 on endothelial alleviates kidney injury (210). Endothelial nitric oxide synthase (eNOS) plays a protective role against kidney injury. S-nitroso-CoA (SNO-CoA)–aldoketo reductase family member (AKR1A1) is highly expressed in renal proximal tubules, where it promotes the activity of eNOS in reprogramming intermediary metabolism which is regulated by PKM2, thus protecting kidneys against acute kidney injury. In addition, podocyte-specific PKM2-knockdown mice with diabetes present with worse albuminuria and glomerular pathology. PKM2 activation protects against diabetic nephropathy by increasing glucose metabolic flux, inhibiting production of toxic glucose metabolites and by inducing mitochondrial biogenesis to restore mitochondrial function (211). Therefore, PKM2 protects organ injury by abrogating oxidative stress in specific cells. Specificity of agents targeting PKM2 should be explored to avoid adverse side effects, however, PKM2 is an attractive target.
7 Conclusion
The cytokine storm is an overreaction of immune system, which excessively activates immune cells, resulting in secretion and release of high levels of proinflammatory cytokines ultimately leading to lethal inflammation. Although the cytokine storm is the leading cause of death in many inflammatory diseases, effective therapy options for treating cytokine storm storms have not been fully explored. A previous study reported that immune cells, but not cancer cells, dominate glucose metabolism in the tumour microenvironment, thus providing anew understanding of the tumour microenvironment and further indicated the importance of immunometabolic reprogramming (212, 213).
PKM2 is a pyruvate kinase with lower enzymatic activity compared with PKM1. PKM2 can promote inflammation by switching glucose metabolic pathways to glycolysis. Therefore, PKM2 is a crucial mediator between inflammation and metabolic disorder. The current study reviewed the role of PKM2-mediated immunometabolic reprogramming in cytokine storm through multiple pathways. Firstly, it was shown that PKM2-mediated Warburg effect activates immune cells and induces pro-inflammatory activity. Secondly, PKM2 directly promotes the secretion of cytokines by inducing metabolic reprogramming in cells, which results in cytokine storm. Finally, PKM2 was implicated in novel forms of pro-inflammatory cell death including pyroptosis and ferroptosis. PKM2-mediated activation of inflammasome and stress granule signalling triggers pyroptosis of cells. In addition, PKM2-dependent glycolysis can indirectly induce ferroptosis. However, the specific mechanisms on how PKM2 modulates pyroptosis or ferroptosis have not been fully elucidated.
Several studies have explored therapeutic methods and drugs targeting PKM2. These findings indicate that PKM2 represents a novel potential target for the development of anti-inflammatory drugs that can inhibit cytokine storm and for treatment of severe inflammatory diseases. However, the potential side effects of PKM2 modulation have not been explored.
Intensive studies and clinical trial should be conducted to validate the effectiveness of targeting PKM2 for treatment of inflammatory diseases. Therefore, the current study states some basics to be explored thus providing a guidance for further research. PKM2 exists in different isoforms in different cells of different species, and their intracellular localization differs across cells (9, 10, 77, 214). The exact roles that PKM2 plays in different cells and the mechanism of action has not been fully elucidated. In addition, the inhibition of PKM2 or activation of PKM2 tetramer is a double-edged sword in treatment of inflammatory disease, as PKM2 dominates metabolism in some cells such as endothelial cells (204). Studies should thus explore the possibility of specifically targeting PKM2 to prevent excessive immune response without injuring PKM2-depedent cells. Moreover, inflammatory responses facilitate innate immunity against infections, whereas overactivation of inflammatory cells leads to excessive inflammation resulting in sepsis and even death. Therefore, if inhibition of PKM2 and activation of PKM2 tetramer can prevent excessive immune response, however, this regulation can limit the function of immune system, thus leading to increase in pathogens in bodies. Therefore, studies should explore modulation of immune system by targeting PKM2, and elucidate if the therapy targeting PKM2 can be achieved in both infectious diseases and non-infectious inflammatory diseases. Further, transformation of PKM2 dimer and tetramer can be modulated by protein modifications such as phosphorylation, acetylation, and succinylation (32–34). Studies should explore if the structure and function of PKM2 can be modulated by designing agents that modify its protein modification processes. Addressing these issues is important in developing agents that target PKM2 for inflammatory diseases.
In summary, this review on PKM2 and immunometabolic reprogramming provide insights into the molecular and metabolic mechanisms resulting in cytokine storm and thus provide a basis for restoring balance in the immune system and for development of potential therapies for inflammatory diseases.
Author Contributions
ZL, YL, and HC researched data and wrote the manuscript. JZ and DL established the scope of the review. All authors made substantial contributions to the discussion and revision of the manuscript before submission.
Funding
The research was supported by the National Natural Science Foundation of China (No.82074079), the Natural Science Foundation of Zhejiang Province (No.LY19H280003), the Opening Project of Key Laboratory of Integrative Chinese and Western Medicine for the Diagnosis and Treatment of Circulatory Diseases of Zhejiang Province (No.2C32005), Zhejiang science and technology innovation program for college students (No.2021R410044) and “the Postgraduate Scientific Research Fund” of Zhejiang Chinese Medical University (No.2020YKJ15).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We appreciated the technical support from the Public Platform of Medical Research Center, Academy of Chinese Medical Science, Zhejiang Chinese Medical University. And we would like to thank Freescience for the helpful suggestions and language polishing on this manuscript.
References
1. Fajgenbaum DC, June CH. Cytokine Storm. N Engl J Med (2020) 383(23):2255–73. doi: 10.1056/NEJMra2026131
2. Palsson-McDermott EM, O’Neill LAJ. The Warburg Effect Then and Now: From Cancer to Inflammatory Diseases. BioEssays [Internet] (2013) 35(11):965–73. doi: 10.1002/bies.201300084
3. Zhang Z, Deng X, Liu Y, Liu Y, Sun L, Chen F. PKM2, Function and Expression and Regulation. Cell Biosci [Internet] (2019) 9(1):52. doi: 10.1186/s13578-019-0317-8
4. Yang L, Xie M, Yang M, Yu Y, Zhu S, Hou W, et al. PKM2 Regulates the Warburg Effect and Promotes HMGB1 Release in Sepsis. Nat Commun [Internet] (2014) 5(1):4436. doi: 10.1038/ncomms5436
5. Xie M, Yu Y, Kang R, Zhu S, Yang L, Zeng L, et al. PKM2-Dependent Glycolysis Promotes NLRP3 and AIM2 Inflammasome Activation. Nat Commun [Internet] (2016) 7(1):13280. doi: 10.1038/ncomms13280
6. McElvaney OJ, McEvoy NL, McElvaney OF, Carroll TP, Murphy MP, Dunlea DM, et al. Characterization of the Inflammatory Response to Severe COVID-19 Illness. Am J Respir Crit Care Med [Internet] (2020) 202(6):812–21. doi: 10.1164/rccm.202005-1583OC
7. Yang W, Lu Z. Pyruvate Kinase M2 at a Glance. J Cell Sci [Internet] (2015) 128(9):1655–60. doi: 10.1242/jcs.166629
8. David CJ, Chen M, Assanah M, Canoll P, Manley JL. Hnrnp Proteins Controlled by C-Myc Deregulate Pyruvate Kinase Mrna Splicing in Cancer. Nat [Internet] (2010) 463(7279):364–8. doi: 10.1038/nature08697
9. Shirai T, Nazarewicz RR, Wallis BB, Yanes RE, Watanabe R, Hilhorst M, et al. The Glycolytic Enzyme PKM2 Bridges Metabolic and Inflammatory Dysfunction in Coronary Artery Disease. J Exp Med [Internet] (2016) 213(3):337–54. doi: 10.1084/jem.20150900
10. Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MAR, Sheedy FJ, Gleeson LE, et al. Pyruvate Kinase M2 Regulates Hif-1α Activity and IL-1β Induction and Is a Critical Determinant of the Warburg Effect in LPS-Activated Macrophages. Cell Metab [Internet] (2015) 21(1):65–80. doi: 10.1016/j.cmet.2015.01.017
11. Dombrauckas JD, Santarsiero BD, Mesecar AD. Structural Basis for Tumor Pyruvate Kinase M2 Allosteric Regulation and Catalysis †, ‡. Biochem [Internet] (2005) 44(27):9417–29. doi: 10.1021/bi0474923
12. Vander Heiden MG, Locasale JW, Swanson KD, Sharfi H, Heffron GJ, Amador-Noguez D, et al. Evidence for an Alternative Glycolytic Pathway in Rapidly Proliferating Cells. Sci (80-) [Internet] (2010) 329(5998):1492–9. doi: 10.1126/science.1188015
13. Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC. Pyruvate Kinase M2 Is a Phosphotyrosine-Binding Protein. Nat [Internet] (2008) 452(7184):181–6. doi: 10.1038/nature06667
14. Lv L, Li D, Zhao D, Lin R, Chu Y, Zhang H, et al. Acetylation Targets the M2 Isoform of Pyruvate Kinase for Degradation Through Chaperone-Mediated Autophagy and Promotes Tumor Growth. Mol Cell [Internet] (2011) 42(6):719–30. doi: 10.1016/j.molcel.2011.04.025
15. Wang Y, Han X, Fu M, Wang J, Song Y, Liu Y, et al. Qiliqiangxin Attenuates Hypoxia-Induced Injury in Primary Rat Cardiac Microvascular Endothelial Cells via Promoting HIF-1α-Dependent Glycolysis. J Cell Mol Med [Internet] (2018) 22(5):2791–803. doi: 10.1111/jcmm.13572
16. Wong N, Ojo D, Yan J, Tang D. PKM2 Contributes to Cancer Metabolism. Cancer Lett [Internet] (2015) 356(2):184–91. doi: 10.1016/j.canlet.2014.01.031
17. Dayton TL, Jacks T, Vander Heiden MG. PKM2, Cancer Metabolism, and the Road Ahead. EMBO Rep [Internet] (2016) 17(12):1721–30. doi: 10.15252/embr.201643300
18. Wang P, Sun C, Zhu T, Xu Y. Structural Insight Into Mechanisms for Dynamic Regulation of PKM2. Protein Cell [Internet] (2015) 6(4):275–87. doi: 10.1007/s13238-015-0132-x
19. Anastasiou D, Yu Y, Israelsen WJ, Jiang J-K, Boxer MB, Hong BS, et al. Pyruvate Kinase M2 Activators Promote Tetramer Formation and Suppress Tumorigenesis. Nat Chem Biol [Internet] (2012) 8(10):839–47. doi: 10.1038/nchembio.1060
20. Jiang J, Walsh MJ, Brimacombe KR, Anastasiou D, Yu Y, Israelsen WJ, et al. ML265: A Potent PKM2 Activator Induces Tetramerization and Reduces Tumor Formation and Size in a Mouse Xenograft Model. In: Probe Reports From the NIH Molecular Libraries Program [Internet]. Bethesda (MD):National Center for Biotechnology Information (US) (2010). Available at: http://www.ncbi.nlm.nih.gov/pubmed/23905203.
21. Jiang J, Boxer MB, Vander Heiden MG, Shen M, Skoumbourdis AP, Southall N, et al. Evaluation of Thieno[3,2-B]Pyrrole[3,2-D]Pyridazinones as Activators of the Tumor Cell Specific M2 Isoform of Pyruvate Kinase. Bioorg Med Chem Lett [Internet] (2010) 20(11):3387–93. doi: 10.1016/j.bmcl.2010.04.015
22. Keller KE, Tan IS, Lee Y-S. SAICAR Stimulates Pyruvate Kinase Isoform M2 and Promotes Cancer Cell Survival in Glucose-Limited Conditions. Sci (80-) [Internet] (2012) 338(6110):1069–72. doi: 10.1126/science.1224409
23. Kung C, Hixon J, Choe S, Marks K, Gross S, Murphy E, et al. Small Molecule Activation of PKM2 in Cancer Cells Induces Serine Auxotrophy. Chem Biol [Internet] (2012) 19(9):1187–98. doi: 10.1016/j.chembiol.2012.07.021
24. Park S-H, Ozden O, Liu G, Song HY, Zhu Y, Yan Y, et al. SIRT2-Mediated Deacetylation and Tetramerization of Pyruvate Kinase Directs Glycolysis and Tumor Growth. Cancer Res [Internet] (2016) 76(13):3802–12. doi: 10.1158/0008-5472.CAN-15-2498
25. Srivastava SP, Li J, Kitada M, Fujita H, Yamada Y, Goodwin JE, et al. SIRT3 Deficiency Leads to Induction of Abnormal Glycolysis in Diabetic Kidney With Fibrosis. Cell Death Dis [Internet] (2018) 9(10):997. doi: 10.1038/s41419-018-1057-0
26. Bhardwaj A, Das S. SIRT6 Deacetylates PKM2 to Suppress its Nuclear Localization and Oncogenic Functions. Proc Natl Acad Sci [Internet] (2016) 113(5):E538–47. doi: 10.1073/pnas.1520045113
27. Wang F, Wang K, Xu W, Zhao S, Ye D, Wang Y, et al. SIRT5 Desuccinylates and Activates Pyruvate Kinase M2 to Block Macrophage IL-1β Production and to Prevent DSS-Induced Colitis in Mice. Cell Rep [Internet] (2017) 19(11):2331–44. doi: 10.1016/j.celrep.2017.05.065
28. Chaneton B, Hillmann P, Zheng L, Martin ACL, Maddocks ODK, Chokkathukalam A, et al. Serine Is a Natural Ligand and Allosteric Activator of Pyruvate Kinase M2. Nat [Internet] (2012) 491(7424):458–62. doi: 10.1073/pnas.1520045113
29. Mor I, Carlessi R, Ast T, Feinstein E, Kimchi A. Death-Associated Protein Kinase Increases Glycolytic Rate Through Binding and Activation of Pyruvate Kinase. Oncogene [Internet] (2012) 31(6):683–93. doi: 10.1038/onc.2011.264
30. Morgan HP, O’Reilly FJ, Wear MA, O’Neill JR, Fothergill-Gilmore LA, Hupp T, et al. M2 Pyruvate Kinase Provides a Mechanism for Nutrient Sensing and Regulation of Cell Proliferation. Proc Natl Acad Sci [Internet] (2013) 110(15):5881–6. doi: 10.1073/pnas.1217157110
31. Nakatsu D, Horiuchi Y, Kano F, Noguchi Y, Sugawara T, Takamoto I, et al. L-Cysteine Reversibly Inhibits Glucose-Induced Biphasic Insulin Secretion and ATP Production by Inactivating PKM2. Proc Natl Acad Sci [Internet] (2015) 112(10):E1067–76. doi: 10.1073/pnas.1417197112
32. Hitosugi T, Kang S, Vander Heiden MG, Chung T-W, Elf S, Lythgoe K, et al. Tyrosine Phosphorylation Inhibits PKM2 to Promote the Warburg Effect and Tumor Growth. Sci Signal [Internet] (2009) 2(97):ra73–3. doi: 10.1126/scisignal.2000431
33. Lv L, Xu Y-P, Zhao D, Li F-L, Wang W, Sasaki N, et al. Mitogenic and Oncogenic Stimulation of K433 Acetylation Promotes PKM2 Protein Kinase Activity and Nuclear Localization. Mol Cell [Internet] (2013) 52(3):340–52. doi: 10.1016/j.molcel.2013.09.004
34. Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang J-k, Shen M, et al. Inhibition of Pyruvate Kinase M2 by Reactive Oxygen Species Contributes to Cellular Antioxidant Responses. Sci (80-) [Internet] (2011) 334(6060):1278–83. doi: 10.1126/science.1211485
35. Ning X, Qi H, Li R, Li Y, Jin Y, McNutt MA, et al. Discovery of Novel Naphthoquinone Derivatives as Inhibitors of the Tumor Cell Specific M2 Isoform of Pyruvate Kinase. Eur J Med Chem [Internet] (2017) 138:343–52. doi: 10.1016/j.ejmech.2017.06.064
36. Chen J, Xie J, Jiang Z, Wang B, Wang Y, Hu X. Shikonin and its Analogs Inhibit Cancer Cell Glycolysis by Targeting Tumor Pyruvate Kinase-M2. Oncogene [Internet] (2011) 30(42):4297–306. doi: 10.1038/onc.2011.137
37. Guo C, He J, Song X, Tan L, Wang M, Jiang P, et al. Pharmacological Properties and Derivatives of Shikonin—A Review in Recent Years. Pharmacol Res [Internet] (2019) 149:104463. doi: 10.1016/j.phrs.2019.104463
38. Liu T, Li S, Wu L, Yu Q, Li J, Feng J, et al. Experimental Study of Hepatocellular Carcinoma Treatment by Shikonin Through Regulating PKM2. J Hepatocell Carcinoma [Internet] (2020) 7:19–31. doi: 10.2147/JHC.S237614
39. Thonsri U, Seubwai W, Waraasawapati S, Wongkham S, Boonmars T, Cha’on U, et al. Antitumor Effect of Shikonin, a PKM2 Inhibitor, in Cholangiocarcinoma Cell Lines. Anticancer Res [Internet] (2020) 40(9):5115–24. doi: 10.21873/anticanres.14515
40. Liu B, Jin J, Zhang Z, Zuo L, Jiang M, Xie C. Shikonin Exerts Antitumor Activity by Causing Mitochondrial Dysfunction in Hepatocellular Carcinoma Through PKM2–AMPK–PGC1α Signaling Pathway. Biochem Cell Biol [Internet] (2019) 97(4):397–405. doi: 10.1139/bcb-2018-0310
41. Wang Z, Yin J, Li M, Shen J, Xiao Z, Zhao Y, et al. Combination of Shikonin With Paclitaxel Overcomes Multidrug Resistance in Human Ovarian Carcinoma Cells in a P-Gp-Independent Manner Through Enhanced ROS Generation. Chin Med [Internet] (2019) 14(1):7. doi: 10.1186/s13020-019-0231-3
42. Vander Heiden MG, Christofk HR, Schuman E, Subtelny AO, Sharfi H, Harlow EE, et al. Identification of Small Molecule Inhibitors of Pyruvate Kinase M2. Biochem Pharmacol [Internet] (2010) 79(8):1118–24. doi: 10.1016/j.bcp.2009.12.003
43. Qi H, Ning X, Yu C, Ji X, Jin Y, McNutt MA, et al. Succinylation-Dependent Mitochondrial Translocation of PKM2 Promotes Cell Survival in Response to Nutritional Stress. Cell Death Dis [Internet] (2019) 10(3):170. doi: 10.1038/s41419-018-1271-9
44. Yang W, Zheng Y, Xia Y, Ji H, Chen X, Guo F, et al. ERK1/2-Dependent Phosphorylation and Nuclear Translocation of PKM2 Promotes the Warburg Effect. Nat Cell Biol [Internet] (2012) 14(12):1295–304. doi: 10.1038/ncb2629
45. Lufei C, Cao X. Nuclear Import of Pin1 Is Mediated by a Novel Sequence in the Ppiase Domain. FEBS Lett [Internet] (2009) 583(2):271–6. doi: 10.1016/j.febslet.2008.12.011
46. Mason DA, Stage DE, Goldfarb DS. Evolution of the Metazoan-Specific Importin α Gene Family. J Mol Evol [Internet] (2009) 68(4):351–65. doi: 10.1007/s00239-009-9215-8
47. Li J, Li S, Guo J, Li Q, Long J, Ma C, et al. Natural Product Micheliolide (MCL) Irreversibly Activates Pyruvate Kinase M2 and Suppresses Leukemia. J Med Chem [Internet] (2018) 61(9):4155–64. doi: 10.1021/acs.jmedchem.8b00241
48. Chen T-J, Wang H-J, Liu J-S, Cheng H-H, Hsu S-C, Wu M-C, et al. Mutations in the PKM2 Exon-10 Region Are Associated With Reduced Allostery and Increased Nuclear Translocation. Commun Biol [Internet] (2019) 2(1):105. doi: 10.1038/s42003-019-0343-4
49. Yang W, Lu Z. Nuclear PKM2 Regulates the Warburg Effect. Cell Cycle [Internet] (2013) 12(19):3343–7. doi: 10.4161/cc.26182
50. Chen S, Youhong T, Tan Y, He Y, Ban Y, Cai J, et al. EGFR-PKM2 Signaling Promotes the Metastatic Potential of Nasopharyngeal Carcinoma Through Induction of FOSL1 and ANTXR2. Carcinogenesis [Internet] (2020) 41(6):723–33. doi: 10.1093/carcin/bgz180
51. Yang W, Xia Y, Ji H, Zheng Y, Liang J, Huang W, et al. Nuclear PKM2 Regulates β-Catenin Transactivation Upon EGFR Activation. Nat [Internet] (2011) 480(7375):118–22. doi: 10.1038/nature10598
52. Yang W, Xia Y, Hawke D, Li X, Liang J, Xing D, et al. PKM2 Phosphorylates Histone H3 and Promotes Gene Transcription and Tumorigenesis. Cell [Internet] (2012) 150(4):685–96. doi: 10.1016/j.cell.2012.07.018
53. Corcoran SE, O’Neill LAJ. Hif1α and Metabolic Reprogramming in Inflammation. J Clin Invest [Internet] (2016) 126(10):3699–707. doi: 10.1172/JCI84431
54. Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, et al. Succinate Links TCA Cycle Dysfunction to Oncogenesis by Inhibiting HIF-α Prolyl Hydroxylase. Cancer Cell [Internet] (2005) 7(1):77–85. doi: 10.1016/j.ccr.2004.11.022
55. Luo W, Hu H, Chang R, Zhong J, Knabel M, O’Meally R, et al. Pyruvate Kinase M2 Is a PHD3-Stimulated Coactivator for Hypoxia-Inducible Factor 1. Cell [Internet] (2011) 145(5):732–44. doi: 10.1016/j.cell.2011.03.054
56. Culver C, Sundqvist A, Mudie S, Melvin A, Xirodimas D, Rocha S. Mechanism of Hypoxia-Induced NF-κb. Mol Cell Biol [Internet] (2010) 30(20):4901–21. doi: 10.1128/MCB.00409-10
57. Xu Q, Liu L-Z, Yin Y, He J, Li Q, Qian X, et al. Regulatory Circuit of PKM2/NF-κb/Mir-148a/152-Modulated Tumor Angiogenesis and Cancer Progression. Oncogene [Internet] (2015) 34(43):5482–93. doi: 10.1038/onc.2015.6
58. Azoitei N, Becher A, Steinestel K, Rouhi A, Diepold K, Genze F, et al. PKM2 Promotes Tumor Angiogenesis by Regulating HIF-1α Through NF-κb Activation. Mol Cancer [Internet] (2016) 15(1):3. doi: 10.1186/s12943-015-0490-2
59. Hasan D, Gamen E, Abu Tarboush N, Ismail Y, Pak O, Azab B. PKM2 and HIF-1α Regulation in Prostate Cancer Cell Lines. Konopleva M, Editor. PloS One [Internet] (2018) 13(9):e0203745. doi: 10.1371/journal.pone.0203745
60. Wang H-J, Hsieh Y-J, Cheng W-C, Lin C-P, Lin Y-S, Yang S-F, et al. JMJD5 Regulates PKM2 Nuclear Translocation and Reprograms HIF-1 -Mediated Glucose Metabolism. Proc Natl Acad Sci [Internet] (2014) 111(1):279–84. doi: 10.1073/pnas.1311249111
61. Liu L, Xu H, Zhao H, Jiang C. Steap4 Inhibits Hif-1α/PKM2 Signaling and Reduces High Glucose-Induced Apoptosis of Retinal Vascular Endothelial Cells. Diabetes Metab Syndr Obes Targets Ther [Internet] (2020) 13:2573–82. doi: 10.2147/DMSO.S251663
62. Feng J, Dai W, Mao Y, Wu L, Li J, Chen K, et al. Simvastatin Re-Sensitizes Hepatocellular Carcinoma Cells to Sorafenib by Inhibiting HIF-1α/Ppar-γ/PKM2-Mediated Glycolysis. J Exp Clin Cancer Res [Internet] (2020) 39(1):24. doi: 10.1186/s13046-020-1528-x
63. Gao X, Wang H, Yang JJ, Liu X, Liu Z-R. Pyruvate Kinase M2 Regulates Gene Transcription by Acting as a Protein Kinase. Mol Cell [Internet] (2012) 45(5):598–609. doi: 10.1016/j.molcel.2012.01.001
64. Gao SP, Mark KG, Leslie K, Pao W, Motoi N, Gerald WL, et al. Mutations in the EGFR Kinase Domain Mediate STAT3 Activation via IL-6 Production in Human Lung Adenocarcinomas. J Clin Invest [Internet] (2007) 117(12):3846–56. doi: 10.1172/JCI31871
65. Yu H, Pardoll D, Jove R. Stats in Cancer Inflammation and Immunity: A Leading Role for STAT3. Nat Rev Cancer [Internet] (2009) 9(11):798–809. doi: 10.1038/nrc2734
66. Wang B, Liu S, Fan B, Xu X, Chen Y, Lu R, et al. PKM2 Is Involved in Neuropathic Pain by Regulating ERK and STAT3 Activation in Rat Spinal Cord. J Headache Pain [Internet] (2018) 19(1):7. doi: 10.1186/s10194-018-0836-4
67. Demaria M, Poli V. PKM2, STAT3 and HIF-1α. JAK-STAT [Internet] (2012) 1(3):194–6. doi: 10.4161/jkst.20662
68. Sola-Penna M, Paixão LP, Branco JR, Ochioni AC, Albanese JM, Mundim DM, et al. Serotonin Activates Glycolysis and Mitochondria Biogenesis in Human Breast Cancer Cells Through Activation of the Jak1/STAT3/ERK1/2 and Adenylate Cyclase/PKA, Respectively. Br J Cancer [Internet] (2020) 122(2):194–208. doi: 10.1038/s41416-019-0640-1
69. Hosios AM, Fiske BP, Gui DY, Vander Heiden MG. Lack of Evidence for PKM2 Protein Kinase Activity. Mol Cell [Internet] (2015) 59(5):850–7. doi: 10.1016/j.molcel.2015.07.013
70. Alves-Filho JC, Pålsson-McDermott EM. Pyruvate Kinase M2: A Potential Target for Regulating Inflammation. Front Immunol [Internet] (2016) 7:145. doi: 10.3389/fimmu.2016.00145
71. Warburg O, Wind F, Negelein E. The Metabolism Oftumors in the Body. J Gen Physiol [Internet] (1927) 8(6):519–30. doi: 10.1085/jgp.8.6.519
73. Fox CJ, Hammerman PS, Thompson CB. Fuel Feeds Function: Energy Metabolism and the T-Cell Response. Nat Rev Immunol [Internet] (2005) 5(11):844–52. doi: 10.1038/nri1710
74. MacIver NJ, Michalek RD, Rathmell JC. Metabolic Regulation of T Lymphocytes. Annu Rev Immunol [Internet] (2013) 31(1):259–83. doi: 10.1146/annurev-immunol-032712-095956
75. Palmer CS, Ostrowski M, Balderson B, Christian N, Crowe SM. Glucose Metabolism Regulates T Cell Activation, Differentiation, and Functions. Front Immunol [Internet] (2015) 6:1. doi: 10.3389/fimmu.2015.00001
76. Kelly B, O’Neill LAJ. Metabolic Reprogramming in Macrophages and Dendritic Cells in Innate Immunity. Cell Res [Internet] (2015) 25(7):771–84. doi: 10.1038/cr.2015.68
77. Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, et al. The M2 Splice Isoform of Pyruvate Kinase Is Important for Cancer Metabolism and Tumour Growth. Nat [Internet] (2008) 452(7184):230–3. doi: 10.1038/nature06734
78. Feng Y, Liu J, Guo W, Guan Y, Xu H, Guo Q, et al. Atg7 Inhibits Warburg Effect by Suppressing PKM2 Phosphorylation Resulting Reduced Epithelial-Mesenchymal Transition. Int J Biol Sci [Internet] (2018) 14(7):775–83. doi: 10.7150/ijbs.26077
79. Samec M, Liskova A, Koklesova L, Samuel SM, Zhai K, Buhrmann C, et al. Flavonoids Against the Warburg Phenotype—Concepts of Predictive, Preventive and Personalised Medicine to Cut the Gordian Knot of Cancer Cell Metabolism. EPMA J [Internet] (2020) 11(3):377–98. doi: 10.1007/s13167-020-00217-y
80. Yan X, Qi M, Li P, Zhan Y, Shao H. Apigenin in Cancer Therapy: Anti-Cancer Effects and Mechanisms of Action. Cell Biosci [Internet] (2017) 7(1):50. doi: 10.1186/s13578-017-0179-x
81. Miao L, Yuanyuan W, Yuxia R, Changsen B, Li Q, Yanfen C, et al. PKM2 Promotes Reductive Glutamine Metabolism. Cancer Biol Med [Internet] (2018) 15(4):389. doi: 10.20892/j.issn.2095-3941.2018.0122
82. Hiscock N, Petersen EW, Krzywkowski K, Boza J, Halkjaer-Kristensen J, Pedersen BK. Glutamine Supplementation Further Enhances Exercise-Induced Plasma IL-6. J Appl Physiol [Internet] (2003) 95(1):145–8. doi: 10.1152/japplphysiol.00471.2002
83. Cruzat V, Macedo Rogero M, Noel Keane K, Curi R, Newsholme P. Glutamine: Metabolism and Immune Function, Supplementation and Clinical Translation. Nutrients [Internet] (2018) 10(11):1564. doi: 10.3390/nu10111564
84. Rodríguez-Prados J-C, Través PG, Cuenca J, Rico D, Aragonés J, Martín-Sanz P, et al. Substrate Fate in Activated Macrophages: A Comparison Between Innate, Classic, and Alternative Activation. J Immunol [Internet] (2010) 185(1):605–14. doi: 10.4049/jimmunol.0901698
85. Sica A, Mantovani A. Macrophage Plasticity and Polarization: In Vivo Veritas. J Clin Invest [Internet] (2012) 122(3):787–95. doi: 10.1172/JCI59643
86. Gordon S, Martinez FO. Alternative Activation of Macrophages: Mechanism and Functions. Immun [Internet] (2010) 32(5):593–604. doi: 10.1016/j.immuni.2010.05.007
87. Biswas SK, Mantovani A. Macrophage Plasticity and Interaction With Lymphocyte Subsets: Cancer as a Paradigm. Nat Immunol [Internet] (2010) 11(10):889–96. doi: 10.1038/ni.1937
88. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage Polarization: Tumor-Associated Macrophages as a Paradigm for Polarized M2 Mononuclear Phagocytes. Trends Immunol [Internet] (2002) 23(11):549–55. doi: 10.1016/S1471-4906(02)02302-5
89. Kasmi KC EL, Stenmark KR. Contribution of Metabolic Reprogramming to Macrophage Plasticity and Function. Semin Immunol [Internet] (2015) 27(4):267–75. doi: 10.1016/j.smim.2015.09.001
90. Xu F, Guo M, Huang W, Feng L, Zhu J, Luo K, et al. Annexin A5 Regulates Hepatic Macrophage Polarization via Directly Targeting PKM2 and Ameliorates NASH. Redox Biol [Internet] (2020) 36:101634. doi: 10.1016/j.redox.2020.101634
91. Kong Q, Li N, Cheng H, Zhang X, Cao X, Qi T, et al. HSPA12A Is a Novel Player in Nonalcoholic Steatohepatitis via Promoting Nuclear PKM2-Mediated M1 Macrophage Polarization. Diabetes [Internet] (2019) 68(2):361–76. doi: 10.2337/db18-0035
92. Das Gupta K, Shakespear MR, Curson JEB, Murthy AMV, Iyer A, Hodson MP, et al. Class Iia Histone Deacetylases Drive Toll-Like Receptor-Inducible Glycolysis and Macrophage Inflammatory Responses via Pyruvate Kinase M2. Cell Rep [Internet] (2020) 30(8):2712–28.e8. doi: 10.1016/j.celrep.2020.02.007
93. Zhu J, Paul WE. CD4 T Cells: Fates, Functions, and Faults. Blood [Internet] (2008) 112(5):1557–69. doi: 10.1182/blood-2008-05-078154
94. O’Neill LAJ, Kishton RJ, Rathmell J. A Guide to Immunometabolism for Immunologists. Nat Rev Immunol [Internet] (2016) 16(9):553–65. doi: 10.1038/nri.2016.70
95. Bettencourt IA, Powell JD. Targeting Metabolism as a Novel Therapeutic Approach to Autoimmunity, Inflammation, and Transplantation. J Immunol [Internet] (2017) 198(3):999–1005. doi: 10.4049/jimmunol.1601318
96. Pearce EL, Pearce EJ. Metabolic Pathways in Immune Cell Activation and Quiescence. Immun [Internet] (2013) 38(4):633–43. doi: 10.1016/j.immuni.2013.04.005
97. Klein Geltink RI, Kyle RL, Pearce EL. Unraveling the Complex Interplay Between T Cell Metabolism and Function. Annu Rev Immunol [Internet] (2018) 36(1):461–88. doi: 10.1146/annurev-immunol-042617-053019
98. Angiari S, Runtsch MC, Sutton CE, Palsson-McDermott EM, Kelly B, Rana N, et al. Pharmacological Activation of Pyruvate Kinase M2 Inhibits CD4+ T Cell Pathogenicity and Suppresses Autoimmunity. Cell Metab [Internet] (2020) 31(2):391–405. doi: 10.1016/j.cmet.2019.10.015
99. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The Transcription Factor Myc Controls Metabolic Reprogramming Upon T Lymphocyte Activation. Immun [Internet] (2011) 35(6):871–82. doi: 10.1016/j.immuni.2011.09.021
100. Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, et al. The Kinase Mtor Regulates the Differentiation of Helper T Cells Through the Selective Activation of Signaling by Mtorc1 and Mtorc2. Nat Immunol [Internet] (2011) 12(4):295–303. doi: 10.1038/ni.2005
101. Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, et al. The Mtor Kinase Differentially Regulates Effector and Regulatory T Cell Lineage Commitment. Immun [Internet] (2009) 30(6):832–44. doi: 10.1016/j.immuni.2009.04.014
102. Shehade H, Acolty V, Moser M, Oldenhove G. Cutting Edge: Hypoxia-Inducible Factor 1 Negatively Regulates Th1 Function. J Immunol [Internet] (2015) 195(4):1372–6. doi: 10.4049/jimmunol.1402552
103. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. Hif1α–Dependent Glycolytic Pathway Orchestrates a Metabolic Checkpoint for the Differentiation of TH17 and Treg Cells. J Exp Med [Internet] (2011) 208(7):1367–76. doi: 10.1084/jem.20110278
104. Lü S, Deng J, Liu H, Liu B, Yang J, Miao Y, et al. PKM2-Dependent Metabolic Reprogramming in CD4+ T Cells Is Crucial for Hyperhomocysteinemia-Accelerated Atherosclerosis. J Mol Med [Internet] (2018) 96(6):585–600. doi: 10.1007/s00109-018-1645-6
105. Lü S, Dang G, Deng J, Liu H, Liu B, Yang J, et al. Shikonin Attenuates Hyperhomocysteinemia-Induced CD4+ T Cell Inflammatory Activation and Atherosclerosis in Apoe–/– Mice by Metabolic Suppression. Acta Pharmacol Sin [Internet] (2020) 41(1):47–55. doi: 10.1038/s41401-019-0308-7
106. Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. Th17: An Effector CD4 T Cell Lineage With Regulatory T Cell Ties. Immun [Internet] (2006) 24(6):677–88. doi: 10.1016/j.immuni.2006.06.002
107. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol [Internet] (2009) 27(1):485–517. doi: 10.1146/annurev.immunol.021908.132710
108. Ohkura N, Kitagawa Y, Sakaguchi S. Development and Maintenance of Regulatory T Cells. Immun [Internet] (2013) 38(3):414–23. doi: 10.1016/j.immuni.2013.03.002
109. Damasceno LEA, Prado DS, Veras FP, Fonseca MM, Toller-Kawahisa JE, Rosa MH, et al. PKM2 Promotes Th17 Cell Differentiation and Autoimmune Inflammation by Fine-Tuning STAT3 Activation. J Exp Med [Internet] (2020) 217(10):e20190613. doi: 10.1084/jem.20190613/151965/PKM2-promotes-Th17-cell-differentiation-and
110. Koga T, Hedrich CM, Mizui M, Yoshida N, Otomo K, Lieberman LA, et al. Camk4-Dependent Activation of AKT/Mtor and CREM-α Underlies Autoimmunity-Associated Th17 Imbalance. J Clin Invest [Internet] (2014) 124(5):2234–45. doi: 10.1172/JCI73411
111. Kono M, Maeda K, Stocton-Gavanescu I, Pan W, Umeda M, Katsuyama E, et al. Pyruvate Kinase M2 Is Requisite for Th1 and Th17 Differentiation. JCI Insight [Internet] (2019) 4(12):e127395. doi: 10.1172/jci.insight.127395
112. Seki SM, Posyniak K, McCloud R, Rosen DA, Fernández-Castañeda A, Beiter RM, et al. Modulation of PKM Activity Affects the Differentiation of T H 17 Cells. Sci Signal [Internet] (2020) 13(655):eaay9217. doi: 10.1126/scisignal.aay9217
113. Kumar S. Natural Killer Cell Cytotoxicity and its Regulation by Inhibitory Receptors. Immunol [Internet] (2018) 154(3):383–93. doi: 10.1111/imm.12921
114. Fauriat C, Long EO, Ljunggren H-G, Bryceson YT. Regulation of Human NK-Cell Cytokine and Chemokine Production by Target Cell Recognition. Blood [Internet] (2010) 115(11):2167–76. doi: 10.1182/blood-2009-08-238469
115. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of Natural Killer Cells. Nat Immunol [Internet] (2008) 9(5):503–10. doi: 10.1038/ni1582
116. Assmann N, O’Brien KL, Donnelly RP, Dyck L, Zaiatz-Bittencourt V, Loftus RM, et al. Srebp-Controlled Glucose Metabolism Is Essential for NK Cell Functional Responses. Nat Immunol [Internet] (2017) 18(11):1197–206. doi: 10.1038/ni.3838
117. Walls JF, Subleski JJ, Palmieri EM, Gonzalez-Cotto M, Gardiner CM, McVicar DW, et al. Metabolic But Not Transcriptional Regulation by PKM2 Is Important for Natural Killer Cell Responses. Elife [Internet] (2020) 9:e59166. doi: 10.7554/eLife.59166
118. Merad M, Sathe P, Helft J, Miller J, Mortha A. The Dendritic Cell Lineage: Ontogeny and Function of Dendritic Cells and Their Subsets in the Steady State and the Inflamed Setting. Annu Rev Immunol [Internet] (2013) 31(1):563–604. doi: 10.1146/annurev-immunol-020711-074950
119. Banchereau J, Steinman RM. Dendritic Cells and the Control of Immunity. Nat [Internet] (1998) 392(6673):245–52. doi: 10.1038/32588
120. Waisman A, Lukas D, Clausen BE, Yogev N. Dendritic Cells as Gatekeepers of Tolerance. Semin Immunopathol [Internet] (2017) 39(2):153–63. doi: 10.1007/s00281-016-0583-z
121. Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, et al. Toll-Like Receptor–Induced Changes in Glycolytic Metabolism Regulate Dendritic Cell Activation. Blood [Internet] (2010) 115(23):4742–9. doi: 10.1182/blood-2009-10-249540
122. Everts B, Amiel E, Huang SC-C, Smith AM, Chang C-H, Lam WY, et al. TLR-Driven Early Glycolytic Reprogramming via the Kinases TBK1-Ikkε Supports the Anabolic Demands of Dendritic Cell Activation. Nat Immunol [Internet] (2014) 15(4):323–32. doi: 10.1038/ni.2833
123. Guak H, Al Habyan S, Ma EH, Aldossary H, Al-Masri M, Won SY, et al. Glycolytic Metabolism Is Essential for CCR7 Oligomerization and Dendritic Cell Migration. Nat Commun [Internet] (2018) 9(1):2463. doi: 10.1038/s41467-018-04804-6
124. Jin X, Zhang W, Wang Y, Liu J, Hao F, Li Y, et al. Pyruvate Kinase M2 Promotes the Activation of Dendritic Cells by Enhancing IL-12p35 Expression. Cell Rep [Internet] (2020) 31(8):107690. doi: 10.1016/j.celrep.2020.107690
125. Petersone L, Edner NM, Ovcinnikovs V, Heuts F, Ross EM, Ntavli E, et al. T Cell/B Cell Collaboration and Autoimmunity: An Intimate Relationship. Front Immunol [Internet] (2018) 9:1941. doi: 10.3389/fimmu.2018.01941
126. Caro-Maldonado A, Wang R, Nichols AG, Kuraoka M, Milasta S, Sun LD, et al. Metabolic Reprogramming Is Required for Antibody Production That Is Suppressed in Anergic But Exaggerated in Chronically BAFF-Exposed B Cells. J Immunol [Internet] (2014) 192(8):3626–36. doi: 10.4049/jimmunol.1302062
127. Doughty CA, Bleiman BF, Wagner DJ, Dufort FJ, Mataraza JM, Roberts MF, et al. Antigen Receptor–Mediated Changes in Glucose Metabolism in B Lymphocytes: Role of Phosphatidylinositol 3-Kinase Signaling in the Glycolytic Control of Growth. Blood [Internet] (2006) 107(11):4458–65. doi: 10.1182/blood-2005-12-4788
128. Deng J, Lü S, Liu H, Liu B, Jiang C, Xu Q, et al. Homocysteine Activates B Cells via Regulating PKM2-Dependent Metabolic Reprogramming. J Immunol [Internet] (2017) 198(1):170–83. doi: 10.4049/jimmunol.1600613
129. Ratajczak MZ, Kucia M. SARS-Cov-2 Infection and Overactivation of Nlrp3 Inflammasome as a Trigger of Cytokine “Storm” and Risk Factor for Damage of Hematopoietic Stem Cells. Leukemia [Internet] (2020) 34(7):1726–9. doi: 10.1038/s41375-020-0887-9
130. Martinon F, Burns K, Tschopp J. The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of Proil-Beta. Mol Cell [Internet] (2002) 10(2):417–26. doi: 10.1016/s1097-2765(02)00599-3
131. Schroder K, Tschopp J. The Inflammasomes. Cell [Internet] (2010) 140(6):821–32. doi: 10.1016/j.cell.2010.01.040
132. Cerretti D, Kozlosky C, Mosley B, Nelson N, Van Ness K, Greenstreet T, et al. Molecular Cloning of the Interleukin-1 Beta Converting Enzyme. Sci (80-) [Internet] (1992) 256(5053):97–100. doi: 10.1126/science.1373520
133. Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, et al. A Novel Heterodimeric Cysteine Protease Is Required for Interleukin-1β Processing in Monocytes. Nat [Internet] (1992) 356(6372):768–74. doi: 10.1038/356768a0
134. Rathinam VAK, Vanaja SK, Fitzgerald KA. Regulation of Inflammasome Signaling. Nat Immunol [Internet] (2012) 13(4):333–42. doi: 10.1038/ni.2237
135. Latz E, Xiao TS, Stutz A. Activation and Regulation of the Inflammasomes. Nat Rev Immunol [Internet] (2013) 13(6):397–411. doi: 10.1038/nri3452
136. Rathinam VAK, Fitzgerald KA. Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell [Internet] (2016) 165(4):792–800. doi: 10.1016/j.cell.2016.03.046
137. Shi C-S, Shenderov K, Huang N-N, Kabat J, Abu-Asab M, Fitzgerald KA, et al. Activation of Autophagy by Inflammatory Signals Limits IL-1β Production by Targeting Ubiquitinated Inflammasomes for Destruction. Nat Immunol [Internet] (2012) 13(3):255–63. doi: 10.1038/ni.2215
138. Lu B, Nakamura T, Inouye K, Li J, Tang Y, Lundbäck P, et al. Novel Role of PKR in Inflammasome Activation and HMGB1 Release. Nat [Internet] (2012) 488(7413):670–4. doi: 10.1038/nature11290
139. Liu L, Sun B. Neutrophil Pyroptosis: New Perspectives on Sepsis. Cell Mol Life Sci [Internet] (2019) 76(11):2031–42. doi: 10.1007/s00018-019-03060-1
140. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by Inflammatory Caspases Determines Pyroptotic Cell Death. Nat [Internet] (2015) 526(7575):660–5. doi: 10.1038/nature15514
141. Shi J, Gao W, Shao F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem Sci [Internet] (2017) 42(4):245–54. doi: 10.1016/j.tibs.2016.10.004
142. Shen H-H, Yang Y-X, Meng X, Luo X-Y, Li X-M, Shuai Z-W, et al. NLRP3: A Promising Therapeutic Target for Autoimmune Diseases. Autoimmun Rev [Internet] (2018) 17(7):694–702. doi: 10.1016/j.autrev.2018.01.020
143. Song L, Pei L, Yao S, Wu Y, Shang Y. NLRP3 Inflammasome in Neurological Diseases, From Functions to Therapies. Front Cell Neurosci [Internet] (2017) 11:63. doi: 10.3389/fncel.2017.00063
144. Zhaolin Z, Guohua L, Shiyuan W, Zuo W. Role of Pyroptosis in Cardiovascular Disease. Cell Prolif [Internet] (2019) 52(2):e12563. doi: 10.1111/cpr.12563
145. Pezuk JA. Pyroptosis in Combinatorial Treatment to Improve Cancer Patients’ Outcome, Is That What We Want? EBioMedicine [Internet] (2019) 41:63. doi: 10.3389/fncel.2017.00063
146. Liu D, Xiao Y, Zhou B, Gao S, Li L, Zhao L, et al. PKM2-Dependent Glycolysis Promotes Skeletal Muscle Cell Pyroptosis by Activating the NLRP3 Inflammasome in Dermatomyositis/Polymyositis. Rheumatol [Internet] (2021) 60(5):2177–89. doi: 10.1093/rheumatology/keaa473/5961496
147. Le S, Zhang H, Huang X, Chen S, Wu J, Chen S, et al. PKM2 Activator TEPP-46 Attenuates Thoracic Aortic Aneurysm and Dissection by Inhibiting NLRP3 Inflammasome-Mediated IL-1β Secretion. J Cardiovasc Pharmacol Ther [Internet] (2020) 25(4):364–76. doi: 10.1177/1074248420919966
148. Samir P, Kesavardhana S, Patmore DM, Gingras S, Malireddi RKS, Karki R, et al. DDX3X Acts as a Live-or-Die Checkpoint in Stressed Cells by Regulating NLRP3 Inflammasome. Nat [Internet] (2019) 573(7775):590–4. doi: 10.1038/s41586-019-1551-2
149. Li Q, Leng K, Liu Y, Sun H, Gao J, Ren Q, et al. The Impact of Hyperglycaemia on PKM2-Mediated NLRP3 Inflammasome/Stress Granule Signalling in Macrophages and its Correlation With Plaque Vulnerability: An In Vivo and In Vitro Study. Metab [Internet] (2020) 107:154231. doi: 10.1016/j.metabol.2020.154231
150. Li J, Cao F, Yin H, Huang Z, Lin Z, Mao N, et al. Ferroptosis: Past, Present and Future. Cell Death Dis [Internet] (2020) 11(2):88. doi: 10.1038/s41419-020-2298-2
151. Cao JY, Dixon SJ. Mechanisms of Ferroptosis. Cell Mol Life Sci [Internet] (2016) 73(11–12):2195–209. doi: 10.1007/s00018-016-2194-1
152. Latunde-Dada GO. Ferroptosis: Role of Lipid Peroxidation, Iron and Ferritinophagy. Biochim Biophys Acta - Gen Subj [Internet] (2017) 1861(8):1893–900. doi: 10.1016/j.bbagen.2017.05.019
153. Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: Process and Function. Cell Death Differ [Internet] (2016) 23(3):369–79. doi: 10.1038/cdd.2015.158
154. Yang WS, Stockwell BR. Synthetic Lethal Screening Identifies Compounds Activating Iron-Dependent, Nonapoptotic Cell Death in Oncogenic-RAS-Harboring Cancer Cells. Chem Biol [Internet] (2008) 15(3):234–45. doi: 10.1016/j.chembiol.2008.02.010
155. Le Y, Zhang Z, Wang C, Lu D. Ferroptotic Cell Death: New Regulatory Mechanisms for Metabolic Diseases. Endocrine Metab Immune Disord - Drug Targets [Internet] (2020) 20:234–45. doi: 10.2174/1871530320666200731175328
156. Mou Y, Wang J, Wu J, He D, Zhang C, Duan C, et al. Ferroptosis, a New Form of Cell Death: Opportunities and Challenges in Cancer. J Hematol Oncol [Internet] (2019) 12(1):34. doi: 10.1186/s13045-019-0720-y
157. Sun Y, Chen P, Zhai B, Zhang M, Xiang Y, Fang J, et al. The Emerging Role of Ferroptosis in Inflammation. BioMed Pharmacother [Internet] (2020) 127:110108. doi: 10.1016/j.biopha.2020.110108
158. Kim EH, Wong S-W, Martinez J. Programmed Necrosis and Disease: We Interrupt Your Regular Programming to Bring You Necroinflammation. Cell Death Differ [Internet] (2019) 26(1):25–40. doi: 10.1038/s41418-018-0179-3
159. Martin-Sanchez D, Ruiz-Andres O, Poveda J, Carrasco S, Cannata-Ortiz P, Sanchez-Niño MD, et al. Ferroptosis, But Not Necroptosis, Is Important in Nephrotoxic Folic Acid–Induced AKI. J Am Soc Nephrol [Internet] (2017) 28(1):218–29. doi: 10.1681/ASN.2015121376
160. Proneth B, Conrad M. Ferroptosis and Necroinflammation, a Yet Poorly Explored Link. Cell Death Differ [Internet] (2019) 26(1):14–24. doi: 10.1038/s41418-018-0173-9
161. Takaoka Y, Konno M, Koseki J, Colvin H, Asai A, Tamari K, et al. Mitochondrial Pyruvate Carrier 1 Expression Controls Cancer Epithelial-Mesenchymal Transition and Radioresistance. Cancer Sci [Internet] (2019) 110(4):1331–9. doi: 10.1111/cas.13980
162. Cui J, Quan M, Xie D, Gao Y, Guha S, Fallon MB, et al. A Novel KDM5A/MPC-1 Signaling Pathway Promotes Pancreatic Cancer Progression via Redirecting Mitochondrial Pyruvate Metabolism. Oncogene [Internet] (2020) 39(5):1140–51. doi: 10.1038/s41388-019-1051-8
163. You JH, Lee J, Roh J-L. Mitochondrial Pyruvate Carrier 1 Regulates Ferroptosis in Drug-Tolerant Persister Head and Neck Cancer Cells via Epithelial-Mesenchymal Transition. Cancer Lett [Internet] (2021) 507:40–54. doi: 10.1016/j.canlet.2021.03.013
164. van Vuren AJ, van Beers EJ, van Wijk R. A Proposed Concept for Defective Mitophagy Leading to Late Stage Ineffective Erythropoiesis in Pyruvate Kinase Deficiency. Front Physiol [Internet] (2021) 11:609103. doi: 10.3389/fphys.2020.609103
165. Wang X, Lu S, He C, Wang C, Wang L, Piao M, et al. RSL3 Induced Autophagic Death in Glioma Cells via Causing Glycolysis Dysfunction. Biochem Biophys Res Commun [Internet] (2019) 518(3):590–7. doi: 10.1016/j.bbrc.2019.08.096
166. Kang R, Zeng L, Zhu S, Xie Y, Liu J, Wen Q, et al. Lipid Peroxidation Drives Gasdermin D-Mediated Pyroptosis in Lethal Polymicrobial Sepsis. Cell Host Microbe [Internet] (2018) 24(1):97–108. doi: 10.1016/j.chom.2018.05.009
167. Russo AJ, Rathinam VAK. Lipid Peroxidation Adds Fuel to Pyr(Optosis). Cell Host Microbe [Internet] (2018) 24(1):8–9. doi: 10.1016/j.chom.2018.06.010
168. Li X-N, Yang S-Q, Li M, Li X-S, Tian Q, Xiao F, et al. Formaldehyde Induces Ferroptosis in Hippocampal Neuronal Cells by Upregulation of the Warburg Effect. Toxicol [Internet] (2021) 448:152650. doi: 10.1016/j.tox.2020.152650
169. Dandekar A, Mendez R, Zhang K. Cross Talk Between ER Stress, Oxidative Stress, and Inflammation in Health and Disease. In: Oslowski C. editor. Stress Responses. Methods in Molecular Biology, vol 1292. New York, NY: Humana Press (2015). p. 205–14. doi: 10.1007/978-1-4939-2522-3_15
170. Senoner T, Dichtl W. Oxidative Stress in Cardiovascular Diseases: Still a Therapeutic Target? Nutrients [Internet] (2019) 11(9):2090. doi: 10.3390/nu11092090
171. Chen Y, Yang M, Huang W, Chen W, Zhao Y, Schulte ML, et al. Mitochondrial Metabolic Reprogramming by CD36 Signaling Drives Macrophage Inflammatory Responses. Circ Res [Internet] (2019) 125(12):1087–102. doi: 10.1161/CIRCRESAHA.119.315833
172. Borish LC, Steinke JW. Cytokines and Chemokines. J Allergy Clin Immunol [Internet] (2003) 111(2):S460–75. doi: 10.1067/mai.2003.108
173. Zhu R, Lei Y-Q, Zhao D-C. Overexpression of CXCL14 Alleviates Ventilator-Induced Lung Injury Through the Downregulation of PKM2-Mediated Cytokine Production. Mediators Inflammation (2020) 2020:1–9. doi: 10.1155/2020/7650978
174. Zhang YY, Liu X, Zhang X, Zhang J. Shikonin Improve Sepsis-Induced Lung Injury via Regulation of Mirna-140-5p/TLR4—A Vitro and Vivo Study. J Cell Biochem [Internet] (2020) 121(3):2103–17. doi: 10.1002/jcb.28199
175. Tsaknis G, Siempos II, Kopterides P, Maniatis NA, Magkou C, Kardara M, et al. Metformin Attenuates Ventilator-Induced Lung Injury. Crit Care [Internet] (2012) 16(4):R134. doi: 10.1186/cc11439
176. Tanaka S, Nishiumi S, Nishida M, Mizushina Y, Kobayashi K, Masuda A, et al. Vitamin K3 Attenuates Lipopolysaccharide-Induced Acute Lung Injury Through Inhibition of Nuclear Factor-κb Activation. Clin Exp Immunol [Internet] (2009) 160(2):283–92. doi: 10.1111/j.1365-2249.2009.04083.x
177. van de Wetering C, Aboushousha R, Manuel AM, Chia SB, Erickson C, MacPherson MB, et al. Pyruvate Kinase M2 Promotes Expression of Proinflammatory Mediators in House Dust Mite–Induced Allergic Airways Disease. J Immunol [Internet] (2020) 204(4):763–74. doi: 10.4049/jimmunol.1901086
178. Hu K, Yang Y, Lin L, Ai Q, Dai J, Fan K, et al. Caloric Restriction Mimetic 2-Deoxyglucose Alleviated Inflammatory Lung Injury via Suppressing Nuclear Pyruvate Kinase M2–Signal Transducer and Activator of Transcription 3 Pathway. Front Immunol [Internet] (2018) 9:426. doi: 10.3389/fimmu.2018.00426
179. Ďuračková Z. Some Current Insights Into Oxidative Stress. Physiol Res [Internet] (2010) 59(4):459–69. doi: 10.33549/physiolres.931844
180. Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative Stress, Inflammation, and Cancer: How Are They Linked? Free Radic Biol Med [Internet] (2010) 49(11):1603–16. doi: 10.1016/j.freeradbiomed.2010.09.006
181. Saleme B, Gurtu V, Zhang Y, Kinnaird A, Boukouris AE, Gopal K, et al. Tissue-Specific Regulation of P53 by PKM2 Is Redox Dependent and Provides a Therapeutic Target for Anthracycline-Induced Cardiotoxicity. Sci Transl Med [Internet] (2019) 11(478):eaau8866. doi: 10.1126/scitranslmed.aau8866
182. Zhou Q, Tang M, He L, Chen S. PKM2: A Crucial Neuroprotective Target Against Oxidative Stress. Acta Biochim Biophys Sin (Shanghai) [Internet] (2020) 52(12):1432–4. doi: 10.1093/abbs/gmaa121
183. Siragusa M, Thöle J, Bibli S, Luck B, Loot AE, Silva K, et al. Nitric Oxide Maintains Endothelial Redox Homeostasis Through PKM2 Inhibition. EMBO J [Internet] (2019) 38(17):e100938. doi: 10.15252/embj.2018100938
184. Liang J, Cao R, Wang X, Zhang Y, Wang P, Gao H, et al. Mitochondrial PKM2 Regulates Oxidative Stress-Induced Apoptosis by Stabilizing Bcl2. Cell Res [Internet] (2017) 27(3):329–51. doi: 10.1038/cr.2016.159
185. Li Q, Qi X, Jia W. 3,3′,5-Triiodothyroxine Inhibits Apoptosis and Oxidative Stress by the PKM2/PKM1 Ratio During Oxygen-Glucose Deprivation/Reperfusion AC16 and HCM-a Cells. Biochem Biophys Res Commun [Internet] (2016) 475(1):51–6. doi: 10.1016/j.bbrc.2016.05.030
186. Hejazian SM, Hosseiniyan Khatibi SM, Barzegari A, Pavon-Djavid G, Razi Soofiyani S, Hassannejhad S, et al. Nrf-2 as a Therapeutic Target in Acute Kidney Injury. Life Sci [Internet] (2021) 264:118581. doi: 10.1016/j.lfs.2020.118581
187. Wei Y, Lu M, Mei M, Wang H, Han Z, Chen M, et al. Pyridoxine Induces Glutathione Synthesis via PKM2-Mediated Nrf2 Transactivation and Confers Neuroprotection. Nat Commun [Internet] (2020) 11(1):941. doi: 10.1038/s41467-020-14788-x
188. Wei X, Jin X-H, Meng X-W, Hua J, Ji F-H, Wang L-N, et al. Platelet-Rich Plasma Improves Chronic Inflammatory Pain by Inhibiting PKM2-Mediated Aerobic Glycolysis in Astrocytes. Ann Transl Med [Internet] (2020) 8(21):1456–6. doi: 10.21037/atm-20-6502
189. Checker R, Sharma D, Sandur SK, Subrahmanyam G, Krishnan S, Poduval TB, et al. Plumbagin Inhibits Proliferative and Inflammatory Responses of T Cells Independent of ROS Generation But by Modulating Intracellular Thiols. J Cell Biochem [Internet] (2010) 110(5):1082–93. doi: 10.1002/jcb.22620
190. Checker R, Patwardhan RS, Sharma D, Menon J, Thoh M, Sandur SK, et al. Plumbagin, a Vitamin K3 Analogue, Abrogates Lipopolysaccharide-Induced Oxidative Stress, Inflammation and Endotoxic Shock via NF-κb Suppression. Inflammation [Internet] (2014) 37(2):542–54. doi: 10.1007/s10753-013-9768-y
191. Wang T, Wu F, Jin Z, Zhai Z, Wang Y, Tu B, et al. Plumbagin Inhibits LPS-Induced Inflammation Through the Inactivation of the Nuclear Factor-Kappa B and Mitogen Activated Protein Kinase Signaling Pathways in RAW 264.7 Cells. Food Chem Toxicol [Internet] (2014) 64:177–83. doi: 10.1016/j.fct.2013.11.027
192. Zhang Z, Deng W, Kang R, Xie M, Billiar T, Wang H, et al. Plumbagin Protects Mice From Lethal Sepsis by Modulating Immunometabolism Upstream of PKM2. Mol Med [Internet] (2016) 22(1):162–72. doi: 10.2119/molmed.2015.00250
193. Lu J, Yan J, Yan J, Zhang L, Chen M, Chen Q, et al. Network Pharmacology Based Research Into the Effect and Mechanism of Xijiao Dihuang Decoction Against Sepsis. BioMed Pharmacother [Internet] (2020) 122:109777. doi: 10.1016/j.biopha.2019.109777
194. Lu J, Zhang L, Cheng L, He S, Zhang Y, Yan J, et al. Xijiao Dihuang Decoction Improves Prognosis of Sepsis via Inhibition of Aerobic Glycolysis. BioMed Pharmacother [Internet] (2020) 129:110501. doi: 10.1016/j.biopha.2020.110501
195. Li X, Zhang J, Huang J, Ma A, Yang J, Li W, et al. A Multicenter, Randomized, Double-Blind, Parallel-Group, Placebo-Controlled Study of the Effects of Qili Qiangxin Capsules in Patients With Chronic Heart Failure. J Am Coll Cardiol [Internet] (2013) 62(12):1065–72. doi: 10.1016/j.jacc.2013.05.035
196. Lin S, Wu X, Tao L, Bei Y, Zhang H, Zhou Y, et al. The Metabolic Effects of Traditional Chinese Medication Qiliqiangxin on H9C2 Cardiomyocytes. Cell Physiol Biochem [Internet] (2015) 37(6):2246–56. doi: 10.1159/000438580
197. Tao L, Shen S, Fu S, Fang H, Wang X, Das S, et al. Traditional Chinese Medication Qiliqiangxin Attenuates Cardiac Remodeling After Acute Myocardial Infarction in Mice. Sci Rep [Internet] (2015) 5(1):8374. doi: 10.1038/srep08374
198. Zhang J, Wei C, Wang H, Tang S, Jia Z, Wang L, et al. Protective Effect of Qiliqiangxin Capsule on Energy Metabolism and Myocardial Mitochondria in Pressure Overload Heart Failure Rats. Evidence-Based Complement Altern Med [Internet] (2013) 2013:1–9. doi: 10.1155/2013/378298
199. Wang J, Zhou J, Ding X, Zhu L, Jiang K, Fu M, et al. Qiliqiangxin Improves Cardiac Function and Attenuates Cardiac Remodeling in Rats With Experimental Myocardial Infarction. Int J Clin Exp Pathol [Internet] (2015) 8(6):6596–606.
200. Ding H, Wang J, Zhang X-Y, Yin L, Feng T. Lycium Barbarum Polysaccharide Antagonizes LPS-Induced Inflammation by Altering the Glycolysis and Differentiation of Macrophages by Triggering the Degradation of PKM2. Biol Pharm Bull [Internet] (2021) 44(3):379–88. doi: 10.1248/bpb.b20-00752
201. Rohlenova K, Veys K, Miranda-Santos I, De Bock K, Carmeliet P. Endothelial Cell Metabolism in Health and Disease. Trends Cell Biol [Internet] (2018) 28(3):224–36. doi: 10.1016/j.tcb.2017.10.010
202. Pircher A, Treps L, Bodrug N, Carmeliet P. Endothelial Cell Metabolism: A Novel Player in Atherosclerosis? Basic Principles and Therapeutic Opportunities. Atheroscl [Internet] (2016) 253:247–57. doi: 10.1016/j.atherosclerosis.2016.08.011
203. De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, et al. Role of PFKFB3-Driven Glycolysis in Vessel Sprouting. Cell [Internet] (2013) 154(3):651–63. doi: 10.1016/j.cell.2013.06.037
204. Kim B, Jang C, Dharaneeswaran H, Li J, Bhide M, Yang S, et al. Endothelial Pyruvate Kinase M2 Maintains Vascular Integrity. J Clin Invest [Internet] (2018) 128(10):4543–56. doi: 10.1172/JCI120912
205. Stone OA, El-Brolosy M, Wilhelm K, Liu X, Romão AM, Grillo E, et al. Loss of Pyruvate Kinase M2 Limits Growth and Triggers Innate Immune Signaling in Endothelial Cells. Nat Commun [Internet] (2018) 9(1):4077. doi: 10.1038/s41467-018-06406-8
206. Zhang Y, Li L, Liu Y, Liu Z-R. PKM2 Released by Neutrophils at Wound Site Facilitates Early Wound Healing by Promoting Angiogenesis. Wound Repair Regener [Internet] (2016) 24(2):328–36. doi: 10.1111/wrr.12411
207. Chen D, Wei L, Liu Z-R, Yang JJ, Gu X, Wei ZZ, et al. Pyruvate Kinase M2 Increases Angiogenesis, Neurogenesis, and Functional Recovery Mediated by Upregulation of STAT3 and Focal Adhesion Kinase Activities After Ischemic Stroke in Adult Mice. Neurother [Internet] (2018) 15(3):770–84. doi: 10.1007/s13311-018-0635-2
208. Kodani A, Kikuchi T, Tohda C. Acteoside Improves Muscle Atrophy and Motor Function by Inducing New Myokine Secretion in Chronic Spinal Cord Injury. J Neurotrauma [Internet] (2019) 36(12):1935–48. doi: 10.1089/neu.2018.6000
209. Kikuchi T, Tohda C, Suyama M. Recovery of Motor Function of Chronic Spinal Cord Injury by Extracellular Pyruvate Kinase Isoform M2 and the Underlying Mechanism. Sci Rep [Internet] (2020) 10(1):19475. doi: 10.1038/s41598-020-76629-7
210. Zhou H-L, Zhang R, Anand P, Stomberski CT, Qian Z, Hausladen A, et al. Metabolic Reprogramming by the s-Nitroso-Coa Reductase System Protects Against Kidney Injury. Nat [Internet] (2019) 565(7737):96–100. doi: 10.1038/s41586-018-0749-z
211. Qi W, Keenan HA, Li Q, Ishikado A, Kannt A, Sadowski T, et al. Pyruvate Kinase M2 Activation may Protect Against the Progression of Diabetic Glomerular Pathology and Mitochondrial Dysfunction. Nat Med [Internet] (2017) 23(6):753–62. doi: 10.1038/nm.4328
212. Reinfeld BI, Madden MZ, Wolf MM, Chytil A, Bader JE, Patterson AR, et al. Cell-Programmed Nutrient Partitioning in the Tumour Microenvironment. Nat [Internet] (2021) 593(7858):282–8. doi: 10.1038/s41586-021-03442-1
213. Boxer MB, Jiang J, Vander Heiden MG, Shen M, Skoumbourdis AP, Southall N, et al. Evaluation of Substituted N, N ′-Diarylsulfonamides as Activators of the Tumor Cell Specific M2 Isoform of Pyruvate Kinase. J Med Chem [Internet] (2010) 53(3):1048–55. doi: 10.1021/jm901577g
Keywords: cytokine storm, pyruvate kinase M2, immunometabolic reprogramming, proinflammatory cytokines, inflammatory diseases
Citation: Liu Z, Le Y, Chen H, Zhu J and Lu D (2021) Role of PKM2-Mediated Immunometabolic Reprogramming on Development of Cytokine Storm. Front. Immunol. 12:748573. doi: 10.3389/fimmu.2021.748573
Received: 28 July 2021; Accepted: 11 October 2021;
Published: 25 October 2021.
Edited by:
Bernahrd Ryffel, Centre National de la Recherche Scientifique (CNRS), FranceReviewed by:
Bin Gong, University of Texas Medical Branch at Galveston, United StatesLuis Eduardo Alves Damasceno, University of São Paulo, Brazil
Copyright © 2021 Liu, Le, Chen, Zhu and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dezhao Lu, ludezhao@126.com
†These authors have contributed equally to this work and share first authorship