 Ryu Watanabe1†
Ryu Watanabe1† Gerald J. Berry
Gerald J. Berry Jörg J. Goronzy
Jörg J. Goronzy Cornelia M. Weyand
Cornelia M. Weyand- 1Department of Medicine, Stanford University School of Medicine, Stanford, CA, United States
- 2Department of Pathology, Stanford University School of Medicine, Stanford, CA, United States
Autoimmune and autoinflammatory diseases of the medium and large arteries, including the aorta, cause life-threatening complications due to vessel wall destruction but also by wall remodeling, such as the formation of wall-penetrating microvessels and lumen-stenosing neointima. The two most frequent large vessel vasculitides, giant cell arteritis (GCA) and Takayasu arteritis (TAK), are HLA-associated diseases, strongly suggestive for a critical role of T cells and antigen recognition in disease pathogenesis. Recent studies have revealed a growing spectrum of effector functions through which T cells participate in the immunopathology of GCA and TAK; causing the disease-specific patterning of pathology and clinical outcome. Core pathogenic features of disease-relevant T cells rely on the interaction with endothelial cells, dendritic cells and macrophages and lead to vessel wall invasion, formation of tissue-damaging granulomatous infiltrates and induction of the name-giving multinucleated giant cells. Besides antigen, pathogenic T cells encounter danger signals in their immediate microenvironment that they translate into disease-relevant effector functions. Decisive signaling pathways, such as the AKT pathway, the NOTCH pathway, and the JAK/STAT pathway modify antigen-induced T cell activation and emerge as promising therapeutic targets to halt disease progression and, eventually, reset the immune system to reestablish the immune privilege of the arterial wall.
Introduction
Arterial blood vessels are categorized according to their diameter into large, medium, and small vessels. Large vessels are the aorta and its major branches, medium vessels are the main visceral arteries and the arteries supplying blood to the brain, and small vessels are intraparenchymal arteries (1). Common elements are the lumen-lining endothelial cells (intimal layer) and the smooth muscle cells enabling dynamic change of diameter and resistance (medial layer). The outermost layer (adventitial layer) contains connective tissue, nerves, and vasa vasorum networks to supply nutrients and blood to the wall (2). Notably, each category of arteries is subject to different disease processes. Atherosclerotic disease, now recognized as a smoldering inflammatory process triggered by subendothelial lipid deposits that spans multiple decades of life, is a major cause of morbidity and mortality (3–5). In contrast, autoimmune inflammation of arteries is a more aggressive process, complicated by hemorrhage, vessel rupture and vessel occlusion. Vasculitides share immunopathologic features with other autoimmune diseases but have specifying immune abnormalities and clinical manifestations that are related to the life-sustaining role of arteries (1).
The two most frequent autoimmune diseases affecting the aorta and its branch vessels are giant cell arteritis (GCA) and Takayasu arteritis (TAK), two vasculitides manifesting with aortitis and wall inflammation in the carotid, subclavian, mesenteric and more peripheral arteries (6–8). In both diseases, CD4+ T cells and macrophages form granulomatous infiltrates in the vessel wall leading to wall vascularization, loss of medial smooth muscle cells, destruction of elastic membranous lamellae and elastin fibers in the medial layer and growth of lumen-stenosing neointima (6–8) (Figures 1, 2). Damage patterns are similar in GCA and TAK, but the individuals at risk are clearly distinct based on geographic distribution and age at disease onset. Recent advances in non-invasive imaging techniques have demonstrated that the blood vessels targeted by GCA and TAK overlap, giving rise to the ongoing debate whether the two diseases are separate or within the same disease spectrum (9–12). Molecular studies have emphasized that pathogenic events rely on cellular signal transduction pathways that are common in the two diseases, particularly when it comes to upstream pathologic effector functions of CD4+ T cells (13–17). However, significant differences mediating disease-relevant processes and more detailed analyses of participating immune cells have supported the proposition that disease-specific activation pathways are potential therapeutic targets (18–20). Here, we will review recent progress in understanding the particular contributions of T cells in disease pathogenesis, how they arrive in the tissue microenvironment of a blood vessel wall, how they function as signal-sending and signal-receiving cells and how their reliance on activating signaling pathways might be exploited therapeutically.
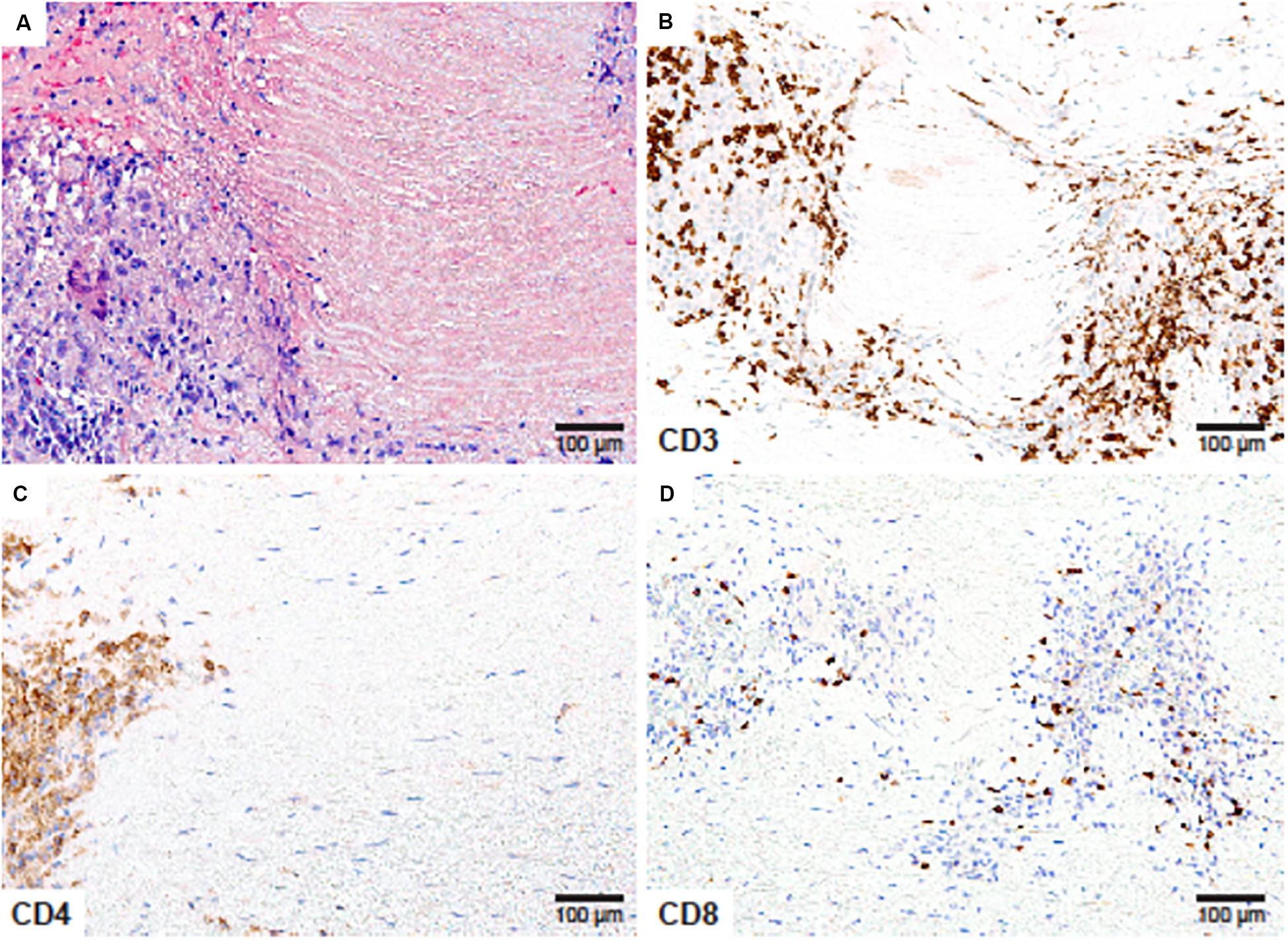
Figure 1. CD4+ and CD8+ T cells in Giant Cell Arteritis. Tissue sections from an ascending aortic aneurysm repair in a 66-year-old woman presenting with back pain in the mid-thoracic region. (A) High power magnification of the medial layer showing granulomatous inflammation surrounding depleted smooth muscle cells and necrotic elastic fibers (Hematoxylin and eosin ×200). (B) CD3 immunostaining showing numerous T cells within the medial infiltrates (×200). (C) CD4 immunostaining of T-helper cells at the edge of the granuloma (×200). (D) CD8 staining showing scarce T-cytotoxic cells within the T-cell rich inflammatory lesions (×200).
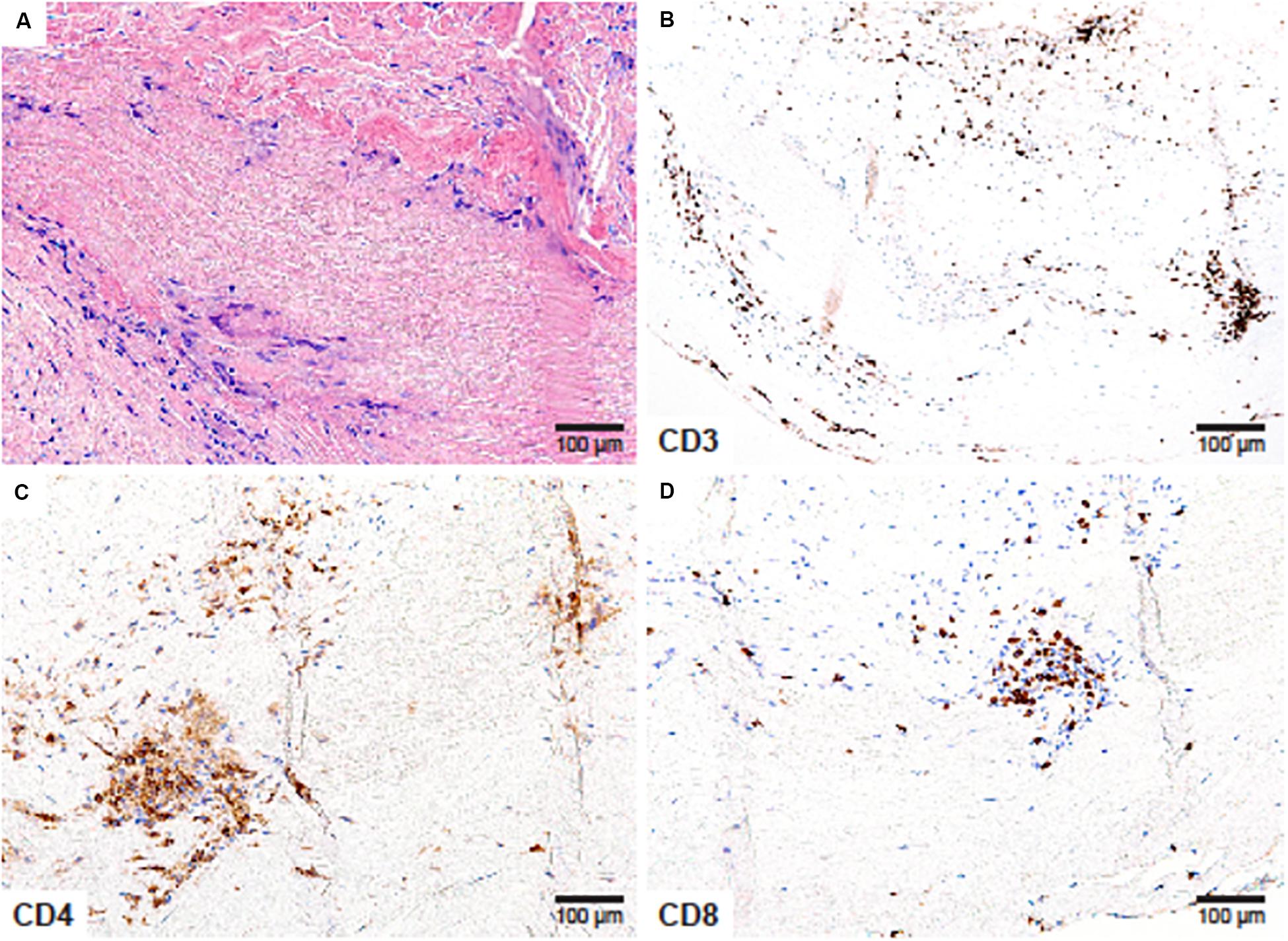
Figure 2. CD4+ and CD8+ T cells in Takayasu arteritis. Aneurysm of the aortic root and the aortic arch, complicated by aortic regurgitation in a 41-year-old female. (A) Necrotizing granulomatous inflammation composed of lymphocytes and multinucleated giant cells surrounding a zone of necrotic medial tissue (H&E ×200); (B) CD3+ T cells create an inflammatory collar around the zone of necrosis (×200); (C) CD4+ T-helper cells (×200) and (D) CD8+ T-cytotoxic cells (×200) comprise the majority of tissue-residing T-cells.
Giant Cell Arteritis
Earlier studies gave rise to the notion that T cells are clonally expanded within the vascular lesions and that human leukocyte antigen (HLA)-DRB1 alleles are genetic risk factors for GCA (21–24). More recent genetic analyses have confirmed the strong correlation between HLA class I and II molecules and susceptibility to GCA (25, 26). These data provide compelling support to the concept that GCA is a disease in which CD4+ T cells react to antigen presented within polymorphic HLA molecules and that antigen recognition and expansion of CD4+ T cells are critical disease mechanisms. Here we review different aspects of T cell biology in GCA, with the intent to utilize that knowledge to design more effective therapeutic interventions.
Effector CD4+ T Cells
Multiple studies have shown that Th1 cells that produce interferon (IFN)-γ and Th17 cells that produce IL-17 participate in the vasculitic infiltrates (27–31). An expansion of Th1 and Th17 cells has also been reported for peripheral blood of GCA patients (27, 32). Interferons (IFNs) have a key role in antiviral immunity. IFN-γ, the sole type II IFN, has weaker antiviral effects than type I IFNs, such as IFN-α and IFN-β, but is a potent regulator of various cell types such as endothelial cells, stromal cells, dendritic cells and macrophages (33). IFN-γ vigorously increases major histocompatibility complex expression, increases antigen presentation and amplifies chemokine production, while suppressing cell proliferation (33). IFN-γ is the prototypic macrophage-activating factor that promotes cytokine and chemokine production, phagocytosis, and intracellular killing of microbial pathogens. By releasing IFN-γ, vasculitogenic T cells can effectively activate macrophages and direct their multiple effector functions. In GCA-affected arteries, stimulated macrophages release vascular endothelial growth factor (VEGF), thus fostering neoangiogenesis (34). A spectrum of macrophage functions depends directly on activating signals deriving from IFN-γ-producing CD4+ T cells, assigning a key role to these T helper cells in granulomatous vasculitis.
The T cell cytokine IL-17 serves a complimentary role in the disease process. Th17 cells utilize the master transcription factor RAR-related orphan receptor gamma (RORγt) and require IL-23 for lineage differentiation and commitment (35). Besides the classical IFN-γ-supplying Th1 lineage, the so-called “IL-23-IL-17” axis makes critical contributions to autoimmune disease (36). IL-17A has been implicated in barrier and surface protection, functions in neutrophil recruitment and contributes to tissue repair. The “IL-23-IL-17” axis appears to be particularly important in psoriasis and spondyloarthritis (37–39). How IL-17 affects the disease process in GCA is not entirely understood.
Another T cell effector cytokine linked to granulomatous vasculitis is IL-21 (32). IL-21 is predominantly produced by follicular helper T (Tfh) and also by Th17 cells. IL-21 balances helper T cell subsets and induces B cell generation and differentiation into plasma cells, thus enhancing the production of immunoglobulins (40, 41).
IL-9-producing Th9 cells are also enriched in GCA lesions (42). IL-9 is believed to be involved in type 2 inflammation, induces activation of T helper cells and affects the function of various tissue resident cells such as mast cells and epithelial cells in the mucosa. T helper cells make a commitment to the Th9 lineage when stimulated with transforming growth factor β and IL-4 (43). IL-4 is distinctly low in GCA lesions and it is unclear whether Th9 cells are recruited as precursors or fully differentiated. Also, the precise role of IL-9 in GCA lesion remains unknown.
Increased expression of IL-22 in temporal artery biopsies from GCA patients has been reported (44). IL-22 is produced by both innate and adaptive immune cells, including innate lymphoid cells, and natural killer (NK) cells as well as T lymphocytes (Th17 and Th22) (45). IL-22 acts synergistically with TNF-α, IL-1β, and IL-17, and overall has pro-inflammatory effects (46). IL-22 is considered as potential therapeutic target in several autoimmune disease (47–49), but its precise role and drug-ability in GCA requires further investigation.
CD4+ Regulatory T Cells
Regulatory T (Treg) cells, characterized by the expression of the lineage-determining transcription factor FOXP3, have a critical role in the maintenance of immune homeostasis and prevention of autoimmunity (50, 51). Patients with GCA are believed to have insufficient CD4+ Treg cell function, eventually resulting in a peripheral tolerance defect (32). Frequencies of CD4+FOXP3+CD25highCD127–Tregs have been reported to be around 3% in GCA patients, as compared to 4–5% in age-matched controls (32). How this reduction in CD4+ Treg cells leads to vasculitis is unresolved. Additional evidence for insufficient Treg cell function came from studies describing enrichment of dysfunctional Treg cells in active GCA patients (52). Specifically, such patients had higher frequencies of IL-17-secreting Tregs, characterized by the expression of an hypofunctional isoform of FOXP3 that lacks exon 2 (52). It has been proposed that tocilizumab, an antibody blocking the IL-6 receptor, may be able to improve the function of Tregs (52). Mechanistic studies, measuring recruitment, stability and suppressive functions of bona fide CD4+ Treg cells in the vasculitic lesions are needed to get a better understanding of FOXP3+CD4+ T cells in GCA.
CD8+ T Cells in GCA
While there is agreement that CD4+ T cells are key drivers of both GCA and TAK, CD8+ T cells appear to play a disease-specific role. The contribution of effector CD8+ T cells to the pathogenesis of GCA is considered to be minor, based on the low number of CD8+ T cells in GCA-affected arteries and a marked decrease of circulating CD8+ T cells in active GCA patients (53, 54). CD8+ T cells in GCA patients have been described to be clonally expanded and to use a restricted T cell repertoire (55, 56). Through which mechanisms effector CD8+ T cells may have an impact on pathogenic events in GCA is unresolved. Recent immunohistochemical analyses have confirmed that overall CD8+ T cells were lower in GCA-affected vascular lesions compared to Takayasu’s arteritis (57).
One specialized CD8+ T cell subset, CD8+FOXP3+ Treg cells, have been assigned a critical role in the breakdown of the vessel wall immune privilege in GCA (58). Like CD4+ Tregs, CD8+CD45RA+CCR7+FOXP3+ regulatory T cells have immunosuppressive potential and can be induced ex vivo from naïve CD8+ T cells by low-affinity T cell receptor signaling combined with IL-15 (59). CD8+ Treg cells localize to secondary lymphoid organs in young, healthy individuals, and suppress effector CD4+ T cells by inhibiting phosphorylation of ZAP-70, a proximal adaptor molecule in the T cell receptor activation cascade (Figure 3) (58). However, in older individuals and in patients with GCA, CD8+ Treg cells are low in numbers and diminished in function. CD8+ Treg cells function by releasing NADHP oxidase 2 (NOX2)-containing exosomes, that transfer reactive oxygen species (ROS) into recipient CD4+ T cells. Inability to secrete NOX-2-containing exosomes has been identified as the underlying defect of CD8+ Treg cells in the old and in the GCA patient (58). Therapeutic targeting of CD8+ Tregs, such as increasing functional CD8+ Treg numbers or restoring NOX2 production in CD8+ Tregs, may control not only GCA but also age-related inflammation or “inflammaging” (58, 60–62).
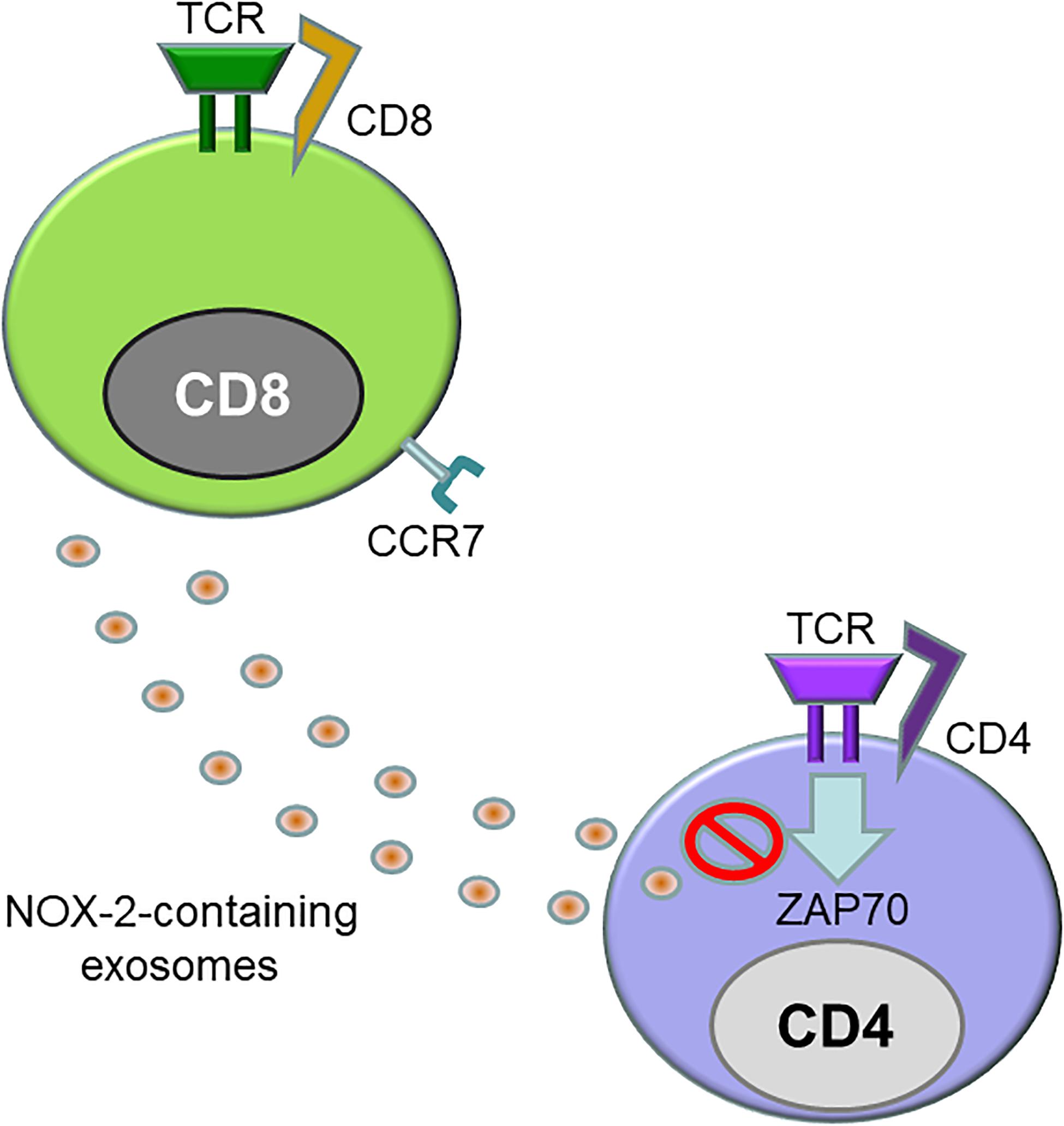
Figure 3. Defective CD8+ regulatory T cells in giant cell arteritis. Like CD4+ regulatory T cells, CD8+ regulatory T cells (CD8+ Treg) express the transcription factor FOXP3 and act as a suppressor of immune responses. CD8+ CCR7+ Tregs inhibit immunity by releasing exosomes that contain the enzyme NADPH oxidase 2 (NOX2). These exosomes are integrated into the membrane of neighboring CD4+ T cells, where they disrupt proximal signaling events, including the phosphorylation of ZAP70. CD8+ Tregs from patients with GCA are reduced in number and are functionally defective.
T Cell-Macrophage Interactions
Macrophages are immune cells of hematopoietic origin that provide fast immune defense (63). They are equipped to sense and respond to danger signals, usually released from dead and dying cells attacked by infectious microorganisms or other noxious stimuli (64, 65). In GCA, macrophages are unequivocal disease drivers and, together with CD4+ T cells, form the pathognomonic granulomatous lesions. They not only produce cytokines (IL-1β, IL-6, and TNF-α) and chemokines (CXCL9, 10, 11, CCL5, and CCL 18) but also contribute to phagocytosis and antigen presentation, and provide co-stimulatory ligands regulating in situ T cell activation and survival (66). In the vasculitic lesions, they differentiate into tissue-destructive effector cells by releasing collagenases and matrix metalloproteases (MMP-2, 7, and 9) (67–69). Notably, MMP-9 is almost exclusively produced by CD68+ macrophages and controls T cell entry into the vessel wall by digesting the structural integrity of the external basement membrane (68). Blocking MMP-9 efficiently suppressed T cell infiltration into the artery and abrogated the remodeling of the vessel wall, including neointima formation and adventitial neovascularization (68). Essentially, CD4+ T cell require MMP-9-releasing macrophages to enter the immune-privileged tissue site and cause vascular inflammation.
While patient-derived macrophages in GCA patients have many features of pro-inflammatory effector cells, their metabolic signature is surprisingly insipid. Expressions of glucose transporters, glycolytic enzymes and transcription factors regulating glycolysis have been described to be indistinguishable from macrophages generated from healthy individuals (66), with healthy and GCA macrophages utilizing glucose as their main substrate. As GCA macrophages enter the tissue microenvironment, they may have access to additional non-glucose energy sources, supporting longevity in the tissue niche. By providing fuel sources adapted to the needs of tissue-invading monocytes and macrophages, the tissue microenvironment attacked by vasculitic immune responses may make a critical contribution to disease pathogenesis.
T Cell-Dendritic Cell Interactions
Dendritic cells (DCs) are defined as cells with a stellate morphology that can efficiently present antigens on MHC molecules and activate naïve T cells (70). DCs initiate and shape both innate and adaptive immune responses. Considering the reactivity to autoantigen, DCs have long been considered important players in the loss of tolerance leading to autoimmunity, but in recent years their quintessential role in anti-tumor immunity has also been recognized (71, 72). Three-layered human arteries contain a population of DCs, so-called vascular DCs, localized at the adventitia-media border, where they can interact with T cells entering the vessel wall from the adventitial vasa vasorum (73). Vascular DCs sense danger-associated molecular patterns released by pathogens and Toll-like receptor (TLR)-stimulated vascular DCs break self-tolerance and induce T- cell recruitment and activation via co-stimulatory molecule expressions and chemokine release (74, 75). Notably, TLR profiles expressed by vascular DCs are unique for each vascular territory (76).
In addition to co-stimulatory molecules, co-inhibitory molecules are expressed on DCs (77), enabling the DCs to control the initiation, duration and robustness of an immune response. Interactions between programmed cell death protein-1 (PD-1) on activated T cells and its ligand (PD-L1) on DCs have attracted much attention recently since blockade of this axis has anticancer potential (78–81). Our recent work has identified a deficiency of PD-L1 expression on vascular DCs in GCA (82). PD-L1 expression on GCA monocyte-derived DCs was diminished compared to healthy monocyte-derived DCs, even under optimal stimulatory conditions with IFN-γ and LPS. This deficiency leads to overactivation of CD4+ T cells, and production of IFN-γ, IL-17, and IL-21 (82). Using a large-vessel vasculitis model, we showed that PD-L1 blockade exacerbates vascular inflammation, promoting infiltration of activated T cells into the arterial wall and wall remodeling with neointima formation and adventitial neovascularization (82). These data provide unequivocal evidence that activated T cells play a central role in vascular remodeling and that anti-tumor therapy with checkpoint inhibitors blocking the PD-1-PD-L1 axis threatens the immune protection of the aorta and its major branches (83). In support of these data, case reports and observational studies have confirmed that patients treated with checkpoint inhibitors are at risk for therapy-induced vasculitis (84, 85). Autoimmunity and tumor immunity emerge as two sides of the same coin. If PD-L1 expression is reduced on DCs, risk for autoimmunity is high, but anti-tumor immunity is effective. In fact, it has been reported that the frequency of malignancy is relatively high in rheumatoid arthritis, Sjogren’s syndrome, inflammatory myositis (86–89); however, the overall risk for cancer in GCA is not increased compared to healthy individuals (90). Understanding why GCA patients have a defect in PD-L1 expression should yield important insights. PD-L1 expression is dependent on glucose uptake and intracellular glycolytic activity (66, 91, 92). Large epidemiological studies demonstrate that body mass index and fasting blood glucose levels were negatively associated with the development of GCA (93, 94), supporting the concept that low glycolytic activity promotes low PD-L1 expression, enabling uncontrolled autoimmunity.
Additional co-inhibitory pathways appear to also be less functional in GCA patients. V-domain immunoglobulin-containing suppressor of T cell activation (VISTA) has been identified as a novel inhibitory receptor expressed on myeloid cells and T cells (95). Expression of VISTA on CD4+ T cell from GCA patients has been reported to be decreased, thereby facilitating T cell differentiation toward Th1, Th17, and Tfh (96). Expression of V-set and immunoglobulin domain containing 3 (VSIG-3), a ligand of VISTA (97), has not been explored in GCA lesions.
Signal Transduction Pathways in GCA CD4+ T Cells
CD28-AKT-mTORC1 Axis
Activation of T cells requires at least two signals; one delivered by the T cell receptor complex and one provided by engagement of co-stimulatory receptors, such as CD28 (98, 99). T cell activation initiates a metabolic program required for cell growth, proliferation and differentiation (100). The demand for glucose uptake and glycolysis during T cell activation is known as the Warburg effect (101, 102). CD28 co-stimulation signals the activation of the phosphatidylinositol 3-kinase (PI3K)-AKT axis and maximizes glycolytic flux (103, 104). The mechanistic target of rapamycin (mTOR) is a serine/threonine protein kinase and forms part of mTOR complex 1 (mTORC1) (105). mTORC1 is targeted by the PI3K-AKT axis, and integrates a variety of environmental cues to regulate cell growth and tissue repair (106). mTORC1 promotes glycolysis through upregulation of the hypoxia inducible factor (HIF) 1α (105). mTORC1 is recognized as a signaling hub in multiple pathological conditions, such as cancer, obesity, neurodegeneration and the aging process (107). The PI3K-AKT-mTORC1 pathway is frequently overactivated in various human cancers (108). Sustained activation of mTORC1 is a signature abnormality of CD4+ T cells in GCA (Figure 4). In the humanized mouse model of GCA, blockade of CD28 co-stimulation by anti-CD28 antibody was highly effective in dampening vascular inflammation. This therapeutic effect depended on inhibiting mTORC1 activity and constraining glycolytic flux in CD4+ T cells (109). Blocking CD28 signaling and preventing mTORC1 activation curtailed mitochondrial respiration and, subsequently, cytokine production (109). Thus, the CD28-AKT-mTORC1 pathway is essential for vasculitic activity and emerges as a promising therapeutic target (13, 15, 109).
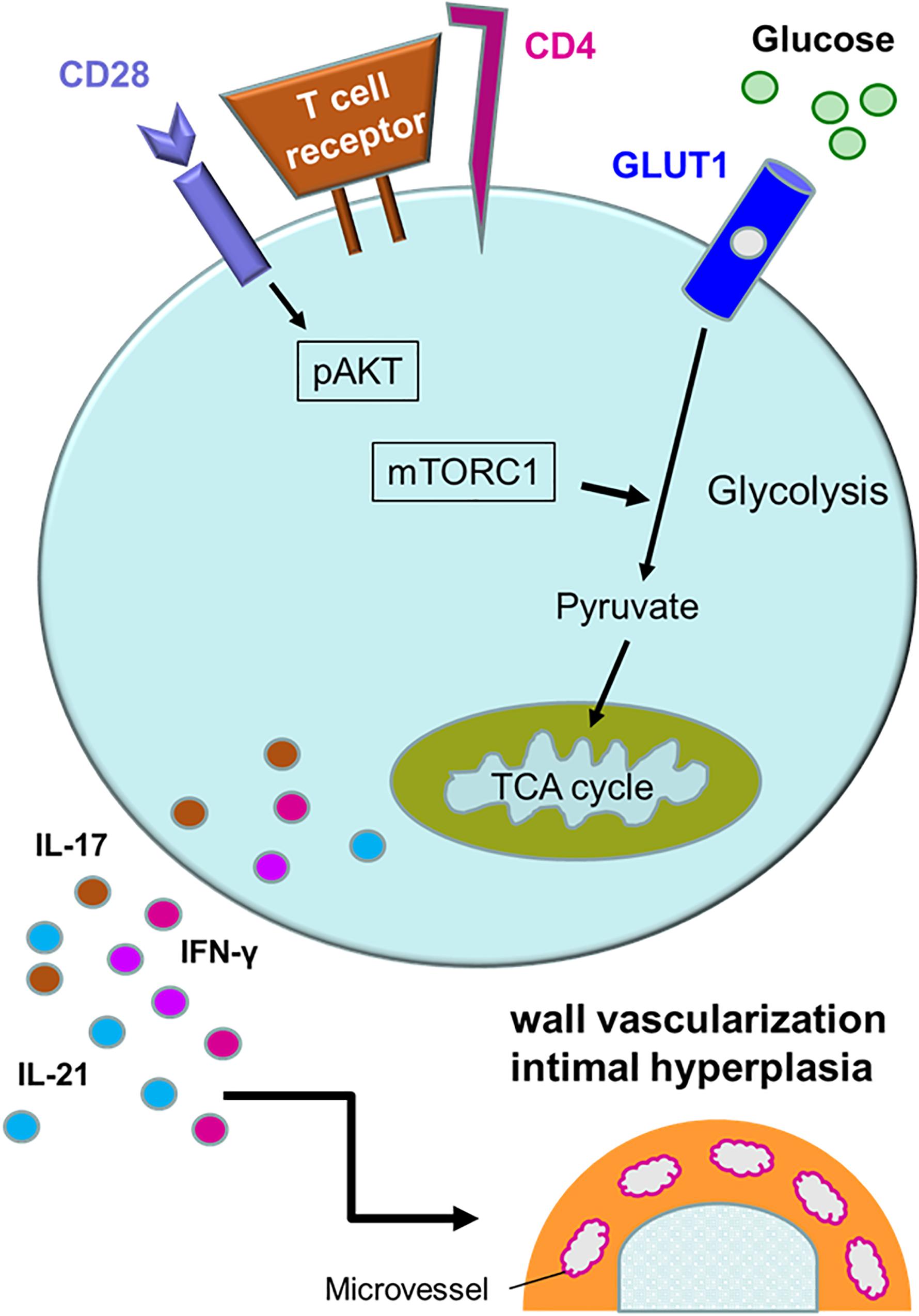
Figure 4. Activation pathways in pathogenic CD4+ T cells. Multiple signaling pathways contribute to the activation of pathogenic CD4+ T cells in giant cell arteritis. Antigen recognition by the T cell receptor triggers rapid division, differentiation and lineage commitment. CD28 receptor engagement co-stimulates, mediated through AKT phosphorylation and downstream activation of the mechanistic target of rapamycin complex 1 (mTORC1). Additional stimuli derive from metabolic signals. Glucose uptake is controlled through the expression of the glucose transporter 1 (GLUT1). Glycolytic breakdown generates pyruvate, which functions as a critical energy carrier for mitochondria, sustaining ATP production and the release of metabolic intermediates. Antigen-stimulated, metabolically active CD4+ T cells differentiate into effector cells secreting IFN-γ, IL-17, and IL-21 and sustain a vessel wall remodeling program resulting in wall vascularization and lumen-occlusive intimal hyperplasia.
NOTCH-mTORC1 Axis
“Classical” mTOR inputs are growth factors, nutrients and cellular energy, whereas “non-classical” mTOR inputs include WNT signaling and NOTCH signaling (110). NOTCH signaling primarily regulates cell proliferation, differentiation and cell fate decisions (111). Binding of cell-surface-bound ligands (Delta and JAGGED) to NOTCH receptors on neighboring cells initiates a biochemical cascade that results in of cleavages of the NOTCH receptor. The NOTCH intracellular domain translocates into the nucleus and acts as a transcriptional co-activator that promotes gene expression (110, 111). Aberrant expression of the NOTCH1 receptor is a signature abnormality in CD4+ T cells of GCA patients (15). Transcriptomic analysis of biopsy material form GCA patients led to the discovery that the NOTCH1 ligand, JAGGED1, is expressed on microvascular endothelial cells, specifically on the vasa vasorum. VEGF, circulating in high amounts in the blood of GCA patients, was identified as an inducer of JAGGED1 on the endothelial cells (15). Activation of the NOTCH1 pathway resulted in the elevation of the HES1 protein, a potent activator of gene transcription (Figure 5) (15). Taken together, not only the CD28-PI3K-AKT axis but also the JAGGED1-NOTCH1 pathway contribute to high mTORC1 activity, leading to Th1 and Th17 differentiation and to equipping lesional T cells with functionality of disease orchestrators. Recognizing the NOTCH pathway as a driving force in medium and large vessel vasculitis will provide new opportunities for immunomodulation (112).
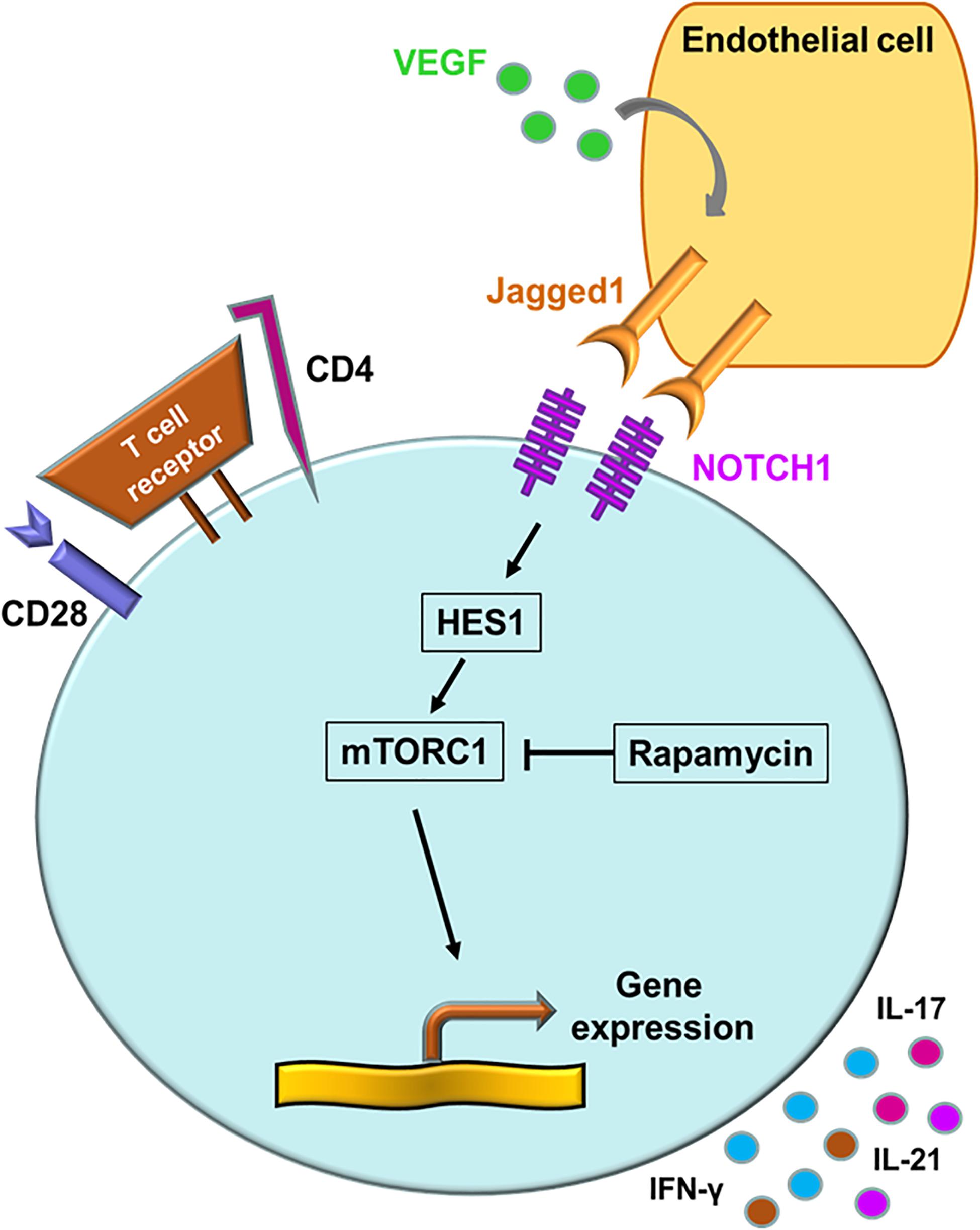
Figure 5. The NOTCH signaling pathway in giant cell arteritis. A signature abnormality of CD4+ T cells in GCA is the aberrant expression of the NOTCH1 receptor. NOTCH1+ CD4+ T cells engage the JAGGED1 ligand on the surface of microvascular endothelial cells. Vascular endothelial growth factor (VEGF) in the circulation functions as the JAGGED1 inducer. Via HES1, the NOTCH signaling pathway activates the mechanistic target of rapamycin complex 1 (mTORC1). Persistent NOTCH signaling promotes T cell proliferation, invasion of CD4+ T cells into the vessel wall, upregulation of the metabolic program and differentiation into cytokine-producing effector cells.
JAK-STAT Pathway
Cytokines represent soluble factors with essential roles in immune response and employ diverse intracellular pathways. A subset of cytokines that bind type I and type II cytokine receptors utilizes the Janus kinase-signal transducer of activators of transcription (JAK-STAT) pathway (113, 114). They include IL-6, common, chain cytokines (IL-2, 4, 7, 9, 13, and 15), granulocyte colony-stimulating factor (G-CSF), granulocyte macrophage colony-stimulating factor (GM-CSF), IL-10, IFN-α, IFN-β, and IFN-γ (115). Since genome-wide association studies have revealed this pathway as highly relevant to human autoimmunity, pharmacological inhibitors of JAK or JAKinibs have created a new paradigm for the treatment of rheumatic disease (115). In the autoimmune disease rheumatoid arthritis, JAK inhibitors are as effective as biological disease-modifying antirheumatic disease (116, 117), indicating that key disease pathways rely on JAK-STAT signaling. JAK-STAT signaling appears to be exceedingly relevant in vasculitis as well, as recent work in GCA-affected tissue lesions and in patients’ T cells suggests (Figure 6) (16). In human artery-SCID chimera mice, tofacitinib, an inhibitor that primarily targets JAK3 and JAK1, efficiently suppressed vasculitis by restraining the activation of T cells and macrophages (16). Analysis of T cells in the vasculitic infiltrates identified a population of highly proliferative tissue-resident memory T cells, that was dependent on JAK-STAT signaling and sensitive to tofacitinib-mediated inhibition. Such tissue-resident T cells are now emerging as the source of self-renewing T cells, that maintain the granulomatous formations in the vessel wall. In a clinical study, re-biopsy of patients after 3, 6, 9, and 12 months of corticosteroid therapy demonstrated that the vascular inflammatory lesions were unexpectedly stable and tissue-occupying T cells persisted in the majority of patients for more than 1 year (118). These “tissue-resident memory T cells” are now recognizes as a specialized T cell subset, that can adapt to the tissue environment and retain the capacity to locally replenish the infiltrate. These T cells express CD103 and CD69 as a phenotypic surface marker (119–121). In a trans-engraftment model, in which human arteries with vasculitis lesions are engrafted into “empty” immunodeficient mice, the survival strategy of human T cells can be analyzed. In these trans-engrafted arteries, tissue-resident memory T cells expand autonomously and their expansion is druggable with JAKinib (16). Tofacitinib was sufficient to diminish cell proliferation and cytokine production from activated CD4+ T cells (16), demonstrating that JAK-STAT signaling contributes to disease-relevant processes in GCA.
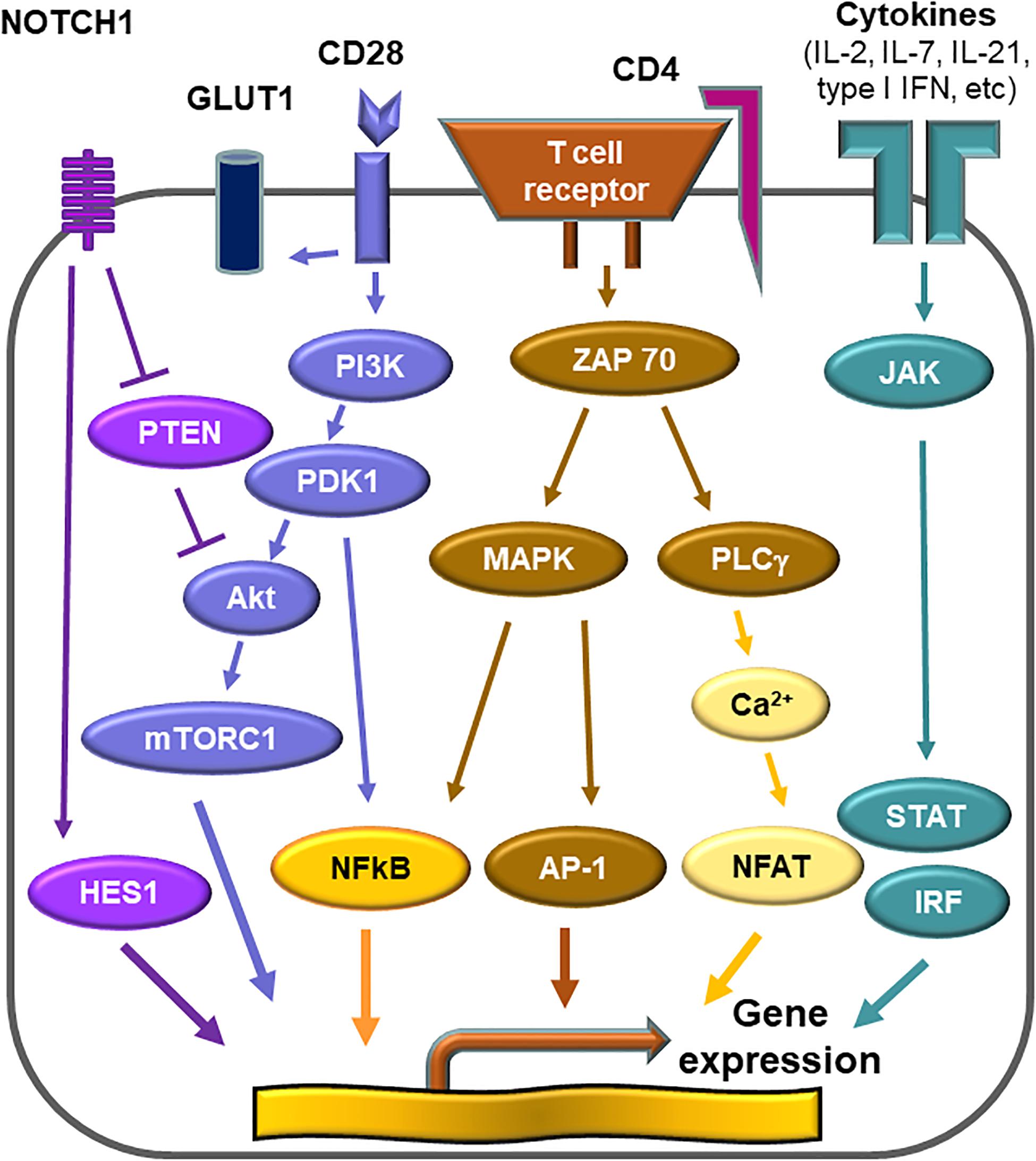
Figure 6. Druggable signaling cascades in vasculitogenic T cells. Numerous signals converge to shape the activation patterns of T cells. In the case of vasculitis-inducing T cells, vasculitogenic antigens trigger the T cell receptor activation cascade. Several co-stimulatory signals adjust signal strength and duration and thus determine differentiation, effector functions and longevity of the T cell. CD28-dependent signaling regulates the metabolic program of vasculitogenic T cells. CD28-mediated signals activate mTORC1, thus determining T cell proliferation and lineage assignment. Persistent NOTCH signaling is a signature abnormality of vasculitogenic CD4+ T cells and regulates tissue invasiveness. Cytokines modulating T cell function utilize the JAK-STAT signaling pathway. Disease-relevant cytokine signals derive from IL-2, IL-7, and, possibly, from type 1 interferon.
Type I IFN Signature
Interferons (IFN) are cytokines that have antiviral, antiproliferative, and immunomodulatory effects (122). Inborn errors or impaired function of IFN-mediated immunity confer predisposition to viral and mycobacterial infection (123). There are two main classes of IFN: IFN-γ is the only type II IFN, and type I IFNs include IFN-α, IFN-β, and others that bind a common cell-surface receptor (122). Type I IFNs activate the JAK-STAT signaling pathway to induce expression of interferon-stimulated genes, called “interferon signature” (Figure 6) (33). Interferons also induce an “interferon epigenomic signature” by activating latent enhancers and chromatin (33). The type I IFN signature is upregulated in several autoimmune diseases, identifying this cellular activation pathway as a prime candidate for immunosuppressive therapy (124, 125). Transcriptome analysis of biopsy material from patients with GCA has demonstrated robust induction of type I and II IFN signatures (16), indicating that both type I and type II IFNs are abundant within the vasculitic lesions. The source of type I IFNs in GCA has not been defined, providing an opportunity to identify key cellular drivers. In the autoimmune disease systemic lupus erythematosus, plasmacytoid DCs are the main source of type I IFN (126, 127). There is no evidence, to date, that plasmacytoid DC are present in the inflamed vessel wall. The effect that IFNs have on T cells is complex, involving direct and indirect interference in T cell functionality (128). Type I IFN receptors are broadly expressed in many tissues and by many cell types, both innate as well as adaptive immune cells are regulated by this cytokine family. Receptors for type II IFN are mostly encountered on granulocytes, monocytes and macrophages, Type II IFN is a product mainly of T cells and NK cells, placing the T cell-monocyte/macrophage axis at center stage in GCA.
Takayasu’s Arteritis
Unlike GCA, that has the highest incidence in elderly women of Northern European descent (8, 129), Takayasu’s arteritis (TAK) is more prevalent in young Asian women (130–132). HLA-B52 is recognized as a susceptibility locus in TAK (20, 133–135). A subset of TAK patients have pre-existent or co-existent inflammatory bowel disease, connecting this vasculitis to systemic immune abnormalities (136, 137). HLA-B52 is detected in a high rate of patients with TAK complicated with ulcerative colitis (138). The vascular lesions of TAK resemble those encountered in GCA and are composed of highly activated T cells and macrophages, arranged in granulomatous formations (6). In the past decade, TAK and GCA have often been considered to represent a spectrum of one disease, supported by similarities in target blood vessel patterning and histopathologic findings (9–12). However, genome-wide associated studies and immunophenotyping of immune cells in the peripheral blood and in the vascular lesions have revealed substantial differences (18–20, 57). Particularly, immune cells other than CD4+ T cells and macrophages have shown to be critical in the pathogenesis of TAK (19, 20, 139, 140). Here, we review the current knowledge implicating CD4+ T cells, macrophages, CD8+ T cells, natural killer (NK) cells as critical players in the disease process leading to TAK. New data demonstrating the production of autoantibodies against endothelial cells in TAK patients shed new light on pathogenic adaptive immunity (141). Special emphasis has been placed on cellular signaling pathways that are active in TAK and GCA, with the goal to define common and disease-specific pathogenic mechanisms.
CD4+ Effector T Cell
Giant cell arteritis and Takayasu arteritis share the predominance of Th1 cells and the participation of Th17 cells in the inflammatory process (141). IFN-γ and IL-17 are markedly increased in the peripheral blood and in the affected aortic tissue (141). The entire signaling pathways mediating the induction of Th1 cells appears to be upregulated. IL-12B is well established as a susceptibility gene in TAK (134), and plasma IL-12 levels are elevated (142), biasing T cell differentiation toward the Th1 lineage (143). IL-23 is also increased in the serum of patients with TAK (141), and IL-23 promotes IL-17 production by CD4+ T cells (144). In essence, the cytokine environment enables the emergence of Th1 and Th17 cells, promoting immune responses associated with granulomatous inflammation.
Differential responses to glucocorticoid therapy have been reported for GCA and TAK patients. In GCA, Th17 cells are susceptible, while Th1 cells are resistant to glucocorticoid therapy; whereas steroid treatment is able to suppress Th1 cytokines, but left Th17 cytokines unaffected in TAK (27, 141). The reason for this strikingly different responsiveness remains unclear, but these data strongly support the hypothesis that the cytokine environment may ultimately be very different in these two disorders.
A shared feature of CD4+ T cells in both GCA and TAK is the strong upregulation of mTORC1 activity (13, 14). mTOR is a signaling hub in T cell fate decisions and mTORC1 activation biases T cell differentiation toward the Th1 and Th17 lineage. Strong evidence for mTORC1’s role as a key decision maker comes from studies using rapamycin to guide T cell differentiation. The mTORC1 blocker was able to prevent differentiation into Th1 and Th17 cells (13, 14). The cause of persistent mTORC1 activation in GCA and TAK T cells is not understood but may be an independent factor in directing disease-relevant immunity. Upregulation of the mTORC1 pathway has also been reported for endothelial cells and vascular smooth muscle cells in the aorta (13, 145), suggesting that the mTORC1 pathway is universally activated in TAK.
In a recent study, a group of French scientists explored whether IFN signatures are activated in isolated CD4+ and CD8+ T cells from patients with active TAK (17). Transcriptome analysis demonstrated that 248 genes were dysregulated in CD4+ T cells and 432 genes in CD8+ T cells. Pathway enrichment analysis identified type I and type II IFN signatures and cytokine/chemokine signaling as highly enriched in both CD4+ and CD8+ T cells from TAK patients. Further analysis pointed toward active signaling in the STAT5 pathway, suggesting that the patients’ T cells might be exposed to elevated levels of the T cell growth factor IL-2. Treatment of two TAK patients with a JAK-STAT inhibitor resulted in a measurable decreased of Th1 and Th17 cell frequencies (17), supporting the premise that T cells activation in TAK is ultimately controlled by cytokines and growths factors utilizing the JAK-STAT pathway. This conclusion is supported by a series of case reports showing the efficacy of JAK inhibitors in refractory TAK patients (146, 147). These studies suggest a potential role of JAK-STAT pathway blockade as a promising approach to dampen disease activity in TAK (148).
CD4+ Regulatory T Cells
Common to most autoimmune diseases, Treg cell dysfunction has been proposed to underlie the chronic immune stimulation in TAK. Recent work has shown a decrease in the function of peripheral blood CD4+ Treg cells in TAK patients (149). Plasticity of CD4+ Tregs acquiring effector cell function has been described (150, 151), and the authors proposed that Th2-like transformed Tregs that secret IL-4 and IL-13 contribute to the development of TAK (149). Th2 cytokines are rarely encountered in GCA (152), and the appearance of this class of cytokines may indeed be a distinguishing features between TAK and GCA. Selective therapeutic manipulation of CD4+ Tregs in autoimmunity has long been an objective (51) and the above-mentioned study reported that the blockade of the JAK-STAT pathway can restore CD4+ Treg cells and increase the ratio of CD4+Treg/CD4+ effector T cells (17). Correction of the aberrantly activated JAK-STAT pathway may, therefore, be effective in modifying several pathogenic domains in TAK. However, this form of treatment in TAK raises a number of safety concerns, all of which need to be addressed before new therapies can be introduced into standard management; including the age of the patients, the chronicity of disease and the risk of immunosuppression during the pandemic spread of new viral agents, promotion of neoplasia, etc.
CD8+ T Cells
Detailed analyses of the inflammatory burden in TAK have demonstrated CD8+ T cells accounting for approximately 15% of the wall-infiltrating cells in TAK-affected aortas (153). CD8+ T cells are able to release perforin directly onto the surface of aortic vascular cells, thus causing direct tissue damage (153). Immunophenotypic studies of peripheral blood immune cells using multiparametric fluorescent techniques have provided evidence that the numbers of total CD8+ T cells and CD8+ effector T cells are both higher in TAK than in GCA (19). Notably, memory CD8+ T cells remained high even during clinical remission and the number of total CD8+ T cells was correlated with TAK, but not GCA relapse (19). In TAK patients, CD8+ T cells are increased not only in the circulation but also in the vessel wall infiltrates (57). Immunohistochemical analysis has shown that, compared with GCA-affected arteries, vascular surgical specimens in TAK had increased proportions of CD8+ T cells and the CD4/CD8 ratio differentiated the two vasculitides (57). A recent GWAS study also supported the critical role of CD8+ T cells in the pathogenesis of TAK (20). Taken together, these results indicate that GCA and TAK have critical differences in immune cell composition in the peripheral blood and within the vasculitic lesions.
CD4+ T cells and CD8+ T cells in TAK patients share sustained mTORC1 activity (14, 154, 155). Upstream signals that drive activation of the mTORC1 pathway have not yet been defined and our understanding of the functional consequences for protective and pathogenic immunity in these patients is limited. Continuous activation of central cellular signaling pathways appears to be an overall theme in patients affected by TAK. Besides chronic mTORC1 activation, T cells from TAK patients have ongoing activation of the JAK-STAT pathway, inducing type I and type II IFN gene expression signatures (17). Comparative studies of CD4+ and CD8+ T cells, which are responsible for fundamentally different effector pathways, may shed light on the state of chronic stimulation of the adaptive immune system in this form of vasculitis.
Natural Killer (NK) Cells
Seko et al., were the first to report that NK cells have a prominent position in the vascular injury leading to TAK (153). CD16+ NK cells account for 20% of the immune cells in the aortic wall infiltrates (153). Like CD8+ T cells, NK cells were positive for perforin immunostaining, providing strong support for a role in inducing cellular damage. Further support for the critical contribution of NK cells has come from the GWAS study by Terao et al., defining NK cells as the most promising target in the pathophysiology of TAK (20). NK cells are regulated by accessory molecules, such as the major histocompatibility complex (MHC) class I chain-related gene (MIC) family, and the leukocyte immunoglobulin-like receptor (LILR) family (139). Overexpression of MIC-related A (MICA) and its receptor natural killer group 2 member D (NKG2D) in aortic tissue from TAK cases has been reported (156). Expression of MHC and MICA on aortic vascular cells allows NK cells to recognize them through the NKG2D receptor and, in turn, attack them (139). LILR family members (LILR A1 to A6 and LILR B1 to B5) are widely expressed on hematopoietic cells and mediate activation as well as inhibition of immune cell function (157–159). Among them, LILRA3 was identified as a novel susceptibility loci in the TAK GWAS study (20). How precisely LILRA3 regulates immune response in TAK remains largely unknown, but it has been proposed that LILRA3 may bind to the major TAK susceptibility molecule HLA-B52 (20, 139). So far, NK-directed therapies are unavailable but TAK may become the signature disease to tap into novel opportunities of immune modulation and preventing immune cell-dependent cellular injury.
Autoantibodies Against Endothelial Cells (ECs)
A recent report on anti-endothelial cell autoantibodies has raised the question of the potential role that B cells participate in TAK disease pathogenesis. B cells are infrequent in the vasculitic infiltrates, which are typically cell admixtures of T cells and macrophages. However, B cells may play a critical role in the breakdown of immune tolerance that precedes the invasion of the vessel wall by inflammatory cells. Anti-endothelial cell antibodies (AECA) are a heterogenous group of autoantibodies against ECs (160). AECA have been detected across the spectrum of systemic vasculitides, ranging from small-vessel vasculitides to medium- and large-vessel vasculitides (161). Classically, AECA bind to ECs and induce apoptosis through direct complement-dependent cytotoxicity or through indirect antibody-dependent cytotoxicity (161), identifying antibodies as regulators of EC survival. In TAK, IgG deposits have been reported in the intima (162) and autoantibodies recognizing ECs have been evaluated as disease activity markers (163, 164). As expected, identification and quantification of AECA varies based on the technique used, e.g., indirect immunofluorescence, enzyme-linked immunosorbent assay, fluorescence-activated cell sorting, and immunoblot assays (160). Accordingly, measurement of AECA has not found its way into routine diagnostic schemes. This may change as a recent publication has defined autoantigens recognized by AECA. Mutoh et al., have identified two autoantigens expressed on ECs by applying an elegant serological identification system based on a retroviral expression system and flow cytometry (165). In this system, a cDNA library of ECs was retrovirally transfected into a rat myeloma cell line and AECA-positive clones were sorted by flow cytometry (165, 166). This yielded endothelial protein C receptor (EPCR) and scavenger receptor class B type 1 (SR-BI) as bona fide autoantigens (165). In a cohort study, the authors found that approximately 1 in 3 patients with TAK produce autoantibodies against either antigen. Remarkably, both molecules that are recognized by autoantibodies negatively regulate endothelial cell function, undermining the protective role of the endothelial layer (165). The authors propose that the autoantibodies disrupt the barrier function of ECs, opening the intimal surface to immune cell infiltration. Autoantibodies recognizing antigen specifically expressed by ECs appears to accompany chronic inflammatory disease (167, 168), but, interestingly, the autoantigens display a disease-specific pattern. Understanding the timing of events such as the emergence of AECA may be informative in assigning AECA-specific pathogenic determinants.
Conclusion
The inaccessibility of the body’s major arteries to tissue sampling have complicated the diagnosis and pathogenic understanding of autoimmunity in blood vessels. With the advent of non-invasive imaging techniques, vasculitis of the aorta and its major branch vessels can now be assessed, classified, and monitored. Comprehensive analysis using DNA, RNA, proteins, cell surface markers, transcription factors, and signaling pathways have greatly contributed to our understanding of the pathophysiology of large vessel vasculitis. A major insight has been that LVV has two principle disease components, the systemic inflammatory response, and the granulomatous vasculitis in the vessel wall. Traditionally, it has been assumed that patients with GCA and TAK develop autoimmunity against vascular antigens, which induces granulomatous vasculitis and, as a spill over, systemic inflammation. Much improved conceptualization of autoimmunity is beginning to question the validity of this traditional disease concept.
Patients with GCA and TAK are born with genetic risk factors that render them susceptible to a disease that will present clinically 2–6 decades later. Genes within and outside of the HLA complex have been identified as risk determinants, but a consistent theme of the association studies is the connection of genetic risk factors with immune cell function. With solid evidence that GCA and TAK are essentially immune-mediated diseases, multiple immune cell types are now recognized as critical disease players, including CD4+ and CD8+ T cells, monocytes and macrophages, NK cells and autoantibody-producing B cells. Emerging data indicate that CD8+ T cells may be more important in TAK than in GCA, opening opportunities to implicate different immune cells in different aspects of pathogenesis (Table 1). Pathogenic studies in TAK have been complicated by multiple hurdles, such as access to diseased tissue, the lack of reliable animal models and the low disease prevalence. Therefore, data on disease mechanisms in TAK have remained less robust. In both, GCA and TAK, immune cell-mediated injury to vascular cells may lie upstream of the chronic granulomatous reaction typifying these autoimmune diseases.
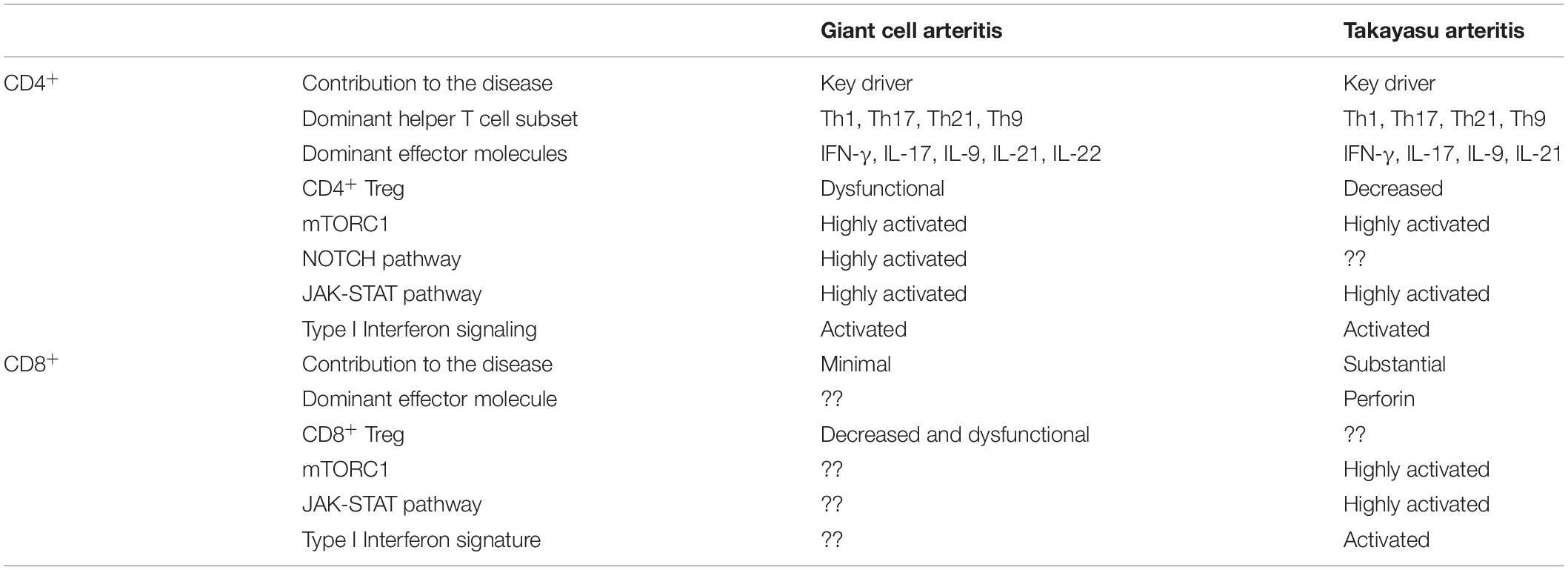
Table 1. CD4+ and CD8+ T cells in giant cell arteritis and in Takayasu arteritis.
As in other autoimmune diseases there is now recognition that pathogenic events leading to GCA and TAK may involve multiple disease components, that are not always coordinated and that require specialized therapeutic interventions. Specifically, the extravascular and the vascular component of LVV seem to follow different trajectories, rely on different mechanisms, and respond differently to current treatments. Extravascular GCA and TAK are characterized by intense systemic inflammation and are measured by elevation of acute phase reactants, such as C-reactive protein and erythrocyte sedimentation rate. They are simple to measure in the peripheral blood and are easy to suppress with corticosteroids or by blocking IL-6 signaling (169, 170). Much more challenging is the vascular component of TAK and GCA; a persistent, refractory granulomatous inflammation positioned in the vessel wall (69, 171). Vascular GCA and TAK are difficult to treat; 50% of patients have persistent vasculitis despite intense therapy for 1 year (118). The resistance to standard immunosuppression is corroborated by recent reports of ongoing disease activity in patients treated with anti-IL-6 therapy (172–176). Underlying molecular mechanisms are those of sustained activation of innate and adaptive immune cells through a plethora of signaling pathways. Most significant are the enduring activation of the mTOR pathway, the NOTCH signaling pathway and the JAK-STAT pathway (Table 1 and Figure 6). In combination, these essential cellular signaling pathways drive lasting immune responses in a tissue site that is intolerant to damage. The unparalleled effectiveness of corticosteroids in treating large vessel vasculitis may well reflect their imprecision in suppressing cellular activation pathways.
Author Contributions
RW, JG, and CW wrote the manuscript. GB contributed the tissue images. DL participated in the concept development. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Institutes of Health (R01 AR042527, R01 HL117913, R01 AI108906, R01 HL142068, and P01 HL129941 to CW and R01 AI108891, R01 AG045779, U19 AI057266, and R01 AI129191 to JG) and Merit Review Award I01 BX001669 from the United States (U.S.) Department of Veterans Affairs to JG.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank all patients who have contributed to our research program. This work would not have been possible without the generous support of the Encrantz Family Fund and the Governor Family Fund. The authors would like to thank the Praespero Foundation for generous support of the program.
References
1. Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised international chapel hill consensus conference nomenclature of vasculitides. Arthritis Rheum. (2013) 65:1–11. doi: 10.1002/art.37715
2. Tellides G, Pober JS. Inflammatory and immune responses in the arterial media. Circ Res. (2015) 116:312–22. doi: 10.1161/CIRCRESAHA.116.301312
3. Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, et al. Atherosclerosis. Nat Rev Dis Primer. (2019) 5:56. doi: 10.1038/s41572-019-0106-z
4. Liberale L, Montecucco F, Tardif J-C, Libby P, Camici GG. Inflamm-ageing: the role of inflammation in age-dependent cardiovascular disease. Eur Heart J. (2020) 1:ehz961. doi: 10.1093/eurheartj/ehz961
5. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
6. Weyand CM, Goronzy JJ. Medium-and large-vessel vasculitis. N Engl J Med. (2003) 349:160–9. doi: 10.1056/NEJMra022694
7. Weyand CM, Goronzy JJ. Immune mechanisms in medium and large-vessel vasculitis. Nat Rev Rheumatol. (2013) 9:731–40. doi: 10.1038/nrrheum.2013.161
8. Weyand CM, Goronzy JJ. Clinical practice. Giant-cell arteritis and polymyalgia rheumatica. N Engl J Med. (2014) 371:50–7. doi: 10.1056/NEJMcp1214825
9. Gribbons KB, Ponte C, Carette S, Craven A, Cuthbertson D, Hoffman GS, et al. Patterns of arterial disease in takayasu’s arteritis and giant cell arteritis. Arthritis Care Res. (2019). doi: 10.1002/acr.24055 [Epub ahead of print].
10. Soriano A, Pazzola G, Boiardi L, Casali M, Muratore F, Pipitone N, et al. Distribution patterns of 18F-fluorodeoxyglucose in large vessels of Takayasu’s and giant cell arteritis using positron emission tomography. Clin Exp Rheumatol. (2018) 36(Suppl. 111):99–106.
11. Kermani TA. Takayasu arteritis and giant cell arteritis: are they a spectrum of the same disease? Int J Rheum Dis. (2019) 22(Suppl. 1):41–8. doi: 10.1111/1756-185X.13288
12. Kermani TA, Crowson CS, Muratore F, Schmidt J, Matteson EL, Warrington KJ. Extra-cranial giant cell arteritis and Takayasu arteritis: How similar are they? Semin Arthritis Rheum. (2015) 44:724–8. doi: 10.1016/j.semarthrit.2015.01.005
13. Maciejewski-Duval A, Comarmond C, Leroyer A, Zaidan M, Le Joncour A, Desbois AC, et al. mTOR pathway activation in large vessel vasculitis. J Autoimmun. (2018) 94:99–109. doi: 10.1016/j.jaut.2018.07.013
14. Zhang J, Zhao L, Wang J, Cheng Z, Sun M, Zhao J, et al. Targeting mechanistic target of rapamycin complex 1 restricts proinflammatory T cell differentiation and ameliorates takayasu arteritis. Arthritis Rheumatol. (2020) 72:303–15. doi: 10.1002/art.41084
15. Wen Z, Shen Y, Berry G, Shahram F, Li Y, Watanabe R, et al. The microvascular niche instructs T cells in large vessel vasculitis via the VEGF-Jagged1-Notch pathway. Sci Transl Med. (2017) 9:3322. doi: 10.1126/scitranslmed.aal3322
16. Zhang H, Watanabe R, Berry GJ, Tian L, Goronzy JJ, Weyand CM. Inhibition of JAK-STAT signaling suppresses pathogenic immune responses in medium and large vessel vasculitis. Circulation. (2018) 137:1934–48. doi: 10.1161/CIRCULATIONAHA.117.030423
17. Régnier P, Le Joncour A, Maciejewski-Duval A, Desbois A-C, Comarmond C, Rosenzwajg M, et al. Targeting JAK/STAT pathway in Takayasu’s arteritis. Ann Rheum Dis. (2020) 79:951–9. doi: 10.1136/annrheumdis-2019-216900
18. De Smit E, Lukowski SW, Anderson L, Senabouth A, Dauyey K, Song S, et al. Longitudinal expression profiling of CD4+ and CD8+ cells in patients with active to quiescent giant cell arteritis. BMC Med Genomics. (2018) 11:61. doi: 10.1186/s12920-018-0376-4
19. Matsumoto K, Suzuki K, Yoshimoto K, Seki N, Tsujimoto H, Chiba K, et al. Significant association between clinical characteristics and changes in peripheral immuno-phenotype in large vessel vasculitis. Arthritis Res Ther. (2019) 21:304. doi: 10.1186/s13075-019-2068-7
20. Terao C, Yoshifuji H, Matsumura T, Naruse TK, Ishii T, Nakaoka Y, et al. Genetic determinants and an epistasis of LILRA3 and HLA-B∗52 in Takayasu arteritis. Proc Natl Acad Sci USA. (2018) 115:13045–50. doi: 10.1073/pnas.1808850115
21. Weyand CM, Hicok KC, Hunder GG, Goronzy JJ. The HLA-DRB1 locus as a genetic component in giant cell arteritis. Mapping of a disease-linked sequence motif to the antigen binding site of the HLA-DR molecule. J Clin Invest. (1992) 90:2355–61. doi: 10.1172/JCI116125
22. Weyand CM, Hunder NN, Hicok KC, Hunder GG, Goronzy JJ. HLA-DRB1 alleles in polymyalgia rheumatica, giant cell arteritis, and rheumatoid arthritis. Arthritis Rheum. (1994) 37:514–20. doi: 10.1002/art.1780370411
23. Weyand CM, Schönberger J, Oppitz U, Hunder NN, Hicok KC, Goronzy JJ. Distinct vascular lesions in giant cell arteritis share identical T cell clonotypes. J Exp Med. (1994) 179:951–60. doi: 10.1084/jem.179.3.951
24. Grunewald J, Andersson R, Rydberg L, Gigliotti D, Schaufelberger C, Hansson GK, et al. CD4+ and CD8+ T cell expansions using selected TCR V and J gene segments at the onset of giant cell arteritis. Arthritis Rheum. (1994) 37:1221–7. doi: 10.1002/art.1780370817
25. Carmona FD, Mackie SL, Martín J-E, Taylor JC, Vaglio A, Eyre S, et al. A large-scale genetic analysis reveals a strong contribution of the HLA class II region to giant cell arteritis susceptibility. Am J Hum Genet. (2015) 96:565–80. doi: 10.1016/j.ajhg.2015.02.009
26. Carmona FD, Vaglio A, Mackie SL, Hernández-Rodríguez J, Monach PA, Castañeda S, et al. A Genome-wide Association study identifies risk alleles in plasminogen and P4HA2 associated with giant cell arteritis. Am J Hum Genet. (2017) 100:64–74. doi: 10.1016/j.ajhg.2016.11.013
27. Deng J, Younge BR, Olshen RA, Goronzy JJ, Weyand CM. Th17 and Th1 T-cell responses in giant cell arteritis. Circulation. (2010) 121:906–15. doi: 10.1161/CIRCULATIONAHA.109.872903
28. Samson M, Audia S, Fraszczak J, Trad M, Ornetti P, Lakomy D, et al. Th1 and Th17 lymphocytes expressing CD161 are implicated in giant cell arteritis and polymyalgia rheumatica pathogenesis. Arthritis Rheum. (2012) 64:3788–98. doi: 10.1002/art.34647
29. Samson M, Ghesquière T, Berthier S, Bonnotte B. Ustekinumab inhibits Th1 and Th17 polarisation in a patient with giant cell arteritis. Ann Rheum Dis. (2018) 77:e6. doi: 10.1136/annrheumdis-2017-211622
30. Weyand CM, Younge BR, Goronzy JJ. IFN-γ and IL-17: the two faces of T-cell pathology in giant cell arteritis. Curr Opin Rheumatol. (2011) 23:43–9. doi: 10.1097/BOR.0b013e32833ee946
31. Watanabe R, Hosgur E, Zhang H, Wen Z, Berry G, Goronzy JJ, et al. Pro-inflammatory and anti-inflammatory T cells in giant cell arteritis. Joint Bone Spine. (2017) 84:421–6. doi: 10.1016/j.jbspin.2016.07.005
32. Terrier B, Geri G, Chaara W, Allenbach Y, Rosenzwajg M, Costedoat-Chalumeau N, et al. Interleukin-21 modulates Th1 and Th17 responses in giant cell arteritis. Arthritis Rheum. (2012) 64:2001–11. doi: 10.1002/art.34327
33. Barrat FJ, Crow MK, Ivashkiv LB. Interferon target-gene expression and epigenomic signatures in health and disease. Nat Immunol. (2019) 20:1574–83. doi: 10.1038/s41590-019-0466-2
34. Kaiser M, Younge B, Björnsson J, Goronzy JJ, Weyand CM. Formation of new vasa vasorum in vasculitis. Production of angiogenic cytokines by multinucleated giant cells. Am J Pathol. (1999) 155:765–74. doi: 10.1016/S0002-9440(10)65175-9
35. Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol. (2010) 10:479–89. doi: 10.1038/nri2800
36. McGeachy MJ, Cua DJ, Gaffen SL. The IL-17 Family of cytokines in health and disease. Immunity. (2019) 50:892–906. doi: 10.1016/j.immuni.2019.03.021
37. Schön MP, Erpenbeck L. The interleukin-23/interleukin-17 Axis links adaptive and innate immunity in psoriasis. Front Immunol. (2018) 9:1323. doi: 10.3389/fimmu.2018.01323
38. Gravallese EM, Schett G. Effects of the IL-23-IL-17 pathway on bone in spondyloarthritis. Nat Rev Rheumatol. (2018) 14:631–40. doi: 10.1038/s41584-018-0091-8
39. Raychaudhuri SP, Raychaudhuri SK. IL-23/IL-17 axis in spondyloarthritis-bench to bedside. Clin Rheumatol. (2016) 35:1437–41. doi: 10.1007/s10067-016-3263-4
40. Long D, Chen Y, Wu H, Zhao M, Lu Q. Clinical significance and immunobiology of IL-21 in autoimmunity. J Autoimmun. (2019) 99:1–14. doi: 10.1016/j.jaut.2019.01.013
41. Gharibi T, Majidi J, Kazemi T, Dehghanzadeh R, Motallebnezhad M, Babaloo Z. Biological effects of IL-21 on different immune cells and its role in autoimmune diseases. Immunobiology. (2016) 221:357–67. doi: 10.1016/j.imbio.2015.09.021
42. Ciccia F, Rizzo A, Guggino G, Cavazza A, Alessandro R, Maugeri R, et al. Difference in the expression of IL-9 and IL-17 correlates with different histological pattern of vascular wall injury in giant cell arteritis. Rheumatol Oxf Engl. (2015) 54:1596–604. doi: 10.1093/rheumatology/kev102
43. Kaplan MH, Hufford MM, Olson MR. The development and in vivo function of T helper 9 cells. Nat Rev Immunol. (2015) 15:295–307. doi: 10.1038/nri3824
44. Zerbini A, Muratore F, Boiardi L, Ciccia F, Bonacini M, Belloni L, et al. Increased expression of interleukin-22 in patients with giant cell arteritis. Rheumatol Oxf Engl. (2018) 57:64–72. doi: 10.1093/rheumatology/kex334
45. Ronacher K, Sinha R, Cestari M. IL-22: an underestimated player in natural resistance to tuberculosis? Front Immunol. (2018) 9:2209. doi: 10.3389/fimmu.2018.02209
46. Wolk K, Witte E, Witte K, Warszawska K, Sabat R. Biology of interleukin-22. Semin Immunopathol. (2010) 32:17–31. doi: 10.1007/s00281-009-0188-x
47. Azizi G, Yazdani R, Mirshafiey A. Th22 cells in autoimmunity: a review of current knowledge. Eur Ann Allergy Clin Immunol. (2015) 47:108–17.
48. Yang X, Zheng SG. Interleukin-22: a likely target for treatment of autoimmune diseases. Autoimmun Rev. (2014) 13:615–20. doi: 10.1016/j.autrev.2013.11.008
49. Watanabe R, Goronzy JJ, Berry G, Liao YJ, Weyand CM. Giant cell arteritis: from pathogenesis to therapeutic management. Curr Treat Options Rheumatol. (2016) 2:126–37. doi: 10.1007/s40674-016-0043-x
50. Ohkura N, Kitagawa Y, Sakaguchi S. Development and maintenance of regulatory T cells. Immunity. (2013) 38:414–23. doi: 10.1016/j.immuni.2013.03.002
51. Wing JB, Tanaka A, Sakaguchi S. Human FOXP3+ regulatory T cell heterogeneity and function in autoimmunity and cancer. Immunity. (2019) 50:302–16. doi: 10.1016/j.immuni.2019.01.020
52. Miyabe C, Miyabe Y, Strle K, Kim ND, Stone JH, Luster AD, et al. An expanded population of pathogenic regulatory T cells in giant cell arteritis is abrogated by IL-6 blockade therapy. Ann Rheum Dis. (2017) 76:898–905. doi: 10.1136/annrheumdis-2016-210070
53. Elling P, Olsson A, Elling H. CD8+ T lymphocyte subset in giant cell arteritis and related disorders. J Rheumatol. (1990) 17:225–7.
54. Dasgupta B, Duke O, Timms AM, Pitzalis C, Panayi GS. Selective depletion and activation of CD8+ lymphocytes from peripheral blood of patients with polymyalgia rheumatica and giant cell arteritis. Ann Rheum Dis. (1989) 48:307–11. doi: 10.1136/ard.48.4.307
55. Martinez-Taboada VM, Goronzy JJ, Weyand CM. Clonally expanded CD8 T cells in patients with polymyalgia rheumatica and giant cell arteritis. Clin Immunol Immunopathol. (1996) 79:263–70. doi: 10.1006/clin.1996.0078
56. Lopez-Hoyos M, Bartolome-Pacheco MJ, Blanco R, Rodriguez-Valverde V, Martinez-Taboada VM. Selective T cell receptor decrease in peripheral blood T lymphocytes of patients with polymyalgia rheumatica and giant cell arteritis. Ann Rheum Dis. (2004) 63:54–60. doi: 10.1136/ard.2003.005900
57. Kurata A, Saito A, Hashimoto H, Fujita K, Ohno S-I, Kamma H, et al. Difference in immunohistochemical characteristics between Takayasu arteritis and giant cell arteritis: It may be better to distinguish them in the same age. Mod Rheumatol. (2019) 29:992–1001. doi: 10.1080/14397595.2019.1570999
58. Wen Z, Shimojima Y, Shirai T, Li Y, Ju J, Yang Z, et al. NADPH oxidase deficiency underlies dysfunction of aged CD8+ Tregs. J Clin Invest. (2016) 126:1953–67. doi: 10.1172/JCI84181
59. Suzuki M, Jagger AL, Konya C, Shimojima Y, Pryshchep S, Goronzy JJ, et al. CD8+CD45RA+CCR7+FOXP3+ T cells with immunosuppressive properties: a novel subset of inducible human regulatory T cells. J Immunol Baltim. (2012) 189:2118–30. doi: 10.4049/jimmunol.1200122
60. Goronzy JJ, Weyand CM. Mechanisms underlying T cell ageing. Nat Rev Immunol. (2019) 19:573–83. doi: 10.1038/s41577-019-0180-1
61. Goronzy JJ, Weyand CM. Successful and maladaptive T cell aging. Immunity. (2017) 46:364–78. doi: 10.1016/j.immuni.2017.03.010
62. Ferrucci L, Fabbri E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol. (2018) 15:505–22. doi: 10.1038/s41569-018-0064-2
63. Lavin Y, Mortha A, Rahman A, Merad M. Regulation of macrophage development and function in peripheral tissues. Nat Rev Immunol. (2015) 15:731–44. doi: 10.1038/nri3920
64. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. (2010) 11:373–84. doi: 10.1038/ni.1863
65. Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol. (2012) 13:1118–28. doi: 10.1038/ni.2419
66. Watanabe R, Hilhorst M, Zhang H, Zeisbrich M, Berry GJ, Wallis BB, et al. Glucose metabolism controls disease-specific signatures of macrophage effector functions. JCI Insight. (2018) 3:e123047. doi: 10.1172/jci.insight.123047
67. Rittner HL, Kaiser M, Brack A, Szweda LI, Goronzy JJ, Weyand CM. Tissue-destructive macrophages in giant cell arteritis. Circ Res. (1999) 84:1050–8. doi: 10.1161/01.res.84.9.1050
68. Watanabe R, Maeda T, Zhang H, Berry GJ, Zeisbrich M, Brockett R, et al. (Matrix Metalloprotease)-9-producing monocytes enable T cells to invade the vessel wall and cause vasculitis. Circ Res. (2018) 123:700–15. doi: 10.1161/CIRCRESAHA.118.313206
69. Weyand CM, Watanabe R, Zhang H, Akiyama M, Berry GJ, Goronzy JJ. Cytokines, growth factors and proteases in medium and large vessel vasculitis. Clin Immunol Orlando Fla. (2019) 206:33–41. doi: 10.1016/j.clim.2019.02.007
70. Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, et al. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol. (2014) 14:571–8. doi: 10.1038/nri3712
71. Ganguly D, Haak S, Sisirak V, Reizis B. The role of dendritic cells in autoimmunity. Nat Rev Immunol. (2013) 13:566–77. doi: 10.1038/nri3477
72. Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol. (2020) 20:7–24. doi: 10.1038/s41577-019-0210-z
73. Weyand CM, Ma-Krupa W, Pryshchep O, Gröschel S, Bernardino R, Goronzy JJ. Vascular dendritic cells in giant cell arteritis. Ann N Y Acad Sci. (2005) 1062:195–208. doi: 10.1196/annals.1358.023
74. Deng J, Ma-Krupa W, Gewirtz AT, Younge BR, Goronzy JJ, Weyand CM. Toll-like receptors 4 and 5 induce distinct types of vasculitis. Circ Res. (2009) 104:488–95. doi: 10.1161/CIRCRESAHA.108.185777
75. Ma-Krupa W, Jeon M-S, Spoerl S, Tedder TF, Goronzy JJ, Weyand CM. Activation of arterial wall dendritic cells and breakdown of self-tolerance in giant cell arteritis. J Exp Med. (2004) 199:173–83. doi: 10.1084/jem.20030850
76. Pryshchep O, Ma-Krupa W, Younge BR, Goronzy JJ, Weyand CM. Vessel-specific Toll-like receptor profiles in human medium and large arteries. Circulation. (2008) 118:1276–84. doi: 10.1161/CIRCULATIONAHA.108.789172
77. Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol. (2005) 23:515–48. doi: 10.1146/annurev.immunol.23.021704.115611
78. Ferris RL, Blumenschein G, Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. (2016) 375:1856–67. doi: 10.1056/NEJMoa1602252
79. Tawbi HA, Forsyth PA, Algazi A, Hamid O, Hodi FS, Moschos SJ, et al. Combined nivolumab and ipilimumab in melanoma metastatic to the brain. N Engl J Med. (2018) 379:722–30. doi: 10.1056/NEJMoa1805453
80. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. (2012) 366:2443–54. doi: 10.1056/NEJMoa1200690
81. Fife BT, Pauken KE, Eagar TN, Obu T, Wu J, Tang Q, et al. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat Immunol. (2009) 10:1185–92. doi: 10.1038/ni.1790
82. Zhang H, Watanabe R, Berry GJ, Vaglio A, Liao YJ, Warrington KJ, et al. Immunoinhibitory checkpoint deficiency in medium and large vessel vasculitis. Proc Natl Acad Sci USA. (2017) 114:E970–9. doi: 10.1073/pnas.1616848114
83. Watanabe R, Zhang H, Berry G, Goronzy JJ, Weyand CM. Immune checkpoint dysfunction in large and medium vessel vasculitis. Am J Physiol Heart Circ Physiol. (2017) 312:H1052–9. doi: 10.1152/ajpheart.00024.2017
84. Betrains A, Blockmans DE. Immune checkpoint inhibitor-associated polymyalgia rheumatica/giant cell arteritis occurring in a patient after treatment with nivolumab. J Clin Rheumatol Pract Rep Rheum Musculoskelet Dis. (2019). doi: 10.1097/RHU.0000000000001012 [Epub ahead of print].
85. Salem J-E, Manouchehri A, Moey M, Lebrun-Vignes B, Bastarache L, Pariente A, et al. Cardiovascular toxicities associated with immune checkpoint inhibitors: an observational, retrospective, pharmacovigilance study. Lancet Oncol. (2018) 19:1579–89. doi: 10.1016/S1470-2045(18)30608-9
86. Simon TA, Thompson A, Gandhi KK, Hochberg MC, Suissa S. Incidence of malignancy in adult patients with rheumatoid arthritis: a meta-analysis. Arthritis Res Ther. (2015) 17:212. doi: 10.1186/s13075-015-0728-9
87. Liang Y, Yang Z, Qin B, Zhong R. Primary Sjogren’s syndrome and malignancy risk: a systematic review and meta-analysis. Ann Rheum Dis. (2014) 73:1151–6. doi: 10.1136/annrheumdis-2013-203305
88. Yang Z, Lin F, Qin B, Liang Y, Zhong R. Polymyositis/dermatomyositis and malignancy risk: a metaanalysis study. J Rheumatol. (2015) 42:282–91. doi: 10.3899/jrheum.140566
89. Ishizuka M, Watanabe R, Ishii T, Machiyama T, Akita K, Fujita Y, et al. Long-term follow-up of 124 patients with polymyositis and dermatomyositis: Statistical analysis of prognostic factors. Mod Rheumatol. (2016) 26:115–20. doi: 10.3109/14397595.2015.1054081
90. Stamatis P, Turesson C, Willim M, Nilsson J-Å, Englund M, Mohammad AJ. Malignancies in giant cell arteritis: a population-based cohort study. J Rheumatol. (2020) 47:400–6. doi: 10.3899/jrheum.190236
91. Shirai T, Nazarewicz RR, Wallis BB, Yanes RE, Watanabe R, Hilhorst M, et al. The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J Exp Med. (2016) 213:337–54. doi: 10.1084/jem.20150900
92. Watanabe R, Shirai T, Namkoong H, Zhang H, Berry GJ, Wallis BB, et al. Pyruvate controls the checkpoint inhibitor PD-L1 and suppresses T cell immunity. J Clin Invest. (2017) 127:2725–38. doi: 10.1172/JCI92167
93. Jakobsson K, Jacobsson L, Warrington K, Matteson EL, Liang K, Melander O, et al. Body mass index and the risk of giant cell arteritis: results from a prospective study. Rheumatol Oxf Engl. (2015) 54:433–40. doi: 10.1093/rheumatology/keu331
94. Wadström K, Jacobsson L, Mohammad AJ, Warrington KJ, Matteson EL, Turesson C. Negative associations for fasting blood glucose, cholesterol and triglyceride levels with the development of giant cell arteritis. Rheumatol Oxf Engl. (2020) 2:keaa080. doi: 10.1093/rheumatology/keaa080
95. Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, Guo Y, et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J Exp Med. (2011) 208:577–92. doi: 10.1084/jem.20100619
96. Hid Cadena R, Reitsema RD, Huitema MG, van Sleen Y, van der Geest KSM, Heeringa P, et al. Decreased expression of negative immune checkpoint VISTA by CD4+ T cells facilitates T helper 1, T helper 17, and T follicular helper lineage differentiation in GCA. Front Immunol. (2019) 10:1638. doi: 10.3389/fimmu.2019.01638
97. Wang J, Wu G, Manick B, Hernandez V, Renelt M, Erickson C, et al. VSIG-3 as a ligand of VISTA inhibits human T-cell function. Immunology. (2019) 156:74–85. doi: 10.1111/imm.13001
98. Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. (2013) 13:227–42. doi: 10.1038/nri3405
99. Alegre ML, Frauwirth KA, Thompson CB. T-cell regulation by CD28 and CTLA-4. Nat Rev Immunol. (2001) 1:220–8. doi: 10.1038/35105024
100. Bantug GR, Galluzzi L, Kroemer G, Hess C. The spectrum of T cell metabolism in health and disease. Nat Rev Immunol. (2018) 18:19–34. doi: 10.1038/nri.2017.99
101. Palsson-McDermott EM, O’Neill LAJ. The warburg effect then and now: from cancer to inflammatory diseases. BioEssays News Rev Mol Cell Dev Biol. (2013) 35:965–73. doi: 10.1002/bies.201300084
102. Palmer CS, Ostrowski M, Balderson B, Christian N, Crowe SM. Glucose metabolism regulates T cell activation, differentiation, and functions. Front Immunol. (2015) 6:1. doi: 10.3389/fimmu.2015.00001
103. Kane LP, Weiss A. The PI-3 kinase/Akt pathway and T cell activation: pleiotropic pathways downstream of PIP3. Immunol Rev. (2003) 192:7–20. doi: 10.1034/j.1600-065x.2003.00008.x
104. Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity. (2002) 16:769–77. doi: 10.1016/s1074-7613(02)00323-0
105. Ben-Sahra I, Manning BD. mTORC1 signaling and the metabolic control of cell growth. Curr Opin Cell Biol. (2017) 45:72–82. doi: 10.1016/j.ceb.2017.02.012
106. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. (2012) 149:274–93. doi: 10.1016/j.cell.2012.03.017
107. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. (2017) 168:960–76. doi: 10.1016/j.cell.2017.02.004
108. Aoki M, Fujishita T. Oncogenic roles of the PI3K/AKT/mTOR Axis. Curr Top Microbiol Immunol. (2017) 407:153–89. doi: 10.1007/82_2017_6
109. Zhang H, Watanabe R, Berry GJ, Nadler SG, Goronzy JJ, Weyand CM. CD28 signaling controls metabolic fitness of pathogenic t cells in medium and large vessel vasculitis. J Am Coll Cardiol. (2019) 73:1811–23. doi: 10.1016/j.jacc.2019.01.049
110. Shimobayashi M, Hall MN. Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat Rev Mol Cell Biol. (2014) 15:155–62. doi: 10.1038/nrm3757
111. Bray SJ. Notch signalling in context. Nat Rev Mol Cell Biol. (2016) 17:722–35. doi: 10.1038/nrm.2016.94
112. Piggott K, Deng J, Warrington K, Younge B, Kubo JT, Desai M, et al. Blocking the NOTCH pathway inhibits vascular inflammation in large-vessel vasculitis. Circulation. (2011) 123:309–18. doi: 10.1161/CIRCULATIONAHA.110.936203
113. Hirahara K, Schwartz D, Gadina M, Kanno Y, O’Shea JJ. Targeting cytokine signaling in autoimmunity: back to the future and beyond. Curr Opin Immunol. (2016) 43:89–97. doi: 10.1016/j.coi.2016.10.001
114. O’Shea JJ, Plenge R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity. (2012) 36:542–50. doi: 10.1016/j.immuni.2012.03.014
115. Schwartz DM, Bonelli M, Gadina M, O’Shea JJ. Type I/II cytokines, JAKs, and new strategies for treating autoimmune diseases. Nat Rev Rheumatol. (2016) 12:25–36. doi: 10.1038/nrrheum.2015.167
116. Smolen JS, Landewé RBM, Bijlsma JWJ, Burmester GR, Dougados M, Kerschbaumer A, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann Rheum Dis. (2020) 79:685–99. doi: 10.1136/annrheumdis-2019-216655
117. Taylor PC, Keystone EC, van der Heijde D, Weinblatt ME, Del Carmen Morales L, Reyes GJ, et al. Baricitinib versus placebo or adalimumab in rheumatoid arthritis. N Engl J Med. (2017) 376:652–62. doi: 10.1056/NEJMoa1608345
118. Maleszewski JJ, Younge BR, Fritzlen JT, Hunder GG, Goronzy JJ, Warrington KJ, et al. Clinical and pathological evolution of giant cell arteritis: a prospective study of follow-up temporal artery biopsies in 40 treated patients. Mod Pathol Off J U S Can Acad Pathol Inc. (2017) 30:788–96. doi: 10.1038/modpathol.2017.10
119. Molodtsov A, Turk MJ. Tissue resident CD8 memory T cell responses in cancer and autoimmunity. Front Immunol. (2018) 9:2810. doi: 10.3389/fimmu.2018.02810
120. Klicznik MM, Morawski PA, Höllbacher B, Varkhande SR, Motley SJ, Kuri-Cervantes L, et al. Human CD4+CD103+ cutaneous resident memory T cells are found in the circulation of healthy individuals. Sci Immunol. (2019) 4:eaav8995. doi: 10.1126/sciimmunol.aav8995
121. Gebhardt T, Palendira U, Tscharke DC, Bedoui S. Tissue-resident memory T cells in tissue homeostasis, persistent infection, and cancer surveillance. Immunol Rev. (2018) 283:54–76. doi: 10.1111/imr.12650
122. Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. (2005) 5:375–86. doi: 10.1038/nri1604
123. Zhang S-Y, Boisson-Dupuis S, Chapgier A, Yang K, Bustamante J, Puel A, et al. Inborn errors of interferon (IFN)-mediated immunity in humans: insights into the respective roles of IFN-alpha/beta, IFN-gamma, and IFN-lambda in host defense. Immunol Rev. (2008) 226:29–40. doi: 10.1111/j.1600-065X.2008.00698.x
124. Higgs BW, Liu Z, White B, Zhu W, White WI, Morehouse C, et al. Patients with systemic lupus erythematosus, myositis, rheumatoid arthritis and scleroderma share activation of a common type I interferon pathway. Ann Rheum Dis. (2011) 70:2029–36. doi: 10.1136/ard.2011.150326
125. Rönnblom L, Eloranta M-L. The interferon signature in autoimmune diseases. Curr Opin Rheumatol. (2013) 25:248–53. doi: 10.1097/BOR.0b013e32835c7e32
126. Kim J-M, Park S-H, Kim H-Y, Kwok S-K. A plasmacytoid dendritic Cells-type I interferon axis is critically implicated in the pathogenesis of systemic lupus erythematosus. Int J Mol Sci. (2015) 16:14158–70. doi: 10.3390/ijms160614158
127. Swiecki M, Colonna M. The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol. (2015) 15:471–85. doi: 10.1038/nri3865
128. Crouse J, Kalinke U, Oxenius A. Regulation of antiviral T cell responses by type I interferons. Nat Rev Immunol. (2015) 15:231–42. doi: 10.1038/nri3806
129. Buttgereit F, Dejaco C, Matteson EL, Dasgupta B. Polymyalgia rheumatica and giant cell arteritis: a systematic review. JAMA. (2016) 315:2442–58. doi: 10.1001/jama.2016.5444
130. Hellmich B, Agueda A, Monti S, Buttgereit F, de Boysson H, Brouwer E, et al. 2018 Update of the EULAR recommendations for the management of large vessel vasculitis. Ann Rheum Dis. (2020) 79:19–30. doi: 10.1136/annrheumdis-2019-215672
131. Salvarani C, Hatemi G. Management of large-vessel vasculitis. Curr Opin Rheumatol. (2019) 31:25–31. doi: 10.1097/BOR.0000000000000561
132. Yoshida M, Watanabe R, Ishii T, Machiyama T, Akita K, Fujita Y, et al. Retrospective analysis of 95 patients with large vessel vasculitis: a single center experience. Int J Rheum Dis. (2016) 19:87–94. doi: 10.1111/1756-185X.12777
133. Numano F, Kishi Y, Tanaka A, Ohkawara M, Kakuta T, Kobayashi Y. Inflammation and atherosclerosis. Atherosclerotic lesions in Takayasu arteritis. Ann N Y Acad Sci. (2000) 902:65–76. doi: 10.1111/j.1749-6632.2000.tb06301.x
134. Terao C, Yoshifuji H, Kimura A, Matsumura T, Ohmura K, Takahashi M, et al. Two susceptibility loci to Takayasu arteritis reveal a synergistic role of the IL12B and HLA-B regions in a Japanese population. Am J Hum Genet. (2013) 93:289–97. doi: 10.1016/j.ajhg.2013.05.024
135. Sahin Z, Bıcakcıgil M, Aksu K, Kamali S, Akar S, Onen F, et al. Takayasu’s arteritis is associated with HLA-B∗52, but not with HLA-B∗51, in Turkey. Arthritis Res Ther. (2012) 14:R27. doi: 10.1186/ar3730
136. Kwon OC, Lee S-W, Park Y-B, Oh JS, Lee SH, Hong S, et al. Extravascular manifestations of Takayasu arteritis: focusing on the features shared with spondyloarthritis. Arthritis Res Ther. (2018) 20:142. doi: 10.1186/s13075-018-1643-7
137. Watanabe R, Ishii T, Nakamura K, Shirai T, Fujii H, Saito S, et al. Ulcerative colitis is not a rare complication of Takayasu arteritis. Mod Rheumatol. (2014) 24:372–3. doi: 10.3109/14397595.2013.854045
138. Terao C, Matsumura T, Yoshifuji H, Kirino Y, Maejima Y, Nakaoka Y, et al. Takayasu arteritis and ulcerative colitis: high rate of co-occurrence and genetic overlap. Arthritis Rheumatol. (2015) 67:2226–32. doi: 10.1002/art.39157
139. Yoshifuji H, Terao C. Roles of cytotoxic lymphocytes and MIC/LILR families in pathophysiology of Takayasu arteritis. Inflamm Regen. (2020) 40:9. doi: 10.1186/s41232-020-00119-6
140. Yoshifuji H. Pathophysiology of large vessel vasculitis and utility of interleukin-6 inhibition therapy. Mod Rheumatol. (2019) 29:287–93. doi: 10.1080/14397595.2018.1546358
141. Saadoun D, Garrido M, Comarmond C, Desbois AC, Domont F, Savey L, et al. Th1 and Th17 cytokines drive inflammation in Takayasu arteritis. Arthritis Rheumatol. (2015) 67:1353–60. doi: 10.1002/art.39037
142. Nakajima T, Yoshifuji H, Shimizu M, Kitagori K, Murakami K, Nakashima R, et al. A novel susceptibility locus in the IL12B region is associated with the pathophysiology of Takayasu arteritis through IL-12p40 and IL-12p70 production. Arthritis Res Ther. (2017) 19:197. doi: 10.1186/s13075-017-1408-8
143. Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. (2003) 3:133–46. doi: 10.1038/nri1001
144. Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol. (2014) 14:585–600. doi: 10.1038/nri3707
145. Hadjadj J, Canaud G, Mirault T, Samson M, Bruneval P, Régent A, et al. mTOR pathway is activated in endothelial cells from patients with Takayasu arteritis and is modulated by serum immunoglobulin G. Rheumatol Oxf Engl. (2018) 57:1011–20. doi: 10.1093/rheumatology/key017
146. Kuwabara S, Tanimura S, Matsumoto S, Nakamura H, Horita T. Successful remission with tofacitinib in a patient with refractory Takayasu arteritis complicated by ulcerative colitis. Ann Rheum Dis. (2020) 79:1125–6. doi: 10.1136/annrheumdis-2019-216606
147. Sato S, Matsumoto H, Temmoku J, Fujita Y, Matsuoka N, Furuya M, et al. A case of Takayasu arteritis complicated by refractory ulcerative colitis successfully treated with tofacitinib. Rheumatol Oxf Engl. (2019) 59:1773–5. doi: 10.1093/rheumatology/kez580
148. Watanabe R. JAK inhibitors as promising agents for refractory Takayasu arteritis. Ann Rheum Dis. (2020). doi: 10.1136/annrheumdis-2020-217577 [Epub ahead of print].
149. Gao N, Cui W, Zhao LM, Li TT, Zhang JH, Pan LL. Contribution of Th2-like Treg cells to the pathogenesis of Takayasu’s arteritis. Clin Exp Rheumatol. (2020) 38(Suppl. 124):48–54.
150. Zhou L, Chong MMW, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity. (2009) 30:646–55. doi: 10.1016/j.immuni.2009.05.001
151. Lee YK, Mukasa R, Hatton RD, Weaver CT. Developmental plasticity of Th17 and Treg cells. Curr Opin Immunol. (2009) 21:274–80. doi: 10.1016/j.coi.2009.05.021
152. Weyand CM, Hicok KC, Hunder GG, Goronzy JJ. Tissue cytokine patterns in patients with polymyalgia rheumatica and giant cell arteritis. Ann Intern Med. (1994) 121:484–91. doi: 10.7326/0003-4819-121-7-199410010-00003
153. Seko Y, Minota S, Kawasaki A, Shinkai Y, Maeda K, Yagita H, et al. Perforin-secreting killer cell infiltration and expression of a 65-kD heat-shock protein in aortic tissue of patients with Takayasu’s arteritis. J Clin Invest. (1994) 93:750–8. doi: 10.1172/JCI117029
154. Liu X, Jiang W, Wang Y, Sun M, Li Z, Wen Z. Reply. Arthritis Rheumatol. (2020) 72:697–8. doi: 10.1002/art.41197
155. Watanabe R. CD4+ T cells as key players in the immunopathology of Takayasu arteritis: comment on the article by Zhang et al. Arthritis Rheumatol. (2020) 72:696–7. doi: 10.1002/art.41198
156. Seko Y, Sugishita K, Sato O, Takagi A, Tada Y, Matsuo H, et al. Expression of costimulatory molecules (4-1BBL and Fas) and major histocompatibility class I chain-related A (MICA) in aortic tissue with Takayasu’s arteritis. J Vasc Res. (2004) 41:84–90. doi: 10.1159/000076437
157. Zhang J, Mai S, Chen H-M, Kang K, Li XC, Chen S-H, et al. Leukocyte immunoglobulin-like receptors in human diseases: an overview of their distribution, function, and potential application for immunotherapies. J Leukoc Biol. (2017) 102:351–60. doi: 10.1189/jlb.5MR1216-534R
158. Favier B. Regulation of neutrophil functions through inhibitory receptors: an emerging paradigm in health and disease. Immunol Rev. (2016) 273:140–55. doi: 10.1111/imr.12457
159. Lewis Marffy AL, McCarthy AJ. Leukocyte Immunoglobulin-Like Receptors (LILRs) on Human Neutrophils: modulators of Infection and Immunity. Front Immunol. (2020) 11:857. doi: 10.3389/fimmu.2020.00857
160. Legendre P, Régent A, Thiebault M, Mouthon L. Anti-endothelial cell antibodies in vasculitis: a systematic review. Autoimmun Rev. (2017) 16:146–53. doi: 10.1016/j.autrev.2016.12.012
161. Guilpain P, Mouthon L. Antiendothelial cells autoantibodies in vasculitis-associated systemic diseases. Clin Rev Allergy Immunol. (2008) 35:59–65. doi: 10.1007/s12016-007-8069-3
162. Gupta S. Surgical and immunological aspects of Takayasu’s disease. Ann R Coll Surg Engl. (1981) 63:325–32.
163. Nityanand S, Mishra K, Shrivastava S, Holm G, Lefvert AK. Autoantibodies against cardiolipin and endothelial cells in Takayasu’s arteritis: prevalence and isotype distribution. Br J Rheumatol. (1997) 36:923–4. doi: 10.1093/rheumatology/36.8.923
164. Eichhorn J, Sima D, Thiele B, Lindschau C, Turowski A, Schmidt H, et al. Anti-endothelial cell antibodies in Takayasu arteritis. Circulation. (1996) 94:2396–401. doi: 10.1161/01.cir.94.10.2396
165. Mutoh T, Shirai T, Ishii T, Shirota Y, Fujishima F, Takahashi F, et al. Identification of two major autoantigens negatively regulating endothelial activation in Takayasu arteritis. Nat Commun. (2020) 11:1253. doi: 10.1038/s41467-020-15088-0
166. Shirai T, Fujii H, Ono M, Watanabe R, Ishii T, Harigae H. An innovative method to identify autoantigens expressed on the endothelial cell surface: serological identification system for autoantigens using a retroviral vector and flow cytometry (SARF). Clin Dev Immunol. (2013) 2013:453058. doi: 10.1155/2013/453058
167. Shirai T, Fujii H, Ono M, Nakamura K, Watanabe R, Tajima Y, et al. novel autoantibody against fibronectin leucine-rich transmembrane protein 2 expressed on the endothelial cell surface identified by retroviral vector system in systemic lupus erythematosus. Arthritis Res Ther. (2012) 14:R157. doi: 10.1186/ar3897
168. Shirai T, Fujii H, Ono M, Watanabe R, Shirota Y, Saito S, et al. novel autoantibody against ephrin type B receptor 2 in acute necrotizing encephalopathy. J Neuroinflammation. (2013) 10:128. doi: 10.1186/1742-2094-10-128
169. Stone JH, Tuckwell K, Dimonaco S, Klearman M, Aringer M, Blockmans D, et al. Trial of tocilizumab in giant-cell arteritis. N Engl J Med. (2017) 377:317–28. doi: 10.1056/NEJMoa1613849
170. Nakaoka Y, Isobe M, Takei S, Tanaka Y, Ishii T, Yokota S, et al. Efficacy and safety of tocilizumab in patients with refractory Takayasu arteritis: results from a randomised, double-blind, placebo-controlled, phase 3 trial in Japan (the TAKT study). Ann Rheum Dis. (2018) 77:348–54. doi: 10.1136/annrheumdis-2017-211878
171. Watanabe R. Challenging therapeutic goal in large vessel vasculitis: comment on “Statins reduce relapse rate in Takayasu arteritis” published in the journal. Int J Cardiol. (2019) 287:120. doi: 10.1016/j.ijcard.2019.04.014
172. Keser G, Aksu K, Direskeneli H. Discrepancies between vascular and systemic inflammation in large vessel vasculitis: an important problem revisited. Rheumatol Oxf Engl. (2018) 57:784–90. doi: 10.1093/rheumatology/kex333
173. Savioli B, Salu BR, de Brito MV, Vilela Oliva ML, de Souza AWS. Silent arterial inflammation during the apparent remission state of Takayasu’s arteritis. What do cytokines tell us? Clin Exp Rheumatol. (2018) 36(Suppl. 111):33–9.
174. Liebling EJ, Peterson R, Victoria T, Burnham JM. Aortic ulceration in a tocilizumab-treated patient with Takayasu arteritis. Ann Rheum Dis. (2019) 78:e116. doi: 10.1136/annrheumdis-2018-214191
175. Johnson LR, Anis A, Olson K, Kusmirek J, Halabi A, Arnason J. Active aortitis and dissection in giant cell arteritis while on tocilizumab monotherapy. J Clin Rheumatol Pract Rep Rheum Musculoskelet Dis. (2020). doi: 10.1097/RHU.0000000000001423 [Epub ahead of print].
Keywords: giant cell arteritis, Takayasu arteritis, large vessel vasculitis, T cells, macrophages, NOTCH, co-stimulation, immune checkpoint
Citation: Watanabe R, Berry GJ, Liang DH, Goronzy JJ and Weyand CM (2020) Cellular Signaling Pathways in Medium and Large Vessel Vasculitis. Front. Immunol. 11:587089. doi: 10.3389/fimmu.2020.587089
Received: 24 July 2020; Accepted: 25 August 2020;
Published: 25 September 2020.
Edited by:
Sara Ferrando-Martinez, NeoImmuneTech Inc., United StatesReviewed by:
Matthew Koster, Mayo Clinic, United StatesPeter C. Grayson, National Institutes of Health (NIH), United States
Copyright © 2020 Watanabe, Berry, Liang, Goronzy and Weyand. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cornelia M. Weyand, cweyand@stanford.edu
†Present address: Ryu Watanabe, Department of Advanced Medicine for Rheumatic Diseases, Kyoto University Graduate School of Medicine, Kyoto, Japan