 Edoardo Errichiello1*
Edoardo Errichiello1* Efthimios Dardiotis2Fiorenza Mannino3
Efthimios Dardiotis2Fiorenza Mannino3 Juha Paloneva4,5
Juha Paloneva4,5 Teresa Mattina3Orsetta Zuffardi1
Teresa Mattina3Orsetta Zuffardi1- 1Unit of Medical Genetics, Department of Molecular Medicine, University of Pavia, Pavia, Italy
- 2Department of Neurology, University Hospital of Larissa, University of Thessaly, Larissa, Greece
- 3Department of Biomedical and Biotechnological Sciences, University of Catania, Catania, Italy
- 4Department of Surgery, Central Finland Hospital, Jyväskylä, Finland
- 5University of Eastern Finland, Kuopio, Finland
Nasu-Hakola disease (NHD) is a rare autosomal recessive disorder characterized by progressive presenile dementia and bone cysts, caused by variants in either TYROBP or TREM2. Despite the well-researched role of TREM2 and TYROBP/DAP12 in immunity, immunological phenotypes have never been reported in NHD patients. We initially diagnosed an Italian patient, using whole exome sequencing, with classical NHD clinical sequelae who additionally showed a decrease in NK cells and autoimmunity features underlined by the presence of autoantibodies. Based on this finding, we retrospectively explored the immunophenotype in another two NHD patients, in whom a low NK cell count and positive autoantibody serology were recorded. Accordingly, Trem2−/− mice show abnormal levels of circulating proinflammatory cytokines and the dysfunction of immune cells, whereas knockout mice for Tyrobp, encoding the adapter for TREM2, exhibit increased levels of autoantibodies and defective NK cell activity. Our findings tend to redefine NHD as a multisystem “immunological” disease, considering that osteoclasts are derived from the fusion of mononuclear myeloid precursors, whereas neurological anomalies in NHD are directly caused by microglia dysfunction.
Background
Nasu-Hakola disease (NHD; OMIM#221770 and #618193), also referred to as polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (PLOSL), is a rare autosomal recessive disease characterized by progressive presenile dementia and bone cysts (1). NHD is caused by loss-of-function variants in TREM2 and TYROBP/DAP12 (2, 3), which encode different proteins of the same receptor signaling complex, involved in the activation of the immune response. Furthermore, heterozygous variants in both genes have been involved in neurodegenerative disorders, such as frontotemporal dementia, Alzheimer's and Parkinson's diseases, and more recently, multiple system atrophy (4–6).
TREM2 (Triggering receptor expressed on myeloid cells 2) is a receptor of the immunoglobulin superfamily expressed on innate immune cells in different tissues, including the central nervous system, where it exerts a major role in regulating microglia, the sensor of neurodegeneration (7). Furthermore, TREM2 is involved in the differentiation of myeloid progenitors toward mature monocyte-derived dendritic cells (DCs), osteoclasts, and the microglia, which may be considered as the key players of NHD pathogenesis. Similarly, TYROBP/DAP12, the intracellular adaptor of TREM2, has been recognized as a key activating signal transduction element in natural killer (NK) cells (8, 9).
In this study, we provided the first evidence that NHD patients carrying biallelic variants in TREM2 or TYROBP may manifest immunological abnormalities, primarily a reduction of the NK cell population.
Clinical Data and Genetic Findings
Case 1 (Index Case)
The index patient, a 43 year-old woman of Italian origin, is the third child of healthy parents; the eldest sister is unaffected, while the second-born died perinatally. The patient developed alopecia universalis at age 8 and painful swollen ankles and recurrent pathological fractures at 25 (Figure 1A). X-rays showed bone cysts and generalized osteopenia (T-score −1.9); abdominal ultrasound imaging identified a mild hepatomegaly with hyperechoic hepatic hemangiomas, and an inhomogeneous uterus texture with cystic areas. A bone biopsy revealed the presence of fibro-adipose tissue surrounded by hemorrhagic foci.
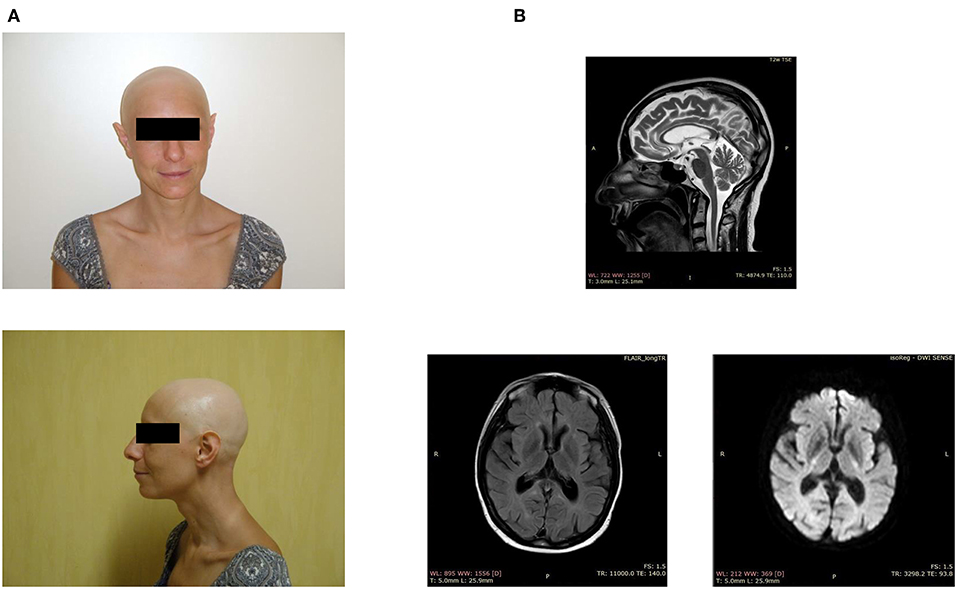
Figure 1. Clinical features of the index case. (A) Facial appearance of the patient, showing alopecia and sharp ears. (B) Brain MRI demonstrates periventricular white matter hyperintensities, ventriculomegaly, and a thin corpus callosum.
When the patient was around 40 years of age, we noticed the first neurological symptoms, manifesting as severe depression. The magnetic resonance imaging showed periventricular white matter hyperintensities, mild ventriculomegaly, and dilation of the subarachnoid spaces, small lacunar infarcts, a thin corpus callosum, and basal ganglia calcification (Figure 1B). The electroencephalogram, performed after the appearance of generalized seizures, revealed the absence of a normal alpha rhythm but persistent diffuse theta activity. During the next 3 years, the patient showed progressive dementia with motor disability, worsening gait impairment, sleep disturbances, spatial, and temporal disorientation, and marked memory loss, leading finally to a vegetative state.
The family history was negative for bone cysts, alopecia, dementia, as well as the manifestation of a dysfunctional immune system (Figure 2A).
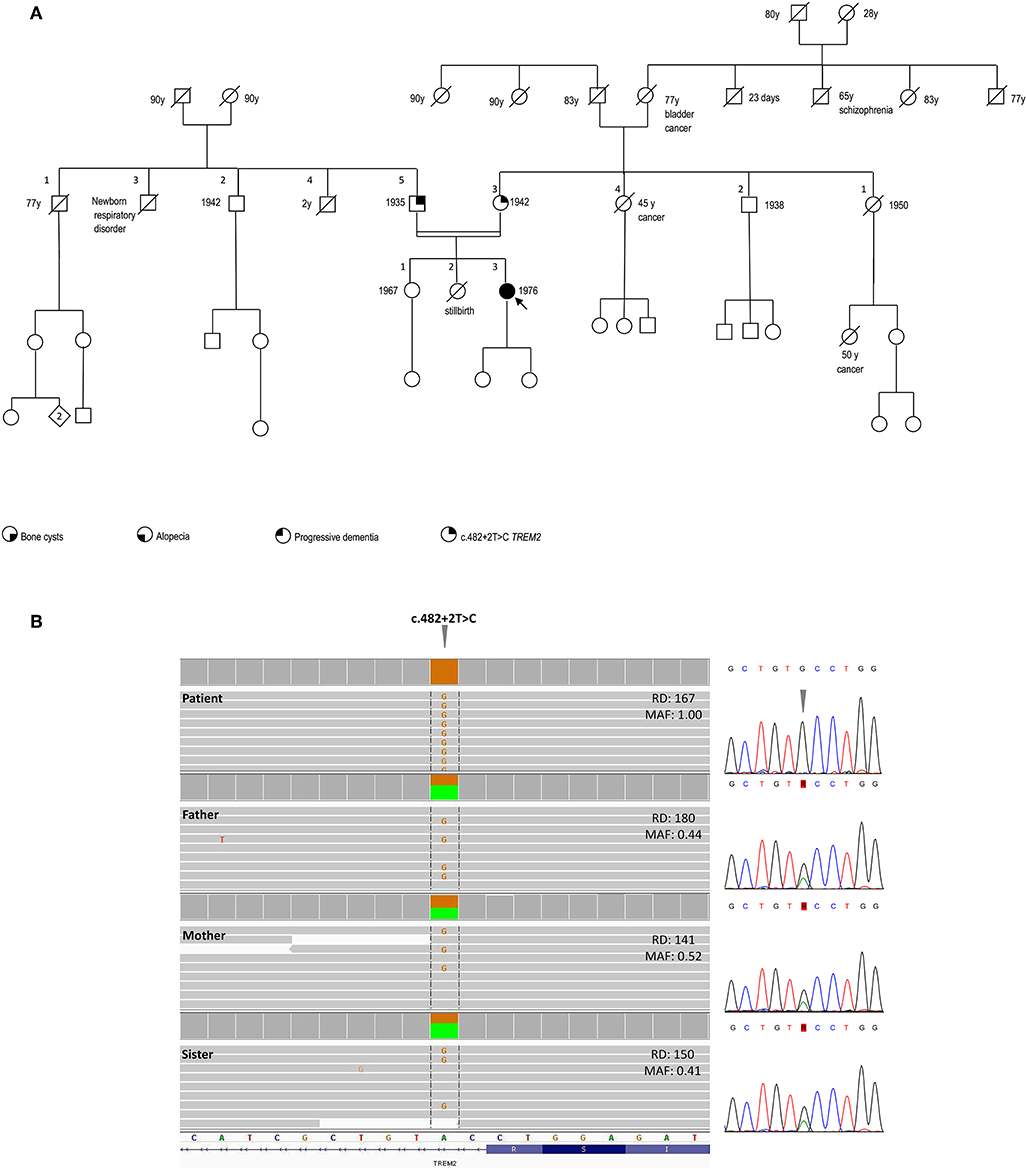
Figure 2. Pedigree and genetic findings of the index case. (A) Pedigree of the family and recorded clinical phenotypes. (B) IGV (Integrative Genomics View) visualization (right panel) of the chr6:41,127,528A>G (GRCh37/hg19) splice-site variant c.482+2T>C in patient (homozygous) and relatives (heterozygous). The variant, disrupting the donor splice site of TREM2 intron 3, is annotated in both ClinVar (#RCV000050138.1) and HGMD (#CS022111) as a disease-causing/likely pathogenic and has an extremely rare allele frequency in population databases (gnomAD: 0.0008126%; ExAC: 0.00165%), with only two reported heterozygous individuals in the European Non-Finnish population and no homozygotes. The variant is included in one of the numerous regions of homozygosity (chr6:24,747,411-42,365,687). Read depths (DP) and Mutant Allele Fraction (MAF) of the variant are shown for each member of the family. Sanger sequencing confirmation of the TREM2 variant is shown in the left panel.
SNP-array unveiled multiple regions of homozygosity (ROHs) in the patient's DNA likely due to the unawareness of parental consanguinity (inbreeding coefficient: 1/64, i.e., second cousins, Supplemental Table 1). Whole-exome sequencing (WES) was performed on DNA samples isolated from the peripheral blood of the patient, her parents, and the healthy sister. Libraries were generated using a commercial target enrichment kit (SureSelect Human All Exome V7, Agilent Technologies, Santa Clara, CA), and sequenced on the HiSeq 2500 platform (paired-end 2 × 100 bp; Illumina, San Diego, CA, USA), as we previously reported (10), obtaining a ~90X mean depth of coverage after mapping and removal of duplicated reads. WES identified a TREM2 homozygous splice-donor variant (NM_018965.3:c.482+2T>C) in the proband, falling in one of the detected ROHs and affecting the splicing of exon 3, as previously demonstrated (11). This variant was at the heterozygous state in the healthy parents and sister (Figure 2B).
Case 2
Case 2 is a 34 year-old female patient of Greek descent, originally reported by Dardiotis et al. (12). She had recurrent pathological fractures from the age of 28 and progressive cognitive and behavioral frontotemporal-like symptoms from the age of 30. At the age of 33, the patient showed a Mini-Mental State Examination (MMSE) score of 14/30, and a Montreal Cognitive Assessment (MoCA) score of 8/30. Computer tomography (CT) scan revealed cerebellar atrophy, diffuse low-density areas of the brain white matter, and subependymal and basal ganglia calcifications. Brain MRI showed hyperintensities of the white matter, brain atrophy, and a thin corpus callosum. Her condition worsened gradually, with gait instability, urinary incontinence, and memory loss. She was unable to stand and walk from the age of 37 and she underwent tracheostomy and gastrostomy at the age of 42.
The patient harbored the homozygous missense c.244G>T (p.Trp50Cys) variant in the exon 2 of TREM2.
Case 3
Case 3 is a Finnish patient, originally reported by Paloneva et al. (8). The patient showed typical neurological findings and clinical evolution of NHD, including bone cysts and recurrent fractures of the ankles and wrists. Although the complete clinical history of the patient is no longer available, obvious clinical autoimmunity signs were not reported in the clinical records.
The patient carried a homozygous deletion, encompassing exons 1–4 of TYROBP.
NK Cells Count and Autoantibody Screening
Flow cytometry analysis in the index case revealed low CD16+56+ NK cell counts (48 cells/μl, n.v. 90–590; 3%, n.v. 5–27%). Autoimmune screening was positive for anti-nuclear (ANA; 1:80), anti-smooth muscle (ASMA; 1:160), anti-parathyroid (1:80), and anti-Saccharomyces cerevisiae (ASCA; 1:80) autoantibodies.
Based on these results, we extended the immunological analysis to a previously diagnosed 34 year-old Greek patient harboring a homozygous missense variant in TREM2 (case 2), in which we detected a low number of NK cells (94 cells/μl; 4.3%) as well as positive anti-parietal cell antibody (APCA; 1:320). No other autoantibodies were detected in cases 1 and 2.
In addition, clinical data of the Finnish patient (case 3) with a homozygous deletion encompassing exons 1–4 of TYROBP, demonstrated a low NK cell count (3.5%).
No impairment of any other cell population was detected in the three patients.
Discussion
TREM2 and its binding partner TYROBP form an immunoreceptor signaling complex that orchestrates the differentiation of myeloid progenitors toward mature monocyte-derived dendritic cells (DCs), osteoclasts, microglia, and possibly a subset of oligodendrocytes originating in the myeloid lineage (13, 14). Curiously, TREM2 is located on 6p21.1, contiguous to other TREM family members—TREM and TREM-like receptors (TREM1, TREML1, TREML2, TREML3, TREML4) expressed on myeloid cells—and relatively close (~8 Mb) to the HLA gene complex, thus configuring interacting genomic clusters on chromosome 6p involved in immune regulation that may have originated during evolution.
Despite the well-researched role of TREM2 and TYROBP in innate and adaptive immunity, immunological phenotypes have never been described in NHD patients, although we cannot completely rule out that subtle immunological impairments have not been sought.
The careful immunological assessment of our index patient revealed autoimmunity signs, such as persistent autoantibodies, alopecia, malabsorption, joint pain, and muscle cramps. Our findings provide the first evidence of the role of TREM2 and TYROBP in the negative regulation of inflammatory responses in patients. Although we cannot completely rule the influence of additional genetic factors on autoimmune pre-disposition out, we excluded pathogenic variants in autoimmunity-associated loci or any other gene prioritized for immune system in the index case, in which the entire exome was analyzed.
Although further studies are needed, our preliminary findings are supported by multiple lines of evidence. First, Trem2 homozygous mutant mice show abnormal levels of circulating proinflammatory cytokines (IL-6, TNF, IFN) and dysfunction of the immune cells, osteoclasts, and microglia, as reported in the Mouse Genome Database (http://www.informatics.jax.org/). Similarly, knockout mice for Tyrobp, the other gene commonly associated with NHD encoding the adapter protein for TREM2, exhibits increased autoantibodies (e.g., anti-double stranded DNA) and IgG2a/3 levels, and defects of NK and T cells' regulatory function. Recently, Qu et al. (15) found an increased production of pro-inflammatory cytokines (IL-1β, IL-18, and CXCL2) and overactivation of the NLRP3 inflammasome pathway in Trem2−/− mice.
Second, TREM2/TYROBP interact with plenty of other autoimmune-related proteins and pathways: Syk/ZAP70 protein tyrosine kinases (9), associated with autosomal recessive infantile-onset multisystem autoimmune disease-2 (OMIM #17006); LCP2/SLP-76, regulating proinflammatory cytokines, and autoantibodies production (16); PLXNA1 and SEMA6D, which modulate T-cell activation and DC maturation (17); and MIF, implicated in numerous autoimmune inflammatory disorders (18). Interestingly, MIF overexpression has been found in the serum and lesioned skin of patients with alopecia (19), a symptom also observed in the index patient. Furthermore, in a proteomic analysis of lymphoblastoid cells from TREM2-NHD patients (20), the authors found differentially expressed proteins consisting in autoantigens for α-enolase/ENO1, as reported in rheumatoid arthritis, Hashimoto's encephalopathy, and Behçet's disease, as well as tuners of immunogenic/tolerogenic balance, such as VDAC (Voltage-dependent anion-selective channel protein), which modulates thymocyte survival.
Third and more importantly, we confirmed a decrease of the NK cell population and positive autoantibody serology/autoimmunity features in additional NHD patients. Interestingly, the 34 year-old Greek female patient with TREM2-associated NHD in this study, has been proven to carry a reduced number of NK cells as well as positive autoantibody serology, specifically APCA. Nevertheless, the patient does not currently show manifesting signs of autoimmune atrophic gastritis nor pernicious anemia (or type 1 diabetes/autoimmune thyroiditis), although it might be considered that APCA-related manifestations may develop over many years in APCA-positive symptomless patients (21). Curiously, phagocytosis and respiratory burst of the peripheral blood cells of the patient were not significantly impaired compared to the parents and healthy controls (12). However, our current immunological findings suggest that TREM2/TYROBP impairment may rather be reflected on the specific NK cell subpopulation.
It is worth noting that regulatory NK cells are crucial for preventing autoimmune and inflammatory diseases, although the underlying mechanism(s) and the contribution of NK education remain largely to be investigated (22). Remarkably, selective NK cell deficiency is extremely rare in humans (23). To date, a few patients have been reported to carry variants in one of the following genes: GATA2 (24), MCM4 (25, 26), RTEL1 (27), GINS1 (28), IRF8 (29), and FCGR3A (30). Frequently, these patients show extra-immune features—such as growth retardation, microcephaly, and adrenal insufficiency in individuals with homozygous variants in the MCM4 gene—that did not overlap with our patients' phenotypes. Furthermore, we excluded pathogenic variants in all genes associated with the aforementioned six monogenic disorders in our index case for which the entire exome was sequenced. Therefore, we speculate that both selective NK cell deficiency and associated autoimmunological signs we documented in our patients are not coincidental events in NHD.
In conclusion, our findings expand the phenotypic spectrum of NHD and further underline the tight interplay between the immune and central nervous systems in neurodegeneration. Recent studies found detectable TREM2 expression in whole blood cells, with higher levels in patients with Alzheimer's disease compared to healthy subjects (31, 32), whereas Galimberti et al. (33) recently identified a specific inflammatory expression profile in peripheral blood mononuclear cells from three NHD patients carrying the pathogenic p.Gln33X variant in TREM2, reinforcing our hypothesis that inflammation may play a role in the pathogenesis of NHD. We hypothesize that NHD could be redefined as a multisystem “immunological” disease, considering that osteoclasts are derived from the fusion of mononuclear myeloid precursors, whereas neurological anomalies in NHD can be caused by microglia dysfunction (“disease-associated microglia”) (34) and reactive gliosis (Figure 3). Anti-inflammatory drugs or repositioning/repurposing of myeloid-specific compounds may therefore, represent a therapeutic option for modulating early stages of NHD pathogenesis, thus possibly preventing the dramatic (and often lethal) progressive neurodegeneration that characterizes patients with NHD. Whether our index patient had benefited from a potential snRNA-based approach aiming to correct mis-splicing of TREM2-mutant transcript, as recently suggested (35), remains a promising speculative hypothesis.
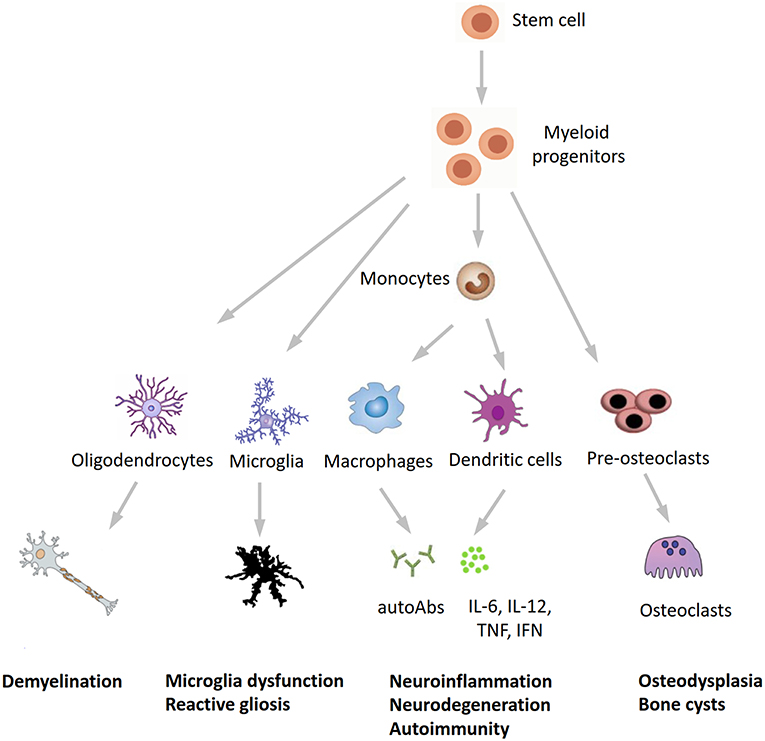
Figure 3. A novel hypothesis for the pathogenesis of Nasu-Hakola disease. TREM2/TYROBP, encoding different proteins of the same receptor signaling complex involved in the activation of the immune response, are expressed by osteoclasts, microglia, oligodendrocytes, dendritic cells, macrophages, and granulocytes. On the immune system side, the inactivation of TREM2 or TYROBP may lead to the overproduction of autoantibodies and pro-inflammatory cytokines by macrophages and dendritic cells, a process which is normally restrained by the physiological presence of TREM2 and TYROBP. Finally, the inactivation of TREM2 and TYROBP is reflected in the dysfunction of different bodily systems, including bones, CNS, and (auto)immunity, all sharing a common myeloid progenitor and pointing to Nasu-Hakola disease as a “myeloid cell disease.” autoAbs, autoantibodies.
Data Availability
All datasets analyzed for this study are included in the manuscript and the Supplementary Files.
Ethics Statement
The study was conducted in accordance with the Declaration of Helsinki and national guidelines. Written informed consent was obtained from the patient's guardian for the publication of this case report and any potentially identifying images/information.
Author Contributions
EE designed and conceptualized the study, analyzed the NGS data, made genotype-phenotype correlations, and drafted the manuscript. ED, FM, JP, and TM collected the clinical data. ED, JP, and TM helped in drafting the manuscript. OZ revised the manuscript for intellectual content and funded the study. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
EE benefits from a research position granted by the University of Pavia in the context of the strategic plan: MIGRAT.IN.G.–MIGRATions: toward an Interdisciplinary Governance model. This research was supported by a Grant of the Italian Ministry of Education, University and Research (MIUR) to the Department of Molecular Medicine of the University of Pavia under the initiative Dipartimenti di Eccellenza (2018–2022). This publication is distributed under the terms of open access policies implemented by the Italian Ministry of Education, University and Research (MIUR).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.01685/full#supplementary-material
Supplemental Table 1. Regions of homozygosity (ROHs) in the index patient's genome.
References
1. Paloneva J, Autti T, Raininko R, Partanen J, Salonen O, Puranen M, et al. CNS manifestations of Nasu-Hakola disease: a frontal dementia with bone cysts. Neurology. (2001) 56:1552–8. doi: 10.1212/WNL.56.11.1552
2. Paloneva J, Manninen T, Christman G, Hovanes K, Mandelin J, Adolfsson R, et al. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am J Hum Genet. (2002) 71:656–62. doi: 10.1086/342259
3. Klünemann HH, Ridha BH, Magy L, Wherrett JR, Hemelsoet DM, Keen RW, et al. The genetic causes of basal ganglia calcification, dementia, and bone cysts: DAP12 and TREM2. Neurology. (2005) 64:1502–7. doi: 10.1212/01.WNL.0000160304.00003.CA
4. Cuyvers E, Bettens K, Philtjens S, Van Langenhove T, Gijselinck I, van der Zee J, et al. Investigating the role of rare heterozygous TREM2 variants in Alzheimer's disease and frontotemporal dementia. Neurobiol Aging. (2014) 35:726.e11–9. doi: 10.1016/j.neurobiolaging.2013.09.009
5. Liu G, Liu Y, Jiang Q, Jiang Y, Feng R, Zhang L, et al. Convergent genetic and expression datasets highlight TREM2 in Parkinson's disease susceptibility. Mol Neurobiol. (2016) 53:4931–8. doi: 10.1007/s12035-015-9416-7
6. Ogaki K, Heckman MG, Koga S, Martens YA, Labbé C, Lorenzo-Betancor O, et al. Association study between multiple system atrophy and TREM2 p.R47H. Neurol Genet. (2018) 4:e257. doi: 10.1212/NXG.0000000000000257
7. Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. (2017) 47:566–81.e9. doi: 10.1016/j.immuni.2017.08.008
8. Paloneva J, Kestilä M, Wu J, Salminen A, Böhling T, Ruotsalainen V, et al. Loss-of-function mutations in TYROBP (DAP12) result in a presenile dementia with bone cysts. Nat Genet. (2000) 25:357–61. doi: 10.1038/77153
9. Hamerman JA, Ni M, Killebrew JR, Chu CL, Lowell CA. The expanding roles of ITAM adapters FcRgamma and DAP12 in myeloid cells. Immunol Rev. (2009) 232:42–58. doi: 10.1111/j.1600-065X.2009.00841.x
10. Errichiello E, Vetro A, Mina T, Wischmeijer A, Berrino E, Carella M, et al. Whole exome sequencing in the differential diagnosis of Diamond-Blackfan anemia: clinical and molecular study of three patients with novel RPL5 and mosaic RPS19 mutations. Blood Cells Mol Dis. (2017) 64:38–44. doi: 10.1016/j.bcmd.2017.03.002
11. Numasawa Y, Yamaura C, Ishihara S, Shintani S, Yamazaki M, Tabunoki H, et al. Nasu-Hakola disease with a splicing mutation of TREM2 in a Japanese family. Eur J Neurol. (2011) 18:1179–83. doi: 10.1111/j.1468-1331.2010.03311.x
12. Dardiotis E, Siokas V, Pantazi E, Dardioti M, Rikos D, Xiromerisiou G, et al. A novel mutation in TREM2 gene causing Nasu-Hakola disease and review of the literature. Neurobiol Aging. (2017) 53:194.e13–22. doi: 10.1016/j.neurobiolaging.2017.01.015
13. Colonna M. TREMs in the immune system and beyond. Nat Rev Immunol. (2003) 3:445–53. doi: 10.1038/nri1106
14. Tomasello E, Vivier E. KARAP/DAP12/TYROBP: three names and a multiplicity of biological functions. Eur J Immunol. (2005) 35:1670–7. doi: 10.1002/eji.200425932
15. Qu W, Wang Y, Wu Y, Liu Y, Chen K, Liu X, et al. Triggering receptors expressed on myeloid cells 2 promotes corneal resistance against pseudomonas aeruginosa by inhibiting caspase-1-dependent pyroptosis. Front Immunol. (2018) 9:1121. doi: 10.3389/fimmu.2018.01121
16. Siggs OM, Miosge LA, Daley SR, Asquith K, Foster PS, Liston A, Goodnow CC. Quantitative reduction of the TCR adapter protein SLP-76 unbalances immunity and immune regulation. J Immunol. (2015) 194:2587–95. doi: 10.4049/jimmunol.1400326
17. Takegahara N, Takamatsu H, Toyofuku T, Tsujimura T, Okuno T, Yukawa K, et al. Plexin-A1 and its interaction with DAP12 in immune responses and bone homeostasis. Nat Cell Biol. (2006) 8:615–22. doi: 10.1038/ncb1416
18. Yoo SA, Leng L, Kim BJ, Du X, Tilstam PV, Kim KH, et al. MIF allele-dependent regulation of the MIF coreceptor CD44 and role in rheumatoid arthritis. Proc Natl Acad Sci USA. (2016) 113:E7917–26. doi: 10.1073/pnas.1612717113
19. Salem SA, Asaad MK, Elsayed SB, Sehsah HM. Evaluation of macrophage migration inhibitory factor (MIF) levels in serum and lesional skin of patients with alopecia areata. Int J Dermatol. (2016) 55:1357–61. doi: 10.1111/ijd.13344
20. Giuliano S, Agresta AM, De Palma A, Viglio S, Mauri P, Fumagalli M, et al. Proteomic analysis of lymphoblastoid cells from Nasu-Hakola patients: a step forward in our understanding of this neurodegenerative disorder. PLoS ONE. (2014) 9:e110073. doi: 10.1371/journal.pone.0110073
21. Neumann WL, Coss E, Rugge M, Genta RM. Autoimmune atrophic gastritis–pathogenesis, pathology and management. Nat Rev Gastroenterol Hepatol. (2013) 10:529–41. doi: 10.1038/nrgastro.2013.101
22. Boudreau JE, Hsu KC. Natural killer cell education in human health and disease. Curr Opin Immunol. (2018) 50:102–11. doi: 10.1016/j.coi.2017.11.003
23. Mace EM, Orange JS. Emerging insights into human health and NK cell biology from the study of NK cell deficiencies. Immunol Rev. (2019) 287:202–25. doi: 10.1111/imr.12725
24. Mace EM, Hsu AP, Monaco-Shawver L, Makedonas G, Rosen JB, Dropulic L, et al. Mutations in GATA2 cause human NK cell deficiency with specific loss of the CD56(bright) subset. Blood. (2013) 121:2669–77. doi: 10.1182/blood-2012-09-453969
25. Gineau L, Cognet C, Kara N, Lach FP, Dunne J, Veturi U, et al. Partial MCM4 deficiency in patients with growth retardation, adrenal insufficiency, and natural killer cell deficiency. J Clin Invest. (2012) 122:821–32. doi: 10.1172/JCI61014
26. Hughes CR, Guasti L, Meimaridou E, Chuang CH, Schimenti JC, King PJ, et al. MCM4 mutation causes adrenal failure, short stature, and natural killer cell deficiency in humans. J Clin Invest. (2012) 122:814–20. doi: 10.1172/JCI60224
27. Hanna S, Béziat V, Jouanguy E, Casanova JL, Etzioni A. A homozygous mutation of RTEL1 in a child presenting with an apparently isolated natural killer cell deficiency. J Allergy Clin Immunol. (2015) 136:1113–4. doi: 10.1016/j.jaci.2015.04.021
28. Cottineau J, Kottemann MC, Lach FP, Kang YH, Vély F, Deenick EK, et al. Inherited GINS1 deficiency underlies growth retardation along with neutropenia and NK cell deficiency. J Clin Invest. (2017) 127:1991–2006. doi: 10.1172/JCI90727
29. Mace EM, Bigley V, Gunesch JT, Chinn IK, Angelo LS, Care MA, et al. Biallelic mutations in IRF8 impair human NK cell maturation and function. J Clin Invest. (2017) 127:306–20. doi: 10.1172/JCI86276
30. Grier JT, Forbes LR, Monaco-Shawver L, Oshinsky J, Atkinson TP, Moody C, et al. Human immunodeficiency-causing mutation defines CD16 in spontaneous NK cell cytotoxicity. J Clin Invest. (2012) 122:3769–80. doi: 10.1172/JCI64837
31. Tan YJ, Ng ASL, Vipin A, Lim JKW, Chander RJ, Ji F, et al. Higher peripheral TREM2 mRNA levels relate to cognitive deficits and hippocampal atrophy in Alzheimer's disease and amnestic mild cognitive impairment. J Alzheimers Dis. (2017) 58:413–23. doi: 10.3233/JAD-161277
32. Casati M, Ferri E, Gussago C, Mazzola P, Abbate C, Bellelli G, et al. Increased expression of TREM2 in peripheral cells from mild cognitive impairment patients who progress into Alzheimer's disease. Eur J Neurol. (2018) 25:805–10. doi: 10.1111/ene.13583
33. Galimberti D, Fenoglio C, Ghezzi L, Serpente M, Arcaro M, D'Anca M, et al. Inflammatory expression profile in peripheral blood mononuclear cells from patients with Nasu-Hakola Disease. Cytokine. (2019) 116:115–9. doi: 10.1016/j.cyto.2018.12.024
34. Deczkowska A, Keren-Shaul H, Weiner A, Colonna M, Schwartz M, Amit I. Disease-associated microglia: a universal immune sensor of neurodegeneration. Cell. (2018) 173:1073–81. doi: 10.1016/j.cell.2018.05.003
Keywords: Nasu-Hakola disease (NHD), TREM2, TYROBP, NK cells, autoantibodies, microglia, neurodegeneration, bone cysts
Citation: Errichiello E, Dardiotis E, Mannino F, Paloneva J, Mattina T and Zuffardi O (2019) Phenotypic Expansion in Nasu-Hakola Disease: Immunological Findings in Three Patients and Proposal of a Unifying Pathogenic Hypothesis. Front. Immunol. 10:1685. doi: 10.3389/fimmu.2019.01685
Received: 17 April 2019; Accepted: 04 July 2019;
Published: 23 July 2019.
Edited by:
Jorge Matias-Guiu, Complutense University of Madrid, SpainReviewed by:
Daniela Galimberti, University of Milan, ItalyRomain Marignier, Hospices Civils de Lyon, France
Copyright © 2019 Errichiello, Dardiotis, Mannino, Paloneva, Mattina and Zuffardi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Edoardo Errichiello, edoardo.errichiello@unipv.it