 Jessica Hoppstädter1*
Jessica Hoppstädter1* Alaina J. Ammit2,3
Alaina J. Ammit2,3- 1Department of Pharmacy, Pharmaceutical Biology, Saarland University, Saarbrücken, Germany
- 2Faculty of Science, School of Life Sciences, University of Technology Sydney, Sydney, NSW, Australia
- 3Woolcock Emphysema Centre, Woolcock Institute of Medical Research, University of Sydney, Sydney, NSW, Australia
Glucocorticoids (GCs) potently inhibit pro-inflammatory responses and are widely used for the treatment of inflammatory diseases, such as allergies, autoimmune disorders, and asthma. Dual-specificity phosphatase 1 (DUSP1), also known as mitogen-activated protein kinase (MAPK) phosphatase-1 (MKP-1), exerts its effects by dephosphorylation of MAPKs, i.e., extracellular-signal-regulated kinase (ERK), p38, and c-Jun N-terminal kinase (JNK). Endogenous DUSP1 expression is tightly regulated at multiple levels, involving both transcriptional and post-transcriptional mechanisms. DUSP1 has emerged as a central mediator in the resolution of inflammation, and upregulation of DUSP1 by GCs has been suggested to be a key mechanism of GC actions. In this review, we discuss the impact of DUSP1 on the efficacy of GC-mediated suppression of inflammation and address the underlying mechanisms.
Introduction
Glucocorticoids (GCs) are steroid hormones with immunosuppressive activity that are used to treat a wide variety of inflammatory conditions, including rheumatoid arthritis, pulmonary diseases, and acute inflammation caused by microbial infection.
Anti-inflammatory properties of GCs are partially dependent on their ability to suppress mitogen-activated protein kinases (MAPKs) (1, 2). MAPKs are a family of protein kinases that respond to a wide variety of extracellular stimuli. They are activated by phosphorylation of tyrosine and threonine residues within their active domains and are inactivated by dephosphorylation of either residue (2–4). MAPK cascades are evolutionary conserved and control a large number of cellular processes, including proliferation, differentiation, apoptosis, motility, and stress responses. The three major signaling cascades either involve extracellular signal-regulated kinase 1/2 (ERK1/2), c-Jun N-terminal kinase (JNK), or p38 MAPK (2–4). Dysregulation of MAPK activity has been suggested to contribute to the onset of many pathologies, including neurodegenerative diseases, diabetes, cancer, and inflammation (4–7).
MAPKs can be dephosphorylated by tyrosine-specific phosphatases, serine-threonine phosphatases, or dual-specificity (Thr/Tyr) phosphatases (DUSPs) (8, 9). GC treatment primarily attenuates MAPK signaling via DUSP1, also known as mitogen-activated protein kinase phosphatase-1 (MKP-1) (10–12). In this review, we discuss the influence of DUSP1 on GC-mediated effects (Figure 1).
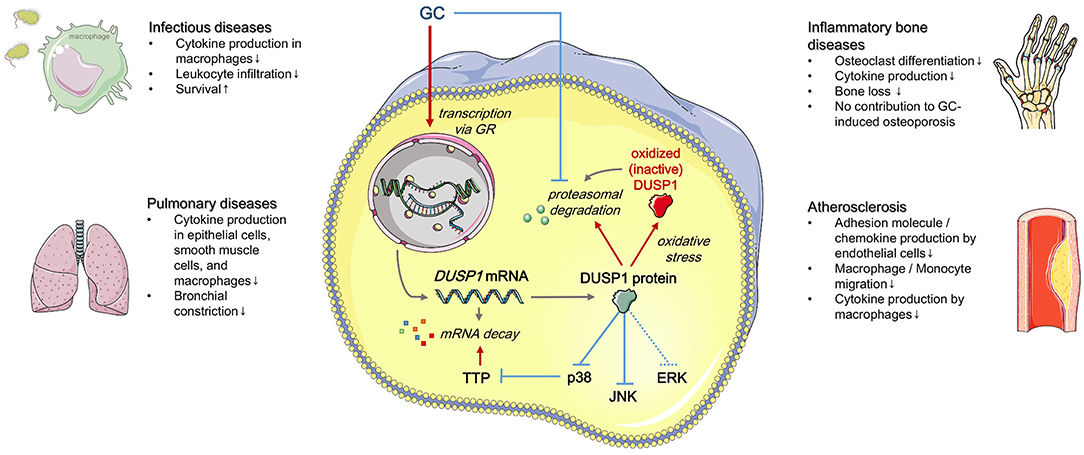
Figure 1. Regulation and effects of GC-induced DUSP1 expression (simplified). Red: positive regulation, blue: negative regulation. See text for details. Templates from Servier Medical Art (http://www.servier.com) were used to generate the figure.
Dual-Specificity Phosphatase 1 (DUSP1)
Although DUSP1 was initially identified as an ERK-specific phosphatase, p38 MAPK and JNK are its preferred substrates in several cell types, including myeloid cells (13–16). Thus, DUSP1 activity limits p38 and JNK-dependent pro-inflammatory gene transcription (17–20).
However, DUSP1 is also involved in the regulation of anti-inflammatory genes. Over-production of IL-10 in Dusp1−/− mice was observed in peritonitis models after lipopolysaccharide (LPS) challenge or infection with Escherichia coli or Staphylococcus aureus, and LPS-treated Dusp1−/− macrophages. This can be explained by the interaction of DUSP1 with the RNA-binding protein tristetraprolin (TTP, gene name Zfp36): increased p38-mediated phosphorylation of TTP results in its inactivation, followed by accumulation of the inactive, but stable, form of TTP and enhanced stability of TTP target mRNAs. These target mRNAs comprise pro-inflammatory chemokines and cytokines, e.g., Tnf, Cxcl1, and Cxcl2, but also the anti-inflammatory Il10. Approximately 50% of the genes dysregulated in Dusp1−/− macrophages are affected by TTP inactivation (21).
The promoter region of the Dusp1 gene contains binding sites for several transcription factors, including activator protein 1 (AP-1), nuclear factor-κB (NF-κB), cAMP response element-binding protein (CREB), and the glucocorticoid receptor (GR) (22–25). Hence, DUSP1 can be induced under various conditions, ranging from inflammatory activation to altered cellular metabolism and GC excess during stress responses. GCs may further enhance DUSP1 expression by inhibiting its proteasomal degradation (12).
DUSP1 has been shown in a few studies to be regulated via the stability of its mRNA. Several mRNA binding proteins can influence Dusp1 mRNA stability. TTP-mediated Dusp1 mRNA decay has been suggested to be a feedback mechanism in inflammatory responses by which TTP limits its own activity: reduced DUSP1 expression enhances p38 MAPK phosphorylation, thereby promoting TTP inactivation (26). Besides, several miRNAs, such as miR-101, have been shown to modulate DUSP1 expression (27). Posttranslational DUSP1 modifications include phosphorylation, acetylation, and oxidation. ERK-mediated phosphorylation of DUSP1 can either lead to increased or decreased DUSP1 protein stability, depending on the phosphorylation site (28, 29). Acetylation of Lys57 results in increased phosphatase activity and more effective suppression of the MAPK signaling cascades (30, 31). In contrast, oxidation of Cys258 within the active site inactivates DUSP1 and leads to its rapid degradation by the proteasome. In this manner, DUSP1 oxidation prolongs MAPK activation, ultimately resulting in enhanced inflammatory responses (32–34). S-glutathionylation of Cys258 has similar effects, indicating that DUSP1 activity is redox-sensitive (35).
Role of DUSP1 in Inflammatory Diseases and Its Influence on GC Treatment Efficacy
Infectious Diseases and Sepsis
In the context of infectious diseases and sepsis, research on the role of DUSP1 focused mainly on macrophage responses. Macrophages are a subtype of innate immune cells with high plasticity that play a crucial role in acute inflammation. They recognize pathogen- or danger-associated molecular patterns via pattern recognition receptors, such as toll-like receptors (TLRs). Stimulation of macrophages initially leads to excessive inflammation, followed by a phenotypic switch toward an anti-inflammatory and wound-healing phenotype that promotes the resolution of inflammation (36, 37).
Early studies on the role of DUSP1 in the response of macrophages to bacterial LPS suggested that DUSP1 is required to balance inflammatory responses in sepsis and infectious diseases. The ectopic expression of DUSP1 in LPS-stimulated macrophages accelerated JNK and p38 inactivation and substantially inhibited the production of TNF-α and IL-6 (38). Moreover, increased cytokine production and elevated expression of the differentiation markers CD86 and CD40 were observed in macrophages from Dusp1−/− mice when activated by TLR ligands. Dusp1−/− macrophages also showed enhanced constitutive and TLR-induced activation of p38 MAPK (39). Moreover, LPS-induced IFN-β production was increased in Dusp1−/− macrophages, both due to elevated JNK-mediated activation of cJun and Ifnb mRNA stabilization by TTP inactivation (20). DUSP1 induction has also been shown to be involved in endogenous feedback loops initiated by either adenosine or prostaglandin E2 signaling that skew macrophages toward an anti-inflammatory phenotype (40, 41).
Several studies confirmed the relevance of these in vitro findings for the in vivo situation. In LPS-treated mice, DUSP1 is upregulated in various tissues and cell types and limits p38 MAPK activation. In accordance, depletion of DUSP1 led to the excessive release of inflammatory cytokines, such as TNF-α, IL-6, CCL3, and CCL4, and increased LPS-induced mortality (14, 15, 39, 42). Likewise, Dusp1−/− mice showed amplified inflammatory responses and lethality after infection with either S. aureus (43) or E. coli (44).
The phenotype of Dusp1−/− mice in two sophisticated models of sepsis, i.e., caecal ligation and puncture (CLP) and colon ascendens stent peritonitis (CASP), strongly resembled those observed after LPS shock, with highly increased levels of IL-6, CCL3, and CCL4 and excess lethality (45).
Glucocorticoids induce DUSP1 in mouse macrophages, and DUSP1 is required for the inhibition of JNK and p38 MAPK by dexamethasone in these cells (10, 38). Consequently, the GC-mediated shift toward an anti-inflammatory macrophage phenotype was attenuated in cells from Dusp1−/− mice (10, 46). In a cutaneous air pouch model, the zymosan-induced production of pro-inflammatory mediators and the infiltration of leukocytes into a pre-formed dorsal cavity were inhibited by oral dexamethasone administration in wild-type, but not in Dusp1−/−, mice, suggesting that DUSP1 is indeed required to unfold the full anti-inflammatory potential of GCs (10). In another study, the reduction of TNF-α-induced mortality caused by pretreatment with dexamethasone was dependent on the presence of DUSP1: whereas wildtype mice were entirely protected by dexamethasone administration, Dusp1−/− animals did not benefit from the GC treatment (1).
In conclusion, both in vitro and in vivo evidence suggests that DUSP1 critically contributes to the resolution of acute inflammatory responses and mediates protective GC effects in this context.
Inflammatory Bone Disorders
The bone mass is subject to constant remodeling orchestrated by osteoblasts and osteoclasts. In inflammatory bone disorders, e.g., autoimmune-driven rheumatoid arthritis or pathogen-induced periodontitis, the balance of osteoblast and osteoclast activity is compromised, resulting in bone loss (47).
DUSP1 was strongly downregulated in synovial biopsies from patients with rheumatoid arthritis and osteoarthritis (GEO datasets GDS5401 and GDS5403; Figure 2A), suggesting that DUSP1 deficiency may contribute to disease progression.
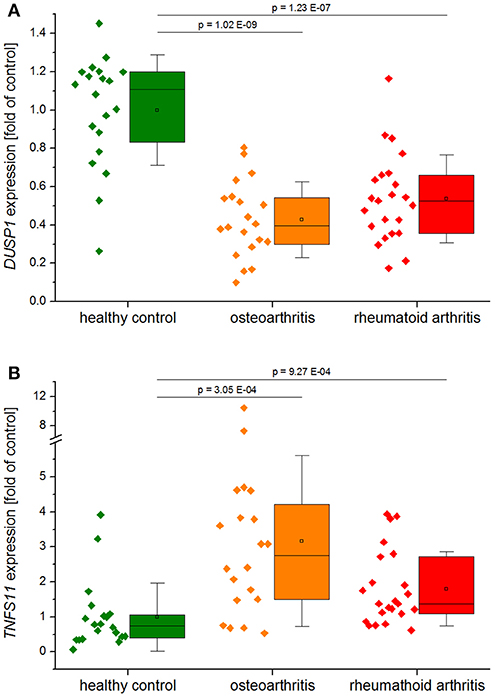
Figure 2. DUSP1 (A) and TNFSF11 (RANKL, B) expression in synovial tissues from healthy controls, patients with rheumatoid arthritis, or osteoarthritis. Data obtained from GEO Datasets GDS5401 (Berlin dataset) and GDS5403 (Jena dataset) were normalized against their respective healthy control values before compilation. Data are shown as individual values per sample, and boxplots show the 25–75th percentiles (box), mean (square), median (line), and standard deviation (whiskers). P-values were generated by one-way ANOVA and Bonferroni's post-hoc test (A, normal distribution) or Mann–Whitney U-test (B, not normally distributed).
DUSP1 indeed effectively reduced osteolysis in studies utilizing mouse models of LPS-induced inflammatory bone loss and collagen-induced arthritis (CIA) (48, 49). Dusp1−/− mice showed excessive bone loss, more inflammatory infiltrates, and an increase in osteoclastogenesis at the site of LPS-injection in a model of experimental periodontitis (49). In line with these findings, adenovirus-mediated overexpression of DUSP1 was shown to protect against bone loss in a similar experimental model of periodontal disease (50). Furthermore, Dusp1−/− mice exhibited higher penetrance, earlier onset, and increased severity of experimental arthritis, accompanied by higher numbers of osteoclasts in inflamed joints and more extensive loss of bone mass. Complementary in vitro experiments showed that DUSP1 acts as a negative regulator of osteoclast formation and activation via suppression of p38 MAPK (48, 51). A recently published study showed that the presence of calcium crystals, which are critical factors in the pathogenesis of osteoarthritis, stimulate receptor activator of NF-κB ligand (RANKL) secretion by osteoblasts via DUSP1 downregulation, thereby promoting osteoclastogenesis (52). RANKL induction was also observed in synovial biopsies from arthritis patients in the GEO datasets mentioned above (Figure 2B). Moreover, overexpression of DUSP1 in fibroblast-like synoviocytes from osteoarthritis patients inhibited the expression of osteoarthritis-associated mediators (53).
However, DUSP1 depletion did not affect age-related spontaneously occurring osteoarthritis, since knockout mice showed a similar disease progression compared to controls at 21 months of age (54). Thus, the modulatory function of DUSP1 in the context of bone homeostasis seems to be most evident in the presence of a potent inflammatory trigger.
Due to their high anti-inflammatory capacity and their ability to decrease radiologic disease progression, GCs are frequently used for the treatment of rheumatoid arthritis. Paradoxically, one common side effect of GC use, primarily when used at high dosages or over prolonged periods, is a loss of bone mass, also known as GC-induced osteoporosis. This adverse effect is associated with increased osteoclastogenesis and depletion of osteoblasts (55, 56).
DUSP1 has been suggested to contribute to GC-induced bone loss since GC-inducible attenuation of osteoblast proliferation involves inhibition of the MAPK/ERK signaling pathway and can be reversed by the protein tyrosine phosphatase (PTP) inhibitor vanadate in vitro and in vivo (57–59). The assumption that the PTP in question might be DUSP1 was, however, not supported by studies with Dusp1−/− mice, which demonstrated that GC-induced bone loss was not prevented upon DUSP1 depletion: after treatment with the GC methylprednisolone for 28 days, both wildtype and Dusp1−/− mice showed a similar reduction of osteoid surfaces, volumes, and osteoblast numbers (60).
In summary, loss of DUSP1 favors bone loss, especially under highly inflammatory conditions. Further studies are required to clarify whether DUSP1 contributes to the beneficial or adverse effects of GCs in the therapy of bone-related diseases.
Pulmonary Diseases
GCs are first line anti-inflammatory medicines in chronic respiratory diseases, including asthma and chronic obstructive pulmonary disease (COPD), and are commonly used therapeutically as inhaled corticosteroids (ICS). ICS effectively control inflammation in asthma but are less effective in COPD. This is thought to be due to corticosteroid insensitivity, where the molecular pathways responsible for the effect of GCs have been modified by oxidative stress or infections (61). Moreover, a subset of asthmatics (~10%) are refractory to ICS and classified as having severe asthma. DUSP1 has been shown to contribute to the effects of GCs in several in vitro, ex vivo, and in vivo studies with relevance to respiratory disease (61–63) and in some key studies, an impact on DUSP1 function has been shown to be responsible for corticosteroid insensitivity/resistance. For instance, an ex vivo study examined the repressive effect of GCs on stimulated production of inflammatory cytokines by alveolar macrophages from patients with severe asthma to those with non-severe asthma. GCs were less effective in macrophages from severe asthma patients, and this GC insensitivity was linked with increased p38 MAPK activation and impaired inducibility of DUSP1 (64).
The first to demonstrate that GCs upregulated DUSP1 in primary airway smooth muscle cells were Issa et al. (65). This was confirmed in a publication by the Ammit group that showed that GC-induced DUSP1 controlled cytokine mRNA stability in a p38 MAPK-mediated manner (66). Notably, knockdown of DUSP1 with siRNA showed that GC-induced DUSP1 was a significant contributor to anti-inflammatory effects at the post-transcriptional level.
Several ex vivo and in vivo studies utilizing Dusp1−/− mice highlighted the contribution of DUSP1 to GC effects in respiratory disease. For example, GC-mediated repression of the contractile response in bronchial rings from mice was abrogated by Dusp1 depletion (67). Interestingly, the anti-inflammatory impact of DUSP1 was lost in ozone-exposed mice in a model that may recapitulate corticosteroid resistance in severe asthma (68). A plausible explanation is that GC-induced DUSP1 in the wild-type mice was oxidized by ozone and rendered non-functional. Oxidization of DUSP1 may prove to be a roadblock to further development of DUSP1 as a therapeutic target in respiratory disease as oxidative stress is a well-appreciated feature of COPD and other conditions where smoking is a risk factor (69, 70).
Finally, there are publications that note that GC-mediated effects in respiratory disease are DUSP1-independent. These include a study that detected gene expression of known GC targets in biopsies from allergen-challenged asthmatic subjects (71). Evidence from studies utilizing Dusp1−/− mice in models with relevance to asthma is somewhat equivocal and does not fully support the assertion that DUSP1 is a significant contributor to the effect of GCs in vivo (72).
Atherosclerosis
GCs are not a therapeutic option for the treatment of atherosclerosis, since side effects of long-term GC treatment include hyperglycemia, hypertension, dyslipidemia, and obesity and may, therefore, promote adverse cardiovascular events (73, 74). However, as inflammation plays a significant role in the pathogenesis in atherosclerosis, GCs may exert some anti-atherosclerotic effects. Early studies demonstrated that dexamethasone reduced the severity of atherosclerosis in experimental rabbit models (75–77). Moreover, vein graft thickening was prevented by short-term dexamethasone treatment in hypercholesterolemic mice (78). The development of a drug-eluting bioadhesive gel that allowed to dissociate the systemic adverse and local anti-inflammatory effects of GC treatment. In atherosclerotic mice, inflamed plaques treated with GC-eluting adhesive gels showed reduced macrophage numbers and developed protective fibrous caps covering the plaque core. This was paralleled by lowered plasma cytokine levels and biomarkers of inflammation in the plaque (79).
The onset of atherosclerosis is triggered by proinflammatory mediators, which induce adhesion molecules in endothelial cells (ECs) by activating MAPKs, particularly p38 MAPK. Dexamethasone-induced DUSP1 upregulation caused inactivation of p38 MAPK in TNF-α-treated ECs and mediated inhibition of E-selectin expression, as shown in murine Dusp1−/− ECs and human ECs upon DUSP1 silencing (80).
The assumption that DUSP1 is atheroprotective via inhibition of EC activation was further supported by studies investigating the influence of shear stress. ECs respond to shear stress via mechanoreceptors that translate mechanical distortions into various molecular signals, including GR translocation (81, 82). Regions of the arterial tree exposed to high shear stress are protected from endothelial activation, inflammation, and atherosclerosis, whereas regions exposed to low or oscillatory shear stress, are susceptible (83, 84). The expression of DUSP1 in cultured ECs was elevated by shear stress, whereas vascular cell adhesion protein (VCAM)-1 levels were reduced; silencing of DUSP1 restored VCAM-1 expression. In vivo, DUSP1 was preferentially expressed by ECs in a high-shear, protected region of the mouse aorta and was necessary for the suppression of EC activation (84).
Apart from its effect on the endothelium, DUSP1 also determines the monocyte/macrophage phenotype in atherosclerosis (35, 85, 86).
Metabolic stress was shown to induce the S-glutathionylation, inactivation, and subsequent degradation of DUSP1 in monocytes. As a result, increased p38 MAPK and ERK activity primed monocytes for chemokine-induced recruitment, thereby promoting monocyte adhesion and migration. In vivo, transplantation of DUSP1-deficient bone marrow into atherosclerosis-prone mice exacerbated atherosclerotic lesion formation by sensitizing monocytes to chemoattractants and polarizing macrophages toward an inflammatory phenotype (35, 86). Thus, monocyte and macrophage dysregulation by metabolic stress may drive the progression of atherosclerosis due to DUSP1 inactivation.
Interestingly, the administration of inhaled GCs has been suggested to be atheroprotective in asthma patients, although plasma levels of the drug were presumed to be very low and were not sufficient to provoke cardiovascular GC side effects (87). Whether this observation might be due to elevated DUSP1 expression or activity in ECs or the monocyte/macrophage compartment presently remains elusive.
DUSP1: a Therapeutic Target?
As underscored by this review, there are several clinical areas where targeting DUSP1 (i.e., increasing its amount and/or activity) would be clinically beneficial. These may also comprise psoriasis or colitis, as a number of studies suggested an involvement of DUSP1 downregulation in the pathogenesis of these diseases (88–91).
Novel ligands to upregulate DUSP1 levels might represent an attractive anti-inflammatory strategy—particularly in atherosclerosis, where GCs cannot be used due to their cardiovascular side effects. Corticosteroid-sparing strategies to reduce the GC dose while achieving effective disease control have always been of clinical importance, and this is also a potential area of focus for DUSP1 upregulators. The failure of p38 MAPK clinical trials, including those recently published in COPD (92) could also bolster the search for DUSP1 modulators. The failure of targeting p38 MAPK is because while pro-inflammatory cytokines are repressed, so are the p38 MAPK-driven anti-inflammatory proteins, including DUSP1 (63, 93, 94).
However, there are challenges to overcome in the drive to develop DUSP as a therapeutic target. First and foremost, it is essential to consider that the overall impact of the MAPK-deactivator DUSP1 within the clinical context will depend on the role played by the MAPK involved. If the rationale is that MAPK needs to be inhibited, then there is a need to upregulate DUSP1 (e.g., in respiratory inflammation). Conversely, in some clinical situations, DUSP1 inhibitors may prove beneficial. For example, in some cancers, DUSP1 is overexpressed and is considered responsible for the failure of JNK-driven apoptotic pathways induced by chemotherapeutics; i.e., adjunct therapeutics with a DUSP1 inhibitor would have merit (95). The challenge in drug discovery would, therefore, be developing targeted therapies that could be delivered to the site of disease without collateral damage. Secondly, DUSP1 is sensitive to oxidative stress, and the phosphatase activity can be reduced. Notably, oxidative stress can be cause or consequence of the disease, and GCs themselves can contribute to the production of oxidative stress (96). Thus, although we may find techniques to increase DUSP1 abundance, it may be non-functional due to oxidation. Reactivation of oxidized DUSP1 function is worthy of further investigation. Thirdly, and perhaps most importantly, we need to get the timing right and ensure that that the temporal kinetics of the impact on DUSP1 on inflammatory pathways are considered. Taken together, the future utility of DUSP1 as a therapeutic strategy depends on it being active (not oxidized) and present at the right place at the right time. Treatment with exogenous DUSP1 upregulators would be akin to the usage of p38 MAPK inhibitors and as they have failed in clinical trials, restoring physiological DUSP1 activity in a manner that fully exploits dynamic regulation exerted by the p38 MAPK/DUSP1/TTP network might even be the better option.
Author Contributions
JH and AA reviewed the literature and drafted the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge support by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) and Saarland University within the funding program Open Access Publishing.
Abbreviations
AP-1, activator protein-1; CASP, colon ascendens stent peritonitis; CCL, CC-chemokine ligand; CLP, caecal ligation and puncture; COPD, chronic obstructive pulmonary disease; CREB, cAMP response element-binding protein; CXCL, C–X–C motif ligand; DUSP1, dual-specificity phosphatase; ERK, extracellular-signal-regulated kinase; EC, endothelial cell; GC, glucocorticoid; GR, GC receptor; GRE, GR responsive element; ICAM1, intercellular adhesion molecule 1; ICS, inhaled corticosteroid; IL, interleukin; INF, interferon; IRFs, interferon regulatory factors; JNK, c-Jun N-terminal kinase; LPS, lipopolysaccharide; NF-κB, nuclear factor-κB; MAPK, mitogen-activated protein kinase; MKP-1, mitogen-activated protein kinase phosphatase-1; RANKL, receptor activator of NF-κB ligand; TNF, tumor necrosis factor; TLR, toll-like receptor; TTP, tristetraprolin; VCAM-1, vascular cell adhesion molecule 1.
References
1. Vandevyver S, Dejager L, Van Bogaert T, Kleyman A, Liu Y, Tuckermann J, et al. Glucocorticoid receptor dimerization induces MKP1 to protect against TNF-induced inflammation. J Clin Investig. (2012) 122:2130–40. doi: 10.1172/JCI60006
2. Plotnikov A, Zehorai E, Procaccia S, Seger R. The MAPK cascades: signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim Biophys Acta. (2011) 1813:1619–33. doi: 10.1016/j.bbamcr.2010.12.012
3. Flores K, Yadav SS, Katz AA, Seger R. The nuclear translocation of mitogen-activated protein kinases: molecular mechanisms and use as novel therapeutic target. Neuroendocrinology. (2019) 108:121–31. doi: 10.1159/000494085
4. Su B, Karin M. Mitogen-activated protein kinase cascades and regulation of gene expression. Curr Opin Immunol. (1996) 8:402–11. doi: 10.1016/S0952-7915(96)80131-2
5. Kim EK, Choi EJ. Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta. (2010) 1802:396–405. doi: 10.1016/j.bbadis.2009.12.009
6. Tanti JF, Jager J. Cellular mechanisms of insulin resistance: role of stress-regulated serine kinases and insulin receptor substrates (IRS) serine phosphorylation. Curr Opin Pharmacol. (2009) 9:753–62. doi: 10.1016/j.coph.2009.07.004
7. Rovida E, Stecca B. Mitogen-activated protein kinases and Hedgehog-GLI signaling in cancer: a crosstalk providing therapeutic opportunities? Semin Cancer Biol. (2015) 35:154–67. doi: 10.1016/j.semcancer.2015.08.003
8. Seternes OM, Kidger AM, Keyse SM. Dual-specificity MAP kinase phosphatases in health and disease. Biochim Biophys Acta Mol Cell Res. (2019) 1866:124–43. doi: 10.1016/j.bbamcr.2018.09.002
9. Dickinson RJ, Keyse SM. Diverse physiological functions for dual-specificity MAP kinase phosphatases. J Cell Sci. (2006) 119(Pt 22):4607–15. doi: 10.1242/jcs.03266
10. Abraham SM, Lawrence T, Kleiman A, Warden P, Medghalchi M, Tuckermann J, et al. Antiinflammatory effects of dexamethasone are partly dependent on induction of dual specificity phosphatase 1. J Exp Med. (2006) 203:1883–9. doi: 10.1084/jem.20060336
11. Vandevyver S, Dejager L, Tuckermann J, Libert C. New insights into the anti-inflammatory mechanisms of glucocorticoids: an emerging role for glucocorticoid-receptor-mediated transactivation. Endocrinology. (2013) 154:993–1007. doi: 10.1210/en.2012-2045
12. Kassel O, Sancono A, Kratzschmar J, Kreft B, Stassen M, Cato AC. Glucocorticoids inhibit MAP kinase via increased expression and decreased degradation of MKP-1. EMBO J. (2001) 20:7108–16. doi: 10.1093/emboj/20.24.7108
13. Abraham SM, Clark AR. Dual-specificity phosphatase 1: a critical regulator of innate immune responses. Biochem Soc Trans. (2006) 34(Pt 6):1018–23. doi: 10.1042/BST0341018
14. Hammer M, Mages J, Dietrich H, Servatius A, Howells N, Cato AC, et al. Dual specificity phosphatase 1 (DUSP1) regulates a subset of LPS-induced genes and protects mice from lethal endotoxin shock. J Exp Med. (2006) 203:15–20. doi: 10.1084/jem.20051753
15. Zhao Q, Wang X, Nelin LD, Yao Y, Matta R, Manson ME, et al. MAP kinase phosphatase 1 controls innate immune responses and suppresses endotoxic shock. J Exp Med. (2006) 203:131–40. doi: 10.1084/jem.20051794
16. Comalada M, Lloberas J, Celada A. MKP-1: a critical phosphatase in the biology of macrophages controlling the switch between proliferation and activation. Eur J Immunol. (2012) 42:1938–48. doi: 10.1002/eji.201242441
17. Franklin CC, Kraft AS. Conditional expression of the mitogen-activated protein kinase (MAPK) phosphatase MKP-1 preferentially inhibits p38 MAPK and stress-activated protein kinase in U937 cells. J Biol Chem. (1997) 272:16917–23. doi: 10.1074/jbc.272.27.16917
18. Franklin CC, Srikanth S, Kraft AS. Conditional expression of mitogen-activated protein kinase phosphatase-1, MKP-1, is cytoprotective against UV-induced apoptosis. Proc Natl Acad Sci USA. (1998) 95:3014–9. doi: 10.1073/pnas.95.6.3014
19. Wu JJ, Bennett AM. Essential role for mitogen-activated protein (MAP) kinase phosphatase-1 in stress-responsive MAP kinase and cell survival signaling. J Biol Chem. (2005) 280:16461–6. doi: 10.1074/jbc.M501762200
20. McGuire VA, Rosner D, Ananieva O, Ross EA, Elcombe SE, Naqvi S, et al. Beta interferon production is regulated by p38 mitogen-activated protein kinase in macrophages via both MSK1/2- and tristetraprolin-dependent pathways. Mol Cell Biol. (2017) 37:e00454–16. doi: 10.1128/MCB.00454-16
21. Smallie T, Ross EA, Ammit AJ, Cunliffe HE, Tang T, Rosner DR, et al. Dual-specificity phosphatase 1 and tristetraprolin cooperate to regulate macrophage responses to lipopolysaccharide. J Immunol. (2015) 195:277–88. doi: 10.4049/jimmunol.1402830
22. Kwak SP, Hakes DJ, Martell KJ, Dixon JE. Isolation and characterization of a human dual specificity protein-tyrosine phosphatase gene. J Biol Chem. (1994) 269:3596–604.
23. Noguchi T, Metz R, Chen L, Mattei MG, Carrasco D, Bravo R. Structure, mapping, and expression of erp, a growth factor-inducible gene encoding a nontransmembrane protein tyrosine phosphatase, and effect of ERP on cell growth. Mol Cell Biol. (1993) 13:5195–205. doi: 10.1128/MCB.13.9.5195
24. Shipp LE, Lee JV, Yu CY, Pufall M, Zhang P, Scott DK, et al. Transcriptional regulation of human dual specificity protein phosphatase 1 (DUSP1) gene by glucocorticoids. PLoS ONE. (2010) 5:e13754. doi: 10.1371/journal.pone.0013754
25. Reddy TE, Pauli F, Sprouse RO, Neff NF, Newberry KM, Garabedian MJ, et al. Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res. (2009) 19:2163–71. doi: 10.1101/gr.097022.109
26. Clark AR, Dean JL. The control of inflammation via the phosphorylation and dephosphorylation of tristetraprolin: a tale of two phosphatases. Biochem Soc Trans. (2016) 44:1321–37. doi: 10.1042/BST20160166
27. Zhu QY, Liu Q, Chen JX, Lan K, Ge BX. MicroRNA-101 targets MAPK phosphatase-1 to regulate the activation of MAPKs in macrophages. J Immunol. (2010) 185:7435–42. doi: 10.4049/jimmunol.1000798
28. Brondello JM, Pouyssegur J, McKenzie FR. Reduced MAP kinase phosphatase-1 degradation after p42/p44MAPK-dependent phosphorylation. Science. (1999) 286:2514–7. doi: 10.1126/science.286.5449.2514
29. Lin YW, Yang JL. Cooperation of ERK and SCFSkp2 for MKP-1 destruction provides a positive feedback regulation of proliferating signaling. J Biol Chem. (2006) 281:915–26. doi: 10.1074/jbc.M508720200
30. Cao W, Bao C, Padalko E, Lowenstein CJ. Acetylation of mitogen-activated protein kinase phosphatase-1 inhibits Toll-like receptor signaling. J Exp Med. (2008) 205:1491–503. doi: 10.1084/jem.20071728
31. Jeong Y, Du R, Zhu X, Yin S, Wang J, Cui H, et al. Histone deacetylase isoforms regulate innate immune responses by deacetylating mitogen-activated protein kinase phosphatase-1. J Leukoc Biol. (2014) 95:651–9. doi: 10.1189/jlb.1013565
32. Tephly LA, Carter AB. Differential expression and oxidation of MKP-1 modulates TNF-alpha gene expression. Am J Respir Cell Mol Biol. (2007) 37:366–74. doi: 10.1165/rcmb.2006-0268OC
33. Liu RM, Choi J, Wu JH, Gaston Pravia KA, Lewis KM, Brand JD, et al. Oxidative modification of nuclear mitogen-activated protein kinase phosphatase 1 is involved in transforming growth factor beta1-induced expression of plasminogen activator inhibitor 1 in fibroblasts. J Biol Chem. (2010) 285:16239–47. doi: 10.1074/jbc.M110.111732
34. Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. (2005) 120:649–61. doi: 10.1016/j.cell.2004.12.041
35. Kim HS, Ullevig SL, Zamora D, Lee CF, Asmis R. Redox regulation of MAPK phosphatase 1 controls monocyte migration and macrophage recruitment. Proc Natl Acad Sci USA. (2012) 109:E2803–12. doi: 10.1073/pnas.1212596109
36. Hamidzadeh K, Christensen SM, Dalby E, Chandrasekaran P, Mosser DM. Macrophages and the recovery from acute and chronic inflammation. Annu Rev Physiol. (2017) 79:567–92. doi: 10.1146/annurev-physiol-022516-034348
37. Hoppstädter J, Kiemer AK. Glucocorticoid-induced leucine zipper (GILZ) in immuno suppression: master regulator or bystander? Oncotarget. (2015) 6:38446–57. doi: 10.18632/oncotarget.6197
38. Chen P, Li J, Barnes J, Kokkonen GC, Lee JC, Liu Y. Restraint of proinflammatory cytokine biosynthesis by mitogen-activated protein kinase phosphatase-1 in lipopolysaccharide-stimulated macrophages. J Immunol. (2002) 169:6408–16. doi: 10.4049/jimmunol.169.11.6408
39. Salojin KV, Owusu IB, Millerchip KA, Potter M, Platt KA, Oravecz T. Essential role of MAPK phosphatase-1 in the negative control of innate immune responses. J Immunol. (2006) 176:1899–907. doi: 10.4049/jimmunol.176.3.1899
40. Koroskenyi K, Kiss B, Szondy Z. Adenosine A2A receptor signaling attenuates LPS-induced pro-inflammatory cytokine formation of mouse macrophages by inducing the expression of DUSP1. Biochim Biophys Acta. (2016) 1863(7 Pt A):1461–71. doi: 10.1016/j.bbamcr.2016.04.003
41. Tang T, Scambler TE, Smallie T, Cunliffe HE, Ross EA, Rosner DR, et al. Macrophage responses to lipopolysaccharide are modulated by a feedback loop involving prostaglandin E2, dual specificity phosphatase 1 and tristetraprolin. Sci Rep. (2017) 7:4350. doi: 10.1038/s41598-017-04100-1
42. Chi H, Barry SP, Roth RJ, Wu JJ, Jones EA, Bennett AM, et al. Dynamic regulation of pro- and anti-inflammatory cytokines by MAPK phosphatase 1 (MKP-1) in innate immune responses. Proc Natl Acad Sci USA. (2006) 103:2274–9. doi: 10.1073/pnas.0510965103
43. Wang X, Meng X, Kuhlman JR, Nelin LD, Nicol KK, English BK, et al. Knockout of Mkp-1 enhances the host inflammatory responses to gram-positive bacteria. J Immunol. (2007) 178:5312–20. doi: 10.4049/jimmunol.178.8.5312
44. Frazier WJ, Wang X, Wancket LM, Li XA, Meng X, Nelin LD, et al. Increased inflammation, impaired bacterial clearance, and metabolic disruption after gram-negative sepsis in Mkp-1-deficient mice. J Immunol. (2009) 183:7411–9. doi: 10.4049/jimmunol.0804343
45. Hammer M, Echtenachter B, Weighardt H, Jozefowski K, Rose-John S, Mannel DN, et al. Increased inflammation and lethality of Dusp1-/- mice in polymicrobial peritonitis models. Immunology. (2010) 131:395–404. doi: 10.1111/j.1365-2567.2010.03313.x
46. Pemmari A, Paukkeri EL, Hamalainen M, Leppanen T, Korhonen R, Moilanen E. MKP-1 promotes anti-inflammatory M(IL-4/IL-13) macrophage phenotype and mediates the anti-inflammatory effects of glucocorticoids. Basic Clin Pharmacol Toxicol. (2018) 124:404–15. doi: 10.1111/bcpt.13163
47. Mbalaviele G, Novack DV, Schett G, Teitelbaum SL. Inflammatory osteolysis: a conspiracy against bone. J Clin Invest. (2017) 127:2030–9. doi: 10.1172/JCI93356
48. Vattakuzhi Y, Abraham SM, Freidin A, Clark AR, Horwood NJ. Dual-specificity phosphatase 1-null mice exhibit spontaneous osteolytic disease and enhanced inflammatory osteolysis in experimental arthritis. Arthritis Rheum. (2012) 64:2201–10. doi: 10.1002/art.34403
49. Sartori R, Li F, Kirkwood KL. MAP kinase phosphatase-1 protects against inflammatory bone loss. J Dental Res. (2009) 88:1125–30. doi: 10.1177/0022034509349306
50. Yu H, Li Q, Herbert B, Zinna R, Martin K, Junior CR, et al. Anti-inflammatory effect of MAPK phosphatase-1 local gene transfer in inflammatory bone loss. Gene Ther. (2011) 18:344–53. doi: 10.1038/gt.2010.139
51. Valerio MS, Herbert BA, Basilakos DS, Browne C, Yu H, Kirkwood KL. Critical role of MKP-1 in lipopolysaccharide-induced osteoclast formation through CXCL1 and CXCL2. Cytokine. (2015) 71:71–80. doi: 10.1016/j.cyto.2014.08.007
52. Choi Y, Yoo JH, Lee Y, Bae MK, Kim HJ. Calcium-phosphate crystals promote RANKL expression via the downregulation of DUSP1. Mol Cells. (2019) 42:183–8. doi: 10.14348/molcells.2018.0382
53. Peng HZ, Yun Z, Wang W, Ma BA. Dual specificity phosphatase 1 has a protective role in osteoarthritis fibroblastlike synoviocytes via inhibition of the MAPK signaling pathway. Mol Med Rep. (2017) 16:8441–7. doi: 10.3892/mmr.2017.7617
54. Pest MA, Pest CA, Bellini MR, Feng Q, Beier F. Deletion of dual specificity phosphatase 1 does not predispose mice to increased spontaneous osteoarthritis. PLoS ONE. (2015) 10:e0142822. doi: 10.1371/journal.pone.0142822
55. Abbasi M, Mousavi MJ, Jamalzehi S, Alimohammadi R, Bezvan MH, Mohammadi H, et al. Strategies toward rheumatoid arthritis therapy; the old and the new. J Cell Physiol. (2019) 234:10018–31. doi: 10.1002/jcp.27860
56. Briot K, Roux C. Glucocorticoid-induced osteoporosis. RMD Open. (2015) 1:e000014. doi: 10.1136/rmdopen-2014-000014
57. Horsch K, de Wet H, Schuurmans MM, Allie-Reid F, Cato AC, Cunningham J, et al. Mitogen-activated protein kinase phosphatase 1/dual specificity phosphatase 1 mediates glucocorticoid inhibition of osteoblast proliferation. Mol Endocrinol. (2007) 21:2929–40. doi: 10.1210/me.2007-0153
58. Engelbrecht Y, de Wet H, Horsch K, Langeveldt CR, Hough FS, Hulley PA. Glucocorticoids induce rapid up-regulation of mitogen-activated protein kinase phosphatase-1 and dephosphorylation of extracellular signal-regulated kinase and impair proliferation in human and mouse osteoblast cell lines. Endocrinology. (2003) 144:412–22. doi: 10.1210/en.2002-220769
59. Conradie MM, de Wet H, Kotze DD, Burrin JM, Hough FS, Hulley PA. Vanadate prevents glucocorticoid-induced apoptosis of osteoblasts in vitro and osteocytes in vivo. J Endocrinol. (2007) 195:229–40. doi: 10.1677/JOE-07-0217
60. Conradie MM, Cato AC, Ferris WF, de Wet H, Horsch K, Hough S. MKP-1 knockout does not prevent glucocorticoid-induced bone disease in mice. Calcif Tissue Int. (2011) 89:221–7. doi: 10.1007/s00223-011-9509-x
61. Ammit AJ. Glucocorticoid insensitivity as a source of drug targets for respiratory disease. Curr Opin Pharmacol. (2013) 13:370–6. doi: 10.1016/j.coph.2013.02.001
62. Korhonen R, Moilanen E. Mitogen-activated protein kinase phosphatase 1 as an inflammatory factor and drug target. Basic Clin Pharmacol Toxicol. (2014) 114:24–36. doi: 10.1111/bcpt.12141
63. Moosavi SM, Prabhala P, Ammit AJ. Role and regulation of MKP-1 in airway inflammation. Respir Res. (2017) 18:154. doi: 10.1186/s12931-017-0637-3
64. Bhavsar P, Hew M, Khorasani N, Torrego A, Barnes PJ, Adcock I, et al. Relative corticosteroid insensitivity of alveolar macrophages in severe asthma compared with non-severe asthma. Thorax. (2008) 63:784–90. doi: 10.1136/thx.2007.090027
65. Issa R, Xie S, Khorasani N, Sukkar M, Adcock IM, Lee KY, et al. Corticosteroid inhibition of growth-related oncogene protein-alpha via mitogen-activated kinase phosphatase-1 in airway smooth muscle cells. J Immunol. (2007) 178:7366–75. doi: 10.4049/jimmunol.178.11.7366
66. Quante T, Ng YC, Ramsay EE, Henness S, Allen JC, Parmentier J, et al. Corticosteroids reduce IL-6 in ASM cells via up-regulation of MKP-1. Am J Respir Cell Mol Biol. (2008) 39:208–17. doi: 10.1165/rcmb.2007-0014OC
67. Li F, Zhang M, Hussain F, Triantaphyllopoulos K, Clark AR, Bhavsar PK, et al. Inhibition of p38 MAPK-dependent bronchial contraction after ozone by corticosteroids. Eur Respir J. (2011) 37:933–42. doi: 10.1183/09031936.00021110
68. Pinart M, Hussain F, Shirali S, Li F, Zhu J, Clark AR, et al. Role of mitogen-activated protein kinase phosphatase-1 in corticosteroid insensitivity of chronic oxidant lung injury. Eur J Pharmacol. (2014) 744:108–14. doi: 10.1016/j.ejphar.2014.10.003
69. Sundar IK, Yao H, Rahman I. Oxidative stress and chromatin remodeling in chronic obstructive pulmonary disease and smoking-related diseases. Antioxid Redox Signal. (2013) 18:1956–71. doi: 10.1089/ars.2012.4863
70. Kirkham PA, Barnes PJ. Oxidative stress in COPD. Chest. (2013) 144:266–73. doi: 10.1378/chest.12-2664
71. Kelly MM, King EM, Rider CF, Gwozd C, Holden NS, Eddleston J, et al. Corticosteroid-induced gene expression in allergen-challenged asthmatic subjects taking inhaled budesonide. Br J Pharmacol. (2012) 165:1737–47. doi: 10.1111/j.1476-5381.2011.01620.x
72. Clark AR, Martins JR, Tchen CR. Role of dual specificity phosphatases in biological responses to glucocorticoids. J Biol Chem. (2008) 283:25765–9. doi: 10.1074/jbc.R700053200
73. Ng MK, Celermajer DS. Glucocorticoid treatment and cardiovascular disease. Heart. (2004) 90:829–30. doi: 10.1136/hrt.2003.031492
74. Ross IL, Marais AD. The influence of glucocorticoids on lipid and lipoprotein metabolism and atherosclerosis. S Afr Med J. (2014) 104:671–4. doi: 10.7196/SAMJ.7979
75. Makheja AN, Bloom S, Muesing R, Simon T, Bailey JM. Anti-inflammatory drugs in experimental atherosclerosis. 7. Spontaneous atherosclerosis in WHHL rabbits and inhibition by cortisone acetate. Atherosclerosis. (1989) 76:155–61. doi: 10.1016/0021-9150(89)90099-3
76. Hagihara H, Nomoto A, Mutoh S, Yamaguchi I, Ono T. Role of inflammatory responses in initiation of atherosclerosis: effects of anti-inflammatory drugs on cuff-induced leukocyte accumulation and intimal thickening of rabbit carotid artery. Atherosclerosis. (1991) 91:107–16. doi: 10.1016/0021-9150(91)90192-6
77. Asai K, Funaki C, Hayashi T, Yamada K, Naito M, Kuzuya M, et al. Dexamethasone-induced suppression of aortic atherosclerosis in cholesterol-fed rabbits. Possible mechanisms. Arterioscler Thromb. (1993) 13:892–9. doi: 10.1161/01.ATV.13.6.892
78. Schepers A, Pires NM, Eefting D, de Vries MR, van Bockel JH, Quax PH. Short-term dexamethasone treatment inhibits vein graft thickening in hypercholesterolemic ApoE3Leiden transgenic mice. J Vasc Surg. (2006) 43:809–15. doi: 10.1016/j.jvs.2005.11.019
79. Kastrup CJ, Nahrendorf M, Figueiredo JL, Lee H, Kambhampati S, Lee T, et al. Painting blood vessels and atherosclerotic plaques with an adhesive drug depot. Proc Natl Acad Sci USA. (2012) 109:21444–9. doi: 10.1073/pnas.1217972110
80. Furst R, Schroeder T, Eilken HM, Bubik MF, Kiemer AK, Zahler S, et al. MAPK phosphatase-1 represents a novel anti-inflammatory target of glucocorticoids in the human endothelium. FASEB J. (2007) 21:74–80. doi: 10.1096/fj.06-6752com
81. Tzima E, Irani-Tehrani M, Kiosses WB, Dejana E, Schultz DA, Engelhardt B, et al. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature. (2005) 437:426–31. doi: 10.1038/nature03952
82. Ji JY, Jing H, Diamond SL. Shear stress causes nuclear localization of endothelial glucocorticoid receptor and expression from the GRE promoter. Circ Res. (2003) 92:279–85. doi: 10.1161/01.RES.0000057753.57106.0B
83. Chaudhury H, Zakkar M, Boyle J, Cuhlmann S, van der Heiden K, Luong le A, et al. c-Jun N-terminal kinase primes endothelial cells at atheroprone sites for apoptosis. Arterioscler Thromb Vasc Biol. (2010) 30:546–53. doi: 10.1161/ATVBAHA.109.201368
84. Zakkar M, Chaudhury H, Sandvik G, Enesa K, Luong le A, Cuhlmann S, et al. Increased endothelial mitogen-activated protein kinase phosphatase-1 expression suppresses proinflammatory activation at sites that are resistant to atherosclerosis. Circ Res. (2008) 103:726–32. doi: 10.1161/CIRCRESAHA.108.183913
85. Kim HS, Asmis R. Mitogen-activated protein kinase phosphatase 1 (MKP-1) in macrophage biology and cardiovascular disease. A redox-regulated master controller of monocyte function and macrophage phenotype. Free Radic Biol Med. (2017) 109:75–83. doi: 10.1016/j.freeradbiomed.2017.03.020
86. Kim HS, Tavakoli S, Piefer LA, Nguyen HN, Asmis R. Monocytic MKP-1 is a sensor of the metabolic environment and regulates function and phenotypic fate of monocyte-derived macrophages in atherosclerosis. Sci Rep. (2016) 6:34223. doi: 10.1038/srep34223
87. Podgorski M, Kupczyk M, Grzelak P, Bochenska-Marciniak M, Polguj M, Kuna P, et al. Inhaled corticosteroids in asthma: promoting or protecting against atherosclerosis? Med Sci Monit. (2017) 23:5337–44. doi: 10.12659/MSM.904469
88. Matta R, Barnard JA, Wancket LM, Yan J, Xue J, Grieves J, et al. Knockout of Mkp-1 exacerbates colitis in Il-10-deficient mice. Am J Physiol Gastrointest Liver Physiol. (2012) 302:G1322–35. doi: 10.1152/ajpgi.00018.2012
89. Kjellerup RB, Johansen C, Kragballe K, Iversen L. The expression of dual-specificity phosphatase 1 mRNA is downregulated in lesional psoriatic skin. Br J Dermatol. (2013) 168:339–45. doi: 10.1111/bjd.12020
90. Yang J, Sun L, Han J, Zheng W, Peng W. DUSP1/MKP-1 regulates proliferation and apoptosis in keratinocytes through the ERK/Elk-1/Egr-1 signaling pathway. Life Sci. (2019) 223:47–53. doi: 10.1016/j.lfs.2019.03.018
91. Chang WJ, Niu XP, Hou RX, Li JQ, Liu RF, Wang Q, et al. LITAF, HHEX, and DUSP1 expression in mesenchymal stem cells from patients with psoriasis. Genet Mol Res. (2015) 14:15793–801. doi: 10.4238/2015.December.1.31
92. Patel NR, Cunoosamy DM, Fageras M, Taib Z, Asimus S, Hegelund-Myrback T, et al. The development of AZD7624 for prevention of exacerbations in COPD: a randomized controlled trial. Int J Chron Obstruct Pulmon Dis. (2018) 13:1009–19. doi: 10.2147/COPD.S150576
93. Manetsch M, Che W, Seidel P, Chen Y, Ammit AJ. MKP-1: a negative feedback effector that represses MAPK-mediated pro-inflammatory signaling pathways and cytokine secretion in human airway smooth muscle cells. Cell Signal. (2012) 24:907–13. doi: 10.1016/j.cellsig.2011.12.013
94. Clark AR, Dean JL. The p38 MAPK pathway in rheumatoid arthritis: a sideways look. Open Rheumatol J. (2012) 6:209–19. doi: 10.2174/1874312901206010209
95. Doddareddy MR, Rawling T, Ammit AJ. Targeting mitogen-activated protein kinase phosphatase-1 (MKP-1): structure-based design of MKP-1 inhibitors and upregulators. Curr Med Chem. (2012) 19:163–73. doi: 10.2174/092986712803414196
Keywords: sepsis, infection, arthritis, bone disease, asthma, COPD, atherosclerosis
Citation: Hoppstädter J and Ammit AJ (2019) Role of Dual-Specificity Phosphatase 1 in Glucocorticoid-Driven Anti-inflammatory Responses. Front. Immunol. 10:1446. doi: 10.3389/fimmu.2019.01446
Received: 15 April 2019; Accepted: 10 June 2019;
Published: 26 June 2019.
Edited by:
Claude Libert, Flanders Institute for Biotechnology, BelgiumReviewed by:
Jens Staal, Flanders Institute for Biotechnology, BelgiumSimona Ronchetti, Università degli Studi di Perugia, Italy
Copyright © 2019 Hoppstädter and Ammit. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jessica Hoppstädter, j.hoppstaedter@mx.uni-saarland.de