 Eirini Pantazi1*
Eirini Pantazi1* Nick Powell2*
Nick Powell2*- 1Kennedy Institute of Rheumatology, University of Oxford, Oxford, United Kingdom
- 2Department of Inflammation Biology, Centre for Inflammation and Cancer Immunology, King's College London, London, United Kingdom
A complex network of interactions exists between the microbiome, the epithelium, and immune cells that reside along the walls of the gastrointestinal tract. The intestinal immune system has been assigned with the difficult task of discriminating between commensal, harmless bacteria, and invading pathogens that translocate across the epithelial monolayer. Importantly, it is trained to maintain tolerance against commensals, and initiate protective immune responses against pathogens to secure intestinal homeostasis. Breakdown of this fine balance between the host and its intestinal microbiota can lead to intestinal inflammation and subsequently to development of inflammatory bowel disease (IBD). A decade since their discovery, innate lymphoid cells (ILCs) are now recognized as important regulators of intestinal homeostasis. ILC3s have emerged as a critical subset in the gut. They are the most phenotypically diverse ILC population and interact directly with numerous different cell types (haematopoietic and non-haematopoeitic), as well as interface with the bacterial flora. In addition to their contribution to intestinal pathogen immunity, they also mitigate against tissue damage occurring following acute injury, by facilitating tissue repair and regeneration, a key function in the maintenance of intestinal homeostasis. However, in chronic inflammation the tables are turned and ILC3s may acquire a pro-inflammatory phenotype in the gut. Chronic ILC activation can lead to persistent inflammation contributing to IBD and/or colorectal cancer. In this review, we discuss current knowledge of group 3 ILCs and their contributions to intestinal homeostasis and disease leading to novel therapeutic targets and clinical approaches that may inform novel treatment strategies for immune-mediated disorders, including IBD.
Introduction
With more than 1013 microorganisms residing in the human gastrointestinal tract (1), our mucosal immune system has excelled in preserving intestinal homeostasis by generating protective immune responses against invading, harmful pathogens whilst maintaining tolerance toward commensals. However, breakdown of this fine balance may lead to excessive immune activation, persistent inflammation and ultimately to the development of inflammatory bowel diseases (IBD). The most common IBD phenotypes, Crohn's disease (CD) and ulcerative colitis (UC) are characterized by alternating phases of clinical relapse and remission. In CD inflammation can occur in any part of the gastrointestinal tract, whereas in UC inflammation is restricted to the colon (2). Despite the increasing incidence of IBD in the Western world (3), its complex etiology is yet to be fully understood. In general, it is thought that IBD is caused by a dysregulated immune response against the commensal bacterial flora in a genetically predisposed host (4). In accordance with this notion, genome-wide association studies (GWAS) have so far associated single nucleotide polymorphisms (SNPs) in more than 200 genetic loci with IBD susceptibility (5) including genes involved in bacterial recognition, epithelial barrier integrity and immune activation (6) highlighting the importance of microbiota-host interactions and the role of the intestinal immune system, in particular the innate one, in IBD pathogenesis.
Innate Lymphoid Cells—A New Recruit to Mucosal Sentinel Duty
Defense against intestinal pathogens is multifaceted. The intestinal epithelium comprises a physical barrier, which together with the mucus layer and production of anti-microbial peptides provides a containment barrier, which confines microbes in the lumen. Cells of the innate immune compartment residing in the lamina propria are key early warning sentinels detecting invading pathogens through conserved pattern recognition receptors, such as toll-like receptors. Pathogen detecting populations, include cells of the mononuclear phagocyte system, including macrophages and dendritic cells (DCs), which engulf and process microbial antigens and then shape adaptive immune responses by providing initial signals to adaptive lymphocytes to engage potent antigen-specific T and B cell responses. Innate lymphoid cells (ILCs) are recent additions to the innate immune cell family (7–9). They are distributed throughout the human body in lymphoid and non-lymphoid tissues, but are especially enriched at the mucosal barrier surfaces (7), where they directly interact with a number of different cell types; hematopoietic or other (10–12). ILCs have lymphoid-like morphology, but lack any antigen-specific receptors. Arising from a common lymphoid progenitor and similarly to T cells, they can be further subdivided into phenotypically and functionally distinct populations that produce different combinations of effector cytokines to mediate their functions (9–11). Their development depends on different transcription factors, which are also used to help divide ILCs into 3 main groups; group 1 ILCs that includes the well characterized NK cells, as well as the non-cytotoxic ILC1s, group 2 ILCs or ILC2s, and finally group 3 ILCs including ILC3s and lymphoid tissue inducer (LTi) cells. However, recently discovered regulatory ILCs or ILCregs (13) may now represent an additional, distinct ILC family member generating a potential new 4th group of ILCs.
Much of the early work describing ILCs focused on their developmental requirements and capacity for plasticity. An early ILC progenitor (EILCP) rising form a common lymphoid progenitor, which has lost T and B potential, gives rise to NK cells, as well as all ILC lineages (14–16). Downstream of EILCP, Id2 expressing common helper-like ILC precursor (CHILP) gives rise to all ILCs, but not to NK cells (14), whereas all ILCs (except lymphoid tissue inducer cells, LTis), arise from an ILC precursor (ILCP) that expresses both Id2, and PLZF (17) (Figure 1). Group 3 ILCs that represent the most diverse and possibly best-characterized ILC populations both in humans and mice (12), require RORγt for their development (18–20), while the transcriptional factor aryl hydrocarbon receptor (AhR) is essential for their maintenance (21, 22). In general, group 3 ILCs, can be further divided into CCR6+ LTi cells that may or may not express CD4, and CCR6low/− ILC3s. Based on whether or not CCR6low/− ILC3s express natural cytotoxicity receptors (NCRs), they are additionally subdivided into NCR+ and NCR− ILC3s (Figure 1). Upon activation, NCR− ILC3s and LTi cells produce mostly interleukin (IL)-17A, IL-17F and IL-22, while NCR+ ILC3s produce mainly IL-22 (20, 23–27). Several independent studies have provided evidence suggesting plasticity within group 3 ILCs (28, 29). NCR− ILC3s can differentiate into NCR+ ILC3s in the presence of increasing expression of T-bet (30, 31), or even acquire an ILC1-like phenotype by completely losing RORγt expression (29, 32). Similarly, ILC1-like ILC3s may also differentiate in vivo into NCR− ILC3s in the presence of a RORγt gradient (32, 33) (Figure 1). Given the enriched numbers of group 3 ILCs in the gut mucosa and their role in preserving intestinal homeostasis, additional studies are required to clarify whether this profound, finely tuned plasticity within group 3 ILCs is a mechanism to regulate intestinal inflammation, and whether this plasticity may also be extended to recently identified ILCregs (13).
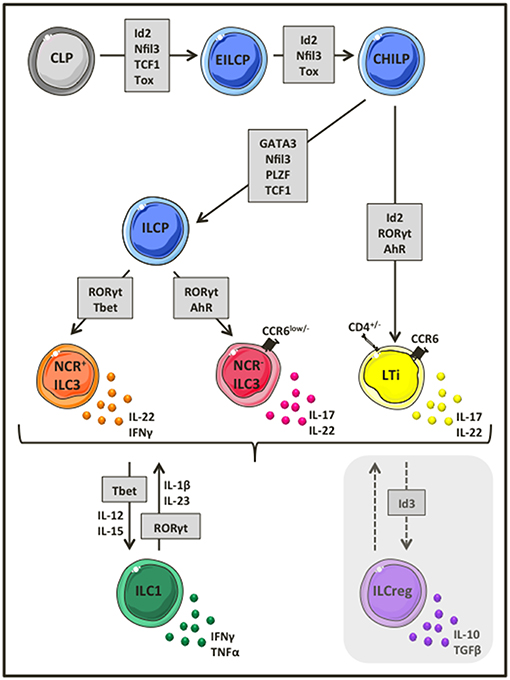
Figure 1. Group 3 ILC development and heterogeneity. Group 3 ILCs represent the most diverse ILC subset, comprising of CCR6+ LTi cells, and ILC3s that can be further divided into NCR− and NCR+ ILC3s. Group 3 ILCs rise from a common lymphoid progenitor that lost T, B and eventually NK cell potential and depend on the transcriptional factor RORγt for their development. Depending on the environmental stimulus, ILC3s produce a variety of distinct cytokines such as IL-17, IL-22, IFNγ, and GM-CSF to mediate their functions. Interestingly, ILC3s are highly plastic cells and as such NCR− ILC3s can acquire NCR expression and became NCR+ ILC3s in the presence of a T-bet gradient or loose completely RORγt expression to differentiate into ILC1 like ILCs and vice versa.
Although group 3 ILCs are primarily known for their role in anti-bacterial immunity (20, 27), they emerge as key effector cells at barrier surfaces. Recent studies describe their involvement in several immune-mediated diseases such as psoriasis (34, 35), multiple sclerosis (36), or cancer (37, 38), improving our understanding of these highly plastic cells in controlling inflammation, while suggesting new ways of therapeutic immune intervention. In this review, we focus on the role of group 3 ILCs in the gut, during intestinal homeostasis and disease.
Group 3 ILCs Promote Intestinal Peace
Microbiota-ILC3 Interactions—A Key Partnership for a Finely Tuned Intestinal Immune System
Group 3 ILCs accumulate in the gastrointestinal tract and gut-associated lymphoid tissues in a microbiota-independent manner (21, 39–41). They directly interact with the bacterial flora, as well as with immune and non-hematopoietic cells creating a dynamic network between the host and its resident microbiota that favors symbiosis and preserves intestinal homeostasis (Figure 2). An important aspect of this partnership is containment of commensals to the lumen allowing for controlled bacterial sampling by lamina propria mononuclear phagocytes. ILC3s are key regulators of this process. Loss of ILC3s in the intestine leads to diminished IL-22 production (a key cytokine produced by ILC3s), and impaired production of antimicrobial peptides by intestinal epithelial cells, culminating in peripheral dissemination of Alcaligenes bacteria and systemic inflammation that could be prevented by exogenous replacement of IL-22 (41). ILC3s also play a key role in shaping adaptive immunity. Importantly, Hepworth et al. showed for the first time that loss of RORγt+ ILCs in immunocompetent mice lead to dysregulated adaptive immune responses against commensals, an effect that was not mediated by known ILC3 associated cytokines such as IL-17A, IL-22 and IL-23, but through MHC II:TCR interactions instead (42). In particular, selective deletion of MHC II expression on RORγt+ ILCs resulted in enhanced antigen-specific Th17 responses against bacterial flora, promoting spontaneous intestinal inflammation (42). Others have also reported contribution of ILC3s in the generation of Th17 responses against commensals (43), while antigen presenting capacities through MHC II and expression of co-stimulatory molecules have also been described for splenic ILC3s using in vitro systems of T cell priming (44). Although ILC3s can uptake, process and present antigen, they don't express co-stimulatory molecules and as such they can potentially result in T cell energy. Thus, when absent there is uncontrolled T cell activation and exaggerated T cell responses to commensals that would otherwise be regulated. Establishing a balanced, two-way relationship between ILC3s and T cells in the gut, T cells have proven to be crucial for keeping ILC3 numbers and functions in check. Absence of CD4+ T cells resulted in elevated ILC numbers, increased IL-22 production and subsequently enhanced secretion of anti-microbial peptides by epithelial cells (45). Other more complex, dynamic interactions between ILC3s, T cells and the microbiota are important at different development stages of the host. While ILC3s have an important role in influencing bacterial composition at early developmental stages, most likely to prevent unnecessary inflammatory responses, as bacteria burden expands, CD4+ T cells accumulate to establish tolerance using distinct mechanisms compared to their innate counterparts, that may become quiescent (46).
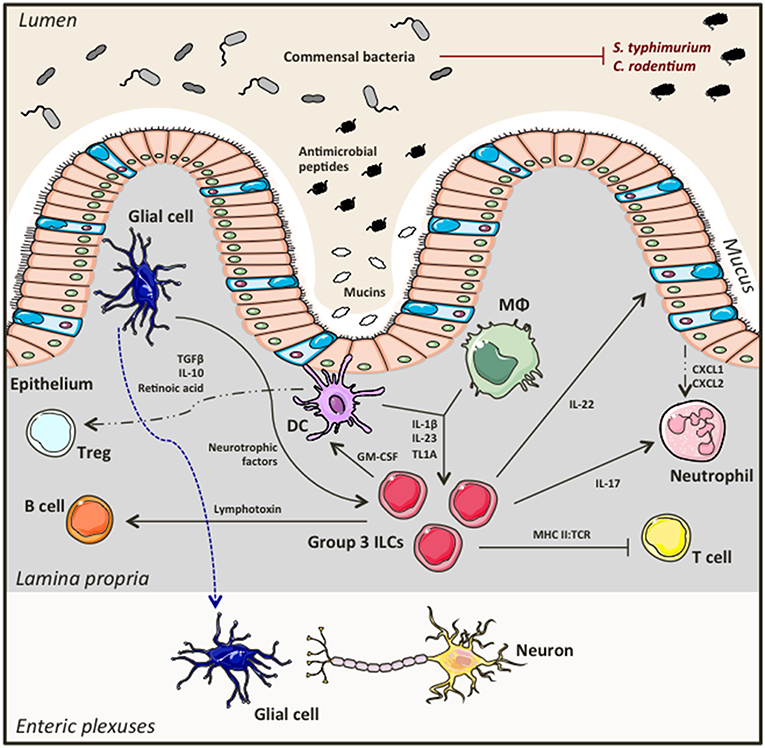
Figure 2. Group 3 ILCs in intestinal homeostasis. Group 3 ILCs including ILC3s and LTi cells directly interact with the commensal bacteria in the lumen, the intestinal epithelium, as well as with other immune cells and neurons in the lamina propria forming a finely tuned network that secures and maintains intestinal homeostasis.
Although IL-22 plays a central role regulating mucosal immunity, the microbiota also play an important role fine-tuning the ILC3/IL-22 axis. Segmented filamentous bacteria (SFB) are commensal bacteria that selectively colonize the terminal ileum of mice, a key inductive site of mucosal immunity. They play a central role in the differentiation of Th17 cells (47, 48), but also regulate innate IL-22 production. Mono-association of germ free mice with SFB results in marked augmentation of IL-22 production by intestinal ILC3s. In this system IL-22 induced production of serum amyloid A proteins 1 and 2 from the intestinal epithelial cells, which in turn played a key role promoting the differentiation of Th17 cells (49). Other commensal bacteria may have an opposing role on IL-22 production, through induction of IL-25 by intestinal epithelial cells, which suppresses IL-22 production by RORγt+ ILCs (50).
The microbiota ILC3 partnership extends its branches to B cells too, the other major adaptive immune cell. In 2008, Tsuji et al. showed that LTi cells are essential for the formation of isolated lymphoid follicles (ILFs) and T cell independent generation of immunoglobulin A (IgA) by B cells in the gut (51). Later that year, a study published in Nature complemented these findings by showing that peptidoglycans from Gram− bacteria activate LTi cells in the gut, which then induce chemokine production by stromal cells resulting in B cell recruitment and subsequently formation of ILFs (52). Kruglov et al. showed that RORγt+ ILCs mediate both T cell dependent and independent IgA production by B cells through secretion of soluble lymphotoxin α (sLTα3) and membrane-bound lymphotoxin β (LTα1β2), respectively (53). Beneficial interactions have also been described between group 3 ILCs and mononuclear phagocytes (54). Bacterial sampling by intestinal macrophages and DCs induces IL-1β secretion that activates ILC3s, which in turn produce GM-CSF that feeds back to mononuclear phagocytes to produce anti-inflammatory mediators such as IL-10 and retinoic acid eventually leading to Treg expansion and immune tolerance (54). Surprisingly, Ibiza et al. showed that group 3 ILCs also interact with the enteric nervous system in an attempt to maintain intestinal homeostasis (55). Interestingly, it was shown that glial cells in the lamina propria sense bacterial presence and secrete neurotrophic factors that induce IL-22 production by ILC3s through the neuroregulatory receptor RET (55), providing the first evidence of direct neuron involvement in innate immune regulation in the gut.
ILC3s, Important Soldiers Fighting Foreign Pathogens
In accordance with studies showing how group 3 ILCs directly interact with the bacterial flora while working closely with other hematopoietic and non-immune cells to secure and maintain intestinal homeostasis, ILC3s have also been described as key effector cells in immunity against pathogens. Even prior to acquiring their official name, group 3 ILCs were associated with protection against Proteobacteria (20). Loss of NKp46+ RORγt+ IL-22 producing innate immune cells is linked to increased susceptibility to Citrobacter rodentium infection, which is a model for enteropathic E.coli infection (20) ILC3s were especially important as early producers of IL-22 (56). Lymphotoxin produced by RORγt expressing innate immune cells was necessary to control C. rodentium infection as lymphotoxin acted on the intestinal epithelium inducing CXCL1 and CXCL2 chemokine production and subsequently neutrophil recruitment at the early stages of infection (57). Sonnenberg et al. showed that infection with C. rodentium induced IL-23 mediated IL-22 production by CD4+ LTi cells, and that this subset of ILC3 was sufficient to promote immunity in immunodeficient hosts (27). More recent studies showed that expression of the G-protein-coupled receptor 183 (GPR183) on LTi cells is not only essential for their migration to cryptopatches and ILFs (58), but also required for ILC3-mediated protection against C. rodentium infection (59). Notably, immunity to Citrobacter is STAT3 dependent. STAT3 deficiency was associated with impaired IL-22 production, and increased disease severity, which could be rescued with exogenous IL-22 (60). STAT3 expression was only required in ILC3s and T cells to induce protection (60).
One of the mechanisms of IL-22 mediated regulation of the microbiota is through regulation of the glycosylation pattern of epithelial cells. IL-22 and lymphotoxin expressed by ILC3s control the expression of fucosyltransferase 2 (Fut2), which triggers fucosylation of epithelial cells, which in turn can be utilized as a nutrient source by luminal commensals (61). IL-22 mediated fucosylation of the epithelium was dependent on colonization of the GI tract with bacteria, and in a positive circuit, fucosylation promoted colonization with symbiotic bacteria, presumably by providing a favorable nutrient supply. Disruption of this system resulted in loss of host-microbe mutualism and rendered the host susceptible to Salmonella typhimurium infection (61). In accordance with these findings, Pickard et al. showed that IL-22 induced fucosylation is mediated by ILC3s upon activation with IL-23 produced by DCs during pathogen-induced stress (62). In this setting, rapid fucosylation could also improve tolerance to C. rodentium infection (62).
Klose et al. demonstrated that Tbet expressing; IFNγ producing RORγt+ ILCs are key drivers of immunity against Salmonella infections (30). Other work supports a role for ILC3s in protective immunity against E. coli, or Klebsiella pneumoniae and Toxoplasma gondi infection (63, 64). ILC3s also contribute to host resistance to Rotavirus. ILC3 derived IL-22 synergizes with IFNλ to minimize viral replication (65).
These studies point to an important role of group 3 ILCs in promoting symbiosis with commensals and protection against pathogens. ILC3s form a dynamic network of interactions with the microbiome, other immune cells, the intestinal epithelium and surprisingly with neuro-glial cells to secure intestinal homeostasis (Figure 2).
Group 3 ILCS in Intestinal Disease: the Other Side of the Coin
In a landmark study, Buonocore et al. showed that chronic infection of immunodeficient mice with Helicobacter hepaticus resulted in RORγt+ ILC3 mediated gut inflammation. ILC3s produced high levels of IL-17 and IFNγ in response to IL-23, thus contributing to the development of T-cell independent inflammation (26). Similarly, ILC3s were crucial for the development of innate mediated colitis using the anti-CD40 model of IBD (26). In support of a pathogenic role of ILC3s in intestinal inflammation (Figure 3), Geremia et al. showed that IL-23 responsive ILC3s were increased in the intestines of patients with CD, where in response to IL-23 they produced high levels of IBD-relevant cytokines such as IL-17 and IL-22 (66). Shedding more light into the mechanisms underlying ILC3-driven intestinal pathology, Pearson et al. showed that ILC3s use GM-CSF to attract inflammatory monocytes on site, whereas mobility of ILC3s in and out of cryptopatches appeared to induce inflammation at non-inflamed sites (67). Recently, Bauché et al. showed that Tregs were able to prevent ILC3-induced colitis (68). In particular, latent activation gene 3 (LAG3) expressing Tregs reduced IL-1β and IL-23 production by CX3CR1+ macrophages resulting in impaired IL-22 production by ILC3s and therefore attenuated disease (68). Moreover, using Tbx21−/−Rag2−/− Ulcerative Colitis (TRUC) mice, which develop spontaneous colitis with clinical features that resemble aspects of human UC (69), Powell et al. showed that IL-23 and TNFα produced by CD103−CD11b+ mononuclear phagocytes activated ILC3s to drive intestinal inflammation through production of IL-17 (70). In particular, NCR− ILC3s, the major ILC subset present in the colon of TRUC mice, were potent producers of IL-17 and IL-22 in response to IL-1α and IL-23, an effect that was more profound in the presence of IL-6 (71). Interestingly, in vivo blockade of IL-6 using neutralizing antibodies significantly attenuated colonic inflammation in TRUC mice (71).
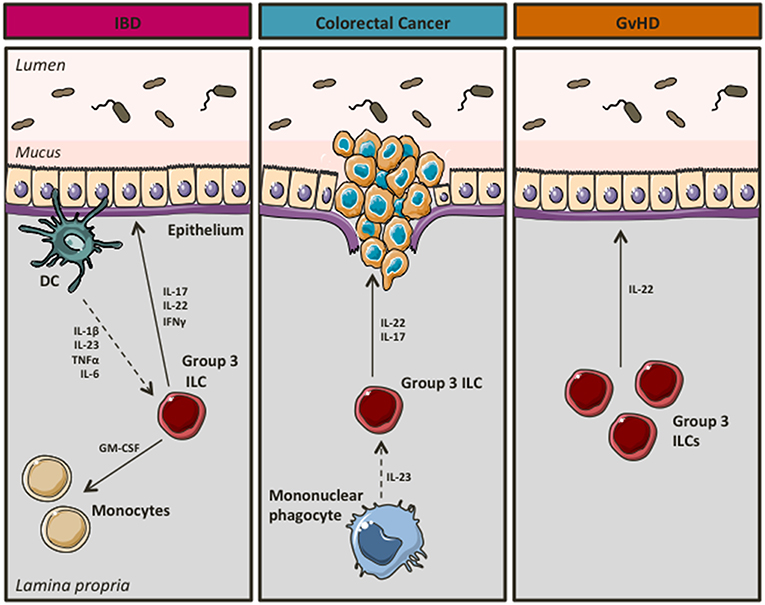
Figure 3. Group 3 ILCs during intestinal disease. In chronic inflammation group 3 ILCs seem to acquire a pro-inflammatory phenotype, thus contributing to the development of IBD and/or colorectal cancer. However, they appear to have a beneficial role in GvHD.
Interestingly, group 3 ILCs have also been associated with a pro-inflammatory, pathogenic role in cancer (Figure 3), a feared complication of unopposed inflammation in patients with UC (72, 73). IL-17+IL-22+ ILCs accumulate in colorectal cancer occurring in Helicobacter hepaticus associated colorectal cancer. ILC depletion alleviated invasive cancer in this model (74). Importantly, IL-22, but not IL-17, was necessary for cancer maintenance (74). In support of ILC3s having a role in promoting colorectal cancer, Chan et al. showed that ILC3s were key drivers of IL-23 induced tumorigenesis, however in this setting ILC3 actions were mediated by IL-17 (75).
Intestinal inflammation can also occur as a result of graft vs. host disease (GvHD) in recipients of allogenic hematopoietic stem cell transplants (AHSCT) as a treatment of hematopoietic cell disorders including blood cancers (76). Although several studies suggest that ILC3s may contribute to intestinal inflammation promoting the development of IBD or colorectal cancer, in GvHD they might be beneficial (Figure 3). Hanash et al. showed that IL-22 production by ILC3s was increased in patients following pretransplant conditioning, whereas IL-22 levels were reduced upon emergence of GvHD (77). Notably, IL-22 deficiency in recipients resulted in significant intestinal inflammation and tissue damage (77). Moreover, Munneke et al. suggested that elevated numbers of NCR+ ILC3s in peripheral blood of leukemia patients following AHSCT were associated with reduced risk of GvHD (78).
Concluding Remarks
Although they are phenotypically diverse, and exist as multiple different subsets group 3 ILCs are probably the best-characterized ILC lineage, and they appear to play an important role regulating the balance between maintenance and loss of intestinal homeostasis. A decade after their discovery, ILC3s have emerged as important regulators of inflammation at mucosal surfaces, and in the gut in particular ILC3s form a dynamic network of interactions with the microbiome, other immune cells, the intestinal epithelium and enteric neurons to secure intestinal homeostasis. However, ILC3s may also promote inflammation leading to the development of IBD and/or colorectal cancer. Additional work is needed to scrutinize ILC3 biology and their contributions to intestinal disease. Targeting ILCs, their key upstream activating mediators (e.g., IL-23, IL-1β, or IL6), their survival factors (e.g., IL-7), or their effector cytokines (IL-22, IL-17, and IFNγ) hold promise for treating inflammatory diseases such as IBD.
Author Contributions
EP wrote the manuscript and designed the figures. NP critically reviewed the manuscript.
Funding
NP is funded by the Wellcome Trust.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, et al. Metagenomic analysis of the human distal gut microbiome. Science. (2006) 312:1355–9. doi: 10.1126/science.1124234
2. Baumgart DC, Sandborn WJ. Inflammatory bowel disease: clinical aspects and established and evolving therapies. Lancet. (2007) 369:1641–57. doi: 10.1016/S0140-6736(07)60751-X
3. Loftus EV Jr. Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology. (2004) 126:1504–17. doi: 10.1053/j.gastro.2004.01.063
4. Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med. (2009) 361:2066–78. doi: 10.1056/NEJMra0804647
5. de Lange KM, Barrett JC. Understanding inflammatory bowel disease via immunogenetics. J Autoimmun. (2015) 64:91–100. doi: 10.1016/j.jaut.2015.07.013
6. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. (2012) 491:119–24. doi: 10.1038/nature11582
7. Spits H, Cupedo T. Innate lymphoid cells: emerging insights in development, lineage relationships, and function. Annu Rev Immunol. (2012) 30:647–75. doi: 10.1146/annurev-immunol-020711-075053
8. Sonnenberg GF, Mjosberg J, Spits H, Artis D. SnapShot: innate lymphoid cells. Immunity. (2013) 39:622.e1. doi: 10.1016/j.immuni.2013.08.021
9. Artis D, Spits H. The biology of innate lymphoid cells. Nature. (2015) 517:293–301. doi: 10.1038/nature14189
10. Spits H, Di Santo JP. The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nat Immunol. (2011) 12:21–7. doi: 10.1038/ni.1962
11. Eberl G. Development and evolution of RORgammat+ cells in a microbe's world. Immunol Rev. (2012) 245:177–88. doi: 10.1111/j.1600-065X.2011.01071.x
12. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells–a proposal for uniform nomenclature. Nat Rev Immunol. (2013) 13:145–9. doi: 10.1038/nri3365
13. Wang S, Xia P, Chen Y, Qu Y, Xiong Z, Ye B, et al. Regulatory innate lymphoid cells control innate intestinal inflammation. Cell. (2017) 171:201–16.e18. doi: 10.1016/j.cell.2017.07.027
14. Klose CSN, Flach M, Mohle L, Rogell L, Hoyler T, Ebert K, et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell. (2014) 157:340–56. doi: 10.1016/j.cell.2014.03.030
15. Possot C, Schmutz S, Chea S, Boucontet L, Louise A, Cumano A, et al. Notch signaling is necessary for adult, but not fetal, development of RORgammat(+) innate lymphoid cells. Nat Immunol. (2011) 12:949–58. doi: 10.1038/ni.2105
16. Cherrier M, Sawa S, Eberl G. Notch, Id2, and RORgammat sequentially orchestrate the fetal development of lymphoid tissue inducer cells. J Exp Med. (2012) 209:729–40. doi: 10.1084/jem.20111594
17. Constantinides MG, McDonald BD, Verhoef PA, Bendelac A. A committed precursor to innate lymphoid cells. Nature. (2014) 508:397–401. doi: 10.1038/nature13047
18. Sun Z, Unutmaz D, Zou YR, Sunshine MJ, Pierani A, Brenner-Morton S, et al. Requirement for RORgamma in thymocyte survival and lymphoid organ development. Science. (2000) 288:2369–73. doi: 10.1126/science.288.5475.2369
19. Eberl G, Marmon S, Sunshine MJ, Rennert PD, Choi Y, Littman DR. An essential function for the nuclear receptor RORgamma(t) in the generation of fetal lymphoid tissue inducer cells. Nat Immunol. (2004) 5:64–73. doi: 10.1038/ni1022
20. Satoh-Takayama N, Vosshenrich CA, Lesjean-Pottier S, Sawa S, Lochner M, Rattis F, et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity. (2008) 29:958–70. doi: 10.1016/j.immuni.2008.11.001
21. Lee JS, Cella M, McDonald KG, Garlanda C, Kennedy GD, Nukaya M, et al. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat Immunol. (2011) 13:144–51. doi: 10.1038/ni.2187
22. Kiss EA, Vonarbourg C, Kopfmann S, Hobeika E, Finke D, Esser C, et al. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science. (2011) 334:1561–5. doi: 10.1126/science.1214914
23. Takatori H, Kanno Y, Watford WT, Tato CM, Weiss G, Ivanov II, et al. Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J Exp Med. (2009) 206:35–41. doi: 10.1084/jem.20072713
24. Cupedo T, Crellin NK, Papazian N, Rombouts EJ, Weijer K, Grogan JL, et al. Human fetal lymphoid tissue-inducer cells are interleukin 17-producing precursors to RORC+ CD127+ natural killer-like cells. Nat Immunol. (2009) 10:66–74. doi: 10.1038/ni.1668
25. Cella M, Fuchs A, Vermi W, Facchetti F, Otero K, Lennerz JK, et al. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature. (2009) 457:722–5. doi: 10.1038/nature07537
26. Buonocore S, Ahern PP, Uhlig HH, Ivanov II, Littman DR, Maloy KJ, et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature. (2010) 464:1371–5. doi: 10.1038/nature08949
27. Sonnenberg GF, Monticelli LA, Elloso MM, Fouser LA, Artis D. CD4(+) lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity. (2011) 34:122–34. doi: 10.1016/j.immuni.2010.12.009
28. Cella M, Otero K, Colonna M. Expansion of human NK-22 cells with IL-7, IL-2, and IL-1beta reveals intrinsic functional plasticity. Proc Natl Acad Sci USA. (2010) 107:10961–6. doi: 10.1073/pnas.1005641107
29. Vonarbourg C, Mortha A, Bui VL, Hernandez PP, Kiss EA, Hoyler T, et al. Regulated expression of nuclear receptor RORgammat confers distinct functional fates to NK cell receptor-expressing RORgammat(+) innate lymphocytes. Immunity. (2010) 33:736–51. doi: 10.1016/j.immuni.2010.10.017
30. Klose CS, Kiss EA, Schwierzeck V, Ebert K, Hoyler T, d'Hargues Y, et al. A T-bet gradient controls the fate and function of CCR6-RORgammat+ innate lymphoid cells. Nature. (2013) 494:261–5. doi: 10.1038/nature11813
31. Rankin LC, Groom JR, Chopin M, Herold MJ, Walker JA, Mielke LA, et al. The transcription factor T-bet is essential for the development of NKp46+ innate lymphocytes via the Notch pathway. Nat Immunol. (2013) 14:389–95. doi: 10.1038/ni.2545
32. Bernink JH, Krabbendam L, Germar K, de Jong E, Gronke K, Kofoed-Nielsen M, et al. Interleukin-12 and−23 control plasticity of CD127(+) Group 1 and Group 3 innate lymphoid cells in the intestinal lamina propria. Immunity. (2015) 43:146–60. doi: 10.1016/j.immuni.2015.06.019
33. Rankin LC, Girard-Madoux MJ, Seillet C, Mielke LA, Kerdiles Y, Fenis A, et al. Complementarity and redundancy of IL-22-producing innate lymphoid cells. Nat Immunol. (2016) 17:179–86. doi: 10.1038/ni.3332
34. Pantelyushin S, Haak S, Ingold B, Kulig P, Heppner FL, Navarini AA, et al. Rorgammat+ innate lymphocytes and gammadelta T cells initiate psoriasiform plaque formation in mice. J Clin Invest. (2012) 122:2252–6. doi: 10.1172/JCI61862
35. Villanova F, Flutter B, Tosi I, Grys K, Sreeneebus H, Perera GK, et al. Characterization of innate lymphoid cells in human skin and blood demonstrates increase of NKp44+ ILC3 in psoriasis. J Invest Dermatol. (2014) 134:984–91. doi: 10.1038/jid.2013.477
36. Perry JS, Han S, Xu Q, Herman ML, Kennedy LB, Csako G, et al. Inhibition of LTi cell development by CD25 blockade is associated with decreased intrathecal inflammation in multiple sclerosis. Sci Transl Med. (2012) 4:145ra106. doi: 10.1126/scitranslmed.3004140
37. Langowski JL, Zhang X, Wu L, Mattson JD, Chen T, Smith K, et al. IL-23 promotes tumour incidence and growth. Nature. (2006) 442:461–5. doi: 10.1038/nature04808
38. Shields JD, Kourtis IC, Tomei AA, Roberts JM, Swartz MA. Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21. Science. (2010) 328:749–52. doi: 10.1126/science.1185837
39. Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol. (2011) 12:1045–54. doi: 10.1038/ni.2131
40. Sawa S, Cherrier M, Lochner M, Satoh-Takayama N, Fehling HJ, Langa F, et al. Lineage relationship analysis of RORgammat+ innate lymphoid cells. Science. (2010) 330:665–9. doi: 10.1126/science.1194597
41. Sonnenberg GF, Monticelli LA, Alenghat T, Fung TC, Hutnick NA, Kunisawa J, et al. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science. (2012) 336:1321–5. doi: 10.1126/science.1222551
42. Hepworth MR, Monticelli LA, Fung TC, Ziegler CG, Grunberg S, Sinha R, et al. Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature. (2013) 498:113–7. doi: 10.1038/nature12240
43. Goto Y, Panea C, Nakato G, Cebula A, Lee C, Diez MG, et al. Segmented filamentous bacteria antigens presented by intestinal dendritic cells drive mucosal Th17 cell differentiation. Immunity. (2014) 40:594–607. doi: 10.1016/j.immuni.2014.03.005
44. von Burg N, Chappaz S, Baerenwaldt A, Horvath E, Bose Dasgupta S, Ashok D, et al. Activated group 3 innate lymphoid cells promote T-cell-mediated immune responses. Proc Natl Acad Sci USA. (2014) 111:12835–40. doi: 10.1073/pnas.1406908111
45. Korn LL, Thomas HL, Hubbeling HG, Spencer SP, Sinha R, Simkins HM, et al. Conventional CD4+ T cells regulate IL-22-producing intestinal innate lymphoid cells. Mucosal Immunol. (2014) 7:1045–57. doi: 10.1038/mi.2013.121
46. Mao K, Baptista AP, Tamoutounour S, Zhuang L, Bouladoux N, Martins AJ, et al. Innate and adaptive lymphocytes sequentially shape the gut microbiota and lipid metabolism. Nature. (2018) 554:255–9. doi: 10.1038/nature25437
47. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. (2006) 126:1121–33. doi: 10.1016/j.cell.2006.07.035
48. Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. (2007) 8:967–74. doi: 10.1038/ni1488
49. Sano T, Huang W, Hall JA, Yang Y, Chen A, Gavzy SJ, et al. An IL-23R/IL-22 circuit regulates epithelial serum amyloid A to promote local effector Th17 responses. Cell. (2016) 164:324. doi: 10.1016/j.cell.2015.12.047
50. Sawa S, Lochner M, Satoh-Takayama N, Dulauroy S, Berard M, Kleinschek M, et al. RORgammat+ innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat Immunol. (2011) 12:320–6. doi: 10.1038/ni.2002
51. Tsuji M, Suzuki K, Kitamura H, Maruya M, Kinoshita K, Ivanov II, et al. Requirement for lymphoid tissue-inducer cells in isolated follicle formation and T cell-independent immunoglobulin A generation in the gut. Immunity. (2008) 29:261–71. doi: 10.1016/j.immuni.2008.05.014
52. Bouskra D, Brezillon C, Berard M, Werts C, Varona R, Boneca IG, et al. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature. (2008) 456:507–10. doi: 10.1038/nature07450
53. Kruglov AA, Grivennikov SI, Kuprash DV, Winsauer C, Prepens S, Seleznik GM, et al. Nonredundant function of soluble LTalpha3 produced by innate lymphoid cells in intestinal homeostasis. Science. (2013) 342:1243–6. doi: 10.1126/science.1243364
54. Mortha A, Chudnovskiy A, Hashimoto D, Bogunovic M, Spencer SP, Belkaid Y, et al. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science. (2014) 343:1249288. doi: 10.1126/science.1249288
55. Ibiza S, Garcia-Cassani B, Ribeiro H, Carvalho T, Almeida L, Marques R, et al. Glial-cell-derived neuroregulators control type 3 innate lymphoid cells and gut defence. Nature. (2016) 535:440–3. doi: 10.1038/nature18644
56. Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. (2008) 14:282–9. doi: 10.1038/nm1720
57. Wang Y, Koroleva EP, Kruglov AA, Kuprash DV, Nedospasov SA, Fu YX, et al. Lymphotoxin beta receptor signaling in intestinal epithelial cells orchestrates innate immune responses against mucosal bacterial infection. Immunity. (2010) 32:403–13. doi: 10.1016/j.immuni.2010.02.011
58. Emgard J, Kammoun H, Garcia-Cassani B, Chesne J, Parigi SM, Jacob JM, et al. Oxysterol sensing through the receptor GPR183 promotes the lymphoid-tissue-inducing function of innate lymphoid cells and colonic inflammation. Immunity. (2018) 48:120–32.e8. doi: 10.1016/j.immuni.2017.11.020
59. Chu C, Moriyama S, Li Z, Zhou L, Flamar AL, Klose CSN, et al. Anti-microbial functions of Group 3 innate lymphoid cells in gut-associated lymphoid tissues are regulated by G-protein-coupled receptor 183. Cell Rep. (2018) 23:3750–8. doi: 10.1016/j.celrep.2018.05.099
60. Guo X, Qiu J, Tu T, Yang X, Deng L, Anders RA, et al. Induction of innate lymphoid cell-derived interleukin-22 by the transcription factor STAT3 mediates protection against intestinal infection. Immunity. (2014) 40:25–39. doi: 10.1016/j.immuni.2013.10.021
61. Goto Y, Obata T, Kunisawa J, Sato S, Ivanov II, Lamichhane A, et al. Innate lymphoid cells regulate intestinal epithelial cell glycosylation. Science. (2014) 345:1254009. doi: 10.1126/science.1254009
62. Pickard JM, Maurice CF, Kinnebrew MA, Abt MC, Schenten D, Golovkina TV, et al. Rapid fucosylation of intestinal epithelium sustains host-commensal symbiosis in sickness. Nature. (2014) 514:638–41. doi: 10.1038/nature13823
63. Deshmukh HS, Liu Y, Menkiti OR, Mei J, Dai N, O'Leary CE, et al. The microbiota regulates neutrophil homeostasis and host resistance to Escherichia coli K1 sepsis in neonatal mice. Nat Med. (2014) 20:524–30. doi: 10.1038/nm.3542
64. Wagage S, Harms Pritchard G, Dawson L, Buza EL, Sonnenberg GF, Hunter CA. The Group 3 innate lymphoid cell defect in aryl hydrocarbon receptor deficient mice is associated with T cell hyperactivation during intestinal infection. PLoS ONE. (2015) 10:e0128335. doi: 10.1371/journal.pone.0128335
65. Hernandez PP, Mahlakoiv T, Yang I, Schwierzeck V, Nguyen N, Guendel F, et al. Interferon-lambda and interleukin 22 act synergistically for the induction of interferon-stimulated genes and control of rotavirus infection. Nat Immunol. (2015) 16:698–707. doi: 10.1038/ni.3180
66. Geremia A, Arancibia-Carcamo CV, Fleming MP, Rust N, Singh B, Mortensen NJ, et al. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. J Exp Med. (2011) 208:1127–33. doi: 10.1084/jem.20101712
67. Pearson C, Thornton EE, McKenzie B, Schaupp AL, Huskens N, Griseri T, et al. ILC3 GM-CSF production and mobilisation orchestrate acute intestinal inflammation. Elife. (2016) 5:e10066. doi: 10.7554/eLife.10066
68. Bauche D, Joyce-Shaikh B, Jain R, Grein J, Ku KS, Blumenschein WM, et al. LAG3(+) regulatory T cells restrain interleukin-23-producing CX3CR1(+) gut-resident macrophages during Group 3 innate lymphoid cell-driven colitis. Immunity. (2018) 49:342–52.e5. doi: 10.1016/j.immuni.2018.07.007
69. Garrett WS, Lord GM, Punit S, Lugo-Villarino G, Mazmanian SK, Ito S, et al. Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell. (2007) 131:33–45. doi: 10.1016/j.cell.2007.08.017
70. Powell N, Walker AW, Stolarczyk E, Canavan JB, Gokmen MR, Marks E, et al. The transcription factor T-bet regulates intestinal inflammation mediated by interleukin-7 receptor+ innate lymphoid cells. Immunity. (2012) 37:674–84. doi: 10.1016/j.immuni.2012.09.008
71. Powell N, Lo JW, Biancheri P, Vossenkamper A, Pantazi E, Walker AW, et al. Interleukin 6 increases production of cytokines by colonic innate lymphoid cells in mice and patients with chronic intestinal inflammation. Gastroenterology. (2015) 149:456–67.e15. doi: 10.1053/j.gastro.2015.04.017
72. Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut. (2001) 48:526–35. doi: 10.1136/gut.48.4.526
73. Rutter MD, Saunders BP, Wilkinson KH, Rumbles S, Schofield G, Kamm MA, et al. Thirty-year analysis of a colonoscopic surveillance program for neoplasia in ulcerative colitis. Gastroenterology. (2006) 130:1030–8. doi: 10.1053/j.gastro.2005.12.035
74. Kirchberger S, Royston DJ, Boulard O, Thornton E, Franchini F, Szabady RL, et al. Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J Exp Med. (2013) 210:917–31. doi: 10.1084/jem.20122308
75. Chan IH, Jain R, Tessmer MS, Gorman D, Mangadu R, Sathe M, et al. Interleukin-23 is sufficient to induce rapid de novo gut tumorigenesis, independent of carcinogens, through activation of innate lymphoid cells. Mucosal Immunol. (2014) 7:842–56. doi: 10.1038/mi.2013.101
76. Ferrara JL, Levine JE, Reddy P, Holler E. Graft-versus-host disease. Lancet. (2009) 373:1550–61. doi: 10.1016/S0140-6736(09)60237-3
77. Hanash AM, Dudakov JA, Hua G, O'Connor MH, Young LF, Singer NV, et al. Interleukin-22 protects intestinal stem cells from immune-mediated tissue damage and regulates sensitivity to graft versus host disease. Immunity. (2012) 37:339–50. doi: 10.1016/j.immuni.2012.05.028
Keywords: group 3 innate lymphoid cells, symbiosis, intestinal inflammation, IBD, Crohn's disease, ulcerative colitis
Citation: Pantazi E and Powell N (2019) Group 3 ILCs: Peacekeepers or Troublemakers? What's Your Gut Telling You?! Front. Immunol. 10:676. doi: 10.3389/fimmu.2019.00676
Received: 31 August 2018; Accepted: 12 March 2019;
Published: 05 April 2019.
Edited by:
Thomas Thornton MacDonald, Queen Mary University of London, United KingdomReviewed by:
Rita Carsetti, Bambino Gesù Children Hospital (IRCCS), ItalyMark Travis, University of Manchester, United Kingdom
Copyright © 2019 Pantazi and Powell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eirini Pantazi, eirini.pantazi@kennedy.ox.ac.uk
Nick Powell, nick.powell@kcl.ac.uk