 Bixin Xi
Bixin Xi Siying Liu1†
Siying Liu1† Aiguo Liu
Aiguo Liu- 1Department of Pediatrics, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 2Department of Gynaecology, Wuhan Children’s Hospital (Wuhan Maternal and Child Healthcare Hospital), Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
Background: Hereditary spherocytosis (HS) is a congenital haemolytic anaemia attributed to dysregulation or abnormal quantities of erythrocyte membrane proteins. Currently, the most common erythrocytic gene, spectrin β (SPTB), variants are located in exons and give rise to mRNA defects. However, the genetic characteristics and pathogenic mechanisms of SPTB intronic variants are not completely understood. This study aimed to analyse a rare intronic inversion variant in the SPTB gene associated with HS, and explore the impact of the variant on SPTB mRNA splicing.
Method: The clinical manifestations of the patient were summarised and analysed for spherocytosis phenotype diagnosis. The pathogenic variant was identified in the proband using targeted next-generation and Sanger sequencing. RNA sequencing was performed to analyse whether SPTB gene splicing and expression were affected.
Results: Targeted next-generation sequencing identified a novel disease-associated intronic inversion variant of the SPTB gene in the proband. The inversion variant was located between intron 19 and 20, and contained the entire exon 20 and partial sequences of adjacent introns. Sanger sequencing confirmed that the intronic inversion variant only appeared in the genome of the proband, not in his parents. RNA sequencing revealed that the variant could result in the skipping of exon 20 and reduced expression of SPTB mRNA.
Conclusion: This study identifies a rare intronic inversion variant in the SPTB gene associated with hereditary spherocytosis. The pathogenic variant can lead to exon 20 skipping and decreased SPTB gene expression. This finding has not been previously reported in any literature. This study can expand the intronic variant spectrum of the SPTB gene, deepen our understanding of HS pathogenesis, and contribute to the genetic diagnosis and clinical management of patients.
1 Introduction
Hereditary spherocytosis is the most common congenital red cell membrane disorder resulting from pathogenic variants in five genes encoding erythrocyte membrane-anchoring and cytoskeleton proteins (Da Costa et al., 2013; Andolfo et al., 2016; Ji et al., 2019). The annual diagnosis of males and females with HS in China is approximately 1.27/100,000 and 1.49/100,000, respectively (Wang et al., 2015). However, the prevalence is likely underestimated given that spherocytosis is sometimes asymptomatic or has mild symptoms (Da Costa et al., 2013; Nieminen et al., 2021; Wu et al., 2021). Hereditary spherocytosis also refers to a group of heterogeneous non-immune anaemia showing a broad spectrum of severity in clinical manifestations that may easily result in misdiagnosis or missed diagnosis in some patients (Wu et al., 2021). Common symptoms of congenital spherocytosis include haemolytic anaemia, jaundice, and splenomegaly. Iron overload and cholelithiasis are the common symptoms of transfusion-dependent forms of congenital spherocytosis (Nieminen et al., 2021).
Pathogenic variants in five genes encoding ankyrin1 (ANK1), spectrin β (SPTB), spectrin alpha 1 (SPTA1), solute carrier family 4 member 1 (SLC4A1), and erythrocyte membrane protein band 4.2 (EPB42) are associated with erythrocyte membrane defects in patients with HS (He et al., 2018). The most common causative genes in Chinese patients with HS are ANK1 (46%) and SPTB (42%), followed by SLC4A1 (11%), whereas SPTA1 and EPB42 variants are less frequently reported (Wang D. et al., 2021). Autosomal dominant (AD) inheritance accounts for 75% of HS cases, mainly through ANK1, SPTB and SLC4A1 gene variants, whereas autosomal recessive (AR) inheritance is described in approximately 25% of cases, frequently involving SPTA1 and EPB42 gene variants (Wang et al., 2020; Wang D. et al., 2021). β-spectrin (encoded by SPTB) plays a key role in the formation and mechanical stability of the erythrocyte membrane (He et al., 2018; Wang et al., 2020; Wang D. et al., 2021; Li et al., 2022). A total of 206 SPTB variants (mainly localised on exons) are reported in the Human Gene Mutation Database (HGMD, http://www.hgmd.cf.ac.uk/ac/index.php, accessed 26 March 2021). The common alterations of SPTB that give rise to mRNA defects and truncated β-spectrin include nonsense, frameshift, and splice site (Maciag et al., 2009).
SPTB gene deficiency is usually inherited in an AD manner (An and Mohandas, 2008). De novo SPTB variants are relatively infrequent in HS, and family investigations provide only limited clinical evidence (Wang et al., 2018; Wang J. F. et al., 2021). Two de novo point variants in the SPTB gene, causing aberrant mRNA splicing and exon skipping, have been identified in HS (Hassoun et al., 1996; Perrotta et al., 2009). One is localized on intron 17 and leads to truncated β-spectrin Winston-Salem. The other is localized on intron 16 and results in aberrant β-spectrin Bari. However, there is a paucity of information on the genetic characteristics and pathogenic mechanisms of SPTB intronic inversion variants.
This study aimed to analyse a rare intronic inversion variant in the SPTB gene associated with HS, and explore the impact of the variant on SPTB mRNA splicing. This study expands the SPTB intronic variant spectrum, deepens our understanding of the pathogenesis of HS, and provides a new perspective for the genetic diagnosis and clinical management.
2 Materials and methods
2.1 Patient and ethical approval
A patient with anaemia was recruited from Tongji Hospital, Wuhan, China. Detailed information, including family medical history, clinical manifestations, progression of HS, and the results of laboratory examinations, was collected. Blood samples were obtained and prior written approval of the subject and his parents were performed. All procedures were performed in accordance with the Declaration of Helsinki, and approved by the Ethics Committee of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology.
2.2 Targeted next-generation sequencing (NGS)
Genomic DNA (1–3 μg) was extracted from peripheral blood samples of the proband and his parents using the QIAamp DNA Mini Kit (Qiagen, Shanghai, China). Each genomic DNA sample was segmented into fragments with an average size of 150 bp using an S220 Focused-ultrasonicator (Covaris, Woburn, MA, United States). Then, the DNA Sample Prep Reagent Set (MyGenostics, Beijing, China) was used to prepare paired-end sequencing libraries. Disease-causing gene variants were identified by customising an NGS panel targeting all exons and adjacent introns of HS-associated genes (ANK1, SPTB, SPTA1, SLC4A1, and EPB42), and a capture strategy was performed using the enrichment kit (MyGenostics Inc.) (Wu et al., 2011). Enriched libraries were sequenced using an Illumina HiSeq X 10 sequencer (Illumina, San Diego, CA, United States) (Song et al., 2019).
2.3 Bioinformatic analysis
Sequencing was followed by quality control to filter adapter sequences and low-quality reads (<80 bp). The filtered sequences were aligned to the human reference genome (hg19) using Burrows–Wheeler Aligner (BWA, http://bio-bwa.sourceforge.net/) (Li and Durbin, 2010). The single nucleotide variants (SNVs) and insertions/deletions (indels) were detected by Genome Analysis Toolkit (GATK, https://software.broadinstitute.org/gatk/) (McKenna et al., 2010). All the SNVs and indels were annotated with Annotate Variation (ANNOVAR, http://annovar.openbioinformatics.org/en/latest/), filtered using GATK Variant Filtration, and estimated via multiple databases (1000 Genomes Project, dbSNP, Exome Aggregation Consortium, Exome Variant Server, and HGMD). Functional prediction software was applied to predict the effect of the intronic inversion variant (Wu et al., 2015). Pathogenic variants were assessed according to the protocols issued by the American College of Medical Genetics and Genomics practice guidelines, in combination with clinical manifestations of the proband, family investigation, and screening tests for HS.
2.4 Sanger sequencing
Sanger sequencing was performed to verify the filtered variants. Specific primers for PCR were designed using Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/). The coding exons and adjacent introns of variant genes were amplified with Ex Taq DNA polymerase (Takara Bio, Dalian, China). Purified PCR products were sequenced on an ABI 3130 Genetic Analyzer (Applied Biosystems, CA), then analysed using the Mutation Surveyor (Softgenetics, PA). The same procedures were conducted in all family members to confirm the pathogenic variants.
2.5 RNA sequencing
Total RNA was extracted from the peripheral blood samples using TRIzol reagent (Invitrogen; Carlsbad, CA). Nanodrop (Thermo Fisher Scientific, Waltham, MA, United States) analysis and gel electrophoresis were performed to examine the RNA quality. The RNA library was constructed according to the manufacturer’s protocol (Illumina). Subsequently, RNA sequencing was performed to detect aberrant RNA expression events and potential changes in the splicing process. OUTRIDER (Outlier in RNA-Seq Finder, http://bioconductor.org/packages/OUTRIDER/) was used to identify aberrantly expressed genes in RNA sequencing data.
3 Results
3.1 Clinical features
The patient was a 7.5-year-old boy delivered full-term by caesarean section without asphyxia, weighing 3.6 kg, with a head circumference and body length of 34.7 cm and 51.3 cm, respectively. At birth, the local hospital detected a reduced haemoglobin level of 94 g/L (normal range [NR], 145–190 g/L) and progressive decline with 92 g/L and 79 g/L on days 3 and 6 after birth, respectively. Haemoglobin levels increased to 133 g/L after the infusion of red blood cells (RBCs). Haemoglobin levels were maintained at 85 g/L after discharge. At 4 months, the patient was readmitted to the hospital for “4 months of anaemia and 1 month of lagging growth”. Haematologic assessment revealed haemoglobin levels at 78.0 g/L (NR, 120–160 g/L), white blood cell count at 7.15 × 109/L (NR, 3.5–9.5 × 109/L), RBC count at 3.29 × 1012/L (NR, 4.0–5.5 × 1012/L), mean corpuscular haemoglobin concentration at 362 g/L (NR, 320–360 g/L), and reticulocytes at 4.95% (NR, 0.5%–1.5%). The indices of iron metabolism showed soluble transferrin receptors at a high level of 8.11 mg/L (NR, 2.2–5.0 mg/L), while other parameters were normal. The direct anti-human globulin test (Coombs test) found no abnormality. The patient had an ABO blood group A, Rh (D)-positive. The erythrocyte osmotic brittleness was elevated. Abdominal ultrasound showed an enlarged spleen. Total serum bilirubin (93.9 μmol/L; NR, <26.0 μmol/L) and indirect bilirubin (86.3 μmol/L; NR, <16.8 μmol/L) levels were elevated. The parents of the patient were not consanguineous and had no history of haematological disease. Their routine blood and serum biochemical tests were normal.
At the age of 7.5 years, clinical symptoms and abnormal laboratory results still existed, but jaundice and splenomegaly were more serious. The patient was readmitted to our hospital. Blood tests revealed a haemoglobin level of 89 g/L, with 3.01 × 1012/L RBCs and 10.69% reticulocytes. Serum biochemical tests revealed that the concentrations of total bilirubin (125.1 μmol/L) and indirect bilirubin (111.1 μmol/L) were higher than previous standards. Abdominal ultrasound showed aggravated splenomegaly. Bone marrow smear analysis showed obvious erythroid hyperplasia (39.2%), with a preponderance of middle and late erythroblasts. RBCs showed a disparity in size, and spherical-shaped erythrocytes (7%) were found in the peripheral blood film (Figure 1A). Glucose-6-phosphate dehydrogenase activity was normal. The α- and β-thalassemia genetic variant screen and haemoglobin electrophoresis were negative. The haematologic examinations are summarised in Table 1.
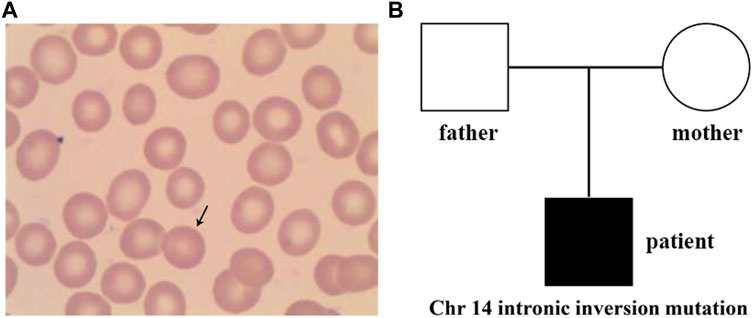
FIGURE 1. Peripheral blood smear and family tree of the patient. (A) Spherical-shaped erythrocytes were found in the peripheral blood film. (B), Family tree of a Chinese family with the patient exhibiting hereditary spherocytosis. Square and circle denote male and female, respectively.
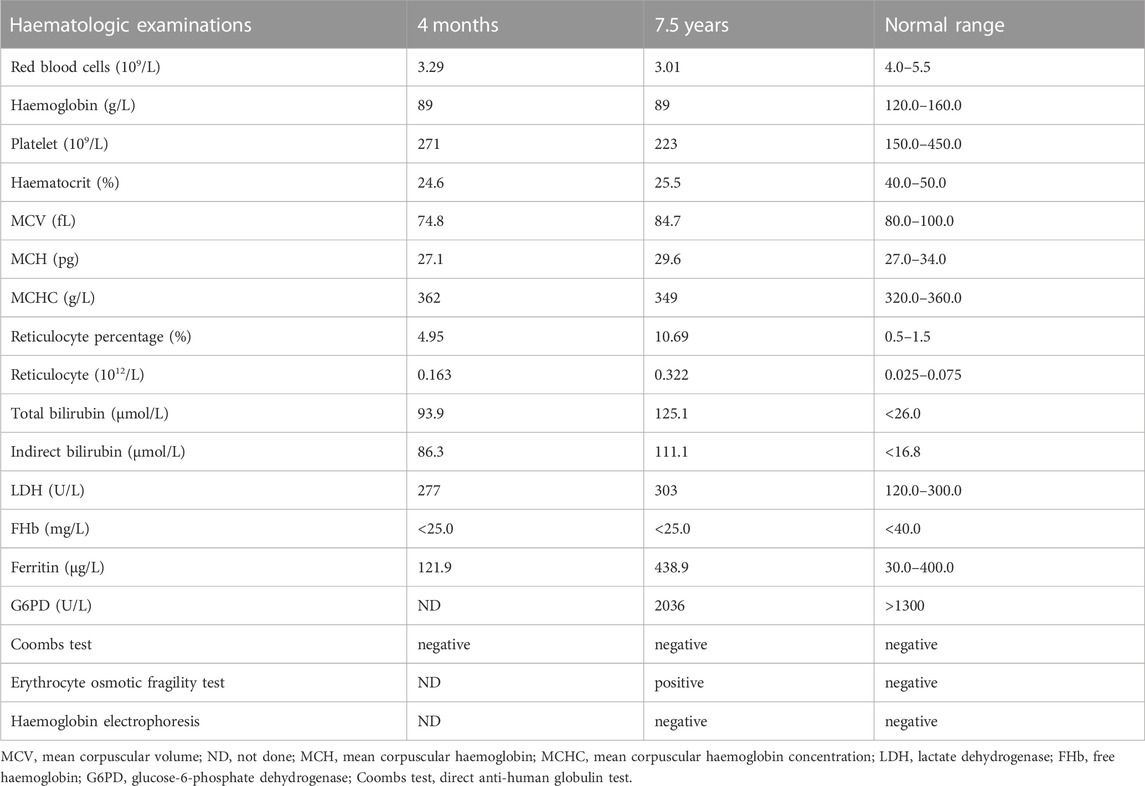
TABLE 1. Results of haematologic examinations of the patient.
3.2 Genetic and phenotype analysis
3.2.1 Next-generation sequencing output and coverage
Next-generation sequencing was performed with a gene panel targeting all coding exons and adjacent introns of genes associated with haematological system disease, including ANK1, SPTA1, EPB42, SLC4A1, and SPTB. Sequencing of the 99.80% targeted amplicons of the proband were covered at least once, with 99.56%, 99.22%, and 98.63% amplicons covered at least 4, 10, and 20 times, respectively. The mean read depth was 531-fold.
3.2.2 Mutation detection
A total of 36 variants in the patient were identified using NGS (Supplementary Table S1), filtered, and annotated to detect pathogenic variants. A novel intronic inversion in the SPTB gene was identified, which was associated with the patient’s phenotype. The inversion variant resided between the 5′-gene breakpoint of intron 19 and 3′-gene breakpoint of intron 20 in the SPTB gene (Figure 2; Figure 3A) that could disrupt the canonical splice site. This inversion variant contained entire exon 20 and partial sequences of adjacent introns. The inversion was represented with a schematic diagram (Figure 3A).
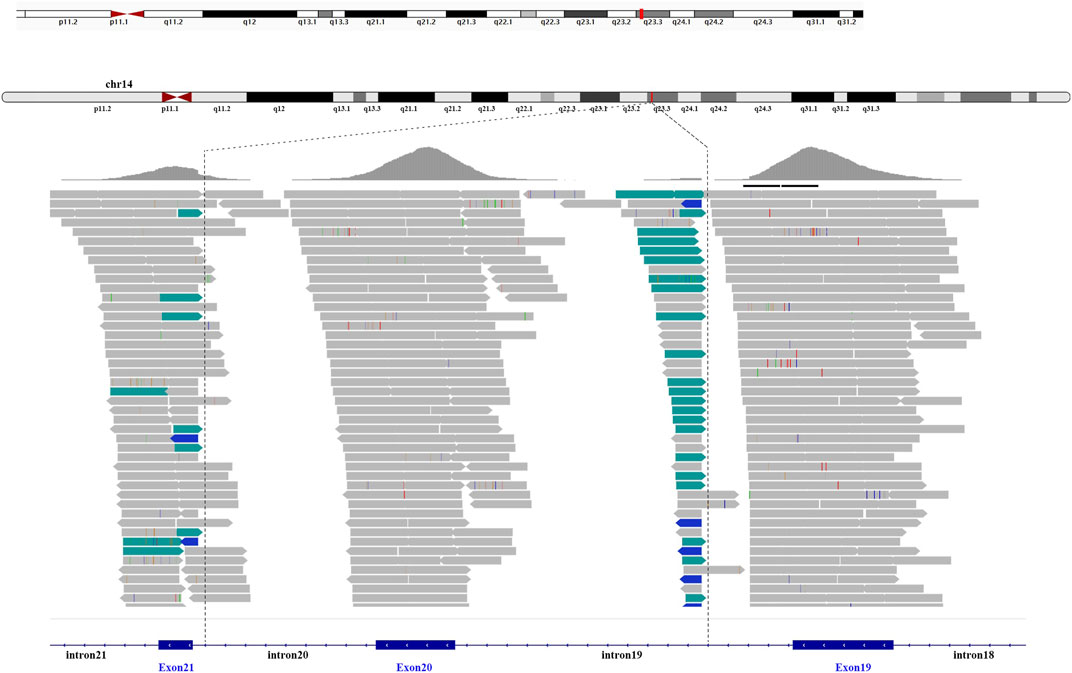
FIGURE 2. Next-generation sequencing. The intronic inversion mutation located between intron 19 and 20 of the SPTB gene was visualized by the integrative genomic viewer. The discordant reads are labelled green and blue.
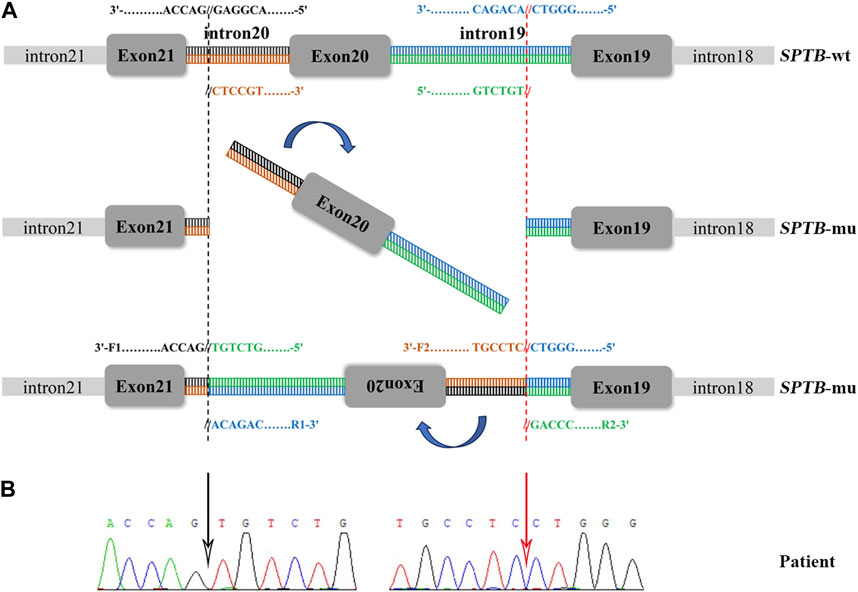
FIGURE 3. Sanger sequencing validation and a schematic diagram of the intronic inversion mutation in SPTB. (A) Schematic diagram of the SPTB gene. F1, forward primer 1; R1, reverse primer 1; F2, forward primer 2; R2, reverse primer 2. (B) The intronic inversion variant carried by the proband was confirmed using Sanger sequencing. The black and red arrow indicates the 5′-gene breakpoint of intron 19 and 3′-gene breakpoint of intron 20 in the SPTB gene, respectively.
3.2.3 Sanger sequencing validation and RNA sequencing analysis
Sanger sequencing was performed on the cDNA samples from all family members to validate the intronic inversion variant in the SPTB gene using the following primers: 3′-gene breakpoint of intron 20 in SPTB: forward primer 1, 5′- AGGACTCACCTGGTTCTTCTTCATG-3′ and reverse primer 1, 3′- TGATGCAGAAACAGAAAACC-5'; 5′-gene breakpoint of intron 19 in SPTB: forward primer 2, 3′-GGAGCAAGTTATTTGGAACCTCAG-5′ and reverse primer 2, 3′- ACAAAGGAGAAGACCCAGCACC-5' (Figure 3A). The PCR product only presented in the proband, not in his parents (Figure 1B; Figure 3B). This confirmed that the intronic inversion was a de novo mutation. RNA sequencing analysis found that the intronic inversion variant was associated with exon 20 skipping in the SPTB mRNA (Figure 4A), and revealed one underexpression outlier, which was the SPTB gene (Figure 4B).
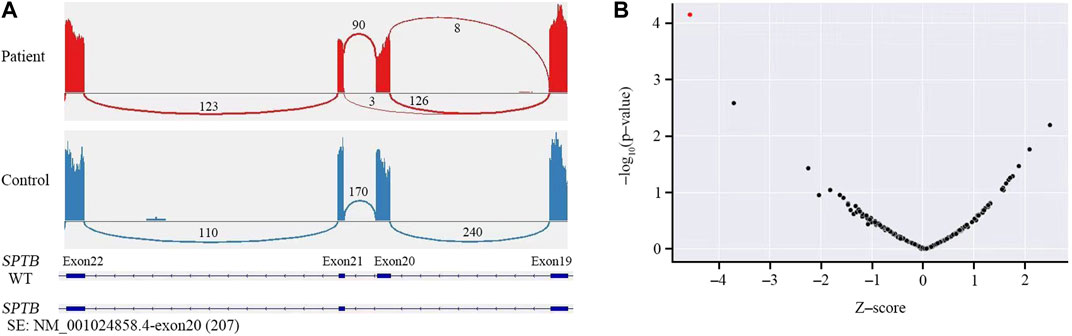
FIGURE 4. RNA sequencing analysis. (A) RNA sequencing showed skipped exon 20 in the SPTB mRNA of the patient, but not the control. (B) Gene-level significance (−log10(P), y-axis) versus Z-score, with SPTB labelled as the expression outlier (red dot) of sample from the proband.
3.2.4 Functional prediction and phenotype analysis
The de novo intronic inversion in SPTB was not found in the 1000G, ExAC, EVS, or HGMD databases. Functional prediction software was applied to predict the effect of the intronic inversion variant. REVEL, SIFT, and MutationTaster predicted the variant to be “disease causing”, while PolyPhen predicted the variant to be “probably damaging.” Additionally, haemolysis, splenomegaly, jaundice, spherically-shaped erythrocytes of peripheral blood film, and increased erythrocyte osmotic brittleness were observed in the patient, which were consistent with the SPTB genetic variant.
4 Discussion
A precise diagnosis of HS is challenging owing to the heterogeneity of clinical manifestations and non-ideal sensitivity or specificity of some diagnostic tests. Misdiagnosis or missed diagnosis of HS still existed in some patients in recent years (Ma et al., 2018; Zhu et al., 2020). The conventional HS diagnosis scheme is mainly based on family history, typical clinical manifestations, and relevant laboratory test results. However, the clinical manifestations of HS in children show obvious heterogeneity. Most patients suffer from well-compensated haemolytic anaemia, while only a minority have severe anaemia requiring blood transfusion (Wu et al., 2021). Routine blood tests, such as low haemoglobin, normal or elevated reticulocytes, normal or increased mean corpuscular haemoglobin concentration, increased number of spherocytes, and elevated indirect bilirubin in the serum are not sufficiently sensitive or specific (Wu et al., 2021). The flow cytometric osmotic fragility test (FOFT) and eosin-5′-maleimide dye-binding test (EMA) have been shown to be superior to conventional osmotic fragility tests for the diagnosis of HS (Arora et al., 2018). In our case, the proband had a negative family history and lack of typical microspherocytes (<10.0%); therefore, he was initially misdiagnosed with autoimmune haemolytic anaemia. Next-generation sequencing facilitated fast and accurate detection of the variant sites of disease-causing genes. This is the first study precisely identifying pathogenic intronic inversion in the SPTB gene using NGS, confirming its clear advantage over conventional inspection methods in diagnosis. Thus, the new HS diagnostic protocol (clinical manifestations, routine laboratory tests, a combination of FOFT and EMA, family investigation, and genetic testing) could improve the clinical efficiency and accuracy of HS diagnosis.
The SPTB gene (OMIM accession number 182870) is located on chromosome 14q23.3, which is over 100 kbp long, and contains 35 exons encoding 2,328 amino acids that make up an actin-binding region, seventeen spectral repeats, and a C-terminus (Ipsaro et al., 2009; Wang D. et al., 2021). In normal human erythrocytes, a-spectrin (encoded by SPTA1) has a synthesis rate that is three to four times faster than β-spectrin (encoded by SPTB). Thus, β-spectrin synthesis is a rate-limiting step for the formation of the α2β2-tetramerisation domain (Perrotta et al., 2008; He et al., 2018). β-spectrin plays a crucial role in maintaining deformability and stability of the erythrocyte membrane. SPTB variants can lead to failures in erythrocyte membrane skeleton formation, decreases in the reversible deformation ability of RBCs, and the destruction of erythrocytes as they traverse the microvasculature and splenic tissues (Tole et al., 2020). ANK1 variants are only identified in HS, while SPTB variants are found in other diseases, such as hereditary elliptocytosis or hereditary pyropoikilocytosis (He et al., 2018). SPTB variants can be distributed throughout the gene; most of the variant sites are located outside the tetramerisation domain (encoded by exons 1–7), followed by spectrin repeats (encoded by exons 8–30) (Park et al., 2016; Wang et al., 2020). A retrospective study of 66 cases of HS showed that all SPTB variants were distributed outside the tetramerisation domain, except four variants associated with the hereditary elliptocytosis phenotype and two variants located in the spectrin repeats domain (Wang D. et al., 2021). These variants in the SPTB gene were mainly nonsense (32/66) and frameshift (15/66), which could lead to the deficiency of SPTB haploids and normal β-spectrin. This study also revealed that most SPTB variants were localised on exons 13, 15, and 18–30. Another study regarding 38 unrelated HS patients found 17 SPTB variants that mainly include nonsense variants (8/17), followed by splicing variants (3/17), duplications (2/17), missense variants (2/17), indel (1/17), and small deletion (1/17) (Wang et al., 2018). In addition, most of these SPTB variants were located on exons (15/17) and are considered as null variants that may result in SPTB haploinsufficiency and β-spectrin deficiency.
To date, most of the gene defects in SPTB (splicing, nonsense variants, and frameshift variants) usually result in exon skipping, mRNA transcript instability, or synthesis of truncated β-spectrin proteins (Hassoun et al., 1997; Perrotta et al., 2009). In a large family with 22 HS patients, researchers found that the intronic variant (SPTB NP_001020029.1: p.Ile294Serfs*35) led to exon 8 skipping, aberrant mRNA splicing, and synthesis of truncated proteins lossing seventeen spectral repeat domains (Nieminen et al., 2021). Another study on intronic SPTB variants showed that the β-spectrin deficiency were also associated with the instability of aberrantly spliced mRNA and the haploinsufficiency of SPTB (Maciag et al., 2009). Our study identified a novel intronic inversion of the SPTB gene in the HS proband. The inversion variant was located between intron 19 and 20, and contained the entirety of exon 20 and partial sequences of adjacent introns. This rare pathogenic inversion was not identified in any other databases. The intronic inversion variant could lead to the skipping of exon 20, a decrease of SPTB gene expression, and the associated spherocytosis phenotype.
However, the specific mechanism through which the pathogenic inversion localized on introns leads to exon skipping, aberrant mRNA splicing and β-spectrin deficiency remains unclear. It is suggested that the intron splice order and speed were crucial factors in the skipping of multiple exons and synthesis of truncated proteins (Takahara et al., 2002). The half-life and stability of truncated β-spectrin protein is worse than that of the wild-type, which contributes to increased proteolytic degradation of the truncated-type (Hassoun et al., 1996; Bogusławska et al., 2017). In addition, it was previously reported that the aberrant SPTB mRNA could not be detected in some HS cases, which may be due to transcript instability (Bassères et al., 2002; Bogusławska et al., 2017). Insufficient sample size and transcript instability were challenges for our study. Further studies on HS pathogenesis are urgently needed.
This study can also provide a theoretical basis for child healthcare, genetic counselling, prenatal diagnosis, and molecular targeted therapy into HS. A structured follow-up was recommended for the patient consisting of blood tests and abdominal ultrasound evaluations in the haematology outpatient clinic, together with visits by the spleen surgeon and child healthcare specialists every 3 months. The patient’s clinical condition has remained stable to date, and all family members are satisfied with the outcomes and agreed to regular follow-ups.
5 Conclusion
This study identified a rare intronic inversion variant localised between intron 19 and 20 of the SPTB gene that associated with the spherocytosis phenotype. It is the first study accurately detecting a pathogenic intronic inversion variant via NGS in China. This improves the clinical efficiency and accuracy of HS diagnosis, and confirms its clear advantage over conventional inspection methods. Moreover, RNA sequencing analysis revealed that the variant could lead to the skipping of exon 20 and decreasing of SPTB gene expression, which provided a new perspective for the pathogenesis of hereditary spherocytosis. These findings can ultimately lead to more informed healthcare and genetic counselling outcomes and better prognosis for patients with HS.
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by the Ethics Committee of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
BX: Data curation, Methodology, Writing–original draft. SL: Data curation, Writing–original draft. YoZ: Data curation, Methodology, Writing–review and editing. DZ: Data curation, Methodology, Writing–review and editing. YuZ: Formal Analysis, Supervision, Writing–review and editing. AL: Formal Analysis, Supervision, Writing–review and editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
We gratefully acknowledge the participation of the patient and his parents for allowing us to develop our knowledge and learn from their stories.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1309040/full#supplementary-material
References
An, X., and Mohandas, N. (2008). Disorders of red cell membrane. Br. J. Haematol. 141, 367–375. doi:10.1111/j.1365-2141.2008.07091.x
Andolfo, I., Russo, R., Gambale, A., and Iolascon, A. (2016). New insights on hereditary erythrocyte membrane defects. Haematologica 101, 1284–1294. doi:10.3324/haematol.2016.142463
Arora, R. D., Dass, J., Maydeo, S., Arya, V., Radhakrishnan, N., Sachdeva, A., et al. (2018). Flow cytometric osmotic fragility test and eosin-5'-maleimide dye-binding tests are better than conventional osmotic fragility tests for the diagnosis of hereditary spherocytosis. Int. J. Lab. Hematol. 40, 335–342. doi:10.1111/ijlh.12794
Bassères, D. S., Tavares, A. C., Costa, F. F., and Saad, S. T. (2002). Beta-Spectrin São PauloII, a novel frameshift mutation of the beta-spectrin gene associated with hereditary spherocytosis and instability of the mutant mRNA. Braz J. Med. Biol. Res. 35, 921–925. doi:10.1590/s0100-879x2002000800009
Bogusławska, D. M., Heger, E., Machnicka, B., Skulski, M., Kuliczkowski, K., and Sikorski, A. F. (2017). A new frameshift mutation of the β-spectrin gene associated with hereditary spherocytosis. Ann. Hematol. 96, 163–165. doi:10.1007/s00277-016-2838-0
Da Costa, L., Galimand, J., Fenneteau, O., and Mohandas, N. (2013). Hereditary spherocytosis, elliptocytosis, and other red cell membrane disorders. Blood Rev. 27, 167–178. doi:10.1016/j.blre.2013.04.003
Hassoun, H., Vassiliadis, J. N., Murray, J., Njolstad, P. R., Rogus, J. J., Ballas, S. K., et al. (1997). Characterization of the underlying molecular defect in hereditary spherocytosis associated with spectrin deficiency. Blood 90, 398–406.
Hassoun, H., Vassiliadis, J. N., Murray, J., Yi, S. J., Hanspal, M., Johnson, C. A., et al. (1996). Hereditary spherocytosis with spectrin deficiency due to an unstable truncated beta spectrin. Blood 87, 2538–2545. doi:10.1182/blood.v87.6.2538.bloodjournal8762538
He, B. J., Liao, L., Deng, Z. F., Tao, Y. F., Xu, Y. C., and Lin, F. Q. (2018). Molecular genetic mechanisms of hereditary spherocytosis: current perspectives. Acta Haematol. 139, 60–66. doi:10.1159/000486229
Ipsaro, J. J., Huang, L., and Mondragón, A. (2009). Structures of the spectrin-ankyrin interaction binding domains. Blood 113, 5385–5393. doi:10.1182/blood-2008-10-184358
Ji, Z., Liu, N., Du, Z., Luo, G., Bing, Z., Xing, Q., et al. (2019). Transcatheter closing atrial septal defect in a child with hereditary spherocytosis. Front. Pediatr. 7, 506. doi:10.3389/fped.2019.00506
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. doi:10.1093/bioinformatics/btp698
Li, S., Guo, P., Mi, L., Chai, X., Xi, K., Liu, T., et al. (2022). A novel SPTB mutation causes hereditary spherocytosis via loss-of-function of β-spectrin. Ann. Hematol. 101, 731–738. doi:10.1007/s00277-022-04773-3
Ma, S., Deng, X., Liao, L., Deng, Z., Qiu, Y., Wei, H., et al. (2018). Analysis of the causes of the misdiagnosis of hereditary spherocytosis. Oncol. Rep. 40, 1451–1458. doi:10.3892/or.2018.6578
Maciag, M., Płochocka, D., Adamowicz-Salach, A., and Burzyńska, B. (2009). Novel beta-spectrin mutations in hereditary spherocytosis associated with decreased levels of mRNA. Br. J. Haematol. 146, 326–332. doi:10.1111/j.1365-2141.2009.07759.x
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi:10.1101/gr.107524.110
Nieminen, T. T., Liyanarachchi, S., Comiskey, D. F., Wang, Y., Li, W., Hendrickson, I. V., et al. (2021). A novel essential splice site variant in SPTB in a large hereditary spherocytosis family. Mol. Genet. Genomic Med. 9, e1641. doi:10.1002/mgg3.1641
Park, J., Jeong, D. C., Yoo, J., Jang, W., Chae, H., Kim, J., et al. (2016). Mutational characteristics of ANK1 and SPTB genes in hereditary spherocytosis. Clin. Genet. 90, 69–78. doi:10.1111/cge.12749
Perrotta, S., Della Ragione, F., Rossi, F., Avvisati, R. A., Di Pinto, D., De Mieri, G., et al. (2009). Beta-spectrinBari: a truncated beta-chain responsible for dominant hereditary spherocytosis. Haematologica 94, 1753–1757. doi:10.3324/haematol.2009.010124
Perrotta, S., Gallagher, P. G., and Mohandas, N. (2008). Hereditary spherocytosis. Lancet 372, 1411–1426. doi:10.1016/S0140-6736(08)61588-3
Song, X., He, Y., Wang, X., and Gu, X. (2019). The complete mitochondrial genome of Cyamophila willieti (Wu) (Hemiptera: psyllidae). Mitochondrial DNA. B. Resour. 4, 3758–3759. doi:10.1080/23802359.2019.1681922
Takahara, K., Schwarze, U., Imamura, Y., Hoffman, G. G., Toriello, H., Smith, L. T., et al. (2002). Order of intron removal influences multiple splice outcomes, including a two-exon skip, in a COL5A1 acceptor-site mutation that results in abnormal pro-alpha1(V) N-propeptides and Ehlers-Danlos syndrome type I. Am. J. Hum. Genet. 71, 451–465. doi:10.1086/342099
Tole, S., Dhir, P., Pugi, J., Drury, L. J., Butchart, S., Fantauzzi, M., et al. (2020). Genotype-phenotype correlation in children with hereditary spherocytosis. Br. J. Haematol. 191, 486–496. doi:10.1111/bjh.16750
Wang, C., Cui, Y., Li, Y., Liu, X., and Han, J. (2015). A systematic review of hereditary spherocytosis reported in Chinese biomedical journals from 1978 to 2013 and estimation of the prevalence of the disease using a disease model. Intractable Rare Dis. Res. 4, 76–81. doi:10.5582/irdr.2015.01002
Wang, D., Song, L., Shen, L., Zhang, K., Lv, Y., Gao, M., et al. (2021a). Mutational characteristics of causative genes in Chinese hereditary spherocytosis patients: a report on fourteen cases and a review of the literature. Front. Pharmacol. 12, 644352. doi:10.3389/fphar.2021.644352
Wang, J. F., Ma, L., Gong, X. H., Cai, C., and Sun, J. J. (2021b). Severe hyperbilirubinemia in a neonate with hereditary spherocytosis due to a de novo ankyrin mutation: a case report. World J. Clin. cases. 9, 5245–5251. doi:10.12998/wjcc.v9.i19.5245
Wang, R., Yang, S., Xu, M., Huang, J., Liu, H., Gu, W., et al. (2018). Exome sequencing confirms molecular diagnoses in 38 Chinese families with hereditary spherocytosis. Sci China. Life Sci. 61, 947–953. doi:10.1007/s11427-017-9232-6
Wang, X., Zhang, A., Huang, M., Chen, L., Hu, Q., Lu, Y., et al. (2020). Genetic and clinical characteristics of patients with hereditary spherocytosis in Hubei Province of China. Front. Genet. 11, 953. doi:10.3389/fgene.2020.00953
Wu, J., Matthaei, H., Maitra, A., Dal Molin, M., Wood, L. D., Eshleman, J. R., et al. (2011). Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development. Sci. Transl. Med. 3, 92ra66. doi:10.1126/scitranslmed.3002543
Wu, L., Schaid, D. J., Sicotte, H., Wieben, E. D., Li, H., and Petersen, G. M. (2015). Case-only exome sequencing and complex disease susceptibility gene discovery: study design considerations. J. Med. Genet. 52, 10–16. doi:10.1136/jmedgenet-2014-102697
Wu, Y., Liao, L., and Lin, F. (2021). The diagnostic protocol for hereditary spherocytosis-2021 update. J. Clin. Lab. Anal. 35, e24034. doi:10.1002/jcla.24034
Keywords: hereditary spherocytosis, SPTB gene, next-generation sequencing, intronic inversion variant, genetic analysis
Citation: Xi B, Liu S, Zhu Y, Zhang D, Zhang Y and Liu A (2023) Case report: Genetic analysis of a novel intronic inversion variant in the SPTB gene associated with hereditary spherocytosis. Front. Genet. 14:1309040. doi: 10.3389/fgene.2023.1309040
Received: 11 October 2023; Accepted: 20 November 2023;
Published: 04 December 2023.
Edited by:
Ammar Husami, Cincinnati Children’s Hospital Medical Center, United StatesReviewed by:
Kshitij Srivastava, Clinical Center (NIH), United StatesImmacolata Andolfo, University of Naples Federico II, Italy
Copyright © 2023 Xi, Liu, Zhu, Zhang, Zhang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aiguo Liu, drliuaiguo@163.com; Yu Zhang, zhangyu080121@163.com
†These authors have contributed equally to this work