 Zou Xin-Yi
Zou Xin-Yi Dai Yang-Li
Dai Yang-Li Zeng Ling-Hui
Zeng Ling-Hui- 1Department of Clinical Medicine, Medical School of Hangzhou City University, Hangzhou, China
- 2Department of Endocrinology, The Children’s Hospital of Zhejiang University School of Medicine, Hangzhou, China
Objective: To analyze the phenotypes, genotypes, and the relationship of phenotypes and genotypes for Chinese patients with Bardet-Biedl syndrome (BBS).
Methods: The Chinese Wanfang and Weipu data, and PubMed were searched up to December 2022. Patients with detailed clinical feature data were involved in the analysis.
Results: A total of 153 Chinese patients, including 87 males, 53 females, and 12 unknown, were enrolled. Their ages ranged from 1.2 to 44 years old with a mean of 16.70 ± 9.90 years old. Among these patients, 80 (52.29%) were reported by ophthalmologists, and only 24 (15.68%) reported by pediatricians. Most patients (132/137, 96.35%) had visual problems; 131/153 (85.62%) had polydactyly; 124/132 (93.93%) were overweight or obese; 63/114 (55.26%) had renal abnormalities; kidney dysfunction was found in 33 (21.57%); 83/104 (79.81%) had hypogonadism and/or genital hypoplasia; and 111/136 (81.62%) had mental retardation. In this series, genetic analysis was performed in 90 (58.82%) patients, including 22 BBS7 (24.71%), 20 BBS2 (22.73%), and 10 BBS10 (11.24%) patients. Moreover, 11 fetuses were diagnosed prenatally in the last 4 years except for one patient in 2004 year. It was noted that BBS7 had higher penetrance. BBS2 had higher hearing impairment and lower renal abnormality penetrance. BBS10 also had lower renal abnormality penetrance as well.
Conclusion: Misdiagnosis or miss diagnosis of BBS may be common in China. In patients with polydactyly, visual impairment, obesity, renal abnormalities, hypogonadism, and mental retardation, or in fetuses with polydactyly and/or renal abnormalities, BBS should be considered in the differential diagnosis. Other deformities should be evaluated carefully and genetic analysis should be performed as early as possible.
1 Introduction
Bardet-Biedl syndrome (BBS, OMIM 209900) is an autosomal recessive disease, caused by a variant of BBS-related genes. It was first described in 1920 by Dr. Georges Bardet. It is a non-motorized ciliary dysfunction that affects multiple systems. The prevalence of BBS is about 1/125,000∼1/160,000 with regional and ethnic differences (Moore et al., 2005; Tsang et al., 2018). A high incidence (around 1/13,500 newborns) among Arab Bedouins in the Arabian Peninsula, North African desert regions, and Newfoundland’s high blood isolation population is related to consanguineous marriage (Farag and Teebi, 1988; Farag and Teebi, 1989; Gouronc et al., 2020). It is characterized by retinitis pigmentosa, obesity, renal abnormalities, polydactyly, mental retardation, and hypogonadism. Other symptoms including ophthalmic diseases (e.g., astigmatism, strabismus, cataracts), hearing impairment, craniofacial congenital anomalies, hypoplastic teeth, toe deformity, short stature, asthma, neurological impairment (e.g., ataxia, hyperspasmia, olfactory abnormalities, behavioral and mental disorders, and poor coordination), digestive system abnormalities (e.g., intestinal atresia, imperforate anus, congenital megacolon, liver fibrillation), metabolic abnormalities (e.g., insulin resistance, hyperglycemia, dyslipidemia, hypothyroidism, hypertension), and cardiovascular abnormalities, have also been reported (Mujahid et al., 2018; Tsang et al., 2018; Guardiola et al., 2021; Zhong et al., 2023).
BBS is a ciliopathy and has been shown to be closely related to dysfunction of immotile cilia. To date, 28 genes have been reported to be associated with the BBS phenotypes, including 2 candidate genes (SCLT1 and SCAPER) and 2 contributors (NPHP1 and TTC21B) (Niederlova et al., 2019; Khan et al., 2023; Gnanasekaran et al., 2023). In comparison, pathogenic variants in the BBS genes involved in encoding the BBSome complex, including BBS1 (OMIM 209901), BBS2 (OMIM 606151), BBS4 (OMIM 600374), BBS5 (OMIM 603650), BBS7 (OMIM 607590), BBS8/TTC8 (OMIM 615985), and BBS9/PTHB1 (OMIM 607968), were the most common, followed by BBS genes involved in encoding BBSome complex “chaperone-like” proteins, including BBS6/MKKS (OMIM 604896), BBS10 (OMIM 610148), and BBS12 (OMIM 610683) (Seongjin et al., 2009; Dai et al., 2022; Melluso et al., 2023). BBS proteins are needed for the maintenance of ciliary structure and function. The BBSome complex is an essential component of cilia differentiation. It mediates the transport of proteins to the membrane structure of cilia and participates in their structure formation and function (Seongjin et al., 2009). BBSome complex chaperone-like proteins are involved in the regulation of the BBSome complex, which together with members of the Rab family of proteins to promote intraflagellar transport (Yan and Shen, 2022; Melluso et al., 2023). Molecular diagnosis can be clarified by genetic testing in about 80% of patients. Most studies reported that BBS1 and BBS10 are the most frequently implicated genes (Mujahid et al., 2018). However, still about 20% of patients currently do not have a definitive molecular diagnosis currently (Forsythe et al., 2018), presenting a significant challenge for diagnosis and genetic counseling.
We aim to highlight the genotype-phenotype relationship and the diagnosis of BBS by summering the BBS patients reported from China. Studies in Chinese and English from China were all reviewed.
2 Materials and methods
2.1 Data collection
According to the PRISMA guidelines, we reviewed the Wanfang and Weipu data in Chinese using “Bardet-Biedl syndrome”, “Laurence-Moon-Biedl syndrome”, “Laurence-Moon-Bardet-Biedl syndrome”, or “Polydactyly-Obesity-Renal-Ocular syndrome” and in Chinese or English, also above keyword and “China or Chinese” in PubMed up to December 2022. Only patients with detailed clinic feature data were involved in the following analysis.
2.2 Subjects
All papers and thesis were reviewed carefully. Laurence-Moon syndrome (OMIM 245800) together with BBS were previously regarded as one disease (named Laurence-Moon-Biedl syndrome or Laurence-Moon-Bardet-Biedl syndrome). Now, Laurence-Moon syndrome has been regarded as another disease. Hence, we removed Laurence-Moon syndrome patients. A total of 61 papers (Xu, 1991; Zhang et al., 1999; Lin et al., 2000; Shao et al., 2000; Shen et al., 2001; Li and Pang, 2003; Tong and Yu, 2003; Wang, 2003; Zhang and Hu, 2004; Zhou and Wang, 2005; Lei et al., 2006; Shou et al., 2006; Yang et al., 2006; Zhang et al., 2007a; Zhang et al., 2007b; Chen et al., 2007; Ke and An, 2007; Lin et al., 2007; Lu and Wang, 2007; Li et al., 2008a; Li et al., 2008b; Chen et al., 2008; Liang and Zhou, 2008; Liu et al., 2008; Si et al., 2008; Ye et al., 2008; Zhang et al., 2009; Shou et al., 2011; Xiao et al., 2011; Zheng et al., 2011; Zheng et al., 2012; Cheng et al., 2013; Jiang, 2013; Li et al., 2013; Lin and Zhang, 2014; Wang et al., 2014; Xia et al., 2014; Yi et al., 2014; Huang et al., 2015; Lou et al., 2015; Qi et al., 2015; Wu and Zhang, 2015; Chen et al., 2017; Li et al., 2017; Ding et al., 2018; Lin et al., 2018; Qu et al., 2018; Shen et al., 2018; Wang et al., 2018; Qiang et al., 2019; Tao et al., 2019; Wu et al., 2019; Li et al., 2020; Xie et al., 2021a; Liu et al., 2021; Li et al., 2022a; Li and Hu, 2022; Lu et al., 2022; Shao and Dong, 2022; Shen et al., 2022; Wei et al., 2022) , 4 thesis in Chinese (Han, 2011; Xin, 2014; Xiu, 2018; Li, 2021), and 24 English papers (Wu, 1956; Wei et al., 1998; Hou, 2004; Yang et al., 2008; Li et al., 2014; Xing et al., 2014; Qi et al., 2017; Li et al., 2019; Shen et al., 2019; Dan et al., 2020; Tao et al., 2020; Huang et al., 2021; Jing et al., 2021; Meng et al., 2021; Tang et al., 2021; Zhang et al., 2021; Li et al., 2022b; Cai et al., 2022; Dong et al., 2022; Shao et al., 2022; Tang et al., 2022; Tao et al., 2022; Yan et al., 2022) in PubMed from China were enrolled. Among these 96 papers and thesis, patients repeated in 6 papers were merged (Lei et al., 2006; Shou et al., 2006; Zhang et al., 2007a; Zhang et al., 2007b; Shou et al., 2011; Shao et al., 2022). Finally, a total of 153 patients with BBS were enrolled in this study. The special field of journals, profession of authors, demographic information, clinical data, and genotype was collected, and compared with previous reports. The description of variants were revised according to the ACMG guidelines.
2.3 Diagnostic criteria
In children, overweight and obesity were defined according to “Body mass index growth curves for Chinese children and adolescents aged 0–18 years” (Li et al., 2009a). In adults, overweight was defined as a BMI between 24.0 and 27.99 kg/m2 while obesity was defined as a BMI ≥28.00 kg/m2.
In children, hypertension was defined according to the “updating blood pressure references for Chinese children aged 3–17 years” (Fan et al., 2017). In adults, hypertension was defined as a diastolic blood pressure >90 mmHg and/or a systolic blood pressure >140 mmHg.
Hyperglycemia included impaired fasting glucose (fasting glucose 5.6–6.9 mmol/L) and impaired glucose tolerance (7.8–11.0 mmol/L). Diabetes was defined as fasting glucose ≥7.0 mmol/L and/or random blood glucose ≥11.1 mmol/L.
Short stature was defined according to the “height and weight standardized growth chats for Chinese children and adolescents aged 0–18 years” (Li et al., 2009b).
2.4 Statistical analysis
Statistical analyses were conducted using SPSS software (version 22). The Pearson’s chi-square test and Fisher’s exact test were used to measure enumeration data between subgroups. Quantitative data with a normal distribution were expressed as the means ± SDs and analyzed by the independent t-test. Quantitative data with skewed distributions were expressed as medians (minimums-maximums). Differences were considered statistically significant at p < 0.05.
3 Results
3.1 Demographics
Among 153 patients (Supplementary Table S1), there were 87 males, 53 females, and 13 unknown (including fetuses) (Li et al., 2019; Qiang et al., 2019; Li et al., 2020; Jing et al., 2021; Cai et al., 2022; Dong et al., 2022; Tang et al., 2022; Yan et al., 2022). Among 124 patients with diagnostic age (also excluding 10 fetuses), their age ranged from 1.2 to 44 years old with a mean of 16.70 ± 9.90 years old. Only 10 (7.93%) were younger than 6 years old; and 69 (54.76%) were 7–18 years old, and 51 (40.48%) were older than 18 years. In our series, 58 patients (37.91%) had a family history of the BBS or similar disease, and the parents of 27 patients (17.65%) were consanguineous.
For the reporters, 80 patients (52.29%) were reported by ophthalmologists, following by pediatricians (24, 15.68%), endocrinologists (20, 13.07%), nephrologists (12, 7.84%), and obstetricians (10, 6.54%). These patients were reported from 23 provinces or regions and mostly from the Eastern region of China, including 34 (22.22%) from Beijing city, 21 (13.73%) from Guangdong province, 12 (7.84%) from Zhejiang province, Sichuan province, and Chongqing city, respectively. This was followed by Fujian province (8, 5.23%), Yunnan province (7, 4.58%), Xinjiang autonomous region (7, 4.58%), and Shanghai city (6, 3.92%).
3.2 Clinical characteristics
3.2.1 Chief complaint
Among 91 patients with proven chief complaints, the most common were visual impairment (37, 38.14%) and obesity (20, 21.97%), followed by polydipsia and/or polyuria (12, 13.17%), abnormal renal image or function (18, 19.78%), polydactyly (12, 13.17%), intellectual disability (9, 9.89%), growth retardation (6, 6.59%), and abnormalities of the reproductive system (4, 4.40%). Malnutrition, anemia, convulsion, dyskinesia, fever and cough, weakness, hypertension, and hyperglycemia were also reported. Among 11 fetuses (Hou, 2004; Li et al., 2019; Qiang et al., 2019; Li et al., 2020; Jing et al., 2021; Cai et al., 2022; Dong et al., 2022; Yan et al., 2022), polydactyly and/or abnormal renal images were found by pregnancy examination, and hydrometrocolpos and vaginal atresia were reported in one fetus (Hou, 2004).
3.2.2 Polydactyly
Among 153 patients, polydactyly was reported in 131 (85.62%), which was similar with Gnanasekaran et al. report (87/108, 80.55%; χ2 = 1.180, p = 0.277), but significantly higher than those in Beales et al. (1999) (75/109, 68.81%; χ2 = 10.707, p = 0.001) or Moore et al. (2005) (29/46, 63.04%; χ2 = 11.441, p = 0.001) reports. Of these, only 2 (1.31%) had postaxial 7-finger deformity, and the other 128 had postaxial 6-finger deformity. At least 97 patients (62.75%) involved fingers and 103 (67.32%) toes, and 85 (55.56%) involved both fingers and toes. Among 110 with detail about polydactyly, bimanual bipedal deformity was reported in 55 (50.00%) patients, with bimanual deformity only in 5 (4.54%), bipedal deformity only in 12 (10.91%), unimanual deformity only in 5 (4.54%), unipedal deformity in 4 (3.64%); unimanual bipedal deformity in 13 (11.82%), bimanual unipedal deformity in 6 (5.45%), and unimanual unipedal deformity 10 (9.09%), as shown in Table 1. Moreover, 25 patients (16.34%) had varying degrees of brachydactyly, 6 (5.45%) had syndactyly, and one had symptoms of thumb contracture. Most patients undergo surgery to remove an extra finger or toe for cosmetic reasons during childhood.

TABLE 1. Clinical features in the current series and previous reports (number, %).
3.2.3 Ocular characteristics
Among 137 patients with recorded ophthalmic examinations, 132 (96.35%) had visual problems (Table 1). The age of onset ranged from 1 to 17 years old with a median age of 5.0 (5.23 ± 3.65) years old in 53 patients with detail. Among these patients, 61 (44.53%) complained of nyctalopia/night blindness which is the initial manifestation of the disease in most people, and 25 (18.25%) blindness or almost blindness (only sense light, hand movement or count fingers) in their age of 20 s. The incidence of blindness was significantly lower than that of Moore et al. (2005) (42/46, 91.30%; χ2 = 97.823, p < 0.001) report. Retinitis pigmentosa was reported in 120 patients (87.59%). Moreover, restricted visual fields (15, 10.95%), nystagmus (22, 16.069%), strabismus (20, 14.59%), optic nerve atrophy (12, 8.76%), cataracts (12, 8.76%), and astigmatism (6, 4.38%) were also reported.
3.2.4 Obesity
Among 132 patients with body mass index (BMI) or body shape, 11 (8.33%) were overweight and 113 (85.61%) were obese. Their BMI ranged from 11.94 to 48.89 kg/m2 with a mean BMI of 29.08 ± 6.40 kg/m2 among 102 patients. The incidence of overweight and obesity in our series (123/132, 93.18%) was higher than those in Gnanasekaran et al. (2023) (77/108, 71.30%; χ2 = 7.375, p = 0.007), Beales et al. (1999) (78/102, 76.47%; χ2 = 14.874, p < 0.001), and Mujahid et al. (2018) (101/131, 77.10%; χ2 = 15.084, p < 0.001) reports, but had no significant difference form and Moore et al. (2005) (45/45, 100%; p = 0.205) report. It was notable that hypertension was reported in 32 patients (20.91%). Moreover, hypertriglyceridemia was reported in 21 (13.73%) patients, with hyperglycemia in 21 (including 9 patients with diabetes mellitus), raised alanine transaminase in 7 (4.57%), fatty liver in 19 (12.42%, ultrasound report), and acanthosis nigricans in 6 (3.92%). Most incidences of these abnormalities were lower than those preorts by Mujahid et al. (2018) and Moore et al. (2005) reports, as shown in Table 1.
3.2.5 Genitourinary system abnormalities
Among 114 patients with urinary data, 63 (55.26%) had varying degrees of renal abnormalities, including cysts in 25 (21.93%, including polycystic kidney in 4), small kidneys (renal atrophy or dysplasia) in 13 (11.40%), large kidneys in 9 (7.89%), and bladder diverticulum and renal calculus, in one patient, respectively. Recurrent urinary tract infections were reported in 2 patients. In this series, kidney dysfunction was found in 33 (21.57%) patients. The incidences of renal cysts was similar to that reported by Beales et al. (1999) (6/57, 10.63%; χ2 = 3.329, p = 0.068), but much lower than that in Moore et al. (2005) report (23/32, 71.88%; χ2 = 28.244, p < 0.001), and much higher than that in Gnanasekaran et al. (Farag and Teebi, 1989) report (5/108, 4.63%; χ2 = 66.916, p < 0.001), as shown in Table 1.
Of the 104 patients with reproductive records, 83 (79.81%) had varying degrees of developmental abnormalities, while 21 patients (20.19%) had no significant abnormalities. Most of the symptoms are hypogonadism and/or genital hypoplasia, such as micropenis, small testicles, cryptorchidism, hypospadias in males, or vulva and breast hypoplasia, uterine and ovary aplasia, and delayed and irregular menstruation in females. The abnormalities of the reproductive system were lower than those in Beales et al. (1999) report for males (60/62, 96.77%; χ2 = 9.368, p = 0.002), but significantly higher than those in Gnanasekaran et al. (Farag and Teebi, 1989) report (29/108, 26.85%; χ2 = 59.621, p < 0.001) and Mujahid et al. (2018) (10/68, 14.7%; χ2 = 70.169, p < 0.001) reports. Polycystic ovary syndrome, hydrometrocolpos and vaginal dysplasia, which were reported by Mujahid et al. (2018), were reported only in one patient, respectively (Hou, 2004; Meng et al., 2021). Notably, precocious puberty was reported in 2 patients (including central precocious puberty in one patient) (Li et al., 2022a; Li et al., 2022b).
3.2.6 Nervous system abnormalities
Mental retardation (including learning difficulties and speech delay) was reported in 111 (81.62%) of 136 patients, which was manifested by an inability to complete school or even to take care of themselves (Table 1). This value was significantly higher than that in reports from Beales et al. (1999) (68/109, 62.39%; χ2 = 11.371, p = 0.001) or Moore et al. (2005) (11/38, 28.94%; χ2 = 39.322, p < 0.001) reports. However, only 13 patients (9.55%) had detailed records of intelligence test results. Motor development delay was alos reported in 4 patients, ataxia/poor coordination in 3 (Huang et al., 2021), and epilepsy was reported in 3 (Wang et al., 2014; Meng et al., 2021).
3.2.7 Other anomalies
Among 91 patients with height and short stature data, 56 (61.54%) were short stature (9 patients ranged from −1 SD to −2 SD, 34 less than −2 SD, 12 without height detail) and 35 (38.46%) were at the normal height for their age. The height of 11 male adults (≥18 years) ranged from 1.45 to 1.75 m with a mean height of 160.91 ± 10.57 m while that of 16 female adults (≥17 years) ranged from 1.36 to 1.56 m with 149.34 ± 8.68 m. Dental dysplasia was noted in 21 patients (13.73%), with hearing impairment in 8 (5.23%), congenital heart disease in 8 (including 4 atrial defects, 2 ventricular defect), osteoarthritis-like changes in 11 (7.19%), hemangioma in 3 (1.96%), short neck in 8 (5.23%), hypothyroidism in 3, Hirschsprung disease, anal stenosis, and anal atresia in one patient, respectively (Table 1).
In this series, 11 fetuses were diagnosed prenatally from 2019 to 2022 years (Li et al., 2019; Qiang et al., 2019; Li et al., 2020; Jing et al., 2021; Cai et al., 2022; Dong et al., 2022; Yan et al., 2022) except for one patient reported in 2004 (Hou, 2004). All fetuses presented abnormal kidney images and/or bilateral enlarged hyperechogenic kidneys that implied renal cysts, and one presented hydrometrocolpos and postaxial polydactyly with vaginal atresia (Hou, 2004). Oligohydramnios was noted in one, and ventricular defect combined with a single atrium, persistent left superior vena cava and ascites were noted in one patient.
3.3 Genotypes and phenotypes
3.3.1 Genotypes
Among 153 patients, genetic analysis was performed in only 90 patients (58.82%), including one negative finding (Tao et al., 2020). Except for 3 patients who were analyzed using linkage analysis (including 2 BBS7 and one BBS5) in 2007 and 2008 (Chen et al., 2007; Liu et al., 2008), other 86 patients with genetic analysis were reported. Most (77, 89.53%) were reported in the last 6 years (from 2017 years). The most common genotypes were BBS7 (22 patients, 24.71%), BBS2 (20, 22.73%), and BBS10 (10, 11.24%). These were followed by BBS12 (8, 8.99%), BBS1 (6, 6.74%), BBS5 (5, 5.62%), BBS6/MKKS (5, 5.62%), BBS9/PTHB1 (5, 5.62%), and BBS4 (4, 4.49%), as shown in Table 2 and Figure 1. The proportions of BBS2, BBS4, BBS7, and BBS9/PTHB1 were higher while the proportion of BBS1 was lower in our Chinese series than those in most reports outside China (Moore et al., 2005; Forsythe and Beales, 2013; Mujahid et al., 2018; Guardiola et al., 2021). Moreover, the affected proteins of 62 patients (69.66%) were involved in the assembly of the BBSome complex while 23 (25.84%) were involved in the chaperone-like protein complexes. Among 81 patients with detailed variants, 42 (51.85%) were compound heterozygous variants, 38 (46.91%) were homozygous (including one uniparental disomy, and one perhaps uniparental disomy), and one was a heterozygous variant with a topical typical phenotype. Among 11 fetuses, 10 patients were reported in the last 4 years except for one reported in 2004 years (Hou, 2004). There were 6 patients (54.55%) with BBS7 (Li et al., 2019; Li et al., 2020; Jing et al., 2021), 3 (27.27%) with BBS1 (Qiang et al., 2019; Cai et al., 2022; Yan et al., 2022), one with BBS6/MKKS (Hou, 2004), and one with BBS10 (Dong et al., 2022).

TABLE 2. The genotypes in the current series and several previous reports (number, %).
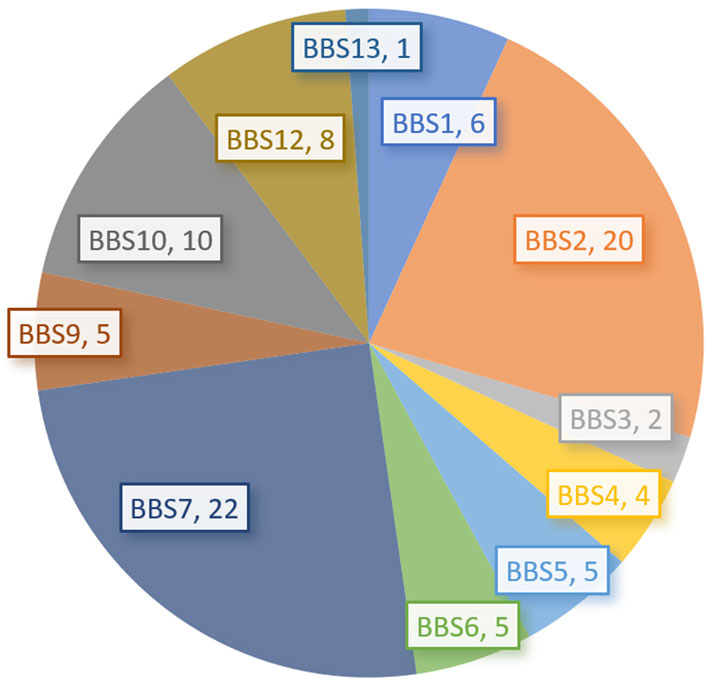
FIGURE 1. The genotypes of BBS in the current series (number).
Except for 2 cases analyzed by linkage analysis without detailed data, 19 BBS7 patients from 13 families were reported (Chen et al., 2007; Yang et al., 2008; Li et al., 2017; Xiu, 2018; Li et al., 2019; Shen et al., 2019; Tao et al., 2019; Li et al., 2020; Tao et al., 2020; Jing et al., 2021; Meng et al., 2021). Homozygous variants were noted in 10 patients (50.00%) and 10 (50.00%) compound heterozygous variants were noted in our series. Unlike a report from the United States (Guardiola et al., 2021), which reported hot spot variants in BBS1 and BBS7, at least 11 variants of BBS7 were reported in this series. The most common variant was the c.1002delT (p. Asn335Ilefs*47) variant, which was found in 7 patients (36.84%) from 5 families, including 2 homozygotes. This was followed by homozygous variant c.389_390delAC (p. Asn130Thrfs*3), which was reported in 5 patients from one family (Shen et al., 2019). The heterozygous variants c.288_289delAG (p. Gly97Lysfs*7) and p.Gly97Lysfs*7 were reported in 4 patients (15.79%) from 4 families, and c.728C>A (p. Cys243Tyr) and p. Cys243Tyr were reported in 4 patients (10.51%) from 4 families. The homozygous variant c.1666A>G (p. Ser556Arg) was reported in 2 patients from one family (Yang et al., 2008). Other variants included c.338C>A (p. Ala113Asp), c.497C>A (p. Ala166Asp), c.718G>A (p. Gly240Ser), c.849+1G>C, c.1395T>A (p. Tyr465*), and p. Ser117Pro. Moreover, the 4q26q27 microdeletion including BBS7 was reported in 2 fetuses from one family.
Except for one case without detailed data of variant, 19 BBS2 patients from 12 families were reported (Xing et al., 2014; Chen et al., 2017; Li et al., 2017; Ding et al., 2018; Dan et al., 2020; Huang et al., 2021; Meng et al., 2021; Tang et al., 2022; Tao et al., 2022; Wei et al., 2022), including 12 (63.16%) compound heterozygous variants and 7 (36.84%) homozygous variants. Unlike report from the United States (Guardiola et al., 2021), which reported hot spot variants in BBS1 and BBS7, a total of 15 variants were reported in our series. The most common was the c.534+1G>T splicing variant in 9 patients (47.37%) from 5 families, including 2 homozygotes. It was notable that one patient with a homozygous c.534+1G>T splicing variant was associated with the paternal uniparental disomy (Wei et al., 2022). The heterozygous variant c.235T>G (p. Thr79Pro) were found in 4 patients (21.05%) from the same family. A c.563delT (p. Ile188Thrfs*13) variant was found in 3 patients (15.79%) from 3 families, including one homozygous. The homozygous variant c.79A>C (p. Thr27Pro) was found in 2 patients (10.53%) from one family. The heterozygous variants c.1278A>G (p. Glu426Glu) and c.2059C+1G>C splicing variant were reported in 2 patients from one and 2 families, respectively. Other variants included c.646C>T (p. Arg216*), c.944G>A (p. Arg315Gln), c.1015C>T (p. Arg339*), c.1148_1149dupTC (p. His384Serfs*34), c.1206dupA (p. Arg403fs*216), c.1237C>T (p. Arg413*), c.1438C>T (p.Arg480*), p. Tyr229*, and p. Arg703*.
Among 10 BBS10 patients (Lin et al., 2018; Wang et al., 2018; Tao et al., 2020; Liu et al., 2021; Li et al., 2022a; Li et al., 2022b; Dong et al., 2022; Lu et al., 2022; Tao et al., 2022; Yan et al., 2022), 7 had compound heterozygous variants and 3 had homozygous variants. A total of 11 variants were also reported. The most common variant was c.539G>A (p. Gly180Glu), which was found in 4 patients (40.0%) in 4 families, including one homozygous. The heterozygous variant c.1391C>G (p. Ser464*) was found in 3 patients (30.0%) in 3 families, and the heterozygous variant c.602G>A (p. Cys201Tyr) was found in 2 patients (20.0%) in 2 families. Other variants included c.184C>T (p. His62Tyr), c.378G>A (p. Trp126*), c.445_446insC (p. Leu149Pfs*3), c.784_785delGA (p. Glu262Asnfs*41), c.891_897delinsTTTGT (p. Met298Leufs*5), 1063C>T (p. Gln355*), c.1812dupT (p. Asn605*), and c.1949delA (p. His650Profs*12). It was notable that 5 (45.45%) of 11 variants were Indel variants.
3.3.2 Genotype-phenotype relationship
We compared the phenotypes among BBS2, BBS7, and BBS10 patients. It was noted that BBS7 had higher penetrance. BBS2 had higher hearing impairment and lower renal abnormality penetrance. Moreover, BBS2 had lower mental retardation and polydactyly penetrance with marginal differneces. BBS10 had higher penetrance except for the lower renal abnormality as well (Table 3).
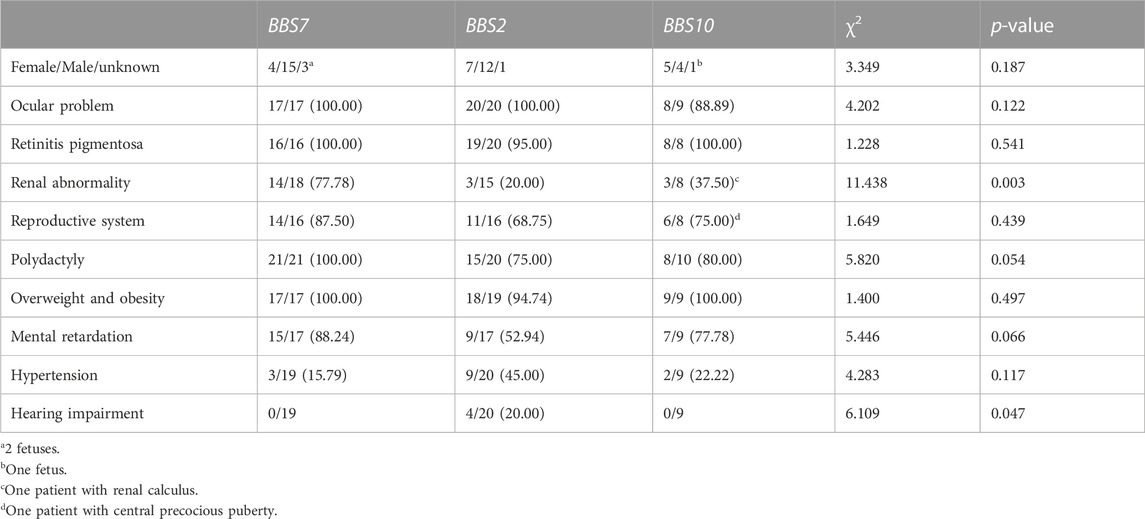
TABLE 3. Genotype and phenotype relationship (number, %).
4 Discussion
BBS is a main cause of syndromic forms of obesity although it is a rare disease. According to the prevalence (about 1/125,000∼1/160,000) reported (Moore et al., 2005; Tsang et al., 2018), these are currently about 10,000 patients in China (about 1.4 billion people) now. However, fewer than 150 patients have been reported to date. Moreover, most cases were reported from the Eastern region. This finding implied that misdiagnosis or miss diagnosis of BBS was common in China, especially in the Midwest region, although it may also be because of the non-register system of BBS in China.
Although it was an autosomal recessive disease, we noted that BBS was predominantly male with the male to female ratio of 1.64:1 (87/53). This result was similar to that in most other studies by Beales et al. (1999) (1.3:1) and Klein and Ammann (1969) (1.4:1), but the reason is still unclear. Whether this is associated with the fact that dysplasia of the vagina, uterus, and ovaries may be more apt to be ignored than cryptorchidism and micropenis needs further investigation. Although the age at diagnosis may be younger than those in other studies from Canada in 2005 (Moore et al., 2005) and the United Kingdom in 2018 (Mujahid et al., 2018), the mean age of these Chinese patients was 16.7 years old. This implies that the delayed diagnosis is still common. It was also notable that while only 15.68% of patients were reported by pediatricians, most (52.29%) were reported by ophthalmologists. In fact, polydactyly, a main feature and early clue of BBS, was noted in 85.62% of patients in this series. Hence, in addition to visual impairment, obesity, and genitourinary system abnormalities, BBS should be considered in infants (even fetuses) with polydactyly by pediatricians and pediatric surgeons.
The phenotype of BBS is heterogeneous. In our Chinese series, the incidences of visual impairment, polydactyly, obesity, genital anomalies, renal anomalies, and mental retardation were higher than those in most previous reports, although the age ass not older than those in previous reports (Beales et al., 1999; Moore et al., 2005; Mujahid et al., 2018). This implies that some atypical patients with few main clinical features may be misdiagnosis in China. It was notable that the incidence of some non-main clinical features (e.g., hearing loss, dental anomalies, anosmia/hyposmia, hydrometrocolpos, vaginal dysplasia, polycystic ovary syndrome in females) and complications (e.g., hypertriglyceridemia, hyperglycemia, diabetes millitus, and hypertension) were significantly lower than those in previous reports (Beales et al., 1999; Moore et al., 2005; Mujahid et al., 2018). These results suggest that careful evaluate of various deformities and congratulations on defects is needed for Chinese patients.
To date, 28 genes have been reported to be associated with BBS phenotypes (Niederlova et al., 2019; Gnanasekaran et al., 2023; Khan et al., 2023). Unlike Caucasian patients with higher proportions of BBS1 and BBS10 (Niederlova et al., 2019; Melluso et al., 2023), we noted that BBS7 was the prominent genotype, followed by BBS2, BBS10, BBS12, and BBS1 in these Chinese seies. This difference may imply the different genotype prominence in different geographic areas. This may also be associated with the fact that poor clinical diagnosis as BBS1 patients tend to have a milder pattern of disease (Niederlova et al., 2019; Guardiola et al., 2021). The analysis for the relationship between genotype and phenotype is difficult, as this rare disease. The BBSome chaperone-like protein is involved in the early synthesis of BBSome, so patients with BBS6/MKKS, BBS10, and BBS12 have more severe symptoms, especially BBS10 (Forsythe et al., 2017; Niederlova et al., 2019). Moreover, patients with BBS2 were reported to have severe symptoms (Florea et al., 2021). The proportion of renal abnormalities in patients with BBS7 types is relatively high (>60%) and relatively lower in BBS2 and BBS10 types (Niederlova et al., 2019; Florea et al., 2021). Patients with BBS2 and BBS4 types have a higher proportion of polydactyly (Niederlova et al., 2019), and BBS10 and BBS12 types are apt to obese (Forsythe and Beales, 2013; Dai et al., 2022; Melluso et al., 2023). We noted that BBS7 had higher penetrance. BBS2 had higher hearing impairment and lower renal abnormality penetrance. BBS10 had higher penetrance except for the lower enal abnormality. It was notable that hearing impairment was reported in BBS2, but not in BBS7 and BBS10. The relationship between genotype and phenotype must be observed with a larger sample size.
The diagnosis of BBS is some difficult due to the heterogeneity of phenotypes. Beales et al. (1999) summarized and amended that BBS clinical diagnosis should meet 4 major symptoms, or 3 major symptoms and 2 secondary symptoms. Fortunately, genetic diagnosis has been improved in recent years in China, and 89 patients had comfirmed diagnosis and genotyping by sequencing in recent years. Unlike reports from the United States (Guardiola et al., 2021), the variant sites are more dispersed in our Chinese series. As most patients have point variants, sequencing, but not karyotype and array comparative genomic hybridization (aCGH), is suggested as the first line genetic analysis. Moreover, we also used next-generation sequencing for newborn screening in China, which may improve the early diagnosis for some rare genetic diseases (Tong et al., 2022).
The diagnosis of BBS based on prenatal findings is still some difficult, as it cannot identify visual impairment, learning difficulties, or obesity in utero. Fortunately, prenatal diagnosis war comfirmed by sequencing for 10 fetuses after renal anomalies and/or polydactyly found by ultrasound in the past 4 years. Hence, in fetuses with genitourinary abnormalities, polydactyly, and/or hydrometrocolpos, BBS should be considered. Further careful evaluation for various deformities and genomic sequencing should be suggested.
Genetic counseling for the family of the proband is needed. As BBS is an autosomal recessive disease, consanguineous marriage should be avoided. The consanguinity rate in our series was 17.65%, which was lower than that in other studies by Beales et al. (1999) (39%) and Klein and Ammann (1969) (48%), and may be associated with more dispersed variant sites in our series. However, this reflected the lack of awareness of the dangers of consanguineous marriage in these Chinese families.
There is still no specific treatment for BBS. Congenital structural abnormalities (e.g., digestive tract abnormalities), obesity and metabolic syndrome, chronic kidney disease, and retinitis pigmentosa are the main influencing factors for the quality of life and longevity of BBS patients. Congenital structural abnormality correction, diet and lifestyle interventions to prevent obesity and metabolic syndrome, relieve chronic kidney disease, and slow down the retinitis pigmentosa progression are important for BBS patients. Drugs (e.g., setmelanotide) for the obesity (Tauber, 2022; Forsythe et al., 2023), and gene therapy for retinitis pigmentosa are in clinical trails (Xie et al., 2021b; Hsu et al., 2023).
There were several limitations. First, some patients were reported having “Laurence-Moon-Biedl syndrome” or “Laurence-Moon-Bardet-Biedl syndrome”. It may not be accurate to exclude some patients without genetic analysis data. Second, as a review analysis, some clinical data of some patients were not provided. Moreover, genetic analysis was not performed in all patients, and most patients reported before 2017 years. Hence, the genotype-phenotype relationship analysis may not be accurate.
In summary, misdiagnosis or miss diagnosis of BBS may be common in China. In patients with polydactyly, visual impairment, obesity, renal abnormalities, hypogonadism, and mental retardation, or in fetuses with polydactyly and/or renal abnormalities, BBS should be considered in the differential diagnosis. Other deformities should be evaluated carefully and genetic analysis should be performed as early as possible.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author contributions
ZL-H supervised the study. ZX-Y and DY-L contributed to the data collection. ZX-Y wrote the original draft. All authors contributed to the article and approved the submitted version.
Funding
The authors declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1247557/full#supplementary-material
References
Beales, P. L., Elcioglu, N., Woolf, A. S., Parker, D., and Flinter, F. A. (1999). New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J. Med. Genet. 36 (6), 437–446. doi:10.1136/jmg.36.6.437
Cai, M., Lin, M., Lin, N., Xu, L., and Huang, H. (2022). Novel homozygous nonsense mutation associated with Bardet-Biedl syndrome in fetuses with congenital renal malformation. Med. Baltim. 101 (32), e30003. doi:10.1097/MD.0000000000030003
Chen, K. C., Yang, Y., Liao, S. H., Hu, J. B., Chen, B., Lin, Y., et al. (2007). A case of non-classical Bardet-Biedl syndrome. Zhongguo Shi Yong Yan Ke Za Zhi 25 (10), 1153. doi:10.3760/cma.j.issn.1006-4443.2007.10.039
Chen, J. C., Li, C. W., Gao, J. S., and Liu, H. G. (2008). Three cases of Bardet-Biedl syndrome. Zhongguo Zhong Yi Yan Ke Za Zhi 18 (1), 38–40.
Chen, Y., Li, J., Wang, J., Li, N., Shen, Y., Huang, X., et al. (2017). Bardet-Biedl syndrome: a case report and literature review. Lin. Chuan Er Ke Za Zhi 35 (1), 28–32. doi:10.3969/j.issn.1000-3606.2017.01.008
Cheng, S. Q., Cao, Y. H., Zhang, J. S., and Qiang, H. (2013). A case report of rare bardet-biedl syndrome. Zhongguo Dang Dai Er Ke Za Zhi 15 (11), 975–976.
Dai, Y. L., Luo, X. P., Gong, C. X., Qiu, Z. Q., Xiong, H., Yang, Y. L., et al. (2022). Chinese expert consensus on the diagnosis and treatment of children with Bardet-Biedl syndrome. Zhongguo Shiyong Erke Zazhi 37 (4), 241–247. doi:10.3760/cma.j.cn112140-20221126-01005
Dan, H., Huang, X., Xing, Y., and Shen, Y. (2020). Application of targeted panel sequencing and whole exome sequencing for 76 Chinese families with retinitis pigmentosa. Mol. Genet. Genomic Med. 8 (3), e1131. doi:10.1002/mgg3.1131
Ding, J. C., Wang, C. L., Fang, Y. L., and Liang, L. (2018). A case report of Bardet Biedl syndrome in children. Zhongguo Xunzheng Yixue Zazhi 13 (2), 155–157. doi:10.3969/j.issn.1673-5501.2018.02.016
Dong, X., Li, Z., Wang, D., Xiong, Y., Li, H., Yang, P., et al. (2022). Prenatal diagnosis of Bardet-Biedl syndrome due to novel variants in the BBS10 gene in a fetus with multiple anomalies: a case report. Exp. Ther. Med. 24 (6), 721. doi:10.3892/etm.2022.11657
Fan, H., Yan, Y. K., and Mi, J. (2017). Updating blood pressure references for Chinese children aged 3-17 years. Zhonghua Gao Xue Ya Za Zhi 25 (5), 428–434.
Farag, T. I., and Teebi, A. S. (1988). Bardet-Biedl and Laurence-Moon syndromes in a mixed Arab population. Clin. Genet. 33 (2), 78–82. doi:10.1111/j.1399-0004.1988.tb03414.x
Farag, T. I., and Teebi, A. S. (1989). High incidence of bardet biedl syndrome among the bedouin. Clin. Genet. 36 (6), 463–464. doi:10.1111/j.1399-0004.1989.tb03378.x
Florea, L., Caba, L., and Gorduza, E. V. (2021). Bardet-Biedl syndrome-multiple kaleidoscope images: insight into mechanisms of genotype-phenotype correlations. Genes (Basel) 12 (9), 1353. doi:10.3390/genes12091353
Forsythe, E., and Beales, P. L. (2013). Bardet-Biedl syndrome. Eur. J. Hum. Genet. 21 (1), 8–13. doi:10.1038/ejhg.2012.115
Forsythe, E., Sparks, K., Best, S., Borrows, S., Hoskins, B., Sabir, A., et al. (2017). Risk factors for severe renal disease in bardet-biedl syndrome. J. Am. Soc. Nephrol. 28 (3), 963–970. doi:10.1681/ASN.2015091029
Forsythe, E., Kenny, J., Bacchelli, C., and Beales, P. L. (2018). Managing bardet-biedl syndrome-now and in the future. Front. Pediatr. 6, 23. doi:10.3389/fped.2018.00023
Forsythe, E., Haws, R. M., Argente, J., Beales, P., Martos-Moreno, G. A., Dollfus, H., et al. (2023). Quality of life improvements following one year of setmelanotide in children and adult patients with Bardet-Biedl syndrome: phase 3 trial results. Orphanet J. Rare Dis. 18 (1), 12. doi:10.1186/s13023-022-02602-4
Gnanasekaran, H., Chandrasekhar, S. P., Kandeeban, S., Periyasamy, P., Bhende, M., Khetan, V., et al. (2023). Mutation profile of Bardet-Biedl syndrome patients from India: implicative role of multiallelic rare variants and oligogenic inheritance pattern. Clin. Genet. 104 (4), 443–460. doi:10.1111/cge.14398
Gouronc, A., Zilliox, V., Jacquemont, M. L., Darcel, F., Leuvrey, A. S., Nourisson, E., et al. (2020). High prevalence of Bardet-Biedl syndrome in La Reunion Island is due to a founder variant in ARL6/BBS3. Clin. Genet. 98 (2), 166–171. doi:10.1111/cge.13768
Guardiola, G. A., Ramos, F., Izquierdo, N. J., and Oliver, A. L. (2021). A genotype-phenotype analysis of the Bardet-Biedl syndrome in Puerto Rico. Clin. Ophthalmol. 15, 3757–3764. doi:10.2147/OPTH.S328493
Han, R. A. (2011). Gene mutation screening of a family with Bardet-Biedl syndrome. Peking Union Medical College.
Hou, J. W. (2004). Bardet-Biedl syndrome initially presenting as McKusick-Kaufman syndrome. J. Formos. Med. Assoc. 103 (8), 629–632.
Hsu, Y., Bhattarai, S., Thompson, J. M., Mahoney, A., Thomas, J., Mayer, S. K., et al. (2023). Subretinal gene therapy delays vision loss in a Bardet-Biedl Syndrome type 10 mouse model. Mol. Ther. Nucleic Acids 31, 164–181. doi:10.1016/j.omtn.2022.12.007
Huang, Q. Y., Yang, C. X., Feng, Z. M., and Jia, W. J. (2015). A case of Bardet-Biedl syndrome with metabolic syndrome. Zhonghua Tang Niao Bing Za Zhi 7 (12), 770–771. doi:10.3760/cma.j.issn.1674-5809.2015.12.011
Huang, L., Sun, L., Wang, Z., Li, S., Chen, C., Luo, X., et al. (2021). Novel compound heterozygous BBS2 and homozygous MKKS variants detected in Chinese families with Bardet-Biedl syndrome. J. Ophthalmol. 2021, 6751857. doi:10.1155/2021/6751857
Jiang, Z. F. (2013). A case of non-classical Laurence-Moon-Bardet-Biedl syndorme. Shi Jie Zui Xin Yi Xue Xin Xi Wen Zhai 13, 349. doi:10.3969/j.issn.1671-3141.2013.19.276
Jing, X. Y., Jiang, F., and Li, D. Z. (2021). Unmasking a recessive allele by a deletion: early prenatal diagnosis of Bardet-Biedl syndrome in a Chinese family. Congenit. Anom. (Kyoto) 61 (4), 138–139. doi:10.1111/cga.12413
Ke, L., and An, Z. H. (2007). Two case of Laurence-Moon-Biedl syndrome. Huaxi Yi Yao 22 (4), 899. doi:10.3969/j.issn.1002-0179.2007.04.138
Khan, S., Focsa, I. O., Budisteanu, M., Stoica, C., Nedelea, F., Bohiltea, L., et al. (2023). Exome sequencing in a Romanian Bardet-Biedl syndrome cohort revealed an overabundance of causal BBS12 variants. Am. J. Med. Genet. A 191 (9), 2376–2391. doi:10.1002/ajmg.a.63322
Klein, D., and Ammann, F. (1969). The syndrome of Laurence-Moon-Bardet-Biedl and allied diseases in Switzerland. Clinical, genetic and epidemiological studies. J. neurological Sci. 9 (3), 479–513. doi:10.1016/0022-510x(69)90091-4
Lei, H., Shou, T., Gao, A. J., and Yan, X. M. (2006). Three cases of Bardet-Biedl syndrome. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 23 (3), 293. doi:10.3969/j.issn.1005-2194.2006.12.019
Li, H., and Hu, Z. (2022). Genetic analysis of novel MKKS variants in a Chinese patient with Bardet-Biedl syndrome. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 39 (7), 754–758. doi:10.3760/cma.j.cn511374-20220427-00285
Li, H. Y., and Pang, G. X. (2003). A case of Laurence-Moon-Bardet-Biedl syndrome. Yan Ke Xin Jin Zan 23 (1), 55. doi:10.3969/j.issn.1003-5141.2003.01.035
Li, S. T., Zhang, D., Liu, G. S., and Yang, F. (2008a). A case of Bardet-Biedl syndrome. Shi Yong Er Ke Lin. Chuan Za Zhi 23 (8), 570–615.
Li, X., Sun, S. J., Liu, Y., and Liu, G. X. (2008b). A case of Laurence-Moon-Biedl-Bardet syndrome presented multiple organ failure. Zhonghua Ji Zhen Yi Xue Za Zhi 18 (3), 231. doi:10.3760/cma.j.issn.1671-0282.2009.03.002
Li, H., Ji, C. Y., Zong, X. N., and Zheang, Y. Q. (2009a). Body mass index growth curves for Chinese children and adolescents aged 0 to 18 years. Clin. J. Pediatr. 47 (7), 493–498. doi:10.3760/cma.j.issn.0578-1310.2009.07.004
Li, H., Ji, C. Y., Zong, X. N., and Zhang, Y. Q. (2009b). Height and weight standardized growth charts for Chinese children and adolescents aged 0 to 18 years. Zhongguo Dang Dai Er Ke Za Zhi 47 (7), 487–492. doi:10.3760/cma.j.issn.0578-1310.2009.07.003
Li, M. D., Li, G. L., Ying, X. B., He, T., and Sun, X. W. (2013). A case of Bardet-Biedl syndrome. Zhonghua Yan Ke Za Zhi 49 (11), 1035–1037. doi:10.3760/cma.j.issn.0412-4081.2013.11.016
Li, Q., Zhang, Y., Jia, L., and Peng, X. (2014). A novel nonsense mutation in BBS4 gene identified in a Chinese family with Bardet-Biedl syndrome. Chin. Med. J. 127 (24), 4190–4196. doi:10.3760/cma.j.issn.0366-6999.20141359
Li, Q., Peng, X., Zhang, Y., Zhu, X., and Zhou, H. (2017). Refinal imaging characteristics of patients with Bardet-Biedl syndrome. Yan ke 26 (3), 179–184. doi:10.13281/j.cnki.issn.1004-4469.2017.03.008
Li, Q. Y., Huang, L. Y., and Li, D. Z. (2019). Early prenatal detection of Bardet-Biedl syndrome in a case with postaxial polydactyly and hyperechoic kidneys confirmed by next generation sequencing. Congenit. Anom. (Kyoto) 59 (4), 142–144. doi:10.1111/cga.12306
Li, B., Xie, J., Geng, Q., Liu, Y., Xu, Z., and Li, S. (2020). Genetic diagnosis of fetal Bardet - biedl syndrome by BBS7 gene mutation: report of two cases. Zhonghua Weichan YIxue Zazhi 23 (6), 380–386. doi:10.3760/cma.j.cn113903-20190605-00364
Li, Y., Luo, Y. F., Sun, G. H., Dilihuma, J., Shen, Y. P., and Mireguli, M. (2022a). Three families with Bardet-Biedl syndrome. Chin. J. Child. Health Care 30 (5), 575–580. doi:10.11852/zgetbjzz2021-0364
Li, H., He, J., Leong, I., Huang, R., and Shi, X. (2022b). Central precocious puberty occurring in Bardet-Biedl syndrome-10 as a method for self-protection of human reproductive function: a case report. Exp. Ther. Med. 24 (3), 574. doi:10.3892/etm.2022.11511
Li, B. H. (2021). Prenatal henotypes and gene diagnosis of Bardet-Biedl syndrome fetuses. Nanfang Medical University.
Liang, J., and Zhou, J. L. (2008). A case of Laurence-Moon-Biedl syndrome. Shi Yong Yi Xue Za Zhi 24 (23), 4048. doi:10.3969/j.issn.1006-5725.2008.23.086
Lin, Q., and Zhang, Q. (2014). A case of Bardet-Biedl syndrome and literature review. Guiyang Yi Xue Yuan Xue Bao 39 (3), 446–447. 449.
Lin, Y. F., Liu, M. Y., and Luo, T. C. (2000). Both siblings suffer from Laurence-Moon-Bardet-Biedl syndrome. Yan Ke Xin Jin Zan 20 (2), 137. doi:10.3969/j.issn.1003-5141.2000.02.044
Lin, X. D., Chen, G. F., and Qu, J. (2007). A case of Bardet-Biedl syndrome. Zhonghua Yan Ke Za Zhi 43 (2), 174–175. doi:10.3760/j:issn:0412-4081.2007.02.019
Lin, B. H., Yang, X. Q., and Shen, T. (2018). A case report of Bardet-Biedl syndrome with recurrent urinary tract infection and literature review. Zhongguo Quan Ke Yi Xue 21 (21), 2643–2646. doi:10.3969/j.issn.1007-9572.2018.00.108
Liu, B., Yang, Y., Lin, Y., Zhang, B., Lu, F., Du, Q., et al. (2008). A Chinese Bardet-Biedl syndrome family and the linkage of BBS5. Xian Dai Yu Fang. Yi Xue 35 (9), 1738–1740. doi:10.3969/j.issn.1003-8507.2008.09.061
Liu, W., Yuan, Z., You, Y., and Han, Q. (2021). A case of infant Bardet-Biedl syndrome and literature review. Jining Yixueyuan Xuebo 44 (5), 378–380. doi:10.3969/j.issn.1000-9760.2021.05.017
Lou, M. J., Hu, Y. Y., and Huang, X. Q. (2015). A case of Bardet-Biedl syndrome and literature review. Shanghai Yi Xue 38 (11), 852–854.
Lu, B. C., and Wang, Y. Y. (2007). A case of Laurence-Moon-Biedl syndrome. Jiangsu Yi Yao 33 (3), 322–323. doi:10.3969/j.issn.0253-3685.2007.03.05
Lu, X., Yuan, L., and Chen, C. (2022). Bardet-Biedl syndrome in a female due to a novel compound herterozygous mutations in BBS10 gene: one case report and literature. Chin. J. Endcrionl Metab. 38 (6), 522–525. doi:10.3760/cma.j.cn311282-20201222-00845
Melluso, A., Secondulfo, F., Capolongo, G., Capasso, G., and Zacchia, M. (2023). Bardet-Biedl syndrome: current perspectives and clinical outlook. Ther. Clin. Risk Manag. 19, 115–132. doi:10.2147/TCRM.S338653
Meng, X., Long, Y., Ren, J., Wang, G., Yin, X., and Li, S. (2021). Ocular characteristics of patients with Bardet-Biedl syndrome caused by pathogenic BBS gene variation in a Chinese cohort. Front. Cell Dev. Biol. 9, 635216. doi:10.3389/fcell.2021.635216
Moore, S. J., Green, J. S., Fan, Y., Bhogal, A. K., Dicks, E., Fernandez, B. A., et al. (2005). Clinical and genetic epidemiology of Bardet-Biedl syndrome in Newfoundland: a 22-year prospective, population-based, cohort study. Am. J. Med. Genet. A 132A (4), 352–360. doi:10.1002/ajmg.a.30406
Mujahid, S., Hunt, K. F., Cheah, Y. S., Forsythe, E., Hazlehurst, J. M., Sparks, K., et al. (2018). The endocrine and metabolic characteristics of a large Bardet-Biedl syndrome clinic population. J. Clin. Endocrinol. Metab. 103 (5), 1834–1841. doi:10.1210/jc.2017-01459
Niederlova, V., Modrak, M., Tsyklauri, O., Huranova, M., and Stepanek, O. (2019). Meta-analysis of genotype-phenotype associations in Bardet-Biedl syndrome uncovers differences among causative genes. Hum. Mutat. 40 (11), 2068–2087. doi:10.1002/humu.23862
Qi, H. F., Tang, X. Y., Li, F., and Fu, R. (2015). A case of Bardet-Biedl syndrome. Yi Nan Bing Za Zhi 14 (11), 1132. doi:10.3969/j.issn.1671-6450.2015.11.01
Qi, Z., Shen, Y., Fu, Q., Li, W., Yang, W., Xu, W., et al. (2017). Whole-exome sequencing identified compound heterozygous variants in MMKS in a Chinese pedigree with Bardet-Biedl syndrome. Sci. China Life Sci. 60 (7), 739–745. doi:10.1007/s11427-017-9085-7
Qiang, W., Wang, Z. R., Zhang, J. Z., Zhu, J. F., Dai, L. G., Liu, X. L., et al. (2019). Genetic analysis and prenatal diagnosis of Bardet Biedl syndrome caused by de novol mutation of BBS1 gene. Zhongguo Xunzheng Yixue Zazhi 14 (6), 467–469. doi:10.3969/j.issn.1673-5501.2019.06.015
Qu, J., Xu, H., Shen, Q., and Wu, B. B. (2018). Novel mutation in the BBS12 gene of Chinese Bardet-Biedl syndrome pedigrees. Zhonghua Shen Zang Bing Za Zhi 34 (8), 592–600. doi:10.3760/cma.j.issn.1001-7097.2018.08.006
Seongjin, S., Lisa, M. B., Nathan, P. S., John, S. B., Qihong, Z., Diane, C., et al. (2009). BBS6, BBS10, and BBS12 form a complex with CCT/TRiC family chaperonins and mediate BBSome assembly. Proc. Natl. Acad. Sci. U. S. A. 107 (4), 1488–1498.
Shao, Y. F., and Dong, B. Z. (2022). Bardet-Biedlsyndrome type 5: a case report and literature review. J. Precis. Med. 37 (5), 430–432. doi:10.13362/j.jpmed.202205013
Shao, W., Fu, Y. F., and Wei, L. Z. (2000). A case of Laurence-Moon-Biedel syndrome. Yi Xue Yan Jiu Sheng Xue Bao 13 (Suppl. p), 68. doi:10.3969/j.issn.1008-8199.2000.z1.032
Shao, Y., An, M., Shi, X., and Shao, L. (2022). Two novel variants in a Bardet-Biedl syndrome type 5 patient with severe renal phenotype. Nephrol. Carlt. 27 (11), 897–900. doi:10.1111/nep.14087
Shen, M., Pang, N., Chen, S. Y., and Liu, P. C. (2001). A case of Bardet-Bied syndrome. Zhonghua Er Ke Za Zhi 39 (8), 1. doi:10.3760/j.issn:0578-1310.2001.08.021
Shen, J. F., Cao, Y., Shuai, L. J., and Wu, X. C. (2018). A novel pathogenic mutation of BBS5 in atypical Bardet-Biedl syndrome patient with prominent clinical manifestation of chronic renal failure: case report. Zhongguo Lin. Chuan Yi Xue 25 (4), 676–678. doi:10.12025/j.issn.1008-6358.2018.20170866
Shen, T., Gao, J. M., Shou, T., Li, L., Zhang, J. P., Zhao, Q., et al. (2019). Identification of a homozygous BBS7 frameshift mutation in two (related) Chinese Miao families with Bardet-Biedl Syndrome. J. Chin. Med. Assoc. 82 (2), 110–114. doi:10.1097/jcma.0000000000000011
Shen, Y. T., Ling, Y., Lu, Z. Q., Li, X. M., Bian, H., Yan, H. M., et al. (2022). Diagnosis and genetic analysis of a case with Bardet-Biedl syndrome caused by compound heterozygous mutations in the BBS12 gene. Yi Chuan 44 (10), 975–982. doi:10.16288/j.yczz.22-182
Shou, T., Lei, H., Gao, A. J., Gao, J. M., and Yan, X. M. (2006). A pedigree survey of 3 cases of Bardet-Biedl syndrome. Zhongguo Wu Zhen Xue Za Zhi 6 (6), 1201–1202. doi:10.3969/j.issn.1009-6647.2006.06.187
Shou, T., Shen, T., Lei, H., Tang, H., Gao, J. M., and Yan, X. M. (2011). A Miao pedigree of Bardet-Biedl syndrome in Yunnan province and their reservation. Zhonghua Nei Fen Mi Dai Xie Za Zhi 27 (2), 137–141. doi:10.3760/cma.j.issn.1000-6699.2011.02.011
Si, H. Y., Jiang, Y., and Jin, Y. L. (2008). One case Bardet-Bied syndrome report and literature review. Lin. Chuan Yi Xue 28 (2), 26–27. doi:10.3969/j.issn.1003-3548.2008.02.014
Tang, H. Y., Xie, F., Dai, R. C., and Shi, X. L. (2021). Novel homozygous protein-truncating mutation of BBS9 identified in a Chinese consanguineous family with Bardet-Biedl syndrome. Mol. Genet. Genomic Med. 9 (8), e1731. doi:10.1002/mgg3.1731
Tang, X., Liu, C., Liu, X., Chen, J., Fan, X., Liu, J., et al. (2022). Phenotype and genotype spectra of a Chinese cohort with nephronophthisis-related ciliopathy. J. Med. Genet. 59 (2), 147–154. doi:10.1136/jmedgenet-2020-107184
Tao, T. C., Wang, L., Cong, W. H., and Li, G. L. (2019). A case of Bardet Biedl syndrome. Zhonghua Shi Yan Yan Ke Za Zhi 37 (1), 28–29. doi:10.3760/cma.j.issn.2095.0160.2019.01.006
Tao, T., Wang, L., Chong, W., Yang, L., and Li, G. (2020). Characteristics of genotype and phenotype in Chinese patients with Bardet-Biedl syndrome. Int. Ophthalmol. 40 (9), 2325–2343. doi:10.1007/s10792-020-01415-3
Tao, T., Liu, J., Wang, B., Pang, J., Li, X., and Huang, L. (2022). Novel mutations in BBS genes and clinical characterization of Chinese families with Bardet-Biedl syndrome. Eur. J. Ophthalmol. 33, 714–722. doi:10.1177/11206721221136324
Tauber, M. (2022). Setmelanotide for controlling weight and hunger in Bardet-Biedl syndrome. Lancet Diabetes Endocrinol. 10 (12), 829–830. doi:10.1016/S2213-8587(22)00309-6
Tong, Y., and Yu, X. R. (2003). A case of Bardet-Biedl syndrome. Yan Ke Xin Jin Zan 23 (2), 110. doi:10.3969/j.issn.1003-5141.2003.02.033
Tong, F., Wang, J., Xiao, R., Wu, B. B., Zou, C. C., Wu, D. W., et al. (2022). Application of next generation sequencing in the screening of monogenic diseases in China, 2021: a consensus among Chinese newborn screening experts. World J. Pediatr. 18 (4), 235–242. doi:10.1007/s12519-022-00522-8
Tsang, S. H., Aycinena, A. R. P., and Sharma, T. (2018). Ciliopathy: bardet-biedl syndrome. Adv. Exp. Med. Biol. 1085, 171–174. doi:10.1007/978-3-319-95046-4_33
Wang, H., Fu, Q., Shen, Y., Liu, X., Zhou, N., Liang, Y., et al. (2014). Clinical features of Bardet-Biedl syndrome with renal abnormalities as initial manifestations. Zhonghua Er Ke Za Zhi 52 (8), 611–615. doi:10.3760/cma.j.issn.0578-1310.2014.08.013
Wang, L., Tao, T. C., Cong, W. H., and Li, G. L. (2018). A case of Bardet-Biedl syndrome with mutation of BBS and USH2A genes. Zhongguo Shi Yong Yan Ke Za Zhi 36 (5), 395–397. doi:10.3760/cma.j.issn.1006-4443.2018.05.014
Wang, S. Y. (2003). Three cases of Laurence-Moon-Biedl syndrome. Zhongguo You Sheng Yu Yi Chuan Za Zhi 11 (1), 126. doi:10.3969/j.issn.1006-9534.2003.01.071
Wei, L. J., Pang, X., Duan, C., and Pang, X. (1998). Bardet-Biedl syndrome: a review of Chinese literature and a report of two cases. Ophthalmic Genet. 19 (2), 107–109. doi:10.1076/opge.19.2.107.2315
Wei, H., Fang, Y., Sun, Y., Zhang, M., Chen, Q., Li, L., et al. (2022). A case of Bardet-Biedl syndrome caused by paternal uniparental disomy of chromosome 16. Chin. J. Ocul. Fundus Dis. 38 (5), 403–404. doi:10.3760/cma.j.cn511434-20210813-00437
Wu, Q. Y., and Zhang, M. X. (2015). A case of Laurence-Moon-Bardet-Biedl syndrome. Yuaxi Yi Xue 30 (6), 1111–1112. doi:10.7507/1002-0179.20150318
Wu, F. L., Wang, S. H., Huang, D. X., Yang, X. F., Li, Q., and Pang, Y. L. (2019). A case of Bardet - biedl syndrome. Anhui Yixue 40 (12), 1411–1422. doi:10.3969/j.issn.1000-0399.2019.12.032
Xia, J. M., Wang, Z. H., and Lin, X. F. (2014). A case of Laurence-Moon-Bardet syndrome combined with stillbirth. Shi Yong Fu Chan Ke Za Zhi 30 (8), 633–634.
Xiao, C. C., Ye, S. D., Ren, A., Hu, Y. Y., Fang, F., and Wang, W. (2011). A case of Bardet-Biedl syndrome. Zhonghua Tang Niao Bing Za Zhi 3 (6), 501–502. doi:10.3760/cma.j.issn.1674-5809.2011.06.013
Xie, H. B., Wang, Y., Zhao, Y. M., Zhang, H. W., Fu, J., Mao, Y. Y., et al. (2021a). Adult Bardet-Biedl syndrome: a case report and literature review. Hanshao Jibing Zazhi 28 (2), 4–5. doi:10.3969/j.issn.1009-3257.2021.02.003
Xie, C., Habif, J. C., Uytingco, C. R., Ukhanov, K., Zhang, L., de Celis, C., et al. (2021b). Gene therapy rescues olfactory perception in a clinically relevant ciliopathy model of Bardet-Biedl syndrome. FASEB J. 35 (9), e21766. doi:10.1096/fj.202100627R
Xin, D. J. (2014). Genetic diagnosis of Bardet-Biedl syndrome and Usher syndrome families. Wenzhou Medical Univeristy.
Xing, D. J., Zhang, H. X., Huang, N., Wu, K. C., Huang, X. F., Huang, F., et al. (2014). Comprehensive molecular diagnosis of Bardet-Biedl syndrome by high-throughput targeted exome sequencing. PloS one 9 (3), e90599. doi:10.1371/journal.pone.0090599
Xiu, Y. H. (2018). Identification of causative gene mutation and pathological mechanism in a Bardet-Biedl Syndrome family. Fujian Medical University.
Xu, S. Q. (1991). A case of generalized Laurence-Moon-Bardet-Biedl syndrome. Zhonghua Yan Ke Za Zhi 28 (2), 82.
Yan, X., and Shen, Y. (2022). Rab-like small GTPases in the regulation of ciliary Bardet-Biedl syndrome (BBS) complex transport. FEBS J. 289 (23), 7359–7367. doi:10.1111/febs.16232
Yan, K., Sun, Y., Yang, Y., Liu, B., and Dong, M. (2022). Case report: identification pathogenic abnormal splicing of BBS1 causing bardet-biedl syndrome type I (BBS1) due to missense mutation. Front. Genet. 13, 849562. doi:10.3389/fgene.2022.849562
Yang, Y. H., Ni, W. F., Chen, S. R., and Yang, S. M. (2006). Two cases of polydactyly-obesity-kidney-eye syndrome and literature review. Zhongguo Shi Yong Nei Ke Za Zhi 26 (12), 931–933. doi:10.3969/j.issn.1005-2194.2006.12.019
Yang, Z., Yang, Y., Zhao, P., Chen, K., Chen, B., Lin, Y., et al. (2008). A novel mutation in BBS7 gene causes Bardet-Biedl syndrome in a Chinese family. Mol. Vis. 14, 2304–2308.
Ye, Z., Li, C. J., Xia, X. Q., and Shen, J. (2008). A case of Bardet-Biedl with type 2 diabete. Zhonghua Nei Fen Mi Dai Xie Za Zhi 24 (3), 340. doi:10.3321/j.issn:1000-6699.2008.03.036
Yi, P., Liu, H., Saipairejing, A., and Niu, H. (2014). Clinical analysis the frisk Uyghur case of Bardet-Biedl syndrome in southern Xinjiang. Guangzhou Yi Yao 45 (5), 44–46.
Zhang, J., and Hu, S. H. (2004). A case of Bardet-Biedl syndrome with hypothyroidism. Lin. Chuan Nei Ke Za Zhi 21 (3), 151. doi:10.3969/j.issn.1001-9057.2004.03.036
Zhang, Y. H., Yuan, X. D., and Wu, X. R. (1999). A case of Laurence-Moon-Biedel syndrome. Shi Yong Er Ke Lin. Chuan Za Zhi 14 (3), 185. doi:10.3969/j.issn.1003-515X.1999.03.048
Zhang, Y. L., Li, S. L., Wang, J., and Shi, B. Y. (2007a). A case report of Bardet-Biedl syndrome. Zhongguo Yi Shi Jin Xiu Za Zhi 30 (7), 26. doi:10.3760/cma.j.issn.1673-4904.2007.19.037
Zhang, Y. L., Li, S. L., Wang, J., and Shi, B. Y. (2007b). A case report of Bardet-Biedl syndrome and literature review. Lin. Chuan Wu Zhen Wu Zhi 20 (10), 18. doi:10.3969/j.issn.1002-3429.2007.10.009
Zhang, Y. Q., Yan, J. H., and Chen, M. G. (2009). A case of Laurence-Moon-Biedl syndrome with chronic kidney disease. Zhongguo Xian Dai Yi Sheng 46 (27), 140. doi:10.3969/j.issn.1673-9701.2008.27.078
Zhang, Y., Xu, M., Zhang, M., Yang, G., and Li, X. (2021). A novel BBS9 mutation identified via whole-exome sequencing in a Chinese family with Bardet-Biedl syndrome. Biomed. Res. Int. 2021, 4514967. doi:10.1155/2021/4514967
Zheng, X. R., Yin, F., Huang, R., and Xiang, Q. L. (2011). A case report of Bardet-Biedl syndrome. Zhongguo Dang Dai Er Ke Za Zhi 13 (7), 602–603.
Zheng, R. D., Zhang, H. Y., Zhuang, Q. Y., Chen, Z. R., and Chen, J. N. (2012). A case of chronic hepatitis B accompanied by Laurence-Moon-Biedel syndrome. Gan Zang 17 (5), 371–372. doi:10.3969/j.issn.1008-1704.2012.05.027
Zhong, J., Xie, Y., Ye, H., Chen, C., Sun, T., Xu, K., et al. (2023). Phenotypic diversity observed in a Chinese patient cohort with biallelic variants in Bardet-Biedl syndrome genes. Eye (Lond). doi:10.1038/s41433-023-02516-w
Keywords: Bardet-Biedl syndrome, cilia dysfunction, genotype, phenotype, diagnosis, China
Citation: Xin-Yi Z, Yang-Li D and Ling-Hui Z (2023) Review of the phenotypes and genotypes of Bardet-Biedl syndrome from China. Front. Genet. 14:1247557. doi: 10.3389/fgene.2023.1247557
Received: 15 July 2023; Accepted: 27 October 2023;
Published: 15 November 2023.
Edited by:
Nagwa Elsayed Afify Gaboon, Ain Shams University, EgyptReviewed by:
Philip L. Beales, University College London, United KingdomJean Muller, INSERM U1112 Laboratoire de Génétique Médicale, France
Copyright © 2023 Xin-Yi, Yang-Li and Ling-Hui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zeng Ling-Hui, zouzouxinyi@163.com