 Jichao Zhou
Jichao Zhou Yuchen Wang
Yuchen Wang Yinghong Zhang2
Yinghong Zhang2 Yi Wang
Yi Wang- 1Department of Ophthalmology, Peking University Third Hospital, Beijing Key Laboratory of Restoration of Damaged Ocular Nerve, Peking University Third Hospital, Beijing, China
- 2Department of Otolaryngology, Peking University Third Hospital, Beijing Key Laboratory of Restoration of Damaged Ocular Nerve, Peking University Third Hospital, Beijing, China
Acro-dermato-ungual-lacrimal-tooth (ADULT) syndrome is a rare autosomal dominant inherited disease caused due to mutations in the TP63 gene. More commonly, mutations in the TP63 gene result in ectodermal dysplasia and/or orofacial cleft. ADULT syndrome is a type of ectoderm-related tissue dysplasia. This case report describes a patient with chronic tearing, congenital atresia, and obstruction of the lacrimal ducts, which are the main clinical manifestations of ADULT syndrome. This patient also presented with some clinical manifestations that were different from those of ADULT syndrome, namely, mild eyelid fusion and abnormal development of the fifth finger (a stiff fifth finger with camptodactyly that was shortened in length). The gene mutation in this patient was also at a site different from those usually reported in the literature. In this patient, c.518G > T resulted in p. G173V (accession number: NM_003722; exon4). We performed successful dacryocystorhinostomy and artificial lacrimal duct implantation. As shown above, we discussed the clinical characteristics and genetics of the disease in detail. In sharing this case, we aim to contribute to the current understanding of the genes and clinical manifestations of ADULT syndrome and to assist clinicians in the clinical diagnosis of TP63 mutation-related diseases.
Introduction
The presence of epiphora early in life is recognized as congenital nasolacrimal duct obstruction, with an incidence ranging from 5% to 20% (Petris and Liu, 2017). When considered as a single disease, obstruction is most often caused by a membrane at the end of the nasolacrimal duct called the valve of Hasner; this manifestation accounts for 73% of this disease, and 96% of the obstructions caused by the valve of Hasner resolve spontaneously (CASSADY, 1952). However, when congenital nasolacrimal duct obstruction or atresia is associated with dysplasia of other systemic organs, ectodermal dysplasia is often suspected. Acro-dermato-ungual-lacrimal-tooth (ADULT) syndrome is a common congenital disease associated with dysplasia of the lacrimal duct. It is a rare autosomal dominant genetic disease, first described in 1993, and is a type of ectodermal dysplasia. Other forms of ectodermal dysplasia include (Rinne et al., 2007) ankyloblepharon-ectodermal dysplasia-clefting syndrome (AEC), limb mammary syndrome (LMS), Rapp–Hodgkin syndrome (RHS), split-hand/split-foot malformation (SHFM), and ectrodactyly ectodermal dysplasia-cleft lip/palate syndrome (EEC). These ectodermal dysplasia types, including ADULT syndrome, are associated with mutations in the TP63 gene (Avitan-Hersh et al., 2010; Prontera et al., 2011), which has a critical role in embryonic development, especially in the development of the limbs, ectodermal tissues, such as hair, skin, teeth, nails, and mammary glands. ADULT syndrome is characterized by sparse hair on the scalp and the axilla, lacrimal duct stenosis or atresia, onychodysplasia, hypodontia or the early loss of permanent teeth, athelia or hypoplastic nipples, and breast hypoplasia (Chan et al., 2004; Slavotinek et al., 2005). Some of the features of ADULT syndrome overlap with those of the other five types (mentioned above) of ectodermal dysplasia. The literature suggests that frequently mutated amino acids including R298Q, R298G, R243W, R227Q, P127L, R337Q, V114M and N6H, may be involved in ADULT syndrome (Slavotinek et al., 2005; Berk et al., 2012). In this report, we describe a patient with ADULT syndrome associated with a rare mutation of the TP63 gene and atypical clinical features including mild symblepharon and a shortened, stiff fifth finger with camptodactyly.
Case report
A 16-year-old Chinese female was referred to our hospital because of epiphora. The patient had experienced continuous and excessive production of tears without any stimuli since childhood, and there has been no significant change over the past 10 years. Over the last 4 years, she experienced sustained swelling, mild tenderness, and a detectable local mass on the right inner canthus that progressively enlarged. The ocular skin became dark and dull, and excessive tearing persisted. The patient’s personal and menstrual history were normal. Both the patient and her parents did not have any significant medical history, including history of carcinomas. Moreover, the patient’s mother and brother also presented with similar features including abnormal hair, nails, teeth, skin, and lacrimal ducts (Table 1). On clinical examination, the patient demonstrated the following features. The puncta were stenotic and bilaterally covered with a membrane. As a result, probing of the nasolacrimal duct was not possible on either side. Thus, aplasia of both lacrimal ducts with chronic tear production and the expansion of the obstructed lacrimal ducts leading to local enlargement around the lower lacrimal tubules were assumed. Furthermore, mild fused lower eyelids were evident.
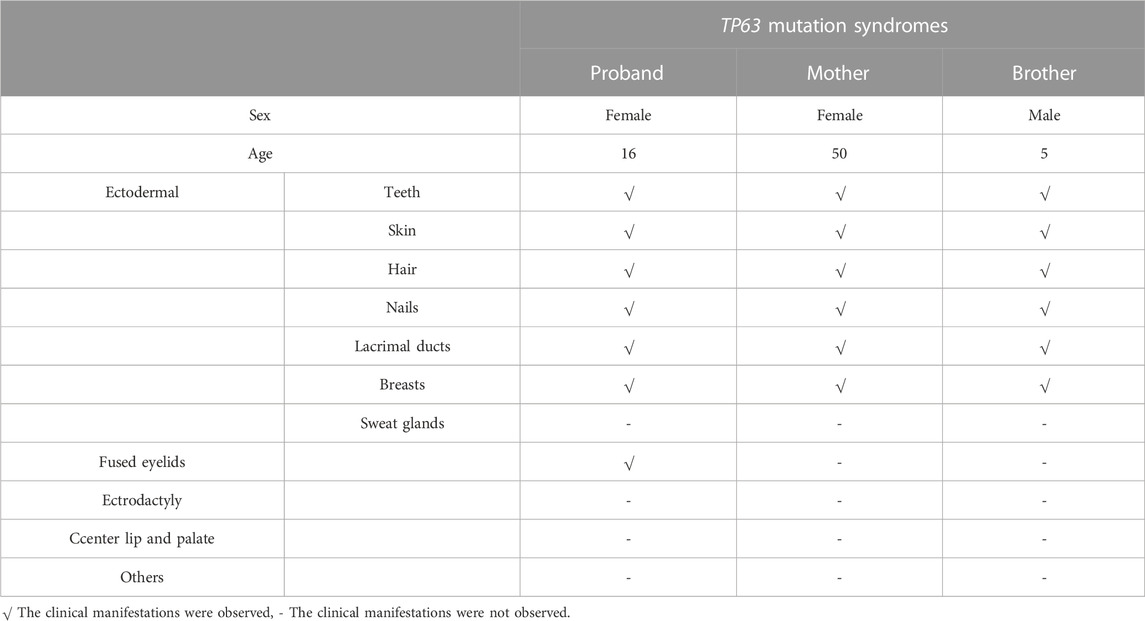
TABLE 1. Clinical features noted in the affected family members.
On physical examination, the following features were observed. (1) Skin: sweaty, pale, and without freckles; (2) Hair: brown and sparse, especially in the front of her scalp; (3) Oral cavity: conical teeth and hypodontia or oligodontia; (4) Nose and ears: small ears and a hooked nose; (5) Mammary glands: absent and bilateral hypoplastic nipples; (6) Hands: brachydactyly, which was most prominent in her fifth fingers, and bilateral fifth finger clinodactyly and camptodactyly; (7) Nails: discolored and irregularly shaped, with short and dystrophic nail plates and horizontal grooves along the length of the nails (Figures 1–3).
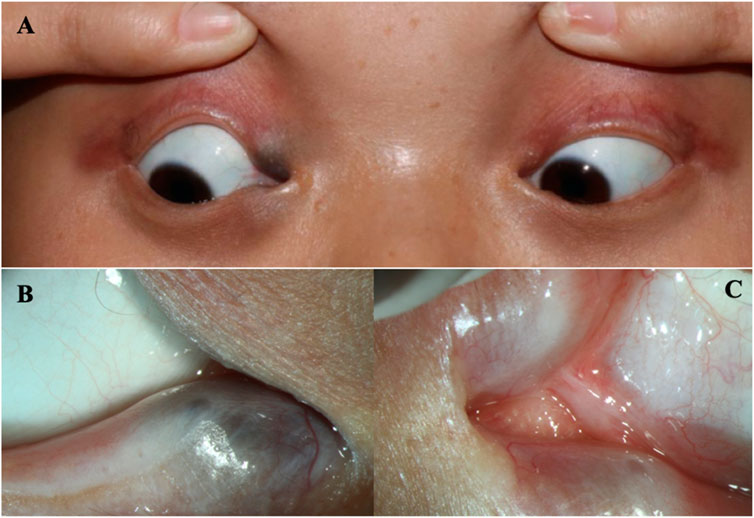
FIGURE 1. The eyes of the patient (A) The clinical appearance of the eyes. (B) The enlargement around the lower lacrimal tubules. (C) Absence of the upper puncta and closure of lower puncta.
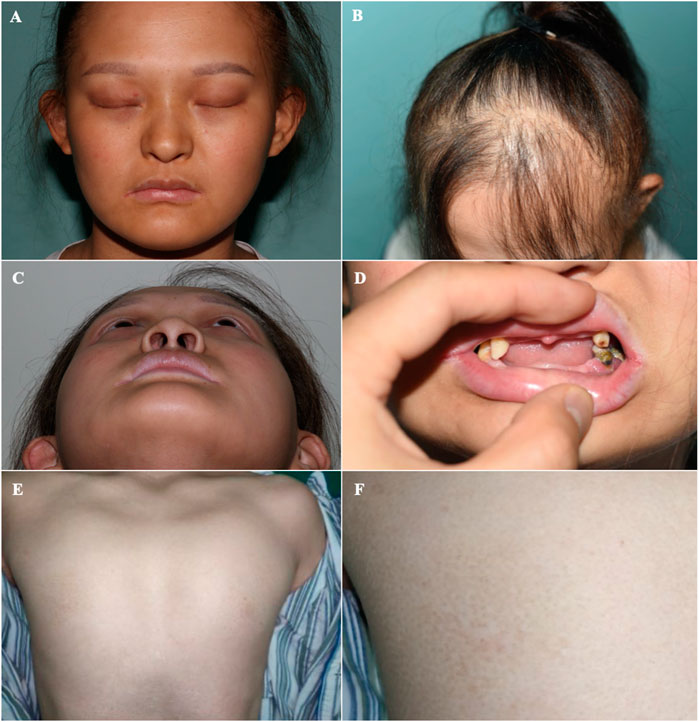
FIGURE 2. Orofacial and mammary glands’ features of the patient (A) Facial phenotype of the proband with sparse eyebrows with tattooing, absent eyelashes, small ears, and a hooked nose. (B) Sparse brown hair, especially in the front of her scalp. (C) A hollow facial appearance. (D) Dental abnormalities including hypodontia or oligodontia and conically shaped teeth. (E, F) Absent mammary glands with bilateral hypoplastic nipples.
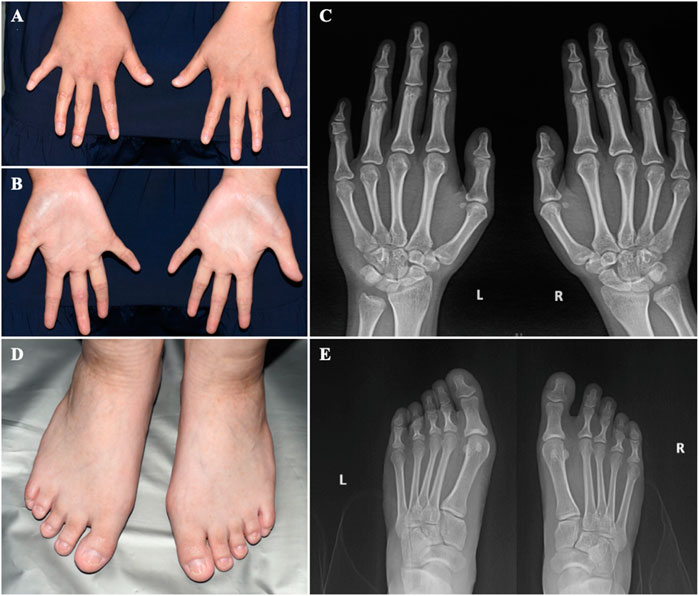
FIGURE 3. The hands and feet of the patient (A) Bilateral clinodactyly of the fifth finger. (B) Palmar hyperlinearity. (C) Radiograph of the hands showing clinodactyly of the fifth fingers. (D) Dystrophic nail plates and horizontal grooves along the length of the nails. (E) Radiograph of the feet.
After obtaining written informed consent from the patient and her parents, peripheral blood samples were collected. Whole-exome sequencing was performed to screen for candidate mutations. Called mutations were validated using Sanger sequencing. We identified a heterozygous G>T transition at cDNA position 518 of TP63 (accession number: NM_003722; exon4; OMIM number 103285) (Figure 4). This mutation is predicted to result in amino acid substitution p. G173V, and its functional effect, analyzed by two prediction tools (SIFT and PolyPhen), was predicted to be deleterious, thus supporting its pathogenicity. In the literature, mutation at cDNA position 518 has been previously reported (Chan et al., 2004) in a patient with ADULT syndrome, with cleft lip and palate. However, the transverse changes of the amino acids in that case were different from those in our patient. To the best of our knowledge, this is the first reported clinical variation of ADULT syndrome with a rare mutation, distinguished by the clinical manifestation of symblepharon and camptodactyly. After confirming the diagnosis, the patient’s chronic epiphora was addressed via binocular dacryocystorhinostomy under general anesthesia, during which artificial tear ducts were placed to drain the tears, and the enlarged lacrimal duct was removed. The surgery was successful, and the patient showed no lacrimal abnormalities on follow-up (Figure 5).
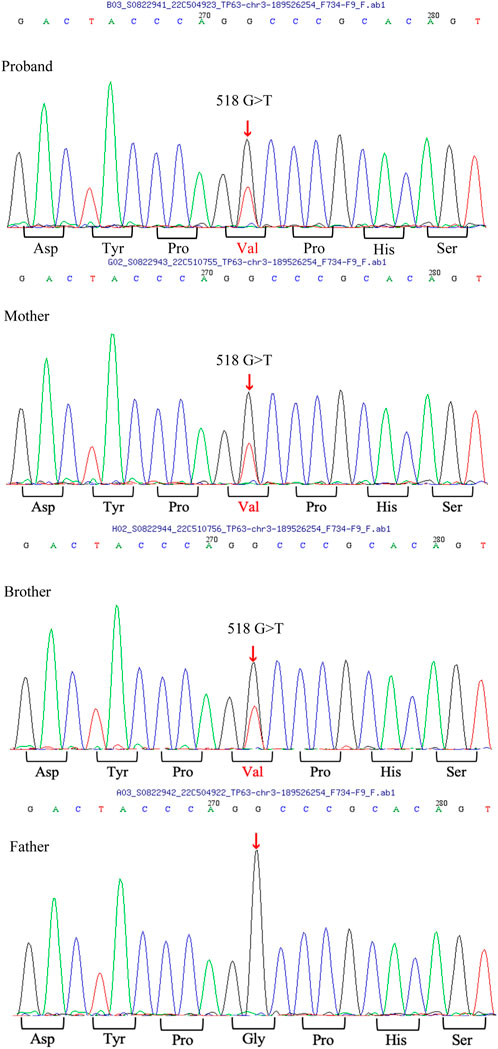
FIGURE 4. TP63 mutation analysis A heterozygous G>T transition at cDNA position 518 of the TP63 gene is found in the patient, as well as in her mother and brother.
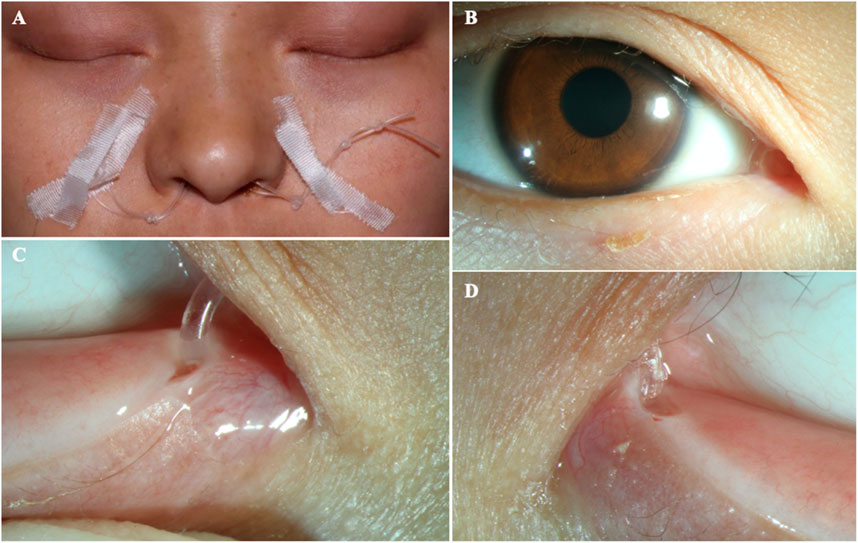
FIGURE 5. The patient after surgery (A–B) The ocular appearance after surgery. (C) The artificial nasolacrimal duct. (D) The close-up appearance of the opened punctum.
Discussion
The TP63 gene is highly expressed in the nuclei of the basal cells of the skin, cervix, tongue, mucosa, esophagus, mammary glands, prostate, and urothelium (Rinne et al., 2007). A crucial transcriptional regulator factor, p63 is often expressed in the epithelial and mesenchymal tissues (Otsuki et al., 2020). It is expressed very early during embryogenesis and epidermal development and plays an essential role in the induction of the ectoderm and the orofacial, limb, and epidermal stratification processes. Moreover, the expression of P-cadherin, which is regulated by p63, acts as a critical regulator of hair development. Previous studies have confirmed that the normal expression of p63 can inhibit the terminal differentiation of keratinocytes, which contributes to maintaining the proliferative potential of the basal cell layer and promoting its formation and integrity (Avitan-Hersh et al., 2010). In view of this, in vitro experiments performed in 1999 confirmed that TP63 gene knockout mice developed ectodermal developmental defects, such as limb defects and the loss of the prostate, mammary glands, epidermis, and other related tissues (Wang et al., 2009). These features were representative of defective ectodermal stem cells and were consistent with the physiological functions of the TP63 gene (Rinne et al., 2007). Since then, syndromes associated with TP63 mutations have been recognized in multiple reports, including the AEC, LMS, ADULT, RHS, SHFM, and EEC. The specific clinical manifestations of these six diseases are summarized in Table 2 (Duijf et al., 2002; Rinne et al., 2006a; Rinne et al., 2006b; Rinne et al., 2007; Otsuki et al., 2016).
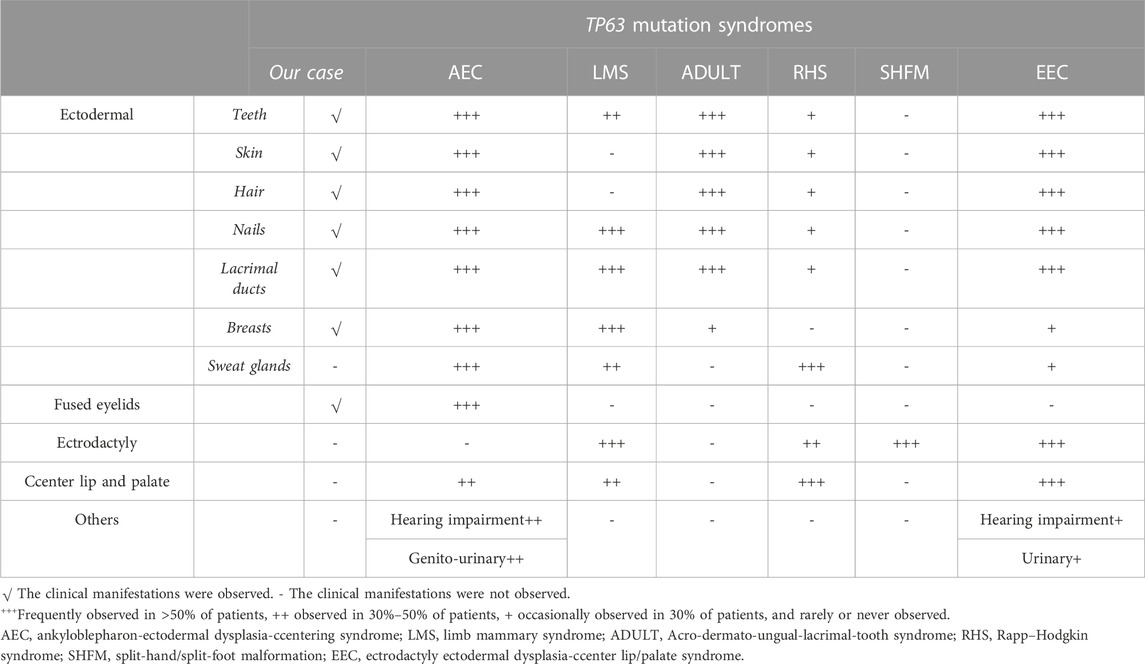
TABLE 2. Clinical features in six overlapping syndromes.
Our patient presented with clinical features of ectodermal dysplasia, with sparse hair, dystrophic nails, small teeth, oligodontia (11 teeth left), lacrimal duct stenosis, and hypoplastic nipples. These clinical features are observed in different syndromes. For EEC, orofacial cleft and ectrodactyly are typical manifestations. In contrast, cleft lip and palate are typically not detected in ADULT syndrome. Hence, this feature can be used to distinguish EEC from ADULT syndrome. On the contrary, it is difficult to distinguish between LMS and ADULT syndrome. LMS also manifests as a form of ectodermal dysplasia with oligodontia, lacrimal atresia, and nail dystrophy, in addition to abnormal development of the mammary glands and hypoplastic nipples. These findings may render a definitive diagnosis challenging. However, most patients with ADULT syndrome present with ectrosyndactylia and hair and skin abnormalities that have not been reported in LMS, and thus, these features may assist with diagnosis. Moreover, our patient had mild symblepharon that can also be observed in AEC, as well as ectodermal-related manifestations, which led us to suspect that the patient may have AEC. However, according to a literature review (Slavotinek et al., 2005; Kawasaki de Araujo et al., 2017), cleft lip and palate are characteristic of most patients with AEC, and more than half of all patients with AEC have hearing impairment and urinary system diseases, which were not consistent with the presentation of our patient. Furthermore, AEC is not typically associated with abnormal limb development. Hence, our patient, with her shortened fifth fingers, was suspected to have ADULT syndrome. Clinically, the six diseases mentioned above share some common manifestations. However, they have different gene inheritance patterns and also some relatively unique features (Rinne et al., 2006a; Rinne et al., 2006b; Rinne et al., 2007).
The literature suggests that the most common site of mutation of the TP63 gene is the DNA binding region, due to a missense point mutation, resulting in the substitution of arginine 298 by glycine or glutamine. In vitro experiments (Chan et al., 2004) confirmed that R298 is not adjacent to the DNA binding domain. Therefore, the mutation of amino acid 298 does not lead to any adverse effects, but results in high transactivation activities of ΔN-p63γ, which may be 25% higher than those of wild-type p63 (Duijf et al., 2002). Missense point mutations in exon 3 can result in the substitution of p. N6H (asparagine to histidine), ultimately resulting in ADULT syndrome. N6H is in the upstream region of the DNA domain of p63 and is only contained in the p63 subtype of the transactivation domain of this protein, which does not affect the activity of the p63 DNA binding domain (DBD) (Slavotinek et al., 2005). However, the above mutations are significantly different from those in EEC. For example, R298G and R298Q increase the activity of p63, while N6H, which is outside the functional domain of p63, does not affect the expression of p63. However, missense mutations in EEC are likely to result in the loss of DNA binding and impaired transactivation activities (Amiel et al., 2001). As a result, an essential difference is detectable at the genetic level, and this can be used to exclude ADULT syndrome.
In our patient, the mutation site was p63, p. G173V, which was consistent with a previously reported mutation. Monti et al. (2013) detected the transactivation abnormality and interfering ability of this mutant protein in yeast and mammalian cells and quantified the protein functional changes after mutation. Monti et al. detected wild-type and p63 p. G173V protein changes by inducing galactosyl-dependent protein expression in yeast through the inducible GAL1, 10 promoter and revealed that the mutation resulted in a 20% reduction in transactivation activities at relatively high galactose concentrations (0.128%). In mammalian cells, the mutation p. G173V retains partial transactivation activity. For example, p. G173V mutants show a high residual transactivation potential on the P21, MDM2, PUMA, and BAX targets, which are regulated by p63, and are involved in the regulation of the cell cycle, protein stability, apoptosis, and epithelial cells.
PERP and COL18A1 are well-known p63-regulated genes involved in skin and epithelial development. As for the interfering ability, the p. G173V mutant clearly interferes only with PERP and COL18A1 targets to lower the transactivation ability compared to wild-types. Considering the corresponding structure and function, the region corresponding to amino acid 173 protrudes on the surface of the protein, so that the protein’s functional structure does not change. The reason for these functional changes may be that amino acid 173 is close to the N-terminus of the DBD, which is involved in the recruitment and assembly of tetramer proteins that affects the machinery of transcription proteins. Meanwhile, amino acid 173 is located in the proline-rich region of the C-terminus, which is important for the structural integrity and apoptosis-inducing function of the transcription protein. Thus, the mutation induces changes in the transactivation and interference ability of the protein.
Different amino acid mutations result in different transactivation abnormalities and interfering abilities, as well as different clinical phenotypes. However, it can be seen from the above discussion that the clinical manifestations of the six TP63-related syndromes overlapped greatly. One amino acid mutation site can cause more than one syndrome. For example, as reported by Avitan-Hersh et al. (2010), the mutated amino acid p. R243W was previously reported to be associated with EEC and LMS. Brunner and Van Bokhoven (2002) also reported a patient with the ADULT syndrome phenotype, but her mutated amino acid (R227Q) was previously related to EEC and LMS. As such, in addition to the overlapping clinical phenotypes, the mutations affecting the amino acids in the TP63 gene also have a certain crossover potential. In other words, even subtle phenotypic differences can represent diversities at the molecular level. For example, the ADULT syndrome mutations can occur in exons 3, 4, 6, or 8, while LMS mutations can occur in exons 4, 13, and 14 (Rinne et al., 2007), and EEC mutations can occur in exon 5–8, 13, or 14 (Rinne et al., 2007; Whittington et al., 2016). For this reason, (Monti et al., 2013), proposed that clinical conditions should be integrated with the functional parameters of p63 mutated proteins, such as the transactivation ability of target genes involved in specific developmental pathways and the interaction and interference ability of mutant isomers, for better genetic diagnosis and differentiation of the overlapping clinical phenotypes (Serra et al., 2011).
At present, there is no treatment for conditions caused by TP63 mutations. However, we can focus on ensuring an accurate diagnosis by differentiating the different types of diseases with TP63 mutations and further improve the accuracy of genetic counseling through gene analysis. In the future, we hope we can assist patients in making reproductive plans and offer therapeutic means to cure the genetic disease from the embryonic stage through the classification of gene variants, such as gain-functional mutations or loss-functional mutations (van Zelst-Stams and van Steensel, 2009; Monti et al., 2013).
Conclusion
In summary, we reported a case of ADULT syndrome caused by a rare amino acid mutation with a rare clinical phenotype, including eyelid fusion and abnormal development of the fifth finger. These findings add to our current understanding of ADULT syndrome and other TP63-related diseases.
All relevant information was explained to the patient and his mother, and written informed consent was obtained from the patient and his mother for publication of this case report and accompanying images.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
Written informed consent was obtained from the individual(s), and minor(s) legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
Author contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by JZ and YW. The first draft of the manuscript was written by JZ and YW, and all authors commented on previous versions of the manuscript. All authors contributed to the article and approved the submitted version.
Acknowledgments
We thank YiW and his team at the Clinical Genetics Center for the DNA analysis. We also thankYinghong Zhang, the otolaryngologist, for her very useful comments, and the otolaryngology department at Peking University Third Hospital for investigating the patients’ hearing and doing the operation with us. We appreciate the kindness of the mother and daughter for allowing us to use their medical and dental information for publication and for the advancement of science. This article is solely dedicated to them.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1150613/full#supplementary-material
References
Amiel, B. G., Francannet, C., Raclin, V., Munnich, A., Lyonnet, S., and Frebourg, T. (2001). TP63 gene mutation in ADULT syndrome. Eur. J. Hum. Genet. 9 (8), 624–625. doi:10.1038/sj.ejhg.5200676
Avitan-Hersh, E., Indelman, M., Bergman, R., and Sprecher, E. (2010). ADULT syndrome caused by a mutation previously associated with EEC syndrome. Pediatr. Dermatol 27 (6), 643–645. doi:10.1111/j.1525-1470.2010.01131.x
Berk, D. R., Shinawi, M., and Whelan, A. J. (2012). ADULT syndrome due to an R243W mutation in TP63. Int. J. Dermatol 51 (6), 693–696. doi:10.1111/j.1365-4632.2011.05375.x
Brunner, H. B., and Van Bokhoven, H. (2002). The p63 gene in EEC and other syndromes. J. Med. Genet. 39 (6), 377–381. doi:10.1136/jmg.39.6.377
Cassady, J. V. (1952). Developmental anatomy of nasolacrimal duct. AMA Arch. Ophthalmol. 47 (2), 141–158. doi:10.1001/archopht.1952.01700030146003
Chan, H. J., Mellerio, J. E., and McGrath, J. A. (2004). ADULT ectodermal dysplasia syndrome resulting from the missense mutation R298Q in the p63 gene. Clin. Exp. Dermatol 29 (6), 669–672. doi:10.1111/j.1365-2230.2004.01643.x
Duijf, P. H., Propping, P., Friedl, W., Krieger, E., McKeon, F., Dötsch, V., et al. (2002). Gain-of-function mutation in ADULT syndrome reveals the presence of a second transactivation domain in p63. Hum. Mol. Genet. 11 (7), 799–804. doi:10.1093/hmg/11.7.799
Kawasaki de Araujo, T., Lustosa-Mendes, E., Dos Santos, A. P., Coelho Molck, M., Mazzariol Volpe-Aquino, R., and Gil-da-Silva-Lopes, V. L. (2017). ADULT phenotype and rs16864880 in the TP63 gene: two new cases and review of the literature. Mol. Syndromol. 8 (4), 201–205. doi:10.1159/000470025
Monti, P., Russo, D., Bocciardi, R., Foggetti, G., Menichini, P., Divizia, M. T., et al. (2013). EEC- and ADULT-associated TP63 mutations exhibit functional heterogeneity toward P63 responsive sequences. Hum. Mutat. 34 (6), 894–904. doi:10.1002/humu.22304
Otsuki, Y., Ueda, K., Nuri, T., Satoh, C., Maekawa, R., and Yoshiura, K. I. (2020). EEC-LM-ADULT syndrome caused by R319H mutation in TP63 with ectrodactyly, syndactyly, and teeth anomaly: A case report. Med. Baltim. 99 (44), e22816. doi:10.1097/MD.0000000000022816
Otsuki, Y., Ueda, K., Satoh, C., Maekawa, R., Yoshiura, K. I., and Iseki, S. (2016). Intermediate phenotype between ADULT syndrome and EEC syndrome caused by R243Q mutation in TP63. Plast. Reconstr. Surg. Glob. Open 4 (12), e1185. doi:10.1097/GOX.0000000000001185
Petris, C., and Liu, D. (2017). Probing for congenital nasolacrimal duct obstruction. Cochrane Database Syst. Rev. 7, CD011109. doi:10.1002/14651858.CD011109.pub2
Prontera, P., Garelli, E., Isidori, I., Mencarelli, A., Carando, A., Silengo, M. C., et al. (2011). Cleft palate and ADULT phenotype in a patient with a novel TP63 mutation suggests lumping of EEC/LM/ADULT syndromes into a unique entity: ELA syndrome. Am. J. Med. Genet. A 155A (11), 2746–2749. doi:10.1002/ajmg.a.34270
Rinne, T., Brunner, H. G., and van Bokhoven, H. (2007). p63-associated disorders. Cell Cycle 6 (3), 262–268. doi:10.4161/cc.6.3.3796
Rinne, T., Hamel, B., van Bokhoven, H., and Brunner, H. G. (2006a). Pattern of p63 mutations and their phenotypes--update. Am. J. Med. Genet. A 140 (13), 1396–1406. doi:10.1002/ajmg.a.31271
Rinne, T., Spadoni, E., Kjaer, K. W., Danesino, C., Larizza, D., Kock, M., et al. (2006b). Delineation of the ADULT syndrome phenotype due to arginine 298 mutations of the p63 gene. Eur. J. Hum. Genet. 14 (8), 904–910. doi:10.1038/sj.ejhg.5201640
Serra, V., Castori, M., Paradisi, M., Bui, L., Melino, G., and Terrinoni, A. (2011). Functional characterization of a novel TP63 mutation in a family with overlapping features of Rapp-Hodgkin/AEC/ADULT syndromes. Am. J. Med. Genet. A 155A (12), 3104–3109. doi:10.1002/ajmg.a.34335
Slavotinek, A. M., Tanaka, J., Winder, A., Vargervik, K., Haggstrom, A., and Bamshad, M. (2005). Acro-dermato-ungual-lacrimal-tooth (ADULT) syndrome: report of a child with phenotypic overlap with ulnar-mammary syndrome and a new mutation in TP63. Am. J. Med. Genet. A 138A (2), 146–149. doi:10.1002/ajmg.a.30900
van Zelst-Stams, W. A., and van Steensel, M. A. (2009). A novel TP63 mutation in family with ADULT syndrome presenting with eczema and hypothelia. Am. J. Med. Genet. A 149A (7), 1558–1560. doi:10.1002/ajmg.a.32881
Wang, X., Tao, A. L., Yang, W. L., and Zhang, H. J. (2009). Mutation analysis of p63 gene in the first Chinese family with ADULT syndrome. Chin. Med. J. Engl. 122 (16), 1867–1871. doi:10.3760/cma.j.issn.0366-6999.2009.16.006
Keywords: TP63 gene, ADULT syndrome, congenital nasolacrimal duct obstruction, ectodermal dysplasia, dacryocystorhinostomy
Citation: Zhou J, Wang Y, Zhang Y, You D and Wang Y (2023) Case report: ADULT syndrome: a rare case of congenital lacrimal duct abnormality. Front. Genet. 14:1150613. doi: 10.3389/fgene.2023.1150613
Received: 24 January 2023; Accepted: 15 September 2023;
Published: 18 October 2023.
Edited by:
Weimin Lin, Sichuan University, ChinaReviewed by:
Dominique Bremond-Gignac, Hôpital Necker-Enfants Malades, FranceHala El-Bassyouni, National Research Centre, Egypt
Copyright © 2023 Zhou, Wang, Zhang, You and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi Wang, wangyieye@126.com