 Giovanni Falcicchio1†
Giovanni Falcicchio1† Antonella Riva2,3†
Antonella Riva2,3† Angela La Neve1
Angela La Neve1 Michele Iacomino4
Michele Iacomino4 Patrizia Lastella5
Patrizia Lastella5 Patrizia Suppressa5Vittorio Sciruicchio6
Patrizia Suppressa5Vittorio Sciruicchio6 Maria Trojano1
Maria Trojano1 Pasquale Striano2,3*
Pasquale Striano2,3*- 1Department of Basic Medical Sciences, Neurosciences and Sense Organs, University of Bari, Bari, Italy
- 2Paediatric Neurology and Muscular Diseases Unit, IRCCS Istituto Giannina Gaslini, Genoa, Italy
- 3Department of Neurosciences, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health, University of Genoa, Genoa, Italy
- 4Unit of Medical Genetics, IRCCS Istituto Giannina Gaslini, Genoa, Italy
- 5Department of Internal Medicine and Rare Diseases Centre “C. Frugoni”, University Hospital of Bari, Bari, Italy
- 6Children Epilepsy and EEG Centre, San Paolo Hospital, Bari, Italy
Background: Malformations of cortical development (MCDs) can lead to peculiar neuroradiological patterns and clinical presentations (i.e., seizures, cerebral palsy, and intellectual disability) according to the specific genetic pathway of the brain development involved; and yet a certain degree of phenotypic heterogeneity exists even when the same gene is affected. Here we report a man with an malformations of cortical development extending beyond occipital lobes associated with a novel stop-gain variant in LAMC3.
Case presentation: The patient is a 28-year-old man suffering from drug-resistant epilepsy and moderate intellectual disability. He underwent a brain magnetic resonance imaging showing polymicrogyria involving occipital and temporal lobes bilaterally. After performing exome sequencing, a novel stop-gain variant in LAMC3 (c.3871C>T; p. Arg1291*) was identified. According to the cortical alteration of the temporal regions, temporal seizures were detected; instead, the patient did not report occipital seizures. Different pharmacological and non-pharmacological interventions (i.e., vagus nerve stimulation) were unsuccessful, even though a partial seizure reduction was obtained after cenobamate administration.
Conclusion: Our case report confirms that variants of a gene known to be related to specific clinical and neuroradiological pictures can unexpectedly lead to new phenotypes involving different areas of the brain.
Background
Malformations of cortical development (MCDs) are a heterogeneous group of abnormal cerebral cortex formation disorders affecting people from early childhood to early adulthood (Severino et al., 2020). The most common clinical presentations of MCDs are epilepsy and/or neurodevelopmental delay (Barkovich et al., 2012). Knowledge about MCDs has greatly increased over the years, leading to the classification of MCDs based on neuroradiological patterns (Severino et al., 2020). Moreover, thanks to the evolution of molecular biology and genetics, more than 100 genes have been identified and associated with different types of MCDs (Kuzniecky, 2015; Accogli et al., 2020). Recessive LAMC3 gene variants cause occipital cortical malformations (Barak et al., 2011), such as pachygyria or polymicrogyria. However, the spectrum of LAMC3-associated cortical malformations has progressively expanded over time, showing that other cortical areas could be affected and that occipital lobes could even be spared in some cases (Zambonin et al., 2018; Kasper et al., 2020; De Angelis et al., 2021).
We report the clinical and neuroradiological features of a patient with a novel homozygous stop-gain variant in LAMC3 (c.3871C>T; p. Arg1291*) identified through whole exome sequencing (WES).
Case presentation
This right-handed, 28-year-old Caucasian man had an unremarkable antenatal and perinatal history. His parents were non-consanguineous and healthy. His older sister did not have known medical conditions. No familial history of genetic, metabolic, or neurological diseases was reported. Educational support was necessary during school years. At the age of six, the patient started to experience daily seizures—often several times a day - with different semeiology: 1) episodes of symmetrical myoclonic spasms of the upper limbs; 2) involuntary movements of both eyes in different directions, followed by behavioural arrest, and impaired awareness with unilateral/bilateral manual automatisms (i.e., manipulating, nose-wiping, and hair-fixing). Multiple antiseizure medications (i.e., lamotrigine, phenobarbital, nitrazepam, vigabatrin, clobazam, topiramate, lacosamide, levetiracetam, oxcarbazepine, brivaracetam) were unsuccessfully administered. The patient came to our attention at the age of 23, when he was taking rufinamide 3,200 mg/day, valproate 1,000 mg/day, and carbamazepine 800 mg/day. Daily seizures with different semeiology (myoclonic, atonic, focal motor, and bilateral tonic-clonic seizures) were reported. Neurological examination was unremarkable except for divergent strabismus in the left eye. No dysmorphic features were identified. Interictal electroencephalogram showed spikes, polyspikes, and polyspike-and-waves localized in the occipital regions. Brain magnetic resonance imaging (MRI) was also performed revealing bilateral temporal and occipital polymicrogyria; atrophy of the pons and cerebellum was also detected (Figure 1). Neuropsychological assessment revealed moderate intellectual disability (ID) with a low intelligence quotient (I.Q. = 48).
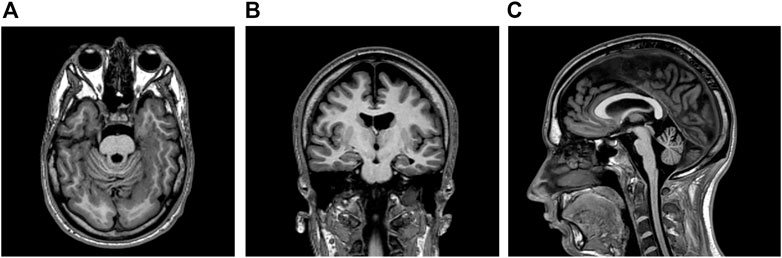
FIGURE 1. (A,B) Brain magnetic resonance imaging (MRI) showing bilateral polymicrogyria in temporal and occipital lobes (T1-weighted axial and coronal sequences respectively) (C) Atrophy in pons and cerebellum was detected in the T1-weighted sagittal sequence.
Vagus nerve stimulation was implanted, without substantial variation neither in seizure intensity nor in frequency. Oral immunotherapy (prednisone) was ineffective as well. Finally, cenobamate was administered and a partial reduction in seizure frequency was registered (from daily to weekly). At the last follow-up, antiseizure treatment included valproate 1,500 mg/day, zonisamide 400 mg/day, and cenobamate 300 mg/day.
Genetic analysis
WES was conducted by Research & Innovation Genetics in Padua, Italy. Proband’s and parents’ genomic DNA was extracted from peripheral blood. The DNA library was constructed using a SureSelect All Exon 6 (Agilent) kit. Whole Exome sequencing runs were performed on the NovaSeq6000 platform (Illumina Inc., San Diego, CA, United States) with 150-base paired-end reads. Paired-end reads were aligned to the reference human genome sequence (GRCh37/hg19) using BWA algorithm (Li and Durbin, 2009). Sequences without specific alignment were excluded. The bioinformatic analysis was performed by GATK, samtools and bcftools packages. Mean sequencing depth of 100× and 99.04% of targeted regions covered at 20× depth were obtained. 273 genes already related to MCDs and epilepsy of suspected genetic etiology were prioritized in the analysis (Supplementary Table S1). Population databases (i.e., The Exome Aggregation Consortium, 1000 Genomes Project, GnomAD), genetic clinical databases (i.e., OMIM, ClinVar, HGMD) and in silico tools (Polyphen2, Sift, CADD, Mutation Taster, Mutation Assessor) were used to screen the variant. A new very rare stop-gain variant c.3871C>T (p.Arg1291*) in LAMC3 was identified. It is a loss-of-function variant, and it is predicted to be pathogenic according to ACMG criteria (PVS1,PM2,PP5) (Richards et al., 2015). Sanger sequencing was performed to validate the heterozygous state in the unaffected parents.
Discussion and conclusions
LAMC3 gene is located on chromosome 9 and encodes the ɣ3 chain of the extracellular laminin family proteins, associated with cell differentiation, migration, and adhesion (Hamill et al., 2009), and it is involved in the organization of the cerebral cortex and its gyration (Radner et al., 2013). Homozygous or compound heterozygous variants in LAMC3 have been described to cause occipital cortical malformations (OMIM #614115) (Barak et al., 2011). So far, eight unrelated individuals with LAMC3 gene variants have been identified worldwide and their clinical, genetic, and neuroradiological characteristics are summarized in Table 1. In nearly all reported cases a frameshift or non-sense LAMC3 variant was detected, except for missense variants in compound heterozygosity in two patients (Barak et al., 2011; De Angelis et al., 2021). Differently from the early literature reporting that only occipital lobes were involved in the cortical malformation process (Barak et al., 2011; Afawi et al., 2016), more recent studies (Zambonin et al., 2018; Kasper et al., 2020; De Angelis et al., 2021; Qian et al., 2021) and the structural analysis of previously reported individuals (Urgen et al., 2019) have demonstrated that LAMC3 variants can cause structural cerebral anomalies going beyond the occipital lobes, which sometimes can even be spared (Kasper et al., 2020; Qian et al., 2021). Awafi et al. identified a consanguineous Arab family in which, out of 14 siblings, all the affected children (n = 6) and one unaffected sibling shared the same LAMC3 homozygous variant; all the affected family members had epilepsy and ID, but their computed tomography scans were unremarkable. Only one of the affected siblings underwent brain MRI, showing occipital dysplasia. In our patient, polymicrogyria was more extended, involving occipital as well temporal lobes.
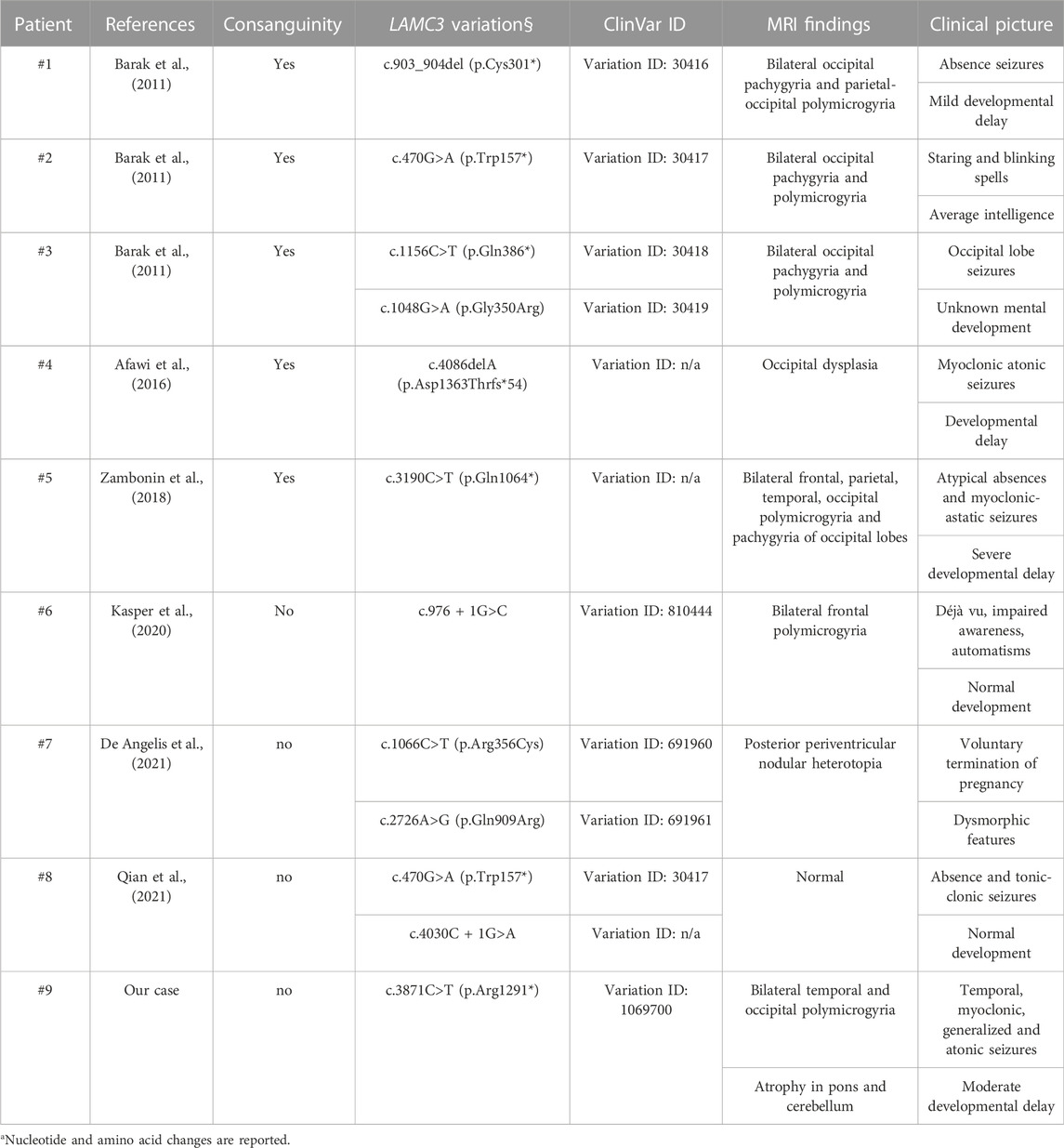
TABLE 1. Summary of previous LAMC3 variations with associated clinical and neuroradiological characteristics.
Polymicrogyria is a heterogeneous MCD which can also be caused by non-genetic factors, with in utero infections, prenatal ischemia, or exposure to medications during gestation as the most frequent alternative aetiologies (Raybaud and Widjaja, 2011). The onset of the neurological and cognitive disturbances in the affected patients is between the age of 2 and 13 years and it is often associated with developmental or cognitive impairment (Barak et al., 2011; Afawi et al., 2016; Zambonin et al., 2018), as in our patient. MCDs caused by variants in a single recessive gene demonstrate how a single gene can influence the development of the cortical structure, as well as the phenotype of the individual carrying the variant with different neurological (epilepsy, cerebral palsy) and cognitive (ID) outcomes. Different seizure types (absences, myoclonic-astatic, tonic-clonic, and occipital lobe seizures) have been reported in patients with epilepsy related to LAMC3 variants (Table 1). In our patient, the involvement of the temporal regions was supported by the suggestive seizure semiology indicating onset in the temporal lobe (i.e., behavioural arrest and impaired awareness and oral and manual automatisms).
In summary, we report a novel LAMC3 variant and broadened the phenotypic spectrum of LAMC3-related MCDs showing involvement outside of the occipital area. The identified, likely pathogenic variant is novel and may result in a loss-of-function mechanism, eventually preventing the formation of the related protein.
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. The patients/participants provided their written informed consent to participate in this study.
Author contributions
GF, AR and VS were responsible for the collection and organization of clinical data. MI, PL and PS were responsible for the collection and analysis of genetic information. PS, MT and AN supervised the work. All authors contributed to the writing of the final manuscript.
Funding
PS received research support from the Italian Ministry of Health (Ricerca Corrente 2022) and Fondazione San Paolo.
Conflict of interest
GF received consultancy fee from Angelini Pharma. AR has received honoraria from Kolfarma s. r.l, Proveca Pharma Ltd., and PTC Therapeutics. PS has served on a scientific advisory board for the Italian Agency of the Drug (AIFA); has received honoraria from GW pharma, Kolfarma s. r.l., Proveca Pharma Ltd., and Eisai Inc.; and has received research support from the Italian Ministry of Health and Fondazione San Paolo. MT has served on scientific Advisory Boards for Biogen, Novartis, Roche, Merck, and Genzyme; has received speaker honoraria from Biogen Idec, Merck, Roche, Teva, Sanofi-Genzyme, and Novartis; and has received research grants for her Institution from Biogen Idec, Merck, Roche, and Novartis. AN has received speaker’s or consultancy fees or travel support from Eisai, Mylan, Sanofi, Bial, GW, UCB Pharma, Arvelle Therapeutics, Angelini Pharma and Neuraxpharma.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.990350/full#supplementary-material
Abbreviations
ACMG, American College of Medical Genetics and Genomics; ID, intellectual disability; IQ, intelligence quotient. MCD, malformation of cortical development. MRI, magnetic resonance imaging.NGS, next-generation sequencing; PCR, polymerase chain reaction; WES, whole exome sequencing.
References
Accogli, A., Severino, M., Riva, A., Madia, F., Balagura, G., Iacomino, M., et al. (2020). Targeted re-sequencing in malformations of cortical development: Genotype-phenotype correlations. Seizure 80, 145–152. doi:10.1016/j.seizure.2020.05.023
Afawi, Z., Oliver, K. L., Kivity, S., Mazarib, A., Blatt, I., Neufeld, M. Y., et al. (2016). Multiplex families with epilepsy: Success of clinical and molecular genetic characterization. Neurology 86 (8), 713–722. doi:10.1212/WNL.0000000000002404
Barak, T., Kwan, K. Y., Louvi, A., Demirbilek, V., Saygı, S., Tüysüz, B., et al. (2011). Recessive LAMC3 mutations cause malformations of occipital cortical development. Nat. Genet. 43 (6), 590–594. doi:10.1038/ng.836
Barkovich, A. J., Guerrini, R., Kuzniecky, R. I., Jackson, G. D., and Dobyns, W. B. (2012). A developmental and genetic classification for malformations of cortical development: Update 2012. Brain 135 (5), 1348–1369. doi:10.1093/brain/aws019
De Angelis, C., Byrne, A. B., Morrow, R., Feng, J., Ha, T., Wang, P., et al. (2021). Compound heterozygous variants in LAMC3 in association with posterior periventricular nodular heterotopia. BMC Med. Genomics 14 (1), 64. doi:10.1186/s12920-021-00911-4
Hamill, K. J., Kligys, K., Hopkinson, S. B., and Jones, J. C. (2009). Laminin deposition in the extracellular matrix: A complex picture emerges. J. Cell Sci. 122 (24), 4409–4417. doi:10.1242/jcs.041095
Kasper, B. S., Kraus, C., Schwarz, M., Rösch, J., Thiel, C. T., Reis, A., et al. (2020). A novel splice variant expands the LAMC3-associated cortical phenotype to frontal only polymicrogyria and adult-onset epilepsy. Am. J. Med. Genet. A 182 (11), 2761–2764. doi:10.1002/ajmg.a.61846
Kuzniecky, R. (2015). Epilepsy and malformations of cortical development: New developments. Curr. Opin. Neurol. 28 (2), 151–157. doi:10.1097/WCO.0000000000000175
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25 (14), 1754–1760. doi:10.1093/bioinformatics/btp324
Qian, X., Liu, X., Zhu, Z., Wang, S., Song, X., Chen, G., et al. (2021). Variants in LAMC3 causes occipital cortical malformation. Front. Genet. 12, 616761. doi:10.3389/fgene.2021.616761
Radner, S., Banos, C., Bachay, G., Li, Y. N., Hunter, D. D., Brunken, W. J., et al. (2013). β2 and γ3 laminins are critical cortical basement membrane components: Ablation of Lamb2 and Lamc3 genes disrupts cortical lamination and produces dysplasia. Dev. Neurobiol. 73 (3), 209–229. doi:10.1002/dneu.22057
Raybaud, C., and Widjaja, E. (2011). Development and dysgenesis of the cerebral cortex: Malformations of cortical development. Neuroimaging Clin. N. Am. 21 (3), 483–543. doi:10.1016/j.nic.2011.05.014
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Severino, M., Geraldo, A. F., Utz, N., Tortora, D., Pogledic, I., Klonowski, W., et al. (2020). Definitions and classification of malformations of cortical development: Practical guidelines. Brain 143 (10), 2874–2894. doi:10.1093/brain/awaa174
Urgen, B. M., Topac, Y., Ustun, F. S., Demirayak, P., Oguz, K. K., Kansu, T., et al. (2019). Homozygous LAMC3 mutation links to structural and functional changes in visual attention networks. Neuroimage 190, 242–253. doi:10.1016/j.neuroimage.2018.03.077
Keywords: cortical malformations, epilepsy, exome sequencing, genetic mutations, LAMC3 case report
Citation: Falcicchio G, Riva A, La Neve A, Iacomino M, Lastella P, Suppressa P, Sciruicchio V, Trojano M and Striano P (2023) Case report: LAMC3-associated cortical malformations: Case report of a novel stop-gain variant and literature review. Front. Genet. 13:990350. doi: 10.3389/fgene.2022.990350
Received: 09 July 2022; Accepted: 01 December 2022;
Published: 06 January 2023.
Edited by:
Anupam Basu, National Institute of Biomedical Genomics (NIBMG), IndiaReviewed by:
Piero Pavone, University of Catania, ItalyJared C. Roach, Institute for Systems Biology (ISB), United States
Copyright © 2023 Falcicchio, Riva, La Neve, Iacomino, Lastella, Suppressa, Sciruicchio, Trojano and Striano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pasquale Striano, strianop@gmail.com
†These authors share first authorship