 Ria Kodariah1,2
Ria Kodariah1,2 Fadilah Fadilah1,3,4*
Fadilah Fadilah1,3,4* Rafika Indah Paramita1,3,4*Linda Erlina1,3,4
Rafika Indah Paramita1,3,4*Linda Erlina1,3,4 Khaerunnisa Anbar Istiadi4,5
Khaerunnisa Anbar Istiadi4,5 Yayi Dwina Billianti1,2Meilania Saraswati1,2
Yayi Dwina Billianti1,2Meilania Saraswati1,2 Sonar Soni Panigoro1,6
Sonar Soni Panigoro1,6- 1Master’s Programme in Biomedical Sciences, Faculty of Medicine, Universitas Indonesia, Jakarta, Indonesia
- 2Department of Anatomical Pathology, Faculty of Medicine, Universitas Indonesia, Jakarta, Indonesia
- 3Department of Medical Chemistry, Faculty of Medicine, Universitas Indonesia, Jakarta, Indonesia
- 4Bioinformatics Core Facilities-IMERI, Faculty of Medicine, Universitas Indonesia, Jakarta, Indonesia
- 5Department of Biology, Institut Teknologi Sumatera, Lampung, Indonesia
- 6Surgical Oncology Division, Department of Surgery, Faculty of Medicine, Universitas Indonesia, Jakarta, Indonesia
1 Introduction
The creation of biobank has steadily developed additional research platforms and created opportunities over the years to learn more about how living systems function in both acute and chronic physiological and pathological circumstances. It involves a process of gathering, preserving, distributing, and utilization of biological samples for prospective research studies. The majority of hospitals and biomedical research facilities in many countries, in Indonesia as well, participate in this activity, which is essential to the development of a successful, effective, and cutting-edge research system. Potential in the new biology era could create fascinating possibilities for comprehending the physiological and pathological mechanisms behind human health by unravelling the more intricate processes (Caenazzo and Tozz, 2020).
Early population-based biobanks generally focused on finding genetic variations linked to disease without taking into account how the information may be relayed back to participants for their own health management. The genetic basis of illness susceptibility varies between ethnicities since many disease-causing variants are uncommon and population-specific, which has contributed to the global creation of biobanks (Wei et al., 2021). One of the disorders linked to an accumulation of somatic mutations, structural variants, epigenetic variables, and changes in copy number is cancer, which frequently arises from a genetic background where hereditary cancer is more prevalent. The application of genomic sequencing in clinical setting have been made possible by advancements in sequencing technology and the creation of computational tools, supporting the therapeutic relevance of genomics to cancer treatment (Rossing et al., 2020). We report raw fastq data for hotspot regions of cancer-related 50 genes using fresh frozen breast carcinoma tissues retrieved from IMERI-FMUI Biobank collection. The data gathered from this study will help understand how breast cancer develops and forecast appropriate treatments based on somatic gene alterations that are associated with it.
2 Materials and methods
2.1 Sample collection and DNA purification
Sixteen freshly frozen breast cancer samples were collected from the IMER-FMUI Biobank, of which the majority were invasive carcinoma (Supplementary Table S1). Purified DNA was extracted from the tissues using the QIAamp DNA Mini Kit® components in accordance with the manufacturer’s instructions (Qiagen Sciences). The DNA input for library preparation is 10 ng. The Nanodrop Thermoscientific 2000 instrument (ThermoFisher) was used to assess DNA purity at a 260/280 absorbance ratio, whereas the Qubit® 3.0 Fluorometer and the Qubit dsDNA BR Assay Kit (Thermo Fisher Scientific) were used to determine ultimate DNA concentration. The findings are displayed in Supplementary Table S2.
2.2 Library preparation and sequencing
DNA libraries were created using AmpliSeq™ for the Illumina Cancer Hotspot Panel v2 (Illumina®, United States). This panel detects somatic variants in a total of 50 cancer-associated genes (Supplementary Table S3). The first step was amplification of target regions of the DNA sample, along with AmpliSeq™ for the Illumina Cancer Hotspot Panel v2 (Illumina®, United States) as well, with 17 cycles of 99°C for 15 s, 60°C for 4 min and then hold at 10°C for up to 24 h at thermal cycler. The indexes were than ligated following LIGATE program on thermal cycler (22°C for 30 min, 68°C for 5 min, 72°C for 5 min and then hold at 10°C for up to 24 h). 30 μl of the Agencourt AMPure XP beads (Beckman Coulter™, United States) was added to the mixtures to clean up the libraries.
The second amplification steps were conducted to ensure sufficient quantity for sequencing on Illumina systems. This step used 7 cycles of 98°C for 15 s, 64°C for 1 min and then hold at 10°C for up to 24 h at thermal cycler. The second cleanup was performed twice to remove high molecular-weight DNA and primer excess by using 25 μl and 60 μl of Agencourt AMPure XP beads (Beckman Coulter™, United States), respectively. The libraries were diluted to the final loading concentration at 7–9 pM and sequenced using the Illumina MiSeq platform.
3 Descriptive analysis
Paired-end libraries (2 × 150 bp) in fastq format were generated by the sequencing operation. Under the BioProject accession number PRJNA820526, the data sequences were submitted to the SRA. FastQC software was used to evaluate the quality of each sample’s paired-end raw readings (Andrews, 2010), and q30 Python programs were used to determine the total number of raw bases and the percentage of Q30 (Chen, 2016). Mosdepth software was used to calculate amplicon mean coverage depth, Coverage Uniformity, and on target rate (Pedersen and Quinlan, 2018).
Illumina sequencing raw read data is stored as a text file in the FASTQ format. Each sequencing read is stored in the FASTQ format on four lines of text, which provide the following information for each nucleotide: 1) identifiers, 2) nucleotide sequences, 3) “+” symbols, and 4) base quality. The first identification line includes useful data, such as the machine name, run ID, lane ID, and flow cell ID, that can be utilized to identify batch effects. The total number of reads sequenced, the GC content, and the overall base quality score are the most frequent metrics to be examined at the raw data level and are all frequently calculated by typical raw data QC programs (Sheng et al., 2017). In Table 1, we provide the descriptive details of the raw data.
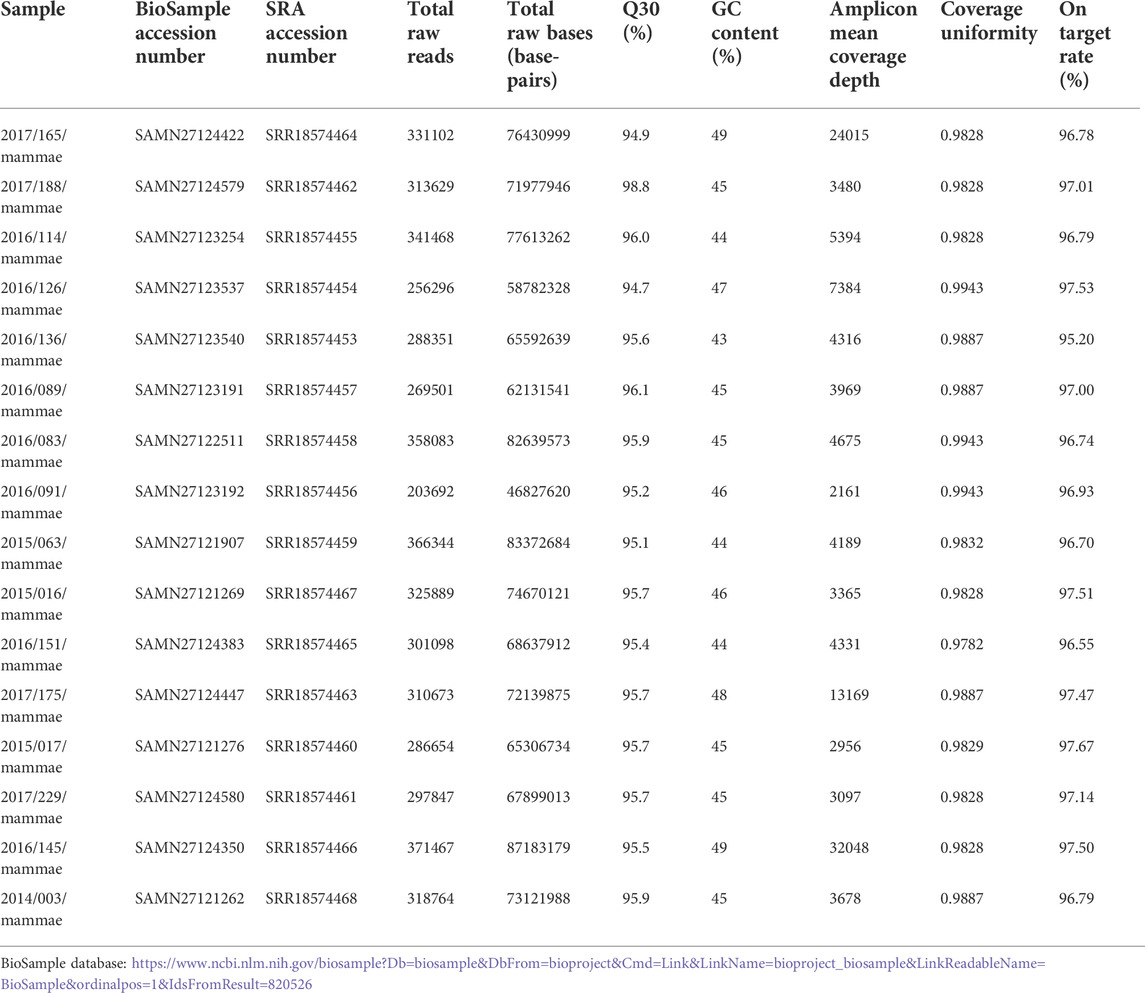
TABLE 1. Descriptive details of the raw data.
Table 1 indicates that all the samples had a Q30 score above 90%, with the sequence quality presented from “per base sequence quality” generated by FastQC software in Figure 1. For example, for the “2016/083/mammae” sample, it showed that all the bases in the reads had a Q score above 32 (p error less than 0.00063), indicating high quality data produced by the illumina instrument.
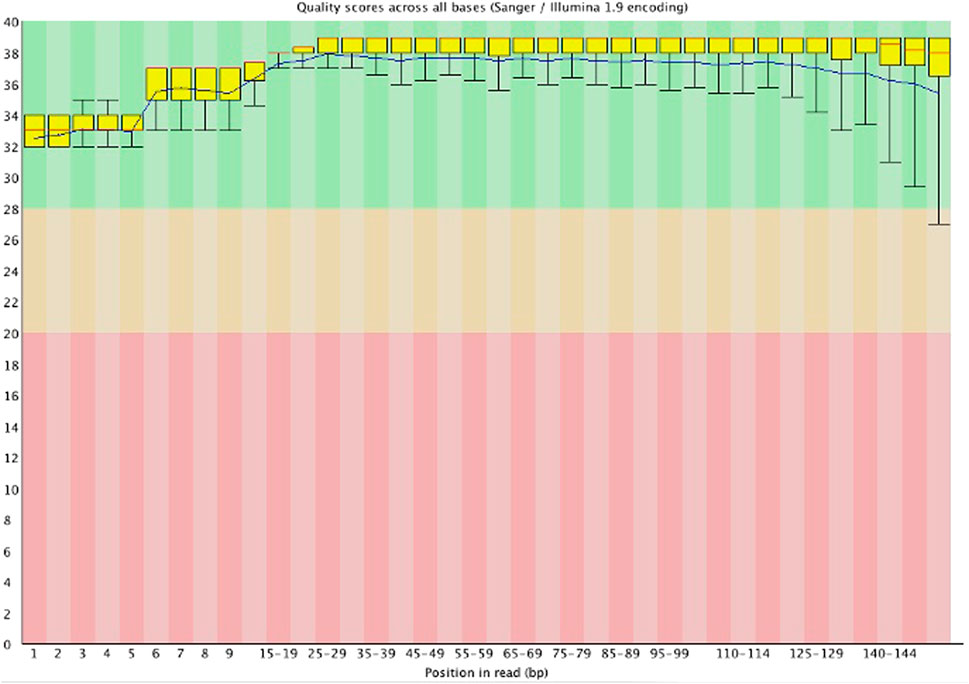
FIGURE 1. Per base sequence quality of “2016/083/mammae” sample (Generated by FastQC software).
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA820526.
Ethics statement
The studies involving human participants were reviewed and approved by the Faculty of Medicine Universitas Indonesia Ethical Committee (approval number: 867/UN2.F1/ETIK/PPM.00.02/2020). The patients/participants provided their written informed consent to participate in this study.
Author contributions
Conceptualization: RK, FF, and RP; methodology: RK, FF, RP, LE, KI, YB, MS, and SP; formal analysis: RP; writing—original draft preparation and editing: RP; supervision: RK, and FF. RK, FF, and RP contributed equally to this work. All authors read and approved the final manuscript.
Funding
This work was supported by a PUTI Q1 Grant year 2020 from Universitas Indonesia (grant number: NKB-1303/UN2.RST/HKP.05.00/2020).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.973453/full#supplementary-material
References
Andrews, S. (2010). FastQC—a quality control tool for high throughput sequence data. Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/(Accessed June 1, 2022).
Caenazzo, L., and Tozz, P. (2020). The future of biobanking: What is next? BioTech. 9 (23), 23–26. doi:10.3390/biotech9040023
Chen, S. (2016). q30 python script. Available at: https://github.com/dayedepps/q30w (Accessed June 1, 2022).
Pedersen, B. S., and Quinlan, A. R. (2018). Mosdepth: Quick coverage calculation for genomes and exomes. Bioinformatics 34 (5), 867–868. doi:10.1093/bioinformatics/btx699
Rossing, M., Sorensen, C. S., Ejlertsen, B., and Nielsen, F. C. (2020). Whole genome sequencing of breast cancer. APMIS 127 (5), 303–315. doi:10.1111/apm.12920
Sheng, Q., Vickers, K., Zhao, S., Wang, J., Samuels, D. C., Koues, O., et al. (2017). Multi-perspective quality control of Illumina RNA sequencing data analysis. Brief. Funct. Genomics 16 (4), 194–204. doi:10.1093/bfgp/elw035
Keywords: biobank IMERI-FMUI, breast cancer, frozen tissues, hot-spot gene sequencing, NGS–next generation sequencing
Citation: Kodariah R, Fadilah F, Paramita RI, Erlina L, Istiadi KA, Billianti YD, Saraswati M and Panigoro SS (2022) Raw fastq data for hotspot regions of cancer-related 50 genes using fresh frozen breast carcinoma tissues obtained from IMERI-FMUI biobank collections. Front. Genet. 13:973453. doi: 10.3389/fgene.2022.973453
Received: 01 July 2022; Accepted: 14 October 2022;
Published: 24 October 2022.
Edited by:
Maritha J. Kotze, Stellenbosch University, South AfricaReviewed by:
Nerina Chrisna Van Der Merwe, National Health Laboratory Service Bloemfontein, South AfricaEri Ogiso-Tanaka, National Museum of Nature and Science, Japan
Copyright © 2022 Kodariah, Fadilah, Paramita, Erlina, Istiadi, Billianti, Saraswati and Panigoro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fadilah Fadilah, fadilah.msi@ui.ac.id; Rafika Indah Paramita, rafikaindah@ui.ac.id