 Mingxiao Han1†
Mingxiao Han1† Zhihao Chen2,3,4†
Zhihao Chen2,3,4† Ping He1,5†Ziyuan Li2
Ping He1,5†Ziyuan Li2 Qi Chen1Zelei Tong2
Qi Chen1Zelei Tong2 Min Wang1
Min Wang1 Hong Du1*
Hong Du1* Haifang Zhang1*
Haifang Zhang1*- 1Department of Clinical Laboratory, The Second Affiliated Hospital of Soochow University, Suzhou, China
- 2Department of Orthopedics, The Second Affiliated Hospital of Soochow University, Suzhou, China
- 3Department of Musculoskeletal Oncology, Sun Yat-sen University Cancer Center, Guangzhou, China
- 4State Key Laboratory of Oncology in Southern China, Collaborative Innovation Center of Cancer Medicine, Guangzhou, China
- 5Department of Clinical Laboratory, Sichuan Province Science City Hospital, Chengdu, China
Sepsis is one of the diseases that can cause serious mortality. In E. coli, an inner membrane protein YgiM encoded by gene ygiM can target the eukaryotic peroxisome. Peroxisome is a membrane-enclosed organelle associated with the ROS metabolism and was reported to play the key role in immune responses and inflammation during the development of sepsis. Klebsiella pneumoniae (K. pneumoniae) is one of the important pathogens causing sepsis. However, the function of gene vk055_4013 which is highly homologous to ygiM of E. coli has not been demonstrated in K. pneumoniae. In this study, we prepared ΔygiM of K. pneumoniae ATCC43816, and found that the deletion of ygiM did not affect bacterial growth and mouse mortality in the mouse infection model. Interestingly, ΔygiM not only resulted in reduced bacterial resistance to macrophages, but also attenuated pathological manifestations in mouse organs. Furthermore, based on the data of Gene Expression Omnibus, the expression profiles of micro RNAs (miRNAs) and messenger RNAs (mRNAs) in the serum of 44 sepsis patients caused by K. pneumoniae infection were analyzed, and 11 differently expressed miRNAs and 8 DEmRNAs associated with the membrane function were found. Finally, the membrane-associated competing endogenous RNAs (ceRNAs) network was constructed. In this ceRNAs network, DEmiRNAs (hsa-miR-7108-5p, hsa-miR-6780a-5p, hsa-miR-6756-5p, hsa-miR-4433b-3p, hsa-miR-3652, hsa-miR-342-3p, hsa-miR-32-5p) and their potential downstream target DEmRNAs (VNN1, CEACAM8, PGLYRP1) were verified in the cell model infected by wild type and ΔygiM of K. pneumoniae, respectively. Taken together, YgiM may trigger the sepsis caused by K. pneumoniae via membrane-associated ceRNAs. This study provided new insights into the role of YgiM in the process of K. pneumoniae induced sepsis.
Introduction
Sepsis is defined as a life-threatening organ dysfunction caused by disorder host response to infection (Singer et al., 2016). In hospital mortality from sepsis has ranged from 25 to 80% over the past few decades (Angus and Wax 2001). The overall mortality rate of sepsis exceeds that of many common cancers and the delaying of appropriate antibiotic treatment for every 1 h will reduce the proportion of survival rate by about 7.6% in the shock of sepsis (Kumar et al., 2006). Due to the lack of understanding about sepsis, there is currently no effective way to mitigate the devastating effects of sepsis on the host (Seymour and Rosengart 2015). During sepsis infection, the immune response is critical for protecting the organism from infection and self-alteration. Peroxisome, a membrane-enclosed organelle associated with the lipid metabolism and ROS metabolism, has recently been recognized as a key regulator of immune function and inflammation during the development of infection, as signaling between cells and immune pathways and may influence the production of inflammatory regulators such as cytokines and antimicrobial peptides (Kumar, Kawalek, and van der Klei 2014; Deb and Nagotu 2017; Di Cara et al., 2017). In addition, peroxisomes can activate innate immunity by triggering phagocytosis, so impaired peroxidase function reduces the body’s resistance to microbial infection (Di Cara et al., 2017). It has been reported that disturbance of peroxisome biogenesis can lead to Zellweger syndrome, manifesting as severe fulminant sepsis in neonates, resulting in neonatal death (Lucaccioni et al., 2020).
MicroRNA (miRNA), a type of non-coding RNA, regulates gene expression by binding to downstream messenger RNA (mRNA) (Carthew and Sontheimer 2009). Recently, it was reported that miR-142-5p can induce an increase in the number of peroxisomes, which is the main source of intracellular ROS. So miR-142-5p can control ROS production by inducing the number of peroxisomes (Houri et al., 2020). MiRNA, as an immunomodulatory, has potentially important significance in sepsis. Funahashi et al. reported that in the spleen, induction of miR-146a expression can reduce multiple organ damage caused by excessive inflammation and sepsis (Funahashi et al., 2019). In the study of Chen et al., miR-133a could aggravate inflammatory responses in lung, liver, and kidney by targeting SIRT1 (Chen et al., 2020). Furthermore, Liu et al. reported that miR-452, induced via NF-ΚB in renal tubular cells in septic, was considered as an effective prognostic predictor for early detection of acute kidney injury in sepsis patients (Liu et al., 2020). Therefore, miRNAs play important roles in sepsis, and also can affect the function of peroxisomes.
Macrophages are professional innate immune cells, and able to engulf microorganisms and trigger immune responses leading to microbial death (Pidwill et al., 2020). Macrophages play indelible roles in sepsis caused by K. pneumoniae infection. For instance, macrophages are highly plastic enabling small-molecule mediators to respond to various triggers, such as inflammation, infection or tissue damage (Ofek et al., 1995; Wynn, Chawla, and Pollard 2013). The early stage of sepsis lead to the over activation of macrophages, which can lead to excessive production of proinflammatory cytokines and is considered to be one of the main reasons for the high early mortality in patients with sepsis (Wang and Deng 2008). It was reported that suppressing hyperactive macrophages can effectively interfere with the occurrence and progression of sepsis caused by bacterial infections (Chen Y. F. et al., 2019). Moreover, in the process of microbial infection, peroxisomes can assist the process of phagocytic cells by activating innate immune signals, thereby promoting survival in the face of microbial challenges (Di Cara et al., 2017). Peroxisomes specialize in several metabolic tasks, including beta-oxidation of very long fatty acids, branched-chain fatty acids, and some polyunsaturated fatty acids (Van Veldhoven 2010; Islinger et al., 2018). Studies have shown that peroxisomal beta-oxidation may be involved in fine-tuning the macrophage phenotype by affecting the dynamic lipid profile during macrophage polarization (Geric et al., 2018).
YgiM, an inner membrane protein, was firstly reported in E. coli, and it was confirmed to be anchored to peroxisomes in yeast and mammalian cells (Maddalo et al., 2011; Lutfullahoglu-Bal et al., 2018). Through homology alignment, we found a homologous gene vk055_4013 with 83% high similarity to ygiM in the genome of K. pneumoniae ATCC 43816. K. pneumoniae is one of the common causes of sepsis, especially in patients with low immunity (Angus and van der Poll 2013). However, the function of YgiM in K. pneumoniae is not clear. Based on the function of YgiM which is associated with the peroxisomes of host cells in E. coli and the key roles of peroxisomes in sepsis, it is suggested that YgiM may be involved in the pathogenesis of sepsis. In recent years, various types of databases and bioinformatics tools were used to study the potential roles of genes or proteins. For example, Yang et al. reported that the MASQC database can perform quality control of N6-methyladenine in eukaryotes and prokaryotes (Yang et al., 2020). Dai and Kong et al. established MTGIpick and 2SigFinder to explore how to reliably identify genomic islands from a single genome (Dai et al., 2018; Kong et al., 2020). In the study of Wang et al., a recursive feature selection with random forest was developed to provide convenient for protein structural class prediction (Wang et al., 2021). Yang et al. constructed HPVMD-C database to provide valuable resources for HPV vaccine research and cervical cancer treatment (Yang et al., 2022). In this study, we also investigated the potential roles of YgiM in sepsis caused by K. pneumoniae through using the data of Gene Expression Omnibus (GEO) to construct and validate the membrane-associated ceRNAs network of host macrophages.
Materials and methods
Plasmids, bacterial strains, primers and growth conditions
Strains and plasmids used in this study are listed in Supplementary Table S1. Primers used in this study were from Sangon Biotech (Shanghai, China) and are listed in Supplementary Tables S2, S3. Bacterial strains were grown in lysogeny broth (LB) medium (1 g yeast extract, 1 g tryptone, 0.5 g NaCl [pH 7.2 to ∼7.4] per 100 ml deionized water). Antibiotics were added at the following concentrations: 30 μg/ml apramycin, 100 μg/ml rifampicin, 100 μg/ml ampicillin for E. coli or K. pneumoniae strains. Unless otherwise noted, all bacteria were cultured at 180 rpm in LB medium at 37°C.
Construction of the ygiM deleted mutant
The ygiM mutant was constructed by using the CRISPR/Cas9 mediated genome-editing system (Wang et al., 2018). The pCas plasmid expresses the Cas9 protein and the λRed recombinant protein under the control of L-arabinose induction, and pSGKP plasmid expresses sgRNA. Furthermore, the pCas and pSGKP plasmids contained temperature sensitive replicon and sucrose-sensitive gene, respectively.
To generate the ygiM mutant, wild-type (WT) gene sequences were analyzed via an online web server (http://crispr.tefor.net/) to identify the appropriate 20-nt base-pairing regions (N20) of sgRNA. Only one of this N20 exists in the whole K. pneumoniae genome. In addition, another feature of the N20 sequence is that it is followed by an “NGG” sequence designed to target the guide RNA to the ygiM. Thereby attracting the Cas9 protein to act as molecular scissors to snip the ygiM in the genome. Then, the sgRNA fragment flanked by SpeI and XbaI restriction sites, along with the N20, was amplified with forward primer SpeI-ΔygiM-gRNA and reverse primer XbaI-gRNA by using the pSGKP-rif plasmid as the template. The PCR product was subsequently inserted into the XbaI and SpeI digested pSGKP-rif plasmid, generating the final pSGKP plasmids with targeted sgRNA (pSGKP-YgiM-N20). Finally, the constructed pSGKP plasmid and homology arms were co-transferred into K. pneumoniae WT strain containing the telegenic pCas-apr plasmid to generate the ygiM deletion. The cultures were plated on LB agar plates containing 30 μg/ml apramycin and 100 μg/ml rifampicin, and the mutants were confirmed in selected colonies by both PCR and Sanger sequencing. The plasmids of pSGKP and pCas-apr deletion were finally depleted by incubation on LB agar medium at 37° and 5% sucrose.
Complementation of the ygiM mutant
Plasmid pBAD24 was used to prepare the complementary strain of ΔygiM. It was generated by PCR amplification of the ygiM from the chromosome of K. pneumoniae WT strain and subsequent cloning into EcoRI-XbaI of the pBAD24 plasmid. The recombinant plasmid pBAD24-ygiM was transformed into E. coli DH5α and then primers YgiM-F and YgiM-R were used to confirm positive colonies on LB agar with 100 μg/ml ampicillin. The ygiM mutant was transformed with the pBAD24-ygiM plasmid and named as C-ygiM.
Bacterial growth curves
Growth curves of K. pneumoniae WT and ΔygiM were determined by subculturing in LB medium and growth. These strains were shaken overnight and then added to 100 ml fresh LB at a ratio of 1:1,000 the next day, shaken at 180 rpm at 37°C. The OD600 of bacteria was measured once every hour, then the bacteria in each time period were diluted ten-fold and the number of colonies (CFU/ml) was calculated after overnight incubation at 37°C.
Cell culture and K. pneumoniae infection in vitro
THP-1 human monocytes were seeded in 6-well tissue culture plates and grown in RPMI1640 containing 10% heat-inactivated fetal bovine serum (Biosharp, China) and treated with 10 ng/μl of phorbol 12-myristate 13-acetate (PMA) for 72 h to differentiate into macrophages. As for bacteria, K. pneumoniae was cultured to 108CFU in 10 ml of LB broth. Macrophages were infected with a MOI of 50 in a final volume of 2 ml RPMI 1640 tissue culture medium supplemented with 10% heat-inactivated FBS. After 2 h, cells were washed 3 times with PBS and then incubated with RPMI1640 containing 10% fetal bovine serum and gentamicin (200 μg/ml) for 1 h to remove extracellular bacteria. Studies have shown that the internalization of K. pneumoniae by THP-1 cells is most obvious at 24 h (Ares et al., 2016; Maisonneuve et al., 2017; Xu et al., 2021). In this study, 40 μg/ml gentamicin was used to maintain the killing concentration of extracellular bacteria to 24 h, and cells were lysed with 0.1% Triton X-100. Then, 10-fold serial dilutions were plated on LB agar plates and total CFU was determined. At the same time, to confirm that K. pneumoniae could be phagocytosed by macrophages, we also constructed a K. pneumoniae strain containing green fluorescent protein. Briefly, GFP+ plasmid was provided by Rao from Third Military Medical University (Rao et al., 2022). This GFP+ plasmid was introduced into K. pneumoniae ATCC43816. The macrophages were fixed using 4% formaldehyde and 0.1% Triton, permeabilized and stained with phalloidin-Alexa 594 (Beyotime, China) and DAPI (Beyotime, China) to visualize the actin filaments and the nuclei acid, respectively. The labelled cells were observed by fluorescence observation (Zeiss, Germany).
Mouse infection assay
To investigate whether ygiM affects bacterial virulence, bacteria were injected into the bloodstream of mouse model via tail vein injection. 6–8 weeks C57BL/6 mice were used with 10 mice in each group, and 106 CFU bacteria was injected into the tail vein, observed for 5 days, and the corresponding survival curve was calculated. The bacteria were shaken overnight, diluted at 1:100 the next day, cultured to 108 CFU/ml, serially diluted 10 times with phosphate buffered saline (PBS), and inoculated on LB agar with appropriate dilution. On the plate, the corresponding CFU number was calculated. In addition, C57BL/6 mice were divided into three groups, each group contained three mice, 48 h after the injection of 106 CFU colonies through the tail vein, the liver and lung tissues of the mice in each group were taken, embedded, stained with hematoxylin-eosin, and the tissues were obtained. Pathological examination to understand the pathological changes. All animal experiments were established by the Soochow University Animal Care and Utilization Committee and complied with all ethics and animal husbandry regulations.
Identification of DEmiRNAs and DEmRNAs by microarray data
The microarray data GSE174507 and GSE13904 were obtained from GEO (https://www.ncbi.nlm.nih.gov/geo/) which is an online public gene data repository for research. In total, the miRNA expression data obtained from GSE174507 included 12 peripheral blood samples from sepsis patients and six peripheral blood samples from control donors. The mRNA raw data obtained from GSE13904 included 32 whole blood samples from sepsis patients adopted at the first day and 18 whole blood samples from healthy individuals. The above raw microarray data was extracted from the GEO database. So, this study was not required any ethical review and informed consent because of the public availability of GEO data.
The DEmiRNAs and DEmRNAs were identified by the expression difference between the sepsis and healthy individuals’ samples of the microarray data. The p-value and the absolute log value of fold-change (log|FC|) were analysis in R language by the limma package. The log|FC|> 1.0 and p-value <0.05 were the selection criteria to define the differentially expressed genes.
Transcription factors and pathway enrichment analysis
Transcription factors, as the direct downstream targets of miRNA, are essential molecules in immunity to bacterial infection. Based on the DEmiRNAs identified, FunRich (Version 3.1.3) was used to analyze and visualize the differential expression transcription factors. FunRich is a stand-alone software tool used mainly for functional enrichment and interaction network analysis of genes and proteins.
Further, we used Gene Ontology (GO) annotation (http://www.geneontology.org) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway to determine the functions of the DEmRNAs. The clusterProfler package in R language was employed.
Construction of the membrane-related interaction network
In the results of the differential analysis of the high-throughput sequencing data information, the DEmiRNAs target genes were predicted by databases of miRDB, miRTarBase and TargetScan. In this study, we use the prediction of DEmiRNAs target genes to intersect with identified differentially expressed downstream mRNA to further screen the prediction results. And we used the prediction results to construct a ceRNA interaction network.
The exploration of protein interactions helps reveal the underlying pathological mechanism in children’s sepsis. In this study, we used the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) database (https://string-db.org/) to construct a PPI network. The clustered sub-networks and hub genes were identified by employing plugins of Cytoscape, Molecular Complex Detection (MCODE), and cytoHubba (version 3.8.0).
Membrane related genes were obtained from the results of GO annotation. The mRNAs annotated in membrane-related pathways were considered candidate mRNAs in the interaction subnetwork. Upstream miRNAs were predicted using the miRDB, miRTarBase, and TargetScan databases. The candidate miRNAs in the membrane-related interaction network were the intersecting mRNAs between the prediction results and the identified DEmiRNAs. The membrane-related interaction network was constructed based on the above results and the protein-protein interaction relationships between DEmRNAs. The results were visualized using Cytoscape (version 3.8.0).
Verification of DEmiRNAs and DEmRNAs expression
Specific validating primers for several differentially expressed RNAs (DERNAs) were designed based on the sequence of linear transcripts. Total RNA was extracted using TRIzol (Invitrogen United States) following the manufacturer’s instructions. The DERNAs complementary DNAs (cDNAs) were synthesized using miScript II RT Kit (QIAGEN, United States). The RT-qPCR was performed using SYBR Green qPCR Master Mix (QIAGEN, United States). The expression level of DEmiRNAs and DEmRNAs were normalized to the endogenous control of human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as the endogenous control. And the expression level of DEmiRNAs was normalized to the endogenous control of U6.
Reverse transcription quantitative polymerase chain reaction
The primers were designed and synthesized by Sangon Biotech (Shanghai, China). RNA template, Primer Mix, dNTP Mix, DTT, RT buffer, HiFi-MMLV, and RNA-free water were dissolved on ice. The reverse transcription system consisted of a volume of 20 μl. The reaction solution was used for fluorescence quantitative PCR according to the instructions of the SYBR® Premix Ex TaqTM II kit (Action-award Biotechnology, China). The 2–ΔΔCt method was used to express the expression relationships of multiple genes between the experimental group and control group. All of the qRT-PCR assays were repeated at least three times.
Statistical analysis
Sepsis children and healthy individuals were compared to evaluate the statistical significance between the two groups. All data analysis was performed using R software (version 4.0.1), FunRich (version 3.1.3), Cytoscape (version 3.8.0) and GraphPad Prism 8. The p value of <0.05 was considered statistically significant.
Results
Two plasmid system pCas-pSGKP for ygiM deleting
The pSGKP plasmid containing ygiM-N20 was confirmed by PCR (Figure 1A), and subsequently the PCR products were confirmed by DNA sequencing (Figure 1B). In addition, the pSGKP plasmid carried the rifampicin gene for screening. As shown in Figure 1C, the ygiM mutant was verified by PCR. The result of DNA sequencing showed that ygiM was successfully knocked out (Figure 1D).

FIGURE 1. Verification of ygiM deletion mutant by PCR and sequencing. (A) The pSGKP plasmid contains YgiM protein N20, and the target band is 220bp after PCR verification. (B) Sanger sequencing indicated that the pSGKP plasmid containing YgiM protein N20 was successfully constructed (red bases represent N20). (C) PCR confirmed that the ygiM mutant was constructed successfully. (D) Sanger sequencing verified that ygiM was successfully knocked out. (Red bases represent upstream of ygiM and blue bases represent downstream of ygiM).
Deletion of ygiM did not affect the bacterial growth
To investigate whether ygiM affects the growth or colony size of K. pneumoniae, we measured the OD600 and specific CFU/ml of WT and ΔygiM strains every hour. The growth curves of these two strains in LB broth for 24 h were similar (Figure 2A). The results showed that the colony morphology of the two strains on LB agar plates was also similar (Figure 2B).
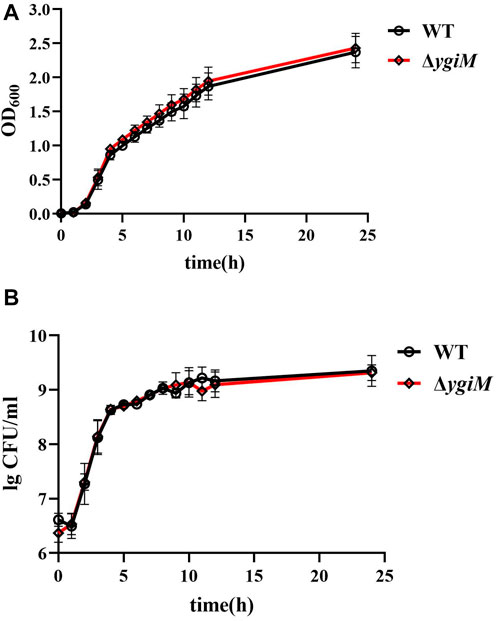
FIGURE 2. Growth curve and bacterial colony number of K. pneumoniae wild-type strain and ygiM mutant strains. (A) Comparison the growth curves of wild-type and ygiM mutant strains. The experiments were independently repeated three times. (B) Comparison the CFU/ml of wild-type and ygiM mutant strains. The experiments were independently repeated three times.
YgiM contributes to bacterial resist against phagocytosis by macrophages
To assess whether ygiM affects pathogenic processes caused by increased phagocytic sensitivity of macrophages, we investigated the antiphagocytic ability of WT and ΔygiM strains. As shown in Figure 3A, THP-1 cells were round with clear nuclei before induction by PMA. After 72 h induction by PMA, the protruding pseudopodia became irregular macrophages with blurred nuclei (Figure 3B). In the fluorescence experiment, we saw that the THP-1-derived macrophages phagocytosed K. pneumoniae (Figure 3C). Our results showed that more ΔygiM strains were recovered from THP-1-derived macrophages than WT at 24 h co-culture of cells and bacteria. It was suggested that ygiM may enhance the bacterial antiphagocytic ability to macrophages (Figure 3D).
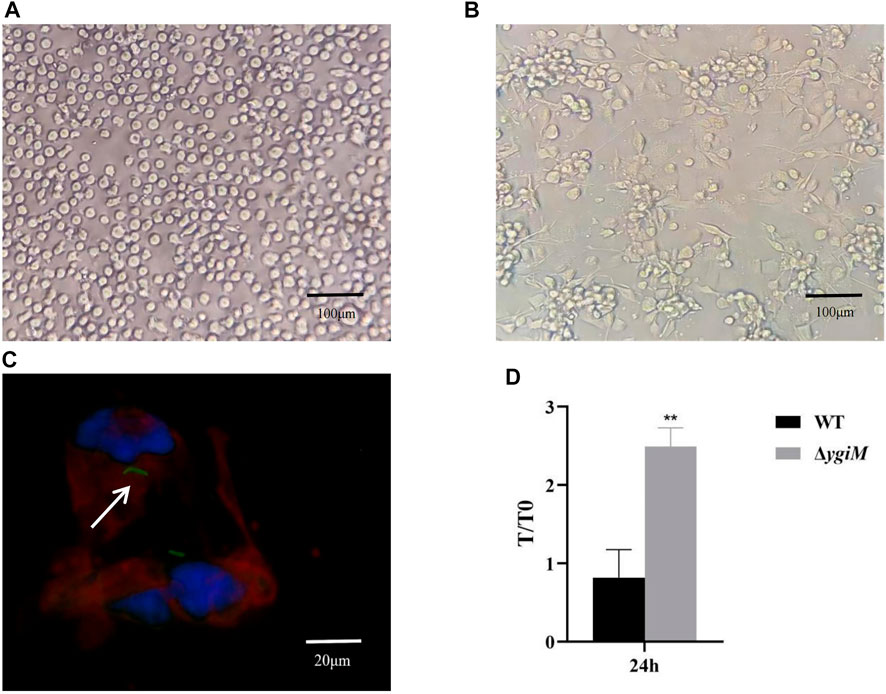
FIGURE 3. Determination of phagocytosis resistance of the wild type and the ygiM gene mutant strains. (A) THP-1 cells before induction (round, clear nuclei). (B) THP-1 cells are induced into macrophages by PMA (with pseudopodia, indistinct nuclei). (C) Fluorescence images of macrophages phagocytosing K. pneumoniae. The arrow indicates fluorescence labelled intracellular K. pneumoniae. (D) Plate to count macrophage phagocytosis. Data is displayed as intracellular bacterial counts of macrophages from three independent experiments (**: p < 0.01).
The potential roles of ygiM of K. pneumoniae in the formation of liver abscesses and pneumonia
To determine whether ygiM is involved in the virulence of K. pneumoniae, C57BL/6 mice were infected with 106 CFU of bacteria by tail vein injection. The number of dead mice was counted for five consecutive days (Figure 4A). The results showed that the survival rate of group infected with WT strains was 20% after infection for 72 h, and that of the group infected with ΔygiM was 10%. So there was no significant difference in the overall survival rate curve between these two groups. We then compared the pathological morphology of the liver and lung of mice infected with PBS, WT and ΔygiM groups. Compared with PBS treated mice, The mice infected by WT strain had damaged lung tissue and severe liver lobule congestion. However, the degree of lung tissue damage and hepatic lobular congestion in mice treated with ΔygiM strain was alleviated (Figure 4B).
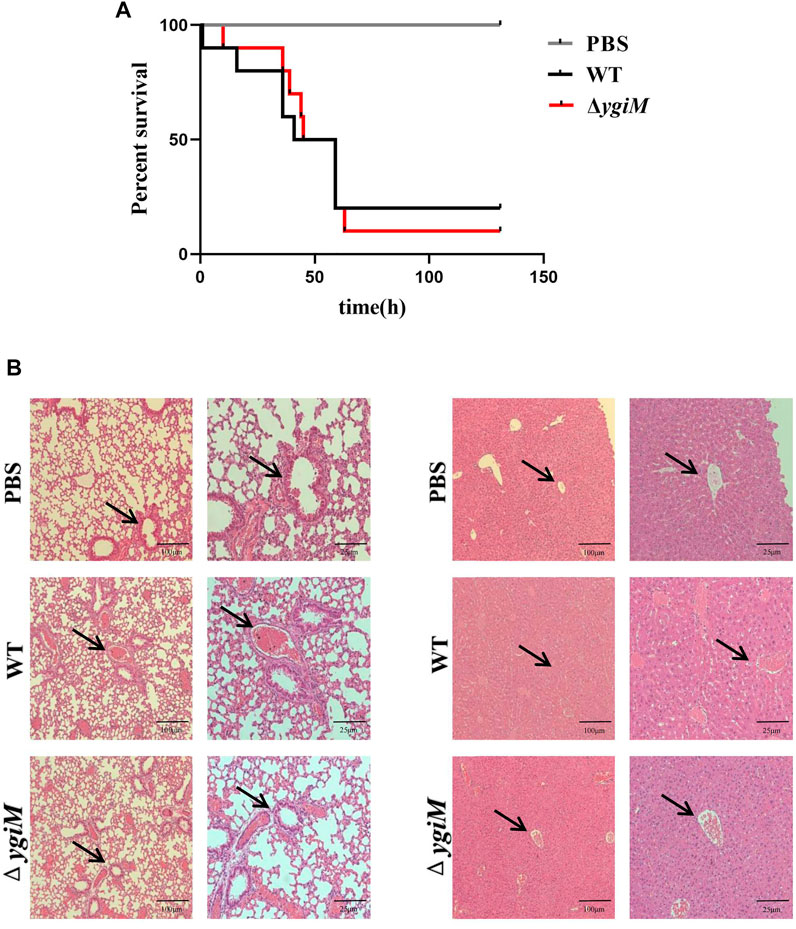
FIGURE 4. Survival curves and pathological morphology of mice. (A) Survival curves of mice in the tail vein infection model. Ten mice each of PBS, WT and ΔygiM were injected with 106 CFU through the tail vein, respectively. The 5 days survival rate of mice was determined. (B) Histopathology of mouse lung and liver after infection with PBS (negative control), WT and ΔygiM. 48 h after infection, the lungs and livers of different groups of mice were dissected and stained. These numbers represent at least three sections from three mice that exhibited similar pathology in their tissues. The arrows indicate the obvious pathological differences.
Identification of DEmiRNAs and DEmRNAs
The log|FC| > 1.0 and p < 0.05 were the selection criteria to define differentially expressed genes. In the miRNA dataset, 21 upregulated miRNAs and 17 downregulated miRNAs were identified (Supplementary Figures S1A,B). In the mRNA dataset, 55 upregulated mRNAs and four downregulated mRNA were identified (Supplementary Figures S2A,B).
Transcription factors and pathway enrichment analysis
We analyzed the differential expression of transcription factors by the 38 identified DEmiRNAs and identified 50 statistically significant transcription factors. HOXD8, EGR1, NKX6-1, LHX3, HOXB4, ONECUT1, SP1, HOXA5, FOXJ2 and MEF2A were the most significant molecules in sepsis (Figure 5A). Among these, HOXD8 was the most statistically significant transcription factor. And SP-1 was the transcription factor with the highest percentage.
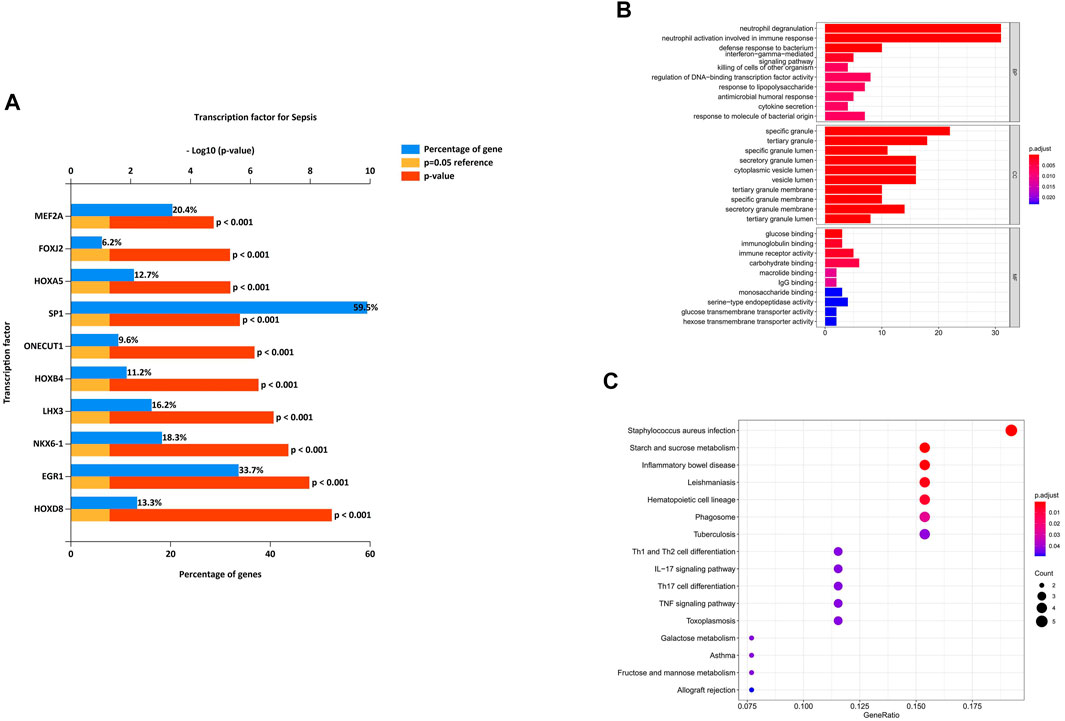
FIGURE 5. Characteristics and functional analysis of differentially expressed genes (DEGs). (A) Differentially expressed transcription factors associated with the identified DEmiRNAs. HOXD8, EGR1, NKX6-1, LHX3, HOXB4, ONECUT1, SP1, HOXA5, FOXJ2 and MEF2A were the most significant molecules in sepsis. (B) GO enrichment analysis of the DEmRNAs in biological process (BP), cellular component (CC) and molecular function (MF) subgroups. (C) KEGG enrichment analysis of DEmRNAs.
To better understand the potential functions of the identified DEmRNAs the initiation of sepsis, GO annotation and KEGG pathway analyses were performed. In GO annotation analysis, we found that the DEmRNAs were significantly enriched in the terms of neutrophil degranulation, neutrophil activation involved in immune response, and defense response to bacterium in the BP subgroup. Specific granule, tertiary granule, and specific granule lumen were the most significant GO terms in the CC subgroup. The top three GO processes included glucose binding, immunoglobulin binding, and immune receptor activity in the MF subgroup for DEmRNAs (Figure 5B).
In KEGG pathway enrichment analysis, starch and sucrose metabolism and inflammatory bowel disease were the most significant pathways enriched for DEmRNAs in the initiation of sepsis (Figure 5C).
Construction of the membrane-related interaction network
To better understand the endogenous regulatory mechanism of differentially expressed genes (DEGs) in the initiation of sepsis, we predicted the downstream target genes of DEmiRNAs by employing the miRDB, miRTarBase, and TargetScan databases. At the miRNA level, 25 miRNAs were predicted, and following a comparison with DEmRNAs, we confirmed 50 intersecting mRNAs in the ceRNA network (Figure 6A).
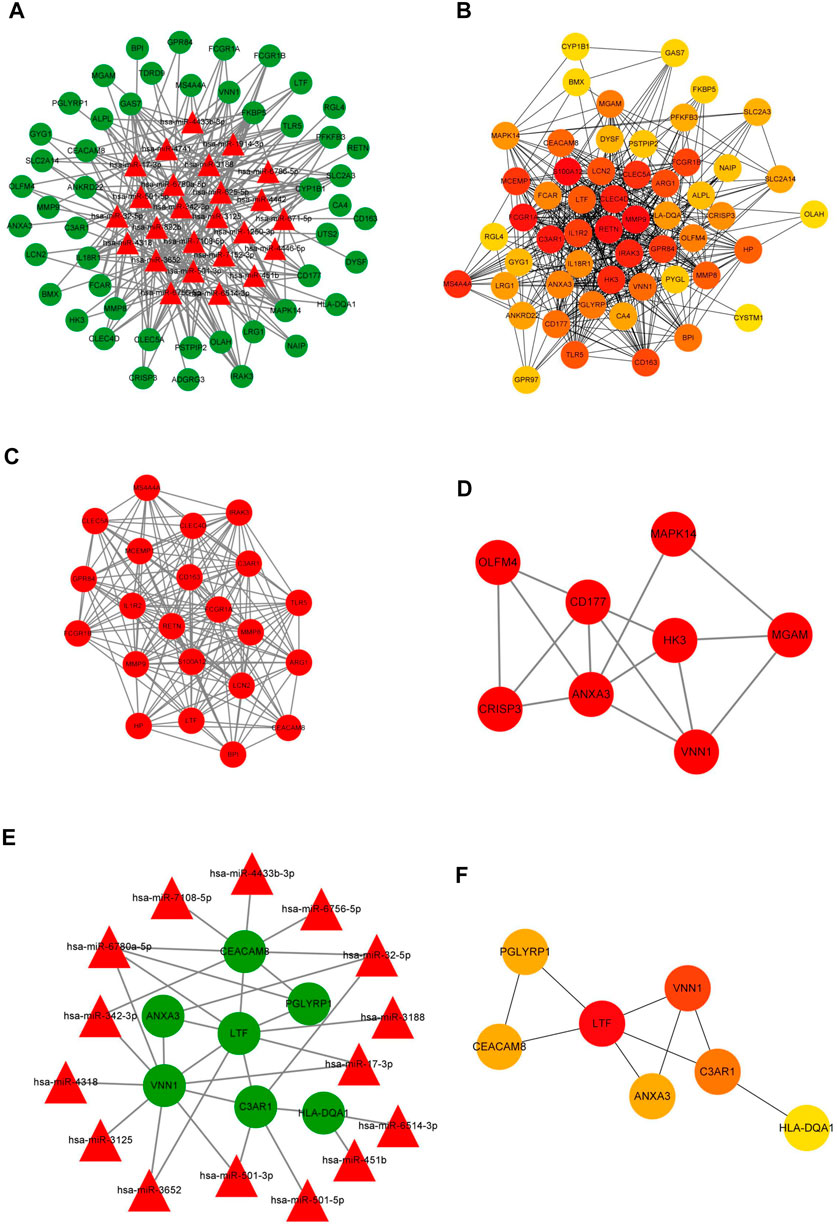
FIGURE 6. Construction of the membrane-related interaction network. (A) The competitive endogenous RNA (ceRNA) network of DEGs in the initiation of sepsis. In total, there were 25 miRNAs and 50 mRNAs in the ceRNA network. (B) The target genes of the identified DEmRNAs were ranked in the PPI network. The depth of red indicates the importance of genes in the network. (C) The most closely-clustered subnetwork was composed of 22 nodes and 178 edges. (D) The clustered subnetwork identified by the MCODE plug-in in Cytoscape had 8 nodes and 15 edges. (E) The membrane-related interaction network in the initiation of sepsis. Based on the results of the prediction of DEmiRNAs and PPI network, we constructed a autophagy interaction network including 15 miRNAs and seven mRNAs. (F) In the membrane-related interaction network, LTF, VNN1, and C3AR1 were the most important hub gene among the identified genes. The depth of red indicates the importance of genes in the network among mRNAs.
Further, we used the identified DEmRNAs to construct a PPI network, which included 54 nodes and 912 edges, under the condition that unconnected points were removed. Among the 54 genes, 40 genes had a score >100 analyzed by the maximal clique centrality (MCC) method in Cytohubba (Figure 6B). The top five hub genes were S100A12, MMP9, RETN, FCGR1A, and C3AR1. We also defined the most closely clustered sub-network by employing the MCODE plug-in in Cytoscape, which consisted of 22 nodes and 178 edges in which FCGR1A, S100A12, RETN were the most important genes (Figure 6C). In addition, we identified other clustered sub-network, with 8 nodes and 15 edges in which ANXA3, CD177, and VNN1 were considered as hub genes in this network (Figure 6D).
Due to the potential interesting roles of ygiM in sepsis, a membrane-related interaction network was constructed based on analysis of the ceRNA and PPI networks. Membrane-related genes were obtained from the results of GO annotation. Seven differentially expressed membrane-related genes were identified in which upstream target genes of these membrane-related genes were identified in sepsis patients at the initial stage. A total of 15 miRNAs and seven mRNAs were involved in network construction (Figure 6E). In the membrane-related interaction network, LTF, VNN1, and C3AR1 were the most important hub gene among the identified genes (Figure 6F).
Verification of autophagy-related genes by using THP-1-derived macrophages
According to the above search conditions, seven potential targets mRNAs of the above DEmiRNAs were found. Of which, three target mRNAs (VNN1, CEACAM8, and PGLYRP1) were shown to be associated with the ygiM. As shown in Figure 7A, compared with the WT group, VNN1 and CEACAM8 in ΔygiM group showed significant downregulation. However, the ΔygiM group of PGLYRP1 showed significant upregulation compared to the wild group. In addition, C3AR1, HLA-DQA1, ANXA3 and LTF had no differential expression in the comparison of the control and WT, the WT and ΔygiM group, ΔygiM and C-ygiM group respectively (Figure 7B).
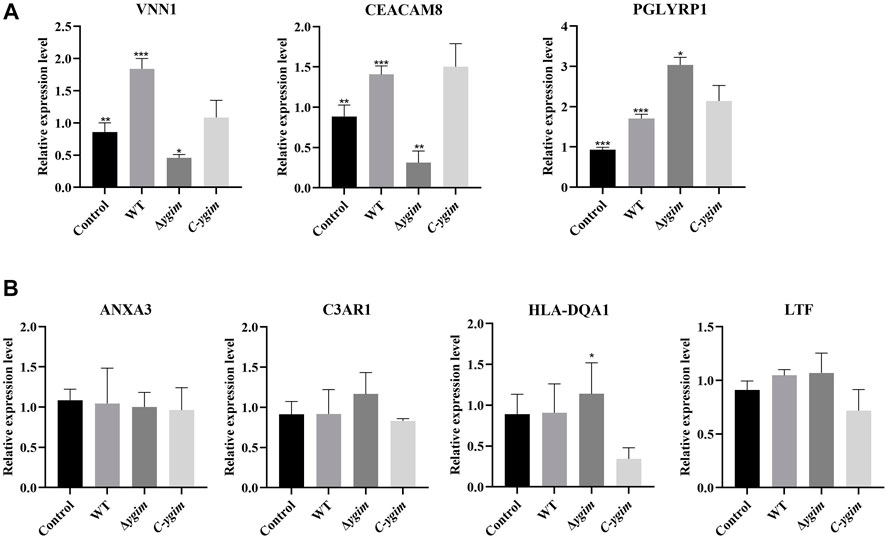
FIGURE 7. The relative expression of mRNA in K. pneumoniae wild-type, ΔygiM and C-ygiM strains. (A) Positive targets of mRNA: VNN1, CEACAM8, PGLYRP1 were considered to be statistically significant in the infection of K. pneumoniae control, wild type and ΔygiM groups (*: p < 0.05, **: p < 0.01, ***: p < 0.001). (B) Negative target of mRNA: C3AR1, HLA-DQA1, ANXA3, LTF were considered to be statistically insignificant in the infection of K. pneumoniae control, wild type and ΔygiM groups (*: p < 0.05).
Based on the results, the miRNAs with differentially expressed downstream mRNAs were further explored. Differential expression of seven miRNAs (hsa-miR-7108-5p, hsa-miR-6780a-5p, hsa-miR-6756-5p, hsa-miR-4433b-3p, hsa-miR-3652, hsa-miR-342-3p, hsa-miR-32-5p) were considered to be statistically significant in the infection of K. pneumoniae WT and ΔygiM (Figure 8A). In addition, hsa-miR-501-3p, hsa-miR-17-3p, hsa-miR-3125 and hsa-miR-4318 had little differential expression in the comparison of the control, WT and ΔygiM mutant groups (Figure 8B).
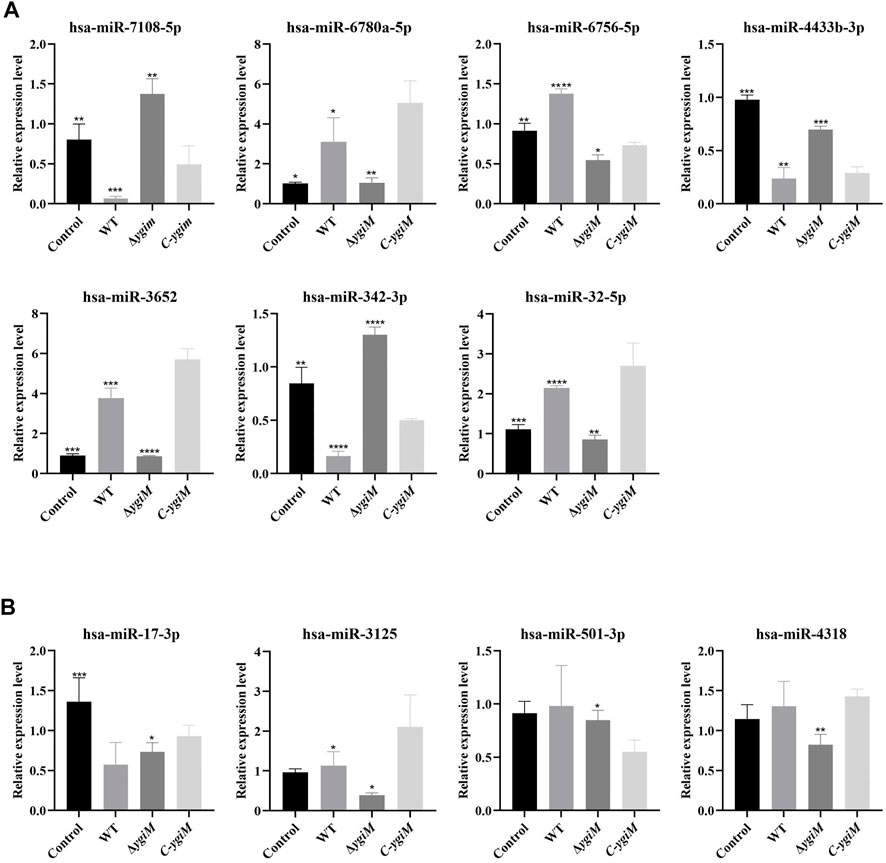
FIGURE 8. The relative expression of miRNA in K. pneumoniae wild-type, ΔygiM and C-ygiM strains. (A) Positive targets of miRNA: hsa-miR-7108-5p, hsa-miR-6780a-5p, hsa-miR-6756-5p, hsa-miR-4433b-3p, hsa-miR-3652, hsa-miR-342-3p and hsa-miR-32-5p were considered to be statistically significant in the infection of K. pneumoniae control, wild type and ΔygiM groups (*: p < 0.05, **: p < 0.01, ***, p < 0.001: ****: p < 0.0001). (B) Negative target of micRNA: hsa-miR-501-3p, hsa-miR-17-3p, hsa-miR-3125 and hsa-miR-4318 were considered to be statistically insignificant in the infection of K. pneumoniae control, wild type and ΔygiM groups (*: p < 0.05, **: p < 0.01, ***: p < 0.001).
In all DEmiRNAs and DEmRNAs, the target expression trend of the C-ygiM group was close to that of the WT group. After comparing the results of miRNAs and mRNAs, we identified hsa-miR-342-3p/VNN1, hsa-miR-7108-5p/CEACAM8, hsa-miR-4433b-3p/CEACAM8, and hsa-miR-342-3p/CEACAM8 as interactive pairs related ygiM that were statistically significant in sepsis.
Discussion
Sepsis can have serious consequences for the host. And can kill 20 to 50 percent of people with severe infections (Felbinger, Suchner, and Goetz 1999; Cohen et al., 2015). Peroxisomes play important roles in the occurrence and development of sepsis. Peroxisomes are multifunctional organelles that rely on reliable substrate and cofactor transport systems to complete various biological processes. Such as fatty acid oxidation (FAO) and hydrogen peroxide (H2O2) metabolism (Platta and Erdmann 2007; Vasko 2016). Also, sepsis leads to disruption of fatty acid oxidation, which is reflected in the downregulation of peroxisome proliferator-activated receptor alpha (PPARα) (Han et al., 2020). Thus peroxisome dysfunction leads to oxidative stress, which interferes with the pathological changes of sepsis. Studies have shown that K. pneumoniae is the major pathogen causing localized infections such as pneumonia and liver abscesses, and disseminated infections that can lead to severe sepsis (Chew, Lin, and Teo 2017). In K. pneumoniae, the inner membrane protein YgiM has been shown to play an important role by localizing peroxisomes in yeast and mammalian cells (Lutfullahoglu-Bal et al., 2018). The present study explored whether YgiM protein deficiency affects the process of K. pneumoniae interfering with the host, and revealed a membrane associated ceRNAs network of YgiM protein in K. pneumoniae induced sepsis. Most importantly, this study has opened up some new targets to a certain extent, which can be used as a reference for the treatment of sepsis.
Since K. pneumoniae has capsule, its resistance to macrophage phagocytosis is one of its virulence mechanisms. The presence of the capsule can act as a protective shield for bacterial antimicrobial peptides, inhibiting the early inflammatory response (Cortes et al., 2002; Al-Awad et al., 2020). Studies have shown that peroxisomes are required for the phagocytosis of bacteria by macrophages and that reduced peroxisome function impairs the response to bacterial challenges (Di Cara et al., 2017). Our results show that ΔygiM results in an enhanced ability of cells to phagocytose bacteria, and thus an enhanced ability of cells to clear bacteria. This further suggests that ygiM can attenuate the host’s attack against K. pneumoniae by targeting the host’s peroxisomes, thereby making it easier for the bacteria to survive and become pathogenic in the host, whereas ΔygiM makes it impossible for the bacteria to target the host’s peroxisomes, thereby increasing the ability of macrophages to clear bacteria. To further test this hypothesis, we established a mouse bloodstream infection model to explore whether the loss of ygiM affects the pathogenicity of bacteria to the host. The results showed that though ΔygiM did not affect the virulence of K. pneumoniae, attenuated congestion and inflammation in the liver and lungs of mice, suggesting that the presence of ygiM enhanced the pathogenicity of K. pneumoniae, but not virulence affecting the entire genome.
Several mRNA targets in sepsis were identified in this study. Based on the results of GO annotation, membrane-related genes were obtained. Further, the membrane-related interaction network was proposed from ceRNA network by the analysis of the miRDB, miRTarBase, and TargetScan databases. It is worth mentioning that eight mRNAs were emerged in the GO annotation in total. Because of the upstream miRNAs of HLA-DRB4 were not annotated, it was not shown in the membrane-related interaction network. But the interesting differential expression of HLA-DRB4 was revealed with ygiM (data show in Supplementary Figure S3). The role of HLA-DRB4 in sepsis also deserves further study in the future. Among the mRNAs related ygiM we verified, VNN1, CEACAM8, and PGLYRP1 were highlighted about sepsis firstly. Among the four mRNAs, VNN1 is an important PPARα target gene, and the production of VNN1 depends on PPARα activity (Rommelaere et al., 2013). PPARα (peroxisome proliferator-activated receptor α) is a ligand-activated transcription factor that plays a major role in different aspects of hepatic lipid metabolism (Kliewer et al., 1997). PPARα expression and activation protects the body from sepsis by promoting an appropriate metabolic response (Paumelle et al., 2019). Our results showed that VNN1 increased after K. pneumoniae infection compared with the control group, but VNN1 decreased to a certain extent due to ygiM deficient, suggesting that it may play a pro-inflammatory role in K. pneumoniae infection. CEACAM8, also known as plasma membrane carcinoembryonic antigen-related cell adhesion molecule, is only expressed in cells of the granulocyte lineage, especially neutrophils (Zhao et al., 2004; Ribon et al., 2019). Neutrophils are not only typical pro-inflammatory cells, but also have immunomodulatory properties, so CEACAM8 may be involved in innate immunity in sepsis (Ribon et al., 2019). Our results showed that WT CEACAM8 was elevated compared with controls, suggesting that K. pneumoniae can promote neutrophil degranulation, thereby promoting the occurrence and development of inflammation. However, the expression of CEACAM8 in the ygiM mutant was significantly decreased, even lower than that in the control group. This further suggests that deletion of the ygiM reduces the inflammatory response induced by K. pneumoniae. PGLYRPs (peptidoglycan recognition proteins) are members of the innate immune system (Yoshida, Kinoshita, and Ashida 1996). As one of the members, PGLYRP1 is mainly present in neutrophil granules, which can be highly bound to the bacterial cell wall, thereby activating the bacterial stress defense response, resulting in bacteria dying (Dziarski et al., 2003; Dziarski, Kashyap, and Gupta 2012). Compared with the K. pneumoniae WT, ygiM resulted in increased expression of PGLYRP1, suggesting that deletion of ygiM enhanced resistance to K. pneumoniae. It may indicate that this protein may provide ideas for the development of new antibacterial agents.
Furthermore, our study noticed that transcription factors play an important role in sepsis. Chen et al. found that EGR1 intervention in vivo reduced host proinflammatory cytokine secretion and rescued survival and tissue damage in a mouse sepsis model (Chen F. et al., 2019). Wu et al. reported that SP1 could be considered a target of miRNA-124-3p to alleviate myocardial injury in sepsis (Wu et al., 2022). And Zhang et al. found that the lncRNA MIR155HG regulates MEF2A to affect apoptosis and inflammation by sponging miR-194-5p in sepsis (Zhang et al., 2021). The above-mentioned molecules were all shown in our transcription factor analysis. And the potential of the results in our transcription factor analysis was highlighted. Studies on other transcription factors in the results deserve further research in the future. Among the miRNAs related ygiM we verified, hsa-miR-7108-5p, hsa-miR-6780a-5p, hsa-miR-6756-5p, hsa-miR-4433b-3p hsa-miR-3652, hsa-miR-32-5p, hsa-miR-342-3p were highlighted about sepsis firstly. As for hsa-miR-342-3p, Fu et al. reported that miR-342-3p regulates the immunity of Mycobacterium tuberculosis by increasing the production of inflammatory cytokines and chemokines (Fu et al., 2021). And our study deepened the understanding of miR-342-3p in infection. MiR-342-3p may have a wide range of abilities to regulate host immune function in intracellular bacterial infections. And ygiM could be considered a key bacterial target that activates host immune targets. In this study, hsa-miR-17-3p, hsa-miR-3652, and hsa-miR-32-5p were verified as ideal targets for the diagnosis of sepsis in the early stage. And the potential of these targets has also attracted the interest of other researchers. Tang et al. reported that miR-3652 is involved in immune regulation in periodontitis (Tang et al., 2021). The study by Li et al. found that miR-3652 was the shared genetic and epigenetic expression target between periodontitis and oral squamous cell carcinoma (Li et al., 2018). As for miRNA-32-5p, Zhang et al. reported that miRNA-32-5p regulates the survival and inflammatory response of mycobacteria in mycobacterium tuberculosis-infected macrophages by targeting FSTL1 (Zhang et al., 2017). Feng et al. reported that miR-32-5p is involved in intestinal epithelial cell apoptosis induced by the activation of transforming growth factor-β-activated kinase 1 (TAK1)-p38 in Helicobacter pylori infection (Feng et al., 2019).
In conclusion, we report the function and role of a novel gene ygiM in K. pneumoniae and construct a network of ceRNAs of ygiM. Several potential novel biomarkers were discovered to understand the pathogenesis of infection by ygiM mutant and WT strains. Furthermore, this study supports that hsa-miR-342-3p/VNN1, hsa-miR-7108-5p/CEACAM8, hsa-miR-4433b-3p/CEACAM8 and hsa-miR-342-3p/CEACAM8 as ygiM-specific interaction networks. Although ygiM did not affect the overall virulence of K. pneumoniae, it enhanced bacterial resistance to macrophages and attenuated organ pathology in mice. This undoubtedly provides a new perspective for future research on sepsis caused by K. pneumoniae.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The animal study was reviewed and approved by The Ethics Committee of Second Affiliated Hospital of Soochow University.
Author contributions
Conception and design: HZ, MH, ZC. Development of methodology: MH, ZC, PH, ZT. Acquisition of data: MH, ZC, PH. Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): ZC, MH, ZL, MW. Writing, review and/or revision of the manuscript: MH, ZC, HZ, HD. Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): MH, ZC, PH. Study supervision: HZ, HD.
Funding
This study was supported by the National Natural Science Foundation of China (82172332), Gusu health youth talent of Suzhou (GSWS2019039, GSWS2020030), the Science and Technology Program of Suzhou (SKY2021007), and Discipline Construction of The Second Affiliated Hospital of Soochow University (XKTJ-TD202001, SDFEYBS 1907).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.973145/full#supplementary-material
Supplementary Figure S1 | Characteristics and functional analysis of differentially expressed genes (DEGs). (A) Unsupervised clustering analysis of DE micro RNAs (miRNAs). Red dots indicate significantly upregulated miRNAs, green dots indicate significantly downregulated miRNAs. (B) Volcano plots of miRNAs. Red dots indicate upregulated DEmiRNAs, green dots indicate downregulated DEmiRNAs, black dots indicate non-differentially expressed miRNAs.
Supplementary Figure S2 | Characteristics and functional analysis of differentially expressed genes (DEGs). (A) Unsupervised clustering analysis of the DE messenger RNAs (mRNAs). Red dots indicate significantly upregulated mRNAs, green dots indicate significantly downregulated mRNAs. (B) Volcano plots of mRNAs. Red dots indicate upregulated DEmRNAs, green dots indicate downregulated DEmRNAs, black dots indicate non-differentially expressed mRNAs.
Supplementary Figure S3 | The HLA-DRB4 expression of mRNA in K.pneumoniae wild-type, ΔygiM and C-ygiM strains. Positive target of HLA-DRB4 was considered to be statistically significant in the infection of K. pneumoniae control, wild type and ΔygiM groups (*, p<0.05, **, p<0.01).
References
Al-Awad, D., Al-Emadi, N., Abu-Madi, M., Al-Thani, A. A., and Zughaier, S. M. (2020). The role of soluble uric acid in modulating autophagy flux and inflammasome activation during bacterial infection in macrophages. Biomedicines 8, E598. doi:10.3390/biomedicines8120598
Angus, D. C., and van der Poll, T. (2013). Severe sepsis and septic shock. N. Engl. J. Med. 369, 2063. doi:10.1056/NEJMc1312359
Angus, D. C., and Wax, R. S. (2001). Epidemiology of sepsis: An update. Crit. Care Med. 29, S109–S116. doi:10.1097/00003246-200107001-00035
Ares, M. A., Fernandez-Vazquez, J. L., Rosales-Reyes, R., Jarillo-Quijada, M. D., von Bargen, K., Torres, J., et al. (2016). H-NS nucleoid protein controls virulence features of Klebsiella pneumoniae by regulating the expression of type 3 pili and the capsule polysaccharide. Front. Cell. Infect. Microbiol. 6, 13. doi:10.3389/fcimb.2016.00013
Carthew, R. W., and Sontheimer, E. J. (2009). Origins and Mechanisms of miRNAs and siRNAs. Cell. 136, 642–655. doi:10.1016/j.cell.2009.01.035
Chen, F., Wang, Y., Zhang, W., Cai, Y., Zhao, T., Mai, H., et al. (2019). A functional polymorphism-mediated disruption of EGR1/ADAM10 pathway confers the risk of sepsis progression. mBio 10, e01663–19. doi:10.1128/mBio.01663-19
Chen, L., Xie, W., Wang, L., Zhang, X., Liu, E., and Kou, Q. (2020). MiRNA-133a aggravates inflammatory responses in sepsis by targeting SIRT1. Int. Immunopharmacol. 88, 106848. doi:10.1016/j.intimp.2020.106848
Chen, Y. F., Chen, G. Y., Chang, C. H., Su, Y. C., Chen, Y. C., Jiang, Y. S., et al. (2019). TRAIL encapsulated to polypeptide-crosslinked nanogel exhibits increased anti-inflammatory activities in Klebsiella pneumoniae-induced sepsis treatment. Mat. Sci. Eng. C Mat. Biol. Appl. 102, 85–95. doi:10.1016/j.msec.2019.04.023
Chew, K. L., Lin, R. T. P., and Teo, J. W. P. (2017). Klebsiella pneumoniae in Singapore: Hypervirulent infections and the carbapenemase threat. Front. Cell. Infect. Microbiol. 7, 515. doi:10.3389/fcimb.2017.00515
Cohen, J., Vincent, J. L., Adhikari, N. K., Machado, F. R., Angus, D. C., Calandra, T., et al. (2015). Sepsis: A roadmap for future research. Lancet. Infect. Dis. 15, 581–614. doi:10.1016/S1473-3099(15)70112-X
Cortes, G., Borrell, N., de Astorza, B., Gomez, C., Sauleda, J., and Alberti, S. (2002). Molecular analysis of the contribution of the capsular polysaccharide and the lipopolysaccharide O side chain to the virulence of Klebsiella pneumoniae in a murine model of pneumonia. Infect. Immun. 70, 2583–2590. doi:10.1128/iai.70.5.2583-2590.2002
Dai, Q., Bao, C., Hai, Y., Ma, S., Zhou, T., Wang, C., et al. (2018). MTGIpick allows robust identification of genomic islands from a single genome. Brief. Bioinform. 19, 361–373. doi:10.1093/bib/bbw118
Deb, R., and Nagotu, S. (2017). Versatility of peroxisomes: An evolving concept. Tissue Cell. 49, 209–226. doi:10.1016/j.tice.2017.03.002
Di Cara, F., Sheshachalam, A., Braverman, N. E., Rachubinski, R. A., and Simmonds, A. J. (2017). Peroxisome-Mediated metabolism is required for immune response to microbial infection. Immunity 47, 93–106. doi:10.1016/j.immuni.2017.06.016
Dziarski, R., Kashyap, D. R., and Gupta, D. (2012). Mammalian peptidoglycan recognition proteins kill bacteria by activating two-component systems and modulate microbiome and inflammation. Microb. Drug Resist. 18, 280–285. doi:10.1089/mdr.2012.0002
Dziarski, R., Platt, K. A., Gelius, E., Steiner, H., and Gupta, D. (2003). Defect in neutrophil killing and increased susceptibility to infection with nonpathogenic gram-positive bacteria in peptidoglycan recognition protein-S (PGRP-S)-deficient mice. Blood 102, 689–697. doi:10.1182/blood-2002-12-3853
Felbinger, T. W., Suchner, U., and Goetz, A. E. (1999). Treating patients with severe sepsis. N. Engl. J. Med. 341, 56–57.
Feng, J., Guo, J., Wang, J. P., and Chai, B. F. (2019). MiR-32-5p aggravates intestinal epithelial cell injury in pediatric enteritis induced by Helicobacter pylori. World J. Gastroenterol. 25, 6222–6237. doi:10.3748/wjg.v25.i41.6222
Fu, B., Lin, X., Tan, S., Zhang, R., Xue, W., Zhang, H., et al. (2021). MiR-342 controls Mycobacterium tuberculosis susceptibility by modulating inflammation and cell death. EMBO Rep. 22, e52252. doi:10.15252/embr.202052252
Funahashi, Y., Kato, N., Masuda, T., Nishio, F., Kitai, H., Ishimoto, T., et al. (2019). miR-146a targeted to splenic macrophages prevents sepsis-induced multiple organ injury. Lab. Invest. 99, 1130–1142. doi:10.1038/s41374-019-0190-4
Geric, I., Tyurina, Y. Y., Krysko, O., Krysko, D. V., De Schryver, E., Kagan, V. E., et al. (2018). Lipid homeostasis and inflammatory activation are disturbed in classically activated macrophages with peroxisomal beta-oxidation deficiency. Immunology 153, 342–356. doi:10.1111/imm.12844
Han, X., Chen, D., Liufu, N., Ji, F., Zeng, Q., Yao, W., et al. (2020). MG53 protects against sepsis-induced myocardial dysfunction by upregulating peroxisome proliferator-activated receptor-α. Oxid. Med. Cell. Longev. 2020, 7413693. doi:10.1155/2020/7413693
Houri, K., Mori, T., Onodera, Y., Tsujimoto, T., Takehara, T., Nakao, S., et al. (2020). miR-142 induces accumulation of reactive oxygen species (ROS) by inhibiting pexophagy in aged bone marrow mesenchymal stem cells. Sci. Rep. 10, 3735. doi:10.1038/s41598-020-60346-2
Islinger, M., Voelkl, A., Fahimi, H. D., and Schrader, M. (2018). The peroxisome: An update on mysteries 2.0. Histochem. Cell. Biol. 150, 443–471. doi:10.1007/s00418-018-1722-5
Kliewer, S. A., Sundseth, S. S., Jones, S. A., Brown, P. J., Wisely, G. B., Koble, C. S., et al. (1997). Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors alpha and gamma. Proc. Natl. Acad. Sci. U. S. A. 94, 4318–4323. doi:10.1073/pnas.94.9.4318
Kong, R., Xu, X., Liu, X., He, P., Zhang, M. Q., and Dai, Q. (2020). 2SigFinder: The combined use of small-scale and large-scale statistical testing for genomic island detection from a single genome. BMC Bioinforma. 21, 159. doi:10.1186/s12859-020-3501-2
Kumar, A., Roberts, D., Wood, K. E., Light, B., Parrillo, J. E., Sharma, S., et al. (2006). Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock. Crit. Care Med. 34, 1589–1596. doi:10.1097/01.CCM.0000217961.75225.E9
Kumar, S., Kawalek, A., and van der Klei, I. J. (2014). Peroxisomal quality control mechanisms. Curr. Opin. Microbiol. 22, 30–37. doi:10.1016/j.mib.2014.09.009
Li, S., Liu, X., Zhou, Y., Acharya, A., Savkovic, V., Xu, C., et al. (2018). Shared genetic and epigenetic mechanisms between chronic periodontitis and oral squamous cell carcinoma. Oral Oncol. 86, 216–224. doi:10.1016/j.oraloncology.2018.09.029
Liu, Z., Yang, D., Gao, J., Xiang, X., Hu, X., Li, S., et al. (2020). Discovery and validation of miR-452 as an effective biomarker for acute kidney injury in sepsis. Theranostics 10, 11963–11975. doi:10.7150/thno.50093
Lucaccioni, L., Righi, B., Cingolani, G. M., Lugli, L., Della Casa, E., Torcetta, F., et al. (2020). Overwhelming sepsis in a neonate affected by zellweger syndrome due to a compound heterozygosis in PEX 6 gene: A case report. BMC Med. Genet. 21, 229. doi:10.1186/s12881-020-01175-y
Lutfullahoglu-Bal, G., Seferoglu, A. B., Keskin, A., Akdogan, E., and Dunn, C. D. (2018). A bacteria-derived tail anchor localizes to peroxisomes in yeast and mammalian cells. Sci. Rep. 8, 16374. doi:10.1038/s41598-018-34646-7
Maddalo, G., Stenberg-Bruzell, F., Götzke, H., Stephen, T., Björkholm, P., Hanna, E., et al. (2011). Systematic analysis of native membrane protein complexes inEscherichia coli. J. Proteome Res. 10, 1848–1859. doi:10.1021/pr101105c
Maisonneuve, E., Cateau, E., Delouche, M., Quellard, N., and Rodier, M. H. (2017). An observational study of phagocytes and Klebsiella pneumoniae relationships: Different behaviors. Microbes Infect. 19, 259–266. doi:10.1016/j.micinf.2016.12.005
Ofek, I., Goldhar, J., Keisari, Y., and Sharon, N. (1995). Nonopsonic phagocytosis of microorganisms. Annu. Rev. Microbiol. 49, 239–276. doi:10.1146/annurev.mi.49.100195.001323
Paumelle, R., Haas, J. T., Hennuyer, N., Bauge, E., Deleye, Y., Mesotten, D., et al. (2019). Hepatic PPARα is critical in the metabolic adaptation to sepsis. J. Hepatol. 70, 963–973. doi:10.1016/j.jhep.2018.12.037
Pidwill, G. R., Gibson, J. F., Cole, J., Renshaw, S. A., and Foster, S. J. (2020). The role of macrophages in Staphylococcus aureus infection. Front. Immunol. 11, 620339. doi:10.3389/fimmu.2020.620339
Platta, H. W., and Erdmann, R. (2007). Peroxisomal dynamics. Trends Cell. Biol. 17, 474–484. doi:10.1016/j.tcb.2007.06.009
Ribon, M., Mussard, J., Semerano, L., Singer, B. B., and Decker, P. (2019). Extracellular chromatin triggers release of soluble CEACAM8 upon activation of neutrophils. Front. Immunol. 10, 1346. doi:10.3389/fimmu.2019.01346
Rao, Y., Peng, H., Shang, W., Hu, Z., Yang, Y., Tan, L., et al. (2022). A vancomycin resistance-associated WalK(S221P) mutation attenuates the virulence of vancomycin-intermediate Staphylococcus aureus. J. Adv. Res. 40, 167–178. doi:10.1016/j.jare.2021.11.015
Rommelaere, S., Millet, V., Gensollen, T., Bourges, C., Eeckhoute, J., Hennuyer, N., et al. (2013). PPARalpha regulates the production of serum Vanin-1 by liver. FEBS Lett. 587, 3742–3748. doi:10.1016/j.febslet.2013.09.046
Seymour, C. W., and Rosengart, M. R. (2015). Septic shock: Advances in diagnosis and treatment. JAMA 314, 708–717. doi:10.1001/jama.2015.7885
Singer, M., Deutschman, C. S., Seymour, C. W., Shankar-Hari, M., Annane, D., Bauer, M., et al. (2016). The Third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 315, 801–810. doi:10.1001/jama.2016.0287
Tang, H., Yuan, S., Chen, T., and Ji, P. (2021). Development of an immune-related lncRNA-miRNA-mRNA network based on competing endogenous RNA in periodontitis. J. Clin. Periodontol. 48, 1470–1479. doi:10.1111/jcpe.13537
Van Veldhoven, P. P. (2010). Biochemistry and genetics of inherited disorders of peroxisomal fatty acid metabolism. J. Lipid Res. 51, 2863–2895. doi:10.1194/jlr.R005959
Vasko, R. (2016). Peroxisomes and kidney injury. Antioxid. Redox Signal. 25, 217–231. doi:10.1089/ars.2016.6666
Wang, T. S., and Deng, J. C. (2008). Molecular and cellular aspects of sepsis-induced immunosuppression. J. Mol. Med. 86, 495–506. doi:10.1007/s00109-007-0300-4
Wang, Y., Wang, S., Chen, W., Song, L., Zhang, Y., Shen, Z., et al. (2018). CRISPR-Cas9 and CRISPR-assisted cytidine deaminase enable precise and efficient genome editing in Klebsiella pneumoniae. Appl. Environ. Microbiol. 84, e01834–18. doi:10.1128/AEM.01834-18
Wang, Y., Xu, Y., Yang, Z., Liu, X., and Dai, Q. (2021). Using recursive feature selection with random forest to improve protein structural class prediction for low-similarity sequences. Comput. Math. Methods Med. 2021, 5529389. doi:10.1155/2021/5529389
Wu, M., Huang, Z., Huang, W., Lin, M., Liu, W., Liu, K., et al. (2022). microRNA-124-3p attenuates myocardial injury in sepsis via modulating SP1/HDAC4/HIF-1α axis. Cell. Death Discov. 8, 40. doi:10.1038/s41420-021-00763-y
Wynn, T. A., Chawla, A., and Pollard, J. W. (2013). Macrophage biology in development, homeostasis and disease. Nature 496, 445–455. doi:10.1038/nature12034
Xu, L., Wang, M., Yuan, J., Wang, H., Li, M., Zhang, F., et al. (2021). The KbvR regulator contributes to capsule production, outer membrane protein biosynthesis, antiphagocytosis, and virulence in Klebsiella pneumoniae. Infect. Immun. 89, e00016–21. doi:10.1128/IAI.00016-21
Yang, S., Wang, Y., Chen, Y., and Dai, Q. (2020). Masqc: Next generation sequencing assists Third generation sequencing for quality control in N6-methyladenine DNA identification. Front. Genet. 11, 269. doi:10.3389/fgene.2020.00269
Yang, Z., Yi, W., Tao, J., Liu, X., Zhang, M. Q., Chen, G., et al. (2022). HPVMD-C: A disease-based mutation database of human papillomavirus in China. Database. 2022, baac018. doi:10.1093/database/baac018
Yoshida, H., Kinoshita, K., and Ashida, M. (1996). Purification of a peptidoglycan recognition protein from hemolymph of the silkworm, Bombyx mori. J. Biol. Chem. 271, 13854–13860. doi:10.1074/jbc.271.23.13854
Zhang, C., Li, J., Li, H., Wang, G., Wang, Q., Zhang, X., et al. (2021). lncRNA MIR155HG accelerates the progression of sepsis via upregulating MEF2A by sponging miR-194-5p. DNA Cell. Biol. 40, 811–820. doi:10.1089/dna.2021.0038
Zhang, Z. M., Zhang, A. R., Xu, M., Lou, J., and Qiu, W. Q. (2017). TLR-4/miRNA-32-5p/FSTL1 signaling regulates mycobacterial survival and inflammatory responses in Mycobacterium tuberculosis-infected macrophages. Exp. Cell. Res. 352, 313–321. doi:10.1016/j.yexcr.2017.02.025
Keywords: sepsis, ygiM, Klebsiella pneumoniae, peroxisome, miRNA, mRNA, ceRNAs network
Citation: Han M, Chen Z, He P, Li Z, Chen Q, Tong Z, Wang M, Du H and Zhang H (2022) YgiM may act as a trigger in the sepsis caused by Klebsiella pneumoniae through the membrane-associated ceRNA network. Front. Genet. 13:973145. doi: 10.3389/fgene.2022.973145
Received: 19 June 2022; Accepted: 07 September 2022;
Published: 23 September 2022.
Edited by:
Jade Ll Teng, The University of Hong Kong, Hong Kong SAR, ChinaReviewed by:
Qi Dai, Zhejiang Sci-Tech University, ChinaHwei Huih Lee, The University of Hong Kong, Hong Kong SAR, China
Copyright © 2022 Han, Chen, He, Li, Chen, Tong, Wang, Du and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haifang Zhang, haifangzhang@sina.com, haifangzhang@suda.edu.cn; Hong Du, hong_du@126.com
†These authors have contributed equally to this work