 Hui-O Chen1,2
Hui-O Chen1,2 Peng-Chan Lin
Peng-Chan Lin Chi-Shiang Wang
Chi-Shiang Wang- 1Department of Computer Science and Information Engineering, College of Electrical Engineering and Computer Science, National Cheng Kung University, Tainan, Taiwan
- 2Institute of Medical Informatics, National Cheng Kung University, Tainan, Taiwan
- 3Department of Oncology, National Cheng Kung University Hospital, College of Medicine, National Cheng Kung University, Tainan, Taiwan
- 4Department of Genomic Medicine, National Cheng Kung University Hospital, College of Medicine, National Cheng Kung University, Tainan, Taiwan
Developing a biomedical-explainable and validatable text mining pipeline can help in cancer gene panel discovery. We create a pipeline that can contextualize genes by using text-mined co-occurrence features. We apply Biomedical Natural Language Processing (BioNLP) techniques for literature mining in the cancer gene panel. A literature-derived 4,679 × 4,630 gene term-feature matrix was built. The EGFR L858R and T790M, and BRAF V600E genetic variants are important mutation term features in text mining and are frequently mutated in cancer. We validate the cancer gene panel by the mutational landscape of different cancer types. The cosine similarity of gene frequency between text mining and a statistical result from clinical sequencing data is 80.8%. In different machine learning models, the best accuracy for the prediction of two different gene panels, including MSK-IMPACT (Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets), and Oncomine cancer gene panel, is 0.959, and 0.989, respectively. The receiver operating characteristic (ROC) curve analysis confirmed that the neural net model has a better prediction performance (Area under the ROC curve (AUC) = 0.992). The use of text-mined co-occurrence features can contextualize each gene. We believe the approach is to evaluate several existing gene panels, and show that we can use part of the gene panel set to predict the remaining genes for cancer discovery.
Introduction
Scientific articles provide text mining (TM) applications in cancer biology (Zhu et al., 2013; Azam et al., 2019; Wang et al., 2020). Several solutions are currently available to meet the growing need for different cancer gene panels. Several commercial gene panels constitute a “one-size-fits-all” solution. In a clinical investigation, we need to design gene panels specifically tailored for particular questions or individual cancers (Hyman et al., 2015). The precision of the designed panel for different tumors plays an important role. They rely on literature reviews and cancer genomics databases. The reason for selecting somatic and germline mutation profiling is also complicated. Emerging TM techniques such as Gene2Vec offer some answers to information interpreting problems. Gene2Vec is a study that explored the idea of gene embedding, distributed representation of genes, in the spirit of word embedding (Demeester et al., 2016; Du et al., 2019). However, we cannot explain the biomedical meaning of the vector in the neural embedding model. The goal of explainability is very important and would be very useful. The ability to provide additional gene suggestions for a gene panel with an explanation would be hugely valuable but also really challenging. Therefore, we developed a biomedical-explainable and validatable text mining pipeline for cancer gene panel discovery.
Firstly, we find a system for predicting genes and interesting applications for a gene panel discovery. The use of text-mined co-occurrences features for each gene can contextualize each gene, and as input for a machine learning system. We extract NER names mentioned in the literature, such as gene NER (Leaman et al., 2013) and disease NER (Wei et al., 2013). The use of PubTator (Wang et al., 2016) along with MeSH (Ikonomakis et al., 2005) is a good way of getting as good enrichment for biomedical relevant terms. The frequency-inverse document frequency (TF-IDF) was used to construct the document-term matrix (Ikonomakis et al., 2005). Machine learning-based and biomedical-explainable approaches have recently become the most popular approaches in the study of the document-term matrix. For example, M. Ikonomakis et al. introduced several machine learning (ML) algorithms applied to text classification such as naïve-Bayes, decision trees, neural networks, nearest neighbors, and support vector machines (Devarajan et al., 2015). Wei Xu et al. proposed a novel document-clustering method based on non-negative matrix factorization (Choo et al., 2013). Choo et al. presented a user-driven topic modeling based on interactive non-negative matrix factorization capable of tuning the topic model result by integrating user interactions (Pedregosa et al., 2011). Summarizing the abovementioned studies, we established a fully integrated text mining pipeline to find the gene term-feature, mutational landscape heatmap, and cancer information topic.
With next-generation sequencing (NGS) technologies (Shabani Azim et al., 2018), many targeted panels have been developed to detect hereditary cancer and monitor somatic mutation changes in progressive cancer (McCabe et al., 2019). The Memorial Sloan Kettering Cancer Center has developed MSK-IMPACT (Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets), a hybridization capture-based next-generation sequencing assay for deep target sequencing of all exons and selected introns of 410 essential cancer genes in tumors (Hyman et al., 2015; Cheng et al., 2015). The MSK-IMPACT panel performed well not only in the above study but also in a large-scale clinical sequencing project with more than 10,000 patients (Zehir et al., 2017). They provided a comprehensive gene panel database including actionable drug targets, cancer susceptibility genes in hematological malignancies, and solid tumors. For solid tumors, the Oncomine Cancer Panel (OCP) is only used for the clinical screening of actionable genetic mutations in solid tumors (Luthra et al., 2017). They significantly provide druggable target databases. We validate the biomedical literature mining through the MSK-IMPACT or OCP cancer gene panel NGS database.
We create a pipeline that can suggest additional genes for a gene panel given an existing set of genes. And we believe the approach is to evaluate several existing gene panels, and show that we can use part of the gene panel set to predict the remaining genes.
Materials and Methods
PUBMED
PubMed, a free database of more than 30 million literature citations for biomedical literature, includes the fields of biomedicine and health. We extracted the abstracts that mentioned genes related to human cancer from PubMed and took the gene’s context by gene window.
Machine Learning Model and Analysis
K nearest neighbors, linear support vector machine (SVM), Gaussian process, decision tree, random forest, neural net, and naive Bayes were used to conduct supervised machine learning. All the models were built by python with the scikit-learn package and used five-fold cross-validation (Wei et al., 2015). The receiver operating characteristic (ROC) curve and the area under the ROC curve (AUC) were used to evaluate the model’s performance.
Biomedical Term Tagging
PubTator
PubTator (Wei et al., 2013) is a web-based PubMed abstract biomedical named entity recognition (NER) system. PubTator can tag the gene, disease, chemical, species, and mutation in PubMed abstracts, and the tagging result could be accessed via the RESTful API. We used PubTator as a part of the biomedical term tagger.
Medical Subject Heading
MeSH is a hierarchically organized medical vocabulary thesaurus used for indexing articles for PubMed. PubMed Articles curated by NLM are indexed with several related MeSH terms; every MeSH term has unique id and hierarchical categories. With these characteristics of MeSH term and our tagging algorithm, we could tag biomedical terms that are not tagged by PubTator. Our algorithm started from the MeSH terms of each PubMed article. For each MeSH term in an article, we first created a MeSH term-mapping set that mapped a MeSH term to another set that contained itself and its lower hierarchy MeSH term. Second, for each MeSH term in the MeSH term-mapping set, we tried matching all of the entry terms, synonyms of a specific MeSH term, to every word in the article. If a word in the article matched any entry names of a MeSH term, we tagged that word as a biomedical term. This way, those terms having the same concepts could be merged and analyzed.
Gene Term-Feature Term Frequency–Inverse Document Frequency Matrix Construction
For a particular gene, considering all of its gene windows in the whole corpus, we calculated the frequency of the co-occurrence of the gene and features (terms) tagged by our algorithm in the window as the term frequency of the feature. The higher the term frequency is, the stronger the association of the gene and feature. In our study, term frequency (TF) was calculated using the following formula:
To calculate the inverse document frequency of each term feature, we simply count the occurrences of the term feature in all genes as document frequency. The inverse document frequency (IDF) was calculated using the following formula:
The higher the IDF, the more specific the term feature is to a particular gene. Finally, by multiplying TF and IDF, the gene term -feature matrix was constructed.
Term Feature Selection by the Hypergeometric Test
We filtered out genes that had less than ten term features. We identified the critical term feature according to the gene panel using the p-values of hypergeometric tests as follows. We input the MSK-IMPACT (Hyman et al., 2015) panel. Ns is the size of the MSK-IMPACT panel set S,
From
The p-value reflects significant phrases in
Topic Modeling
Our topic modeling was based on the algorithms of non-negative matrix factorization (NMF) (Yeganova et al., 2014). Given a nonnegative matrix
The objective function is shown as the following formula:
The function is the most commonly used formulation based on the Frobenius norm. K represents the number of topics we expected,
Gene Window
We take the gene’s context as its gene window. Each gene window contains three sentences. The sentence contains the gene, the previous sentence, and the next sentence. We want to eliminate the redundant part. Using the gene window algorithm, we could iterate through the full abstracts containing specific genes in the text and grip the most critical section for further analysis. We pick three sentences based on the concept that the sentence that is closer to the gene is more relevant to it. Since the closest ones are previous and the next one, so we picked three.
Results
Study Design and Workflow
This study develops a gene panel analysis framework that can discover the characteristics of a gene panel based on biomedical literature mining. The method overview is shown in Figure 1. First, we extracted the PubMed abstracts, which mentioned genes related to humans. The method is shown as Figure 2. In this step, approximately 430,000 PubMed abstracts regarding genes were filtered out from all of the current PubMed corpus (approximately 30 million articles). Second, we performed biomedical named entity recognition (NER) on the extracted PubMed abstracts using PubTator (Wang et al., 2016) and MeSH (Medical Subject Headings). Third, we used the biomedical term to construct the gene term-feature matrix, which has a concept similar to that of the document-term matrix. Fourth, we performed term feature selection according to individual gene panels to make the term feature generated by the previous step stronger and correspond to the target gene panel.
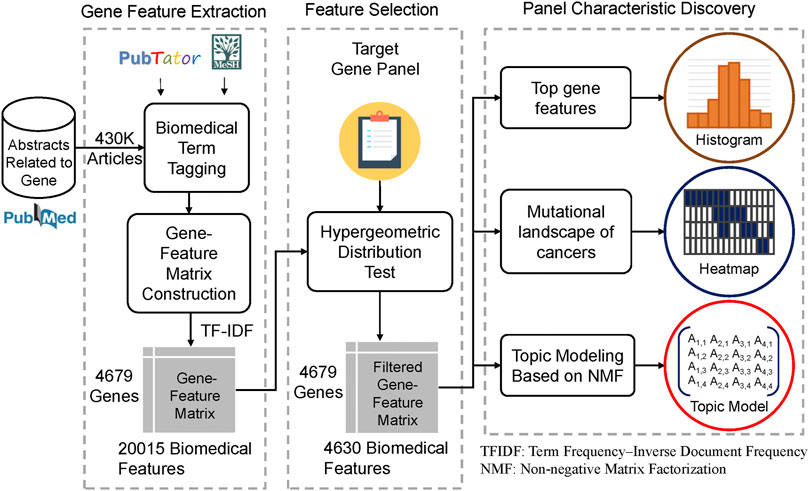
FIGURE 1. Study design and workflow The flowchart shows the overall analysis framework of this study. We first extracted 430,000 abstracts that mentioned genes related to humans in the PubMed corpus. Second, biomedical named entity recognition (NER) was performed to obtain biomedical terms, such as gene name, disease name, and drug name, using PubTator and MeSH. Third, we used the biomedical term tagged by the previous step to construct the gene term-feature matrix whose concept was similar to the document-term matrix. Fourth, we performed term feature selection according to a particular gene panel. We took the MSK-IMPACT panel as an example and made the term features generated by the previous step correspond more to the target gene panel using the hypergeometric distribution. Finally, several analyses, including identifying the top gene term features, creating the mutational landscape of cancers, and topic modeling based on nonnegative matrix factorization, were conducted to determine and interpret the biomedical characteristics of the target gene panel.

FIGURE 2. An example displays how the term “lung cancer”, being tagged in MeSH hierarchical structure. The way “lung cancer” being tagged is as follows. First, we iterate through the MeSH terms of the index of PMID: 27823967 and found “Lung Neoplasms” was one of the MeSH terms, which its synonyms contain “Lung Cancer.” Second, if the term “Lung Cancer” also appeared in the article, the MeSH tagging algorithm would tag this word and take its MeSH ID for further analysis.
Here, we explored the idea of the hypergeometric distribution. For each term feature, by comparing the distribution of occurrences in the target gene set and the whole gene set, the term features that correlated more with the target gene panel would be enriched. This approach is flexible in regard to different target gene sets, such as the Oncomine Cancer Panel or cardiovascular gene panels. Finally, we filtered out 4,630 term features from 20,015 term features. The filtered gene term-feature matrix, whose size is 4,679 (genes) x 4,630 (term features), will be used in the following analysis. Thus, we can discover the top 20 gene term features, the mutational landscape of the cancer genome, and topic modeling of cancer information. In this way, we can find the potential characteristics of the gene panel.
Biomedical Term Extraction by Hypergeometric Test
In the field of biomedical literature mining, tagging the biomedical term is an important issue. For an abstract of the biomedical literature, only biomedical words are what we are interested in, such as drug name, disease name, or gene name. PubTator was capable of tagging the gene, disease, chemical, species, and mutation in PubMed abstracts. Figure 3A shows the term feature extraction result of an EGFR-related abstract compared to the term features extracted by raw text TF-IDF scoring without biomedical term tagging. The biomedical term features were filled with redundant words, such as “with”, “for”, and “after”.
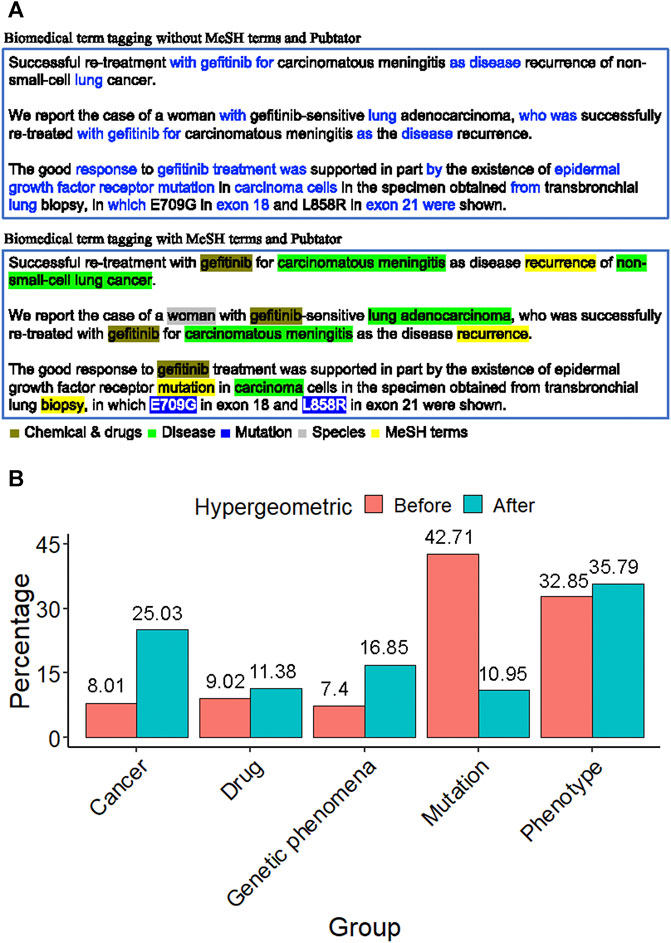
FIGURE 3. Biomedical term extraction (A) The term feature of an EGFR-related abstract. The former was filled with many redundant words, such as with, for, and after. The latter contains lots of biologically meaningful terms, such as gefitinib (chemical), non-small cell lung cancer (disease), L858R (mutation), the woman (species), and recurrence (MeSH). This phenomenon shows that the tagging approach with MeSH and PubTator terms is essential to gene term-feature extraction. (B) The proportion distribution bar chart of the MSK-IMPACT panel in each term feature group before and after the hypergeometric distribution test. It shows that after term feature selection, the proportion of the term feature groups of interest increases, such as cancer, drug, genetic phenomena, and phenotype.
On the other hand, the term feature extraction approach with MeSH terms and PubTator resulted in term features that contained lots of biologically meaningful terms, such as gefitinib (chemical), non-small cell lung cancer (disease), L858R (mutation), the woman (species), and recurrence (MeSH). This phenomenon shows that the tagging approach is essential for gene term feature extraction.
To discover the characteristics of a gene panel, we used the hypergeometric distribution test. According to MeSH terms and PubTator categories, all the term features can be divided into five groups: cancer, drug, genetic phenomena, mutation, and phenotype (Supplementary Table S1). Take the MSK-IMPACT panel as a target gene panel, for example. The distribution of the MSK-IMPACT panel shows that the percentage increases in some term feature groups after using the hypergeometric distribution test (Figure 3B). We filtered out the unimportant genes and found the critical term features according to the gene panel using a hypergeometric distribution test. The proportion of term feature groups in our interest increases, such as cancer, drug, genetic phenomena, and phenotype. The percentage after using the hypergeometric distribution test showed a noticeable improvement from 8.01 to 25.03% in the cancer group. The proportion increased from 9.02 to 11.38% in the drug group and grew from 7.4 to 16.85% in the genetic phenomenon group. There was a slight increase from 32.85 to 35.79% in the phenotype group. After the term feature selection, the proportion decreased from 42.71 to 10.95% in the mutation group. The MSK-IMPACT panel stands for integrated mutation profiling of actionable cancer targets, so the percentage in these groups increases after the hypergeometric distribution test.
3.3 Literature-derived Gene Term Features
The biomedical term features extracted from the literature were directly or indirectly related to each gene. Here, we took some cancer-related genes as examples for further demonstration. Figures 4A,B show the top twenty biomedical term features with the highest TF-IDF scores for EGFR (the score range from 8.02 to 13.49) and BRAF (the score range from 5.35 to 18.66). For EGFR, which has been recognized for its importance in lung cancer (Paez et al., 2004; Shepherd et al., 2005), most of the term features directly represent lung cancer or its subtypes, such as “Adenocarcinoma of the lung,” “Carcinoma, small cell,” and “Carcinoma, Non-Small Cell Lung.” “T790M” is a drug resistance mutation frequently observed in patients with lung cancer (Zhou et al., 2009). “Erlotinib” is an effective tyrosine kinase inhibitor (TKI) targeting EGFR for non-small cell lung carcinoma (NSCLC). “Lapatinib” is a dual EGFR/ERBB2 TKI for metastatic breast cancer (Burris, 2004). Some term features were indirectly relevant to EGFR, such as “Platinum” and “cisplatin,” which are both standard chemotherapy in NSCLC (Arriagada et al., 2004). EGFR TKIs are commonly compared with conventional platinum-based therapies. Another example is BRAF, whose mutations are widely detected in melanoma, thyroid cancer, and colorectal cancer (Chapman et al., 2011). “V600E” is a crucial mutation that causes the constitutive activation of the cellular signaling pathway (Chapman et al., 2011). “Vemurafenib” and “dabrafenib” are competitive inhibitors designed for BRAF with the V600E mutation (Hauschild et al., 2012). The other examples, such as BRCA1, BRCA2, MLH1, and ERBB2, are shown in Supplementary Figure S1. Nearly all of the biomedical term features relevant to these genes were consistent with current knowledge.

FIGURE 4. Top term features in EGFR and BRAF genes (A) The bar chart shows the TF-IDF scores of term features related to EGFR. Most of the identified term features for EGFR were associated with syndromes (e.g., lung adenocarcinoma and non-small cell lung carcinoma), mutations (e.g., T790M), and therapies (e.g., erlotinib and lapatinib) for lung cancer. (B) The bar chart shows the TF-IDF scores of term features related to BRAF. Biomedical term features, including cancer types (e.g., melanoma and thyroid cancer), mutations (e.g., V600E), and inhibitors (e.g., vemurafenib and dabrafenib) for BRAF, were consistent with known findings.
Mutational Landscape of the Actionable Cancer Genome From Biomedical Literature Mining Validated by NGS Database
We constructed the gene-cancer association matrix from the filtered gene term -feature matrix to understand the associations between cancer types and gene mutations. The recurrent common cancer-associated genes are shown in Figure 5A. The most common cancer-associated genes were TP53, EGFR, CTNNB1, NOTCH1, and PTEN, as shown in Figure 5B. Using two genes, EGFR and BRAF, as examples, we found that EGFR L858R and T790M and BRAF V600E were important mutation term features in text mining and were frequently mutated in MSK samples (Figure 5C). The cosine similarity of gene frequency between text mining and a statistical result from clinical sequencing data (Demeester et al., 2016) is 80.8% (Figure 5B). To understand the time series of the association between gene mutations and cancer types in the last decade, we constructed the gene-cancer TF-IDF matrixes of the years from 2011 to 2015 and the years from 2016 to 2019. As shown in Supplementary Figure S2A and S2B, we found that cancer immunotherapy was a major issue in the past 5 years. The rank of CD274 was increased, and CTLA4 first appeared (Seidel et al., 2018). In addition, the TF-IDF value of BRAF mutation in colorectal cancers increased because of the better outcomes of the BRAF-mutant CRC tumors with microsatellite instability (MSI) in immunotherapy (Rosenbaum et al., 2016). The results indicate that we can design a series of cancer gene panels by updating the literature mining time frame.
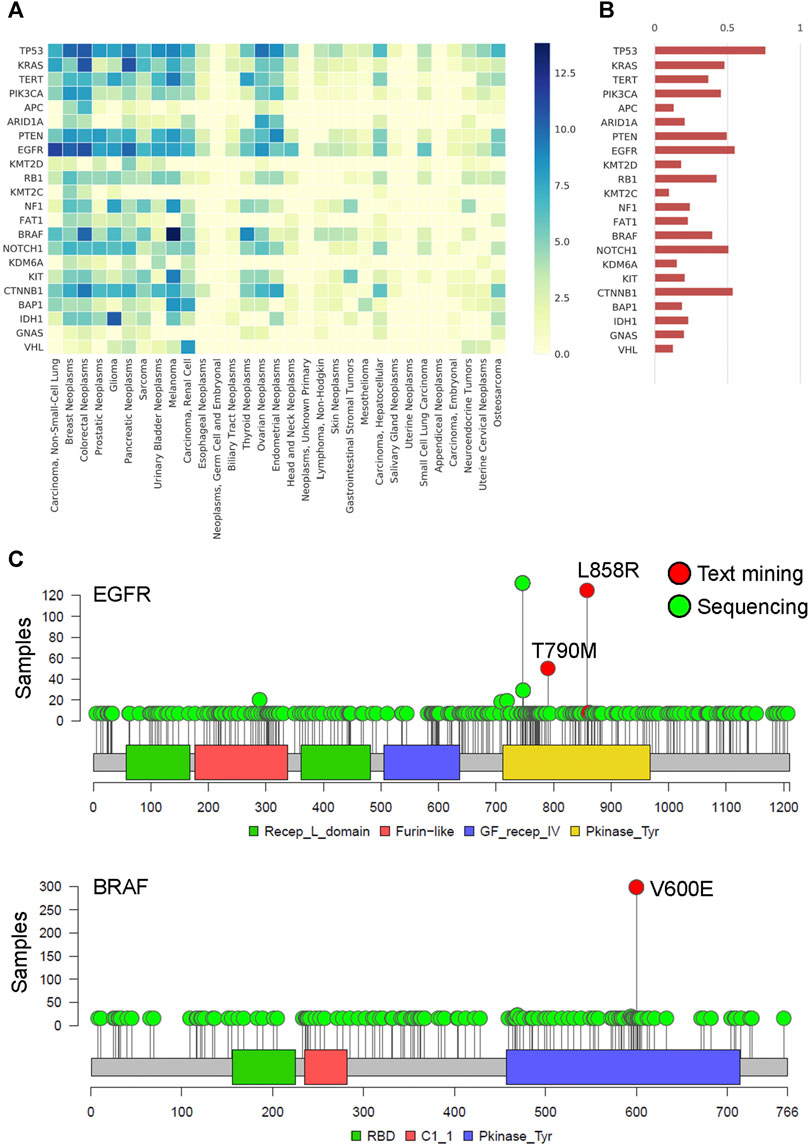
FIGURE 5. The spectrum and frequency of actionable genetic mutation by literature mining (A) Heatmap of cancer genomics by the TF-IDF matrix. The X-axis represents the 31 common cancer types, and the y-axis represents the recurrent somatic genes. The darker color indicates a higher association between genes and cancer. (B) The bar plot shows the gene frequency within all of the cancer types. The data is validated by the MSK-IMPACT Clinical Sequencing Cohort, which is targeted sequencing of 10,000 clinical cases using the MSK-IMPACT assay. The cosine similarity of gene frequency between text mining and a statistical result from clinical sequencing data is 80.8%. (C) Lollipop plot of EGFR and BRAF in the MSK-IMPACT pan-cancer cohort. The critical gene mutation term features found by text mining are shown and labeled in red. Other gene mutations are labeled in green.
Gene Panel Prediction by Machine Learning Models
Seven machine learning prediction models, including nearest neighbors, linear support vector machine (SVM), Gaussian process, decision tree, random forest, neural net, and Naive Bayes (Wei et al., 2015), were used to verify the specific gene panel (Figure 6A). The MSK-IMPACT, Oncomine Comprehensive Assay (Rhodes et al., 2007), and cardiovascular gene panels (Paige et al., 2018) represent different gene characteristics. There are 410 essential cancer genes in the MSK-IMPACT panel. The Oncomine Comprehensive Assay includes 161 cancer-related genes. We used the congenital heart defect focus panel of 115 genes associated with congenital heart defects (CHDs) as the cardiovascular gene panels.
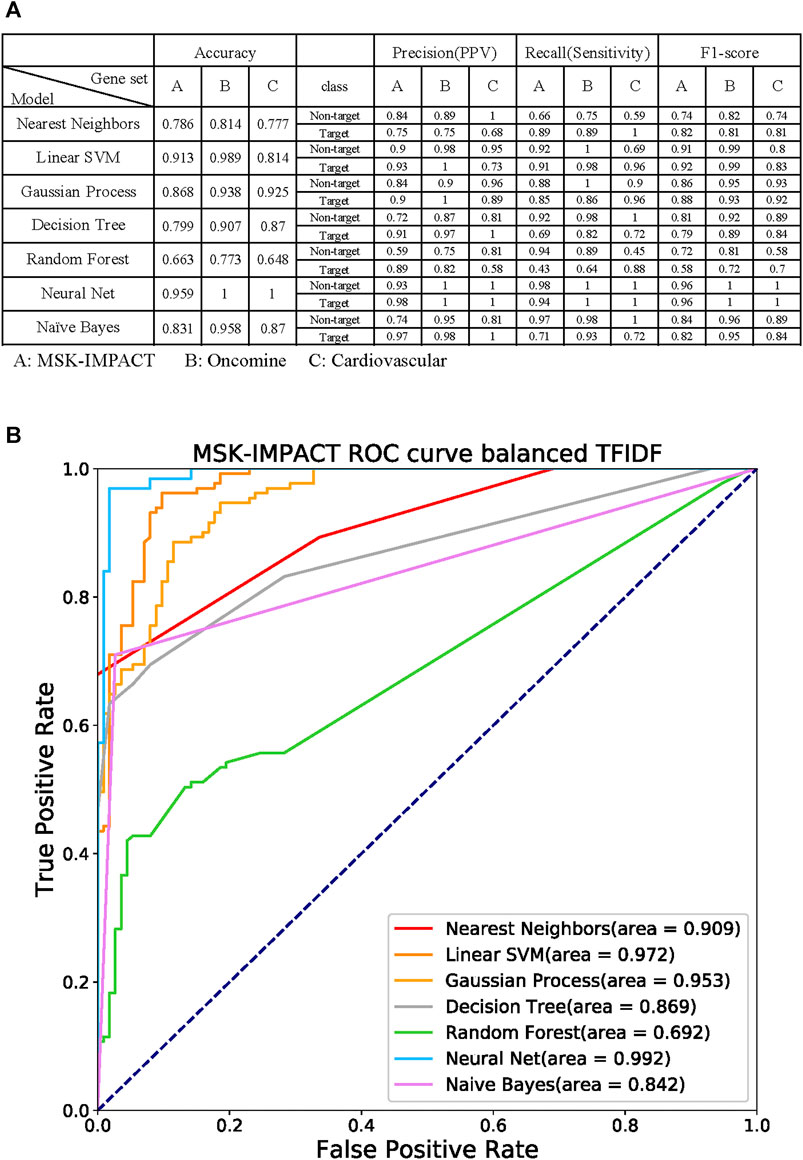
FIGURE 6. Performance of the machine learning models with the gene panel (A) Evaluation of the overall accuracy, precision (positive predictive value, PPV), recall (sensitivity), and F1-score of every prediction model. Each gene could be labeled a target or non-target, indicating whether the gene is in the given target panel. The following seven prediction models were used: nearest neighbors, linear support vector machine (SVM), Gaussian process, decision tree, random forest, neural net, and Naive Bayes. The target gene panels were MSK-IMPACT, Oncomine Comprehensive Assay, and cardiovascular gene panels. (B) Receiver operating characteristic (ROC) curves of the models with the MSK-IMPACT 410-cancer gene panel. The neural net model had the highest area under the ROC curve (AUC), which was 0.992.
Each gene can be labeled as a target or non-target, which indicates whether the gene is in the given target panel. We performed five-fold cross-validation on our dataset to evaluate the models’ efficiency and evaluate the overall accuracy of each prediction model. We measured the target and non-target genes in each prediction model separately with precision (positive predictive value, PPV), recall (sensitivity), and F1-score. The accuracies for nearest neighbors, linear SVM, Gaussian process, decision tree, random forest, neural net, and naive Bayes in the MSK-IMPACT panel were 0.786, 0.913, 0.868, 0.799, 0.663, 0.959, and 0.831, respectively; the accuracies for all models in the OCP gene panel were 0.814, 0.989, 0.938, 0.907, 0.773, 1 and 0.958; and the accuracies for all the models in the cardiovascular gene panel were 0.777, 0.814, 0.925, 0.87, 0.648, 1, and 0.87. The receiver operating characteristic (ROC) curve analysis confirmed that the neural net model had a better prediction performance; the area under the ROC curve (AUC) was 0.992 (Figure 6B). The AUCs of nearest neighbors, linear SVM, Gaussian process, decision tree, random forest, and naive Bayes were 0.909, 0.972, 0.953, 0.869, 0.692, and 0.842, respectively. The results of the biomedical term feature set prediction models are good, and the performance can reach up to 0.9. This means that the term feature sets can contain most of the information in the gene panel.
Design of Cancer-Related Gene Panels Based on Topic Modeling
To understand the MSK-IMPACT panel characteristics, we generated thirty topics that potentially represented different biomedical meanings. The following are some examples of issues relevant to genes in the MSK-IMPACT panel. Figure 7 shows the text features, genes, and related pathways derived from the Reactome of topics 2, 7, and 14, including hematologic, and malignancies. In topic two, leukemia subtypes and targeted inhibitors (e.g., imatinib, dasatinib, and decitabine) were mined. Heart arrest, a common side effect of inhibitors for leukemia, was also been reported (Hochhaus et al., 2009). The related MSK-IMPACT panel in topic two was involved in the signaling of interleukin-4 and interleukin-13 (p = 5.27e-5), which was associated with the apoptosis of leukemia cells (Chaouchi et al., 1996; Peña-Martínez et al., 2018) (Figure 7A). These results indicated that topic two was associated with leukemia, a hematological malignancy. In topic seven, key text features such as kidney neoplasms, carcinoma, renal cell, and Wilms tumor implied the relationship between topic seven and kidney cancer. Inhibitors for kidney cancer, such as sorafenib and everolimus, were also identified (Martín-Aguilar et al., 2021; Ren et al., 2021). The hypoxia pathway enriched by VHL, VEGFA, and PBRM1 (p = 5.41e-11) played a crucial role in the governance of cancer stem cells of renal cancer (Myszczyszyn et al., 2015) (Figure 7B). In topic 14, colorectal neoplasms, hereditary nonpolyposis, adenomatous polyposis coli, oxaliplatin, and cetuximab were associated with colon cancer. Related genes (e.g., MLH1, MSH2, and MSH6) in topic 14 were involved in mismatch repair (p = 5.72e-8), which has clinical importance in Lynch syndrome (Truninger et al., 2005) (Figure 7C). Other examples of different cancers, including brain cancer, gynecologic cancer, and breast cancer, are shown in Supplementary Figure S3. These results indicated that most of the genes in the MSK-IMPACT panel were collected for either therapeutic usage or biological relevance to various cancer types. In the future, we could design a small subset of multiple-gene groups by cancer topic.
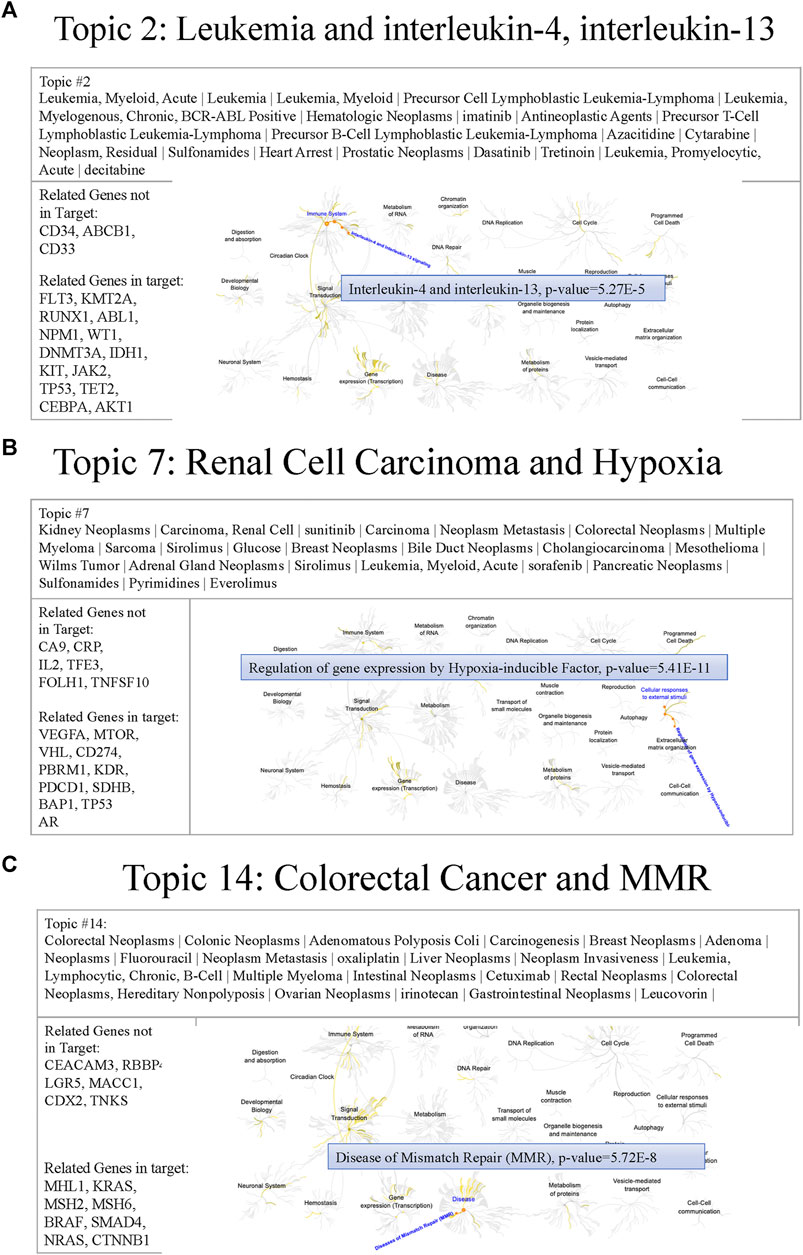
FIGURE 7. Examples of cancer topics containing relevant text features, genes, and pathways (A) Figure showing the text features, genes, and pathways of topic 2. Cancer types (e.g., leukemia) and inhibitors (e.g., imatinib) were reported in this topic. Reactome pathway analysis revealed that the related genes of the MSK-IMPACT panel in topic 2 (e.g., FLT3) were involved in interleukin-4 and interleukin-13 signaling (p = 5.27e-5). (B) Figure showing the text features, genes, and pathways of topic 7. Text features including cancer types (e.g., kidney neoplasms) and inhibitors (e.g., sorafenib) implied the relationship between topic seven and kidney cancer. The hypoxia pathway enriched by related genes (e.g., VHL) of the MSK-IMPACT panel in topic 7 (p = 5.41e-11) played a crucial role in the governance of cancer stem cells of renal cancer. (C) Figure showing the text features, genes, and pathways of topic 14. Many text features containing cancer types (e.g., colorectal neoplasms) and inhibitors (e.g., oxaliplatin) indicated the association between topic 14 and colon cancer. Related genes of the MSK-IMPACT panel in topic 14 (e.g., MLH1) were involved in the mismatch repair pathway (p = 5.72e-8).
Disscussion
It is helpful to gain insight into the field that bridges the knowledge gap between valuable biomedical information and free text by text mining (Sachin Kumar Deshmukh, 2020). With biomedical text mining advances and its applications in cancer research, we can design cancer gene panels by the semantic interpretation of comprehensive cancer narratives. Here, we used a biomedical literature mining model to discover the characteristics of a gene panel. Importantly, we demonstrated and validated the performance of the machine learning approach in text mining of cancer information. Our results highlight the following important points. 1) We developed a gene panel analysis framework based on a biomedical text mining pipeline. 2) Our pipeline can enrich the term features of cancer gene panels. 3) We demonstrated and validated the patterns of the cancer mutational landscape by NGS database. 4) The non-negative matrix factorization (NMF) method and topic modeling are useful for generating cancer information. Biomedical literature mining is valuable for discovering the inherent characteristics of gene panels. These results could be applied to the classification of cancer-related information and strategies for novel cancer gene panel designs.
The hypergeometric distribution test is one of the practical machine learning tools in TM. It can be used to select and extract term features from various genomic characterizations (Pal, 2017). We identified the critical term features according to the gene panel using p-values based on a hypergeometric test. Our term feature selection methods can distinguish in different gene panels. This implicates a high-performance prediction model for different datasets, including the MSK-IMPACT panel, Oncomine Cancer Panel, and cardiovascular gene panels. Although many gene recommendation algorithms have been developed, little is known about gene panel design.
Our biomedical term tagging algorithm provides a compressive cancer gene panel and related information. With our tagging algorithm, most of the essential biomedical terms in the text have been tagged. The construction of a gene term-feature matrix in different categories provides useful profiling for the characteristics of the genes. In this study, we constructed a biologically meaningful platform to analyze gene panels in terms of the diseases, chemicals, mutations, and MeSH terms related to genes. We can implement more biomedical term feature matrixes, such as a drug-feature matrix and disease-feature matrix. These different types of forms can provide strategies to analyze biology. With NMF topic modeling, we can capture cancer gene-drug information compatible with our knowledge. It will be useful to design a small subset of cancer gene panels by interpreting the topic model.
For the discovery of cancer gene panels, Figure 5A and Figure 7C illustrate an example of a cancer gene panel design for colorectal cancer. The most frequent genes are KRAS, EGFR, BRAF, PTEN, TP53, MLH1, PIK3CA, CTNNB1 in colorectal cancer by the heatmap. Hereditary nonpolyposis colon cancer (HNPCC) is caused by inherited mismatch repair genetic mutations, including MLH1, MSH2, and MSH6. The lifetime ovarian cancer risk increased in HNPCC. We can find ovarian cancer and a gene panel including MLH1, MSH2, MSH6, BRAF, KRAS, SMAD4, NRAS, CTNNB1 by topic model. In our study, we can design the two different cancer panels by phenotype. These results indicated the platform could provide an opportunity to construct a cancer gene panel recommendation by different cancer subtypes. There are some text mining limitations in our study. The entity-term based features are based only on co-occurrence in three sentences. However, related entities may have distinct relationships, which are not necessarily co-occurred. The features were obtained from only one resource, PubMed abstracts. Many curated databases have many useful biological features of genes or diseases or drugs; for example, Gene Ontology (GO) (Ashburner et al., 2000; The Gene Ontology Consortium., 2017) contains GO terms that describe genes by the functions of genes or cellular components. It may provide a benefit to the cancer researcher. Unfortunately, the TF-IDF table is going to weight toward common diseases and omit those that are critical in identifying rare diseases. The gene panels are not useful for the identification of unknown or rare gene mutations that are important for treatment. Simultaneously, the manuscripts and supplementary materials may also provide more critical results, but the lack of standardization in accessing this information is a significant problem. The text mining method often focuses on a few sentences due to the challenges of creating a complicated relationship between several critical keywords.
As we know, the random forest algorithm performed well than the decision tree in most of pattern classification cases. However, we found that the random forest approach presented a worse ability for cancer gene panel prediction in the experiments. Several reasons may cause this situation in the model training and evaluation, such as whether or not we specify the maximum number of features to be included at each node split. One of the reasons is that the random forest builds subtrees by randomly choosing features from amounts of features in our study. Unlike the other methods, they calculated the weights for each feature by determining the importance of all features. Thus, the performance might be increased when we increase the number of trees in the random forest. Because the subtrees increased, the model will be seen more features to build more diverse trees. Therefore, the model will become robust and make an excellent performance. Nevertheless, in this paper, we are focusing on a pipeline that can contextualize genes. We used the default parameter in most of the methods in our study. Although we are not emphasizing the methods and parameters optimization, it is also an important issue that we will study in our future works.
Several text mining systems have been developed for mutation-disease association (Erdogmus and Sezermen., 2007; Yeniterzi and Sezerman., 2009; Singhal et al., 2016). An automated pipeline using the full-length biomedical literature was recently established and validated by evidence-based gene panels (Saberian et al., 2020). All these methods focus on mutation-disease associations. In contrast, we contextualized the genes for clinical precision medicine. We provide information about druggable targets, mutations in hereditary cancer syndrome, and disease subtypes.
Although many text mining-based gene panel algorithms were developed, there is still little known to validate the gene panel characteristics. This study provides a biomedical literature mining pipeline in gene panel discovery and interpretation. The platform validated by NGS database could provide an opportunity to construct a gene recommendation and annotation system for precision medicine.
Conclusions
In conclusion, this study highlights the importance of biomedical literature mining in gene panel discovery and interpretation. The platform could provide an opportunity to construct a gene recommendation and annotation system for precision medicine.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author Contributions
Conception and study design: H-OC, P-CL, C-RL, C-SW, and J-HC; Development of methodology: H-OC, P-CL, J-HC; Acquisition of data: H-OC; C-RL, and C-SW; Statistical and computational analysis: P-CL, H-OC, C-RL, and J-HC; Writing, review, and revision of the manuscript: H-OC, P-CL, C-RL, and J-HC; Study supervision: J-HC; All authors have read and approved the manuscript. All authors agree for publication.
Funding
This work was supported in part by the Ministry of Science and Technology (MOST), Taiwan under Research Grant of MOST 110-2634-F-006-014 and MOST 110-2634-F-006-020, Ministry of Health and Welfare (MOHW110-TDU-B-211-144018). All authors have read and approved the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors gratefully acknowledge the significant contribution of Kimforest LTD. Taiwan, KD Yang, CC Pan, and CJ Lee for the bioinformatics support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.771435/full#supplementary-material
References
Arriagada, R., Bergman, B., Dunant, A., Le Chevalier, T., Pignon, J. P., and Vansteenkiste, J. (2004). & International Adjuvant Lung Cancer Trial Collaborative GroupCisplatin-Based Adjuvant Chemotherapy in Patients with Completely Resected Non-small-cell Lung Cancer. N. Engl. J. Med. 350 (4), 351–360. doi:10.1056/NEJMoa031644
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene Ontology: Tool for the Unification of Biology. The Gene Ontology Consortium. Nat. Genet. 25 (1), 25–29. doi:10.1038/75556
Azam, F., Musa, A., Dehmer, M., Yli-Harja, O. P., and Emmert-Streib, F. (2019). Global Genetics Research in Prostate Cancer: A Text Minning and Computational Network Theory Approach. Front. Genet. 10, 70. doi:10.3389/fgene.2019.00070
Burris, H. A. (2004). Dual Kinase Inhibition in the Treatment of Breast Cancer: Initial Experience with the EGFR/ErbB-2 Inhibitor Lapatinib. Oncologist 9 (Suppl. 3), 10–15. doi:10.1634/theoncologist.9-suppl_3-10
Chaouchi, N., Wallon, C., Goujard, C., Tertian, G., Rudent, A., Caput, D., et al. (1996). Interleukin-13 Inhibits Interleukin-2-Induced Proliferation and Protects Chronic Lymphocytic Leukemia B Cells from In Vitro Apoptosis. Blood 87 (3), 1022–1029.
Chapman, P. B., Hauschild, A., Robert, C., Haanen, J. B., Ascierto, P., Larkin, J., et al. BRIM-3 Study Group (2011). Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N. Engl. J. Med. 364 (26), 2507–2516. doi:10.1056/NEJMoa1103782
Cheng, D. T., Mitchell, T. N., Zehir, A., Shah, R. H., Benayed, R., Syed, A., et al. (2015). Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J. Mol. Diagn. 17 (3), 251–264. doi:10.1016/j.jmoldx.2014.12.006
Choo, J., Lee, C., Reddy, C. K., and Park, H. (2013). UTOPIAN: User-Driven Topic Modeling Based on Interactive Nonnegative Matrix Factorization. IEEE Trans. Vis. Comput. Graph. 19 (12), 1992–2001. doi:10.1109/TVCG.2013.212
Demeester, T., Sutskever, I., Chen, K., Dean, J., and Corado, G. (2016). Distributed Representations of Words and Phrases and Their Compositionality. EMNLP 2016 – Conf. Empir. Methods Nat. Lang. Process. Proc., 1389–1399. arXiv: 1606.08359.
Devarajan, K., Wang, G., and Ebrahimi, N. (2015). A Unified Statistical Approach to Non-negative Matrix Factorization and Probabilistic Latent Semantic Indexing. Mach. Learn. 99 (1), 137–163. doi:10.1007/s10994-014-5470-z
Du, J., Jia, P., Dai, Y., Tao, C., Zhao, Z., and Zhi, D. (2019). Gene2vec: Distributed Representation of Genes Based on Co-expression. BMC Genomics 20 (Suppl. 1), 82. doi:10.1186/s12864-018-5370-x
Erdogmus, M., and Sezerman, O. U. (2007). Application of Automatic Mutation-Gene Pair Extraction to Diseases. J. Bioinform. Comput. Biol. 5 (6), 1261–1275. doi:10.1142/s021972000700317x
Hauschild, A., Grob, J. J., Demidov, L. V., Jouary, T., Gutzmer, R., Millward, M., et al. (2012). Dabrafenib in BRAF-Mutated Metastatic Melanoma: a Multicentre, Open-Label, Phase 3 Randomised Controlled Trial. Lancet 380 (9839), 358–365. doi:10.1016/S0140-6736(12)60868-X
Hochhaus, A., O'Brien, S. G., Guilhot, F., Druker, B. J., Branford, S., Foroni, L., et al. IRIS Investigators (2009). Six-year Follow-Up of Patients Receiving Imatinib for the First-Line Treatment of Chronic Myeloid Leukemia. Leukemia 23 (6), 1054–1061. doi:10.1038/leu.2009.38
Hyman, D. M., Solit, D. B., Arcila, M. E., Cheng, D. T., Sabbatini, P., Baselga, J., et al. (2015). Precision Medicine at Memorial Sloan Kettering Cancer Center: Clinical Next-Generation Sequencing Enabling Next-Generation Targeted Therapy Trials. Drug DiscovToday 20 (12), 1422–1428. doi:10.1016/j.drudis.2015.08.005
Ikonomakis, M., Kotsiantis, S., and Tampakas, V. (2005). Text Classification Using Machine Learning Techniques. WSEAS Trans. Comput. 4.
Kumar Deshmukh, S. (2020). Machine Learning for Precision Medicine in Cancer- Transforming Drug Discovery and Treatment. J. Cancer Biol. 1, 20–22. doi:10.46439/cancerbiology.1.005
Leaman, R., Islamaj Dogan, R., and Lu, Z. (2013). DNorm: Disease Name Normalization with Pairwise Learning to Rank. Bioinformatics 29 (22), 2909–2917. doi:10.1093/bioinformatics/btt474
Luthra, R., Patel, K. P., Routbort, M. J., Broaddus, R. R., Yau, J., Simien, C., et al. (2017). A Targeted High-Throughput Next-Generation Sequencing Panel for Clinical Screening of Mutations, Gene Amplifications, and Fusions in Solid Tumors. J. Mol. Diagn. 19 (2), 255–264. doi:10.1016/j.jmoldx.2016.09.011
Martín-Aguilar, A. E., Núñez-López, H., and Ramirez-Sandoval, J. C. (2021). Sorafenib as a Second-Line Treatment in Metastatic Renal Cell Carcinoma in Mexico: a Prospective Cohort Study. BMC Cancer 21, 1–9. doi:10.1186/s12885-020-07720-5
McCabe, M. J., Gauthier, M. A., Chan, C. L., Thompson, T. J., De Sousa, S., Puttick, C., et al. (2019). Development and Validation of a Targeted Gene Sequencing Panel for Application to Disparate Cancers. Sci. Rep. 9 (1), 17052. doi:10.1038/s41598-019-52000-3
Myszczyszyn, A., Czarnecka, A. M., Matak, D., Szymanski, L., Lian, F., Kornakiewicz, A., et al. (2015). The Role of Hypoxia and Cancer Stem Cells in Renal Cell Carcinoma Pathogenesis. Stem Cel Rev. Rep. 11 (6), 919–943. doi:10.1007/s12015-015-9611-y
Paez, J. G., Jänne, P. A., Lee, J. C., Tracy, S., Greulich, H., Gabriel, S., et al. (2004). EGFR Mutations in Lung Cancer: Correlation with Clinical Response to Gefitinib Therapy. Science 304 (5676), 1497–1500. doi:10.1126/science.1099314
Paige, S. L., Saha, P., and Priest, J. R. (2018). Beyond Gene Panels: Whole Exome Sequencing for Diagnosis of Congenital Heart Disease. Circ. Genom. Precis. Med. 11 (3), e002097. doi:10.1161/CIRCGEN.118.002097
Pal, R. (2017). Feature Selection and Extraction from Heterogeneous Genomic Characterizations. Predictive Modeling of Drug Sensitivity, 45–81. doi:10.1016/b978-0-12-805274-7.00003-8
Pedregosa, F., Varoquaux, G., Gramfort, A., Michel, V., Thirion, B., and Grisel, O. (2011). Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 12, 2825–2830. arXiv:201.0490.
Peña-Martínez, P., Eriksson, M., Ramakrishnan, R., Chapellier, M., Högberg, C., Orsmark-Pietras, C., et al. (2018). Interleukin 4 Induces Apoptosis of Acute Myeloid Leukemia Cells in a Stat6-dependent Manner. Leukemia 32 (3), 588–596. doi:10.1038/leu.2017.261
Ren, Z., Niu, Y., Fan, B., Wei, S., Ma, Y., Zhang, X., et al. (2021). Clinical Analysis of Everolimus in the Treatment of Metastatic Renal Cell Carcinoma. Ann. Palliat. Med. 10. doi:10.21037/apm-20-2465
Rhodes, D. R., Kalyana-Sundaram, S., Mahavisno, V., Varambally, R., Yu, J., Briggs, B. B., et al. (2007). Oncomine 3.0: Genes, Pathways, and Networks in a Collection of 18,000 Cancer Gene Expression Profiles. Neoplasia 9 (2), 166–180. doi:10.1593/neo.07112
Rosenbaum, M. W., Bledsoe, J. R., Morales-Oyarvide, V., Huynh, T. G., and Mino-Kenudson, M. (2016). PD-L1 Expression in Colorectal Cancer Is Associated with Microsatellite Instability, BRAF Mutation, Medullary Morphology and Cytotoxic Tumor-Infiltrating Lymphocytes. Mod. Pathol. 29 (9), 1104–1112. doi:10.1038/modpathol.2016.95
Saberian, N., Shafi, A., Peyvandipour, A., and Draghici, S. (2020). MAGPEL: an autoMated Pipeline for Inferring vAriant-Driven Gene PanEls from the Full-Length Biomedical Literature. Sci. Rep. 10 (1), 12365. doi:10.1038/s41598-020-68649-0
Seidel, J. A., Otsuka, A., and Kabashima, K. (2018). Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 8, 86. doi:10.3389/fonc.2018.00086
Shabani Azim, F., Houri, H., Ghalavand, Z., and Nikmanesh, B. (2018). Next Generation Sequencing in Clinical Oncology: Applications, Challenges and Promises: A Review Article. Iran. J. Public Health 47, 1453–1457. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6277731/.
Shepherd, F. A., Rodrigues Pereira, J., Ciuleanu, T., Tan, E. H., Hirsh, V., Thongprasert, S., et al. (2005). Erlotinib in Previously Treated Non-small-cell Lung Cancer. N. Engl. J. Med. 353 (2), 123–132. doi:10.1056/NEJMoa050753
Singhal, A., Simmons, M., and Lu, Z. (2016). Text Mining for Precision Medicine: Automating Disease-Mutation Relationship Extraction from Biomedical Literature. J. Am. Med. Inform. Assoc. 23 (4), 766–772. doi:10.1093/jamia/ocw041
The Gene Ontology Consortium (2017). Expansion of the Gene Ontology Knowledgebase and Resources. Nucleic Acids Res. 45 (D1), D331–D338. doi:10.1093/nar/gkw1108
Truninger, K., Menigatti, M., Luz, J., Russell, A., Haider, R., Gebbers, J. O., et al. (2005). Immunohistochemical Analysis Reveals High Frequency of PMS2 Defects in Colorectal Cancer. Gastroenterology 128 (5), 1160–1171. doi:10.1053/j.gastro.2005.01.056
Wang, C. C. N., Jin, J., Chang, J. G., Hayakawa, M., Kitazawa, A., Tsai, J. J. P., et al. (2020). Identification of Most Influential Co-occurring Gene Suites for Gastrointestinal Cancer Using Biomedical Literature Mining and Graph-Based Influence Maximization. BMC Med. Inform. Decis. Mak. 20, 1–12. doi:10.1186/s12911-020-01227-6
Wang, Y., Wu, S., Li, D., Mehrabi, S., and Liu, H. (2016). A Part-Of-Speech Term Weighting Scheme for Biomedical Information Retrieval. J. Biomed. Inform. 63, 379–389. doi:10.1016/j.jbi.2016.08.026
Wei, C. H., Kao, H. Y., and Lu, Z. (2015). GNormPlus: An Integrative Approach for Tagging Genes, Gene Families, and Protein Domains. Biomed. Res. Int. 918710. doi:10.1155/2015/918710
Wei, C. H., Kao, H. Y., and Lu, Z. (2013). PubTator: a Web-Based Text Mining Tool for Assisting Biocuration. Nucleic Acids Res. 41, W518. doi:10.1093/nar/gkt441
Westlake, A. J., and Larson, H. J. (1970). Introduction to Probability Theory and Statistical Inference. Stat 19, 352.
Yeganova, L., Kim, W., Kim, S., and Wilbur, W. J. (2014). Retro: Concept-Based Clustering of Biomedical Topical Sets. Bioinformatics 30 (22), 3240–3248. doi:10.1093/bioinformatics/btu514
Yeniterzi, S., and Sezerman, U. (2009). EnzyMiner: Automatic Identification of Protein Level Mutations and Their Impact on Target Enzymes from PubMed Abstracts. BMC bioinformatics 10 (Suppl. 8Suppl 8), S2. doi:10.1186/1471-2105-10-S8-S2
Zehir, A., Benayed, R., Shah, R. H., Syed, A., Middha, S., Kim, H. R., et al. (2017). Mutational Landscape of Metastatic Cancer Revealed from Prospective Clinical Sequencing of 10,000 Patients. Nat. Med. 23 (6), 703–713. doi:10.1038/nm.4333
Zhou, W., Ercan, D., Chen, L., Yun, C. H., Li, D., Capelletti, M., et al. (2009). Novel Mutant-Selective EGFR Kinase Inhibitors against EGFR T790M. Nature 462 (7276), 1070–1074. doi:10.1038/nature08622
Keywords: biomedical natural language processing, machine learning, topic modeling, cancer gene panel, text mining
Citation: Chen H-O, Lin P-C, Liu C-R, Wang C-S and Chiang J-H (2021) Contextualizing Genes by Using Text-Mined Co-Occurrence Features for Cancer Gene Panel Discovery. Front. Genet. 12:771435. doi: 10.3389/fgene.2021.771435
Received: 06 September 2021; Accepted: 11 October 2021;
Published: 25 October 2021.
Edited by:
Ying Li, Zhejiang University, ChinaReviewed by:
Rafael Rosell, Catalan Institute of Oncology, SpainEhsan Nazemalhosseini-Mojarad, Shahid Beheshti University of Medical Sciences, Iran
Ruan Maomei, Shanghai Chest Hospital, Shanghai Jiaotong University, China
Copyright © 2021 Chen, Lin, Liu, Wang and Chiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peng-Chan Lin, pengchanlin@gmail.com; Jung-Hsien Chiang, jchiang@mail.ncku.edu.tw