 Felipe Cabarcas1,3
Felipe Cabarcas1,3 Ana Luz Galvan-Diaz4Laura M. Arias-Agudelo1Gisela María García-Montoya1,2,5Juan M. Daza6
Ana Luz Galvan-Diaz4Laura M. Arias-Agudelo1Gisela María García-Montoya1,2,5Juan M. Daza6 Juan F. Alzate1,2,5*
Juan F. Alzate1,2,5*- 1Centro Nacional de Secuenciación Genómica CNSG, Sede de Investigación Universitaria-SIU, Medellín, Colombia
- 2Grupo SISTEMIC, Departamento de Ingeniería Electrónica, Facultad de Ingeniería, Universidad de Antioquia, Medellín, Colombia
- 3Environmental Microbiology Group, School of Microbiology, Universidad de Antioquia, Medellín, Colombia
- 4Departamento de Microbiología y Parasitología, Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia
- 5Grupo Pediaciencias, Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia
- 6Grupo Herpetológico de Antioquia, Institute of Biology, Universidad de Antioquia, Medellín, Colombia
Cryptosporidium is a leading cause of waterborne outbreaks globally, and Cryptosporidium hominis and C. parvum are the principal cause of human cryptosporidiosis on the planet. Thanks to the advances in Next-Generation Sequencing (NGS) sequencing and bioinformatic software development, more than 100 genomes have been generated in the last decade using a metagenomic-like strategy. This procedure involves the parasite oocyst enrichment from stool samples of infected individuals, NGS sequencing, metagenomic assembly, parasite genome computational filtering, and comparative genomic analysis. Following this approach, genomes of infected individuals of all continents have been generated, although with striking different quality results. In this study, we performed a thorough comparison, in terms of assembly quality and purity, of 100+ de novo assembled genomes of C. hominis. Remarkably, after quality genome filtering, a comprehensive phylogenomic analysis allowed us to discover that C. hominis encompasses two lineages with continental segregation. These lineages were named based on the observed continental distribution bias as C. hominis Euro-American (EA) and the C. hominis Afro-Asian (AA) lineages.
Introduction
Cryptosporidium is a ubiquitous apicomplexan parasite with gastrointestinal habitat and a broad range of vertebrate hosts, including humans, other mammals, birds, fish, and reptiles (Zahedi and Ryan, 2020). Cryptosporidium hominis and C. parvum are the preponderant cause of human cryptosporidiosis around the world, with the former being more frequently found in developing nations (Gilchrist et al., 2018; Tichkule et al., 2021). Cryptosporidiosis is usually a self-limiting infection in immunocompetent individuals. However, vulnerable populations, like immunocompromised individuals (especially those with T cell impairment) and children (particularly those below 5 years old), develop persistent to chronic syndromes, with diarrhea as the principal symptom (Innes et al., 2020). According to the Global Enteric Multicenter Study (GEMS), Cryptosporidium was the second cause of moderate-to-severe diarrhea in children from sub-Saharan Africa and South Asia countries; and this infection increased the risk of death in children aged 12–23 months (Kotloff et al., 2013).
Cryptosporidium oocysts are excreted in feces by symptomatic hosts. In this stage, the parasite is highly resistant to common disinfection methods like chlorination (Cacciò and Chalmers, 2016). These characteristics might explain why Cryptosporidium is a leading cause of waterborne outbreaks globally (Efstratiou et al., 2017). Furthermore, depending on the species, the parasite can be transmitted via direct person-to-person contact, indirect (food, water, or fomites), or zoonotic routes (Cacciò and Chalmers, 2016).
Currently, 42 species of Cryptosporidium are recognized (Zahedi and Ryan, 2020), with C. parvum and C. hominis being responsible for greater than 90% of human infections (Feng et al., 2018). Geographic differences in species distribution have been reported, with C. hominis as the leading species in human cases in developing countries (Xiao and Feng, 2008; Ryan et al., 2014; Xiao and Feng, 2017). While C. hominis is associated with a predominant anthroponotic transmission, C. parvum presents a zoonotic transmission route with livestock as the primary source of infection (Nader et al., 2019). In developed European nations, C. parvum infections are more common in rural areas with low human population density and activities related to livestock and agriculture (Lake et al., 2007; Pollock et al., 2010). By contrast, C. hominis has a narrow host range showing a specialization trend toward human hosts. Although it can successfully infect other mammals, it produces mild and asymptomatic infections (Widmer et al., 2020). Subtyping characterization through gp 60 gene analysis reveals more than ten subtype families, with six of them as the most common in humans (Ia, Ib, Id, Ie, If, and Ig), being the IbA10G2 subtype the predominant and most virulent, widely distributed in both developing and developed countries, and frequently associated with outbreaks worldwide (Feng et al., 2018).
Cryptosporidium hominis is an unculturable parasite. This condition implies that the only possibility to obtain genomic DNA of the parasite is to extract it from stool samples of infected hosts. Cryptosporidium oocysts in human feces are usually present at low numbers, requiring specific procedures to capture them. Despite the purification step, DNA of the intestinal microbiota is often abundant, requiring a metagenomic bioinformatics strategy to study the Cryptosporidium genome. A metagenome assembly is performed to start the genome reconstruction. Then, C. hominis genome scaffolds should be selected using informatics tools. Due to the risk of Cryptosporidium coinfections, different species, or isolates, additional quality control steps are needed to avoid contaminated/chimeric genome reconstructions (Isaza et al., 2015).
In this study, we conducted a comparative analysis of the publicly available genomic data of C. hominis under normalized conditions with the aim to have a better understanding of the quality of the generated data in the last decade. Since all these genomes come from a methodology more like a metagenomic approach, additional analyses were performed to detect Cryptosporidium genome mixtures present in one individual.
We also used phylogenetic tools to reconstruct the evolutionary history of ninety-nine C. hominis genomes collected from human individuals in five continents: America, Europe, Asia, Africa, and Oceania. Our phylogenomic analysis showed that C. hominis species encompasses two lineages with phylogeographic structure, with one clade composed mainly of European and American isolates, while the other mainly with African and Asian isolates. The two Oceania representatives were grouped into the Euro-American lineage.
Materials and Methods
Genome Data From Sequence Read Archive-SRA Public Database
One hundred nineteen C. hominis Next-Generation Sequencing (NGS) genome projects with shotgun reads were selected and downloaded (Supplementary Table S1) from the Sequence Read Archive-SRA database. Genome projects based only in 454 Technology were excluded. The raw read data were directly download from the SRA database using the fastq-dump tool with the split option activated.
As outgroups, another 15 genomes were used: C. cuniculus (UKCU5: PRJNA492839, UKCU2: PRJNA315496), C. parvum (UKP2:PRJNA253836, UKP3:PRJNA253840, UKP4:PRJNA253843, UKP5:PRJNA253845, UKP6:PRJNA253846, UKP7:PRJNA253847, UKP8:PRJNA253848, UKP14:PRJNA315506, UKP15:PRJNA315507), and C. meleagridis (UKMEL1:PRJNA222838, UKMEL3:PRJNA315502, UKMEL4:PRJNA315503).
Generation of the New Colombian Cryptosporidium hominis Genome Reference UDEAa567
C. hominis oocysts were purified from a fecal sample collected from an HIV Colombian female patient by flotation in a saturated sodium chloride solution (Kar et al., 2011). Parasite diagnosis was previously confirmed by Kinyoun stain. Species and subtype identification was done by a nested PCR and sequence analysis of the small-subunit (SSU) rDNA gene and 60 kDa glycoprotein gene (IbA10G2), respectively. Purified oocysts were resuspended in PBS and quantified using a Neubauer chamber slide and light microscopy. The sample was stored at 4°C until DNA was extracted. DNA extraction was performed using the kit NORGEN Stool DNA Isolation (CAT 27600), following the manufacturer’s instructions, with previous freeze-thaw cycles (each for 2 min) in liquid nitrogen and a water bath at 37°C. A solution with approximately 1.85 × 108 oocyst was used for DNA purification. DNA obtained was quantified by light absorption at 260 nm (NanoDropTM, Thermo Scientific) and with PicoGreen® reagent. The genome was sequenced using Illumina NOVASEQ 6000 instrument at Macrogen (Seoul, Korea). Paired-end reads of 150 bases were generated and deposited at the SRA database under the accession number SRR14522748.
Cryptosporidium hominis De Novo Genome Assembly
Genomic reads from each experiment were independently assembled using SPADES v3.14.1. First, reads ends were cleaned with rapifilt (homebrew program) filtering at Q30 and only keeping reads with a minimum of 50 bases. Then, assembly was performed using SPADES parameters: -careful -t 40 -m 160 -k 33,55,77,99.
Each assembly was filtered to excluded contaminating sequences using BLASTN (v2.10.1+). Only those longer than 1,000 bases and with a bit score greater than 300 were kept in each C. hominis assembled genome dataset. The bit score was obtained mapping the scaffolds to the C. parvum IOWAII genome reference (CryptoDB v 52), using BLASTN with parameters -evalue 1e-30 -num_alignments 5.
The genomes stats (Total length of sequence, Largest contig, N50 stats, Total number of sequences) were obtained using an in-house python script (see Supplementary Material). The “AltSNPs” (alternate single nucleotide polymorphisms) and “Coverage” of the assembly, were obtained by mapping the cleaned reads of each experiment against the assembled genome, using bowtie2 (version 2.4.1) with default parameters and using samtools (v1.10) view -F 3584 to keep only mapped reads. The coverage was obtained using samtools coverage and obtaining the mean and median of the mean depth of each scaffold. While the SNVs were obtained by counting the number of variants from the Variant Call Format (VCF) created as bcftools mpileup--redo-BAQ--in-BQ 30--per-sample-mF--skip-indels, then bcftools call--multiallelic-caller--variants-only-Ov and then bcftools view -i “%QUAL ≥ 30.”
The SNVs and Identity vs. C. hominis UDEA01 were obtained comparing each genome with the C. hominis UDEA01 using DNAdiff (version 1.3) and getting the TotalSNPs and AvgIdentity from the.report file. The C. hominis genomes that had less than 8,167,000 assembled bases and with more than 3,825 AltSNPs were dropped as they did not meet basic quality parameters, to obtain the selected genomes that are further analyzed. We run BUSCO v5.2.2 (Simão et al., 2015) with parameters “busco-m geno-l coccidia_odb10 -i” for each selected genomes, including outgroups.
The indels vs C. parvum IOWAII were obtained using nucdiff (v2.0.3), with default parameters for each selected genome and the additional C. parvum, C. meleagridis, and C. cuniculus genomes. Using an in-house python script the indels were extracted from “_query_snps.gff” files. The principal component analysis (PCA) analysis was created with python’s sklearn package and plotted with matplotlib.
Phylogenomic Analysis
The selected C. hominis genomes, together with the C. parvum, C. meleagridis and C. cuniculus genomes were used to infer a phylogenetic tree. The sixty one neutrally evolving genes of C. parvum described by Nader et al. (2019) were extracted from each genome. Each gene from all genomes were aligned using MAFFT (version v7.475) with parameters--inputorder--adjustdirection--anysymbol--auto. All the aligned genes were concatenated using catsequences. These aligned sequences were used to infer a maximum likelihood tree using IQTREE2 (2.1.2 COVID-edition for Linux 64-bit) with parameters -B 5000-T AUTO -m MFP+MERGE -rcluster 10 (Lanfear et al., 2012; Lanfear et al., 2014).
Best-fit models selected according to BIC for each coding gene were: K3Pu + F + G4:cgd1_1450 + cgd4_4440 + cgd6_2560 + cgd7_1810 + cgd7_2600 + cgd8_3560,TPM3u + F +G4:cgd1_1730 + cgd1_3650 + cgd1_640 + cgd6_2720 + cgd6_5300 + cgd7_1270 + cgd7_340 + cgd7_890,TIM2 + F + G4:cgd1_2000 + cgd1_3790 + cgd2_2470 + cgd3_4230 + cgd4_2820 + cgd5_2250 + cgd5_4240 + cgd7_2340 + cgd8_2850,TN + F + G4:cgd1_3780 + cgd3_1720 + cgd4_3800 + cgd4_4360 + cgd6_4090 + cgd6_4280 + cgd6_5370 + cgd8_2080 + cgd8_830,TN + F + G4:cgd2_180 + cgd2_3630 + cgd8_1960 + cgd8_3030 + cgd8_5310,HKY + F:cgd2_2060 + cgd5_1340,TN + F + G4:cgd2_3110 + cgd3_1010,K3Pu + F + G4:cgd2_3810 + cgd2_940 + cgd3_380 + cgd4_2620 + cgd5_2890 + cgd7_1330 + cgd7_3550,TPM3 + F + G4:cgd3_2600 + cgd5_3600 + cgd6_2100,HKY + F + I:cgd3_3070 + cgd3_3650 + cgd4_2210 + cgd5_2730,TPM3 + F + G4:cgd3_3310 + cgd5_2700,HKY + F + G4:cgd4_2180 + cgd4_370 + cgd5_1860,TPM2 + F + G4:cgd8_140
We built a haplotype network using the minimum spanning network algorithm (Bandelt et al., 1999) and implemented in PopART (Leigh and Bryant, 2015). We included 95 genomes and excluded four genomes (Chom_EU_SWEH8, Chom_EU_SWEH5, Chom_AFRICA_SWEH3, Chom_AFRICA_SWEH4) because their long branches inferred in the phylogeny, resulting most likely from sequencing errors instead of true genetic variation. The dataset included 118 segregating sites across the 61 genes. The complete step by step analysis pipeline can be found in Supplementary Material.
Statistical and Graphical Analysis
Statistical analysis and graphics were done in R [R version 4.0.4 64, x86_64-apple-darwin17.0 (64-bit)] and R studio (Version 1.2.1335, Macintosh; Intel Mac OS X 10_16_0). Boxplots were generated in R with the function “boxplot.” Central tendency measures and inter-quartile ranges (IQR) were calculated with R functions “mean,” “median,” “quantile,” and “IQR.”
Results
Cryptosporidium hominis Genome Quality Analysis
One hundred and nineteen Cryptosporidium hominis genomes were included in the present study. A new Colombian C. hominis genome with the code UDEAa567 was also generated following C. hominis UdeA01 strategy (Isaza et al., 2015): the oocysts were enriched using a flotation protocol from a stool sample from an HIV + infected individual and then the NGS sequencing was performed using an Illumina Novaseq 6,000 instrument. C. hominis UDEAa567 assembly genome size was 9,052,438 bp. with an N50 value of 93,658 bp. The mean and median read depths were 10.2X and 10.5X, respectively. The UDEAa567 has 99.93% genome identity and 2,655 Single nucleotide variants (SNVs) respect C. hominis UDEA01.
To normalize the analytical conditions for all C. hominis genomes, the processing started with the original raw read data, and then they were all assembled using the same strategy. The C. parvum IOWA II genome (version 52) was downloaded from CryptoDB. All the read sequences used (C. hominis, C. parvum, C. cuniculus, C. meleagridis) are publicly available at the NCBI/SRA website (see Materials and Methods).
The C. hominis genome projects were executed in the last 10 years with different Illumina instruments, different library preparation kits, and were conducted in different laboratories around the world. A small set of these genomes were generated on the ION TORRENT platform. To assess the quality of the assemblies, their general metrics were compared in Figure 1: total assembled bases, median sequencing depth, assembly N50 values, genome nucleotide identity with C. hominis UDEA01, alternate allele count, BUSCO genome completeness, and BUSCO single-copy genes (Supplementary Table S1). The Cryptosporidium assembled genome sizes ranged between 1,148,020 and 10,587,153 Mbp. Despite the broad range of the assembled bases metric, a very narrow dispersion range (an IQR of 21,974 bases) was observed around the median, 9,073,830 bp (Figure 1A). By contrast, the observed median sequencing depth and its IQR were both very variable. The depth ranged from 1.1× to 714×, with a median value of 183× and an IQR of 197 (Figure 1B).
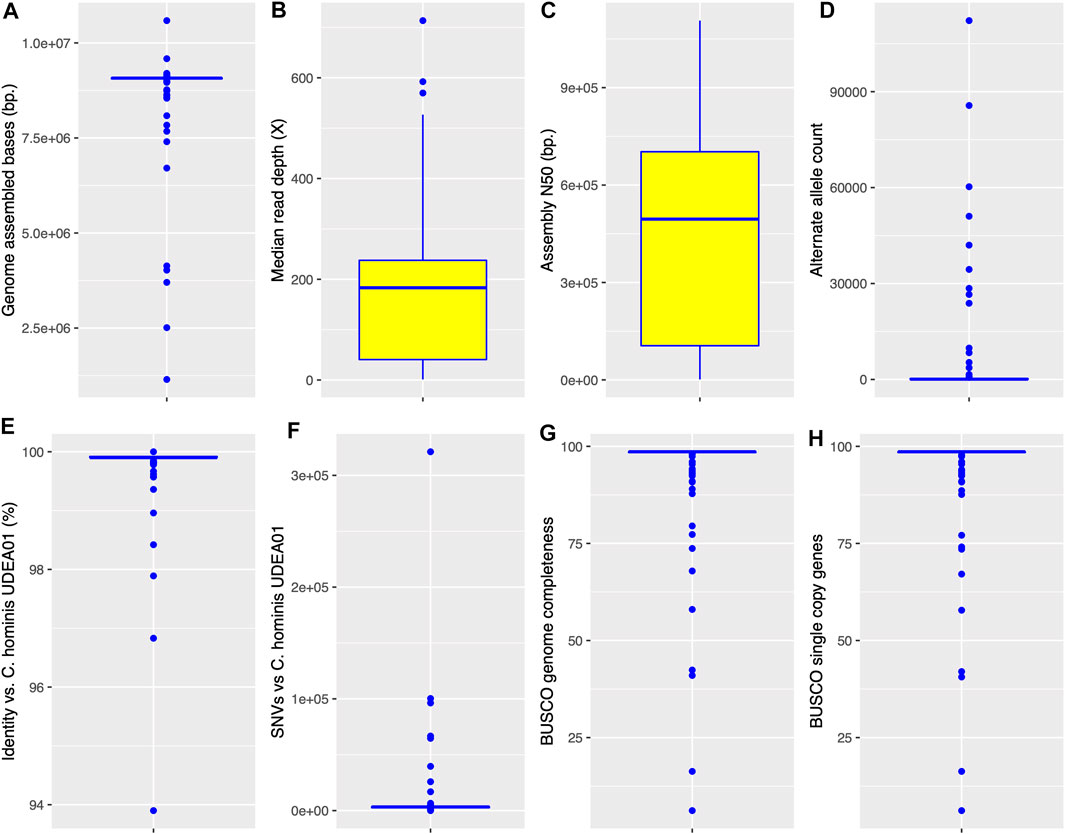
FIGURE 1. Boxplots representing the descriptive statistics of the de novo assembled C. hominis genomes. The box represents the interquartile range, IQR 25th -75th percentile. The line represents the 50th percentile. Whiskers denote the 0th and 100th percentile. (A) Cryptosporidium genome assembled bases. (B) Scaffold median read depth. (C) Cryptosporidium assembly N50 value. D. Scaffold positions with an alternate allele. (E) Assembly genomic nucleotide identity against C. hominis UDEA01 reference. (F) Single nucleotide variants (SNVs) detected in each C. hominis assembly against C. hominis UDEA01 reference. (G) BUSCO genome completeness. (H) BUSCO single-copy genes.
The assembly N50 value also showed a very broad range, from 1,302 to 1,106,561 bp. The median N50 value was 495,148 bases with an IQR of 597,468 (Figure 1C). Furthermore, the global nucleotide identity of all genomes against the C. hominis UDEA01 reference was calculated. They have a median identity of 99.9% and an IQR of 0.02% (Figure 1E). Only 10 genomes had a global identity value below 99.7% (Chom_EU_4127, Chom_AFRICA_SWEH7, Chom_EU_SWEH9, Chom_EU_6940, Chom_EU_6946, Chom_EU_6925, Chom_EU_6934, Chom_EU_4120, Chom_EU_4128, Chom_EU_4118). These genomes are poor-quality references since they are outliers in either assembled genome size or alternative allele count metrics (Supplementary Table S1).
To assess the possible Cryptosporidium coinfections, the reads of each isolate were mapped to its respective assembly and alternate alleles were counted. The observed median number of genomic positions with an alternate allele was 83, with an IQR of 196 (Figure 1D). Eighty-five percent of the C. hominis isolates showed alternate allele counts below 470. Then, to estimate the genetic variations among the C. hominis genomes, we counted the number of SNVs against the C. hominis UDEA01 reference. This analysis showed a median value of 3,324 SNVs with an IQR of 831 (Figure 1F). Ninety percent of the C. hominis genomes had less than 4,000 SNVs regardless its continental origin.
We analyzed the C. hominis genomes with BUSCO using the coccidia ODB10 database. The BUSCO analysis shows the same median and IQR values for both completeness and single-copy metrics, 98.6 and 0.2%, respectively (Figures 1G,H). The duplicated BUSCO median value was 0 with an IQR of 0. All C. hominis genomes, except 1 (Chom_EU_6925), showed duplicated values below 1%. Finally, the BUSCO fragmented metrics showed a median value of 0.4% with an IQR of 0.2% (Supplementary Table S1).
The genomic descriptive measurements showed outliers in several statistic indices, indicating the presence of poor-quality genomes. To filter out them, we set two thresholds: assembled genome size and alternate allele count. The first parameter is directly related to the coverage achieved for each genome and failing to reach a minimum threshold will affect the assembly size and also the accuracy of the assembly. The second parameter, alternate allele count, might indicate the presence of more than one Cryptosporidium genome in the human stool sample. The proposed thresholds rationale are: 1) Assembly size > 8,166,447 bp., which corresponds to 90% of the median value of C. hominis assembled genome size. We think that a representation of 90% of the expected assembly genome size is enough for subsequent comparative or phylogenetic analysis and is equivalent to a BUSCO genome completeness score of >90% (Supplementary Table S1); and 2) AltSNVs threshold of 3,992 SNVs, which corresponds to the percentile 90th of the observed HQ SNVs of the analyzed C. hominis genomes (Figure 2). Applying these two thresholds, 20 genomes were excluded for further analysis, 18 with European origin and 2 with African origin (Chom_EU_SWEH9, Chom_EU_UKH6, Chom_EU_SWEH13, Chom_EU_SWEH11, Chom_EU_H58, Chom_EU_6946, Chom_EU_6945, Chom_EU_6942, Chom_EU_6940, Chom_EU_6939, Chom_EU_6935, Chom_EU_6934, Chom_EU_6925, Chom_EU_6919, Chom_EU_4128, Chom_EU_4127, Chom_EU_4120, Chom_EU_4118, Chom_AFRICA_SWEH7, and Chom_AFRICA_SWEH2). After filtering, the number of genomes per continent was Africa, 33; America, 6; Asia, 29; European Union, 49; Oceania, 2.
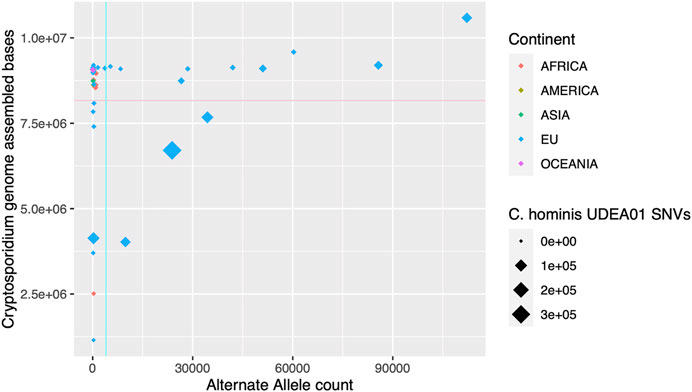
FIGURE 2. Graphical representation of the de novo assembled C. hominis genome size and alternate allele detection. In the x-axis, alternate allele count for each genome. In the y-axis, Cryptosporidium assembly size in bases pairs. The continent where the parasite was isolated is represented in colors. The size of the rhombus depicts the single nucleotide variants (SNVs) counts against the C. hominis UDEA01 genome reference. The cyan and pink lines indicate the thresholds established for low-quality genomes, alternate allele count = 3,992, and assembled genome size = 8,166,447 bases, respectively.
Phylogenomic Analysis of the Cryptosporidium hominis Species
Following the previous work published by Nader et al. (2019), 61 genes identified as neutrally evolving in C. parvum and C. hominis were used for the phylogenomic analysis. After filtering out the low-quality C. hominis genomes, ninety-nine genomes were kept for the phylogenetic reconstruction. As outgroups, 15 genomes were used: two C. cuniculus (UKCU5 and UKCU2), 8 C. parvum parvum (UKP2, UKP3, UKP4, UKP5, UKP6, UKP7, and UKP8), three C. parvum anthroponosum (UKP14 and UKP15), and three C. meleagridis (UKMEL1, UKMEL3, and UKMEL4).
A maximum-likelihood tree was constructed encompassing 114 tips. The total matrix of the 61 aligned genes encompasses 140,425 sites. BIC partitions were reduced to 13 (see Materials and Methods). The consensus tree has a Log-likelihood of −266,302.24. The basal branches recreate the previously described topology for the analyzed species, with C. meleagridis defined as the outgroup. The C. parvum subspecies clades C. parvum parvum and C. parvum anthroponosum are reciprocally monophyletic with 100% bootstrap support. The clade C. hominis comprises two well-supported lineages with strong continental bias: One lineage representing isolates from Asia and Africa (100% nodal support), and the other lineage representing genomes isolated from European or American infected humans (97% nodal support). The two isolates from Oceania are part of the European-American clade (Figure 3A). From now on, these two lineages will be referred to as the C. hominis Euro-American lineage (EA) and the Afro-Asian lineage (AA). The consensus tree shows very well-supported basal nodes but low support at the terminal branches due to low divergence among closely related genomes (Figure 3B). It is also noteworthy that the C. hominis EA lineage is populated by Ib genotypes, mostly IbA10G2/IbA12G2, except for C. hominis UdeA01, which has been classified with the genotype Ie.
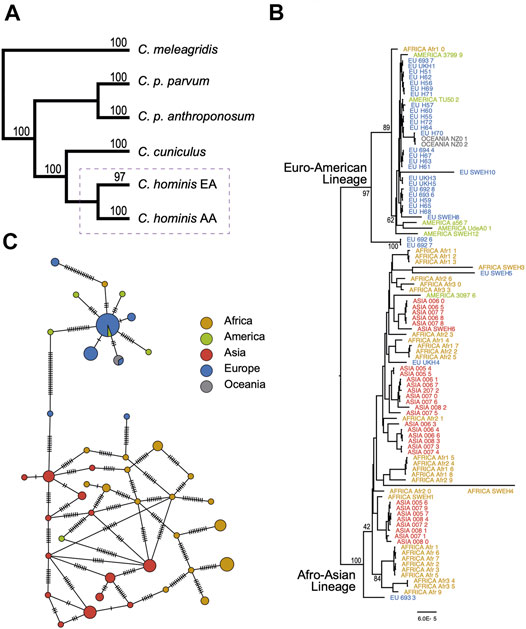
FIGURE 3. Evolutionary relationships among species and populations of Cryptosporidium hominis. (A) Summary of main phylogenetic relationships among the main human-infecting Cryptosporidium species. The complete tree is provided in the Supplementary Material. The dashed rectangle is expanded in (B,C). (B) Subtree depicting the phylogenetic relationships among C. hominis isolates from five continents. Tree inference based on 61 coding genes and 140,425 sites. (C) Haplotype network based on 116 segregating sites across 61 coding genes (see methods for details). The size of the circles is proportional to the number of individuals with each haplotype. The cross lines represent the number of expected mutations among haplotypes.
Four terminal branches showed exceptional longer branch lengths, one in the AA lineage, and three in the EA lineage. It is also particular to see that these four samples come from the same study and were sequenced with the same instrument, the ION TORRENT.
To further confirm the lineage segregation in C. hominis, haplotype analysis was performed based on the 61 neutrally evolving genes. Haplotype network is congruent with the two main lineages (Euro-American and Afro-Asian lineages, Figure 3C). However, we did not find structure within these two lineages, and haplotype diversity appears to be higher in the Afro-Asian lineage.
Genomic Deletions Analysis
Indel events are common in Cryptosporidium parasites (Arias-Agudelo et al., 2020). To assess the genomic deletion and insertion profiles within C. hominis species, genomic alignment of the assemblies against the C. parvum IOWA II reference was performed and analyzed. Insertion and deletion events were treated independently to give more clarity.
Deletion events were more common than insertions and spanned from 1 to 78 bases. The most frequent loss harbored a single base and was followed in frequency by the loss of three bases (Figure 4A). All the C. hominis genomes displayed a set of 3,958 shared deletion events. Furthermore, there are 609 specific deletion events in C. hominis EA lineage, and 2,338 specific deletion events in AA lineages.
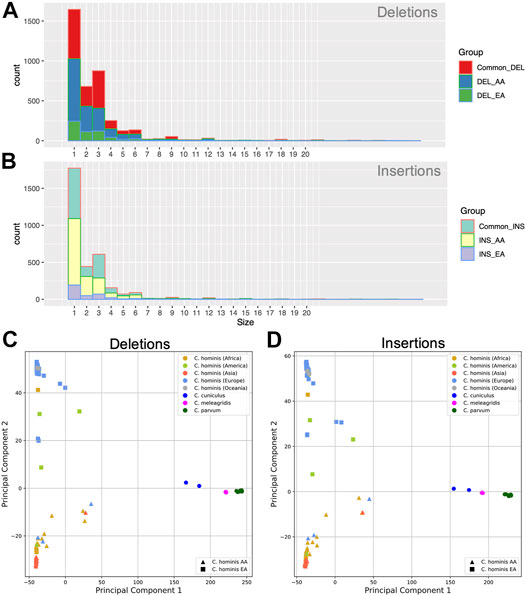
FIGURE 4. Genomic insertion and deletion events in C. hominis lineages. (A) Genomic deletion event length (base pair) frequency common to both lineages (Common_DEL), EA and AA. DEL_AA: Specific to the AA lineage. DEL_EA: specific for the EA lineage. (B) Genomic Insertion events length (base pair) frequency common to both lineages (Common_INS), EA and AA. INS_AA: Specific to the AA lineage. INS_EA: specific for the EA lineage. (C) Principal component analysis (PCA) of the genomic deletion events detected in C. hominis. (D) Principal component analysis (PCA) of the genomic insertion events detected in C. hominis.
Like the deletions, the insertion events spanned from 1 to 68 bases, being, in order, more frequent those that involve one and three bases (Figure 4B). Most of the insertion events were shared between both lineages (n = 3,272). Nonetheless, each lineage displayed specific insertion events. The AA lineage has 1,996 common insertions, while the EA lineage has 380.
Indel events may help to understand the relationship among the different parasite clades. With the aim to test the informative signal of the indel event profile of each genome, a Principal component analysis (PCA) analysis of the studied genomes was performed. As can be seen in Figures 4C,D, deletions or insertion event profiles segregate the parasites according to species and the lineage of C. hominis.
Discussion
We are at the dawn of a new era in microbiology. Nowadays, genomes of pathogens are analyzed directly from infected tissues or other human-derived samples like feces. Next-generation sequencing technologies not only fostered, in general, the high-resolution analysis of life on Earth but also prompted the genomic analysis of non-culturable pathogens like C. hominis. In this apicomplexan, we can observe how metagenomic analyses nurtured the advance in comparative and evolutionary genomics. Hundreds of reference genomes were generated in the last decade using the oocysts enrichment and metagenomic shotgun sequencing strategy. (Guo et al., 2015; Hadfield et al., 2015; Isaza et al., 2015; Ifeonu et al., 2016; Sikora et al., 2017; Gilchrist et al., 2018; Morris et al., 2019; Nader et al., 2019; Xu et al., 2019; Arias-Agudelo et al., 2020; Tichkule et al., 2021).
This metagenomic strategy poses challenges like the need for new standardized informatics methods to validate genome quality and purity since the infected individual could harbor a mixture of Cryptosporidium parasites: either different lineages of the same species or even multispecies infections. Furthermore, this strategy offers a great opportunity to study pathogens without the bias of artificial selection in vitro cultures and observe their genomes in natural settings under the complex conditions where they must co-exist and compete with other microorganisms.
The C. hominis genomes, sequenced so far, were generated in very dissimilar conditions, with different oocysts isolation protocols, and sequenced with different instruments in the last decade (Hadfield et al., 2015; Isaza et al., 2015; Ifeonu et al., 2016; Sikora et al., 2017; Gilchrist et al., 2018; Morris et al., 2019; Nader et al., 2019; Tichkule et al., 2021). The results presented in this work show that descriptive genome assembly statistics differ significantly, although some metrics have very narrow dispersion values enabling a rapid way to detect outliers. For instance, C. hominis assembled genome size has a narrow dispersion around the median value of 9,074,262 bp (IQR 20688 bp.), regardless of its continental origin or the NGS platform used. Additionally, the global nucleotide identity and SNVs count against the C. hominis UDEA01 also have a narrow dispersion close to their respective median values. This might be used as a marker for intraspecies boundaries for C. hominis genome around 99.7% identity and 4000 SNVs.
The median sequencing depth and the N50 values show the highest dispersion ranges. Median sequencing depth depends mainly on two main factors: The efficiency of the Cryptosporidium oocysts enrichment process, and the sequencing scale (number of reads generated). Even after an efficient enrichment process, C. hominis oocysts will be accompanied by intestinal microbiota, mainly bacteria. The more bacteria present in the sample, the fewer reads that will be available to support the target Cryptosporidium genome.
Insufficient sequencing depth might generate smaller assembled genome sizes, less accurate scaffolds, lower N50 values, and more fragmented genome models. Another situation that should be considered is that mixed parasite populations in one infected individual might lead, even under sufficient sequencing depth conditions, to more fragmented assemblies. In the case of samples with mixed Cryptosporidium species, it might be possible to observe outlier assemblies with larger-than-expected genome sizes and higher counts of alternate alleles.
It is noteworthy to mention that the phylogenomic analysis also enables to detection of outlier genome models since suspicious genomes with long branches can be spotted. In this work, four genomes showed extreme branch lengths, albeit they passed the genome quality thresholds. These four genomes come from different continents but have in common the instrument used for NGS data generation, the ION TORRENT. This instrument has shown to have higher mutation rates than Illumina instruments, supporting the hypothesis that these longer branches are more likely associated with sequencing artifacts rather than real higher evolution rates (Quail et al., 2012). This observation implies that genome assembly statistics alone are not enough for quality control. Complementary phylogenomic analyses help to detect genome assemblies that could accumulate higher error rates.
In general, the analyzed C. hominis genomes have low alternate allele counts, 85% with less than 470 events, suggesting that most of the infected individuals might harbor one clonal Cryptosporidium genome with minor nucleotide variants (0.005%).
The highest number of alternate alleles went as high as 112,225, nearly 34 times higher than the observed median value. In this specific patient, we might be dealing with a case of Cryptosporidium mixed species infection. This observation is supported by the facts that the global genome identity went down to 98.96% and that the Cryptosporidium assembled genome size was exceptionally large, exceeding 11 Mbp.
Several research works have described the presence of mixed C. hominis genotypes or even species in one infected individual (Cama et al., 2006; Gilchrist et al., 2018; Korpe et al., 2019; Sannella et al., 2019). These observations can be reconciled with our results considering that coinfecting C. hominis genotypes of other Cryptosporidium species could be below the detection threshold of the actual metagenomic approaches.
The phylogenomic analysis allowed us to discover two C. hominis lineages with continental segregation. These lineages were named based on the observed continental distribution bias as C. hominis Euro-American (EA) lineage and the C. hominis Afro-Asian (AA) lineage.
The reference isolate UDEA01 is part of the EA lineage, as well as the other Latin-American isolates UdeAa567 and SWEH12. This clade also encompasses isolates of the United States, the United Kingdom, Spain, and Greece. Only one genome belongs to a different continental region, Afr10, which was isolated from a human in Madagascar, Africa. All the genomes in the EA clade are classified as genotypes IbA10G2/IbA12G2 except for C. hominis UDEA01, which has been classified as genotype IeA11G3T3. Using an SNVs phylogenomic approach, Sikora et al. (2017) made a previous observation that supports our findings. In their study, C. hominis genomes of the genotype IbA10G2 separated in a monophyletic clade independent of other studied C. hominis genotypes. Another line of evidence that supports our finding was published recently by Tichkule et al. In their work, the researchers observed a geographic structuring of the C. hominis isolates in African countries (Tichkule et al., 2021).
Haplotype network analysis supported the geographical isolation of the EA and AA C. hominis lineages. Additionally, the AA lineage showed a higher haplotype diversity. Nevertheless, it must be highlighted that the sampling was higher in the AA lineage.
INDEL events are frequent in Cryptosporidium genomes (Arias-Agudelo et al., 2020). PCA analysis of the INDEL profiles showed that they can segregate C. hominis of the other studied Cryptosporidium species: C. parvum, C. meleagridis, and C cuniculus. Additionally, INDEL events profile showed to be taxonomically informative in C. hominis since they were able to segregate the two lineages, adding support to the previous observe results.
Summarizing, three independent lines of evidence support our observation that C. hominis encompasses two separate lineages. These two monophyletic lineages have a differential continental distribution pattern with a clear dominance of one lineage in Eastern Europe and the Americas, while the other is the main lineage in Africa and Asia.
Previous research works on infectious diseases have demonstrated a phylogeographic relationship among European and American human populations. This is the case for the etiological agent of human tuberculosis, Mycobacterium tuberculosis. This pathogen presents a specific lineage, called the LAM family (L4.3), which is more common in the Mediterranean and Latin-American countries, and showed the spread of the bacteria associated with human migrations from Europe to America (Gagneux et al., 2006; Brynildsrud et al., 2018).
By the beginning of the 2000s C. parvum and C. hominis were considered a single species (Sulaiman et al., 2000), with the name of the former as the type species. Two years later, Morgan-Ryan et al., using complementary molecular analysis showed that, in fact, two closely related species could be discriminated and C. hominis and C. parvum were separated as independent species (Morgan-Ryan et al., 2002).
Ending the next decade, in 2019, within the C. parvum clade, a new subspecies was discovered, C. parvum antroponosum (Nader et al., 2019). Cryptosporidium parvum parvum is described as a zoonotic parasite while the new subspecies C. parvum antroponosum has an anthroponotic transmission scheme. These discoveries were achieved thanks to the incorporation of more resolutive molecular analytical methods, like phylogenomic tools.
In this work, we present a similar observation for C. hominis, this new lineage may represent a new subspecies, but its confirmation and biological implications should be further investigated. Previous evolutionary works have shown an evolution model in Cryptosporidium parasites where speciation events are related to new host adaptations. In almost every major vertebrate lineage, an adapted Cryptosporidium species have been described (Garcia-R and Hayman, 2016). In the case of C. hominis, a particularly interesting question raises since C. hominis has been already adapted to our human ancestors around 6 million years ago (Garcia-R and Hayman, 2016). Additional work should be performed in order to establish the origin of the separation of the C. hominis lineages.
Data Availability Statement
The datasets presented in this study can be found in online repositories (NCBI SRA). See Supplementary Table S1. New Colombian C hominis genome read data were deposited in the NCBI SRA database accession number SRR14522748 (https://dataview.ncbi.nlm.nih.gov/object/SRR14522748).
Author Contributions
FC: Bioinformatic analysis design, Data processing, and statistical analysis. Article writing and review. AG-D: Parasite sample collection and enrichment. Patient enrollment and bioethics follow-up. Literature review. Articles writing and review. LA: Parasite wet-lab experiments. Parasite enrichment and DNA extraction and QC. Literature review. GG-M: Parasite sample collection and enrichment. Patient enrollment and bioethics follow-up. Articles review. JD: Phylogenetic and haplotype analysis. Articles review. JA: Study design. Bioinformatic analysis design, Data processing, and statistical analysis. Article writing and review.
Funding
COLCIENCIAS Convocatoria 777-2017 Para Proyectos de Ciencia, Tecnología e Innovación en Salud 2017, project name: “Estudio de prevalencia, diversidad genética y genomica de Cryptosporidium en población VIH positiva de Antioquia'.” 111577757608. Vicerrectoría de Investigación (CODI), Universidad de Antioquia, grant “Finalización del genoma de referencia de C. hominis aislado UdeA01'”: 2017-16-171.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.740940/full#supplementary-material
References
Arias-Agudelo, L. M., Garcia-Montoya, G., Cabarcas, F., Galvan-Diaz, A. L., and Alzate, J. F. (2020). Comparative Genomic Analysis of the Principal Cryptosporidium Species that Infect Humans. PeerJ 8, e10478. doi:10.7717/peerj.10478
Bandelt, H. J., Forster, P., and Röhl, A. (1999). Median-Joining Networks for Inferring Intraspecific Phylogenies. Mol. Biol. Evol. 16 (1), 37–48. doi:10.1093/oxfordjournals.molbev.a026036
Brynildsrud, O. B., Pepperell, C. S., Suffys, P., Grandjean, L., Monteserin, J., Debech, N., et al. (2018). Global Expansion of Mycobacterium Tuberculosis Lineage 4 Shaped by Colonial Migration and Local Adaptation. Sci. Adv. 4 (10), eaat5869. doi:10.1126/sciadv.aat5869
Cacciò, S. M., and Chalmers, R. M. (2016). Human Cryptosporidiosis in Europe. Clin. Microbiol. Infect. 22 (6), 471–480. doi:10.1016/j.cmi.2016.04.021
Cama, V., Gilman, R. H., Vivar, A., Ticona, E., Ortega, Y., Bern, C., et al. (2006). MixedCryptosporidiumInfections and HIV. Emerg. Infect. Dis. 12 (6), 1025–1028. doi:10.3201/eid1206.060015
Efstratiou, A., Ongerth, J. E., and Karanis, P. (2017). Waterborne Transmission of Protozoan Parasites: Review of Worldwide Outbreaks - an Update 2011-2016. Water Res. 114, 14–22. doi:10.1016/j.watres.2017.01.036
Feng, Y., Ryan, U. M., and Xiao, L. (2018). Genetic Diversity and Population Structure of Cryptosporidium. Trends Parasitol. 34, 997–1011. doi:10.1016/j.pt.2018.07.009
Gagneux, S., Deriemer, K., Van, T., Kato-Maeda, M., de JongDe Jong, B. C., Narayanan, S., et al. (2006). Variable Host-Pathogen Compatibility in Mycobacterium Tuberculosis. Proc. Natl. Acad. Sci. U S A. 103 (8), 2869–2873. Available at: www.pnas.org. doi:10.1073/pnas.0511240103
Garcia-R, J. C., and Hayman, D. T. S. (2016). Origin of a Major Infectious Disease in Vertebrates: The Timing of Cryptosporidium Evolution and its Hosts. Parasitology 143 (13), 1683–1690. doi:10.1017/S0031182016001323
Gilchrist, C. A., Cotton, J. A., Burkey, C., Arju, T., Gilmartin, A., Lin, Y., et al. (2018). Genetic Diversity of Cryptosporidium Hominis in a Bangladeshi Community as Revealed by Whole-Genome Sequencing. J. Infect. Dis. 218 (2), 259–264. doi:10.1093/infdis/jiy121
Guo, Y., Tang, K., Rowe, L. A., Li, N., Roellig, D. M., Knipe, K., et al. (2015). Comparative Genomic Analysis Reveals Occurrence of Genetic Recombination in Virulent Cryptosporidium Hominis Subtypes and Telomeric Gene Duplications in Cryptosporidium Parvum. BMC Genomics 16 (1), 320. doi:10.1186/s12864-015-1517-1
Hadfield, S. J., Pachebat, J. A., Swain, M. T., Robinson, G., Cameron, S. J., Alexander, J., et al. (2015). Generation of Whole Genome Sequences of New Cryptosporidium Hominis and Cryptosporidium Parvum Isolates Directly from Stool Samples. BMC Genomics 16 (1), 650. doi:10.1186/s12864-015-1805-9
Ifeonu, O. O., Chibucos, M. C., Orvis, J., Qi, S., Elwin, K., Guo, F., et al. (2016). Annotated Draft Genome Sequences of Three Species of Cryptosporidium: Cryptosporidium Meleagridis Isolate UKMEL1, C. Baileyi Isolate TAMU-09Q1 and C. Hominis Isolates TU502 2012 and UKH1. Pathog. Dis. 74 (7), 1–5. doi:10.1093/femspd/ftw080
Innes, E. A., Chalmers, R. M., Wells, B., and Pawlowic, M. C. (2020). A One Health Approach to Tackle Cryptosporidiosis. Trends Parasitol. 36, 290–303. doi:10.1016/j.pt.2019.12.016
Isaza, J. P., Galván, A. L., Polanco, V., Huang, B., Matveyev, A. V., Serrano, M. G., et al. (2015). Revisiting the Reference Genomes of Human Pathogenic Cryptosporidium Species: Reannotation of C. Parvum Iowa and a New C. Hominis Reference. Sci. Rep. 5, 16324. doi:10.1038/srep16324
Kar, S., Gawlowska, S., Daugschies, A., and Bangoura, B. (2011). Quantitative Comparison of Different Purification and Detection Methods for Cryptosporidium Parvum Oocysts. Vet. Parasitol. 177 (3), 366–370. doi:10.1016/j.vetpar.2010.12.005
Korpe, P. S., Gilchrist, C., Burkey, C., Taniuchi, M., Ahmed, E., Madan, V., et al. (2019). Case-Control Study of Cryptosporidium Transmission in Bangladeshi Households. Clin. Infect. Dis. 68 (7), 1073–1079. doi:10.1093/cid/ciy593
Kotloff, K. L., Nataro, J. P., Blackwelder, W. C., Nasrin, D., Farag, T. H., Panchalingam, S., et al. (2013). Burden and Aetiology of Diarrhoeal Disease in Infants and Young Children in Developing Countries (The Global Enteric Multicenter Study, GEMS): A Prospective, Case-Control Study. Lancet 382 (9888), 209–222. doi:10.1016/S0140-6736(13)60844-2
Lake, I. R., Harrison, F. C. D., Chalmers, R. M., Bentham, G., Nichols, G., Hunter, P. R., et al. (2007). Case-Control Study of Environmental and Social Factors Influencing Cryptosporidiosis. Eur. J. Epidemiol. 22 (11), 805–811. doi:10.1007/s10654-007-9179-1
Lanfear, R., Calcott, B., Ho, S. Y. W., and Guindon, S. (2012). PartitionFinder: Combined Selection of Partitioning Schemes and Substitution Models for Phylogenetic Analyses. Mol. Biol. Evol. 29 (6), 1695–1701. doi:10.1093/molbev/mss020
Lanfear, R., Calcott, B., Kainer, D., Mayer, C., and Stamatakis, A. (2014). Selecting Optimal Partitioning Schemes for Phylogenomic Datasets. BMC Evol. Biol. 14 (1), 82–14. doi:10.1186/1471-2148-14-82
Leigh, J. W., and Bryant, D. (2015). Popart : Full‐Feature Software for Haplotype Network Construction. Methods Ecol. Evol. 6 (9), 1110–1116. doi:10.1111/2041-210X.12410
Morgan-Ryan, U. M., Fall, A., Ward, L. A., Hijjawi, N., Sulaiman, I., Payer, R., et al. (2002). Cryptosporidium Hominis N. Sp. (Apicomplexa: Cryptosporidiidae) from Homo Sapiens. J. Eukaryot. Microbiol. 49 (6), 433–440. doi:10.1111/j.1550-7408.2002.tb00224.x
Morris, A., Robinson, G., Swain, M. T., and Chalmers, R. M. (2019). Direct Sequencing of Cryptosporidium in Stool Samples for Public Health. Front. Public Health 7, 360. doi:10.3389/fpubh.2019.00360
Nader, J. L., Mathers, T. C., Ward, B. J., Pachebat, J. A., Swain, M. T., Robinson, G., et al. (2019). Evolutionary Genomics of Anthroponosis in Cryptosporidium. Nat. Microbiol. 4 (5), 826–836. doi:10.1038/s41564-019-0377-x
Pollock, K. G. J., Ternent, H. E., Mellor, D. J., Chalmers, R. M., Smith, H. V., Ramsay, C. N., et al. (2010). Spatial and Temporal Epidemiology of Sporadic Human Cryptosporidiosis in Scotland. Zoonoses and Public Health 57 (7–8), 487–492. doi:10.1111/j.1863-2378.2009.01247.x
Quail, M., Smith, M. E., Coupland, P., Otto, T. D., Harris, S. R., Connor, T. R., et al. (2012). A Tale of Three Next Generation Sequencing Platforms: Comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq Sequencers. BMC Genomics 13, 341. doi:10.1186/1471-2164-13-341
Ryan, U., Fayer, R., and Xiao, L. (2014). Cryptosporidiumspecies in Humans and Animals: Current Understanding and Research Needs. Parasitology 141 (13), 1667–1685. doi:10.1017/S0031182014001085
Sannella, A. R., Suputtamongkol, Y., Wongsawat, E., and Cacciò, S. M. (2019). A Retrospective Molecular Study of Cryptosporidium Species and Genotypes in HIV-Infected Patients from Thailand. Parasites Vectors 12 (1), 91. doi:10.1186/s13071-019-3348-4
Sikora, P., Andersson, S., Winiecka-Krusnell, J., Hallström, B., Alsmark, C., Troell, K., et al. (2017). Genomic Variation in IbA10G2 and Other Patient-Derived Cryptosporidium Hominis Subtypes. J. Clin. Microbiol. 55 (3), 844–858. doi:10.1128/JCM.01798-16
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: Assessing Genome Assembly and Annotation Completeness with Single-Copy Orthologs. Bioinformatics 31 (19), 3210–3212. doi:10.1093/bioinformatics/btv351
Sulaiman, I. M., Morgan, U. M., Thompson, R. C. A., Lal, A. A., and Xiao, L. (2000). Phylogenetic Relationships of Cryptosporidium Parasites Based on the 70-Kilodalton Heat Shock Protein (HSP70) Gene. Appl. Environ. Microbiol. 66 (6), 2385–2391. doi:10.1128/AEM.66.6.2385-2391.2000
Tichkule, S., Jex, A. R., Oosterhout, C. V., Rosa Sannella, A., Krumkamp, R., Aldrich, C., et al. (2021). Comparative Genomics Revealed Adaptive Admixture in Cryptosporidium Hominis in Africa. Microb. Genomics 7 (1), 1–14. doi:10.1099/mgen.0.000493
Widmer, G., Köster, P. C., and Carmena, D. (2020). Cryptosporidium Hominis Infections in Non-human Animal Species: Revisiting the Concept of Host Specificity. Int. J. Parasitol. 50, 253–262. doi:10.1016/j.ijpara.2020.01.005
Xiao, L., and Feng, Y. (2017). Molecular Epidemiologic Tools for Waterborne Pathogens Cryptosporidium Spp. And Giardia Duodenalis. Food Waterborne Parasitol. 8-9, 14–32. doi:10.1016/j.fawpar.2017.09.002
Xiao, L., and Feng, Y. (2008). Zoonotic Cryptosporidiosis. FEMS Immunol. Med. Microbiol. 52, 309–323. doi:10.1111/j.1574-695X.2008.00377.x
Xu, Z., Guo, Y., Roellig, D. M., Feng, Y., and Xiao, L. (2019). Comparative Analysis Reveals Conservation in Genome Organization Among Intestinal Cryptosporidium Species and Sequence Divergence in Potential Secreted Pathogenesis Determinants Among Major Human-Infecting Species. BMC Genomics 20 (1), 1–15. doi:10.1186/s12864-019-5788-9
Keywords: Cryptosporidium hominis, comparative genomics, de novo genome assembly, evolution, lineage
Citation: Cabarcas F, Galvan-Diaz AL, Arias-Agudelo LM, García-Montoya GM, Daza JM and Alzate JF (2021) Cryptosporidium hominis Phylogenomic Analysis Reveals Separate Lineages With Continental Segregation. Front. Genet. 12:740940. doi: 10.3389/fgene.2021.740940
Received: 13 July 2021; Accepted: 27 September 2021;
Published: 14 October 2021.
Edited by:
Jun-Hu Chen, National Institute of Parasitic Diseases, ChinaReviewed by:
Karen Luisa Haag, Federal University of Rio Grande do Sul, BrazilAdam Sateriale, Francis Crick Institute, United Kingdom
Alejandro Castellanos-Gonzalez, University of Texas Medical Branch at Galveston, United States
Copyright © 2021 Cabarcas, Galvan-Diaz, Arias-Agudelo, García-Montoya, Daza and Alzate. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan F. Alzate, jfernando.alzate@udea.edu.co