Combined approaches for increasing fetal hemoglobin (HbF) and de novo production of adult hemoglobin (HbA) in erythroid cells from β-thalassemia patients: treatment with HbF inducers and CRISPR-Cas9 based genome editing
 Alessia Finotti1,2*
Alessia Finotti1,2*  Roberto Gambari1,2*
Roberto Gambari1,2*- 1Center “Chiara Gemmo and Elio Zago” for the Research on Thalassemia, University of Ferrara, Ferrara, Italy
- 2Department of Life Sciences and Biotechnology, University of Ferrara, Ferrara, Italy
Genome editing (GE) is one of the most efficient and useful molecular approaches to correct the effects of gene mutations in hereditary monogenetic diseases, including β-thalassemia. CRISPR-Cas9 gene editing has been proposed for effective correction of the β-thalassemia mutation, obtaining high-level “de novo” production of adult hemoglobin (HbA). In addition to the correction of the primary gene mutations causing β-thalassemia, several reports demonstrate that gene editing can be employed to increase fetal hemoglobin (HbF), obtaining important clinical benefits in treated β-thalassemia patients. This important objective can be achieved through CRISPR-Cas9 disruption of genes encoding transcriptional repressors of γ-globin gene expression (such as BCL11A, SOX6, KLF-1) or their binding sites in the HBG promoter, mimicking non-deletional and deletional HPFH mutations. These two approaches (β-globin gene correction and genome editing of the genes encoding repressors of γ-globin gene transcription) can be, at least in theory, combined. However, since multiplex CRISPR-Cas9 gene editing is associated with documented evidence concerning possible genotoxicity, this review is focused on the possibility to combine pharmacologically-mediated HbF induction protocols with the “de novo” production of HbA using CRISPR-Cas9 gene editing.
1 Introduction
The β-thalassemias are a genetically heterogenous group of hereditary hematological diseases caused by hundreds of mutations of the adult β-globin gene, leading to low or absent production of adult hemoglobin (HbA) in erythroid cells (Weatherall, 2001; Galanello and Origa, 2010; Fucharoen and Weatherall, 2016; Origa, 2017). The thalassemia syndromes, together with sickle-cell disease (SCD), are impactful diseases especially in developing countries, where they maintain a very high frequency within the population, due to the lack of genetic counselling and prenatal diagnosis (Weatherall, 2001; Origa, 2017). Regular blood transfusion, chelation therapy and bone marrow transplantation (Fucharoen and Weatherall, 2016) are currently employed for the clinical management of β-thalassemia patients. Alternatively, induction of fetal hemoglobin (HbF) can be considered (Forget, 1998; Musallam et al., 2012; Liu et al., 2014; Sripichai and Fucharoen, 2016a), taking into account the increasing number of laboratory and clinical evidences demonstrating that reactivation of HbF production in adult life can be beneficial for β-thalassemia, leading in some cases to transfusion-independency (Sripichai and Fucharoen, 2016a). As for other human pathologies and rare diseases, most of the new innovative approaches for developing protocols of possible interest for future treatments of β-thalassemias are focusing on personalized treatments on one hand, and precise targeting on the other. In order to reach these objectives, an exciting strategy recently proposed for β-thalassemia (and other genetic diseases) is genome editing of human hematopoietic stem and progenitor cells (HSPC) (Boulad et al., 2018; Magrin et al., 2019; Ernst et al., 2020; Ali et al., 2021; Ferrari et al., 2021; Karamperis et al., 2021; Rosanwo and Bauer, 2021; Quintana-Bustamante et al., 2022; Eckrich and Frangoul, 2023; Khiabani et al., 2023). In this respect, the Clustered Regularly Interspaced Palindromic Repeats (CRISPR)-Cas9 nuclease system should be considered among the most studied gene editing strategies (Dever et al., 2016; Hu, 2016; Lau, 2018; Papasavva et al., 2019).
In addition to possible applications in the therapeutic field, gene editing and the most recent base editing and prime editing approaches, are powerful tools to identify novel druggable targets. For instance, Ravi NS et al. reported an example describing how CRISPR based editing can be applied to mapping gene regulatory elements in highly homologous loci (in this case the γ-globin gene promoter) (Ravi et al., 2022), strongly supporting the concept that this approach will be in the future a prominent therapeutic strategy for monogenetic disorders, such as β-thalassemia, as demonstrated by the recently published clinical study performed by Frangoul et al., who reported the results on two patients (one with transfusion-dependent thalassemia, the other with sickle-cell disease) who received autologous CD34+ cells edited with CRISPR-Cas9 targeting the BCL11A enhancer (ClinicalTrials.gov numbers, NCT03655678 and NCT03745287) (Frangoul et al., 2021). Interestingly, after 1 year follow-up, the β-thalassemia patient exhibited pancellular increases in HbF production and transfusion independence, while in the SCD patient elimination of vaso-occlusive episodes was found (Frangoul et al., 2021).
The extremely high variety of CRISPR-Cas9 based tailored approaches (Figure 1) opens new avenues in the management of β-thalassemias, also based on combined treatments with the aim to rescue HbA production and, in parallel, reactivate the expression of γ-globin genes, leading to HbF production.
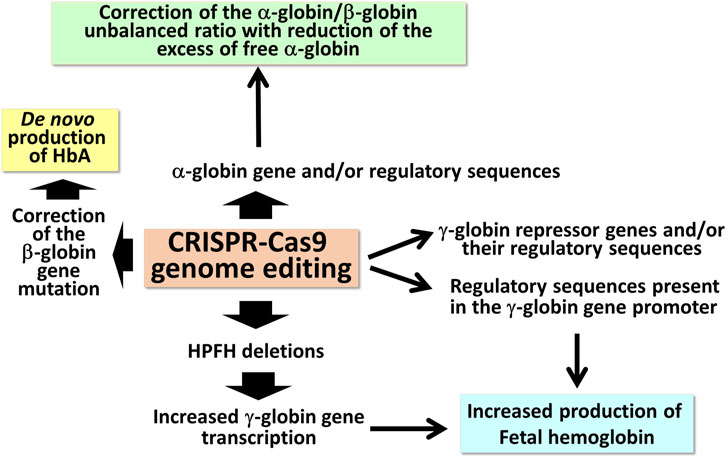
FIGURE 1. Possible applications of CRISPR-Cas9 based gene editing to achieve end-points of interest in the management of β-thalassemia patients.
In this short review, we will focus on gene editing based on the CRISPR-Cas9 technology (for which a large number of studies are available in the literature), trying to discuss whether this approach can be suitable for multiple interventions, such as combined treatments to reach “de novo” production of HbA together with increased HbF content.
2 CRISPR-Cas9 based approaches to reactivate γ-globin gene expression
The first observations strongly suggesting that reactivation of the silent γ-globin genes in adult β-thalassemic patients might be highly beneficial to the patients, ameliorating their clinical phenotype, were reported in studies focusing on rare forms of β0-thalassemia, associated with large genomic deletions causing HPFH (hereditary persistence of fetal hemoglobin) and identified as “HPFH deletions” in Figure 1; these patients are characterized by absence of β-globin production, but presence of high levels of γ-globin chains, resulting in high levels of HbF associated with a relatively benign clinical course (Forget, 1998; Sharma et al., 2020). More recent clinical studies support the concept that naturally higher production of HbF improves the clinical phenotype of a variety of β-thalassemia patients (Uda et al., 2008; Galanello et al., 2009; Nuinoon et al., 2010; Badens et al., 2011; Danjou et al., 2012). Accordingly, these observations have prompted the activity of a large number of research groups in performing studies on inducers of HbF that can be proposed in clinical trials, as they reproduce to some extent what occurs in β-thalassemia patients with a natural persistence of higher levels of HbF (Gambari and Fibach, 2007; Finotti and Gambari, 2014; Lavelle et al., 2018; Nuamsee et al., 2021). In addition to HbF inducers, reactivation of HbF production can be obtained using different CRISPR-Cas9-based gene editing approaches (Figure 2).
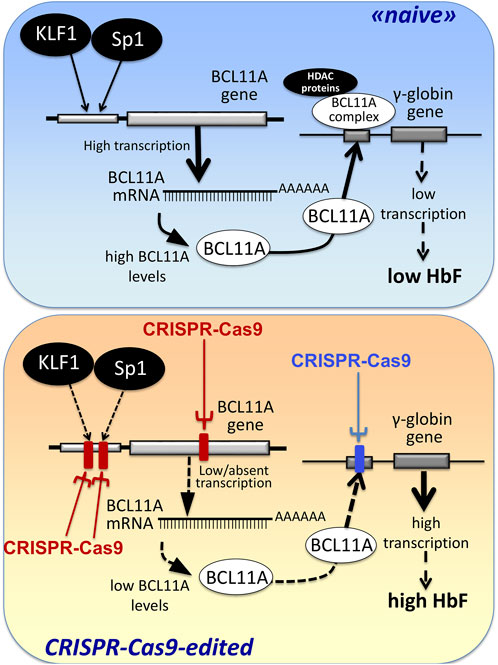
FIGURE 2. Alternative CRISPR-Cas9 based protocols for the reactivation of the expression of γ-globin genes and the increased production of HbF.
One very interesting strategy has been proposed by several groups trying to reproduce genetic alterations leading to an HPFH in phenotype. For instance, Ye et al. (2016) proposed genome editing using CRISPR-Cas9 to create an HPFH genotype in hematopoietic stem/progenitor cells (HSPCs), obtaining an increase of expression of γ-globin genes. In fact, these gene edited regions were known (or were hypothesized) to contain putative fetal hemoglobin (HbF) silencers, as also pro-posed by Antoniani et al. (2018) and more recently by Venkatesan et al. (2023a).
In the context of gene deletions for increasing expression of γ-globin genes, an interesting study was published by Topfer et al. (2022), who found that CRISPR-Cas9 disruption of the adult β-globin (HBB) gene promoter was associated with reactivation of γ-globin gene expression; in these experimental conditions the γ-globin gene outcompetes the HBB gene for binding to the LCR (Topfer et al., 2022). The experimental gene editing strategy designed by Topfer and collaborators was based on the observation that in all the deletion-associated HPFH conditions the proximal adult β-globin (HBB) promoter was deleted (Topfer et al., 2022). This study is an excellent validation of the recently proposed model of the switch from fetal γ-globin gene expression to transcription of the adults β-globin gene (Deng et al., 2014). This model is based on the hypothesis that both HBB and HBG promoters compete for the LCR (Carter et al., 2002a; Okamura et al., 2009; Deng et al., 2014). The regulatory mechanisms operating in this model are fundamental for tissue-specific transcriptional control of globin gene expression. The interplay between local DNA regulatory elements and remodeling of chromatin and transcription at the globin gene cluster have been extensively discussed (Carter et al., 2002b; Chakalova et al., 2005) focusing also on the requirement of LCR to open the chromatin. In this context, the disruption of the adult β-globin (HBB) gene promoter leads to deep changes in promoter interactions with the LCR enhancer.
An alternative view has been followed by several studies based on the established concept that γ-globin genes are under the control of transcriptional repressors. The objective, in this case, is a CRISPR-Cas9 based gene editing finalized at a) the knock-out of repressors of γ-globin gene transcription (such as BCL11A) or b) the disruption of their binding sites present within regulatory regions of the β-like gene cluster (including those present within the γ-globin gene). A pictorial representation of these strategies is shown in Figure 2, while a summary of interesting studies in this research field is reported in Table 1 (Canver et al., 2015; Shariati et al., 2016; Martyn et al., 2018; Psatha et al., 2018; Shariati et al., 2018; Khosravi et al., 2019; Weber et al., 2020; Fu et al., 2022; Han et al., 2022; Wakabayashi et al., 2022).
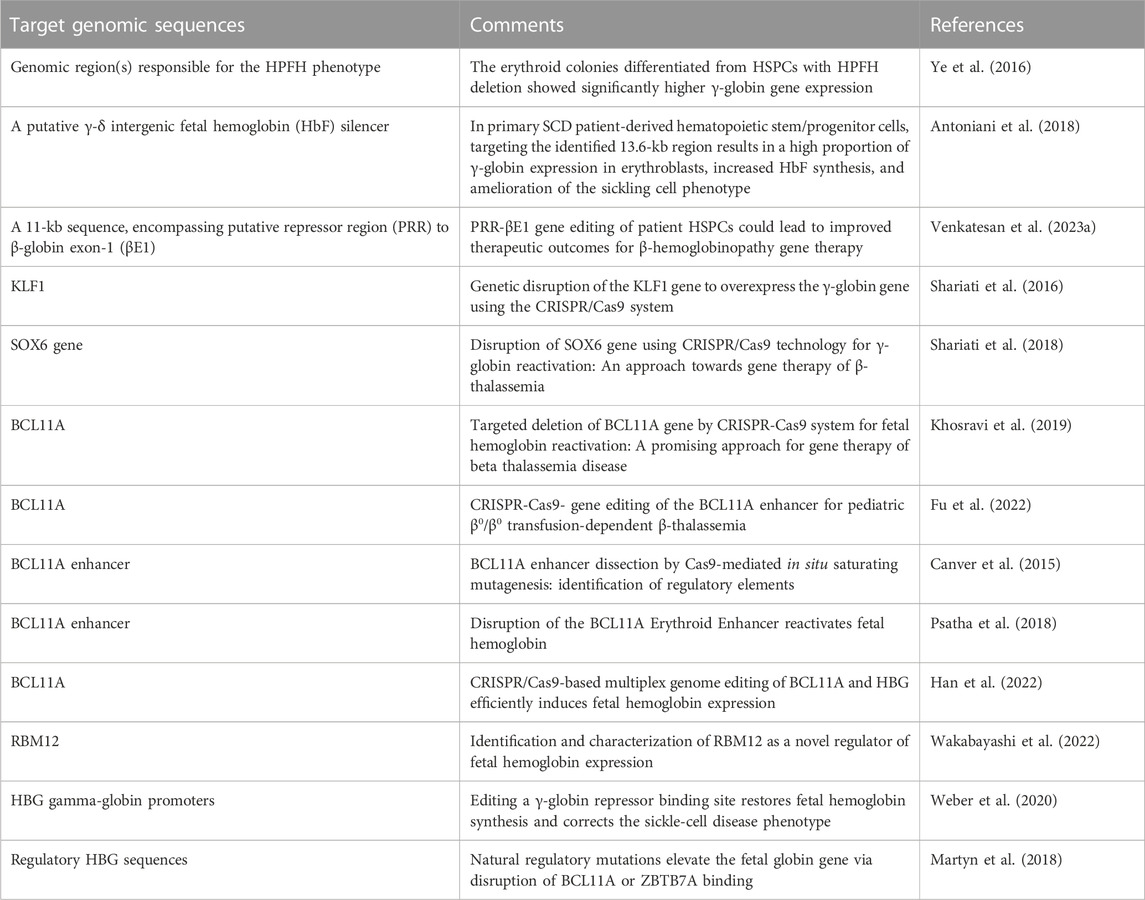
TABLE 1. CRISPR-Cas9 gene editing for Fetal Hemoglobin (HbF) reactivation.
2.1 Deletion of repressors of γ-globin genes or their enhancer regions
A key study in this field of investigation was reported by Canver et al. (2015), who performed a BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. They used the HUDEP-2 cell line as screening system and primary human CD34+ hematopoietic stem and progenitor cells (HSPCs) for validation of the results obtained (Canver et al., 2015). This important study generated a detailed enhancer map, very useful to design therapeutic genome editing strategies. These results were confirmed using another experimental model system by Khosravi et al., who supported the proof-of-principle that deletion(s) of part of the BCL11A gene (including relevant regulatory regions present within the BCL11A enhancer) is (are) associated with the reactivation of HbF production (Khosravi et al., 2019). Using the CRISPR-Cas9 genome-editing strategy on human K562 cells, they deleted a 200 bp genomic region within the BCL11A enhancer, obtaining a strong induction of γ-hemoglobin expression.
Along with the same approach, the association between disruption of γ-globin transcriptional repressor genes by gene editing, and reactivation of expression of the γ-globin genes and production of HbF is sustained also by the study performed by Psatha et al. (2018) on BCL11A and by other research groups using CRISPR-Cas9 gene editing on other repressor genes, such as KLF-1 (Shariati et al., 2016), SOX6 (Shariati et al., 2018) and RBM12 (Wakabayashi et al., 2022).
Fu et al. (2022) applied the study to a CRISPR-Cas9-mediated gene editing protocol designed to alter the BCL11A erythroid enhancer by disrupting the GATA1-binding site at position +58. Preliminary results of an ongoing phase 1/2 trial (NCT04211480) have been presented in this report, evaluating safety and efficacy of this gene editing therapeutic approach in children with blood transfusion-dependent β-thalassemia (TDT). Concerning safety, when BCL11A enhancer-edited, autologous, hematopoietic stem and progenitor cells were transplanted, engraftment was obtained with high efficiency, and adverse events (AEs) were negligible, considered unrelated to gene editing and resolved after specific treatments (Fu et al., 2022). Concerning efficacy, transfusion independency was reached for >18 months after treatment, and Hb increased (Fu et al., 2022). The study by Fu et al. is strongly in agreement with the results obtained by the clinical trials NCT03655678 and NCT03745287 and reported by Frangoul et al. (2021).
Overall, these studies support the concept that disruption of genes encoding a transcriptional repressor of γ-globin genes and/or disruption of their enhancers can be applied and translated into clinical protocols aimed at reactivation of HbF production.
2.2 Mutagenesis of transcriptional repressor binding sites of the γ-globin gene promoter: reconstitution of the HPFH phenotype using CRISPR-Cas9 based approaches
Martyn et al. (2018) reported a key study focusing on the binding sites of the γ-globin gene repressors BCL11A and ZBTB7A (also known as LRF), located at −115 and −200 bp from the start of transcription of γ-globin genes. These sites are bound directly by the BCL11A and ZBTB7A repressors. The results obtained demonstrated that the CRISPR-Cas9 based disruption of the repressor binding raised γ-globin gene expression in erythroid cells (Martyn et al., 2018).
3 Gene editing for precise correction of the β-globin gene mutations
Table 2 summarizes published gene editing studies aiming at demonstrating highly efficient corrections of the mutated β-globin gene in β-thalassemias (Xu et al., 2015; Liu et al., 2017; Gabr et al., 2020; Cosenza et al., 2021; Lu et al., 2022; Trakarnsanga et al., 2022). The target cells (or cell lines) have been several and heterogenous, confirming the efficacy and reproducibility of the gene editing approach and protocols. These include primary erythroid progenitors isolated from β-thalassemia patients, induced pluripotent stem cells, established, patient-derived cell lines and erythroid cells from β-thalassemia mice carrying selective β-thalassemia mutations. For instance, Trakarnsanga et al. (2022) reported the genetic correction of hemoglobin E in an immortalized hemoglobin E/β-thalassemia cell line using the CRISPR/Cas9 system. This study demonstrated that the HbE-corrected clones restored β-globin production with reduced levels of HbE upon erythroid differentiation. Patient-derived pluripotent stem cells were employed by Liu et al. (2017) to correct a β41-42 (TCTT) deletion mutation. Thalassemic mice were employed by Lu et al. (2022) to demonstrate in vivo effects of the correction of the defect of RNA splicing in β654-thalassemia mice using CRISPR/Cas9 gene editing technology. Interestingly, hematologic parameters of all of the edited β654 founders and their offspring were found to be significantly improved compared to those of the control non-edited mice, consistent with the restoration of wild-type β-globin RNA expression (Lu et al., 2022). As a final example, Cosenza et al. reported a CRISPR based approach to obtain efficient correction of the β039-thalassemia mutation in erythroid cells isolated from homozygous β039-thalassemia patients (Cosenza et al., 2021).

TABLE 2. Gene editing procedures for HBB mutations.
In Table 2 a few examples are also reported discussing the newly developed prime and base editing of β-thalassemia mutations (Liang et al., 2017; Zhang et al., 2022; Badat et al., 2023; Hardouin et al., 2023). These novel approaches are expected to limit genotoxic of the gene editing procedures, especially those due to homology-directed repair (HDR), activated following the introduction of DNA double-strand breaks (DSB) during the conventional CRISPR approach (Liang et al., 2017; Komor et al., 2018; Zeng et al., 2020; Collantes et al., 2021; Zhang et al., 2022; Badat et al., 2023; Carusillo et al., 2023; Hardouin et al., 2023).
In respect to the limitations of the CRISPR-Cas9 based procedures, HDR is inherently genotoxic in somatic cells; therefore, the recent development of base editing procedures that edit a target base without requiring the generation of DSB or HDR offers an alternative and very appealing approach (Liang et al., 2017; Zhang et al., 2022; Badat et al., 2023; Hardouin et al., 2023). For instance, Hardouin et al. developed a strategy to correct one of the most prevalent BT mutations (IVS1-110 [G>A]) using the SpRY-ABE8e base editor (Hardouin et al., 2023). These and similar approaches leads to gene editing without double-stranded DNA breaks [64–67, and https://www.biorxiv.org/content/10.1101/2022.06.01.494256v1].
Besides limitations due to possible genotoxicity and applications of the protocols in clinical settings, all these studies concurrently demonstrated the possibility to use these highly efficient gene-editing protocols for personalized treatment and precision medicine of β-thalassemia.
4 CRISPR-Cas9 protocols for decreasing the excess of free α-globin
The pathophysiology of β-Thalassemia is strongly associated with an excess of free α-globin chains caused by the imbalance between α- and β-globin chains, especially in the case of β0-Thalassemia; this is the major factor leading to ineffective erythropoiesis and hemolysis (Schrier, 2002; Xie et al., 2007; Voon et al., 2008; Mettananda et al., 2015; Mettananda et al., 2017a). The reduction of free α-globin chains has a clear clinically beneficial impact, as suggested by studies demonstrating that when α-thalassemia is co-inherited with β-thalassemia, the excess free α-globin chains is in most cases significantly reduced, ameliorating the clinical severity. Furthermore, Lechauve et al. (2019) experimentally validated this hypothesis, demonstrating the role of the autophagy-activating kinase ULK-1 (Unc-51-like kinase 1) in promoting autophagy-associated controlled clearance of the free α-globin chains. Using β-thalassemic mice, it was found that the loss of the ULK1 gene reduced autophagy, exacerbating the disease phenotypes, in association with a lack in the clearance of α-globin in red blood cell precursors. On the contrary, rapamycin-mediated ULK1 and autophagy reduce free α-globin accumulation in erythroid cells.
The expression of the α-globin genes was reduced by Mettananda et al. (2017b) using CRISPR/Cas9 genome editing to mimic a natural mutation causing α-thalassemia in association with a deletion of the MCS-R2 α-globin enhancer (Mettananda et al., 2017b). This CRISPR-Cas9 based approach might be of clinical relevance, as this strategy caused a reduction in α-globin expression and a correction of the globin chain imbalance, in erythroid-differentiated, gene-edited edited CD34+ cells from β-thalassemia patients.
A second study concerning this issue was published by Pavani et al. (2021), who were able to demonstrate a correction of the pathological phenotype of β-thalassemia by CRISPR/Cas9 editing of the α-globin locus in human hematopoietic stem cells. In this study, α-globin was downregulated, by HBA2 gene deletion, in order to generate an α-thalassemia trait, with associated correction of α/β globin imbalance (Pavani et al., 2021).
5 Combined protocols based on gene therapy and gene editing
The translation of the gene editing procedures from pre-clinical studies to clinical therapeutic application has been sustained by the excellent results obtained by clinical trials based on the use of therapeutic lentivirus (LV) carrying a normal β-globin gene (Boulad et al., 2022; Locatelli et al., 2022; Magrin et al., 2022; Tucci et al., 2022; Segura et al., 2023). For instance, Locatelli and collaborators reported a Phase 3 clinical study (HGB-207, NCT02906202) carried on 23 patients receiving “betibeglogene autotemcel or beti-cel” treatment with a median follow-up of 29.5 months. This clinical trial was conducted on adult and pediatric patients with transfusion-dependent β-thalassemia and a non-β0/β0 genotype (Locatelli et al., 2022). The efficacy of the treatment was demonstrated by the finding that transfusion independence occurred in 20 of 22 patients, with an average hemoglobin level of 11.7 g per deciliter (range, 9.5–12.8) (Locatelli et al., 2022). Similar promising results were obtained by Magrin et al. (2022), reporting the data originating by the HGB-205 (NCT02151526) clinical trial.
An interesting development of LV vectors proposed for gene therapy of β-thalassemia is the linkage of the therapeutic β-globin gene to other useful gene sequences. An example of such bifunctional vectors has been described by Brusson et al., using as experimental model system hematopoietic stem and progenitor cells (HSPCs) isolated from sickle-cells disease (SCD) patients (Brusson et al., 2023). In this case, the employed bifunctional LV vector was expressing an anti-sickling β-globin (βAS3-globin) and an artificial microRNA specifically downregulating βS-globin expression (Brusson et al., 2023). The aim of this approach was to obtain a miRNA-mediated reduction of the HbS levels, favoring at the same time the incorporation of βAS3 into Hb tetramers. This study is an excellent proof-of-principle that clinically relevant end-points can be reached by bifunctional LV vectors combining gene addition and gene silencing strategies; furthermore, this study sustain the concept of combined treatments for β-thalassemia.
In this respect, the combination of gene therapy and gene editing was studied by Ramadier et al., who described two therapeutic approaches combining LV-based gene addition therapy ansd CRISPR-Cas9 gene editing (Ramadier et al., 2022). In both cases, the protocols were based on base-editing knock down the sickle β-globin in association with de novo expression of anti-sickling globin (AS3) or anti-sickling fetal γ-globins. While in this study expression of anti-sickling fetal γ-globins was obtained by gene addition (Ramadier et al., 2022), this can be also obtained by gene editing procedures for induction of γ-globin gene expression, as already described in Figure 2 and recently reported in the study by Venkatesan et al. 2023b). These studies confirmed also the possible use of combined protcols based on complementary gene editing procedures.
6 Multiplex CRISPR-Cas9 protocols
Han et al. (2022) recently published an interesting report demonstrating that CRISPR-Cas9-based gene editing approaches can be combined. This was also found by Psatha et al. (2021), who described a very interesting multiplex gene editing protocol based on the combination of two single gene editing strategies, one aimed at silencing the BCL11A repressor, the other aimed at disrupting the BCL11A binding sites of the γ-globin gene promoter (Psatha et al., 2021). The results obtained demonstrated that this CRISPR-Cas9 based multiplex genomic editing efficiently induced fetal hemoglobin expression.
7 Combining de novo production of HbA with HbF induction
In order to obtain increased levels of HbF together with de novo production of HbA, the possibility to perform CRISPR-Cas9 based multiplex genomic editing for BCL11A silencing (Frangoul et al.; Bjurström et al., 2016) and for correction of the primary mutation (Liang et al., 2017) might be considered. The advantage of this strategy is that both protocols use the same target cells (CD34+ cells) and, with respect to the expected therapeutic protocol, the same clinical steps for collecting the CD34+ cells to be gene-edited and for preparing the patients for the infusion of gene-edited cells (Bjurström et al., 2016; Fu et al., 2022). On the other hand, major drawbacks are expected, i.e., higher off-targeting effects and genotoxicity (Bothmer et al., 2017; Samuelson et al., 2021).
In this respect, while Han et al. reported no major increase of off-target effects of multiplex genomic editing (Han et al., 2022), on the contrary, Samuelson et al. have recently found that multiplex CRISPR-Cas9 genome editing in hematopoietic stem cells for HbF reinduction generates chromosomal translocations (Samuelson et al., 2021). This is not unexpected, since genomic instability is one of the major and still unresolved concerns in using gene editing strategies, including CRISPR-Cas9 based approaches (Bothmer et al., 2017). Recent reports described that gene editing might be associated to chromosomal aberrations with formation of micronuclei and chromosome bridges leading to copy number variation, telomeric portion loss, and chromotripsis (Cullot et al., 2019; Blattner et al., 2020; Leibowitz et al., 2021). Moreover, it should be considered that the identification of these alterations is not a simple task. In this respect, Holgersen et al. reported data strongly suggesting that in silico methods are of limited use for predicting the off-target effects of oligonucleotides, and RNA-seq based experimental screening should be instead considered the preferred approach (Holgersen et al., 2021). This raises further concerns in the transfer of gene editing technology from laboratory investigations to clinical settings.
In summary, the issue of genotoxicity of multiplex CRISPR/Cas9 based gene editing should be considered still an open issue, and caution should be suggested in using these multiplexed approaches. For these reasons, for HbF induction in combination with gene-editing approaches, it has been suggested to employ pharmacological HbF induction, using as first-choice repositioned drugs, or drugs already validated and used in clinical trials.
8 Induction of fetal hemoglobin: use of small-molecular weight molecules
The search for fetal hemoglobin inducers is a fast-moving field aiming to bring this approach from the laboratory to clinical settings. Review articles on HbF inducers are available discussing updates and the most recent findings (Gambari and Fibach, 2007; Sripichai and Fucharoen, 2016b; Lohani et al., 2018; Langer and Esrick, 2021; Bou-Fakhredin et al., 2022; Hashemi and Ebrahimzadeh, 2022; Prosdocimi et al., 2022). For preliminary screening of fetal hemoglobin inducers several in vitro cellular systems have been employed, such as In this case, several cellular model systems for the screening of HbF inducers are available, such as the K562 erythroleukemia cell line (Gambari and Fibach, 2007) or the HUDEP-1 cell line derived from umbilical cord blood cells (Papasavva et al., 2021). Of course, the effects of the identified HbF inducers should be confirmed and further characterized using primary cells isolated from β-thalassemia patients, such as erythroid precursor/progenitor cells (Gambari and Fibach, 2007). Table 3 reports a partial list of HbF inducers (Fucharoen et al., 1996; Marianna et al., 2001; Fibach et al., 2003; Lampronti et al., 2003; Fibach et al., 2006; Rönndahl et al., 2006; Aerbajinai et al., 2007; Zuccato et al., 2007; Moutouh-de Parseval et al., 2008; Fibach et al., 2012; Lulli et al., 2013; Shi et al., 2013; Reid et al., 2014; Bianchi et al., 2015; Gasparello et al., 2017; Kalantri et al., 2018; Iftikhar et al., 2019; Mettananda et al., 2019; Gholampour et al., 2020; Guo et al., 2020; Cheng et al., 2021; Zuccato et al., 2021; Iftikhar et al., 2022; Li et al., 2022; Zuccato et al., 2022; Takase et al., 2023), some of them presently considered in clinical trials (such as hydroxyurea and sirolimus). Remarkably, the proposed mechanisms of action (and the molecular/biochemical targets) are highly heterogenous. For instance, HbF inducers can inhibit DNA methylation, inhibit histone lysine methyltransferases, inhibit HDAC activity, activate the p38 MAPK pathway, inhibit the mTOR pathway, inhibit the binding of transcription factors (for instance Sp1, BCL11A) to the target DNA, inhibit the expression of γ-globin gene repressors (such as BCL11A and KLF6). Interestingly, several HbF inducers (for instance thalidomide, rapamycin, hydroxyurea) are repurposed/repositioned drugs, thus strongly facilitating the transfer of laboratory investigations to the clinical practice. Examples of clinical trials based on the HbF inducers enlisted in Table 3 are hydroxyurea (clinical trials NCT00001958 and NCT03183375), sirolimus (clinical trials NCT03877809 and NCT04247750), thalidomide (NCT05132270) and 2,2-dimethylbutyrate (HQK-1001) (NCT00790127).
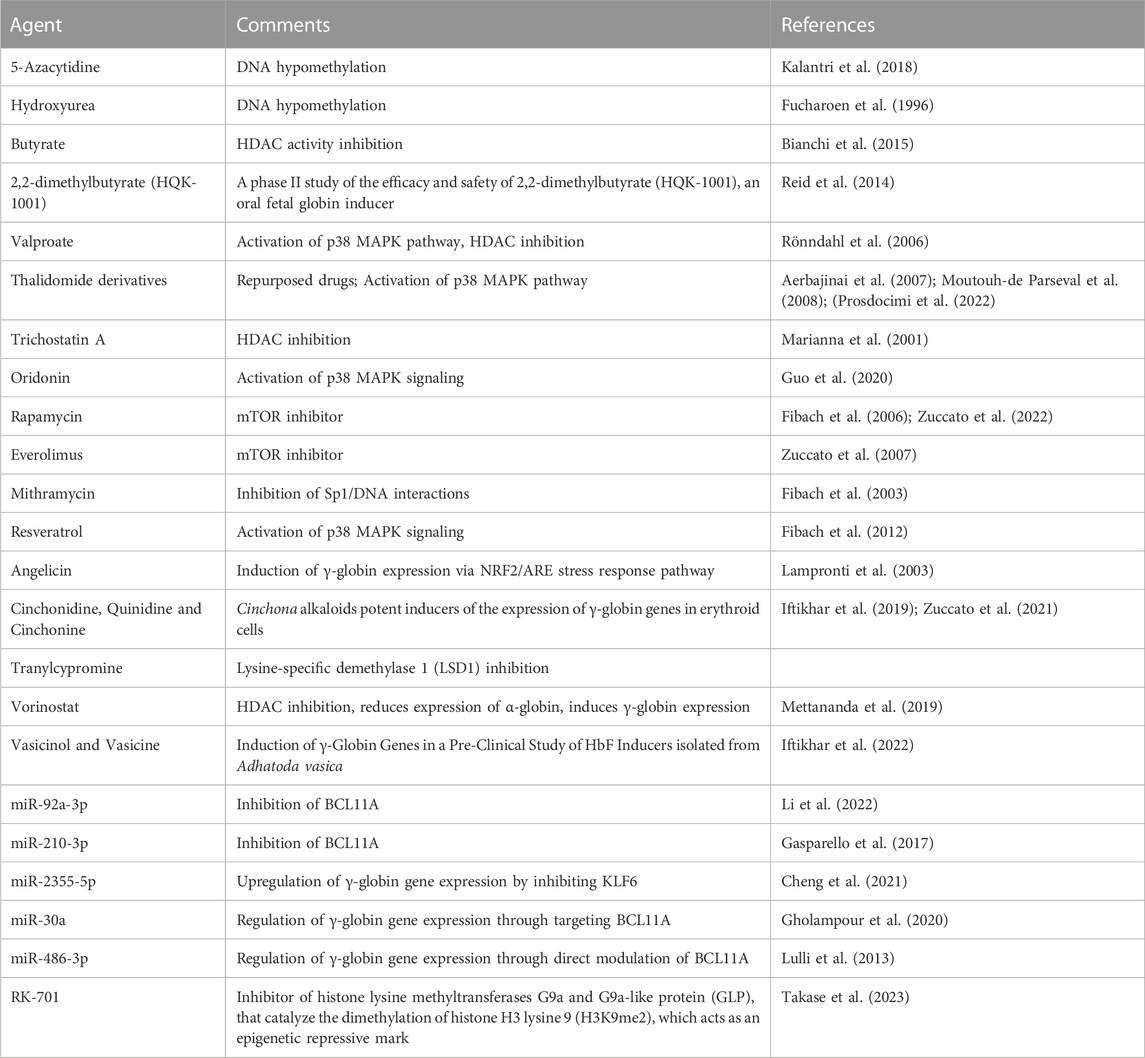
TABLE 3. Fetal hemoglobin inducers.
9 Combining CRISPR-Cas9 based correction of the β-thalassemia mutation with the treatment of erythroid cells with HbF inducers
The idea of a combined approach for developing a therapeutic protocol for β-thalassemia based on gene therapy-mediated adult hemoglobin (HbA) production and fetal hemoglobin (HbF) induction is not new (Breda et al., 2013; Finotti et al., 2015). This is an important point since gene therapy might fail in reaching the complete reversion of the β-thalassemic phenotype (Breda et al., 2013; Finotti et al., 2015). In fact, following gene transfer, all or a large proportion of erythroid cells might express suboptimal levels of β-globin, significantly lowering the therapeutic potential of the gene therapy approach. These limitations might also be present in CRISPR-Cas9 based approaches aimed to reconstitute adult hemoglobin (HbA) production following gene editing (Cosenza et al., 2021). In this research field, Zuccato et al. were able to demonstrate the usefulness of the combination between gene therapy and HbF induction (Zuccato et al., 2012). Their end-point was the reduction in the treated ErPCs of the excess of free α-globin aggregates. Zuccato et al. demonstrated that gene therapy, performed with the lentiviral β-globin vector T9W, and HbF induction with mithramycin could be combined (Zuccato et al., 2012). In fact, the combination of T9W β-globin gene transfer together with fetal hemoglobin induction was far more efficacious than single treatments in removing the excess of α-globin proteins in β-thalassemic erythroid cells. This strategy has been reviewed by Finotti et al. (2015) and Breda et al. (2013). As observed in gene-therapy of β-thalassemia (Breda et al., 2013; Dong et al., 2013; Finotti et al., 2015; Karponi and Zogas, 2019; Thuret et al., 2022), the efficiency and duration of the correction of HbA production might fall below therapeutic levels also in gene editing (Cosenza et al., 2021); in this case, combined treatment with HbF inducers might be considered a useful fallback strategy to reach durable therapeutic hemoglobin levels in treated cells, with contribution of an hemoglobin (HbF) firmly established to be beneficial for β-thalassemia patients (Forget, 1998; Gambari and Fibach, 2007; Musallam et al., 2012; Sripichai and Fucharoen, 2016a).
The first example of combined treatment using HbF inducers and gene editing was recently reported by Cosenza et al. (2022), who previously developed a protocol for CRISPR-Cas9-based gene correction of the β039-thalassemia mutation, one of the most frequent in the Mediterranean area (Origa, 2017). The study was aimed at determining whether pharmacologic induction of HbF could be combined with de novo production of HbA obtained by the correction of the β0-thalassemia mutation using the developed CRISPR-Cas9 protocol (Cosenza et al., 2022) (Figure 3). As an inducer of HbF rapamycin (also known as sirolimus) (Sehgal, 2003), was selected. This lipophilic macrolide has been reported to be a strong inducer of HbF in vitro (Mischiati et al., 2004; Fibach et al., 2006; Zuccato et al., 2007; Pecoraro et al., 2015; Zuccato et al., 2022), in vivo animal model systems (Zhang et al., 2014; Khaibullina et al., 2015; Wang et al., 2016), in some patients affected by sickle-cell disease (SCD) (Gaudre et al., 2017; Al-Khatti and Alkhunaizi, 2019), and in a cohort of β-thalassemia patient participating to the NCT03877809 clinical trial (A Personalized Medicine Approach for β-thalassemia Transfusion Dependent Patients: Testing sirolimus in a First Pilot Clinical Trial) (Zuccato et al., 2022). The data obtained by Cosenza et al. support the concept that CRISPR-Cas9 mediated gene editing restores HbA production in erythroid precursor cells from homozygous β039/β039 homozygous patients, together with pharmacological induction of HbF. Remarkably, no decrease of HbF in gene edited cells occurs (Cosenza et al., 2021; Cosenza et al., 2022) and no evidence of a decrease of HbA following HbF induction in gene edited cells has been reported (Cosenza et al., 2022), suggesting that this might be considered an interesting protocol to minimize side effects (single plex CRISPR-Cas9 intervention is performed) together with maximization of total hemoglobin production (de novo production of HbA and increased production of HbF).
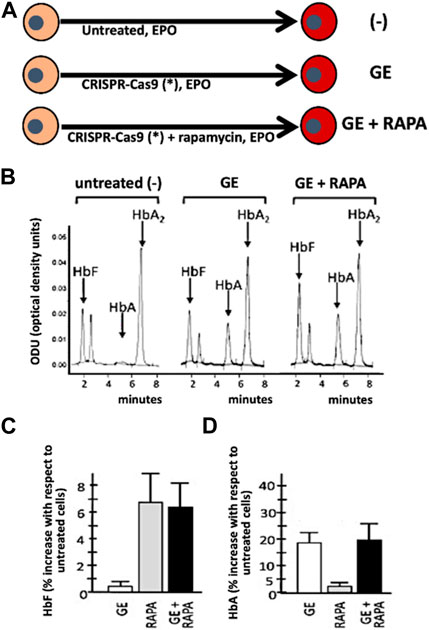
FIGURE 3. Co-treatment of ErPCs from β-thalassemia patients with the CRISPR-Cas9 based approach to correct the β039-globin gene mutation (GE) and fetal hemoglobin induction following treatment with rapamycin (RAPA). (A) Pictorial representation of the experimental protocols. (B–D) Demonstration that in (GE + RAPA) treated cells both HbF (B,C) and HbA (B,D) are increased. Remarkably, in (GE + RAPA) treated cells HbF increase is similar to RAPA treated cells (C) and HbA increase is similar to GE-treated cells. (B–D) of this Figure are modified from Cosenza et al., with permission (copyright can be found at https://www.mdpi.com/2073-4425/13/10/1727) (Cosenza et al., 2022).
Furthermore, it is expected that the pharmacologic induction of HbF can be employed in co-treatment protocols together with gene editing procedures correcting other β-thalassemia mutations. These pre-clinical studies might be facilitated by the availability of β-thalassemia cellular bio banks, avoiding the need of new recruitment of β-thalassemia patients (Cosenza et al., 2016).
10 Conclusion and future perspectives
In our opinion, the possible combination between CRISPR-Cas9 gene editing and HbF induction deserves further studies to determine the real impact in real life for the management of β-thalassemia patients. While it is likely that the use of gene editing will be considered with high caution, due to the possible genotoxic effects of this strategy (Bothmer et al., 2017; Cullot et al., 2019; Blattner et al., 2020; Leibowitz et al., 2021; Samuelson et al., 2021), we would like to remind that clinical trials based on the use of CRISPR-Cas9 are ongoing. Examples are β-thalassemia and SCD patients infused with CRISPR-Cas9 modified autologous CD34+ HSPCs (NCT03655678; NCT03745287). Encouraging results were obtained, including the fact that a) some transfusion-dependent β-thalassemia patients became transfusion-independent already from the first months after infusion and b) one SCD patient was free of vaso-occlusive crises after treatment (Frangoul et al., 2021). With respect to possible combined co-treatment with HbF inducers, it should be underlined that both gene editing and HbF induction are expected to be carried on at sub-optimal conditions, trying to avoid unwanted side effects.
In addition, we have to mention that novel gene-editing strategies (for instance base-priming and base-editing), characterized by lower level of genotoxicity, are available and are expected to be extensively studied and validated in the next future (Liang et al., 2017; Zipkin, 2019; Collantes et al., 2021; Zhang et al., 2022; Badat et al., 2023; Hardouin et al., 2023). In our opinion, this last development of gene editing protocols is of great interest to propose combined treatments based on gene editing (or multiplexed gene editing) with HbF inducers.
A limit of the review is that it is based on few key papers reporting gene-therapy and CRISPR-CAS9-based gene-editing in combination with the use of HbF inducers (Zuccato et al., 2012; Breda et al., 2013; Cosenza et al., 2022). Our review might stimulate further research on this field, in order to verify the real impact of this approach.
In these studies, the choice of the HbF inducer is critical. We suggest employing repurposed drugs that a) increase HbF in cultures from β-thalassemia patients with different basal HbF levels; b) increase the overall Hb content per cell; c) selectively induce γ-globin mRNA accumulation, with only minor effects on β-globin and α-globin mRNAs; d) are currently under investigations in clinical trials, such as hydroxyurea (FDA approved for sickle-cell disease) (Yasara et al., 2020; Yasara et al., 2022), sirolimus (Zuccato et al., 2022), thalidomide (Chen et al., 2021) and 2,2-dimethylbutyrate (HQK-1001) (Reid et al., 2014).
The interest in combined therapies based on the use of gene editing and HbF induction is also due to the fact that HbF inducers might exhibit mechanism(s) of action enforcing erythroid cells to be activate biological processes of great importance for clinical treatments. For instance, in the case of β-thalassemia, a key end point is represented by the reduction of the excess of α-globin chains, as reported in several studies (Schrier, 2002; Xie et al., 2007; Voon et al., 2008; Mettananda et al., 2015; Mettananda et al., 2017a). In this respect, rapamycin has been shown to induce with high efficiency (in addition to increased HbF production) autophagy, thereby decreasing the excess of free α-globin chains (Lechauve et al., 2019; Buffa et al., 2023; Zurlo et al., 2023). In our opinion this double effect of rapamycin, might be used in pre-clinical studies also in combination with gene editing strategies to maximize the decrease expression of α-globin genes in treated cells.
Author contributions
AF and RG conceived and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was sponsored by the EU THALAMOSS Project (Thalassemia Modular Stratification System for Personalized Therapy of Βeta-Thalassemia, No. 306201, FP7-HEALTH-2012, INNOVATION-1), Wellcome Trust (innovator award 208872/Z/17/Z), AIFA (AIFA-2016-02364887), and from FIR and FAR funds from the University of Ferrara. This research was also supported by the Interuniversity Consortium for Biotechnology (CIB), Italy.
Acknowledgments
We thank the Association Tutti per Chiara Onlus and the Veneta Association for the Fight Against Thalassemia (AVLT) for support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aerbajinai, W., Zhu, J., Gao, Z., Chin, K., and Rodgers, G. P. (2007). Thalidomide induces gamma-globin gene expression through increased reactive oxygen species-mediated p38 MAPK signaling and histone H4 acetylation in adult erythropoiesis. Blood 110 (8), 2864–2871. doi:10.1182/blood-2007-01-065201
Al-Khatti, A. A., and Alkhunaizi, A. M. (2019). Additive effect of sirolimus and hydroxycarbamide on fetal haemoglobin level in kidney transplant patients with sickle cell disease. Br. J. Haematol. 185, 959–961. doi:10.1111/bjh.15665
Ali, G., Tariq, M. A., Shahid, K., Ahmad, F. J., and Akram, J. (2021). Advances in genome editing: The technology of choice for precise and efficient β-thalassemia treatment. Gene Ther. 28 (1-2), 6–15. doi:10.1038/s41434-020-0153-9
Antoniani, C., Meneghini, V., Lattanzi, A., Felix, T., Romano, O., Magrin, E., et al. (2018). Induction of fetal hemoglobin synthesis by CRISPR/Cas9-mediated editing of the human β-globin locus. Blood 131 (17), 1960–1973. doi:10.1182/blood-2017-10-811505
Badat, M., Ejaz, A., Hua, P., Rice, S., Zhang, W., Hentges, L. D., et al. (2023). Direct correction of haemoglobin E β-thalassaemia using base editors. Nat. Commun. 14 (1), 2238. doi:10.1038/s41467-023-37604-8
Badens, C., Joly, P., Agouti, I., Thuret, I., Gonnet, K., Fattoum, S., et al. (2011). Variants in genetic modifiers of β-thalassemia can help to predict the major or intermedia type of the disease. Haematologica 96, 1712–1714. doi:10.3324/haematol.2011.046748
Bianchi, N., Chiarabelli, C., Zuccato, C., Lampronti, I., Borgatti, M., Amari, G., et al. (2015). Erythroid differentiation ability of butyric acid analogues: Identification of basal chemical structures of new inducers of foetal haemoglobin. Eur. J. Pharmacol. 752, 84–91. doi:10.1016/j.ejphar.2015.02.018
Bjurström, C. F., Mojadidi, M., Phillips, J., Kuo, C., Lai, S., Lill, G. R., et al. (2016). Reactivating fetal hemoglobin expression in human adult erythroblasts through BCL11A knockdown using targeted endonucleases. Mol. Ther. Nucleic Acids 5 (8), e351. doi:10.1038/mtna.2016.52
Blattner, G., Cavazza, A., Thrasher, A. J., and Turchiano, G. (2020). Gene editing and genotoxicity: Targeting the off-targets. Front. Genome Ed. 2, 613252. doi:10.3389/fgeed.2020.613252
Bothmer, A., Phadke, T., Barrera, L. A., Margulies, C. M., Lee, C. S., Buquicchio, F., et al. (2017). Characterization of the interplay between DNA repair and CRISPR/Cas9-induced DNA lesions at an endogenous locus. Nat. Commun. 8, 13905. doi:10.1038/ncomms13905
Bou-Fakhredin, R., De Franceschi, L., Motta, I., Cappellini, M. D., and Taher, A. T. (2022). Pharmacological induction of fetal hemoglobin in β-thalassemia and sickle cell disease: An updated perspective. Pharm. (Basel) 15 (6), 753. doi:10.3390/ph15060753
Boulad, F., Maggio, A., Wang, X., Moi, P., Acuto, S., Kogel, F., et al. (2022). Lentiviral globin gene therapy with reduced-intensity conditioning in adults with β-thalassemia: A phase 1 trial. Nat. Med. 28 (1), 63–70. doi:10.1038/s41591-021-01554-9
Boulad, F., Mansilla-Soto, J., Cabriolu, A., Rivière, I., and Sadelain, M. (2018). Gene therapy and genome editing. Hematol. Oncol. Clin. North Am. 32, 329–342. doi:10.1016/j.hoc.2017.11.007
Breda, L., Rivella, S., Zuccato, C., and Gambari, R. (2013). Combining gene therapy and fetal hemoglobin induction for treatment of β-thalassemia. Expert Rev. Hematol. 6 (3), 255–264. doi:10.1586/ehm.13.24
Brusson, M., Chalumeau, A., Martinucci, P., Romano, O., Felix, T., Poletti, V., et al. (2023). Novel lentiviral vectors for gene therapy of sickle cell disease combining gene addition and gene silencing strategies. Mol. Ther. Nucleic Acids 32, 229–246. doi:10.1016/j.omtn.2023.03.012
Buffa, V., Alvarez Vargas, J. R., Galy, A., Spinozzi, S., and Rocca, C. J. (2023). Hematopoietic stem and progenitors cells gene editing: Beyond blood disorders. Front. Genome Ed. 4, 997142. doi:10.3389/fgeed.2022.997142
Canver, M. C., Smith, E. C., Sher, F., Pinello, L., Sanjana, N. E., Shalem, O., et al. (2015). BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 527 (7577), 192–197. doi:10.1038/nature15521
Carter, D., Chakalova, L., Osborne, C. S., Dai, Y. F., and Fraser, P. (2002a). Long-range chromatin regulatory interactions in vivo. Nat. Genet. 32 (4), 623–626. doi:10.1038/ng1051
Carter, D., Chakalova, L., Osborne, C. S., Dai, Y. F., and Fraser, P. (2002b). Long-range chromatin regulatory interactions in vivo. Nat. Genet. 32 (4), 623–626. doi:10.1038/ng1051
Carusillo, A., Haider, S., Schäfer, R., Rhiel, M., Türk, D., Chmielewski, K. O., et al. (2023). A novel Cas9 fusion protein promotes targeted genome editing with reduced mutational burden in primary human cells. Nucleic Acids Res. 51 (9), 4660–4673. doi:10.1093/nar/gkad255
Chakalova, L., Carter, D., Debrand, E., Goyenechea, B., Horton, A., Miles, J., et al. (2005). Developmental regulation of the beta-globin gene locus. Prog. Mol. Subcell. Biol. 38, 183–206. doi:10.1007/3-540-27310-7_8
Chen, J. M., Zhu, W. J., Liu, J., Wang, G. Z., Chen, X. Q., Tan, Y., et al. (2021). Safety and efficacy of thalidomide in patients with transfusion-dependent β-thalassemia: A randomized clinical trial. Signal Transduct. Target Ther. 6 (1), 405. doi:10.1038/s41392-021-00811-0
Cheng, Y., Shang, X., Chen, D., Pang, D., Zhao, C., and Xu, X. (2021). MicroRNA-2355-5p regulates γ-globin expression in human erythroid cells by inhibiting KLF6. Br. J. Haematol. 193 (2), 401–405. doi:10.1111/bjh.17134
Collantes, J. C., Tan, V. M., Xu, H., Ruiz-Urigüen, M., Alasadi, A., Guo, J., et al. (2021). Development and characterization of a modular CRISPR and RNA aptamer mediated base editing system. CRISPR J. 4 (1), 58–68. doi:10.1089/crispr.2020.0035
Cosenza, L. C., Breda, L., Breveglieri, G., Zuccato, C., Finotti, A., Lampronti, I., et al. (2016). A validated cellular biobank for β-thalassemia. J. Transl. Med. 14 (1), 255. doi:10.1186/s12967-016-1016-4
Cosenza, L. C., Gasparello, J., Romanini, N., Zurlo, M., Zuccato, C., Gambari, R., et al. (2021). Efficient CRISPR-Cas9-based genome editing of β-globin gene on erythroid cells from homozygous β039-thalassemia patients. Mol. Ther. Methods Clin. Dev. 21, 507–523. doi:10.1016/j.omtm.2021.03.025
Cosenza, L. C., Zuccato, C., Zurlo, M., Gambari, R., and Finotti, A. (2022). Co-treatment of erythroid cells from β-thalassemia patients with CRISPR-cas9-based β039-globin gene editing and induction of fetal hemoglobin. Genes. (Basel) 13 (10), 1727. doi:10.3390/genes13101727
Cullot, G., Boutin, J., Toutain, J., Prat, F., Pennamen, P., Rooryck, C., et al. (2019). CRISPR-Cas9 genome editing induces megabase-scale chromosomal truncations. Nat. Commun. 10 (1), 1136. doi:10.1038/s41467-019-09006-2
Danjou, F., Anni, F., Perseu, L., Satta, S., Dessì, C., Lai, M. E., et al. (2012). Genetic modifiers of β-thalassemia and clinical severity as assessed by age at first transfusion. Haematologica 97, 989–993. doi:10.3324/haematol.2011.053504
Deng, W., Rupon, J. W., Krivega, I., Breda, L., Motta, I., Jahn, K. S., et al. (2014). Reactivation of developmentally silenced globin genes by forced chromatin looping. Cell. 158 (4), 849–860. doi:10.1016/j.cell.2014.05.050
Dever, D. P., Bak, R. O., Reinisch, A., Camarena, J., Washington, G., Nicolas, C. E., et al. (2016). CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature 539, 384–389. doi:10.1038/nature20134
Dong, A., Rivella, S., and Breda, L. (2013). Gene therapy for hemoglobinopathies: Progress and challenges. Transl. Res. 161 (4), 293–306. doi:10.1016/j.trsl.2012.12.011
Eckrich, M. J., and Frangoul, H. (2023). Gene editing for sickle cell disease and transfusion dependent thalassemias- A cure within reach. Semin. Hematol. 60 (1), 3–9. doi:10.1053/j.seminhematol.2022.12.001
Ernst, M. P. T., Broeders, M., Herrero-Hernandez, P., Oussoren, E., van der Ploeg, A. T., and Pijnappel, W. W. M. P. (2020). Ready for repair? Gene editing enters the clinic for the treatment of human disease. Mol. Ther. Methods Clin. Dev. 18, 532–557. doi:10.1016/j.omtm.2020.06.022
Ferrari, G., Thrasher, A. J., and Aiuti, A. (2021). Gene therapy using haematopoietic stem and progenitor cells. Nat. Rev. Genet. 22 (4), 216–234. doi:10.1038/s41576-020-00298-5
Fibach, E., Bianchi, N., Borgatti, M., Prus, E., and Gambari, R. (2003). Mithramycin induces fetal hemoglobin production in normal and thalassemic human erythroid precursor cells. Blood 102 (4), 1276–1281. doi:10.1182/blood-2002-10-3096
Fibach, E., Bianchi, N., Borgatti, M., Zuccato, C., Finotti, A., Lampronti, I., et al. (2006). Effects of rapamycin on accumulation of alpha-beta- and gamma-globin mRNAs in erythroid precursor cells from beta-thalassaemia patients. Eur. J. Haematol. 77 (5), 437–441. doi:10.1111/j.1600-0609.2006.00731.x
Fibach, E., Prus, E., Bianchi, N., Zuccato, C., Breveglieri, G., Salvatori, F., et al. (2012). Resveratrol: Antioxidant activity and induction of fetal hemoglobin in erythroid cells from normal donors and β-thalassemia patients. Int. J. Mol. Med. 29 (6), 974–982. doi:10.3892/ijmm.2012.928
Finotti, A., Breda, L., Lederer, C. W., Bianchi, N., Zuccato, C., Kleanthous, M., et al. (2015). Recent trends in the gene therapy of β-thalassemia. J. Blood Med. 6, 69–85. doi:10.2147/JBM.S46256
Finotti, A., and Gambari, R. (2014). Recent trends for novel options in experimental biological therapy of β-thalassemia. Expert Opin. Biol. Ther. 14, 1443–1454. doi:10.1517/14712598.2014.927434
Forget, B. G. (1998). Molecular basis of hereditary persistence of fetal hemoglobin. Ann. N. Y. Acad. Sci. 850, 38–44. doi:10.1111/j.1749-6632.1998.tb10460.x
Frangoul, H., Altshuler, D., Cappellini, M. D., Chen, Y. S., Domm, J., Eustace, B. K., et al. (2021). CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. N. Engl. J. Med. 384 (3), 252–260. doi:10.1056/NEJMoa2031054
Frangoul, H., Bobruff, Y., Cappellini, M. D., Corbacioglu, S., Fernandez, C. M., de la Fuente, J., et al. (2020). Safety and efficacy of CTX001 in patients with transfusion-dependent β-thalassemia and sickle cell disease: Early results from the climb THAL-111 and climb SCD-121 studies of autologous CRISPR-CAS9-modified CD34+ hematopoietic stem and progenitor cells (abstract) blood 136, 3–4. doi:10.1182/blood-2020-139575
Fu, B., Liao, J., Chen, S., Li, W., Wang, Q., Hu, J., et al. (2022). CRISPR-Cas9-mediated gene editing of the BCL11A enhancer for pediatric β0/β0 transfusion-dependent β-thalassemia. Nat. Med. 28 (8), 1573–1580. doi:10.1038/s41591-022-01906-z
Fucharoen, S., Siritanaratkul, N., Winichagoon, P., Chowthaworn, J., Siriboon, W., Muangsup, W., et al. (1996). Hydroxyurea increases hemoglobin F levels and improves the effectiveness of erythropoiesis in beta-thalassemia/hemoglobin E disease. Blood 87 (3), 887–892. doi:10.1182/blood.v87.3.887.bloodjournal873887
Fucharoen, S., and Weatherall, D. J. (2016). Progress toward the control and management of the thalassemias. Hematol. Oncol. Clin. North Am. 30, 359–371. doi:10.1016/j.hoc.2015.12.001
Gabr, H., El Ghamrawy, M. K., Almaeen, A. H., Abdelhafiz, A. S., Hassan, A. O. S., and El Sissy, M. H. (2020). CRISPR-mediated gene modification of hematopoietic stem cells with beta-thalassemia IVS-1-110 mutation. Stem Cell. Res. Ther. 11 (1), 390. doi:10.1186/s13287-020-01876-4
Galanello, R., and Origa, R. (2010). Beta-thalassemia. Orphanet J. Rare Dis. 5, 11. doi:10.1186/1750-1172-5-11
Galanello, R., Sanna, S., Perseu, L., Sollaino, M. C., Satta, S., Lai, M. E., et al. (2009). Amelioration of Sardinian beta0 thalassemia by genetic modifiers. Blood 114, 3935–3937. doi:10.1182/blood-2009-04-217901
Gambari, R., and Fibach, E. (2007). Medicinal chemistry of fetal hemoglobin inducers for treatment of β-thalassemia. Curr. Med. Chem. 14, 199–212. doi:10.2174/092986707779313318
Gasparello, J., Fabbri, E., Bianchi, N., Breveglieri, G., Zuccato, C., Borgatti, M., et al. (2017). BCL11A mRNA targeting by miR-210: A possible network regulating γ-globin gene expression. Int. J. Mol. Sci. 18 (12), 2530. doi:10.3390/ijms18122530
Gaudre, N., Cougoul, P., Bartolucci, P., Dörr, G., Bura-Riviere, A., Kamar, N., et al. (2017). Improved fetal hemoglobin with mTOR inhibitor-based immunosuppression in a kidney transplant recipient with sickle cell disease. Am. J. Transpl. 17, 2212–2214. doi:10.1111/ajt.14263
Gholampour, M. A., Asadi, M., Naderi, M., Azarkeivan, A., Soleimani, M., and Atashi, A. (2020). miR-30a regulates γ-globin expression in erythoid precursors of intermedia thalassemia through targeting BCL11A. Mol. Biol. Rep. 47 (5), 3909–3918. doi:10.1007/s11033-020-05483-7
Guo, L., Chen, J., Wang, Q., Zhang, J., and Huang, W. (2020). Oridonin enhances γ-globin expression in erythroid precursors from patients with β-thalassemia via activation of p38 MAPK signaling. Mol. Med. Rep. 21 (2), 909–917. doi:10.3892/mmr.2019.10848
Han, Y., Tan, X., Jin, T., Zhao, S., Hu, L., Zhang, W., et al. (2022). CRISPR/Cas9-based multiplex genome editing of BCL11A and HBG efficiently induces fetal hemoglobin expression. Eur. J. Pharmacol. 918, 174788, 174788. doi:10.1016/j.ejphar.2022.174788
Hardouin, G., Antoniou, P., Martinucci, P., Felix, T., Manceau, S., Joseph, L., et al. (2023). Adenine base editor-mediated correction of the common and severe IVS1-110 (G>A) β-thalassemia mutation. Blood 141 (10), 1169–1179. doi:10.1182/blood.2022016629
Hashemi, Z., and Ebrahimzadeh, M. A. (2022). Hemoglobin F (HbF) inducers; history, structure and efficacies. Mini Rev. Med. Chem. 22 (1), 52–68. doi:10.2174/1389557521666210521221615
Holgersen, E. M., Gandhi, S., Zhou, Y., Kim, J., Vaz, B., Bogojeski, J., et al. (2021). Transcriptome-Wide off-target effects of steric-blocking oligonucleotides. Nucleic Acid. Ther. 31 (6), 392–403. doi:10.1089/nat.2020.0921
Hu, X. (2016). CRISPR/Cas9 system and its applications in human hematopoietic cells. Blood Cells Mol. Dis. 62, 6–12. doi:10.1016/j.bcmd.2016.09.003
Iftikhar, F., Ali, H., and Musharraf, S. G. (2019). Cinchona alkaloids as natural fetal hemoglobin inducing agents in human erythroleukemia cells. RSC Adv. 9 (31), 17551–17559. doi:10.1039/c9ra01744e
Iftikhar, F., Rahman, S., Khan, M. B. N., Khan, K., Khan, M. N., Uddin, R., et al. (2022). In vitro and in vivo studies for the investigation of γ-globin gene induction by Adhatoda vasica: A pre-clinical study of HbF inducers for β-thalassemia. Front. Pharmacol. 13, 797853. doi:10.3389/fphar.2022.797853
Kalantri, S. A., Ray, R., Chattopadhyay, A., Bhattacharjee, S., Biswas, A., and Bhattacharyya, M. (2018). Efficacy of decitabine as hemoglobin F inducer in HbE/β-thalassemia. Ann. Hematol. 97 (9), 1689–1694. doi:10.1007/s00277-018-3357-y
Karamperis, K., Tsoumpeli, M. T., Kounelis, F., Koromina, M., Mitropoulou, C., Moutinho, C., et al. (2021). Genome-based therapeutic interventions for β-type hemoglobinopathies. Hum. Genomics 15 (1), 32. doi:10.1186/s40246-021-00329-0
Karponi, G., and Zogas, N. (2019). Gene therapy for beta-thalassemia: Updated perspectives. Appl. Clin. Genet. 12, 167–180. doi:10.2147/TACG.S178546
Khaibullina, A., Almeida, L. E., Wang, L., Kamimura, S., Wong, E. C. C., Nouraie, M., et al. (2015). Rapamycin increases fetal hemoglobin and ameliorates the nociception phenotype in sickle cell mice. Blood Cells Mol. Dis. 55, 363–372. doi:10.1016/j.bcmd.2015.08.001
Khiabani, A., Kohansal, M. H., Keshavarzi, A., Shahraki, H., Kooshesh, M., Karimzade, M., et al. (2023). CRISPR/Cas9, a promising approach for the treatment of β-thalassemia: A systematic review. Mol. Genet. Genomics 298 (1), 1–11. doi:10.1007/s00438-022-01978-z
Khosravi, M. A., Abbasalipour, M., Concordet, J. P., Berg, J. V., Zeinali, S., Arashkia, A., et al. (2019). Targeted deletion of BCL11A gene by CRISPR-cas9 system for fetal hemoglobin reactivation: A promising approach for gene therapy of beta thalassemia disease. Eur. J. Pharmacol. 854, 398–405. doi:10.1016/j.ejphar.2019.04.042
Komor, A. C., Badran, A. H., and Liu, D. R. (2018). Editing the genome without double-stranded DNA breaks. ACS Chem. Biol. 13 (2), 383–388. doi:10.1021/acschembio.7b00710
Lampronti, I., Bianchi, N., Borgatti, M., Fibach, E., Prus, E., and Gambari, R. (2003). Accumulation of gamma-globin mRNA in human erythroid cells treated with angelicin. Eur. J. Haematol. 71 (3), 189–195. doi:10.1034/j.1600-0609.2003.00113.x
Langer, A. L., and Esrick, E. B. (2021). β-Thalassemia: Evolving treatment options beyond transfusion and iron chelation. Hematol. Am. Soc. Hematol. Educ. Program 2021 (1), 600–606. doi:10.1182/hematology.2021000313
Lau, C. H. (2018). Applications of CRISPR-cas in bioengineering, Biotechnology, and translational research. CRISPR J. 1, 379–404. doi:10.1089/crispr.2018.0026
Lavelle, D., Engel, J. D., and Saunthararajah, Y. (2018). Fetal hemoglobin induction by epigenetic drugs. Semin. Hematol. 55, 60–67. doi:10.1053/j.seminhematol.2018.04.008
Lechauve, C., Keith, J., Khandros, E., Fowler, S., Mayberry, K., Freiwan, A., et al. (2019). The autophagy-activating kinase ULK1 mediates clearance of free α-globin in β-thalassemia. Sci. Transl. Med. 11 (506), eaav4881. doi:10.1126/scitranslmed.aav4881
Leibowitz, M. L., Papathanasiou, S., Doerfler, P. A., Blaine, L. J., Sun, L., Yao, Y., et al. (2021). Chromothripsis as an on-target consequence of CRISPR-Cas9 genome editing. Nat. Genet. 53 (6), 895–905. doi:10.1038/s41588-021-00838-7
Li, H., Lin, R., Li, H., Ou, R., Wang, K., Lin, J., et al. (2022). MicroRNA-92a-3p-mediated inhibition of BCL11A upregulates γ-globin expression and inhibits oxidative stress and apoptosis in erythroid precursor cells. Hematology 27 (1), 1152–1162. doi:10.1080/16078454.2022.2128258
Liang, P., Ding, C., Sun, H., Xie, X., Xu, Y., Zhang, X., et al. (2017). Correction of β-thalassemia mutant by base editor in human embryos. Protein Cell. 8 (11), 811–822. doi:10.1007/s13238-017-0475-6
Liu, D., Zhang, X., Yu, L., Cai, R., Ma, X., Zheng, C., et al. (2014). KLF1 mutations are relatively more common in a thalassemia endemic region and ameliorate the severity of β-thalassemia. Blood 124, 803–811. doi:10.1182/blood-2014-03-561779
Liu, Y., Yang, Y., Kang, X., Lin, B., Yu, Q., Song, B., et al. (2017). One-step biallelic and scarless correction of a β-thalassemia mutation in patient-specific iPSCs without drug selection. Mol. Ther. Nucleic Acids 6, 57–67. doi:10.1016/j.omtn.2016.11.010
Locatelli, F., Thompson, A. A., Kwiatkowski, J. L., Porter, J. B., Thrasher, A. J., Hongeng, S., et al. (2022). Betibeglogene autotemcel gene therapy for non-β0/β0 genotype β-thalassemia. N. Engl. J. Med. 386 (5), 415–427. doi:10.1056/NEJMoa2113206
Lohani, N., Bhargava, N., Munshi, A., and Ramalingam, S. (2018). Pharmacological and molecular approaches for the treatment of β-hemoglobin disorders. J. Cell. Physiol. 233 (6), 4563–4577. doi:10.1002/jcp.26292
Lu, D., Gong, X., Fang, Y., Guo, X., Chen, Y., Yang, F., et al. (2022). Correction of RNA splicing defect in β654-thalassemia mice using CRISPR/Cas9 gene-editing technology. Haematologica 107 (6), 1427–1437. doi:10.3324/haematol.2020.278238
Lulli, V., Romania, P., Morsilli, O., Cianciulli, P., Gabbianelli, M., Testa, U., et al. (2013). MicroRNA-486-3p regulates γ-globin expression in human erythroid cells by directly modulating BCL11A. PLoS One 8 (4), e60436. doi:10.1371/journal.pone.0060436
Magrin, E., Miccio, A., and Cavazzana, M. (2019). Lentiviral and genome-editing strategies for the treatment of β-hemoglobinopathies. Blood 134 (15), 1203–1213. doi:10.1182/blood.2019000949
Magrin, E., Semeraro, M., Hebert, N., Joseph, L., Magnani, A., Chalumeau, A., et al. (2022). Long-term outcomes of lentiviral gene therapy for the β-hemoglobinopathies: The HGB-205 trial. Nat. Med. 28 (1), 81–88. doi:10.1038/s41591-021-01650-w
Marianna, P., Kollia, P., Akel, S., Papassotiriou, Y., Stamoulakatou, A., and Loukopoulos, D. (2001). Valproic acid, trichostatin and their combination with hemin preferentially enhance gamma-globin gene expression in human erythroid liquid cultures. Haematologica 86 (7), 700–705.
Martyn, G. E., Wienert, B., Yang, L., Shah, M., Norton, L. J., Burdach, J., et al. (2018). Natural regulatory mutations elevate the fetal globin gene via disruption of BCL11A or ZBTB7A binding. Nat. Genet. 50 (4), 498–503. doi:10.1038/s41588-018-0085-0
Mettananda, S., Fisher, C. A., Hay, D., Badat, M., Quek, L., Clark, K., et al. (2017b). Editing an α-globin enhancer in primary human hematopoietic stem cells as a treatment for β-thalassemia. Nat. Commun. 8 (1), 424. doi:10.1038/s41467-017-00479-7
Mettananda, S., Fisher, C. A., Sloane-Stanley, J. A., Taylor, S., Oppermann, U., Gibbons, R. J., et al. (2017a). Selective silencing of α-globin by the histone demethylase inhibitor IOX1: A potentially new pathway for treatment of β-thalassemia. Haematologica 102 (3), e80–e84. doi:10.3324/haematol.2016.155655
Mettananda, S., Gibbons, R. J., and Higgs, D. R. (2015). α-Globin as a molecular target in the treatment of β-thalassemia. Blood 125 (24), 3694–3701. doi:10.1182/blood-2015-03.633594
Mettananda, S., Yasara, N., Fisher, C. A., Taylor, S., Gibbons, R., and Higgs, D. (2019). Synergistic silencing of α-globin and induction of γ-globin by histone deacetylase inhibitor, vorinostat as a potential therapy for β-thalassaemia. Sci. Rep. 9 (1), 11649. doi:10.1038/s41598-019-48204-2
Mischiati, C., Sereni, A., Lampronti, I., Bianchi, N., Borgatti, M., Prus, E., et al. (2004). Rapamycin-mediated induction of gamma-globin mRNA accumulation in human erythroid cells. Br. J. Haematol. 126, 612–621. doi:10.1111/j.1365-2141.2004.05083.x
Moutouh-de Parseval, L. A., Verhelle, D., Glezer, E., Jensen-Pergakes, K., Ferguson, G. D., Corral, L. G., et al. (2008). Pomalidomide and lenalidomide regulate erythropoiesis and fetal hemoglobin production in human CD34+ cells. J. Clin. Investig. 118 (1), 248–258. doi:10.1172/JCI32322
Musallam, K. M., Sankaran, V. G., Cappellini, M. D., Duca, L., Nathan, D. G., and Taher, A. T. (2012). Fetal hemoglobin levels and morbidity in untransfused patients with β-thalassemia intermedia. Blood 119 (2), 364–367. doi:10.1182/blood-2011-09-382408
Nuamsee, K., Chuprajob, T., Pabuprapap, W., Jintaridth, P., Munkongdee, T., Phannasil, P., et al. (2021). Trienone analogs of curcuminoids induce fetal hemoglobin synthesis via demethylation at Gγ-globin gene promoter. Sci. Rep. 11, 8552. doi:10.1038/s41598-021-87738-2
Nuinoon, M., Makarasara, W., Mushiroda, T., Setianingsih, I., Wahidiyat, P. A., Sripichai, O., et al. (2010). A genome-wide association identified the common genetic variants influence disease severity in beta0-thalassemia/hemoglobin E. Hum. Genet. 127, 303–314. doi:10.1007/s00439-009-0770-2
Okamura, E., Matsuzaki, H., Campbell, A. D., Engel, J. D., Fukamizu, A., and Tanimoto, K. (2009). All of the human beta-type globin genes compete for LCR enhancer activity in embryonic erythroid cells of yeast artificial chromosome transgenic mice. FASEB J. 23 (12), 4335–4343. doi:10.1096/fj.09-137778
Papasavva, P., Kleanthous, M., and Lederer, C. W. (2019). Rare opportunities: CRISPR/Cas-Based therapy development for rare genetic diseases. Mol. Diagn. Ther. 23, 201–222. doi:10.1007/s40291-019-00392-3
Papasavva, P. L., Papaioannou, N. Y., Patsali, P., Kurita, R., Nakamura, Y., Sitarou, M., et al. (2021). Distinct miRNA signatures and networks discern fetal from adult erythroid differentiation and primary from immortalized erythroid cells. Int. J. Mol. Sci. 22 (7), 3626. doi:10.3390/ijms22073626
Pavani, G., Fabiano, A., Laurent, M., Amor, F., Cantelli, E., Chalumeau, A., et al. (2021). Correction of β-thalassemia by CRISPR/Cas9 editing of the α-globin locus in human hematopoietic stem cells. Blood Adv. 5 (5), 1137–1153. doi:10.1182/bloodadvances.2020001996
Pecoraro, A., Troia, A., Calzolari, R., Scazzone, C., Rigano, P., Martorana, A., et al. (2015). Efficacy of rapamycin as inducer of Hb F in primary erythroid cultures from sickle cell disease and β-thalassemia patients. Hemoglobin 39, 225–229. doi:10.3109/03630269.2015.1036882
Prosdocimi, M., Zuccato, C., Cosenza, L. C., Borgatti, M., Lampronti, I., Finotti, A., et al. (2022). A rational approach to drug repositioning in β-thalassemia: Induction of fetal hemoglobin by established drugs. Wellcome Open Res. 7, 150. doi:10.12688/wellcomeopenres.17845.2
Psatha, N., Georgakopoulou, A., Li, C., Nandakumar, V., Georgolopoulos, G., Acosta, R., et al. (2021). Enhanced HbF reactivation by multiplex mutagenesis of thalassemic CD34+ cells in vitro and in vivo. Blood 138 (17), 1540–1553. doi:10.1182/blood.2020010020
Psatha, N., Reik, A., Phelps, S., Zhou, Y., Dalas, D., Yannaki, E., et al. (2018). Disruption of the BCL11A erythroid enhancer reactivates fetal hemoglobin in erythroid cells of patients with β-thalassemia major. Mol. Ther. Methods Clin. Dev. 10, 313–326. doi:10.1016/j.omtm.2018.08.003
Quintana-Bustamante, O., Fañanas-Baquero, S., Dessy-Rodriguez, M., Ojeda-Pérez, I., and Segovia, J. C. (2022). Gene editing for inherited red blood cell diseases. Front. Physiol. 13, 848261. doi:10.3389/fphys.2022.848261
Ramadier, S., Chalumeau, A., Felix, T., Othman, N., Aknoun, S., Casini, A., et al. (2022). Combination of lentiviral and genome editing technologies for the treatment of sickle cell disease. Mol. Ther. 30 (1), 145–163. doi:10.1016/j.ymthe.2021.08.019
Ravi, N. S., Wienert, B., Wyman, S. K., Bell, H. W., George, A., Mahalingam, G., et al. (2022). Identification of novel HPFH-like mutations by CRISPR base editing that elevate the expression of fetal hemoglobin. Elife 11, e65421. doi:10.7554/eLife.65421
Reid, M. E., El Beshlawy, A., Inati, A., Kutlar, A., Abboud, M. R., Haynes, J., et al. (2014). A double-blind, placebo-controlled phase II study of the efficacy and safety of 2,2-dimethylbutyrate (HQK-1001), an oral fetal globin inducer, in sickle cell disease. Am. J. Hematol. 89 (7), 709–713. doi:10.1002/ajh.23725
Rönndahl, G., Mönkemeyer, S., Schulze, S., Pekrun, A., Eikel, D., Nau, H., et al. (2006). Novel valproic acid derivatives with hemoglobin F inducing activity. Am. J. Hematol. 81 (5), 374–376. doi:10.1002/ajh.20575
Rosanwo, T. O., and Bauer, D. E. (2021). Editing outside the body: Ex vivo gene-modification for β-hemoglobinopathy cellular therapy. Mol. Ther. 29 (11), 3163–3178. doi:10.1016/j.ymthe.2021.10.002
Samuelson, C., Radtke, S., Zhu, H., Llewellyn, M., Fields, E., Cook, S., et al. (2021). Multiplex CRISPR/Cas9 genome editing in hematopoietic stem cells for fetal hemoglobin reinduction generates chromosomal translocations. Mol. Ther. Methods Clin. Dev. 23, 507–523. doi:10.1016/j.omtm.2021.10.008
Schrier, S. L. (2002). Pathophysiology of thalassemia. Curr. Opin. Hematol. 9 (2), 123–126. doi:10.1097/00062752-200203000-00007
Segura, E. E. R., Ayoub, P. G., Hart, K. L., and Kohn, D. B. (2023). Gene therapy for β-hemoglobinopathies: From discovery to clinical trials. Viruses 15 (3), 713. doi:10.3390/v15030713
Sehgal, S. N. (2003). Sirolimus: Its discovery, biological properties, and mechanism of action. Transpl. Proc. 35 (3), 7S–14S. doi:10.1016/s0041-1345(03)00211-2
Shariati, L., Khanahmad, H., Salehi, M., Hejazi, Z., Rahimmanesh, I., Tabatabaiefar, M. A., et al. (2016). Genetic disruption of the KLF1 gene to overexpress the γ-globin gene using the CRISPR/Cas9 system. J. Gene Med. 18 (10), 294–301. doi:10.1002/jgm.2928
Shariati, L., Rohani, F., Heidari Hafshejani, N., Kouhpayeh, S., Boshtam, M., Mirian, M., et al. (2018). Disruption of SOX6 gene using CRISPR/Cas9 technology for gamma-globin reactivation: An approach towards gene therapy of β-thalassemia. J. Cell. Biochem. 119 (11), 9357–9363. doi:10.1002/jcb.27253
Sharma, D. C., Singhal, S., Woike, P., Rai, S., Yadav, M., and Gaur, R. (2020). Hereditary persistence of fetal hemoglobin. Asian J. Transfus. Sci. 14 (2), 185–186. doi:10.4103/ajts.AJTS_71_16
Shi, L., Cui, S., Engel, J. D., and Tanabe, O. (2013). Lysine-specific demethylase 1 is a therapeutic target for fetal hemoglobin induction. Nat. Med. 19 (3), 291–294. doi:10.1038/nm.3101
Sripichai, O., and Fucharoen, S. (2016a). Fetal hemoglobin regulation in β-thalassemia: Heterogeneity, modifiers and therapeutic approaches. Expert Rev. Hematol. 9, 1129–1137. doi:10.1080/17474086.2016.1255142
Sripichai, O., and Fucharoen, S. (2016b). Fetal hemoglobin regulation in β-thalassemia: Heterogeneity, modifiers and therapeutic approaches. Expert Rev. Hematol. 9 (12), 1129–1137. doi:10.1080/17474086.2016.1255142
Takase, S., Hiroyama, T., Shirai, F., Maemoto, Y., Nakata, A., Arata, M., et al. (2023). A specific G9a inhibitor unveils BGLT3 lncRNA as a universal mediator of chemically induced fetal globin gene expression. Nat. Commun. 14 (1), 23. doi:10.1038/s41467-022-35404-0
Thuret, I., Ruggeri, A., Angelucci, E., and Chabannon, C. (2022). Hurdles to the adoption of gene therapy as a curative option for transfusion-dependent thalassemia. Stem Cells Transl. Med. 11 (4), 407–414. doi:10.1093/stcltm/szac007
Topfer, S. K., Feng, R., Huang, P., Ly, L. C., Martyn, G. E., Blobel, G. A., et al. (2022). Disrupting the adult globin promoter alleviates promoter competition and reactivates fetal globin gene expression. Blood 139 (14), 2107–2118. doi:10.1182/blood.2021014205
Trakarnsanga, K., Thongsin, N., Metheetrairut, C., Tipgomut, C., Poldee, S., and Wattanapanitch, M. (2022). Genetic correction of haemoglobin E in an immortalised haemoglobin E/beta-thalassaemia cell line using the CRISPR/Cas9 system. Sci. Rep. 12 (1), 15551. doi:10.1038/s41598-022-19934-7
Tucci, F., Galimberti, S., Naldini, L., Valsecchi, M. G., and Aiuti, A. (2022). A systematic review and meta-analysis of gene therapy with hematopoietic stem and progenitor cells for monogenic disorders. Nat. Commun. 13 (1), 1315. doi:10.1038/s41467-022-28762-2
Uda, M., Galanello, R., Sanna, S., Lettre, G., Sankaran, V. G., Chen, W., et al. (2008). Genome wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of β-thalassemia. Proc. Natl. Acad. Sci. U. S. A. 105, 1620–1625. doi:10.1073/pnas.0711566105
Venkatesan, V., Christopher, A. C., Rhiel, M., Azhagiri, M. K. K., Babu, P., Walavalkar, K., et al. (2023a). Editing the core region in HPFH deletions alters fetal and adult globin expression for treatment of β-hemoglobinopathies. Mol. Ther. Nucleic Acids 32, 671–688. doi:10.1016/j.omtn.2023.04.024
Venkatesan, V., Christopher, A. C., Rhiel, M., Azhagiri, M. K. K., Babu, P., Walavalkar, K., et al. (2023b). Editing the core region in HPFH deletions alters fetal and adult globin expression for treatment of β-hemoglobinopathies. Mol. Ther. Nucleic Acids 32, 671–688. doi:10.1016/j.omtn.2023.04.024
Voon, H. P., Wardan, H., and Vadolas, J. (2008). siRNA-mediated reduction of alpha-globin results in phenotypic improvements in beta-thalassemic cells. Haematologica 93 (8), 1238–1242. doi:10.3324/haematol.12555
Wakabayashi, A., Kihiu, M., Sharma, M., Thrasher, A. J., Saari, M. S., Quesnel-Vallieres, M., et al. (2022). Identification and characterization of RBM12 as a novel regulator of fetal hemoglobin expression. Blood Adv. 6, 5956–5968. doi:10.1182/bloodadvances.2022007904
Wang, J., Tran, J., Wang, H., Guo, C., Harro, D., Campbell, A. D., et al. (2016). mTOR Inhibition improves anaemia and reduces organ damage in a murine model of sickle cell disease. Br. J. Haematol. 174, 461–469. doi:10.1111/bjh.14057
Weatherall, D. J. (2001). Phenotype-genotype relationships in monogenic disease: Lessons from the thalassaemias. Nat. Rev. Genet. 2, 245–255. doi:10.1038/35066048
Weber, L., Frati, G., Felix, T., Hardouin, G., Casini, A., Wollenschlaeger, C., et al. (2020). Editing a γ-globin repressor binding site restores fetal hemoglobin synthesis and corrects the sickle cell disease phenotype. Sci. Adv. 6 (7), eaay9392. doi:10.1126/sciadv.aay9392
Xie, S. Y., Ren, Z. R., Zhang, J. Z., Guo, X. B., Wang, Q. X., Wang, S., et al. (2007). Restoration of the balanced alpha/beta-globin gene expression in beta654-thalassemia mice using combined RNAi and antisense RNA approach. Hum. Mol. Genet. 16 (21), 2616–2625. doi:10.1093/hmg/ddm218
Xu, P., Tong, Y., Liu, X. Z., Wang, T. T., Cheng, L., Wang, B. Y., et al. (2015). Both TALENs and CRISPR/Cas9 directly target the HBB IVS2-654 (C > T) mutation in β-thalassemia-derived iPSCs. Sci. Rep. 5, 12065. doi:10.1038/srep12065
Yasara, N., Wickramarathne, N., Mettananda, C., Manamperi, A., Premawardhena, A., and Mettananda, S. (2020). Efficacy and safety of oral hydroxyurea in transfusion-dependent β-thalassaemia: A protocol for randomised double-blind controlled clinical trial. BMJ Open 10 (10), e041958. doi:10.1136/bmjopen-2020-041958
Yasara, N., Wickramarathne, N., Mettananda, C., Silva, I., Hameed, N., Attanayaka, K., et al. (2022). A randomised double-blind placebo-controlled clinical trial of oral hydroxyurea for transfusion-dependent β-thalassaemia. Sci. Rep. 12 (1), 2752. doi:10.1038/s41598-022-06774-8
Ye, L., Wang, J., Tan, Y., Beyer, A. I., Xie, F., Muench, M. O., et al. (2016). Genome editing using CRISPR-Cas9 to create the HPFH genotype in HSPCs: An approach for treating sickle cell disease and β-thalassemia. Proc. Natl. Acad. Sci. U. S. A. 113 (38), 10661–10665. doi:10.1073/pnas.1612075113
Zeng, J., Wu, Y., Ren, C., Bonanno, J., Shen, A. H., Shea, D., et al. (2020). Therapeutic base editing of human hematopoietic stem cells. Nat. Med. 26 (4), 535–541. doi:10.1038/s41591-020-0790-y
Zhang, H., Sun, R., Fei, J., Chen, H., and Lu, D. (2022). Correction of beta-thalassemia IVS-II-654 mutation in a mouse model using prime editing. Int. J. Mol. Sci. 23 (11), 5948. doi:10.3390/ijms23115948
Zhang, X., Campreciós, G., Rimmelé, P., Liang, R., Yalcin, S., Mungamuri, S. K., et al. (2014). FOXO3-mTOR metabolic cooperation in the regulation of erythroid cell maturation and homeostasis. Am. J. Hematol. 89, 954–963. doi:10.1002/ajh.23786
Zipkin, M. (2019). CRISPR's "magnificent moment" in the clinic. Nat. Biotechnol. doi:10.1038/d41587-019-00035-2
Zuccato, C., Bianchi, N., Borgatti, M., Lampronti, I., Massei, F., Favre, C., et al. (2007). Everolimus is a potent inducer of erythroid differentiation and gamma-globin gene expression in human erythroid cells. Acta Haematol. 117 (3), 168–176. doi:10.1159/000097465
Zuccato, C., Breda, L., Salvatori, F., Breveglieri, G., Gardenghi, S., Bianchi, N., et al. (2012). A combined approach for β-thalassemia based on gene therapy-mediated adult hemoglobin (HbA) production and fetal hemoglobin (HbF) induction. Ann. Hematol. 91 (8), 1201–1213. doi:10.1007/s00277-012-1430-5
Zuccato, C., Cosenza, L. C., Zurlo, M., Gasparello, J., Papi, C., D’Aversa, E., et al. (2022). Expression of γ-globin genes in β-thalassemia patients treated with sirolimus: Results from a pilot clinical trial (sirthalaclin). Ther. Adv. Hematol. 13, 20406207221100648. doi:10.1177/20406207221100648
Zuccato, C., Cosenza, L. C., Zurlo, M., Lampronti, I., Borgatti, M., Scapoli, C., et al. (2021). Treatment of erythroid precursor cells from β-thalassemia patients with cinchona alkaloids: Induction of fetal hemoglobin production. Int. J. Mol. Sci. 22 (24), 13433. doi:10.3390/ijms222413433
Zurlo, M., Gasparello, J., Cosenza, L. C., Breveglieri, G., Papi, C., Zuccato, C., et al. (2023). Production and characterization of K562 cellular clones hyper-expressing the gene encoding α-globin: Preliminary analysis of biomarkers associated with autophagy. Genes. (Basel) 14 (3), 556. doi:10.3390/genes14030556
Keywords: β-thalassemia, gene editing, CRISPR-Cas9, fetal hemoglobin (HbF), adult hemoglobin
Citation: Finotti A and Gambari R (2023) Combined approaches for increasing fetal hemoglobin (HbF) and de novo production of adult hemoglobin (HbA) in erythroid cells from β-thalassemia patients: treatment with HbF inducers and CRISPR-Cas9 based genome editing. Front. Genome Ed. 5:1204536. doi: 10.3389/fgeed.2023.1204536
Received: 12 April 2023; Accepted: 30 June 2023;
Published: 17 July 2023.
Edited by:
Claudio Mussolino, University of Freiburg Medical Center, GermanyReviewed by:
Marios Phylactides, The Cyprus Institute of Neurology and Genetics, CyprusSaravanabhavan Thangavel, Center for Stem Cell Research (CSCR), India
Copyright © 2023 Finotti and Gambari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alessia Finotti, alessia.finotti@unife.it; Roberto Gambari, gam@unife.it