 Min Yin
Min Yin Jiaxin Yang
Jiaxin Yang Qinjie Tian
Qinjie Tian- Department of Obstetrics and Gynecology, National Clinical Research Center for Obstetric and Gynecologic Diseases, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China
17α−hydroxylase/17,20−lyase deficiency (17-OHD), caused by mutations in the gene of the cytochrome P450 family 17 subfamily A member 1 (CYP17A1), is a rare type of congenital adrenal hyperplasia (CAH), usually characterized by cortisol and sex steroid deficiency combined with excessive mineralocorticoid. Gonadoblastoma is a relatively rare ovarian tumor that is frequently seen among patients with 46,XY gonadal dysgenesis. Rarely have they been reported in female patients with normal 46,XX karyotype. Here, we report an interesting case of an 11-year-old Chinese girl who presented acute abdominal pain that was later attributed to tumor rupture of right ovarian gonadoblastoma with dysgerminoma. Further evaluations revealed hypertension and hypokalemia. Hormonal findings showed increased progesterone, hypergonadotropic hypogonadism, and low cortisol levels. Her chromosome karyotype was 46,XX without Y chromosome material detected. Genetic analysis revealed that the patient had a homozygous pathogenic variant c.985_987delTACinsAA (p.Y329Kfs*90) in exon 6 of the CYP17A1 gene and that her parents were all heterozygous carriers of this pathogenic variant. Due to the variable clinical manifestations of 17-OHD, meticulous assessment including genetic analysis is necessary. Further study is warranted to unravel the mechanism of gonadoblastoma in a patient with normal karyotypes.
Introduction
Congenital adrenal hyperplasia (CAH) refers to a group of syndromes caused by inherited deficiencies in one of five enzymes involved in the biosynthesis of cortisol from cholesterol (1). The majority of CAH cases (90–95%) are caused by 21α-hydroxylase deficiency, whereas 17α-hydroxylase/17,20-lyase deficiency (17-OHD) is the least frequent form of the condition and only accounts for 1% of all CAH cases (2). The cytochrome P450c17 enzyme catalyzes both 17α-hydroxylase activity and 17,20-lyase activity, which play a pivotal role in the production of cortisol and sex hormones. This enzyme is encoded by the CYP17A1 gene, located on chromosome 10q24.3. Mutations in the CYP17A1 gene lead to a deficiency in 17α-hydroxylase/17,20-lyase. Due to blockage of sex hormone synthesis, 17-OHD causes differences/disorder of sex development (DSD) in 46,XY, while sexual infantilism and primary amenorrhea in 46,XX. Moreover, insufficient glucocorticoids in turn increase adrenocorticotropic hormone (ACTH) secretion, leading to adrenal hyperplasia and excessive mineralocorticoid, causing hypertension and hypokalemia (3).
Gonadoblastomas are relatively rare ovarian tumors consisting of sex cord and primitive germ cell components (4). Although the great majority of gonadoblastomas occur in individuals with 46,XY gonadal dysgenesis, a substantial number arise in individuals with normal 46,XX karyotype (5). In this study, we reported a case of an 11-year-old girl with 46,XX karyotype who initially presented with ovarian gonadoblastoma with dysgerminoma and was finally diagnosed with 17-OHD caused by p.Y329Kfs*90 homozygous pathogenic variant.
Case report
An 11-year-old girl was admitted to the local hospital for abdominal pain in April 2022. CT revealed a large pelvic mass measuring 10 × 8 cm in diameter, with suspicion of adnexal torsion or tumor rupture. Serum tumor markers, including carbohydrate antigen (CA) 125, CA199, carcinogenic embryonic antigen (CEA), alpha-fetoprotein (AFP) and β-human chorionic gonadotropin (β-HCG) were in the normal range. An emergent laparoscopy showed a ruptured ovarian tumor measuring 10 × 8 × 6 cm on the right side, along with approximately 50 ml of intra-abdominal bleeding. The root of the right adnexa was twisted 720 degrees. The uterus and the left ovary were small in size, and no enlarged retroperitoneal lymph nodes were noted. Right salpingo-oophorectomy was performed. The sample was placed in an endo bag and exteriorized through the incision. Microscopically, a mixture of germ cells, sex cord elements, and hyaline bodies were arranged in large nests and lobules. The sex cord cells were arranged at the periphery of these nests (Figure 1A). The dysgerminoma component showed positive immunohistochemical staining (IHC) for OCT4 (Figure 1B), SALL4 and CD117 (Figure 1C). An α-inhibin IHC staining showed positivity in the component of the sex cord of the nests (Figure 1D). Other IHC markers, including estrogen receptor (ER), progesterone receptor (PR), AFP, CEA, β-HCG, testis-specific protein Y-encoded (TSPY), and epithelial membrane antigen (EMA), were negative. Based on these findings, the diagnosis of “gonadoblastoma with dysgerminoma” was made.
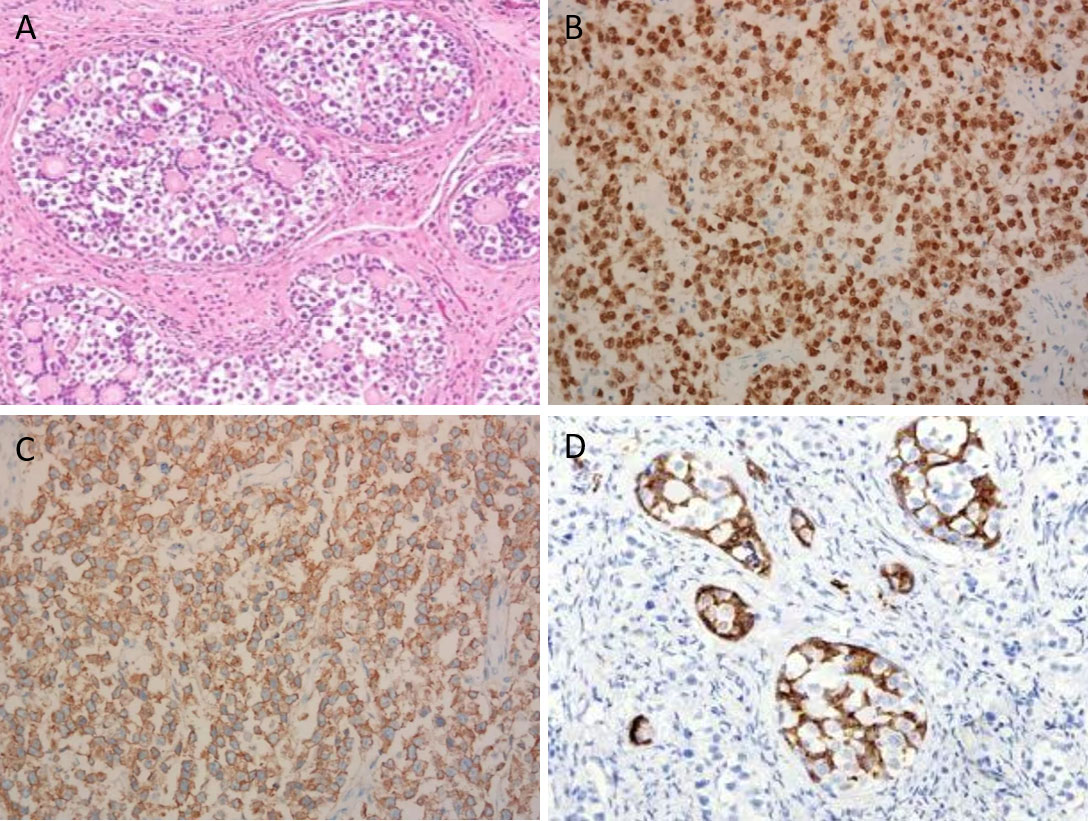
Figure 1 Microscopic view of the ovarian gonadoblastoma with coexisting dysgerminoma (×200). (A) A mixture of germ cells, elements of the sex cord, and hyaline bodies were placed in large nests and lobes. (B) Dysgerminoma component demonstrating positive nuclear staining for OCT4. (C) Dysgerminoma component demonstrating positive membranous staining for CD117. (D) Sex cord component demonstrating positive cytoplasmic staining for α-inhibin.
The patient was then referred to our hospital’s outpatient in May 2022 for further evaluation and treatment. Menstruation has not yet begun. Physical examination showed that her blood pressure was 148/106 mmHg, her body weight was 40 kg, and her height was 160 cm. Her breasts were in Tanner stage I, without pubic or axillary hair. There was no clitoromegaly and the external genitalia were phenotypically female and infantile. Blood biochemistry tests showed hypokalemia and normal plasma sodium and chlorine level. The following hormonal tests revealed low estradiol (E2) and testosterone (T), along with increased levels of progesterone (P), follicle stimulating hormone (FSH) and luteinizing hormone (LH). Furthermore, decreased cortisol and elevated ACTH levels were observed (Table 1). Serum tumor markers were still within the normal range.
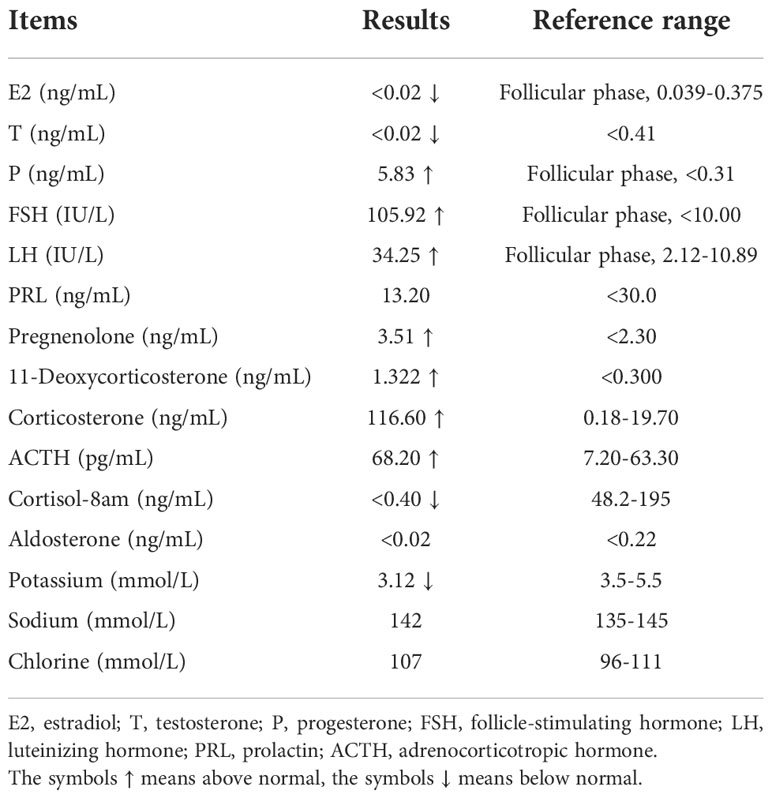
Table 1 Hormone levels and biochemical parameters of the patient.
An abdominal CT scan revealed no apparent hyperplasia in the bilateral adrenal gland (Figure 2A). Based on the X-ray, her bone age was 3 years younger than her actual age (Figure 2B). Karyotype analysis from the peripheral blood sample revealed a normal female 46,XX pattern and ruled out the possibility of mosaicism. To further exclude the possibility of a low level of Y chromosome material, fluorescence in situ hybridization (FISH) analysis using a probe specific for the sex-determining region of Y-chromosome (SRY) gene and zinc-finger Y (ZFY) gene locus at the Y chromosome was performed and no signal was detected for the SRY and ZFY locus. To exclude the possibility of somatic mosaicism, additional molecular analyzes were conducted in both the gonadal stroma and tumor tissue. Quantitative fluorescence-polymerase chain reaction (QF-PCR) results showed no amplification of the SRY in both the gonadal stroma and tumor tissue. Further whole-exome sequencing of the patient indicated that she was homozygous for c.985_987delTACinsAA (p.Y329Kfs*90) in exon 6 of the CYP17A1 gene (Figure 3A). No other genetic variant in gonadal development was found. She has no siblings and peripheral blood samples from her parents were also sent to analyze the genetic sequence of CYP17A1. Although her parents were apparently not consanguineous, they were all heterozygous carriers of this pathogenic variant (Figures 3B, C).
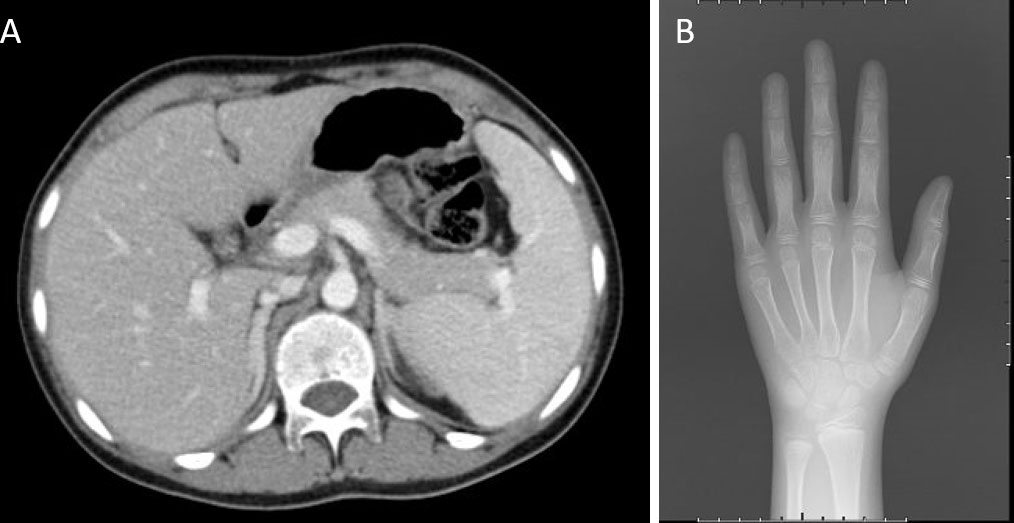
Figure 2 Imaging examinations. (A) An abdominal CT scan revealed no apparent hyperplasia in the bilateral adrenal gland. (B) X-ray showed her bone age was 8 years old.
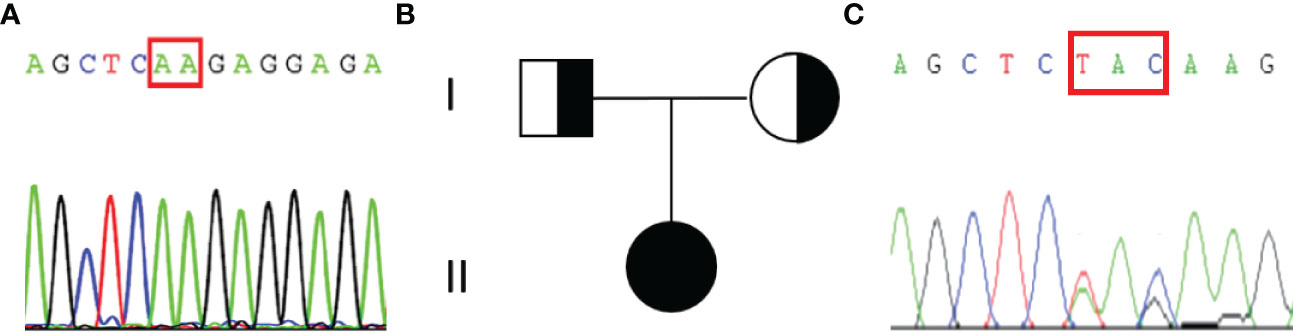
Figure 3 Results of the sequencing of CYP17A1 gene and the pedigree of the patient’s family. (A)The patient was homozygous for the pathogenic variant c.985_987delTACinsAA (p.Y329Kfs*90). The red rectangular represents the altered nuclear acid. (B) Pedigree of the patient’s family. (C) Her parents were all heterozygous for c.985_987delTACinsAA (p.Y329Kfs*90) pathogenic variant.
As no Y-chromosome material was identified in peripheral blood, gonadal stroma, and tumor, the risk of malignancy of the left ovary was estimated to be low. Therefore, the contralateral ovary was preserved. According to the International Federation of Gynecology and Obstetrics (FIGO) staging classification for cancer of the ovary, the pathological staging was stage I gonadoblastoma with dysgerminoma, and no adjuvant chemotherapy was needed based on the National Comprehensive Cancer Network (NCCN) guidelines. Glucocorticoid supplementary treatment was initiated. The patient was prescribed prednisone starting at 5 mg/day. Three months later, her blood pressure varied between 117-130/72-85 mmHg, and plasma levels of potassium, cortisol, and ACTH returned to the normal range. After communicating with the patient and her parents about her condition, supplementary treatment with sex hormones will be used later to promote the development of secondary sexual characteristics and induce an artificial menstrual cycle. During the follow-up period, physical examination, blood parameters, including sex hormone, cortisol, ACTH, aldosterone, blood electrolyte levels, and pelvic ultrasound will be regularly monitored. Currently, the follow-up is still ongoing.
Discussion
17-OHD is a rare kind of congenital adrenal hyperplasia (CAH) characterized by the failure to synthesize cortisol, adrenal androgens, and gonadal steroids (6). Defects in 17α-hydroxylase/17,20-lyase activities result in low cortisol levels and a reduction in dehydroepiandrosterone (DHEA) and androstenedione, which in turn results in low estradiol and testosterone levels (7). Negative feedback is diminished by lower levels of cortisol, and consequentially induces ACTH overproduction, adrenal hyperplasia, and excessive synthesis of mineralocorticoid precursors, such as 11-deoxycorticosterone and corticosterone, resulting in low-renin hypertension and hypokalaemia. A deficiency of sex hormones causes primary amenorrhea in females, and 46,XY DSD in males, which manifests with small testicles, small penis, and mammary gland development.
Biglieri et al. described the first 17-OHD case in 1966, which involved a 35-year-old woman with hypertension and delayed menstruation as the primary symptoms (8). To date, more than 100 pathogenic variants have been reported in the CYP17A1 gene, and most are associated with a classic phenotype of combined 17α-hydroxylase/17,20-lyase deficiency. A smaller number of CYP17A1 missense variants have been reported to exhibit partial impairment of 17α-hydroxylase/17,20-lyase activity. Based on the degree of enzyme deficiency, 17-OHD is classified chiefly into complete and partial type deficiency. Specific CYP17A1 gene variants have been reported to cause a partial loss of 17a-hydroxylase/17,20-lyase activities or dissociation of the 17a-hydroxylase and 17,20-lyase functions (9). Hypergonadotropic hypogonadism and high serum levels of ACTH and mineralocorticoids are found in complete and partial 17-OHD patients. Patients with partial 17-OHD have different clinical characteristics such as the development of breasts and pubic hair, and oligomenorrhea or secondary amenorrhea, due to the various degrees of estrogenic and androgenic impacts (10).
Until now, numerous pathogenic variants in the CYP17A1 gene have been identified in 17-OHD patients, and differ among racial and geographic areas. Tyrosine at amino acid position 329 is particularly critical for the appropriate functioning of the P450c17 enzyme (11). Tyrosine (Y)-329-Lysine (K) is the first impacted amino acid due to a frameshift in exon 6 of the CYP17A1 gene, which occurs when the nucleotide 985 to 987 (TAC) was changed by AA. This frameshift pathogenic variant finally leads to the premature stop codon of 418TGA and generates a truncated protein containing only 417 amino acids without the heme-binding region of codons 435 and 455, which plays a key role in the catalytic function of the enzyme. Therefore, the loss of the pivotal functional domain results in the complete loss of 17-hydroxylase/17,20-lyase activity.
In 2003, a Korean compound heterozygote patient with primary amenorrhea, hypokalemia, and hypertension, was first diagnosed with this pathogenic variant (12). Subsequently, this pathogenic variant was found in many Chinese 17-OHD patients. Tian et al. reported cases with the pathogenic variant p.Y329Kfs*90 and assumed that this was common in Asian people (13). Another study reported that 10 out of 15 (66.7%) Chinese 17α-OHD patients carried the pathogenic variant p.Y329Kfs*90 (14). To clarify the prevalence of 17-OHD pathogenic variants in China, Wang et al. (15) searched PubMed for all published English papers about 17-OHD and summarized all reported CYP17A1 gene pathogenic variants. In total, genetic variants were reported in 181 Chinese cases; of them, 70 (38.6%) had a pathogenic variant p.Y329Kfs*90.
Gonadoblastoma, an uncommon ovarian tumor, occurs almost always in patients with dysgenetic gonads associated with DSD and an aberrant karyotype (4). Although gonadoblastomas are benign themselves, they are frequently associated with invasive germ-cell malignant tumors. The most common malignancy is pure dysgerminoma, whereas other variants include immature teratoma, embryonal carcinoma, yolk sac tumor, and choriocarcinoma (16). In 1970, Scully reviewed 74 cases of ovarian gonadoblastoma and more than 90% of the reviewed cases had a Y chromosome. As a result, the Y chromosome is believed to be the locus of an oncogene necessary for the development of a gonadoblastoma (17). Subsequently, it was postulated that GBY (gonadoblastoma locus on the Y chromosome) exerts oncogenic effects in dysgenetic gonads and the TSPY gene was established as the putative gene for GBY (18). Y chromosome increases the risk of gonadal malignancy by 15% to 50% in patients who have partial or total gonadal dysgenesis, therefore, preventive bilateral gonadectomy is usually recommended (19). Due to the young age at diagnosis, it is crucial to rule out the presence of Y chromosomal material. Typically, peripheral blood lymphocyte karyotyping is performed to check for Y chromosomes in the germline; however, a small chromosomal fragment containing Y chromosome material may be missed (20). The unique approach of incorporating molecular techniques, such as FISH and QF-PCR, were used to confirm the absence of Y chromosome material in peripheral blood and tumor tissue to exclude gonadal mosaicism or the presence of a fragment of the Y chromosome too small to be detected using routine cytogenetic analysis, therefore accurately determining risks to the contralateral gonad (21).
Although most cases of gonadoblastoma occur in sexually abnormal patients with a Y chromosome, a substantial number of cases arise in females with a normal 46,XX karyotype. We reviewed the literature since 1990 and identified 13 female cases of gonadoblastoma with a normal 46,XX karyotype (22–34). Table 2 summarized the clinical characteristics of the 13 cases that we found in the literature. The age at diagnosis varied between 9 to 34 years old. Of the 14 cases, 8 (57%) occurred in women older than 18 years, 4 of whom were fertile. Most (86%) gonadoblastomas were unilateral. Preoperatively, most of them reported abnormally elevated serum tumor markers, such as β-HCG, AFP, and lactic dehydrogenase (LDH). Only one patient reported elevated levels of testosterone, while others did not report hormone levels. Kanaga et al. (29) reported a 14-year-old girl who presented with abdominal distension, excessive hair growth over the body, and hoarseness of voice. An elevation of LDH, β-HCG, and testosterone levels suggested the probable diagnosis of germ cell tumor, mostly dysgerminoma. Laparotomy revealed a large left ovarian gonadoblastoma with dysgerminoma. After surgery and combination chemotherapy, the levels of β-HCG, LDH, and testosterone were reduced to normal. Patients with gonadoblastoma and dysgerminoma generally have an excellent prognosis. When gonadoblastoma is accompanied by more aggressive germ cell tumors, such as yolk sac tumor, embryonal carcinoma, immature teratoma, and choriocarcinoma, the prognosis is different. Even though no hypothesis for the occurrence of these neoplasms has yet been put out in the literature, several pathological studies suggested that they likely developed via a completely distinct molecular pathway from those that arose in people who have a DSD (4, 5).
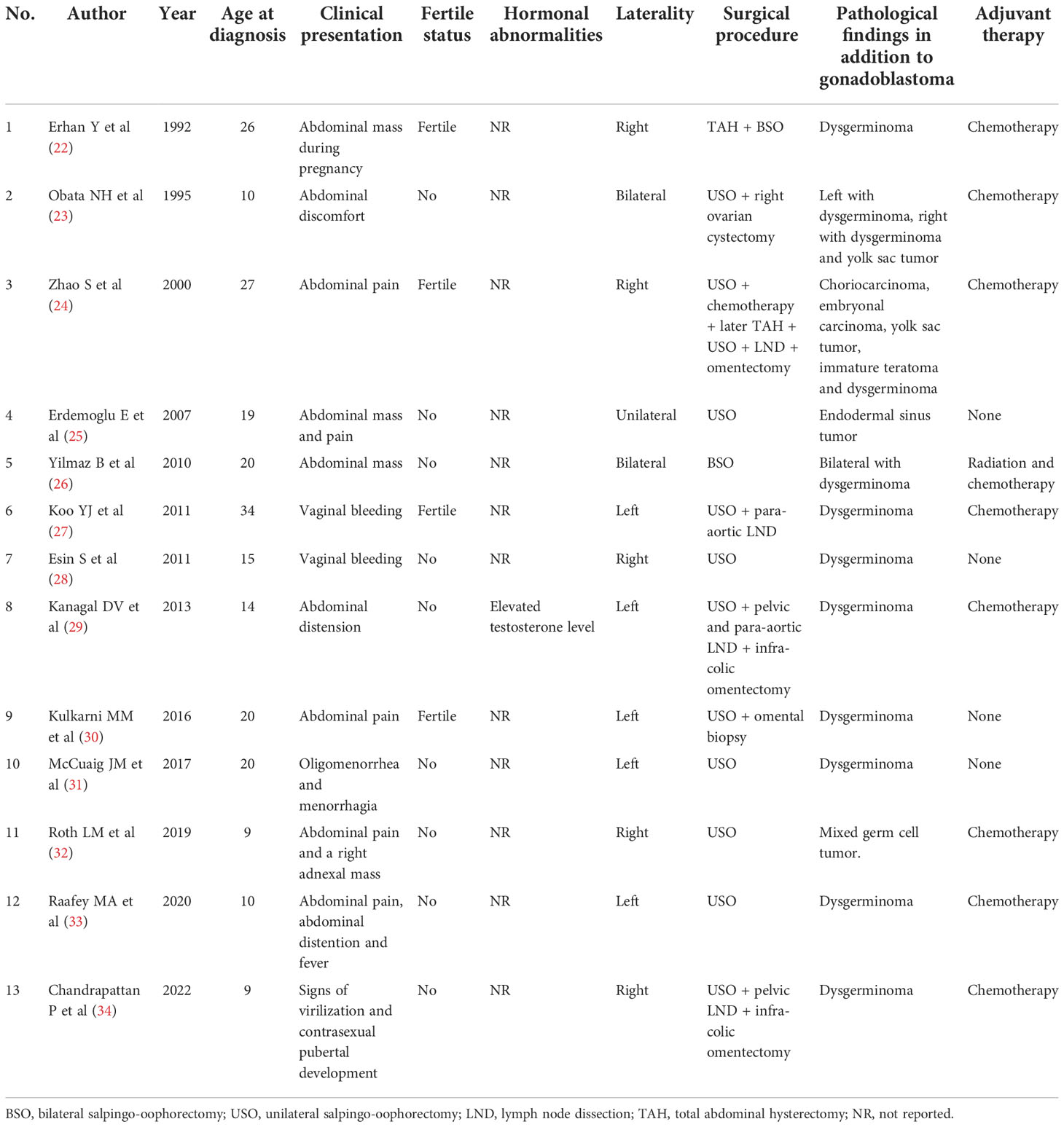
Table 2 Summary of published female cases of gonadoblastoma with normal 46, XX karyotype.
At present, there is limited evidence on the gonadal-malignancy risk of patients with 17-OHD, and there is no clear guidance regarding prophylactic gonadectomies. According to our experience, of the 20 female 17-OHD patients with 46,XY, two (10%) patients had gonadal tumors, one with a Leydig cell tumor and the other with a Sertoli cell tumor (35). The gonads of these two patients were located in the abdomen. Genetic analysis of the CYP17A1 gene showed that one patient had a homozygous pathogenic variant p.T390R in exon 7 of CYP17A1, and another had a homozygous pathogenic variant p.Y329Kfs*90, whose variant type was the same as the case we reported here. Given the fact that the malignancy rate for these patients is quite high, 17-OHD patients with Y chromosome material are recommended to undergo prophylactic gonadectomy to prevent gonadal malignancy. In our case, no Y chromosome material was identified in peripheral blood, gonadal stroma, and tumor, and the risk of malignancy of the contralateral ovary was estimated to be low. So, the second surgery for contralateral ovary resection was avoided.
In the management of 17-OHD, glucocorticoid administration is the key therapy to suppress adrenal hyperplasia and normalize blood pressure as well as plasma potassium level. Treatment should be individualized and the dose should be adjusted according to the patient’s blood pressure, plasma potassium, and hormone levels (36). In cases where glucocorticoids alone cannot control blood pressure and potassium levels well, mineralocorticoid receptor antagonists could be added. Supplementation therapy of sex steroid hormones should be initiated in adolescent female patients to promote the development of secondary sex characteristics. Additionally, cyclical estrogen and progestin therapy are required in patients with an intact uterus to induce menstruation (37).
Conclusion
In summary, 17-OHD is uncommon and challenging in clinical practice. We describe the extremely rare case of a 17-OHD female patient with normal 46,XX karyotype accompanied by ovarian gonadoblastoma with dysgerminoma. Due to the variable clinical characteristics of 17-OHD patients, a meticulous assessment is necessary, including genetic analysis. Further study is warranted to unravel the mechanism of gonadoblastoma in patient with normal karyotypes.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by Peking Union Medical College Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
MY: Writing and literature search. JY and QT: Concept, design, and medical practice. XZ: Analysis and interpretation. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Chinese Academy of Medical Sciences Initiative for Innovative Medicine (CAMS-2017-I2M-1-002) and National Key Research and Development Plan of China (2022YFC2704202).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor SL is currently organizing a research topic with the author QT.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. El-Maouche D, Arlt W, Merke DP. Congenital adrenal hyperplasia. Lancet (2017) 390(10108):2194–210. doi: 10.1016/S0140-6736(17)31431-9
2. Kardelen AD, Toksoy G, Baş F, Yavaş Abalı Z, Gençay G, Poyrazoğlu Ş, et al. A rare cause of congenital adrenal hyperplasia: Clinical and genetic findings and follow-up characteristics of six patients with 17-hydroxylase deficiency including two novel mutations. J Clin Res Pediatr Endocrinol (2018) 10(3):206–15. doi: 10.4274/jcrpe.0032
3. Xu Y, Jiang S, Yan Z, Niu Y, Du W, Liu B, et al. Phenotypic heterogeneity and fertility potential of patients with 17-Hydroxylase/17,20-lyase deficiency. J Clin Endocrinol Metab (2022) 107(6):e2610–8. doi: 10.1210/clinem/dgac029
4. Roth LM, Cheng L. Gonadoblastoma: origin and outcome. Hum Pathol (2020) 100:47–53. doi: 10.1016/j.humpath.2019.11.005
5. Roth LM, Czernobilsky B, Mann SA, Cheng L. Gonadoblastoma versus ovarian mixed germ cell-sex cord stromal tumor in women or girls with no evidence of a disorder of sex development: A problem in differential diagnosis. Pathol Res Pract (2020) 216(11):153198. doi: 10.1016/j.prp.2020.153198
6. Kim YM, Kang M, Choi JH, Lee BH, Kim GH, Ohn JH, et al. A review of the literature on common CYP17A1 mutations in adults with 17-hydroxylase/17,20-lyase deficiency, a case series of such mutations among koreans and functional characteristics of a novel mutation. Metabolism (2014) 63(1):42–9. doi: 10.1016/j.metabol.2013.08.015
7. Unal E, Yıldırım R, Taş FF, Tekin S, Ceylaner S, Haspolat YK. A rare cause of delayed puberty in two cases with 46,XX and 46,XY karyotype: 17 α-hydroxylase deficiency due to a novel variant in CYP17A1 gene. Gynecol Endocrinol (2020) 36(8):739–42. doi: 10.1080/09513590.2019.1707798
8. Biglieri EG, Herron MA, Brust N. 17-hydroxylation deficiency in man. J Clin Invest (1966) 45(12):1946–54. doi: 10.1172/JCI105499
9. Xia Y, Shi P, Xia J, Zhang H, Xu L, Kong X. Novel mutations of the CYP17A1 gene in four Chinese 46,XX cases with partial 17a-hydroxylase/17,20-lyase deficiency. Steroids (2021) 173:108873. doi: 10.1016/j.steroids.2021.108873
10. Tian Q, Zhang Y, Lu Z. Partial 17alpha-hydroxylase/17,20-lyase deficiency-clinical report of five Chinese 46,XX cases. Gynecol Endocrinol (2008) 24(7):362–7. doi: 10.1080/09513590802194051
11. Qiao J, Han B, Liu BL, Liu W, Wu JJ, Pan CM, et al. A unique exonic splicing mutation in the CYP17A1 gene as the cause for steroid 17{alpha}-hydroxylase deficiency. Eur J Endocrinol (2011) 164(4):627–33. doi: 10.1530/EJE-10-0933
12. Hahm JR, Kim DR, Jeong DK, Chung JH, Lee MS, Min YK, et al. A novel compound heterozygous mutation in the CYP17 (P450 17alpha-hydroxylase) gene leading to 17alpha-hydroxylase/17,20-lyase deficiency. Metabolism (2003) 52(4):488–92. doi: 10.1053/meta.2003.50080
13. Tian Q, Yao F, Sha G, Huang S, Tseng H, Schindler AE. Genotyping of a Chinese family with 46,XX and 46,XY 17-hydroxylase deficiency. Gynecol Endocrinol (2009) 25(8):485–90. doi: 10.1080/09513590902898239
14. Han B, Xue L, Fan M, Zhao S, Liu W, Zhu H, et al. Clinical and molecular manifestation of fifteen 17OHD patients: a novel mutation and a founder effect. Endocrine (2016) 53(3):784–90. doi: 10.1007/s12020-016-0957-y
15. Wang M, Wang H, Zhao H, Li L, Liu M, Liu F, et al. Prevalence of CYP17A1 gene mutations in 17α-hydroxylase deficiency in the Chinese han population. Clin Hypertens (2019) 25:23. doi: 10.1186/s40885-019-0128-6
16. Losada DM, Benetti-Pinto CL, Andrade LALA. Gonadoblastoma-associated mixed gonadal germ cell tumor with dysgerminoma and hepatoid yolk sac tumor components in 46XY gonadal dysgenesis. J Pediatr Adolesc Gynecol (2019) 32(5):558–60. doi: 10.1016/j.jpag.2019.05.014
17. Scully RE. Gonadoblastoma. a review of 74 cases. Cancer (1970) 25(6):1340–56. doi: 10.1002/1097-0142(197006)25:6<1340::aid-cncr2820250612>3.0.co;2-n
18. Lau YF, Li Y, Kido T. Gonadoblastoma locus and the TSPY gene on the human y chromosome. Birth Defects Res C Embryo Today (2009) 87(1):114–22. doi: 10.1002/bdrc.20144
19. Arya S, Kumar S, Lila AR, Sarathi V, Memon SS, Barnabas R, et al. Exonic WT1 pathogenic variants in 46,XY DSD associated with gonadoblastoma. Endocr Connect (2021) 10(12):1522–30. doi: 10.1530/EC-21-0289
20. Looijenga LHJ, Kao CS, Idrees MT. Predicting gonadal germ cell cancer in people with disorders of sex development; insights from developmental biology. Int J Mol Sci (2019) 20(20):5017. doi: 10.3390/ijms20205017
21. Kwon A, Hyun SE, Jung MK, Chae HW, Lee WJ, Kim TH, et al. Risk of gonadoblastoma development in patients with turner syndrome with cryptic y chromosome material. Horm Cancer (2017) 8(3):166–73. doi: 10.1007/s12672-017-0291-8
22. Erhan Y, Toprak AS, Ozdemir N, Tiras B. Gonadoblastoma and fertility. J Clin Pathol (1992) 45(9):828–9. doi: 10.1136/jcp.45.9.828
23. Obata NH, Nakashima N, Kawai M, Kikkawa F, Mamba S, Tomoda Y. Gonadoblastoma with dysgerminoma in one ovary and gonadoblastoma with dysgerminoma and yolk sac tumor in the contralateral ovary in a girl with 46XX karyotype. Gynecol Oncol (1995) 58(1):124–8. doi: 10.1006/gyno.1995.1195
24. Zhao S, Kato N, Endoh Y, Jin Z, Ajioka Y, Motoyama T. Ovarian gonadoblastoma with mixed germ cell tumor in a woman with 46,XX karyotype and successful pregnancies. Pathol Int (2000) 50(4):332–5. doi: 10.1046/j.1440-1827.2000.01041.x
25. Erdemoglu E, Ozen S. Ovarian gonodoblastoma with yolk sac tumor in a young 46,XX female: case report. Eur J Gynaecol Oncol (2007) 28(6):516–8. doi: 10.1016/j.earlhumdev.2006.05.007
26. Yilmaz B, Gungor T, Bayramoglu H, Soysal S, Mollamahmutoglu L. Bilateral ovarian gonadoblastoma with coexisting dysgerminoma in a girl with 46,XX karyotype. J Obstet Gynaecol Res (2010) 36(3):697–700. doi: 10.1111/j.1447-0756.2010.01225.x
27. Koo YJ, Chun YK, Kwon YS, Lee IH, Kim TJ, Lee KH, et al. Ovarian gonadoblastoma with dysgerminoma in a woman with 46XX karyotype. Pathol Int (2011) 61(3):171–3. doi: 10.1111/j.1440-1827.2010.02636.x
28. Esin S, Baser E, Kucukozkan T, Magden HA. Ovarian gonadoblastoma with dysgerminoma in a 15-year-old girl with 46,XX karyotype: case report and review of the literature. Arch Gynecol Obstet (2012) 285(2):447–51. doi: 10.1007/s00404-011-2073-9
29. Kanagal DV, Prasad K, Rajesh A, Kumar RG, Cherian S, Shetty H, et al. Ovarian gonadoblastoma with dysgerminoma in a young girl with 46,XX karyotype: A case report. J Clin Diagn Res (2013) 7(9):2021–2. doi: 10.7860/JCDR/2013/6412.3393
30. Kulkarni MM, Sinai Khandeparkar SG, Joshi AR, Bhayekar PV. Unilateral gonadoblastoma with dysgerminoma in normal fertile woman having a child: Extremely rare occurrence with characteristic immunohistomorphology. Indian J Pathol Microbiol (2016) 59(4):527–9. doi: 10.4103/0377-4929.191815
31. McCuaig JM, Noor A, Rosen B, Casper RF, Mitri F, Colgan T, et al. Case report: Use of tumor and germline y chromosomal analysis to guide surgical management in a 46,XX female presenting with gonadoblastoma with dysgerminoma. Int J Gynecol Pathol (2017) 36(5):466–70. doi: 10.1097/PGP.0000000000000349
32. Roth LM, Davis MM, Czernobilsky B. Classic and "Dissecting" gonadoblastoma in a phenotypic girl with a 46,XX peripheral karyotype and no evidence of a disorder of sex development. Int J Gynecol Pathol (2019) 38(6):581–7. doi: 10.1097/PGP.0000000000000551
33. Raafey MA, Abdulwaasey M, Fatima SS, Uddin Z, Tariq MU. Bilateral gonadoblastoma with dysgerminoma in a phenotypically normal female with 46XX karyotype: Report of a rare case and literature review. Cureus (2020) 12(7):e8990. doi: 10.7759/cureus.8990
34. Chandrapattan P, Jena A, Patnayak R, Mangaraj S, Naik S, Panda S. Gonadoblastoma with dysgerminoma presenting as virilizing disorder in a young child with 46,XX karyotype: A case report and review of the literature. Case Rep Endocrinol (2022) 2022:5666957. doi: 10.1155/2022/5666957
35. Jiang JF, Xue W, Deng Y, Tian QJ, Sun AJ. Gonadal malignancy in 202 female patients with disorders of sex development containing y-chromosome material. Gynecol Endocrinol (2016) 32(4):338–41. doi: 10.3109/09513590.2015.1116509
36. Zhang D, Sun JR, Xu J, Xing Y, Zheng M, Ye SD, et al. 17α-hydroxylase/17,20 carbon chain lyase deficiency caused by p.Tyr329fs homozygous mutation: Three case reports. World J Clin Cases (2021) 9(8):1923–30. doi: 10.12998/wjcc.v9.i8.1923
Keywords: congenital adrenal hyperplasia, CYP17A1 gene, ovarian gonadoblastoma, dysgerminoma, 17α-hydroxylase/17,20-lyase deficiency
Citation: Yin M, Yang J, Tian Q and Zhang X (2022) Ovarian gonadoblastoma with dysgerminoma in a girl with 46,XX karyotype 17a-hydroxylase/17, 20-lyase deficiency: A case report and literature review. Front. Endocrinol. 13:989695. doi: 10.3389/fendo.2022.989695
Received: 08 July 2022; Accepted: 30 November 2022;
Published: 15 December 2022.
Edited by:
Sarantis Livadas, Metropolitan Hospital, GreeceReviewed by:
Katja Teerds, Wageningen University and Research, NetherlandsMagnus R. Dias da Silva, Federal University of São Paulo, Brazil
Copyright © 2022 Yin, Yang, Tian and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiaxin Yang, yjxpumch@163.com