 Jingru Yang1,2
Jingru Yang1,2 Xianquan Zhan
Xianquan Zhan- 1Shandong Key Laboratory of Radiation Oncology, Shandong Cancer Hospital and Institute, Shandong First Medical University and Shandong Academy of Medical Sciences, Jinan, China
- 2Medical Science and Technology Innovation Center, Shandong First Medical University, Jinan, China
Protein acetylation is a reversible post-translational modification, and is involved in many biological processes in cells, such as transcriptional regulation, DNA damage repair, and energy metabolism, which is an important molecular event and is associated with a wide range of diseases such as cancers. Protein acetylation is dynamically regulated by histone acetyltransferases (HATs) and histone deacetylases (HDACs) in homeostasis. The abnormal acetylation level might lead to the occurrence and deterioration of a cancer, and is closely related to various pathophysiological characteristics of a cancer, such as malignant phenotypes, and promotes cancer cells to adapt to tumor microenvironment. Therapeutic modalities targeting protein acetylation are a potential therapeutic strategy. This article discussed the roles of protein acetylation in tumor pathology and therapeutic drugs targeting protein acetylation, which offers the contributions of protein acetylation in clarification of carcinogenesis, and discovery of therapeutic drugs for cancers, and lays the foundation for precision medicine in oncology.
Introduction
Cancer is a malignant disease with heterogeneity, and its occurrence and development are affected by a variety of factors (1). It has strong ability to migrate, proliferate, and invade, and can adhere to the surrounding normal tissues. There are many factors to affect cancers, including genetic, epigenetic, and environmental factors, which all enhance tumor malignancy (2).
Epigenetics is the change in the level of gene expression without changes in the gene sequence (3). Abnormal changes in epigenetics may lead to the occurrence and development of various malignant diseases. Epigenetic research mainly includes DNA covalent modifications such as DNA methylation and poly-methylation, chromatin remodeling, and the regulation of gene expression levels by non-coding RNAs (3). Proteins are the ultimate executors of biological functions. Studies have shown that many abnormal post-translational modifications are closely associated with malignant tumors, such as acetylation, ubiquitination, and phosphorylation (4). Of them, protein acetylation was discovered in the 1960s, but acetylation has not been extensively studied until recent years (5). Acetylation occurs on histones and non-histones, and most of the current research focuses on acetylation on histones (6). Histone is an octamer that constitutes the ribosome, consisting of four core histones (H3\H4\H2A\H2B), which combine with surrounding DNA fragments to form subunits of the ribosome, and the histone tail is easily translated by different post-translational modification to affect chromatin state and gene expression (6). Histones are prone to be acetylated (6). Studies have shown that protein acetylation is closely related to transcriptional regulation (7).
Acetylation modification is the process of covalently binding acyl-CoA compounds to specific amino acid sites of proteins under the action of acetyltransferase, generally binding to lysine residues (8). This process can also be reversed by deacetylases. This process is reversible and plays an important role in chromatin remodeling, gene expression, and regulation of protein function (9). Acetylation processes in different organelles are independent of each other. For example, acetyl-CoA in mitochondria and acetyl-CoA outside mitochondria are independent of each other (8). Acetylation in mitochondria plays an important role in biological processes such as the tricarboxylic acid cycle and fatty acid oxidation (9, 10). Moreover, protein acetylation is involved in the transcriptional regulation of genes, and some transcriptional co-activators have acetylase activity and some transcriptional co-repressors have deacetylase activity (7). Protein acetylation is associated with novel drug targets for a variety of diseases such as cancer (11). Thereby, it emphasizes the important scientific merits of protein acetylation in carcinogenesis and targeted drug discovery.
This article reviews (i) the component and process of protein acetylation system in cancers, including types of acetylation (N-acetylation, O-acetylation, and K-acetylation), regulators of acetylation (writers-acetyltransferases, erasers-deacetylases, acetyl coenzyme A, and readers), (ii) biological role of acetylation in cancer pathophysiology, including apoptosis, autophagy, cellular metabolism, cell cycle, proliferation, migration, and invasion, and (iii) acetylation system-based targeted drugs in cancer, including HAT inhibitors, HAT activators, HDAC inhibitors, and BET inhibitors. Also, we proposed the future perspectives about the roles of protein acetylation in carcinogenesis and targeted drug discovery. In this review, we focus on the classification of acetylation and its impact on pathophysiological processes in tumorigenesis. We link protein acetylation with epigenetic drugs for tumor treatment to promote the development of cancer precision medicine.
The components and process of acetylation system in cancers
Types of acetylation in cancers
Protein acetylation is the process of covalently binding acyl-CoA class A compound to protein-specific amino acid sites under the action of acetyltransferases. Vincent Allfrey and his colleagues discovered histone lysine acetylation modification in 1964 (7). In subsequent studies, they gradually discovered the mechanism of acetylation modification, the discovery and identification of HAT and HDAC, and the discovery and identification of reader domains, which laid the foundation for protein acetylation. With the development of mass spectrometry and proteomics, non-histone acetylation was discovered and the regulatory process of non-histone acetylation was revealed (7). More and more studies have proved that histone acetylation and non-histone acetylation have the same importance in the regulation of biological processes in organisms (7). After the discovery of non-histone acetylation, histone acetyltransferases were also renamed lysine acetyltransferases and histone deacetylases were renamed lysine deacetylases (7). Histone acetylation occurs in the nucleus and is a type of epigenetic regulation that regulates chromatin structure to regulate transcription and DNA repair. Histone hyperacetylation by histone acetyltransferase is associated with transcriptional activation, while histone deacetylation by histone deacetylase is associated with transcriptional repression. Histone acetylation promotes transcription by remodeling higher-level chromatin structure, attenuating histone-DNA interactions, and providing binding sites for transcription activation complexes (12). Histone deacetylation inhibits transcription, and histone deacetylation and acetylation maintain homeostasis by opposing mechanisms, including the assembly of higher-order chromatin structures and the exclusion of bromo domain-containing transcriptional activation complexes (12). Histone acetylation and tumorigenesis are also closely related, and histone acetylation promotes the expression of certain genes that can lead to tumors (13). For example, P300 is a histone lysine acetyltransferase that catalyzes the attachment of acetyl groups to lysine residues, which leads to the activation of several genes, including several oncogenes. Study finds elevated expression of p300 in breast cancer (13).
Non-histone acetylation is involved in most biological processes in organisms and occurs with very high frequency. Non-histone acetylation is involved in key cellular processes related to organism physiology and tumors, such as gene transcription, DNA damage repair, cell division, protein folding, autophagy, cell signaling, and metabolism. For example, HDAC6 acts not only on histones, but also on non-histone substrates to maintain the balance of non-histone acetylation (14). α-Tubulin, the first non-histone substrate of HDAC6, reversibly modulates its homeostasis and in turn affects MT stability and function (15). The α-tubulin acetylation affects intracellular trafficking events through the protein encoded by the cylindromatosis gene, thereby participating in mitosis and affecting the development of the cell cycle (14). Non-histone acetylation modifies protein expression through various mechanisms and affects protein function. For example, regulating protein stability, regulating protease activity, affecting subcellular localization, and regulating protein-protein interactions, etc. Protein acetylation can be classified into three types (N-acetylation, O-acetylation, and K-acetylation) according to acetylation site in a protein amino acid sequence.
N-acetylation
N-terminal acetylation in a protein is one of the most common modifications in mammals, which transfers the acetyl group to the N terminus of the protein, the amino group of the first residue in the protein (4). Unlike O-acetylation and K-acetylation, N-acetylation is an irreversible post-translational modification. N-acetylation occurs in 80%-90% of human proteins and is controlled by N-acetyltransferases. The addition of the acetyl group to N-terminus changes the charge carried by the amino acid, neutralizes the positive charge of the amino acid residue itself, changes the molecular weight of amino acid residue, changes the properties of the protein, and then affects the biological function of the protein. Studies have shown that N-acetylation mainly affects protein-membrane binding and protein stability (16). N-acetylation is also one of many factors contributing to tumor progression; for example, slow N-acetylation is a factor in bladder carcinogenesis and muscle invasiveness, and NAT1 is recognized as a biomarker candidate in bladder cancer and a potential target for drug development point (17).
O-acetylation
O-acetylation was detected less frequently than N-acetylation and K-acetylation. O-acetylation occurred mainly on the hydroxyl group at the serine or threonine terminal. O-acetylation was discovered in 2006 by Orth while studying YopJ, a bacterial virulence factor that acts as an acetyltransferase during acetylation (18). Studies have shown that YopJ transfers acetyl groups to the hydroxyl residues of serine or threonine, which inhibits the activation of MAPKK6, thereby inhibits the activation of MAPK and NF-κB pathways, inhibites the response of immune responses, and promotes the occurrence and development of malignant diseases (19). The discovery of O-acetylation adds to the complexity of the study of the regulation of gene expression by acetylation. Some studies have found that O-acetylation can compete with phosphorylation at some modification sites (20). Although there are few studies on O-acetylation, it has been found that O-acetylation is closely related to tumorigenesis in recent years (21). GD2 O-acetylation is elevated in neuroblastoma and glioblastoma, which is a potential biomarker of therapeutic target (21). In childhood acute lymphoblastic leukemia, the expression of 9-O-acetylated sialoglycoprotein was enhanced, decreased with the remission of clinical symptoms, and increased again when the disease relapsed (22). These studies indicate that O-acetylation might be a potential biomarker and target for drug-targeted therapy (22).
K-acetylation
Lysine acetylation is currently the most extensive research field of acetylation. Protein deacetylation is very extensive in the human body, with more than 3600 acetylation sites in more than 1750 proteins (23). Lysine acetylation mainly occurs on the histones of ribosomes and is jointly regulated by lysine acetyltransferase and lysine deacetylase to maintain the dynamic balance of lysine acetylation in cells (9). Lysine acetylation also occurs in non-histone proteins in the nucleus, cytoplasm, and mitochondria, and regulates various biological functions of cells (9). For example, DNA repair enzymes can be carried out in the nucleus through acetylation (24). The dynamic balance of lysine acetylation affects multiple functions in the cell, such as gene replication, gene transcription, stability of protein structure, interaction between proteins and proteins, cell cycle, cellular self-regulation, phagocytosis, and cell apoptosis (25). For example, there is a large amount of tubulin in the cytoplasm. Tubulin acts as a cytoskeletal component to maintain the stability of cells. The acetylation of α-tubulin is a significant marker of microtubule stability (26). Studies have shown that the acetylation of cytoskeleton is related to the occurrence of tumors, and tubulin is the target of many anti-tumor drugs. Lysine acetylation is one of the most important post-translational modifications in cell signaling pathways (10). The occurrence and development of many malignant tumors are closely related to lysine acetylation (27). For example, most metabolic enzymes are targets for lysine acetylation, such as ATM, ABL1, CDK9, BTK, CDK1 (25, 28–32), and a large number of acetylated proteins mediated abnormal changes in cell signaling pathways (33). For instance, acetylated phosphoglycerate kinase 1 is involved in glycolysis and amino acid biosynthesis in nonfunctional pituitary neuroendocrine tumors (NF-PitNETs) (34).
Regulators of acetylation in cancers
Acetylation in eukaryotic cells is in a dynamic equilibrium, which is jointly participated by writer-acetyltransferase, eraser-deacetylase, acetyl coenzyme A, and reader (4) (Figure 1).
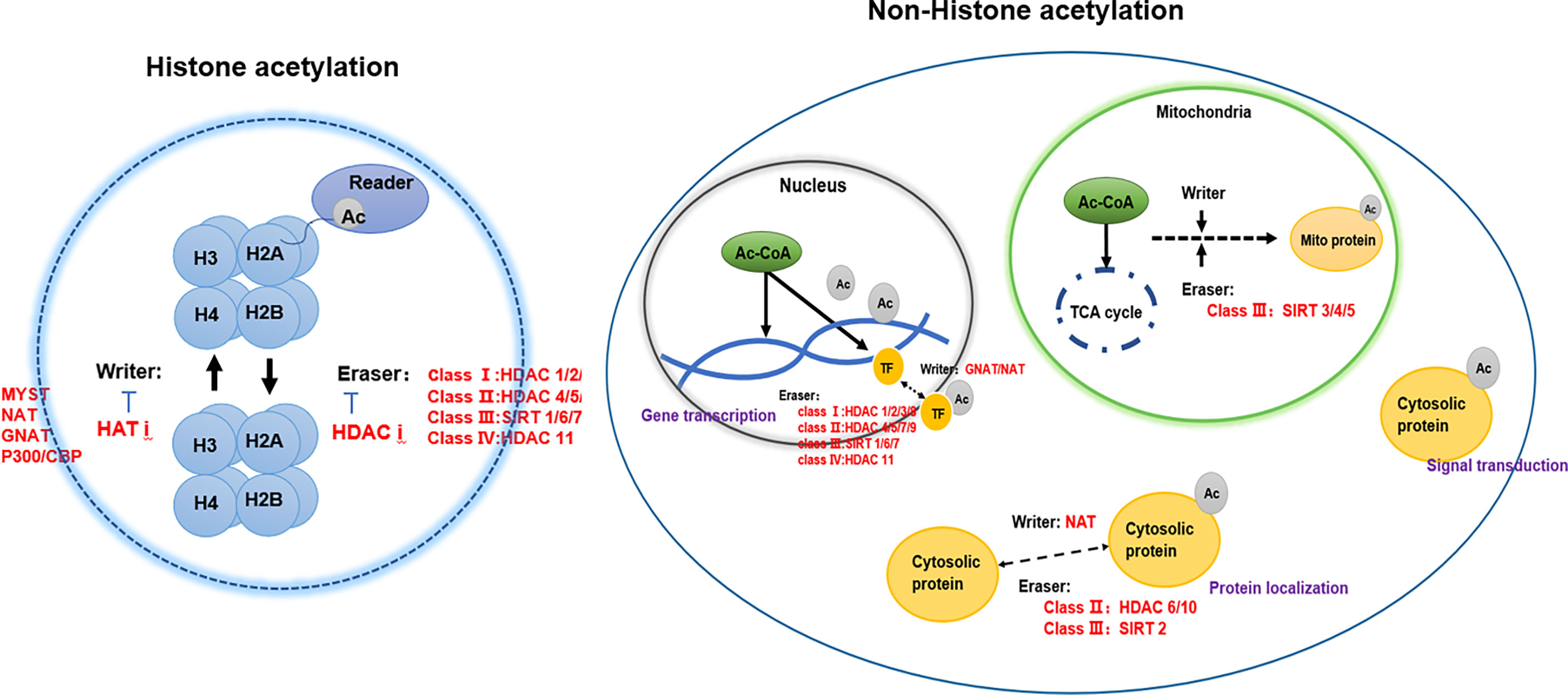
Figure 1 The regulators of protein acetylation-writers, erasers, and readers.
Writer-acetyltransferases
Protein acetylation is a dynamic process by the joint action of acetyltransferases and deacetylases, including N-acetylation, O-acetylation, and K-acetylation (18). Most of the current studies focus on the acetylation of histones (6). Histone acetylation mainly occurs on lysine residues of histones (K-acetylation) in eukaryotic cells. The group is transferred to the side chain of the lysine residue, which in turn changes the R group of the lysine residue, neutralizes the positive charge on the lysine residue, and then affects the properties of the protein, and affects the structure and regulation of chromatin gene expression. According to structural and sequence similarity, mammalian lysine acetyltransferases are mainly divided into three categories: GCN5-related enzymes, p300-related enzymes, and MYST19-related enzymes (35). These acetyltransferases are present in the nucleus, and there are also acetyltransferases such as ESCO1, ESCO2, and HAT1 present in the nucleus (7). In addition to acetyltransferase in the nucleus, tubulin also contains acetyltransferase TAT1 (36). Acetylation of α-tubulin is a prominent marker of microtubule stability, and p27 promotes microtubule acetylation by binding and stabilizing ATAT-1 in glucose-deficient cells (37). Acetyltransferases have substrate specificity, which can regulate the structure of chromatin and thus regulate gene expression (7). For example, MOZ has a plant homeodomain-linked (PHD) type zinc finger that regulates chromatin by binding to trimethylated lysine 4 of histone 3 Structure (38). Acetyltransferases are also closely associated with transcriptional activators. For example, loss of Kat2a affects transcription factor binding and reduces transcriptional burst frequency in a subset of gene promoters, thereby enhancing variability at the transcriptional level (39). CBP/p300 blocks the role of estrogen receptor alpha (ERα) in luminal breast cancer by inhibiting enhancer H3K27 acetylation (40). The mechanism of action of acetyltransferase depends on oncogene activation, which is closely related to the occurrence and development of tumors through signal transduction (41). Both Tip60 expression and ABCE1 acetylation were up-regulated in lung cancer cells (42). Downregulation of Tip60 reduced ABCE1 acetylation levels and inhibited cell proliferation, invasion and migration (42). In addition, downregulation of Tip60 activates the apoptotic pathway, thereby achieves its inhibitory effect (42). Naa10 can acetylate and stabilize TSC2, thereby inhibiting mTOR activity and inhibiting cancer development (43). Acetyltransferase can also control the occurrence and development of tumors by regulating kinases in tumor cells. Naa10 inhibits tumor cell migration by inhibiting MYLK kinase activity through acetylation (44). ESCO2 inhibits the nuclear translocation of hnRNPA1 and increases the binding of hnRNPA1 (heterogeneous nuclear ribonucleoprotein A1) to the intron sequence flanking exon 9 (EI9) of PKM RNA, which ultimately inhibites the formation of PKM1 isoforms and induces the formation of PKM2 isoforms to promote glycolysis of tumor cells, and accelerate the metabolism of tumor cells (45).
The abnormal expression of HATs is usually associated with the occurrence and development of several malignant tumors and poor prognosis, which indicates that HATs may be potential tumor therapy targets and potential biomarkers (46). It is still necessary to in-depth study the effect mechanism of HATs on tumors to clarify the applicability and effectiveness of HATs in the clinical treatment of tumors (Table 1).
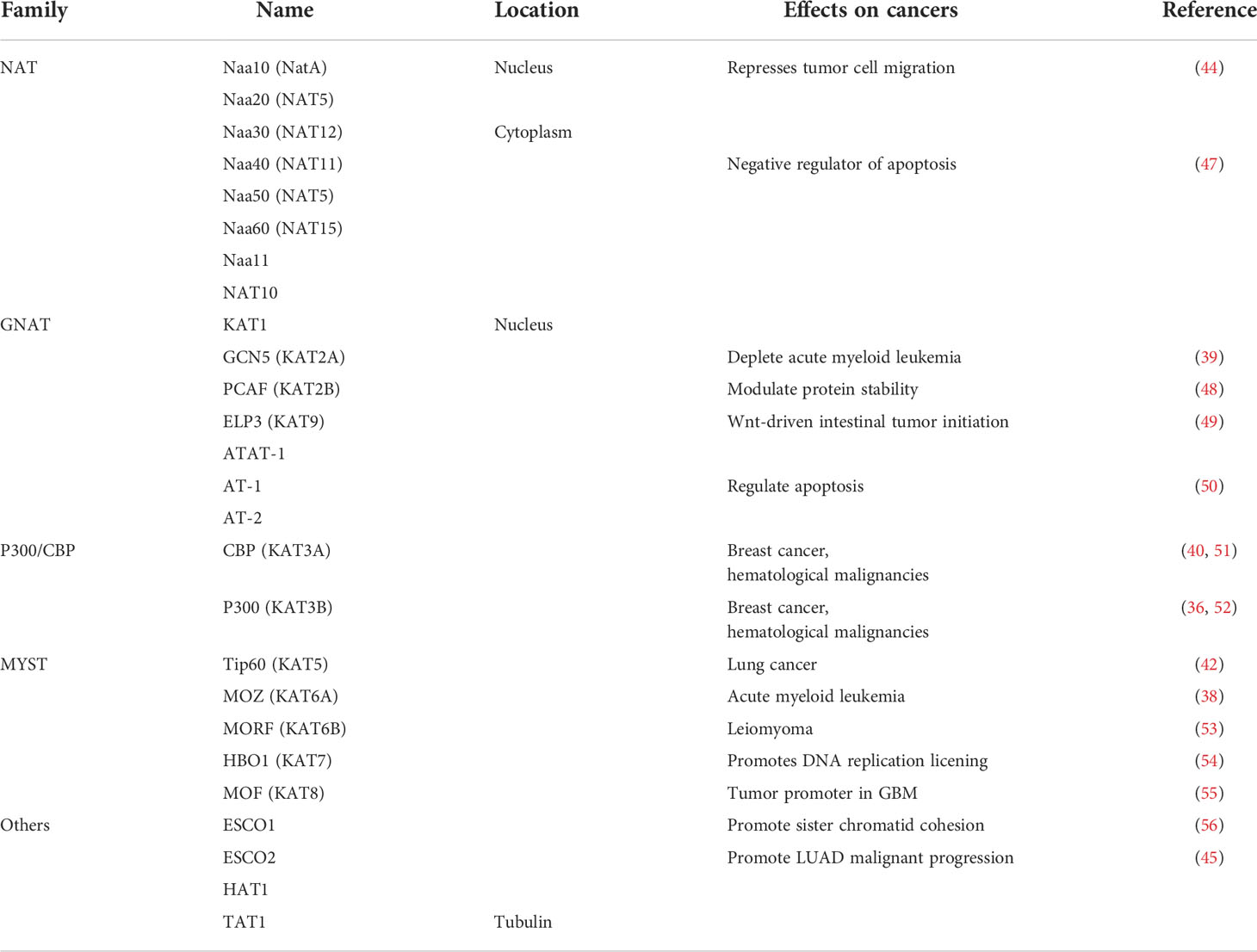
Table 1 Classification, localization and role of acetylases in cancers.
Eraser-deacetylases
Protein deacetylase is called the “eraser” of acetyl group, which reduces the acetyl group attached to the amino acid residue to acetate, affects the R group structure of the amino acid residue, and then reduces the positive charge of the amino acid, and facilitates its binding to negatively charged DNA (52). Protein deacetylases are involved in regulating gene replication, gene transcription, protein structure stability, DNA damage repair, and other cellular functions (7). Mammalian genes encode 18 deacetylases, which act on histone and non-histone proteins in cells to remove their acetyl groups (7). For example, sirtuin enzymes are divided into four classes and localized in different locations of cells (57). Class III belongs to NAD+-dependent sirtuin enzymes, which are localized in mitochondria, cytoplasm and nucleus (57). Zn2+-dependent HDACs have a highly conserved deacetylase domain, including classes I (HDAC 1, 2, 3, and 8), II (HDAC 4, 5, 7, and 9), and IV (HDAC 11) localized in the nucleus (58). Studies have found that HDAC can not only act on histone deacetylation, but also play other roles on histones, such as decrotonylation, and desumoylation (9). HDACs have been found to be abnormally expressed or altered in localization in a variety of cancers (59). Studies have shown that the abnormal expression of HDAC in cancer patients is closely related to the dynamic imbalance of acetylation in the human body (60). In addition, the specific domains of individual sirtuins also have their own specific functions, such as maintaining protein stability (52) (Table 2).
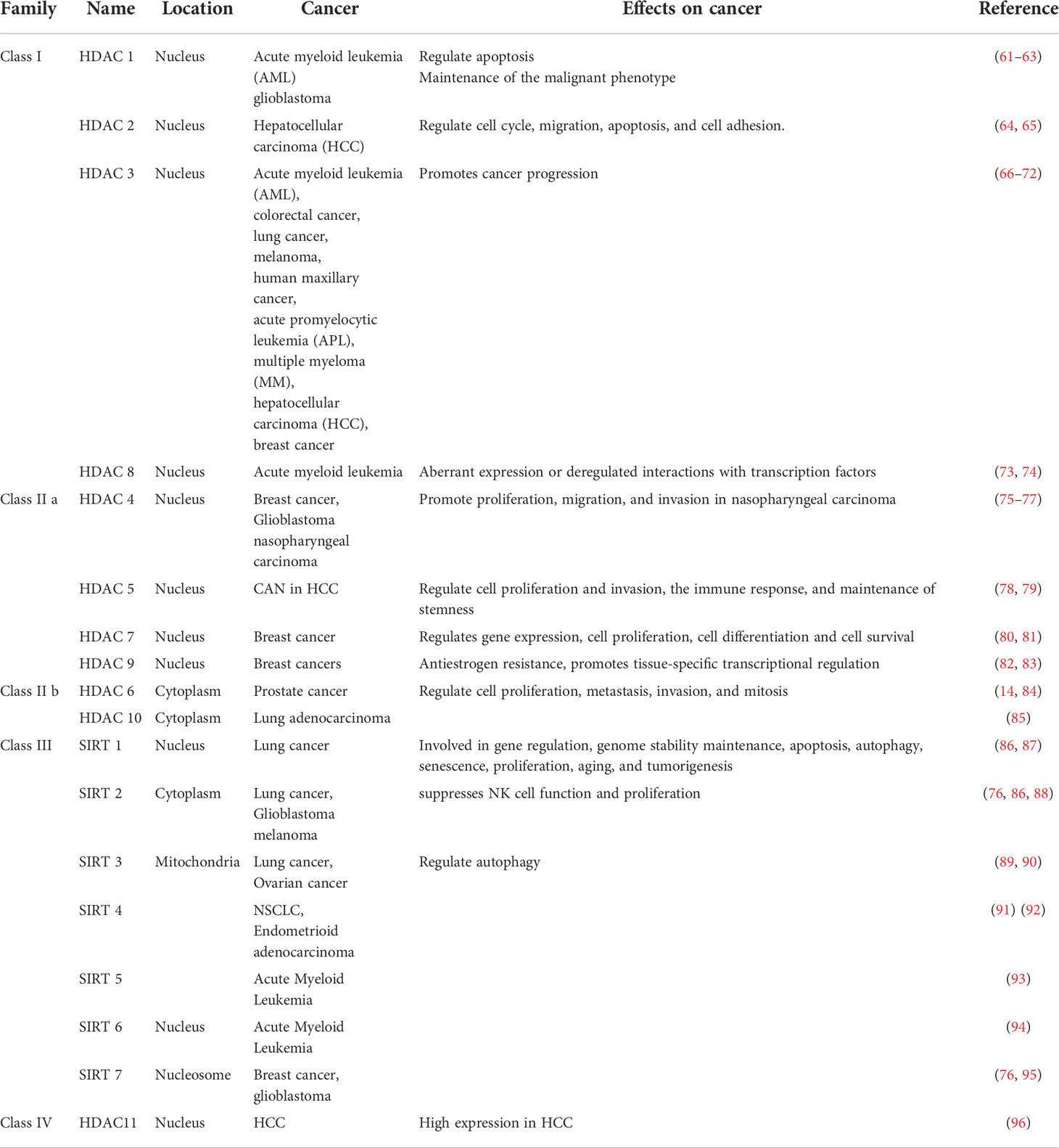
Table 2 Classification, localization and role of deacetylases in cancers.
Transcription factors are a kind of non-histone proteins, and protein deacetylases regulate gene transcription activity by deacetylating transcription factors (27). For example, HDAC7 regulates the acetylation of H3K27 and the transcriptional activity of super-enhancer-related genes in breast cancer stem cells (80). A common mutation in AML is a chromosome 16 inversion that fuses the core-binding factor beta (CBFB) gene with the smooth muscle myosin heavy chain gene (MYH11) to form the oncogene CBFB-MYH11 (61). The expressed protein CBFbeta-SMMHC forms a heterodimer with the key hematopoietic transcription factor RUNX1, and CBFbeta-SMMHC acts together with RUNX1 to activate the transcription of specific target genes (61). HDAC1 promotes transcriptional activation as a cofactor for the leukemic fusion protein CBFbeta-SMMHC (61). HDACs also act directly on proteins involved in tumor migration, metastasis and growth (97). For example, Api5 is a known anti-apoptotic and nuclear protein responsible for inhibiting cell death under conditions of serum starvation (97). The only known post-translational modification of Api5 is the acetylation of lysine 251 (K251) (97). p300 interacts with HDAC1 to regulate cell proliferation by regulating Api5 acetylation and stability (97). Inactivation of SIRT6 in cancer cells results in the accumulation of nuclear ACLY protein, increasing nuclear acetyl-CoA, which in turn drives site-specific histone acetylation and the expression of cancer cell adhesion and migration genes that promote tumor aggressiveness (98). Novel mechanism by which SIRT6 suppresses aggressive cancer cell phenotypes revealed and acetyl-CoA-responsive cell migration and adhesion genes identified as downstream targets of SIRT6 (98). Therefore, the regulatory mechanism of HDACs in tumors is difficult to be clearly described.
Class III HDACs are mainly located in mitochondria, which are the center of cellular energy metabolism (57). Acylated mitochondrial proteins are involved in many functions related to cellular metabolism, including TCA cycle, oxidative phosphorylation, nucleotide metabolism, amino acid metabolism, and urea cycle (99). SIRT can regulate energy production by regulating the acetylation and deacetylation of organisms involved in energy metabolism in mitochondria, thereby affecting cellular metabolism (7). For example, sirtuin 3 (Sirt3) is a key player in maintaining mitochondrial function and is involved in ATP production by regulating the acetyl and pyruvate dehydrogenase complex (PDH) (89). The underlying mechanism of SIRT is also related to the metabolic reprogramming of tumors (9). SIRT5 disruption-induced apoptosis is caused by a decrease in oxidative phosphorylation and glutamine utilization and an increase in mitochondrial superoxide, which is attenuated by ectopic superoxide dismutase 2 (93). SIRT5 controls and orchestrates key metabolic pathways in AML, so SIRT5 may be a potential therapeutic target in AML (93).
Class IV HDACs only contain HDAC11, which is highly expressed in HCC and is closely related to disease prognosis (96). Loss of HDAC11 promotes histone acetylation in the LKB1 promoter region, thereby activating the AMPK signaling pathway and inhibiting the glycolysis pathway, thereby increasing the transcription of LKB1, thereby inhibiting tumorigenesis and HCC progression (96). Histone deacetylases are abnormally expressed in clinical tumor patients and are associated with poor prognosis and survival (59). HDAC9 expression is positively associated with up-regulated genes in endocrine therapy-resistant breast cancer, and high HDAC9 levels are associated with poorer prognosis in patients treated with OHTam (82). HDAC10 regulates tumor stem cell properties in KRAS-driven lung adenocarcinoma, and HDAC10 regulates the stem-like properties of kras-expressing tumor cells by targeting SOX9 (85). The expression of SOX9 is significantly increased in HDAC10-depleted tumor cells, TGFβ pathway-related genes are enriched in HDAC10 knocked out tumor cells, and activation of TGFβ signaling contributes to the induction of SOX9 in HDAC10 knocked out lung adenocarcinoma cells (85). However, HDACs show activating activity in some tumors and inhibitory activity in some tumors, which suggests that their mechanism of action might not be a single one. SIRT1 may exert oncogenic effects by inactivating other tumor suppressors (eg, HIC1) and/or activating tumor-promoting genes (eg, via N-Myc stabilization or p53) or other proteins (cortatin) (100–102). There are interactions between HDACs. Studies have shown that inhibition or knockdown of HDAC1 and HDAC3 results in downregulation of HDAC7, which is associated with reduced histone 3 lysine 27 acetylation (H3K27ac) at transcription start sites (TSS) and super-enhancers (SEs), this is particularly evident in stem-like BrCa cells (80). Inactivation of HDAC7 can lead to suppression of the CSC phenotype, either directly or through the inhibition of HDAC1 and HDAC3, by downregulating multiple se-related oncogenes (80). HDAC7 may be a potential drug target (80).
HDACs inhibitors also have many adverse reactions in clinical application, such as drug resistance and toxic side effects (59). Aberrant expression of HDACs has also been shown to correlate with tumor resistance. HDAC8 increases the expression of p65, a key component of the NF-κB complex, and promotes the expression of IL-6 and IL-8 (103). This may be because HDAC8 can directly bind to the promoter of p65, increasing its transcription and expression. Thus, HDAC8 promotes DNR resistance in human AML cells by regulating IL-6 and IL-8 (103).
Acetyl coenzyme A
Acetyl Coenzyme A is a key precursor that used to synthesize acetyl. The progression of lysine acetylation can be controlled by regulating the concentration of acetyl-CoA. Acetyl-CoA is an important metabolite in cellular biological processes and is the only donor of acetyl groups during acetylation (104). Acetyl-CoA has different production pathways in different organelles. Acetyl-CoA produced in different organelles can be locally utilized in organelles, produced by decarboxylation of pyruvate in mitochondria, and produced by fatty acid β-oxidation in cytoplasm (105). ACLY, ACSS2, PDC can produce acetyl-CoA in organelles to regulate lysine acetylation (106). The interaction between lysine acetylation and acetyl-CoA is influenced by many factors, including the kinds of HATs, the acetyl-CoA/CoA ratio and intracellular pH gradient (107, 108).
Acetyl-CoA is derived from glycolysis and β-oxidation in the mitochondrial matrix, which ultimately leads to the production of cytoplasmic pyruvate,and enters the mitochondria for decarboxylation to form acetyl-CoA (109, 110). Branched-chain amino acids (i.e., valine, leucine, and isoleucine) can also be used to produce acetyl-CoA (111). Most of the acetyl-CoA in the cytoplasm comes from glutamine reductive carboxylation, which generates acetyl-CoA through the TCA cycle (112). Acetyl-CoA also has compartmentalized effects on protein acetylation. Acetyl-CoA exists in mitochondria, nucleus, and cytoplasm (105). Acetyl-CoA in mitochondria has a specific source pathway. Acetyl-CoA can pass through nuclear pores in the nucleus and cytoplasm. During the shuttle, acetyl-CoA has different abundances of acetyl-CoA in the nucleus and cytoplasm, and the occurrence of protein acetylation is also different (105). At the same time, studies have shown that the acetyl-CoA/CoA ratio may be a relevant regulator of HAT enzyme activity, rather than the absolute level of acetyl-CoA (105). This establishes a link between the nuclear and cytoplasmic abundance of acetyl-CoA and the epigenetic regulation of genes (105). In the process of tumorigenesis, abnormal expression of acetyl-CoA was also found. Acetyl-CoA can affect the proliferation, invasion and migration of tumor cells directly or by affecting protein acetylation (113). Acetyl-CoA induces cell growth and proliferation by promoting acetylation of histones at growth genes (113), and increase the levels of acetyl-CoA and acetylated histones to maintain the accelerated proliferation of cancer cells (105).
Reader
For histone acetylation to exert their biological functions, they also need to be combined with specific recognition proteins. Acetylated lysine in a protein will provide a reading site, recruit proteins with special structural domains, affect biological functions such as gene replication, gene transcription, and repair after DNA damage, and jointly participate in the regulation of gene expression (8). Recognition proteins can contain multiple different recognition domains that cooperate with PTM sites. Studies have shown that lysine-containing acetylation modification sites can be specifically recognized by proteins such as bromodomains, dual-PHD finger domains, and YEATS domains (8).
Four BET proteins have been identified in humans, BRD2, BRD3, BRD4 and the testis-specific protein BRDT (114). BRDT is only present in male germ cells (115). The BET family controls the transcription of various proinflammatory and immunoregulatory genes by recognizing acetylated histones (mainly H3 and H4) and recruiting transcription factors (such as RELA) and transcription elongation complexes (such as P-TEFb) to chromatin, thereby promoting the phosphorylation of RNA polymerase II and subsequent transcription initiation and elongation (116).
Localized in the nucleus, BRD2 can bind to hyperacetylated chromatin and play a role in transcriptional regulation through chromatin remodeling (115). BRD2 can regulate the transcription of the CCND1 gene and play a role in nucleosome assembly (117). Abnormal expression of BRD2 affects the development of various malignant tumors (118). For example, Runx3 forms a complex with BRD2 in a KRas-dependent manner in the early stages of the cell cycle, resulting in the inactivation of Runx3 and promoting the development of lung adenocarcinoma (118). Studies have shown that OCCC cells are susceptible to knockdown of epigenetic gene targets such as bromopseudomin and the extraterminal domain (BET) proteins BRD2 and BRD3, and targeting the BET proteins BRD2 and BRD3 in combination with PI3K-AKT inhibition may as a therapeutic strategy for ovarian clear cell carcinoma (119). The abnormal expression of BRD2 is also closely related to the drug resistance of patients. Studies have shown that BRD2 promotes drug resistance in adult T-LBL through the RasGRP1/Ras/ERK signaling pathway (120). Targeting BRD2 may be a new strategy to improve treatment efficacy and prolong survival in adults with T-LBL (120).
Localized to the nucleus, BRD3 is a chromatin reader that recognizes and binds hyperacetylated chromatin and plays a role in transcriptional regulation, possibly through chromatin remodeling and interactions with transcription factors (121). BRD3 regulates transcription by promoting the binding of the transcription factor GATA1 to its targets (122). The study found that BRD3 directly interacts with BCL6 and maintains the negative feedback regulatory loop of BCL6 (123). BRD2 and BRD3 preferentially associate with hyperacetylated chromatin throughout the length of transcribed genes in vivo (121). BRD2- and BRD3-associated chromatin was significantly enriched in H4K5, H4K12, and H3K14 acetylation reactions, and contained relatively less dimethylated H3K9 (121). Both BRD2 and BRD3 allow RNA polymerase II transcription by nucleosomes in a defined transcription system (121).
Localized in the nucleus, BRD4 is currently the most widely studied chromatin reader protein that recognizes and binds acetylated histones and plays a key role in the transmission of epigenetic memory across cell division and transcriptional regulation (124). Remains associated with acetylated chromatin throughout the cell cycle, and by preserving acetylated chromatin state and maintaining higher-order chromatin structure (125). Studies have shown that BRD4 is a transcriptional repressor of autophagy and lysosomal function (126). BRD4 plays a key role in regulating the transcription of signal-induced genes by binding to the P-TEFb complex and recruiting it to promoters. The P-TEFb complex is also recruited to the distal enhancer, an anti-pause enhancer that cooperates with JMJD6 (125). BRD4 and JMJD6 are required to form the transcriptionally active P-TEFb complex by replacing negative regulators such as HEXIM1 and the 7SK snRNA complex from P-TEFb, thereby converting it to the active form, which can then phosphorylate the C-terminal structure of RNA polymerase II Domain (CTD) (125). MYC regulates its own transcription by restricting its site for BRD4-mediated chromatin remodeling (127). The MYC-stabilizing kinase ERK1 regulates MYC levels directly or indirectly by inhibiting BRD4 kinase activity. These findings suggest that BRD4 negatively regulates MYC levels, which is counteracted by ERK1 activation (127).
BRD4 has three isoforms, BRD4 short isoform and BRD4 long isoform (128). There are two BRD4 short isoforms, which are spliced from other mRNAs. The short isoform of BRD4 promotes tumor metastasis, and the long isoform of BRD4 inhibits tumor metastasis and spread (128). Study shows BRD4 isoforms have opposing functions in breast cancer (128). The role of BRD4 in cancer is largely dependent on the long isoform (BRD4-L), and we demonstrated by isoform-specific knockdown and endogenous protein detection as well as transgene expression that the less abundant short isoform of BRD4 (BRD4-L) S) is oncogenic and BRD4-L has a tumor suppressor role in breast cancer cell proliferation and migration as well as breast tumor formation and metastasis (128). An isoform of BRD4 that acts as a chromatin insulator in DNA damage response pathways (129). Inhibits DNA damage response signaling by recruiting condensin-2 complexes to acetylated histones, leading to remodeling of chromatin structure, shielding this region from DNA by limiting the spread of histone H2AX/H2A.x phosphorylation injury response (129).
Due to the abnormal expression of BRD4 in various tumors, targeting BRD4 has emerged as a potential therapeutic strategy (130). For example, the expression of BRD4 in glioma was significantly higher than that in adjacent normal brain tissue (130). BRD4 inhibitors effectively penetrate the blood-brain barrier and target glioma tumor tissue, but have little effect on normal brain tissue (130). BRD4 is overexpressed in NFPA and GHPA, and the effects of BRD4 inhibitors on PA cells in vitro and in vivo were evaluated, so BRD4 is a promising therapeutic target for NFPA and GHPA (131).
BRD4 promotes the progression and metastasis of gastric cancer, and the abundance of BRD4 in human gastric cancer tissue is associated with shorter survival in patients with non-metastatic gastric cancer (132). BRD4 recognizes acetylated K146 and K187 on snails in an acetylation-dependent manner to prevent snails from recognition by their E3 ubiquitin ligases FBXL14 and β-Trcp1, thereby inhibiting snail polyubiquitination and proteases body degradation (132).
The mode of action of I-BET151 is due to the repression of transcription of key genes (BCL2, C-MYC and CDK6) by displacing BRD3/4, PAFc and SEC components from chromatin (133). This suggests that replacing BET proteins from chromatin is a potential epigenetic therapy for aggressive leukemia. BRDT (Bromodomain testis-specific protein), localized in the nucleus, exists only in male germ cells, and not often studied in tumors (115).
YEATS family proteins include YAF9, ENL, AF9, TAF14, SAS5 proteins (4). As the “readers” of protein acetylation, YEATS family proteins can combine with proteins to form various chromatin-related complexes with different complex functions, and play a role in chromatin remodeling and gene expression (4). YEATS family proteins are closely related to the occurrence of various malignant tumors. For example, ENL binds to acetylated histone H3, and co-localizes with H3K27ac and H3K9ac on the promoters of actively transcribed genes that are critical for leukemia (134). ENL is a regulator of leukemia. oncogenic transcriptional program (134), and an intact YEATS chromatin-reader domain was essential for ENL-dependent leukemic growth (135). YEATS4 overexpression enhances the malignant features of breast cancer cells, especially inducing epithelial-to-mesenchymal transition, and YEATS4 is associated with poor prognosis in breast cancer (136). YEATS protein promotes the proliferation of gastric cancer cells and affects tumor development by activating the Wnt/β-catenin signaling pathway (137). GAS41 is abundantly expressed in non-small cell lung cancer and is closely related to the proliferation of lung cancer cells (138). YEATS2, a target gene of HIF1α, promotes pancreatic cancer development under hypoxia (139).
Complex post-translational modifications are affected by many factors, one of which is the way the recognition site binds to the recognition protein. Initial studies believed that a post-translational modification recognition site can only bind to one recognition protein. The researchers found that a PTM recognition site can interact with multiple recognition proteins, eg. At the same time, a single recognition domain can also bind to multiple different protein PTMs, eg. Also, since recognition proteins include multiple distinct domains, synergy is extremely common in recognition proteins.
Biological role of acetylation in cancer pathophysiology
Acetylation of proteins is related to various kinds of cellular processes and human cancer (140). Here, we address the roles of acetylation in cancer cell apoptosis, autophagy, cell metabolism, cell cycle, proliferation, and migration and invasion, which will offer the basis for acetylation enzymes and BETs in reader as the important therapeutic targets (Figure 2).
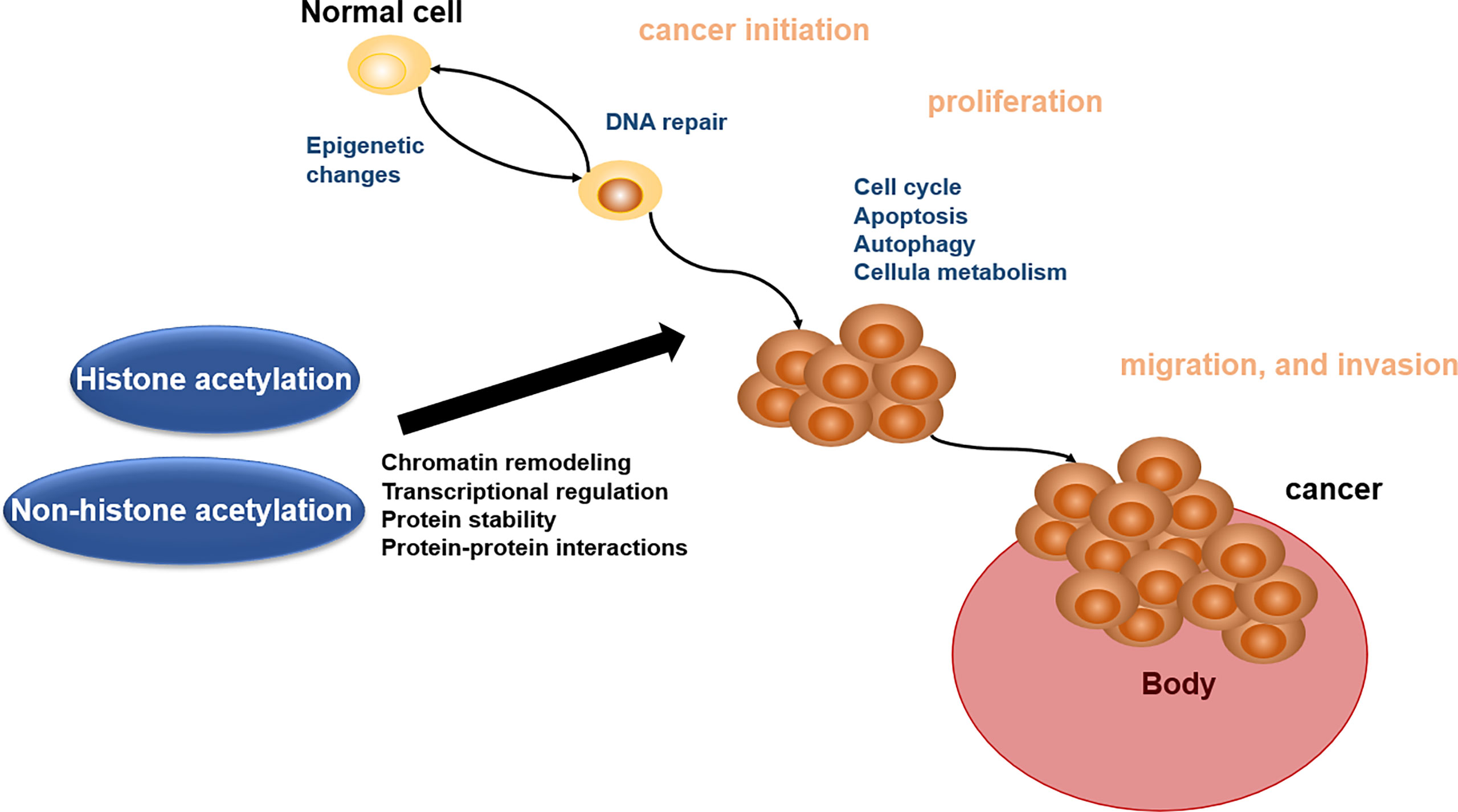
Figure 2 Biological role of histone acetylation and non-histone acetylation in cancer pathophysiology.
Role of acetylation in apoptosis
Apoptosis refers to the orderly death of cells controlled by genes, which is a normal programmed death in order to maintain the stability of the internal environment. In the process of cell apoptosis, it can be divided into the initiation stage, which receives apoptosis signals, interacts with apoptosis regulators, and then activates proteolytic enzymes, resulting in apoptosis (141). However, tumors have the characteristics of avoiding apoptosis, and abnormal apoptosis leads to abnormal tumor growth (142). The abnormal expressions of acetyltransferase and deacetylase can affect the normal apoptosis of cells. For example, PDCD5, a protein associated with apoptosis in human cells, binds to Tip60 and enhances the stability of Tip60 protein under stress-free conditions (143). PDCD5 increases Tip60-dependent K120 acetylation of p53 and is involved in p53-dependent expressions of apoptosis-related genes such as Bax (143). The combination of PDCD5 and Tip60 accelerates DNA damage-induced apoptosis, whereas knockdown of PDCD5 or Tip60 inhibits apoptosis-accelerating activity (143). HDAC1 and HDAC2 double knockout cells show significant activation of apoptosis (144). HDAC6 negatively regulates pro-apoptotic acetylation of p53 at K120 in mesenchymal stem cells (MSCs) (145). Studies have shown that targeting histone acetyltransferases and histone deacetylases can regulate tumor cell apoptosis, thereby affecting tumor growth and development (146). For example, histone deacetylase inhibitors induce apoptosis and autophagy in human neuroblastoma cells (147). Valproic acid induces cell cycle arrest and apoptosis via Hsp70 acetylation and inhibits proliferation of HER2-expressing breast cancer cells (148). When rRNA transcription was inhibited, nucleolar RNA content was reduced. The nucleolar protein Myb-binding protein 1A (MYBBP1A) translocates to the nucleoplasm and increases p53 acetylation as the level of nucleolar RNA content decreases (149). Acetylated p53 enhances p21 and BAX expression and induces apoptosis (149). Targeting protein acetylation to regulate tumor apoptotic activity can provide new therapeutic ideas for the clinical treatment of malignant tumors.
Role of acetylation in autophagy
Autophagy is a special substance degradation pathway in cells, which depends on lysosomes for its action (10). The degradation substrates of autophagy include proteins and organelles. The probability of autophagy occurring in normal cells is low, and it mainly occurs in cells under abnormal conditions, such as starvation, hypoxia or organelle damage (150). There are three main types of autophagy. (i) The first type is microautophagy, in which lysosomes wrap a part of the cytoplasm into lysosomes and degrade them. (ii) The second is macroautophagy, which first generates an autophagosome (151). The double-membrane structure of the phagosome, the fruiting body contains the substances that need to be degraded in the cytoplasm, the autophagosome and the lysosome are combined to generate the autophagolysosome, and the acidic substances in the lysosome are used to degrade the autophagosome (151). Substances are degraded. (iii) The third is chaperone-mediated autophagy. Molecular chaperone-mediated autophagy uses heat shock protein 70 to bind to substrates with specific amino acid sequences and transport the substrates to lysosomes for further development (152). In 2004, Shao et al. found that HDAC inhibitor suberoylanilide hydroxamic acid β-D-glucur onide could induce autophagic death of cancer cells, and researchers gradually began to pay attention to the relationship between protein acetylation and autophagy (153). There is a close relationship between histone acetylation and cell autophagy. Histone acetylation can induce the occurrence of cell autophagy in the face of long-term stress, starvation and other harsh environments (154). The most widely studied is the relationship between H4K16ac and H3K56ac and autophagy. In eukaryotic cells, H4K16ac affects chromatin condensation and promotes gene transcription (155).
There is also a close link between non-histone acetylation and cell autophagy. Non-histone protein associated with autophagy that can be acetylated include transcription factors, autophagy-related proteins, and cytoskeletal proteins (7). The Fox O protein family is a transcriptional activator in eukaryotic cells, and acetylation can affect its biological activity. K on Fox O protein can be acetylated by HAT, and the activity of Fox O protein after acetylation is reduced, inhibiting DNA and its interaction, binding to regulate transcription (156). SIRT1 can also affect autophagy by regulating Fox O activity (157). TFEB protein is also a transcription factor that regulates the transcription of autophagy-related genes, such as LC3, and plays an important role in biological processes such as lysosomal biosynthesis and autophagy activation (158). The biological activity of the TFEB protein family is also affected by acetylation modification, and TFEB deacetylation can significantly enhance the autophagy and lysosomal function of cells (159). The TFEB-specific lysine acetylase is GCN5, which can acetylate the K276 and K279 sites of TFEB, affect the formation of TFEB dimers, interfere with the binding of TFEB and its targets, and inhibit autophagy happened (160). Acetylations affect subcellular localization, thereby affecting autophagy (161). In general, BmP300-mediated acetylation sequesters components of the BmAtg8-PE ubiquitin-like system in the nucleus, leading to inhibition of autophagy. Conversely, BmHDAC1-mediated deacetylation leads to nuclear-to-cytoplasmic transfer of components of the BmAtg8-PE ubiquitin-like system, promoting autophagy (161).
Protein acetylation is an important process regulating autophagy and plays an important role in the development of malignant diseases. The phosphorylation of ATG5 (T101) in the lesion tissue of glioblastoma patients is positively regulated by the acetylation modification of the hypoxia-induced autophagy regulator PAK1, which plays an important role in hypoxia-induced autophagy and promotes the occurrence and development of tumors (162). Targeting protein acetylation modification to regulate autophagy activity can provide new therapeutic ideas for clinical treatment of malignant tumors.
Role of acetylation in cell metabolism
A major feature of tumors is uncontrolled proliferation, fueled by corresponding metabolic dysregulation (2). Tumors undergo metabolic reprogramming to promote tumor cell growth, division, invasion and migration. An abnormal response of tumor cell energy metabolism is called the Warburg effect (89). In the presence of oxygen, tumor cells reprogram glucose metabolism by limiting energy metabolism mainly to glycolysis, thereby generating energy for tumor growth (163). Lysine acetylation is a ubiquitous modification in enzymes that catalyze intermediate metabolism. Almost every enzyme in glycolysis, gluconeogenesis, tricarboxylic acid (TCA) cycle, urea cycle, fatty acid metabolism and glycogen metabolism is found to be acetylated in human liver tissue (10). All seven enzymes in the TCA cycle are acetylated (10). Acetylation occurs in most intermediate metabolic enzymes, and acetylation can directly affect the activity or stability of the enzyme (10). The bioenergetic preference of cancer cells promotes tumor acidosis, which in turn results in a marked reduction in glycolysis and glucose-derived acetyl-CoA (164). Protein acetylation affects tumor metabolism by affecting the TCA cycle. CBP acetylates STAT3 to undergo mitochondrial translocation, and STAT3 associates with pyruvate dehydrogenase complex E1, which in turn accelerates the conversion of pyruvate to acetyl-CoA, increases mitochondrial membrane potential and promotes ATP synthesis (165). SIRT5 removes the STAT3 acetyl group, thereby inhibiting its function in mitochondrial pyruvate metabolism (165). The protein also affects lipid metabolism in tumor cells and thus affects tumor development (166). Dynamic regulation of ME1 phosphorylation and acetylation affects lipid metabolism and colorectal tumorigenesis (166). The manner in which SIRT6 deacetylase antagonizes ACAT1 function involves mutually exclusive ME1 S336 phosphorylation and K337 acetylation (166). ACAT1 acetylates GNPAT at K128, which inhibits TRIM21-mediated GNPAT ubiquitination and degradation (167). GNPAT deacetylation by SIRT4 antagonizes the function of ACAT1. GNPAT inhibits TRIM21-mediated degradation of FASN and promotes lipid metabolism. promote the occurrence of liver cancer (167). Studies have shown that lysine acetylation controls metabolic activity by directly blocking the active site of the enzyme (168).
Role of acetylation in cell cycle
Protein acetylation is closely related to gene transcription. Hyperacetylation promotes gene transcription and expression, while hypoacetylation inhibits gene transcription and expression (12). A large number of proteins involved in chromatin remodeling and cell cycle are acetylated (169). The cell cycle of tumor cells is greatly shortened and disordered. Studies have found that acetylation of tumor cells is also closely related to cell cycle progression (170). Protein acetylation affects tumor cell cycle progression by affecting chromatin remodeling, SIRT2 regulates H4K20me1 deposition through deacetylation of H4K16Ac (acetylation of H4K16), regulates chromatin localization, and affects cell cycle progression (169). Protein acetylation also has effects through the regulation of various factors in the cell cycle. For example, CDC2, a major cyclin-dependent kinase and regulator of S-phase progression and mitosis, is acetylated at residues K6 and K33 in CDC2 (25). SIRT1 interacts with CHK2 and is deacetylated at residure lysine 520, which inhibits CHK2 phosphorylation, dimerization, and thus activation (171). SIRT1 depletion induces CHK2 hyperactivation-mediated cell cycle arrest and subsequent cell death (171). Transcription factor Sp1 is a target of acetylation and is closely associated with cell cycle arrest in colon cancer cell lines (172). Simultaneous regulation of Api5 acetylation and deacetylation is an important factor in cell cycle progression (97).
Role of acetylation in cell proliferation
Cancer cells have unlimited replicative potential with continuous proliferative signals (2). Normal cells and tissues release growth signals in an orderly manner, and these growth signals instruct cells to grow, divide and differentiate in an orderly manner, thereby ensuring the stability of cell numbers and the homeostasis of the internal environment, thereby maintaining normal tissue structure and function (2). However, tumor cell proliferation signals are abnormal and can continuously obtain proliferation signals from a variety of different pathways. In the abnormal proliferation of tumor cells, protein acetylation plays an important role. For example, acetylation at the K323 site of PGK1 is an important regulatory mechanism that promotes its enzymatic activity and cancer cell metabolism (173).
Acetyltransferase and deacetylase dynamically regulate the balance of acetylation, affecting the apoptosis and autophagy of tumor cells and other death methods, thereby affecting the proliferation of tumor cells. For example, Api5 is a known anti-apoptotic and nuclear protein responsible for inhibiting cell death under serum starvation conditions (97). The only known post-translational modification of Api5 is acetylation at K251. The K251 acetylation in Api5 is responsible for its stability, whereas the deacetylated form of Api5 is unstable (97). Inhibition of acetylation by p300 results in a decrease in Api5 levels, whereas inhibition of deacetylation by HDAC1 results in an increase in Api5 levels (97). Acetylation also affects the proliferation of tumor cells by affecting the activities of various metabolic enzymes in cells. For example, PKM2 K305 acetylation reduces PKM2 enzymatic activity and promotes its lysosome-dependent degradation through chaperone-mediated autophagy (CMA) (174). Degrade and promote tumor growth through chaperone-mediated autophagy (174). Ribonucleotide reductase (RNR) catalyzes the de novo synthesis of deoxyribonucleoside diphosphates (dNDPs), which provide dNTP precursors for DNA synthesis (175). Acetylation at residue K95 in RRM2 results in a reduction of the dNTP pool, DNA replication fork arrest, and inhibition of tumor cell growth in vitro and in vivo (175). P300 acetylates MAT IIα at K81 and destabilizes MAT IIα by promoting its ubiquitination and subsequent proteasomal degradation, inhibits tumor cell growth, and is reduced in human hepatocellular carcinoma (176). Inactivation of HDAC2 leads to elevated TPD52 acetylation, which impairs the interaction between TPD52 and HSPA8, resulting in impaired CMA function and tumor growth in vivo (177). Acetylation-dependent regulation of CMA oncogenic function in PCa by TPD52 suggests the possibility of targeting the TPD52-mediated CMA pathway to control PCa progression (177). p21 depletion converts KLF4 from a cell cycle inhibitor to a promoter of bladder cancer cell proliferation (178). Furthermore, KLF4 is acetylated in a p21-dependent manner to inhibit bladder cancer cell growth as a tumor suppressor (178). Since tumor cell proliferation is affected by acetylation modifications, drugs targeting acetylation can be used to treat abnormal tumor growth. For example, Rg3 extracted from ginsenosides has antiproliferative activity against melanoma by reducing HDAC3 and increasing p53 acetylation in vitro and in vivo (179). Therefore, Rg3 may serve as a potential therapeutic agent for the treatment of melanoma (179). Therapeutic modalities targeting acetyltransferases and deacetylases are also a potentially effective tumor treatment modality.
Role of acetylation in migration and invasion
The development of tumor is divided into multiple stages. In the early stage, the primary lesion proliferates indefinitely, and after the formation of an obvious primary lesion, the function of the organ in which it is located is affected (2). Although the primary tumor is extremely malignant, the cause of death in most patients is the abnormal growth of metastatic tumors in sites other than the primary tumor (180). The reasons for these metastases are also unresolved and need to be discovered and solved urgently. Studies have found that protein acetylation is one of the important factors affecting tumor cell metastasis (6). For example, isocitrate dehydrogenase 1 (IDH1) is hyperacetylated in CRC primary tumors and liver metastases (181), sirtuin-2 is the deacetylase of IDH1, and SIRT2 overexpression significantly inhibits CRC cell proliferation, migration and invasion (181). COL6A1 is dysregulated in several human malignancies, and upregulation of H3K27 acetylation-activated COL6A1 promotes cell migration and invasion by inhibiting the STAT1 pathway in OS cells and promotes osteosarcoma lung metastasis (182). ZMYND8 acetylation of P300 at residues K1007 and K1034 is required for HIF activation and breast cancer progression and metastasis (183). TGF-β-activated kinase 1 (TAK1) stimulates phosphorylation by TGF-β and then induces acetylation of tubulin through αTAT1 activation, which subsequently activates AB cell migration and invasion (184). AFP acetylation promotes its oncogenic effects by blocking binding to the phosphatase PTEN and the pro-apoptotic protein caspase-3, thereby increasing signaling of proliferation, migration and invasion and reducing apoptosis (185). In HCC cells, hepatitis B virus X protein (HBx) and palmitic acid (PA) increased the levels of acetylated AFP by disrupting SIRT1-mediated deacetylation (185). AFP acetylation plays an important role in hepatocellular carcinoma progression (185). miR-15a-5p reduces histone H4 acetylation by inhibiting ACSS2 expression, inhibiting acetyl-CoA activity (186). miR-15a-5p inhibits lipid metabolism by inhibiting ACSS2-mediated acetyl-CoA activity and histone acetylation, thereby inhibiting a novel mechanism of lung cancer cell metastasis (186). In addition to histone acetylation affecting tumor cell invasion and migration, non-histone acetylation also affects tumor metastasis. For example, elevated levels of alpha-tubulin acetylation are sufficient reasons for the metastatic potential of breast cancer (187). Metastatic breast cancer cells exhibit high levels of alpha-tubulin acetylation, extending along microantenna (McTN) protrusions (187). Mutation of acetylation sites on α-tubulin and enzymatic regulation of this post-translational modification had a dramatic effect on McTN frequency and reattachment of suspended tumor cells (187). Reducing alpha-tubulin acetylation significantly inhibited migration but not proliferation (187). Targeting protein acetylation to affect tumor invasion and migration may serve as a potentially effective therapeutic strategy.
Acetylation system-based targeted drugs in cancer
Research on abnormal protein acetylation in cancer mainly focuses on the mechanism of tumorigenesis, identification and prediction of new biomarkers for tumor invasion and migration, and tumor therapy. Since the process of protein acetylation is reversible, treating tumors can restore the acetylation process to normal levels for treatment. Therefore, some inhibitors of protein acetylation have also been approved for clinical treatment (59). For example, HAT inhibitors, HDAC inhibitors, HAT activators, and HDAC activators (59, 188, 189). HDAC activators are currently less studied.
Epigenetic regulation is an extremely promising strategy for the treatment of tumors, so many HAT- and HDAC-related modulatory drugs have been clinically tested (190). A research of NEO2734 in clinical trial revealed that there is an ongoing clinical trial. NEO2734 is a dual BET and CBP/p300 inhibitor targeting patients with advanced solid tumors and is in phase 1 clinical trials. Curcumin, a natural product-derived epigenetic modulator, the effect of curcumin on HDAC activity is variable and likely cell-line specific (190). Multiple clinical trials of curcumin have been completed, and other clinical trials are ongoing.
HDAC is considered to be a potential next-generation tumor therapy because HDAC inhibitors have been shown to have significant efficacy in a variety of tumor treatments (191, 192). Among them, vorinostat, romidepsin, panobinostat and belinostat have been approved by the US FDA for cancer treatment and are used in peripheral T-cell lymphoma, cutaneous T-cell lymphoma, and multiple myeloma (191, 192).
Vorinostat has been shown to be effective in the treatment of cutaneous T-cell lymphoma and is already in clinical use (192). Romidepsin regulates the expression of the immune checkpoint ligand PD-L1, and suppresses cellular immune function in colon cancer (193). Romidepsin has antitumor effects on several types of solid tumors (193). Romidepsin is used in clinical treatment of T-cell lymphoma (194). The safety and activity of panobinostat in relapsed/refractory Hodgkin lymphoma was also demonstrated in a multicenter phase II trial, and showed a significant reduction in tumor size (195). Belinostat has been found to be effective and well tolerated in patients with peripheral T-cell lymphoma (PTCL) or cutaneous T-cell lymphoma (CTCL) (196). Abexinostat is an extremely promising new HDAC inhibitor. Clinical trials have been carried out simultaneously in the United States and China. The main indications include hematological tumors (197, 198), metastatic sarcoma (199), breast cancer (200). There are also a number of drugs in clinical trials. Trichostatin A, for example, is in phase I clinical trials and is being tested in the clinic for tolerability in relapsed or refractory hematological malignancies. Ricolinostat is in phase II clinical trials for the treatment of multiple myeloma. The clinical development of HDAC inhibitors illustrates an extremely promising avenue for the treatment of tumors through epigenetic modulation.
HAT inhibitors
HAT is one of the important targets of tumor therapy. HAT inhibitors are inhibitors of protein acetyltransferase, which can inhibit its activity and reduce the level of protein acetylation. Three types of HAT inhibitors have been reported, dual substrate inhibitors, natural compounds and synthetic compounds (201). HAT inhibitors are widely used in tumor treatment. Currently, the main researches are drug inhibitors targeting CBP/P300 and small molecule inhibitors of HAT domain (201) (Table 3).
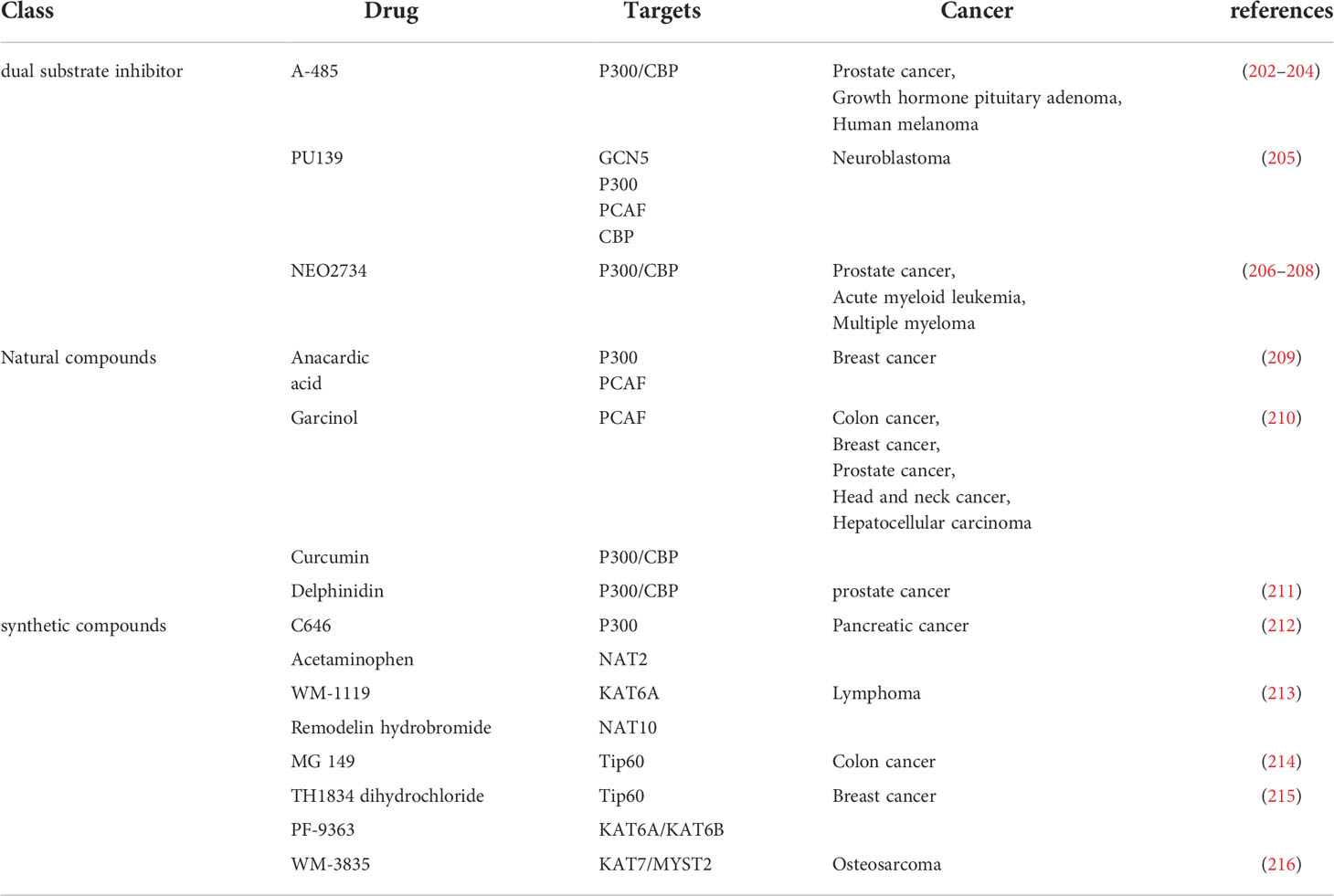
Table 3 Classification and targets of HAT inhibitors in cancers.
Anacardiic acid, a natural compound extracted from natural plants, is a p300/CBP histone acetyltransferase inhibitor, significantly reduces the viability of PTEN-/- cells not in PTEN+/+ cells by inducing apoptosis (209). Delphinoside induces p53-mediated apoptosis in human prostate cancer LNCaP cells by inhibiting HDAC activity and activating p53 acetylation (211). Therefore, delphinidin may have a role in the prevention of prostate cancer (211). There are also synthetic compounds acting on HAT, targeting HAT as inhibitors to regulate intracellular acetylation homeostasis (210). A-485 competes with acetyl-CoA. A-485 selectively inhibits proliferation of lineage-specific tumor types, including several hematological malignancies and androgen receptor-positive prostate cancer (202). WM-3835 is a potent and highly specific HBO1 (KAT7 or MYST2) inhibitor that directly binds to the acetyl-CoA binding site of HBO1 33 WM-3835 activates apoptosis while inhibiting osteosarcoma (OS) cells proliferation, migration and invasion (216). WM-3835 has antitumor activity and potently inhibits the growth of osteosarcoma xenografts in mice (216). TH1834 dihydrochloride is a specific Tip60 (KAT5) histone acetyltransferase inhibitor (215). TH1834 dihydrochloride induces apoptosis and increases DNA damage in breast cancer cells. TH1834 dihydrochloride does not affect the activity of the related histone acetyltransferase MOF. Anticancer activity (215). Combination therapy of CK1 inhibitor SR3029 and Tip60 inhibitor MG149 had stronger inhibitory effects on β-catenin acetylation, transcription of Wnt target genes, and viability and proliferation of colon cancer cells (214). Transcriptional activity of β-catenin can be regulated through the CK1δ/ϵ-β-catenin-Tip60 axis, which may be a potential therapeutic target for colon cancer (214).
HAT activators
HAT activators are activators that act on protein acetyltransferases and can activate acetyltransferases to increase the level of protein acetylation. For example, CTB can induce acetylation of P53 protein by increasing the expression of P300, thereby inducing significant cell death in MCF-7, but it may be well tolerated in MRC-5 (217). Therefore, CTB can be applied in cancer treatment (217). The research on HAT activators is not very extensive, and most of them are activators targeting the CBP/P300 complex (217) (Table 4).

Table 4 Targets of HAT activators and associated cancers.
HDAC inhibitor
HDACs are found to be abnormally expressed in malignant tumors (219). The expression of HDACs is closely related to clinical treatment prognosis and tumor occurrence and development. In liver cancer, inhibition of HDAC2 expression can promote histone acetylation in the promoter region of MIR22HG, thereby upregulating the expression of MIR22HG, promoting the production of miR-22-5p, and ultimately increasing the sensitivity to radiotherapy (64). In acute B lymphocytic leukemia, inhibits the activity of HDAC3, which enhances the sensitivity of acute B lymphocytic leukemia cells to drugs by inhibiting the JAK/signal transducer and activator of transcription 3 signaling pathway (220). Inhibition of HDAC8 activity causes cytotoxic effects, cell cycle arrest in human monocytic leukemia followed by apoptosis, and cytostatic effects in p53-deficient human myelocytic leukemia cells (73). SIRT1/2 inhibition results in HSPA5 acetylation and dissociation from EIF2AK3, leading to endoplasmic reticulum stress response, which in turn upregulates ATF4 and dit4, triggering autophagy (86). Sirtuins have become a promising target for a novel class of anti-cancer drugs. HDAC inhibitor can reverse this phenomenon and reactivate the expression of tumor suppressors, and HDAC inhibitor can act on histone acetylation and non-histone acetylation to inhibit tumor growth, invasion and metastasis, and has become a clinically effective anti-tumor drug (221) (Table 5).
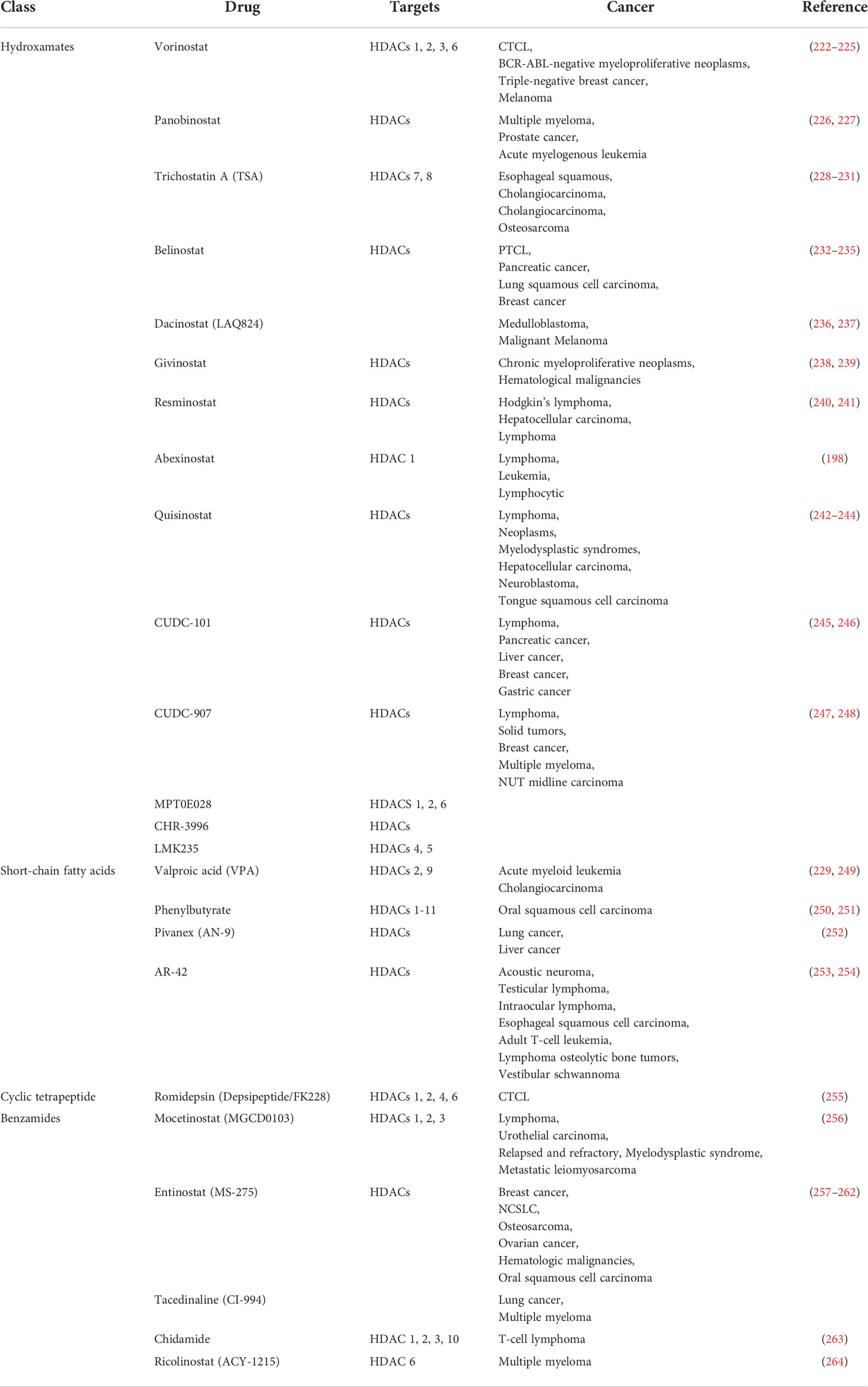
Table 5 Classification and targets of sirtuins in cancers.
Studies have shown that HDAC inhibitor has a significant inhibitory effect on P53, HSP90, NF-κB factors and multiple dephosphorylation enzymes, and a variety of HDAC inhibitors have been developed (59). The FDA has developed and approved several HDAC inhibitors for clinical cancer treatment. HDAC inhibitors are mainly divided into five categories according to different structures, including short-chain fatty acids, amides, hydroxamic acids, cyclic peptides, and chemical substances extracted from plants (265). Among histone deacetylase inhibitors, fatty acids are one of the less commonly used inhibitors. Valproic acid is an anticonvulsant drug that has been used clinically in bipolar disorder (266). The study found that valproic acid can also inhibit histone deacetylase 9, affect Notch cell signaling, and inhibit the activity of human neuroblastoma cells (267). The HDAC inhibitor of the benzamide class is the first inhibitor that selectively targets class I HDACs. There are also a large number of benzamide drugs in clinical trials for tumor treatment (59). The enzyme kinetics study of aminobenzamide-based HDAC inhibitors shows that the aminobenzamide motif has a tight binding mechanism (slow start/slow shutdown) unlike the classical fast-on/fast-off kinetics of hydroxamic acid-based HDAC inhibitors (268).
Hydroxamic acid HDAC inhibitors are the first class of HDAC inhibitors to be developed (59). Vorinostat is the first HDAC inhibitor on the market. At appropriate concentrations, vorinostat can inhibit HDAC1, 2, 3, 6, inhibit the activity of HDAC, and lead to significant hyperacetylation of H4 at residues lysine 5, 8, 12, 1, and 6 (269). These hyperacetylation are closely related to transcriptional changes, and vorinistat can simultaneously increase or decrease the transcription of specific genes in tumor cells, suggesting that HDAC inhibitor can have completely opposite effects throughout the genome (265). Virinostat is currently approved for the treatment of cutaneous T-cell lymphoma (CTCL). Studies have shown that vorinostat has activity in the treatment of recurrent glioblastoma multiforme (270). Clinically, it can be used in combination with other drugs to treat tumors (270). Vorinostat is clinically used in combination with gefitinib in the treatment of lung cancer to enhance the induction of apoptosis of lung cancer cells (271). Panobinostat is involved in many biological processes, including DNA replication and repair, chromatin remodeling, gene transcription, cell cycle progression, protein degradation and cytoskeleton reorganization (226). For example, in prostate cancer, Panobinostat reverses HepaCAM gene expression and inhibits proliferation by increasing histone acetylation (226). Panobinostat can also be used in combination with other drugs to improve treatment efficiency, such as in acute myeloid leukemia, studies have shown that the combination of panobinostat differentiation and arsenic trioxide apoptosis can significantly improve survival (272). Another HDAC inhibitor is SIRT inhibitor, inhibition of SIRT1 and SIRT2 induces cancer cell apoptosis and plays multiple roles in regulating autophagy (86). Salermide in NSCLC cells, inhibiting SIRT1 and 2 by acetylating HSPA5, and then activating ATF4 and dit4 to inhibit the mTOR signaling pathway, thereby inducing pro-survival autophagy (86). Ginsenoside Rg1 inhibits cell proliferation and induces cellular senescence in acute myeloid leukemia cells CD34+CD38- leukemia stem cells by activating Sirtuin 1 (SIRT1)/tuberous sclerosis complex 2 (TSC2) signaling pathway (273). Capsaicin attenuates cell migration by enhancing corticosteroid and -catenin acetylation in bladder cancer cells through SIRT1 targeting and inhibition (274). Capsaicin-reduced cell migration is associated with downregulation of sirtuin 1 (SIRT1) deacetylase, possibly through proteasome-mediated protein degradation (274). Combination therapy of SIRT1/2 inhibitor and drug autophagy inhibitor is an effective therapeutic strategy (86). Some studies have found that synthetic HDAC inhibitors may have toxic side effects such as atrial fibrillation, researchers turned their attention to natural inhibitors extracted from plants (59). Plant-derived inhibitors also showed good activity in inhibiting tumors. For example, hawthorn polyphenol extract (HPE) can significantly reduce ROS levels, apoptosis and inflammation-related factor expression in cells, and also inhibit AMPK/SIRT1/NF-κB and miR-34a/SIRT1/p53 pathways by regulating acetylation (275). Pathway is involved in hyperglycemia-induced inflammation and apoptosis of human retinal epithelial cells (275). These inhibitors can significantly inhibit tumor proliferation, migration and invasion, and can induce apoptosis and induce autophagy (59). However, the application of these inhibitor drugs in clinical practice requires more in-depth research.
BET inhibitor
As a scaffold protein, BET can read epigenetic code, recognize histone acetylation or non-histone acetylation, and regulate gene expression, and play an important role in cell function (115). However, abnormal expression of BET leads to abnormal gene expression, resulting in abnormal cell function, which is related to the development of many malignant diseases. The study found that the abnormal expression of BRD4 is related to glioma, and the expression in glioma is significantly higher than that in normal tissue (130); BRD4 inhibitors effectively penetrated the blood-brain barrier and targeted glioma tumor tissue, but had little effect on normal brain tissue (130). Therefore, BRD4 is a target for the treatment of glioma (130). Targeting BET protein therapy is a very promising tumor treatment strategy. The BET-bromodomain-specific inhibitors JQ1, I-BET and I-BET151 represent initial successes in the development of BET inhibitors (276). The small molecule BET inhibitor drug, JQ1, is a potent growth inhibitor for many cancers and holds promise for cancer therapy (276). However, studies have found that JQ1 can activate other oncogenic pathways and may affect epithelial-to-mesenchymal transition (EMT) (276). That is to say, JQ1 has an unexpected role in promoting prostate cancer invasion (276). In the application of tumor treatment, attention should be paid to the possible toxic and side effects of JQ1. BET inhibitor treatment in HCC cell lines reduces cell migration by downregulating SMARCA4 (277). GS-5829 inhibits CLL cell proliferation and induces leukemia cell apoptosis by deregulating key signaling pathways such as BLK, AKT, ERK1/2, and MYC (278). BRD2 supports borderline activity and raises the possibility that pharmacological BET inhibitors may partially affect gene expression by interfering with regional borderline function (279). Disruption of negative autoregulation by BET inhibitor (BETi) leads to a marked increase in BCL6 transcription, which further activates the mTOR signaling pathway by inhibiting tumor suppressor death-associated protein kinase 2 (123).
The effectiveness of BET-specific targeted inhibitors is often affected by tumor drug resistance (280). There is also an urgent need to address the issue of BET inhibitor resistance. Prostate cancer-associated SPOP mutations confer resistance to BET inhibitors by stabilizing BRD4 (281). Tumor-suppressive effects of SPOP in prostate cancer, where it acts as a negative regulator of BET protein stability, and also provides a molecular mechanism for resistance to BET inhibitors in individuals with prostate cancer carrying SPOP mutations (281). Prostate cancer-associated SPOP mutants display impaired binding to BET proteins, leading to reduced proteasomal degradation and accumulation of the protein in prostate cancer cell lines and patient specimens, and causing resistance to BET inhibitors (282). Transcriptomic and BRD4 enzymatic analysis revealed enhanced expression of GTPase RAC1 and cholesterol biosynthesis-related genes, and activation of AKT-mTORC1 signaling due to BRD4 stabilization (282). Resistance to BET inhibitors in SPOP-mutant prostate cancer can be overcome by combination with AKT inhibitors and further supports the evaluation of SPOP mutations as biomarkers to guide BET inhibitor-directed therapy in prostate cancer patients (282).
Although research on BET inhibitors is still a research focus, the combination use of BET inhibitors with other drugs is also being explored. BET inhibitors can be used in combination with other types of inhibitors in order to promote the therapeutic effect or reduce adverse reactions (283). For example, the combination of BET inhibitor I-BET762 and PARP inhibitor Talazoparib Synergy is used in the treatment of SCLC and has a synergistic effect (283). At the same time, a strategy of combined application of HDAC inhibitor and JQ1 inhibitor has shown good efficacy in the treatment of AML (284). Based on the combination drug strategy, dual-target inhibitors of HDAC and BET are also being developed, and have shown more significant efficacy than single-target inhibitors in the treatment of pancreatic cancer (285). This multi-targeted drug can ensure the efficacy and durability of the anti-cancer effect, and this combination approach also reduces the possibility of tumor resistance (285). This provides a new scope of research for BET inhibitors in the treatment of tumors. BET and HDAC inhibitors are synergistic at reduced doses, suggesting a potential approach to avoid overlapping toxicities of the two drug classes (280). The combination of CPI-0610 with a PRAP inhibitor has been found to better address PRAP inhibitor resistance in ovarian cancer patients (286). It also proposes new therapeutic strategies to address PARP inhibitor resistance using drugs already approved or in clinical development that have the potential to rapidly transform and benefit a broad range of ovarian cancer patients (286) (Table 6).

Table 6 Targets of BET inhibitors and related cancers.
Future perspectives
Tumor is currently the most troublesome problem in human life and seriously affects human health. The development of tumors is affected by many factors, including genetic factors and epigenetic factors (6). The development of tumor is the result of the joint influence of many factors (6). Protein acetylation is at the junction of genetics, epigenetics and tumor microenvironment (9). Protein acetylation is affected by many aspects to promote the occurrence and development of tumors (9). For example, protein acetylation writer, eraser, and reader may be abnormally expressed (7). Regulatory factor or regulatory factors aberrantly promote tumorigenesis and are associated with multiple malignant phenotypes of tumors. The study of protein acetylation provides a deeper understanding of tumor-related mechanisms, facilitates the discovery of potentially effective biomarkers and therapeutic targets, and facilitates the discovery and application of therapeutic drugs (11). At the same time, it is beneficial to solve the drug resistance and recurrence of tumors. At the same time, we also emphasize the strengthening of these studies on protein acetylation in different cancers, combined with PPPM in clinical practice for the treatment of malignant tumors (301).
Conclusions
This review summarized current studies about the role of protein acetylation in tumors and related targeted therapy drugs, including the classification of protein acetylation, related regulators of protein acetylation, the pathological role of protein acetylation in tumors, and targeted proteins acetylated drugs. Protein acetylation affects various physiological functions of tumors and is therefore associated with tumor development and progression. Protein acetylation plays an important role in the link between cancer pathology and post-translational modifications. Therefore, protein acetylation plays an important role in tumor therapy. Drugs about protein acetylation have been extensively studied. Drugs targeting protein acetylation have promising applications in tumor therapy, and combined use with other pathway drugs is a potential therapeutic strategy.
Author contributions
JY collected and analyzed literature, and wrote the manuscript. CS participated in partial literature analysis. XZ conceived the concept, designed the manuscript, coordinated and critically revised manuscript, and was responsible for its financial supports and the corresponding works. All authors contributed to the article and approved the submitted version.
Acknowledgments
The authors acknowledge the financial supports from the Shandong First Medical University Talent Introduction Funds (to XZ), Shandong First Medical University High-level Scientific Research Achievement Cultivation Funding Program (to XZ), the Shandong Provincial Natural Science Foundation (ZR202103020356/ZR2021MH156 to XZ), and the Academic Promotion Program of Shandong First Medical University (2019ZL002).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary

References
1. Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med (2012) 366(10):883–92. doi: 10.1056/NEJMoa1113205
2. Hanahan D. Hallmarks of cancer: New dimensions. Cancer Discovery (2022) 12(1):31–46. doi: 10.1158/2159-8290.CD-21-1059
3. Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell (2012) 150(1):12–27. doi: 10.1016/j.cell.2012.06.013
4. Verdin E, Ott M. 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat Rev Mol Cell Biol (2015) 16(4):258–64. doi: 10.1038/nrm3931
5. Phillips DM. The presence of acetyl groups of histones. Biochem J (1963) 87:258–63. doi: 10.1042/bj0870258
6. Audia JE, Campbell RM. Histone modifications and cancer. Cold Spring Harb Perspect Biol (2016) 8(4):a019521. doi: 10.1101/cshperspect.a019521
7. Narita T, Weinert BT, Choudhary C. Functions and mechanisms of non-histone protein acetylation. Nat Rev Mol Cell Biol (2019) 20(3):156–74. doi: 10.1038/s41580-018-0081-3
8. Hu M, He F, Thompson EW, Ostrikov KK, Dai X. Lysine acetylation, cancer hallmarks and emerging onco-therapeutic opportunities. Cancers (Basel) (2022) 14(2):346. doi: 10.3390/cancers14020346
9. Harachi M, Masui K, Cavenee WK, Mischel PS, Shibata N. Protein acetylation at the interface of genetics, epigenetics and environment in cancer. Metabolites (2021) 11(4):216. doi: 10.3390/metabo11040216
10. Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, et al. Regulation of cellular metabolism by protein lysine acetylation. Science (2010) 327(5968):1000–4. doi: 10.1126/science.1179689
11. Wen S, Li J, Yang J, Li B, Li N, Zhan X. Quantitative acetylomics revealed acetylation-mediated molecular pathway network changes in human nonfunctional pituitary neuroendocrine tumors. Front Endocrinol (Lausanne) (2021) 12:753606. doi: 10.3389/fendo.2021.753606
12. Albaugh BN, Arnold KM, Denu JM. KAT(ching) metabolism by the tail: insight into the links between lysine acetyltransferases and metabolism. Chembiochem Eur J Chem Biol (2011) 12(2):290–8. doi: 10.1002/cbic.201000438
13. Yang H, Pinello CE, Luo J, Li D, Wang Y, Zhao LY, et al. Small-molecule inhibitors of acetyltransferase p300 identified by high-throughput screening are potent anticancer agents. Mol Cancer Ther (2013) 12(5):610–20. doi: 10.1158/1535-7163.MCT-12-0930
14. Li T, Zhang C, Hassan S, Liu X, Song F, Chen K, et al. Histone deacetylase 6 in cancer. J Hematol Oncol (2018) 11(1):111. doi: 10.1186/s13045-018-0654-9
15. Deakin NO, Turner CE. Paxillin inhibits HDAC6 to regulate microtubule acetylation, golgi structure, and polarized migration. J Cell Biol (2014) 206(3):395–413. doi: 10.1083/jcb.201403039
16. Starheim KK, Gevaert K, Arnesen T. Protein n-terminal acetyltransferases: when the start matters. Trends Biochem Sci (2012) 37(4):152–61. doi: 10.1016/j.tibs.2012.02.003
17. El Kawak M, Dhaini HR, Jabbour ME, Moussa MA, El Asmar K, Aoun M. Slow n-acetylation as a possible contributor to bladder carcinogenesis. Mol Carcinog (2020) 59(9):1017–27. doi: 10.1002/mc.23232
18. Diallo I, Seve M, Cunin V, Minassian F, Poisson JF, Michelland S, et al. Current trends in protein acetylation analysis. Expert Rev Proteomics (2019) 16(2):139–59. doi: 10.1080/14789450.2019.1559061
19. Mittal R, Peak-Chew SY, McMahon HT. Acetylation of MEK2 and I kappa b kinase (IKK) activation loop residues by YopJ inhibits signaling. Proc Natl Acad Sci U.S.A. (2006) 103(49):18574–9. doi: 10.1073/pnas.0608995103
20. Kouzarides T. Acetylation: a regulatory modification to rival phosphorylation? EMBO J (2000) 19(6):1176–9. doi: 10.1093/emboj/19.6.1176
21. Cavdarli S, Schroter L, Albers M, Baumann AM, Vicogne D, Le Doussal JM, et al. Role of sialyl-O-Acetyltransferase CASD1 on GD2 ganglioside O-acetylation in breast cancer cells. Cells (2021) 10(6):1468. doi: 10.3390/cells10061468
22. Chowdhury S, Chandra S, Mandal C. 9-o-acetylated sialic acids differentiating normal haematopoietic precursors from leukemic stem cells with high aldehyde dehydrogenase activity in children with acute lymphoblastic leukaemia. Glycoconj J (2014) 31(6-7):523–35. doi: 10.1007/s10719-014-9550-x
23. Choudhary J, Grant SG. Proteomics in postgenomic neuroscience: the end of the beginning. Nat Neurosci (2004) 7(5):440–5. doi: 10.1038/nn1240
24. Li S, Shi B, Liu X, An HX. Acetylation and deacetylation of DNA repair proteins in cancers. Front Oncol (2020) 10:573502. doi: 10.3389/fonc.2020.573502
25. Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science (2009) 325(5942):834–40. doi: 10.1126/science.1175371
26. Eshun-Wilson L, Zhang R, Portran D, Nachury MV, Toso DB, Löhr T, et al. Effects of α-tubulin acetylation on microtubule structure and stability. Proc Natl Acad Sci USA (2019) 116(21):10366–71. doi: 10.1073/pnas.1900441116
27. Gil J, Ramirez-Torres A, Encarnacion-Guevara S. Lysine acetylation and cancer: A proteomics perspective. J Proteomics (2017) 150:297–309. doi: 10.1016/j.jprot.2016.10.003
28. Lin H, Su X, He B. Protein lysine acylation and cysteine succination by intermediates of energy metabolism. ACS Chem Biol (2012) 7(6):947–60. doi: 10.1021/cb3001793
29. Sun Y, Jiang X, Chen S, Fernandes N, Price BD. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci U.S.A. (2005) 102(37):13182–7. doi: 10.1073/pnas.0504211102
30. di Bari MG, Ciuffini L, Mingardi M, Testi R, Soddu S, Barila D. C-abl acetylation by histone acetyltransferases regulates its nuclear-cytoplasmic localization. EMBO Rep (2006) 7(7):727–33. doi: 10.1038/sj.embor.7400700
31. Fu J, Yoon HG, Qin J, Wong J. Regulation of p-TEFb elongation complex activity by CDK9 acetylation. Mol Cell Biol (2007) 27(13):4641–51. doi: 10.1128/MCB.00857-06
32. Liu Z, Mai A, Sun J. Lysine acetylation regulates bruton's tyrosine kinase in b cell activation. J Immunol (2010) 184(1):244–54. doi: 10.4049/jimmunol.0902324
33. Valiuliene G, Vitkeviciene A, Navakauskiene R. The epigenetic treatment remodel genome-wide histone H4 hyper-acetylation patterns and affect signaling pathways in acute promyelocytic leukemia cells. Eur J Pharmacol (2020) 889:173641. doi: 10.1016/j.ejphar.2020.173641
34. Zhan X, Desiderio DM. Mass spectrometric identification of in vivo nitrotyrosine sites in the human pituitary tumor proteome. Methods Mol Biol (2009) 566:137–63. doi: 10.1007/978-1-59745-562-6_10
35. Sheikh BN, Akhtar A. The many lives of KATs - detectors, integrators and modulators of the cellular environment. Nat Rev Genet (2019) 20(1):7–23. doi: 10.1038/s41576-018-0072-4
36. Li G, Chen S, Zhang Y, Xu H, Xu D, Wei Z, et al. Matrix stiffness regulates alpha-TAT1-mediated acetylation of alpha-tubulin and promotes silica-induced epithelial-mesenchymal transition via DNA damage. J Cell Sci (2021) 134(2):jcs243394. doi: 10.1242/jcs.243394
37. Nowosad A, Creff J, Jeannot P, Culerrier R, Codogno P, Manenti S, et al. p27 controls autophagic vesicle trafficking in glucose-deprived cells via the regulation of ATAT1-mediated microtubule acetylation. Cell Death Dis (2021) 12(5):481. doi: 10.1038/s41419-021-03759-9
38. Zhou C, Liu W, Duan Y. MOZ/KAT6A: a promising target for acute myeloid leukemia therapy. Future Med Chem (2020) 12(9):759–61. doi: 10.4155/fmc-2020-0047
39. Domingues AF, Kulkarni R, Giotopoulos G, Gupta S, Vinnenberg L, Arede L, et al. Loss of Kat2a enhances transcriptional noise and depletes acute myeloid leukemia stem-like cells. Elife (2020) 9:e51754. doi: 10.7554/eLife.51754
40. Waddell A, Mahmud I, Ding H, Huo Z, Liao D. Pharmacological inhibition of CBP/p300 blocks estrogen receptor alpha (ERalpha) function through suppressing enhancer H3K27 acetylation in luminal breast cancer. Cancers (Basel) (2021) 13(11):2799. doi: 10.3390/cancers13112799
41. Butler JS, Koutelou E, Schibler AC, Dent SY. Histone-modifying enzymes: regulators of developmental decisions and drivers of human disease. Epigenomics (2012) 4(2):163–77. doi: 10.2217/epi.12.3
42. Liang Z, Yu Q, Ji H, Tian D. Tip60-siRNA regulates ABCE1 acetylation to suppress lung cancer growth via activation of the apoptotic signaling pathway. Exp Ther Med (2019) 17(4):3195–202. doi: 10.3892/etm.2019.7302
43. Kuo HP, Lee DF, Chen CT, Liu M, Chou CK, Lee HJ, et al. ARD1 stabilization of TSC2 suppresses tumorigenesis through the mTOR signaling pathway. Sci Signal (2010) 3(108):ra9. doi: 10.1126/scisignal.2000590
44. Shin DH, Chun YS, Lee KH, Shin HW, Park JW. Arrest defective-1 controls tumor cell behavior by acetylating myosin light chain kinase. PloS One (2009) 4(10):e7451. doi: 10.1371/journal.pone.0007451
45. Zhu HE, Li T, Shi S, Chen DX, Chen W, Chen H. ESCO2 promotes lung adenocarcinoma progression by regulating hnRNPA1 acetylation. J Exp Clin Cancer Res (2021) 40(1):64. doi: 10.1186/s13046-021-01858-1
46. Di Martile M, Del Bufalo D, Trisciuoglio D. The multifaceted role of lysine acetylation in cancer: prognostic biomarker and therapeutic target. Oncotarget (2016) 7(34):55789–810. doi: 10.18632/oncotarget.10048
47. Pavlou D, Kirmizis A. Depletion of histone n-terminal-acetyltransferase Naa40 induces p53-independent apoptosis in colorectal cancer cells via the mitochondrial pathway. Apoptosis (2016) 21(3):298–311. doi: 10.1007/s10495-015-1207-0
48. Dang F, Jiang C, Zhang T, Inuzuka H, Wei W. PCAF and SIRT1 modulate betaTrCP1 protein stability in an acetylation-dependent manner. J Genet Genomics (2021) 48(7):652–5. doi: 10.1016/j.jgg.2021.07.004
49. Ladang A, Rapino F, Heukamp LC, Tharun L, Shostak K, Hermand D, et al. Elp3 drives wnt-dependent tumor initiation and regeneration in the intestine. J Exp Med (2015) 212(12):2057–75. doi: 10.1084/jem.20142288
50. Mak AB, Pehar M, Nixon AM, Williams RA, Uetrecht AC, Puglielli L, et al. Post-translational regulation of CD133 by ATase1/ATase2-mediated lysine acetylation. J Mol Biol (2014) 426(11):2175–82. doi: 10.1016/j.jmb.2014.02.012
51. Hogg SJ, Motorna O, Cluse LA, Johanson TM, Coughlan HD, Raviram R, et al. Targeting histone acetylation dynamics and oncogenic transcription by catalytic P300/CBP inhibition. Mol Cell (2021) 81(10):2183–200.e13. doi: 10.1016/j.molcel.2021.04.015
52. Seto E, Yoshida M. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb Perspect Biol (2014) 6(4):a018713. doi: 10.1101/cshperspect.a018713
53. Moore SD, Herrick SR, Ince TA, Kleinman MS, Dal Cin P, Morton CC, et al. Uterine leiomyomata with t(10;17) disrupt the histone acetyltransferase MORF. Cancer Res (2004) 64(16):5570–7. doi: 10.1158/0008-5472.CAN-04-0050
54. Lan R, Wang Q. Deciphering structure, function and mechanism of lysine acetyltransferase HBO1 in protein acetylation, transcription regulation, DNA replication and its oncogenic properties in cancer. Cell Mol Life Sci (2020) 77(4):637–49. doi: 10.1007/s00018-019-03296-x
55. Dong Z, Zou J, Li J, Pang Y, Liu Y, Deng C, et al. MYST1/KAT8 contributes to tumor progression by activating EGFR signaling in glioblastoma cells. Cancer Med (2019) 8(18):7793–808. doi: 10.1002/cam4.2639
56. Kawasumi R, Abe T, Arakawa H, Garre M, Hirota K, Branzei D. ESCO1/2's roles in chromosome structure and interphase chromatin organization. Genes Dev (2017) 31(21):2136–50. doi: 10.1101/gad.306084.117
57. Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol (2012) 13(4):225–38. doi: 10.1038/nrm3293
58. Wang P, Wang Z, Liu J. Role of HDACs in normal and malignant hematopoiesis. Mol Cancer (2020) 19(1):5. doi: 10.1186/s12943-019-1127-7
59. McClure JJ, Li X, Chou CJ. Advances and challenges of HDAC inhibitors in cancer therapeutics. Adv Cancer Res (2018) 138:183–211. doi: 10.1016/bs.acr.2018.02.006
60. Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov (2014) 13(9):673–91. doi: 10.1038/nrd4360
61. Richter LE, Wang Y, Becker ME, Coburn RA, Williams JT, Amador C, et al. HDAC1 is a required cofactor of CBFbeta-SMMHC and a potential therapeutic target in inversion 16 acute myeloid leukemia. Mol Cancer Res (2019) 17(6):1241–52. doi: 10.1158/1541-7786.MCR-18-0922
62. Bandyopadhyay D, Mishra A, Medrano EE. Overexpression of histone deacetylase 1 confers resistance to sodium butyrate-mediated apoptosis in melanoma cells through a p53-mediated pathway. Cancer Res (2004) 64(21):7706–10. doi: 10.1158/0008-5472.CAN-03-3897
63. Song Y, Jiang Y, Tao D, Wang Z, Wang R, Wang M, et al. NFAT2-HDAC1 signaling contributes to the malignant phenotype of glioblastoma. Neuro Oncol (2020) 22(1):46–57. doi: 10.1093/neuonc/noz136
64. Jin Q, Hu H, Yan S, Jin L, Pan Y, Li X, et al. lncRNA MIR22HG-derived miR-22-5p enhances the radiosensitivity of hepatocellular carcinoma by increasing histone acetylation through the inhibition of HDAC2 activity. Front Oncol (2021) 11:572585. doi: 10.3389/fonc.2021.572585
65. Gediya P, Parikh PK, Vyas VK, Ghate MD. Histone deacetylase 2: A potential therapeutic target for cancer and neurodegenerative disorders. Eur J Medicinal Chem (2021) 216:113332. doi: 10.1016/j.ejmech.2021.113332
66. Chen Z, Huo D, Li L, Liu Z, Li Z, Xu S, et al. Nuclear DEK preserves hematopoietic stem cells potential via NCoR1/HDAC3-Akt1/2-mTOR axis. J Exp Med (2021) 218(5):e20201974. doi: 10.1084/jem.20201974
67. Adhikari N, Amin SA, Trivedi P, Jha T, Ghosh B. HDAC3 is a potential validated target for cancer: An overview on the benzamide-based selective HDAC3 inhibitors through comparative SAR/QSAR/QAAR approaches. Eur J Medicinal Chem (2018) 157:1127–42. doi: 10.1016/j.ejmech.2018.08.081
68. Zhang L, Chen Y, Jiang Q, Song W, Zhang L. Therapeutic potential of selective histone deacetylase 3 inhibition. Eur J Medicinal Chem (2019) 162:534–42. doi: 10.1016/j.ejmech.2018.10.072
69. Sarkar R, Banerjee S, Amin SA, Adhikari N, Jha T. Histone deacetylase 3 (HDAC3) inhibitors as anticancer agents: A review. Eur J Medicinal Chem (2020) 192:112171. doi: 10.1016/j.ejmech.2020.112171
70. Tong L, Liang H, Zhuang H, Liu C, Zhang Z. The relationship between HDAC3 and malignant tumors: A mini review. Crit Rev Eukaryot Gene Expr (2020) 30(3):279–84. doi: 10.1615/CritRevEukaryotGeneExpr.2020034380
71. Adhikari N, Jha T, Ghosh B. Dissecting histone deacetylase 3 in multiple disease conditions: Selective inhibition as a promising therapeutic strategy. J Medicinal Chem (2021) 64(13):8827–69. doi: 10.1021/acs.jmedchem.0c01676
72. Ma L, Qi L, Li S, Yin Q, Liu J, Wang J, et al. Aberrant HDAC3 expression correlates with brain metastasis in breast cancer patients. Thorac Cancer (2020) 11(9):2493–505. doi: 10.1111/1759-7714.13561
73. Spreafico M, Gruszka AM, Valli D, Mazzola M, Deflorian G, Quinte A, et al. HDAC8: A promising therapeutic target for acute myeloid leukemia. Front Cell Dev Biol (2020) 8:844. doi: 10.3389/fcell.2020.00844
74. Chakrabarti A, Melesina J, Kolbinger FR, Oehme I, Senger J, Witt O, et al. Targeting histone deacetylase 8 as a therapeutic approach to cancer and neurodegenerative diseases. Future Medicinal Chem (2016) 8(13):1609–34. doi: 10.4155/fmc-2016-0117
75. Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, et al. The consensus coding sequences of human breast and colorectal cancers. Science (2006) 314(5797):268–74. doi: 10.1126/science.1133427
76. Kunadis E, Lakiotaki E, Korkolopoulou P, Piperi C. Targeting post-translational histone modifying enzymes in glioblastoma. Pharmacol Ther (2021) 220:107721. doi: 10.1016/j.pharmthera.2020.107721
77. Cheng C, Yang J, Li S-W, Huang G, Li C, Min W-P, et al. HDAC4 promotes nasopharyngeal carcinoma progression and serves as a therapeutic target. Cell Death Dis (2021) 12(2):137. doi: 10.1038/s41419-021-03417-0
78. Lachenmayer A, Toffanin S, Cabellos L, Alsinet C, Hoshida Y, Villanueva A, et al. Combination therapy for hepatocellular carcinoma: additive preclinical efficacy of the HDAC inhibitor panobinostat with sorafenib. J Hepatol (2012) 56(6):1343–50. doi: 10.1016/j.jhep.2012.01.009
79. Yang J, Gong C, Ke Q, Fang Z, Chen X, Ye M, et al. Insights into the function and clinical application of HDAC5 in cancer management. Front In Oncol (2021) 11:661620. doi: 10.3389/fonc.2021.661620
80. Caslini C, Hong S, Ban YJ, Chen XS, Ince TA. HDAC7 regulates histone 3 lysine 27 acetylation and transcriptional activity at super-enhancer-associated genes in breast cancer stem cells. Oncogene (2019) 38(39):6599–614. doi: 10.1038/s41388-019-0897-0
81. Wang Y, Abrol R, Mak JYW, Das Gupta K, Ramnath D, Karunakaran D, et al. Histone deacetylase 7: a signalling hub controlling development, inflammation, metabolism and disease. FEBS J (2022). doi: 10.1111/febs.16437
82. Linares A, Assou S, Lapierre M, Thouennon E, Duraffourd C, Fromaget C, et al. Increased expression of the HDAC9 gene is associated with antiestrogen resistance of breast cancers. Mol Oncol (2019) 13(7):1534–47. doi: 10.1002/1878-0261.12505
83. Yang C, Croteau S, Hardy P. Histone deacetylase (HDAC) 9: versatile biological functions and emerging roles in human cancer. Cell Oncol (Dordrecht) (2021) 44(5):997–1017. doi: 10.1007/s13402-021-00626-9
84. Ai J, Wang Y, Dar JA, Liu J, Liu L, Nelson JB, et al. HDAC6 regulates androgen receptor hypersensitivity and nuclear localization via modulating Hsp90 acetylation in castration-resistant prostate cancer. Mol Endocrinol (2009) 23(12):1963–72. doi: 10.1210/me.2009-0188
85. Li Y, Zhang X, Zhu S, Dejene EA, Peng W, Sepulveda A, et al. HDAC10 regulates cancer stem-like cell properties in KRAS-driven lung adenocarcinoma. Cancer Res (2020) 80(16):3265–78. doi: 10.1158/0008-5472.CAN-19-3613
86. Mu N, Lei Y, Wang Y, Wang Y, Duan Q, Ma G, et al. Inhibition of SIRT1/2 upregulates HSPA5 acetylation and induces pro-survival autophagy via ATF4-DDIT4-mTORC1 axis in human lung cancer cells. Apoptosis (2019) 24(9-10):798–811. doi: 10.1007/s10495-019-01559-3
87. Alves-Fernandes DK, Jasiulionis MG. The role of SIRT1 on DNA damage response and epigenetic alterations in cancer. Int J Mol Sci (2019) 20(13):3153. doi: 10.3390/ijms20133153
88. Zhang M, Acklin S, Gillenwater J, Du W, Patra M, Yu H, et al. SIRT2 promotes murine melanoma progression through natural killer cell inhibition. Sci Rep (2021) 11(1):12988. doi: 10.1038/s41598-021-92445-z
89. Chen X, Hao B, Li D, Reiter RJ, Bai Y, Abay B, et al. Melatonin inhibits lung cancer development by reversing the warburg effect via stimulating the SIRT3/PDH axis. J Pineal Res (2021) 71(2):e12755. doi: 10.1111/jpi.12755
90. Shi Y, He R, Yang Y, He Y, Zhan L, Wei B. Potential relationship between Sirt3 and autophagy in ovarian cancer. Oncol Lett (2020) 20(5):162. doi: 10.3892/ol.2020.12023
91. Fu L, Dong Q, He J, Wang X, Xing J, Wang E, et al. SIRT4 inhibits malignancy progression of NSCLCs, through mitochondrial dynamics mediated by the ERK-Drp1 pathway. Oncogene (2017) 36(19):2724–36. doi: 10.1038/onc.2016.425
92. Chen X, Lai X, Wu C, Tian Q, Lei T, Pan J, et al. Decreased SIRT4 protein levels in endometrioid adenocarcinoma tissues are associated with advanced AJCC stage. Cancer biomark (2017) 19(4):419–24. doi: 10.3233/CBM-160419
93. Yan D, Franzini A, Pomicter AD, Halverson BJ, Antelope O, Mason CC, et al. Sirt5 is a druggable metabolic vulnerability in acute myeloid leukemia. Blood Cancer Discov (2021) 2(3):266–87. doi: 10.1158/2643-3230.bcd-20-0168
94. Zhang Y, Huang Z, Sheng F, Yin Z. MYC upregulated LINC00319 promotes human acute myeloid leukemia (AML) cells growth through stabilizing SIRT6. Biochem Biophys Res Commun (2019) 509(1):314–21. doi: 10.1016/j.bbrc.2018.12.133
95. Tang X, Shi L, Xie N, Liu Z, Qian M, Meng F, et al. SIRT7 antagonizes TGF-beta signaling and inhibits breast cancer metastasis. Nat Commun (2017) 8(1):318. doi: 10.1038/s41467-017-00396-9
96. Bi L, Ren Y, Feng M, Meng P, Wang Q, Chen W, et al. HDAC11 regulates glycolysis through the LKB1/AMPK signaling pathway to maintain hepatocellular carcinoma stemness. Cancer Res (2021) 81(8):2015–28. doi: 10.1158/0008-5472.CAN-20-3044
97. Sharma VK, Lahiri M. Interplay between p300 and HDAC1 regulate acetylation and stability of Api5 to regulate cell proliferation. Sci Rep (2021) 11(1):16427. doi: 10.1038/s41598-021-95941-4
98. Zheng W, Tasselli L, Li TM, Chua KF. Mammalian SIRT6 represses invasive cancer cell phenotypes through ATP citrate lyase (ACLY)-dependent histone acetylation. Genes (Basel) (2021) 12(9):1460. doi: 10.3390/genes12091460
99. Williams AS, Koves TR, Davidson MT, Crown SB, Fisher-Wellman KH, Torres MJ, et al. Disruption of acetyl-lysine turnover in muscle mitochondria promotes insulin resistance and redox stress without overt respiratory dysfunction. Cell Metab (2020) 31(1):131–47. doi: 10.1016/j.cmet.2019.11.003
100. Yao J, Yang J, Yang Z, Wang X-P, Yang T, Ji B, et al. FBXW11 contributes to stem-cell-like features and liver metastasis through regulating HIC1-mediated SIRT1 transcription in colorectal cancer. Cell Death Dis (2021) 12(10):930. doi: 10.1038/s41419-021-04185-7
101. Ong ALC, Ramasamy TS. Role of Sirtuin1-p53 regulatory axis in aging, cancer and cellular reprogramming. Ageing Res Rev (2018) 43:64–80. doi: 10.1016/j.arr.2018.02.004
102. Chang H-C, Guarente L. SIRT1 and other sirtuins in metabolism. Trends Endocrinol Metab (2014) 25(3):138–45. doi: 10.1016/j.tem.2013.12.001
103. Wu J, Zhang L, Feng Y, Khadka B, Fang Z, Liu J. HDAC8 promotes daunorubicin resistance of human acute myeloid leukemia cells via regulation of IL-6 and IL-8. Biol Chem (2021) 402(4):461–8. doi: 10.1515/hsz-2020-0196
104. Choudhary C, Weinert BT, Nishida Y, Verdin E, Mann M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol Cell Biol (2014) 15(8):536–50. doi: 10.1038/nrm3841
105. Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F, Kroemer G. Acetyl coenzyme a: a central metabolite and second messenger. Cell Metab (2015) 21(6):805–21. doi: 10.1016/j.cmet.2015.05.014
106. Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol (2007) 14(11):1025–40. doi: 10.1038/nsmb1338
107. Wagner GR, Payne RM. Widespread and enzyme-independent nϵ-acetylation and nϵ-succinylation of proteins in the chemical conditions of the mitochondrial matrix. J Biol Chem (2013) 288(40):29036–45. doi: 10.1074/jbc.M113.486753
108. Denisov IG, Sligar SG. A novel type of allosteric regulation: functional cooperativity in monomeric proteins. Arch Biochem Biophysics (2012) 519(2):91–102. doi: 10.1016/j.abb.2011.12.017
109. Rufer AC, Thoma R, Hennig M. Structural insight into function and regulation of carnitine palmitoyltransferase. Cell Mol Life Sci CMLS (2009) 66(15):2489–501. doi: 10.1007/s00018-009-0035-1
110. Herzig S, Raemy E, Montessuit S, Veuthey J-L, Zamboni N, Westermann B, et al. Identification and functional expression of the mitochondrial pyruvate carrier. Sci (New York NY) (2012) 337(6090):93–6. doi: 10.1126/science.1218530
111. Harris RA, Joshi M, Jeoung NH, Obayashi M. Overview of the molecular and biochemical basis of branched-chain amino acid catabolism. J Nutr (2005) 135(6 Suppl):1527S–30S. doi: 10.1093/jn/135.6.1527S
112. Zaidi N, Swinnen JV, Smans K. ATP-citrate lyase: a key player in cancer metabolism. Cancer Res (2012) 72(15):3709–14. doi: 10.1158/0008-5472.CAN-11-4112
113. Cai L, Sutter BM, Li B, Tu BP. Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol Cell (2011) 42(4):426–37. doi: 10.1016/j.molcel.2011.05.004
114. Shi J, Vakoc CR. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell (2014) 54(5):728–36. doi: 10.1016/j.molcel.2014.05.016
115. Taniguchi Y. The bromodomain and extra-terminal domain (BET) family: Functional anatomy of BET paralogous proteins. Int J Mol Sci (2016) 17(11):1849. doi: 10.3390/ijms17111849
116. Wang N, Wu R, Tang D, Kang R. The BET family in immunity and disease. Signal Transduction Targeted Ther (2021) 6(1):23. doi: 10.1038/s41392-020-00384-4
117. Li X, Li S, Li B, Li Y, Aman S, Xia K, et al. Acetylation of ELF5 suppresses breast cancer progression by promoting its degradation and targeting CCND1. NPJ Precis Oncol (2021) 5(1):20. doi: 10.1038/s41698-021-00158-3
118. Lee Y-S, Lee J-W, Jang J-W, Chi X-Z, Kim J-H, Li Y-H, et al. Runx3 inactivation is a crucial early event in the development of lung adenocarcinoma. Cancer Cell (2013) 24(5):603–16. doi: 10.1016/j.ccr.2013.10.003
119. Shigeta S, Lui GYL, Shaw R, Moser R, Gurley KE, Durenberger G, et al. Targeting BET proteins BRD2 and BRD3 in combination with PI3K-AKT inhibition as a therapeutic strategy for ovarian clear cell carcinoma. Mol Cancer Ther (2021) 20(4):691–703. doi: 10.1158/1535-7163.MCT-20-0809
120. Tian X-P, Cai J, Ma S-Y, Fang Y, Huang H-Q, Lin T-Y, et al. BRD2 induces drug resistance through activation of the RasGRP1/Ras/ERK signaling pathway in adult T-cell lymphoblastic lymphoma. Cancer Commun (London England) (2020) 40(6):245–59. doi: 10.1002/cac2.12039
121. LeRoy G, Rickards B, Flint SJ. The double bromodomain proteins Brd2 and Brd3 couple histone acetylation to transcription. Mol Cell (2008) 30(1):51–60. doi: 10.1016/j.molcel.2008.01.018
122. Lamonica JM, Deng W, Kadauke S, Campbell AE, Gamsjaeger R, Wang H, et al. Bromodomain protein Brd3 associates with acetylated GATA1 to promote its chromatin occupancy at erythroid target genes. Proc Natl Acad Sci USA (2011) 108(22):E159–E68. doi: 10.1073/pnas.1102140108
123. Guo J, Liu Y, Lv J, Zou B, Chen Z, Li K, et al. BCL6 confers KRAS-mutant non-small-cell lung cancer resistance to BET inhibitors. J Clin Invest (2021) 131(1):e133090. doi: 10.1172/JCI133090
124. Slaughter MJ, Shanle EK, Khan A, Chua KF, Hong T, Boxer LD, et al. HDAC inhibition results in widespread alteration of the histone acetylation landscape and BRD4 targeting to gene bodies. Cell Rep (2021) 34(3):108638. doi: 10.1016/j.celrep.2020.108638
125. Patel MC, Debrosse M, Smith M, Dey A, Huynh W, Sarai N, et al. BRD4 coordinates recruitment of pause release factor p-TEFb and the pausing complex NELF/DSIF to regulate transcription elongation of interferon-stimulated genes. Mol Cell Biol (2013) 33(12):2497–507. doi: 10.1128/MCB.01180-12
126. Sakamaki J-I, Wilkinson S, Hahn M, Tasdemir N, O'Prey J, Clark W, et al. Bromodomain protein BRD4 is a transcriptional repressor of autophagy and lysosomal function. Mol Cell (2017) 66(4):517–32. doi: 10.1016/j.molcel.2017.04.027
127. Devaiah BN, Mu J, Akman B, Uppal S, Weissman JD, Cheng D, et al. MYC protein stability is negatively regulated by BRD4. Proc Natl Acad Sci USA (2020) 117(24):13457–67. doi: 10.1073/pnas.1919507117
128. Wu S-Y, Lee C-F, Lai H-T, Yu C-T, Lee J-E, Zuo H, et al. Opposing functions of BRD4 isoforms in breast cancer. Mol Cell (2020) 78(6):1114–32. doi: 10.1016/j.molcel.2020.04.034
129. Floyd SR, Pacold ME, Huang Q, Clarke SM, Lam FC, Cannell IG, et al. The bromodomain protein Brd4 insulates chromatin from DNA damage signalling. Nature (2013) 498(7453):246–50. doi: 10.1038/nature12147
130. Yang H, Wei L, Xun Y, Yang A, You H. BRD4: An emerging prospective therapeutic target in glioma. Mol Ther Oncolytics (2021) 21:1–14. doi: 10.1016/j.omto.2021.03.005
131. Shi C, Ye Z, Han J, Ye X, Lu W, Ji C, et al. BRD4 as a therapeutic target for nonfunctioning and growth hormone pituitary adenoma. Neuro Oncol (2020) 22(8):1114–25. doi: 10.1093/neuonc/noaa084
132. Qin Z-Y, Wang T, Su S, Shen L-T, Zhu G-X, Liu Q, et al. BRD4 promotes gastric cancer progression and metastasis through acetylation-dependent stabilization of snail. Cancer Res (2019) 79(19):4869–81. doi: 10.1158/0008-5472.CAN-19-0442
133. Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan W-I, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature (2011) 478(7370):529–33. doi: 10.1038/nature10509
134. Wan L, Wen H, Li Y, Lyu J, Xi Y, Hoshii T, et al. ENL links histone acetylation to oncogenic gene expression in acute myeloid leukaemia. Nature (2017) 543(7644):265–9. doi: 10.1038/nature21687
135. Erb MA, Scott TG, Li BE, Xie H, Paulk J, Seo H-S, et al. Transcription control by the ENL YEATS domain in acute leukaemia. Nature (2017) 543(7644):270–4. doi: 10.1038/nature21688
136. Li Y, Li L, Wu J, Qin J, Dai X, Jin T, et al. YEATS4 is associated with poor prognosis and promotes epithelial-to-mesenchymal transition and metastasis by regulating ZEB1 expression in breast cancer. Am J Cancer Res (2021) 11(2):416–40.
137. Ji S, Zhang Y, Yang B. YEATS domain containing 4 promotes gastric cancer cell proliferation and mediates tumor progression via activating the wnt/β-catenin signaling pathway. Oncol Res (2017) 25(9):1633–41. doi: 10.3727/096504017X14878528144150
138. Hsu C-C, Shi J, Yuan C, Zhao D, Jiang S, Lyu J, et al. Recognition of histone acetylation by the GAS41 YEATS domain promotes H2A.Z deposition in non-small cell lung cancer. Gene Dev (2018) 32(1):58–69. doi: 10.1101/gad.303784.117
139. Zeng Z, Lei S, He Z, Chen T, Jiang J. YEATS2 is a target of HIF1α and promotes pancreatic cancer cell proliferation and migration. J Cell Physiol (2021) 236(3):2087–98. doi: 10.1002/jcp.29995
140. Zaib S, Rana N, Khan I. Histone modifications and their role in epigenetics of cancer. Curr Med Chem (2021) 29(14):2399–411. doi: 10.2174/0929867328666211108105214
141. Wyllie AH. "Where, O death, is thy sting?" a brief review of apoptosis biology. Mol Neurobiol (2010) 42(1):4–9. doi: 10.1007/s12035-010-8125-5
142. Wong RSY. Apoptosis in cancer: from pathogenesis to treatment. J Exp Clin Cancer Res CR (2011) 30:87. doi: 10.1186/1756-9966-30-87
143. Xu L, Chen Y, Song Q, Xu D, Wang Y, Ma D. PDCD5 interacts with Tip60 and functions as a cooperator in acetyltransferase activity and DNA damage-induced apoptosis. Neoplasia (New York NY) (2009) 11(4):345–54. doi: 10.1593/neo.81524
144. Lin C-L, Tsai M-L, Lin C-Y, Hsu K-W, Hsieh W-S, Chi W-M, et al. HDAC1 and HDAC2 double knockout triggers cell apoptosis in advanced thyroid cancer. Int J Mol Sci (2019) 20(2):454. doi: 10.3390/ijms20020454
145. Park S-Y, Phorl S, Jung S, Sovannarith K, Lee S-I, Noh S, et al. HDAC6 deficiency induces apoptosis in mesenchymal stem cells through p53 K120 acetylation. Biochem Biophys Res Commun (2017) 494(1-2):51–6. doi: 10.1016/j.bbrc.2017.10.087
146. Bao L, Diao H, Dong N, Su X, Wang B, Mo Q, et al. Histone deacetylase inhibitor induces cell apoptosis and cycle arrest in lung cancer cells via mitochondrial injury and p53 up-acetylation. Cell Biol Toxicol (2016) 32(6):469–82. doi: 10.1007/s10565-016-9347-8
147. Francisco R, Pérez-Perarnau A, Cortés C, Gil J, Tauler A, Ambrosio S. Histone deacetylase inhibition induces apoptosis and autophagy in human neuroblastoma cells. Cancer Lett (2012) 318(1):42–52. doi: 10.1016/j.canlet.2011.11.036
148. Mawatari T, Ninomiya I, Inokuchi M, Harada S, Hayashi H, Oyama K, et al. Valproic acid inhibits proliferation of HER2-expressing breast cancer cells by inducing cell cycle arrest and apoptosis through Hsp70 acetylation. Int J Oncol (2015) 47(6):2073–81. doi: 10.3892/ijo.2015.3213
149. Kumazawa T, Nishimura K, Katagiri N, Hashimoto S, Hayashi Y, Kimura K. Gradual reduction in rRNA transcription triggers p53 acetylation and apoptosis via MYBBP1A. Sci Rep (2015) 5:10854. doi: 10.1038/srep10854
150. Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Differentiation (2005) 12 Suppl 2:1542–52. doi: 10.1038/sj.cdd.4401765
151. Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res (2014) 24(1):24–41. doi: 10.1038/cr.2013.168
152. Kaushik S, Cuervo AM. The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol (2018) 19(6):365–81. doi: 10.1038/s41580-018-0001-6
153. Shao Y, Gao Z, Marks PA, Jiang X. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc Natl Acad Sci USA (2004) 101(52):18030–5. doi: 10.1073/pnas.0408345102
154. Bánréti A, Sass M, Graba Y. The emerging role of acetylation in the regulation of autophagy. Autophagy (2013) 9(6):819–29. doi: 10.4161/auto.23908
155. Füllgrabe J, Klionsky DJ, Joseph B. The return of the nucleus: transcriptional and epigenetic control of autophagy. Nat Rev Mol Cell Biol (2014) 15(1):65–74. doi: 10.1038/nrm3716
156. Brown AK, Webb AE. Regulation of FOXO factors in mammalian cells. Curr Top Dev Biol (2018) 127:165–92. doi: 10.1016/bs.ctdb.2017.10.006
157. Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, et al. FoxO3 controls autophagy in skeletal muscle. vivo. Cell Metab (2007) 6(6):458–71. doi: 10.1016/j.cmet.2007.11.001
158. Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, et al. TFEB links autophagy to lysosomal biogenesis. Sci (New York NY) (2011) 332(6036):1429–33. doi: 10.1126/science.1204592
159. Bao J, Zheng L, Zhang Q, Li X, Zhang X, Li Z, et al. Deacetylation of TFEB promotes fibrillar aβ degradation by upregulating lysosomal biogenesis in microglia. Protein Cell (2016) 7(6):417–33. doi: 10.1007/s13238-016-0269-2
160. Wang Y, Huang Y, Liu J, Zhang J, Xu M, You Z, et al. Acetyltransferase GCN5 regulates autophagy and lysosome biogenesis by targeting TFEB. EMBO Rep (2020) 21(1):e48335. doi: 10.15252/embr.201948335
161. Wu W, Li K, Guo S, Xu J, Ma Q, Li S, et al. P300/HDAC1 regulates the acetylation/deacetylation and autophagic activities of LC3/Atg8-PE ubiquitin-like system. Cell Death Discov (2021) 7(1):128. doi: 10.1038/s41420-021-00513-0
162. Feng X, Zhang H, Meng L, Song H, Zhou Q, Qu C, et al. Hypoxia-induced acetylation of PAK1 enhances autophagy and promotes brain tumorigenesis via phosphorylating ATG5. Autophagy (2021) 17(3):723–42. doi: 10.1080/15548627.2020.1731266
163. Li N, Li H, Wang Y, Cao L, Zhan X. Quantitative proteomics revealed energy metabolism pathway alterations in human epithelial ovarian carcinoma and their regulation by the antiparasite drug ivermectin: data interpretation in the context of 3P medicine. EPMA J (2020) 11(4):661–94. doi: 10.1007/s13167-020-00224-z
164. Corbet C, Pinto A, Martherus R, Santiago de Jesus JP, Polet F, Feron O. Acidosis drives the reprogramming of fatty acid metabolism in cancer cells through changes in mitochondrial and histone acetylation. Cell Metab (2016) 24(2):311–23. doi: 10.1016/j.cmet.2016.07.003
165. Xu YS, Liang JJ, Wang Y, Zhao X-ZJ, Xu L, Xu Y-Y, et al. STAT3 undergoes acetylation-dependent mitochondrial translocation to regulate pyruvate metabolism. Sci Rep (2016) 6:39517. doi: 10.1038/srep39517
166. Zhu Y, Gu L, Lin X, Liu C, Lu B, Cui K, et al. Dynamic regulation of ME1 phosphorylation and acetylation affects lipid metabolism and colorectal tumorigenesis. Mol Cell (2020) 77(1):138–49. doi: 10.1016/j.molcel.2019.10.015
167. Gu L, Zhu Y, Lin X, Tan X, Lu B, Li Y. Stabilization of FASN by ACAT1-mediated GNPAT acetylation promotes lipid metabolism and hepatocarcinogenesis. Oncogene (2020) 39(11):2437–49. doi: 10.1038/s41388-020-1156-0
168. Nakayasu ES, Burnet MC, Walukiewicz HE, Wilkins CS, Shukla AK, Brooks S, et al. Ancient regulatory role of lysine acetylation in central metabolism. mBio (2017) 8(6):e01894–17. doi: 10.1128/mBio.01894-17
169. Serrano L, Martínez-Redondo P, Marazuela-Duque A, Vazquez BN, Dooley SJ, Voigt P, et al. The tumor suppressor SirT2 regulates cell cycle progression and genome stability by modulating the mitotic deposition of H4K20 methylation. Gene Dev (2013) 27(6):639–53. doi: 10.1101/gad.211342.112
170. Wang C, Fu M, Mani S, Wadler S, Senderowicz AM, Pestell RG. Histone acetylation and the cell-cycle in cancer. Front Biosci (2001) 6:D610–D29. doi: 10.2741/1wang1
171. Zhang W, Feng Y, Guo Q, Guo W, Xu H, Li X, et al. SIRT1 modulates cell cycle progression by regulating CHK2 acetylation-phosphorylation. Cell Death Differentiation (2020) 27(2):482–96. doi: 10.1038/s41418-019-0369-7
172. Waby JS, Chirakkal H, Yu C, Griffiths GJ, Benson RSP, Bingle CD, et al. Sp1 acetylation is associated with loss of DNA binding at promoters associated with cell cycle arrest and cell death in a colon cell line. Mol Cancer (2010) 9:275. doi: 10.1186/1476-4598-9-275
173. Hu H, Zhu W, Qin J, Chen M, Gong L, Li L, et al. Acetylation of PGK1 promotes liver cancer cell proliferation and tumorigenesis. Hepatol (Baltimore Md) (2017) 65(2):515–28. doi: 10.1002/hep.28887
174. Lv L, Li D, Zhao D, Lin R, Chu Y, Zhang H, et al. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol Cell (2011) 42(6):719–30. doi: 10.1016/j.molcel.2011.04.025
175. Chen G, Luo Y, Warncke K, Sun Y, Yu DS, Fu H, et al. Acetylation regulates ribonucleotide reductase activity and cancer cell growth. Nat Commun (2019) 10(1):3213. doi: 10.1038/s41467-019-11214-9
176. Yang H-B, Xu Y-Y, Zhao X-N, Zou S-W, Zhang Y, Zhang M, et al. Acetylation of MAT IIα represses tumour cell growth and is decreased in human hepatocellular cancer. Nat Commun (2015) 6:6973. doi: 10.1038/ncomms7973
177. Fan Y, Hou T, Gao Y, Dan W, Liu T, Liu B, et al. Acetylation-dependent regulation of TPD52 isoform 1 modulates chaperone-mediated autophagy in prostate cancer. Autophagy (2021) 17(12):4386–400. doi: 10.1080/15548627.2021.1917130
178. Jia Z-M, Ai X, Teng J-F, Wang Y-P, Wang B-J, Zhang X. p21 and CK2 interaction-mediated HDAC2 phosphorylation modulates KLF4 acetylation to regulate bladder cancer cell proliferation. Tumour Biol J Int Soc For Oncodevelopmental Biol Med (2016) 37(6):8293–304. doi: 10.1007/s13277-015-4618-1
179. Shan X, Fu Y-S, Aziz F, Wang X-Q, Yan Q, Liu J-W. Ginsenoside Rg3 inhibits melanoma cell proliferation through down-regulation of histone deacetylase 3 (HDAC3) and increase of p53 acetylation. PloS One (2014) 9(12):e115401. doi: 10.1371/journal.pone.0115401
180. Ganesh K, Massagué J. Targeting metastatic cancer. Nat Med (2021) 27(1):34–44. doi: 10.1038/s41591-020-01195-4
181. Wang B, Ye Y, Yang X, Liu B, Wang Z, Chen S, et al. SIRT2-dependent IDH1 deacetylation inhibits colorectal cancer and liver metastases. EMBO Rep (2020) 21(4):e48183. doi: 10.15252/embr.201948183
182. Zhang Y, Liu Z, Yang X, Lu W, Chen Y, Lin Y, et al. H3K27 acetylation activated-COL6A1 promotes osteosarcoma lung metastasis by repressing STAT1 and activating pulmonary cancer-associated fibroblasts. Theranostics (2021) 11(3):1473–92. doi: 10.7150/thno.51245
183. Chen Y, Zhang B, Bao L, Jin L, Yang M, Peng Y, et al. ZMYND8 acetylation mediates HIF-dependent breast cancer progression and metastasis. J Clin Invest (2018) 128(5):1937–55. doi: 10.1172/JCI95089
184. Yoshimoto S, Morita H, Okamura K, Hiraki A, Hashimoto S. αTAT1-induced tubulin acetylation promotes ameloblastoma migration and invasion. Lab Investigation; J Tech Methods Pathol (2022) 102(1):80–9. doi: 10.1038/s41374-021-00671-w
185. Xue J, Cao Z, Cheng Y, Wang J, Liu Y, Yang R, et al. Acetylation of alpha-fetoprotein promotes hepatocellular carcinoma progression. Cancer Lett (2020) 471:12–26. doi: 10.1016/j.canlet.2019.11.043
186. Ni Y, Yang Y, Ran J, Zhang L, Yao M, Liu Z, et al. miR-15a-5p inhibits metastasis and lipid metabolism by suppressing histone acetylation in lung cancer. Free Radical Biol Med (2020) 161:150–62. doi: 10.1016/j.freeradbiomed.2020.10.009
187. Boggs AE, Vitolo MI, Whipple RA, Charpentier MS, Goloubeva OG, Ioffe OB, et al. α-tubulin acetylation elevated in metastatic and basal-like breast cancer cells promotes microtentacle formation, adhesion, and invasive migration. Cancer Res (2015) 75(1):203–15. doi: 10.1158/0008-5472.CAN-13-3563
188. Feng L, Wang G, Chen Y, He G, Liu B, Liu J, et al. Dual-target inhibitors of bromodomain and extra-terminal proteins in cancer: A review from medicinal chemistry perspectives. Med Res Rev (2022) 42(2):710–43. doi: 10.1002/med.21859
189. Oike T, Komachi M, Ogiwara H, Amornwichet N, Saitoh Y, Torikai K, et al. C646, a selective small molecule inhibitor of histone acetyltransferase p300, radiosensitizes lung cancer cells by enhancing mitotic catastrophe. Radiother Oncol (2014) 111(2):222–7. doi: 10.1016/j.radonc.2014.03.015
190. Meja KK, Rajendrasozhan S, Adenuga D, Biswas SK, Sundar IK, Spooner G, et al. Curcumin restores corticosteroid function in monocytes exposed to oxidants by maintaining HDAC2. Am J Respir Cell Mol Biol (2008) 39(3):312–23. doi: 10.1165/rcmb.2008-0012OC
191. Fan W, Zhang L, Jiang Q, Song W, Yan F, Zhang L. Histone deacetylase inhibitor based prodrugs. Eur J Medicinal Chem (2020) 203:112628. doi: 10.1016/j.ejmech.2020.112628
192. Siegel D, Hussein M, Belani C, Robert F, Galanis E, Richon VM, et al. Vorinostat in solid and hematologic malignancies. J Hematol Oncol (2009) 2:31. doi: 10.1186/1756-8722-2-31
193. Shi Y, Fu Y, Zhang X, Zhao G, Yao Y, Guo Y, et al. Romidepsin (FK228) regulates the expression of the immune checkpoint ligand PD-L1 and suppresses cellular immune functions in colon cancer. Cancer Immunol Immunother CII (2021) 70(1):61–73. doi: 10.1007/s00262-020-02653-1
194. Smolewski P, Robak T. The discovery and development of romidepsin for the treatment of T-cell lymphoma. Expert Opin On Drug Discov (2017) 12(8):859–73. doi: 10.1080/17460441.2017.1341487
195. Younes A, Sureda A, Ben-Yehuda D, Zinzani PL, Ong T-C, Prince HM, et al. Panobinostat in patients with relapsed/refractory hodgkin's lymphoma after autologous stem-cell transplantation: results of a phase II study. J Clin Oncol Off J Am Soc Clin Oncol (2012) 30(18):2197–203. doi: 10.1200/JCO.2011.38.1350
196. Foss F, Advani R, Duvic M, Hymes KB, Intragumtornchai T, Lekhakula A, et al. A phase II trial of belinostat (PXD101) in patients with relapsed or refractory peripheral or cutaneous T-cell lymphoma. Br J Haematology (2015) 168(6):811–9. doi: 10.1111/bjh.13222
197. Vey N, Prebet T, Thalamas C, Charbonnier A, Rey J, Kloos I, et al. Phase 1 dose-escalation study of oral abexinostat for the treatment of patients with relapsed/refractory higher-risk myelodysplastic syndromes, acute myeloid leukemia, or acute lymphoblastic leukemia. Leukemia Lymphoma (2017) 58(8):1880–6. doi: 10.1080/10428194.2016.1263843
198. Ribrag V, Kim WS, Bouabdallah R, Lim ST, Coiffier B, Illes A, et al. Safety and efficacy of abexinostat, a pan-histone deacetylase inhibitor, in non-Hodgkin lymphoma and chronic lymphocytic leukemia: results of a phase II study. Haematologica (2017) 102(5):903–9. doi: 10.3324/haematol.2016.154377
199. Choy E, Flamand Y, Balasubramanian S, Butrynski JE, Harmon DC, George S, et al. Phase 1 study of oral abexinostat, a histone deacetylase inhibitor, in combination with doxorubicin in patients with metastatic sarcoma. Cancer (2015) 121(8):1223–30. doi: 10.1002/cncr.29175
200. Salvador MA, Wicinski J, Cabaud O, Toiron Y, Finetti P, Josselin E, et al. The histone deacetylase inhibitor abexinostat induces cancer stem cells differentiation in breast cancer with low xist expression. Clin Cancer Res an Off J Am Assoc For Cancer Res (2013) 19(23):6520–31. doi: 10.1158/1078-0432.CCR-13-0877
201. Huang MY, Huang JK, Zheng YC, Sun Q. Histone acetyltransferase inhibitors: An overview in synthesis, structure-activity relationship and molecular mechanism. Eur J Medicinal Chem (2019) 178:259–86. doi: 10.1016/j.ejmech.2019.05.078
202. Lasko LM, Jakob CG, Edalji RP, Qiu W, Montgomery D, Digiammarino EL, et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage specific tumours. Nature (2018) 558(7710):E1–E. doi: 10.1038/s41586-018-0111-5. (vol 550, pg 128, 2017).
203. Ji C, Xu W, Ding H, Chen Z, Shi C, Han J, et al. The p300 inhibitor a-485 exerts antitumor activity in growth hormone pituitary adenoma. J Clin Endocrinol Metab (2022) 107(6):e2291–e300. doi: 10.1210/clinem/dgac128
204. Wang R, He Y, Robinson V, Yang Z, Hessler P, Lasko LM, et al. Targeting lineage-specific MITF pathway in human melanoma cell lines by a-485, the selective small-molecule inhibitor of p300/CBP. Mol Cancer Ther (2018) 17(12):2543–50. doi: 10.1158/1535-7163.MCT-18-0511
205. Gajer JM, Furdas SD, Grunder A, Gothwal M, Heinicke U, Keller K, et al. Histone acetyltransferase inhibitors block neuroblastoma cell growth. vivo. Oncogenesis (2015) 4:e137. doi: 10.1038/oncsis.2014.51
206. Spriano F, Gaudio E, Cascione L, Tarantelli C, Melle F, Motta G, et al. Antitumor activity of the dual BET and CBP/EP300 inhibitor NEO2734. Blood Adv (2020) 4(17):4124–35. doi: 10.1182/bloodadvances.2020001879
207. van Gils N, Martiañez Canales T, Vermue E, Rutten A, Denkers F, van der Deure T, et al. The novel oral BET-CBP/p300 dual inhibitor NEO2734 is highly effective in eradicating acute myeloid leukemia blasts and Stem/Progenitor cells. HemaSphere (2021) 5(8):e610. doi: 10.1097/HS9.0000000000000610
208. Ryan KR, Giles F, Morgan GJ. Targeting both BET and CBP/EP300 proteins with the novel dual inhibitors NEO2734 and NEO1132 leads to anti-tumor activity in multiple myeloma. Eur J Haematology (2021) 106(1):90–9. doi: 10.1111/ejh.13525
209. Liu WJ, Cui Y, Ren W, Irudayaraj J. Epigenetic biomarker screening by FLIM-FRET for combination therapy in ER plus breast cancer. Clin Epigenet (2019) 11:1–9. doi: 10.1186/s13148-019-0620-6
210. Kopytko P, Piotrowska K, Janisiak J, Tarnowski M. Garcinol-a natural histone acetyltransferase inhibitor and new anti-cancer epigenetic drug. Int J Mol Sci (2021) 22(6):2828. doi: 10.3390/ijms22062828
211. Jeong M-H, Ko H, Jeon H, Sung G-J, Park S-Y, Jun WJ, et al. Delphinidin induces apoptosis via cleaved HDAC3-mediated p53 acetylation and oligomerization in prostate cancer cells. Oncotarget (2016) 7(35):56767–80. doi: 10.18632/oncotarget.10790
212. Ono H, Kato T, Murase Y, Nakamura Y, Ishikawa Y, Watanabe S, et al. C646 inhibits G2/M cell cycle-related proteins and potentiates anti-tumor effects in pancreatic cancer. Sci Rep (2021) 11(1):10078. doi: 10.1038/s41598-021-89530-8
213. Baell JB, Leaver DJ, Hermans SJ, Kelly GL, Brennan MS, Downer NL, et al. Inhibitors of histone acetyltransferases KAT6A/B induce senescence and arrest tumour growth. Nature (2018) 560(7717):253–7. doi: 10.1038/s41586-018-0387-5
214. Ning J, Sun Q, Su Z, Tan L, Tang Y, Sayed S, et al. The CK1δ/∈-Tip60 axis enhances wnt/β-catenin signaling regulating β-catenin acetylation in colon cancer. Front In Oncol (2022) 12:844477. doi: 10.3389/fonc.2022.844477
215. Idrissou M, Judes G, Daures M, Sanchez A, El Ouardi D, Besse S, et al. TIP60 inhibitor TH1834 reduces breast cancer progression in xenografts in mice. OMICS (2019) 23(9):457–9. doi: 10.1089/omi.2019.0126
216. Gao Y-Y, Ling Z-Y, Zhu Y-R, Shi C, Wang Y, Zhang X-Y, et al. The histone acetyltransferase HBO1 functions as a novel oncogenic gene in osteosarcoma. Theranostics (2021) 11(10):4599–615. doi: 10.7150/thno.55655
217. Dastjerdi MN, Salahshoor MR, Mardani M, Hashemibeni B, Roshankhah S. The effect of CTB on P53 protein acetylation and consequence apoptosis on MCF-7 and MRC-5 cell lines. Adv BioMed Res (2013) 2:24. doi: 10.4103/2277-9175.108005
218. Picaud S, Fedorov O, Thanasopoulou A, Leonards K, Jones K, Meier J, et al. Generation of a selective small molecule inhibitor of the CBP/p300 bromodomain for leukemia therapy. Cancer Res (2015) 75(23):5106–19. doi: 10.1158/0008-5472.CAN-15-0236
219. Ishihama K, Yamakawa M, Semba S, Takeda H, Kawata S, Kimura S, et al. Expression of HDAC1 and CBP/p300 in human colorectal carcinomas. J Clin Pathol (2007) 60(11):1205–10. doi: 10.1136/jcp.2005.029165
220. Guo Y, Li X, He Z, Ma D, Zhang Z, Wang W, et al. HDAC3 silencing enhances acute b lymphoblastic leukaemia cells sensitivity to MG-132 by inhibiting the JAK/Signal transducer and activator of transcription 3 signaling pathway. Chemotherapy (2020) 65(3-4):85–100. doi: 10.1159/000500713
221. Yang T, Wang P, Yin X, Zhang J, Huo M, Gao J, et al. The histone deacetylase inhibitor PCI-24781 impairs calcium influx and inhibits proliferation and metastasis in breast cancer. Theranostics (2021) 11(5):2058–76. doi: 10.7150/thno.48314
222. Tiffon C, Adams J, van der Fits L, Wen S, Townsend P, Ganesan A, et al. The histone deacetylase inhibitors vorinostat and romidepsin downmodulate IL-10 expression in cutaneous T-cell lymphoma cells. Br J Pharmacol (2011) 162(7):1590–602. doi: 10.1111/j.1476-5381.2010.01188.x
223. Cardoso BA, Ramos TL, Belo H, Vilas-Boas F, Real C, Almeida AM. Vorinostat synergizes with antioxidant therapy to target myeloproliferative neoplasms. Exp Hematol (2019) 72:60–71. doi: 10.1016/j.exphem.2019.02.002
224. Palczewski MB, Kuschman HP, Bovee R, Hickok JR, Thomas DD. Vorinostat exhibits anticancer effects in triple-negative breast cancer cells by preventing nitric oxide-driven histone deacetylation. Biol Chem (2021) 402(4):501–12. doi: 10.1515/hsz-2020-0323
225. Wang L, Leite de Oliveira R, Huijberts S, Bosdriesz E, Pencheva N, Brunen D, et al. An acquired vulnerability of drug-resistant melanoma with therapeutic potential. Cell (2018) 173(6):1413–25. doi: 10.1016/j.cell.2018.04.012
226. Chen E, Liu N, Zhao Y, Tang M, Ou L, Wu X, et al. Panobinostat reverses HepaCAM gene expression and suppresses proliferation by increasing histone acetylation in prostate cancer. Gene (2022) 808:145977. doi: 10.1016/j.gene.2021.145977
227. Morabito F, Voso MT, Hohaus S, Gentile M, Vigna E, Recchia AG, et al. Panobinostat for the treatment of acute myelogenous leukemia. Expert Opin On Investigational Drugs (2016) 25(9):1117–31. doi: 10.1080/13543784.2016.1216971
228. Ma J, Guo X, Zhang S, Liu H, Lu J, Dong Z, et al. A histone deacetylase inhibitor, suppresses proliferation and promotes apoptosis of esophageal squamous cell lines. Mol Med Rep (2015) 11(6):4525–31. doi: 10.3892/mmr.2015.3268
229. Wang JH, Lee EJ, Ji M, Park SM. HDAC inhibitors, trichostatin a and valproic acid, increase e−cadherin and vimentin expression but inhibit migration and invasion of cholangiocarcinoma cells. Oncol Rep (2018) 40(1):346–54. doi: 10.3892/or.2018.6441
230. Geng Y, Liu J, Xie Y, Jiang H, Zuo K, Li T, et al. Trichostatin a promotes GLI1 degradation and P21 expression in multiple myeloma cells. Cancer Manage Res (2018) 10:2905–14. doi: 10.2147/CMAR.S167330
231. Bai Y, Chen Y, Chen X, Jiang J, Wang X, Wang L, et al. Trichostatin a activates FOXO1 and induces autophagy in osteosarcoma. Arch Med Sci AMS (2019) 15(1):204–13. doi: 10.5114/aoms.2018.73860
232. Lee H-Z, Kwitkowski VE, Del Valle PL, Ricci MS, Saber H, Habtemariam BA, et al. FDA Approval: Belinostat for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma. Clin Cancer Res an Off J Am Assoc For Cancer Res (2015) 21(12):2666–70. doi: 10.1158/1078-0432.CCR-14-3119
233. Wang B, Wang X-b, Chen L-y, Huang L, Dong R-z. Belinostat-induced apoptosis and growth inhibition in pancreatic cancer cells involve activation of TAK1-AMPK signaling axis. Biochem Biophys Res Commun (2013) 437(1):1–6. doi: 10.1016/j.bbrc.2013.05.090
234. Kong LR, Tan TZ, Ong WR, Bi C, Huynh H, Lee SC, et al. Belinostat exerts antitumor cytotoxicity through the ubiquitin-proteasome pathway in lung squamous cell carcinoma. Mol Oncol (2017) 11(8):965–80. doi: 10.1002/1878-0261.12064
235. Zuo Y, Xu H, Chen Z, Xiong F, Zhang B, Chen K, et al. 17−AAG synergizes with belinostat to exhibit a negative effect on the proliferation and invasion of MDA−MB−231 breast cancer cells. Oncol Rep (2020) 43(6):1928–44. doi: 10.3892/or.2020.7563
236. Zhang S, Gong Z, Oladimeji PO, Currier DG, Deng Q, Liu M, et al. A high-throughput screening identifies histone deacetylase inhibitors as therapeutic agents against medulloblastoma. Exp Hematol Oncol (2019) 8:30. doi: 10.1186/s40164-019-0153-x
237. Garmpis N, Damaskos C, Garmpi A, Dimitroulis D, Spartalis E, Margonis G-A, et al. Targeting histone deacetylases in malignant melanoma: A future therapeutic agent or just great expectations? Anticancer Res (2017) 37(10):5355–62. doi: 10.21873/anticanres.11961
238. Ganai SA. Histone deacetylase inhibitor givinostat: the small-molecule with promising activity against therapeutically challenging haematological malignancies. J Chemother (Florence Italy) (2016) 28(4):247–54. doi: 10.1080/1120009X.2016.1145375
239. Amaru Calzada A, Pedrini O, Finazzi G, Leoni F, Mascagni P, Introna M, et al. Givinostat and hydroxyurea synergize in vitro to induce apoptosis of cells from JAK2(V617F) myeloproliferative neoplasm patients. Exp Hematol (2013) 41(3):253–60. doi: 10.1016/j.exphem.2012.10.013
240. Bitzer M, Horger M, Giannini EG, Ganten TM, Wörns MA, Siveke JT, et al. Resminostat plus sorafenib as second-line therapy of advanced hepatocellular carcinoma - the SHELTER study. J Hepatol (2016) 65(2):280–8. doi: 10.1016/j.jhep.2016.02.043
241. Streubel G, Schrepfer S, Kallus H, Parnitzke U, Wulff T, Hermann F, et al. Histone deacetylase inhibitor resminostat in combination with sorafenib counteracts platelet-mediated pro-tumoral effects in hepatocellular carcinoma. Sci Rep (2021) 11(1):9587. doi: 10.1038/s41598-021-88983-1
242. Wang X, Liu K, Gong H, Li D, Chu W, Zhao D, et al. Death by histone deacetylase inhibitor quisinostat in tongue squamous cell carcinoma via apoptosis, pyroptosis, and ferroptosis. Toxicol Appl Pharmacol (2021) 410:115363. doi: 10.1016/j.taap.2020.115363
243. Kommalapati VK, Kumar D, Tangutur AD. Quisinostat mediated autophagy is associated with differentiation in neuroblastoma SK-N-SH cells. Mol Biol Rep (2021) 48(5):4973–9. doi: 10.1007/s11033-021-06481-z
244. He B, Dai L, Zhang X, Chen D, Wu J, Feng X, et al. The HDAC inhibitor quisinostat (JNJ-26481585) supresses hepatocellular carcinoma alone and synergistically in combination with sorafenib by G0/G1 phase arrest and apoptosis induction. Int J Biol Sci (2018) 14(13):1845–58. doi: 10.7150/ijbs.27661
245. Li H, Cui R, Ji M, Jin S-Y. CUDC-101 enhances the chemosensitivity of gemcitabine-treated lymphoma cells. Leukemia Res (2021) 106:106575. doi: 10.1016/j.leukres.2021.106575
246. Ji M, Li Z, Lin Z, Chen L. Antitumor activity of the novel HDAC inhibitor CUDC-101 combined with gemcitabine in pancreatic cancer. Am J Cancer Res (2018) 8(12):2402–18.
247. Li X, Su Y, Hege K, Madlambayan G, Edwards H, Knight T, et al. The HDAC and PI3K dual inhibitor CUDC-907 synergistically enhances the antileukemic activity of venetoclax in preclinical models of acute myeloid leukemia. Haematologica (2021) 106(5):1262–77. doi: 10.3324/haematol.2019.233445
248. Li Z-J, Hou Y-J, Hao G-P, Pan X-X, Fei H-R, Wang F-Z. CUDC-907 enhances TRAIL-induced apoptosis through upregulation of DR5 in breast cancer cells. J Cell Commun Signal (2020) 14(4):377–87. doi: 10.1007/s12079-020-00558-3
249. Rucker FG, Lang KM, Futterer M, Komarica V, Schmid M, Dohner H, et al. Molecular dissection of valproic acid effects in acute myeloid leukemia identifies predictive networks. Epigenetics (2016) 11(7):517–25. doi: 10.1080/15592294.2016.1187350
250. Al-Keilani MS, Al-Sawalha NA. Potential of phenylbutyrate as adjuvant chemotherapy: An overview of cellular and molecular anticancer mechanisms. Chem Res Toxicol (2017) 30(10):1767–77. doi: 10.1021/acs.chemrestox.7b00149
251. Qian K, Sun L, Zhou G, Ge H, Meng Y, Li J, et al. Sodium phenylbutyrate inhibits tumor growth and the epithelial-mesenchymal transition of oral squamous cell carcinoma In vitro and In vivo. Cancer Biotherapy Radiopharmaceuticals (2018) 33(4):139–45. doi: 10.1089/cbr.2017.2418
252. Gridelli C, Rossi A, Maione P. The potential role of histone deacetylase inhibitors in the treatment of non-small-cell lung cancer. Crit Rev In Oncol/hematol (2008) 68(1):29–36. doi: 10.1016/j.critrevonc.2008.03.002
253. Zhu Y, Yuan T, Zhang Y, Shi J, Bai L, Duan X, et al. AR-42: A pan-HDAC inhibitor with antitumor and antiangiogenic activities in esophageal squamous cell carcinoma. Drug Design Dev Ther (2019) 13:4321–30. doi: 10.2147/DDDT.S211665
254. Elshafae SM, Kohart NA, Breitbach JT, Hildreth BE, Rosol TJ. The effect of a histone deacetylase inhibitor (AR-42) and zoledronic acid on adult T-cell Leukemia/Lymphoma osteolytic bone tumors. Cancers (2021) 13(20):16. doi: 10.3390/cancers13205066
255. Pojani E, Barlocco D. Romidepsin (FK228), a histone deacetylase inhibitor and its analogues in cancer chemotherapy. Curr Med Chem (2021) 28(7):1290–303. doi: 10.2174/0929867327666200203113926
256. Boumber Y, Younes A, Garcia-Manero G. Mocetinostat (MGCD0103): a review of an isotype-specific histone deacetylase inhibitor. Expert Opin Investig Drugs (2011) 20(6):823–9. doi: 10.1517/13543784.2011.577737
257. Ruiz R, Raez LE, Rolfo C. Entinostat (SNDX-275) for the treatment of non-small cell lung cancer. Expert Opin Investig Drugs (2015) 24(8):1101–9. doi: 10.1517/13543784.2015.1056779
258. Trapani D, Esposito A, Criscitiello C, Mazzarella L, Locatelli M, Minchella I, et al. Entinostat for the treatment of breast cancer. Expert Opin Investig Drugs (2017) 26(8):965–71. doi: 10.1080/13543784.2017.1353077
259. Kiany S, Harrison D, Gordon N. The histone deacetylase inhibitor Entinostat/Syndax 275 in osteosarcoma. Adv In Exp Med Biol (2020) 1257:75–83. doi: 10.1007/978-3-030-43032-0_7
260. Gupta VG, Hirst J, Petersen S, Roby KF, Kusch M, Zhou H, et al. Entinostat, a selective HDAC1/2 inhibitor, potentiates the effects of olaparib in homologous recombination proficient ovarian cancer. Gynecol Oncol (2021) 162(1):163–72. doi: 10.1016/j.ygyno.2021.04.015
261. Knipstein J, Gore L. Entinostat for treatment of solid tumors and hematologic malignancies. Expert Opin On Investigational Drugs (2011) 20(10):1455–67. doi: 10.1517/13543784.2011.613822
262. Marques AEM, do Nascimento Filho CHV, Marinho Bezerra TM, Guerra ENS, Castilho RM, Squarize CH. Entinostat is a novel therapeutic agent to treat oral squamous cell carcinoma. J Oral Pathol Med Off Publ Int Assoc Oral Pathol Am Acad Oral Pathol (2020) 49(8):771–9. doi: 10.1111/jop.13039
263. Sun Y, Xu Z, Jiang J, Xu T, Xu J, Liu P. High expression of succinate dehydrogenase subunit a which is regulated by histone acetylation, acts as a good prognostic factor of multiple myeloma patients. Front Oncol (2020) 10:563666. doi: 10.3389/fonc.2020.563666
264. Richardson PG, Moreau P, Laubach JP, Maglio ME, Lonial S, San-Miguel J. Deacetylase inhibitors as a novel modality in the treatment of multiple myeloma. Pharmacol Res (2017) 117:185–91. doi: 10.1016/j.phrs.2016.11.020
265. Zhao C, Dong H, Xu Q, Zhang Y. Histone deacetylase (HDAC) inhibitors in cancer: a patent review (2017-present). Expert Opin Ther Pat (2020) 30(4):263–74. doi: 10.1080/13543776.2020.1725470
266. Romoli M, Mazzocchetti P, D'Alonzo R, Siliquini S, Rinaldi VE, Verrotti A, et al. Valproic acid and epilepsy: From molecular mechanisms to clinical evidences. Curr Neuropharmacol (2019) 17(10):926–46. doi: 10.2174/1570159X17666181227165722
267. Stockhausen MT, Sjolund J, Manetopoulos C, Axelson H. Effects of the histone deacetylase inhibitor valproic acid on notch signalling in human neuroblastoma cells. Br J Cancer (2005) 92(4):751–9. doi: 10.1038/sj.bjc.6602309
268. Chou CJ, Herman D, Gottesfeld JM. Pimelic diphenylamide 106 is a slow, tight-binding inhibitor of class I histone deacetylases. J Biol Chem (2008) 283(51):35402–9. doi: 10.1074/jbc.M807045200
269. Bantscheff M, Hopf C, Savitski MM, Dittmann A, Grandi P, Michon A-M, et al. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat Biotechnol (2011) 29(3):255–65. doi: 10.1038/nbt.1759
270. Bezecny P. Histone deacetylase inhibitors in glioblastoma: pre-clinical and clinical experience. Med Oncol (Northwood London England) (2014) 31(6):985. doi: 10.1007/s12032-014-0985-5
271. Park SE, Kim DE, Kim MJ, Lee JS, Rho JK, Jeong S-Y, et al. Vorinostat enhances gefitinib−induced cell death through reactive oxygen species−dependent cleavage of HSP90 and its clients in non−small cell lung cancer with the EGFR mutation. Oncol Rep (2019) 41(1):525–33. doi: 10.3892/or.2018.6814
272. Salmon JM, Bots M, Vidacs E, Stanley KL, Atadja P, Zuber J, et al. Correction to: Combining the differentiating effect of panobinostat with the apoptotic effect of arsenic trioxide leads to significant survival benefit in a model of t (8,21) acute myeloid leukemia. Clin Epigenet (2020) 12(1):178. doi: 10.1186/s13148-020-00964-9
273. Tang YL, Zhang CG, Liu H, Zhou Y, Wang YP, Li Y, et al. Ginsenoside Rg1 inhibits cell proliferation and induces markers of cell senescence in CD34+CD38- leukemia stem cells derived from KG1alpha acute myeloid leukemia cells by activating the sirtuin 1 (SIRT1)/Tuberous sclerosis complex 2 (TSC2) signaling pathway. Med Sci Monit (2020) 26:e918207. doi: 10.12659/MSM.918207
274. Islam A, Yang YT, Wu WH, Chueh PJ, Lin MH. Capsaicin attenuates cell migration via SIRT1 targeting and inhibition to enhance cortactin and beta-catenin acetylation in bladder cancer cells. Am J Cancer Res (2019) 9(6):1172–82.
275. Liu S, Fang Y, Yu J, Chang X. Hawthorn polyphenols reduce high glucose-induced inflammation and apoptosis in ARPE-19 cells by regulating miR-34a/SIRT1 to reduce acetylation. J Food Biochem (2021) 45(2):e13623. doi: 10.1111/jfbc.13623
276. Wang L, Xu M, Kao C-Y, Tsai SY, Tsai M-J. Small molecule JQ1 promotes prostate cancer invasion via BET-independent inactivation of FOXA1. J Clin Invest (2020) 130(4):1782–92. doi: 10.1172/JCI126327
277. Choi HI, An GY, Baek M, Yoo E, Chai JC, Lee YS, et al. BET inhibitor suppresses migration of human hepatocellular carcinoma by inhibiting SMARCA4. Sci Rep (2021) 11(1):11799. doi: 10.1038/s41598-021-91284-2
278. Kim E, Ten Hacken E, Sivina M, Clarke A, Thompson PA, Jain N, et al. The BET inhibitor GS-5829 targets chronic lymphocytic leukemia cells and their supportive microenvironment. Leukemia (2020) 34(6):1588–98. doi: 10.1038/s41375-019-0682-7
279. Hsu SC, Gilgenast TG, Bartman CR, Edwards CR, Stonestrom AJ, Huang P, et al. The BET protein BRD2 cooperates with CTCF to enforce transcriptional and architectural boundaries. Mol Cell (2017) 66(1):102–16. doi: 10.1016/j.molcel.2017.02.027
280. Doroshow DB, Eder JP, LoRusso PM. BET inhibitors: a novel epigenetic approach. Ann Oncol Off J Eur Soc For Med Oncol (2017) 28(8):1776–87. doi: 10.1093/annonc/mdx157
281. Dai X, Gan W, Li X, Wang S, Zhang W, Huang L, et al. Prostate cancer-associated SPOP mutations confer resistance to BET inhibitors through stabilization of BRD4. Nat Med (2017) 23(9):1063–71. doi: 10.1038/nm.4378
282. Zhang P, Wang D, Zhao Y, Ren S, Gao K, Ye Z, et al. Intrinsic BET inhibitor resistance in SPOP-mutated prostate cancer is mediated by BET protein stabilization and AKT-mTORC1 activation. Nat Med (2017) 23(9):1055–62. doi: 10.1038/nm.4379
283. Fiorentino FP, Marchesi I, Schröder C, Schmidt R, Yokota J, Bagella L. BET-inhibitor I-BET762 and PARP-inhibitor talazoparib synergy in small cell lung cancer cells. Int J Mol Sci (2020) 21(24):9595. doi: 10.3390/ijms21249595
284. Braun T, Gardin C. Investigational BET bromodomain protein inhibitors in early stage clinical trials for acute myelogenous leukemia (AML). Expert Opin On Investigational Drugs (2017) 26(7):803–11. doi: 10.1080/13543784.2017.1335711
285. He S, Dong G, Li Y, Wu S, Wang W, Sheng C. Potent dual BET/HDAC inhibitors for efficient treatment of pancreatic cancer. Angewandte Chemie (International Ed In English) (2020) 59(8):3028–32. doi: 10.1002/anie.201915896
286. Lui GYL, Shaw R, Schaub FX, Stork IN, Gurley KE, Bridgwater C, et al. BET, SRC, and BCL2 family inhibitors are synergistic drug combinations with PARP inhibitors in ovarian cancer. EBioMedicine (2020) 60:102988. doi: 10.1016/j.ebiom.2020.102988
287. Sun Y, Han J, Wang Z, Li X, Sun Y, Hu Z. Safety and efficacy of bromodomain and extra-terminal inhibitors for the treatment of hematological malignancies and solid tumors: A systematic study of clinical trials. Front In Pharmacol (2020) 11:621093. doi: 10.3389/fphar.2020.621093
288. Lewin J, Soria J-C, Stathis A, Delord J-P, Peters S, Awada A, et al. Phase ib trial with birabresib, a small-molecule inhibitor of bromodomain and extraterminal proteins, in patients with selected advanced solid tumors. J Clin Oncol Off J Am Soc Clin Oncol (2018) 36(30):3007–14. doi: 10.1200/JCO.2018.78.2292
289. Zhang Y, Duan S, Jang A, Mao L, Liu X, Huang G. JQ1, a selective inhibitor of BRD4, suppresses retinoblastoma cell growth by inducing cell cycle arrest and apoptosis. Exp Eye Res (2021) 202:108304. doi: 10.1016/j.exer.2020.108304
290. Bagratuni T, Mavrianou N, Gavalas NG, Tzannis K, Arapinis C, Liontos M, et al. JQ1 inhibits tumour growth in combination with cisplatin and suppresses JAK/STAT signalling pathway in ovarian cancer. Eur J Cancer (Oxford Engl 1990) (2020) 126:125–35. doi: 10.1016/j.ejca.2019.11.017
291. Xie F, Huang M, Lin X, Liu C, Liu Z, Meng F, et al. The BET inhibitor I-BET762 inhibits pancreatic ductal adenocarcinoma cell proliferation and enhances the therapeutic effect of gemcitabine. Sci Rep (2018) 8(1):8102. doi: 10.1038/s41598-018-26496-0
292. Liu M, Zhou J, Liu X, Feng Y, Yang W, Wu F, et al. Targeting monocyte-intrinsic enhancer reprogramming improves immunotherapy efficacy in hepatocellular carcinoma. Gut (2020) 69(2):365–79. doi: 10.1136/gutjnl-2018-317257
293. Liu A, Fan D, Wang Y. The BET bromodomain inhibitor i-BET151 impairs ovarian cancer metastasis and improves antitumor immunity. Cell Tissue Res (2018) 374(3):577–85. doi: 10.1007/s00441-018-2906-y
294. Guo N-H, Zheng J-F, Zi F-M, Cheng J. I-BET151 suppresses osteoclast formation and inflammatory cytokines secretion by targetting BRD4 in multiple myeloma. Bioscience Rep (2019) 39(5):12. doi: 10.1042/BSR20181245
295. Siu KT, Ramachandran J, Yee AJ, Eda H, Santo L, Panaroni C, et al. Preclinical activity of CPI-0610, a novel small-molecule bromodomain and extra-terminal protein inhibitor in the therapy of multiple myeloma. Leukemia (2017) 31(8):1760–9. doi: 10.1038/leu.2016.355
296. Hupe MC, Hoda MR, Zengerling F, Perner S, Merseburger AS, Cronauer MV. The BET-inhibitor PFI-1 diminishes AR/AR-V7 signaling in prostate cancer cells. World J Urol (2019) 37(2):343–9. doi: 10.1007/s00345-018-2382-8
297. Liu Z, Li P, Yang Y-Q, Cai S, Lin X, Chen M-B, et al. I-BET726 suppresses human skin squamous cell carcinoma cell growth in vitro and in vivo. Cell Death Dis (2020) 11(5):318. doi: 10.1038/s41419-020-2515-z
298. Healy JR, Hart LS, Shazad AL, Gagliardi ME, Tsang M, Elias J, et al. Limited antitumor activity of combined BET and MEK inhibition in neuroblastoma. Pediatr Blood Cancer (2020) 67(6):e28267. doi: 10.1002/pbc.28267
299. Zhang L, Cai T, Lin X, Huang X, Bui MH, Plotnik JP, et al. Selective inhibition of the second bromodomain of BET family proteins results in robust antitumor activity in preclinical models of acute myeloid leukemia. Mol Cancer Ther (2021) 20(10):1809–19. doi: 10.1158/1535-7163.MCT-21-0029
300. Faivre EJ, McDaniel KF, Albert DH, Mantena SR, Plotnik JP, Wilcox D, et al. Selective inhibition of the BD2 bromodomain of BET proteins in prostate cancer. Nature (2020) 578(7794):306–10. doi: 10.1038/s41586-020-1930-8
Keywords: acetylation, HAT, HDAC, post-translational modification, cancer, HDAC inhibitor
Citation: Yang J, Song C and Zhan X (2022) The role of protein acetylation in carcinogenesis and targeted drug discovery. Front. Endocrinol. 13:972312. doi: 10.3389/fendo.2022.972312
Received: 18 June 2022; Accepted: 23 August 2022;
Published: 12 September 2022.
Edited by:
Tadashi Nakagawa, Tohoku University, JapanReviewed by:
Fengchao Lang, National Institutes of Health (NIH), United StatesIsabel Castro-Piedras, Texas Tech University Health Sciences Center, United States
Copyright © 2022 Yang, Song and Zhan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xianquan Zhan, yjzhan2011@gmail.com