Corrigendum: Physiology and Pharmacology of DPP-4 in Glucose Homeostasis and the Treatment of Type 2 Diabetes
 Carolyn F. Deacon
Carolyn F. Deacon- Department of Biomedical Sciences, University of Copenhagen, Copenhagen, Denmark
Dipeptidyl peptidase-4 (DPP-4), also known as the T-cell antigen CD26, is a multi-functional protein which, besides its catalytic activity, also functions as a binding protein and a ligand for a variety of extracellular molecules. It is an integral membrane protein expressed on cells throughout the body, but is also shed from the membrane and circulates as a soluble protein in the plasma. A large number of bioactive molecules can be cleaved by DPP-4 in vitro, but only a few of these have been demonstrated to be physiological substrates. One of these is the incretin hormone, glucagon-like peptide-1 (GLP-1), which plays an important role in the maintenance of normal glucose homeostasis, and DPP-4 has been shown to be the key enzyme regulating its biological activity. This pathway has been targeted pharmacologically through the development of DPP-4 inhibitors, and these are now a successful class of anti-hyperglycaemic agents used to treat type 2 diabetes (T2DM). DPP-4 may additionally influence metabolic control via its proteolytic effect on other regulatory peptides, but it has also been reported to affect insulin sensitivity, potentially mediated through its non-enzymatic interactions with other membrane proteins. Given that altered expression and activity of DPP-4 are associated with increasing body mass index and hyperglycaemia, DPP-4 has been proposed to play a role in linking obesity and the pathogenesis of T2DM by functioning as a local mediator of inflammation and insulin resistance in adipose and hepatic tissue. As well as these broader systemic effects, it has also been suggested that DPP-4 may be able to modulate β-cell function as part of a paracrine system involving GLP-1 produced locally within the pancreatic islets. However, while it is evident that DPP-4 has the potential to influence glycaemic control, its overall significance for the normal physiological regulation of glucose homeostasis in humans and its role in the pathogenesis of metabolic disease remain to be established.
Introduction
Dipeptidyl peptidase-4 (DPP-4; EC 3.4.14.5) is a member of the prolyl oligopeptidase family of related proteins, which, besides prolyl oligopeptidase (a.k.a. prolyl endopeptidase), contains a number of exopeptidases, including quiescent cell proline dipeptidase (QPP, a.k.a. DPP-II or DPP-7), and attractin (a.k.a. mahogany peptide). Within this family, the DPP-4 gene family belongs to the subclan S9B, and comprises six proteins, of which four [fibroblast activation protein (FAPα, a.k.a. seprase), DPP-4, -8, and -9] are enzymatically active, but also exert effects via protein-protein interactions, and two [DPP-6, known also as DPP4-like protein (DPL)-1] and DPP-10 (a.k.a. DPL-2) are catalytically inactive, exerting their effects by modulating voltage-gated potassium channels (Table 1) (1–3). DPP-4 is an amino-peptidase, which liberates a dipeptide from the N-terminal of its substrates. Like other members of the family, it prefers proline or alanine in the penultimate position, although peptides with other residues (glycine, serine, valine) in this position can be cleaved, albeit more slowly (Figure 1). DPP-4 is an integral membrane protein which has a widespread distribution, being expressed in numerous tissues including intestinal and renal brush border membranes, vascular endothelium, the liver and pancreas, glandular epithelial cells, and by cells of the immune system (where it is also known as the T-cell differentiation antigen, CD26). The DPP-4 protein consists of a large extracellular domain, anchored in the cell membrane by a flexible segment coupled to a trans-membrane sequence, with a short intracellular tail at the N-terminus (Figure 2). It can, however, be cleaved at the stalk to release the extracellular domain, which circulates in the plasma as “soluble” DPP-4 (aa 49-766), but is also present in other bodily fluids such as seminal fluid and cerebrospinal fluid. The catalytic site resides in the C-terminal region of the extracellular part of the protein, but the extracellular domain also contains a cysteine-rich region and a region rich in glycosylation sites. These regions are believed to be involved in many of the non-enzymatic functions of DPP-4, interacting with other proteins, such as adenosine deaminase, caveolin-1, streptokinase and plasminogen, and with components of the extracellular matrix (collagen, fibronectin), as well as functioning as binding sites for the chemokine CXCR4 receptor, the T-cell differentiation antigen, CD45, and the sodium-hydrogen exchanger-3, among others (1, 4).
TABLE 1
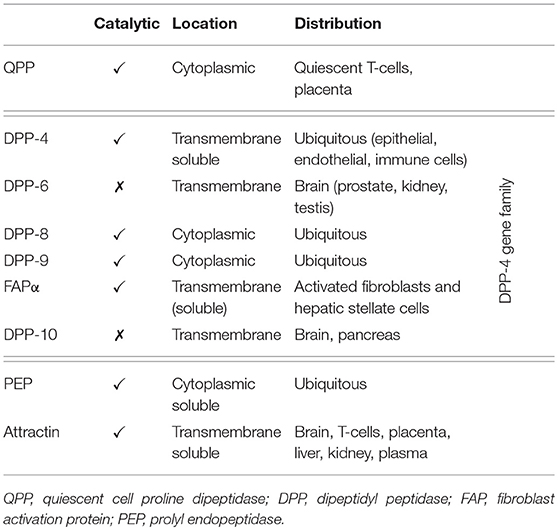
Table 1. The proline-specific peptidases.
FIGURE 1
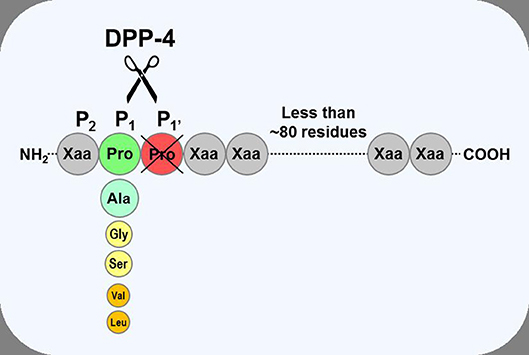
Figure 1. Substrate specificity of DPP-4. DPP-4 is an amino peptidase which liberates a dipeptide from its substrates. It prefers peptides or small proteins (below 80–100 residues) with proline or alanine as the penultimate N-terminal residue, although some substrates with glycine, serine, valine, or leucine can be cleaved at a slower rate. The enzyme is unable to cleave substrates with proline in position three.
FIGURE 2
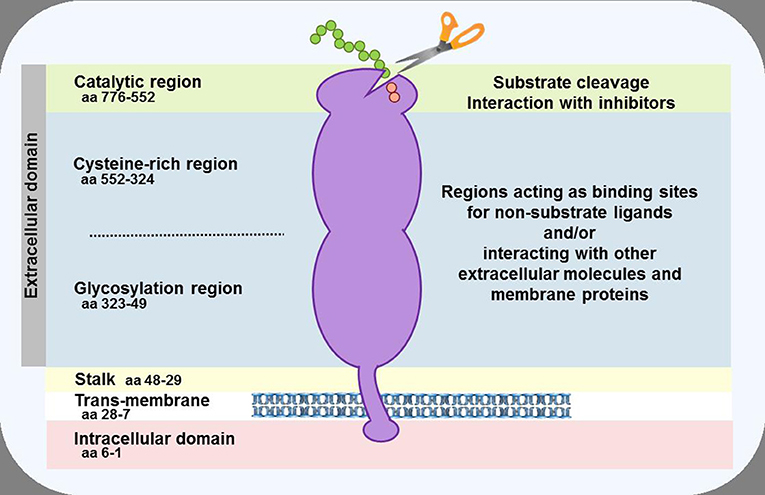
Figure 2. Schematic representation of the DPP-4 protein. In the cell membrane, two DPP-4 monomers dimerise to form a homodimer. The monomers can be cleaved at the stalk to release the soluble form of DPP-4, which circulates in the plasma. The enzymatic activity resides in the catalytic pocket, formed by residues (including serine at position 630) located in the C-terminal portion of the protein. Sites within the cysteine-rich and glycosylation regions serve as a receptor or ligand for different molecules, including adenosine deaminase, caveolin-1, collagen, fibronectin, chemokine CXCR4 receptor, CD45, and the sodium-hydrogen exchanger-3, to mediate the non-enzymatic functions of the protein.
Accordingly, DPP-4 can be viewed as a multi-functional protein with a spectrum of actions which go beyond its role as a proteolytic enzyme. There is a wealth of data identifying DPP-4 as a receptor or ligand for a variety of different molecules which, either alone or in combination with its enzymatic activity, allow it affect physiological processes such as the interaction between cells and the extracellular matrix, cell migration, and proliferation. Within the immune system, DPP-4/CD26 is one of several proteins which can act as co-stimulatory molecules. These molecules cannot directly stimulate T-cells, but they are involved in amplifying the signal derived from the interaction with an antigen via their interaction with other cell surface molecules, thereby leading to T-cell activation (5). For DPP-4, this immune function appears to be independent of its catalytic activity, since the co-stimulatory activity of the molecule is retained by mutant proteins lacking the catalytic region (6) and is unaffected by inhibitors of the enzymatic activity of DPP-4 (7). Consequently, given its various roles, altered expression, and/or activity of DPP-4 have been implicated in several pathological processes, including inflammation, viral entry, immune-mediated diseases, and tumor biology (1, 3, 4). More recently, DPP-4 has been found to have effects on metabolic control, raising the possibility that it may play a role in metabolic diseases such as diabetes and obesity. However, the relative importance of the soluble vs. the membrane-bound form of the molecule for the various functions of DPP-4 has not been clarified, nor is it clear whether (or to what extent) changes in circulating levels of soluble DPP-4 might be regulated (e.g., whether they simply reflect alterations in non-specific cleavage or shedding of the membrane-bound form or whether this is a process which might be modulated in response to pathological processes or changes in metabolic status).
DPP-4 and Glucose Homeostasis
The link between DPP-4 and glucose homeostasis was only identified after the intestinal hormone, glucagon-like peptide-1 (GLP-1) was found to be a DPP-4 substrate. A role for GLP-1 in regulating glycaemia had been noted in 1986, when this, then newly described (8, 9), peptide was found to have profound effects upon the endocrine pancreas. In studies using perfused pig and rat pancreata, researchers in Denmark and the US described potent insulinotropic (10, 11), and glucagonostatic (12) effects in vitro, while studies in healthy humans revealed that circulating levels of GLP-1 increased after nutrient intake and confirmed GLP-1 to be an incretin hormone in vivo (13). However, it was when the insulinotropic and glucose-lowering effects were shown to be preserved in patients with type 2 diabetes (T2DM) (14, 15) that real interest in GLP-1 took off. In a now landmark study, Michael Nauck demonstrated that hyperglycaemic fasting glucose levels could be brought into the normal range by intravenous GLP-1 infusion in individuals with T2DM (16). Moreover, this study also nicely illustrated the glucose dependency of GLP-1's anti-diabetic actions—insulin secretion was stimulated, and glucagon secretion suppressed, only at elevated glucose levels, with the effects of GLP-1 becoming progressively less as euglycaemia was approached. However, whilst intravenous infusion of GLP-1 was effective (16), the insulinotropic effect of a single subcutaneous injection was surprisingly short-lived, with insulin levels peaking at 30 min before returning toward baseline within 90 min, even though glucose levels were still well within the hyperglyacemic range and circulating immunoreactive GLP-1 levels were significantly elevated for several hours (17). The reason for this paradox was initially unclear. However, when it was reported that GLP-1 was a substrate for DPP-4 in pharmacological in vitro kinetic studies (18), this was quickly followed by the demonstration that the metabolite generated by DPP-4 cleavage was the major circulating component of GLP-1-like immunoreactivity in healthy individuals (19) and that the same metabolite formed rapidly following exogenous administration of GLP-1 in both healthy subjects and those with T2DM (20). In line with these results, similar findings were reported after exogenous GLP-1 administration in rats (21). These studies, therefore, indicated that GLP-1 was a true physiological substrate of DPP-4, and led to the suggestion that blocking this route of degradation may be a way to increase endogenous intact (active) GLP-1 concentrations and enhance its anti-hyperglycaemic actions (20). Accordingly, and using the analogy of angiotensin-converting enzyme inhibitors for treating hypertension, DPP-4 inhibition was proposed as a novel approach to the treatment of T2DM (20, 22).
Inhibiting DPP-4 as a Therapy for T2DM
In order for this approach to be viable, DPP-4 cleavage would need to be the initial and primary route of metabolism of GLP-1. If this was not the case, other clearance pathways would simply take over once the DPP-4 pathway had been blocked, and levels of the intact peptide would not be increased. In those early days, several DPP-4 inhibitors had been described (23), but none were suitable for human use, so proof-of-hypothesis came from preclinical studies. In anesthetized pigs, a prototype DPP-4 inhibitor, valine pyrrolidide, was demonstrated to reduce plasma DPP-4 activity sufficiently to fully protect intravenously infused GLP-1 from degradation. Moreover, this was associated with an enhanced insulin response, confirming that DPP-4-mediated degradation played a significant role in limiting the insulinotropic effect of GLP-1 (24). The pivotal role of DPP-4 in endogenous GLP-1 metabolism was highlighted using the isolated perfused porcine small intestine, which revealed that over half of newly released GLP-1 was degraded even before it left the splanchnic bed. Again, this could be completely prevented by DPP-4 inhibition with valine pyrrolidide, confirming that cleavage by DPP-4 was the key initial step in GLP-1 degradation, and that other enzymatic pathways played a limited role (25). This study also investigated the expression of DPP-4 and found it to be present on the vascular endothelium, including the local capillaries in the lamina propria adjacent to the GLP-1 producing L-cells, thus providing an explanation for the rapid degradation of the peptide once it had been released. An acute glucose-lowering effect of DPP-4 inhibition in a rodent model of T2DM (the obese Zucker rat) was demonstrated when it was shown that a different inhibitor (isoleucine thiazolidide) reduced plasma DPP-4 activity and was associated with a larger insulin response and improved glucose tolerance after an oral glucose load (when endogenous GLP-1 secretion would be stimulated) (26). No effects were seen when glucose was not administered, compatible with the idea that DPP-4 inhibition exerts an anti-hyperglycaemic effect by preventing degradation of endogenously released GLP-1 (20). Although GLP-1 levels were not actually measured in that study (26), subsequent studies in glucose-intolerant rodent models did show that the improved glycaemic control following in vivo DPP-4 inhibition was associated with enhanced intact GLP-1 responses to glucose challenges (27–29). The final step in establishing preclinical proof-of-hypothesis was made when the results of these acute studies were recapitulated with chronic dosing, showing that the beneficial pancreatic islet and glucose-lowering effects of DPP-4 inhibition persisted over several months of treatment (30, 31). Collectively, these studies paved the way for clinical investigation into the feasibility of using pharmacological inhibition of DPP-4 to improve glycaemic control in patients with T2DM. The first two reports, both 4 week studies in drug naïve patients with relatively mild T2DM, showed that DPP-4 inhibition was well-tolerated in humans and was associated with significantly lower fasting and meal-related glucose concentrations (32, 33). Absolute insulin responses were not augmented, but they were sustained in face of the improved glycaemia which, together with the lower glucagon levels could be suggestive of an improved islet function related to the observed increase in intact GLP-1 levels (33). The longer-term efficacy of DPP-4 inhibition was examined in a third study published later in the same year, this time undertaken in patients with T2DM on metformin monotherapy. This 12 week study with a 40 week extension period revealed that the addition of vildagliptin significantly lowered HbA1c levels by week 12, with the improvement being sustained to the end of the study period, whereas glycaemic control deteriorated in the placebo recipients (34). As in the shorter-term studies, DPP-4 inhibition was well-tolerated with no increased risk of hypoglycaemia, despite the improvement in glycaemic control, and the overall adverse event profile was similar in both arms of the study. These key studies clearly provided clinical proof-of-concept that blocking the catalytic activity of DPP-4 could be a viable approach to treat T2DM, whilst at the same time helped to allay concern over whether the metabolism of any other potential DPP-4 substrates might be affected to cause unwanted off-target effects. These studies were soon followed by a number of phase 2 and phase 3 clinical trials with several different DPP-4 inhibitors, all essentially showing that DPP-4 inhibition was a safe and well-tolerated means of improving glycaemic control in patients with T2DM. The first DPP-4 inhibitor received marketing authorization in 2006, and now, world-wide, there are currently at least 11 different inhibitors approved for therapy of T2DM (35).
Metabolic Effects of DPP-4 Related to Its Enzymatic Activity
Despite DPP-4 having a relatively restricted substrate preference (Figure 1), a large number of peptides, proteins, and chemokines still fulfill the criteria and could be predicted to be suitable substrates, and indeed, many have been shown to be cleaved by DPP-4 in pharmacological kinetic, in vitro or animal studies, often using high substrate concentrations (1, 4, 36–38). However, simply showing that the enzyme can cleave a substrate in vitro does not necessarily mean that DPP-4 plays an important physiological role in regulating concentrations and/or modulating biological activity of the endogenous molecules, particularly in humans and, for many of these putative substrates, there is little, if any experimental evidence to show that DPP-4 does actually cleave them in vivo and/or whether this has any physiological consequence. Thus, for a number of them, even if they are good substrates in vitro, other clearance pathways may be more important in vivo or, if they are cleaved by DPP-4, removal of the N-terminal dipeptide may have no effect upon their biological activity. However, it should also be recognized that it may be difficult to definitively demonstrate whether DPP-4 does play a physiologically relevant role in cleaving some endogenous substrates in vivo because it can be difficult to distinguish between the intact substrate and the truncated metabolite (because assays are not specific or sensitive enough) and/or because it may be difficult to obtain appropriate samples (e.g., from synapses [neurotransmitters] or interstitial fluid [paracrine signallers]). Accordingly, the list of molecules which have been shown unequivocally to be physiological substrates of DPP-4 is short (37, 38). Moreover, given that there are now millions of patient-years of clinical experience with DPP-4 inhibitors since they were given regulatory approval in 2006, their benign, placebo-like side effect profile (39) also suggests that the number of physiologically relevant substrates is likely to be limited.
GLP-1 and GIP
With regard to metabolic effects, the best characterized DPP-4 substrates are the two incretin hormones, GLP-1, and glucose-dependent insulinotropic polypeptide (GIP). As discussed above, endogenous GLP-1 is rapidly degraded by DPP-4, whereby it loses its insulinotropic activity, and preventing this degradation results in increased intact GLP-1 levels, improved pancreatic islet responses (enhanced insulin and suppressed glucagon levels) and beneficial effects on glucose homeostasis. Similarly, the other incretin hormone, GIP is efficiently cleaved by DPP-4 in vivo (40), and DPP-4 inhibition increases intact GIP levels and enhances its effects (41). Accordingly, it is well-established that DPP-4 does play a pivotal role in the initial inactivation of both endogenous incretin hormones. It is, however, less clear whether this is a process which can be regulated, or whether it is simply a consequence of DPP-4's ubiquitous distribution. Nevertheless, the fact that DPP-4 is a key player in the clearance of both endogenous peptides has been exploited pharmacologically and, as mentioned above, DPP-4 inhibitors are now an established therapy of T2DM.
It was initially thought that the anti-diabetic effects of DPP-4 inhibition were predominantly due to the enhanced levels of intact GLP-1 [because the insulinotropic effect of GIP is substantially impaired in subjects with T2DM (42)], but this now seems not to be the case. Thus, in mice, significant antihyperglycaemic effects of DPP-4 inhibition are still apparent in animals lacking the GLP-1 receptor (43), while in clinical studies, GLP-1 receptor antagonism (using exendin 9-39) eliminates only approximately half of the glucose-lowering effect in patients with T2DM treated with DPP-4 inhibitors (44, 45). It is possible that GIP may still be involved in spite of the observations that its actions are impaired in patients with T2DM (42), because it appears that these can be at least partially restored. Thus, treatment to improve glycaemic control, using insulin (46) or a DPP-4 inhibitor (47) led to improved insulin responses to exogenous GIP. This would be in line with preclinical observations that incretin receptor expression is down-regulated by exposure to high glucose, and improves once normoglycaemia is restored (48), and that dys-regulation of glycaemic control in otherwise healthy humans leads to impaired incretin actions (49). Thus, GIP is a likely candidate for mediating some of the therapeutic actions of DPP-4 inhibition, and the recent development of a GIP receptor antagonist suitable for use in clinical studies (50) should allow this hypothesis to be tested.
Peptide Tyrosine Tyrosine
Peptide tyrosine tyrosine (PYY) is another gastrointestinal peptide which is released after food ingestion and which also fulfills the criteria to be classified as a physiological DPP-4 substrate. Both exogenous and endogenous PYY have been shown to be degraded in vivo in animal and clinical studies. Moreover, the truncated peptide, PYY 3-36, is the major circulating endogenous form in humans (51), and its formation is reduced by DPP-4 inhibition (52). In contrast to the incretin hormones, where cleavage by DPP-4 results in a loss of their insulinotropic activity, cleavage of PYY results in altered receptor selectivity. Thus, intact PYY 1-36 binds with equal affinity to the four Y receptor subtypes, while PYY 3-36 is a selective high affinity agonist for the Y2 receptor. Although there is no evidence that PYY can directly influence insulin secretion in humans (53), activation of Y1 receptors is associated with increased intestinal motility and orexigenic effects, while conversely, Y2 receptor activation mediates the enterogastrone and anorectic effects of PYY (54). Accordingly, DPP-4 may influence metabolic control indirectly by modulating PYY effects on food intake and body weight. Again, whether this is a process which is regulated is unknown, but it may help to explain why pharmacological use of DPP-4 inhibitors is generally not associated with weight loss (the appetite suppressive effects of increased intact GLP-1 levels being counter-balanced by the loss of the anorectic effects of PYY 3-36).
Oxyntomodulin
Surprisingly, given that glucagon is not a physiological DPP-4 substrate (37, 38), there is some evidence that the C-terminally extended peptide, oxyntomodulin might be. Oxyntomodulin is generated by post-translational processing of the glucagon precursor, proglucagon, in the intestine. A specific oxyntomodulin receptor has not been identified, and oxyntomodulin is believed to exert its effects by behaving as a weak agonist at both the GLP-1 and glucagon receptors (55). Accordingly, it has several metabolic effects consistent with the actions of GLP-1 and glucagon (stimulation of insulin secretion, reduced appetite, increased energy expenditure), although whether these are important physiologically is uncertain (55). Nevertheless, oxyntomodulin can be cleaved by DPP-4 in vitro (36, 56), and in animal studies, the anorectic effects of exogenous oxyntomodulin are increased after DPP-4 inhibition (56), while DPP-4-resistant oxyntomodulin analogs have been associated with improved glucose tolerance, reduced food intake, greater energy expenditure, and weight loss (57, 58). Nevertheless, because endogenous oxyntomodulin is currently not believed to play a major role in regulating metabolic processes (55), it seems unlikely that cleavage of oxyntomodulin by DPP-4 would be associated with significant effects under normal physiological circumstances. However, whether pharmacological inhibition of the enzyme could elevate oxyntomodulin levels sufficiently to contribute to the therapeutic effect of DPP-4 inhibitor therapy is not known.
Stromal Cell-Derived Factor-1α
There is fairly strong evidence that stromal cell-derived factor-1 (SDF-1α) also qualifies as a physiological DPP-4 substrate (37, 38). SDF-1α, also referred to as CXCL12, functions as a chemotactic cytokine and proangiogenic chemokine, which enhances hematopoietic and endothelial progenitor cell recruitment to sites of cell injury. However, it is also expressed in the pancreas, where in vitro studies have indicated that it can improve β-cell survival (59, 60) and enhance α-cell GLP-1 biosynthesis (60). Moreover, transgenic mice over-expressing SDF-1α become resistant to streptozotocin-induced diabetes (61), further suggesting that SDF-1α may have some metabolic effects. The involvement of DPP-4 in metabolism of endogenous SDF-1α is compatible with the finding that the truncated metabolite, SDF-1α 3-67, was present in wild-type mice, but not in animals lacking DPP-4 (62), with this observation being extended in subsequent studies in mice and Rhesus monkeys (63) and in patients with T2DM (64) showing that levels of SDF-1α 3-67 were reduced while those of the intact 1-67 form increased concomitantly when DPP-4 activity was inhibited. Cleavage of SDF-1α by DPP-4 results in loss of activity (65), and consistent with this, DPP-4 inhibition in humans has been associated with enhancement of some effects which are associated with SDF-1α [increased levels of circulating endothelial progenitor cells (66) and increased distal tube sodium excretion (64)]. However, although the preclinical studies cited above point toward potential metabolic effects of SDF-1α, it is unknown whether there are any physiological or pharmacological consequences of DPP-4-mediated cleavage of SDF-1α for metabolic control in humans.
A Potential Role for DPP-4 in the Pathogenesis of T2DM?
Given that DPP-4 clearly plays a role in the clearance of a number of hormones involved in the regulation of glucose homeostasis, the question arises of whether changes in DPP-4 activity might play a role in the pathogenesis of T2DM. Indeed, a number of reports have included assessment of DPP-4 activity, albeit with conflicting results. While some studies found no difference between individuals with T2DM and matched non-diabetic controls (67–69), others found plasma DPP-4 activity to be reduced in the patients, and suggested this might reflect a compensatory response to the reduced insulin sensitivity and hyperglycaemia, in an attempt to increase intact incretin hormone concentrations and improve insulin secretion (70, 71). However, increased levels have also been reported (68, 72, 73), raising speculation that elevated plasma DPP-4 activity might constitute a risk factor associated with the deterioration of glycaemic control. In this scenario, greater degradation of GLP-1 (and the other incretin, GIP), with a consequent impairment of the incretin effect would contribute to worsening of glycaemic control. This might arise early in the pathogenesis of T2DM, as one of many risk factors contributing to the initial onset of hyperglycaemia in susceptible individuals, or later, as a consequence of already existing metabolic changes causing increased DPP-4 activity and further exacerbating the deterioration of glycaemic control. Accordingly, several studies have reported a positive correlation between plasma DPP-4 activity and fasting plasma glucose or HbA1c (68, 72), although it should be borne in mind that an association does not necessarily reflect a causative relationship. Moreover, it appears that it is the membrane-bound form of the enzyme, on sites such as the luminal surface of capillary endothelial cells, which is more important for GLP-1 degradation than the circulating (soluble) enzyme (74, 75), questioning the relevance of any association between the plasma activity per se and glycaemic control, since it is unclear whether changes in soluble DPP-4 activity in the circulation accurately reflect any changes in total DPP-4 activity. Glucose itself does not seem to directly modulate the catalytic activity of DPP-4, as shown in vitro, by incubating the enzyme with high concentrations of glucose (76). Moreover, plasma DPP-4 activity is not lowered when glycaemic control is improved in patients with T2DM, thereby also suggesting that hyperglycaemia is not a direct determinant of levels of DPP-4 activity (73). However, exposure to high glucose conditions did increase mRNA expression and membrane-bound DPP-4 activity in human glomerular (76) and dermal (77) microvascular endothelial cells in vitro, although, interestingly, not in macrovascular (human aortic) endothelial cells (77). Changes in levels of plasma DPP-4 activity could, therefore, be related to overall changes in the total expression of DPP-4 and/or simply altered shedding from cell membranes. Nonetheless, the underlying reasons for the inconsistencies in the reported associations between T2DM and levels of plasma DPP-4 activity (lower, unaltered, higher) have not been adequately explained, although one study did suggest that it might be related to the populations studied, with changes only becoming evident once moderate or severe hyperglycaemia becomes established (68).
On the other hand, T2DM is often associated with increased body mass index (BMI), and other studies have suggested that levels of plasma DPP-4 activity actually correlate better with clinical parameters of obesity rather than of T2DM and glucose per se. Thus, a number of studies have shown a positive association between plasma DPP-4 activity and BMI in non-diabetic individuals (78–80), and it has been proposed that the release of DPP-4 from adipose tissue may provide one explanation for the increased plasma DPP-4 activity measured in obese subjects. Accordingly, the DPP-4 protein has been identified in both subcutaneous and visceral adipose tissue, with its expression, particularly in visceral adipocytes, being more pronounced in obese subjects (81). In another study, both circulating DPP-4 levels and the amount released from adipocyte explants in vitro were found to be positively correlated with adipocyte size as well as with anthropometric parameters such as BMI, waist circumference and percent body fat (79). Moreover, following surgically-induced weight loss in morbid obesity, the elevated adipocyte DPP-4 release, and circulating DPP-4 levels were both substantially reduced, further suggesting that the adipose tissue is likely to be a major source of plasma DPP-4 (79). On the other hand, Dpp4 expression is reported to be relatively low in mouse adipocytes and not to be markedly increased after high fat feeding (82), with expression within adipose tissue being predominantly associated with cells of the stromal vascular fraction (82, 83).
However, there is also evidence pointing toward a hepatic origin of circulating DPP-4, and it has been suggested that the inclusion of some patients with unidentified liver disease may have contributed to the conflicting data regarding the association of plasma DPP-4 activity and T2DM. In mice, hepatocyte Dpp4 expression increases with obesity (82), and selective elimination of Dpp4 in the liver is associated with reduced levels of soluble DPP-4 in the plasma (82, 83). In humans, plasma DPP-4 activity is higher in patients with non-alcoholic fatty liver disease irrespective of their glucose tolerance, whereas it was not elevated in patients with T2DM in whom liver disease had been excluded (84). This association between liver disease and DPP-4 has been confirmed in other studies, showing higher plasma levels (85, 86) as well as higher hepatic Dpp4 expression (87) in the patients compared to healthy individuals.
Protease-Independent Metabolic Effects of DPP-4
While some of the effects of DPP-4—and, arguably, most, if not all of the pharmacological effects of DPP-4 inhibitors (38)—on glucose homeostasis are mediated primarily by modulating the activity of the incretin hormones, it has also been proposed that DPP-4 may influence metabolic control via mechanisms which do not necessarily involve its catalytic activity, with several studies noting a positive correlation between DPP-4 and insulin resistance (79, 81, 88). Accordingly, DPP-4 has been proposed to function as a locally acting adipokine with influence on insulin sensitivity. In vitro studies have shown that addition of DPP-4 is associated with impaired insulin signaling in primary human adipocytes, skeletal muscle, and smooth muscle cells (79), while conversely, silencing Dpp4 (using short interfering RNA) resulted in improved adipocyte insulin responsiveness (89). In both cases, the mechanism was suggested to involve the insulin receptor and downstream signaling pathways including protein kinase B (Akt), although how DPP-4 might influence this process was not clarified. The catalytic activity of the molecule was suggested to be, at least partially, involved (since insulin signaling was improved following inclusion of a DPP-4 inhibitor), but the DPP-4 substrate(s) mediating the effect were not identified. DPP-4 can also have signaling functions via interactions with the extracellular matrix (90), and it was therefore speculated that the DPP-4 inhibitor might have improved insulin sensitivity by interfering with this process in some way rather than by blocking the enzymatic activity per se (79, 89). However, DPP-4 can interact with the integral membrane protein, caveolin-1 (91), which is expressed in various cell types, including adipocytes (92), and may be upregulated in obesity (93). In adipocytes, caveolin-1 plays a role in the insulin transduction pathway by modulating the activity of down-stream signaling proteins including Akt (94), raising the possibility that DPP-4 may be able to influence adipocyte insulin sensitivity by modulating the activity of caveolin-1 (Figure 3).
FIGURE 3
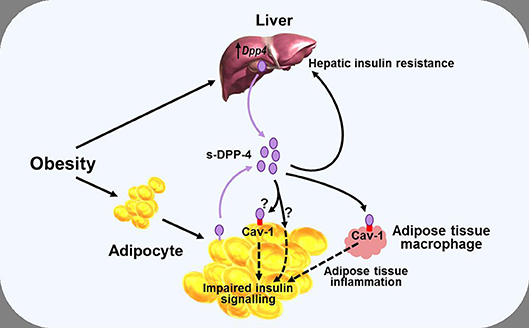
Figure 3. Diagram illustrating a potential role of DPP-4 as a mechanism linking obesity with inflammation and insulin resistance. Obesity is associated with increased levels of soluble DPP-4 (s-DPP-4), derived from up-regulated hepatocyte Dpp4 expression and larger adipocytes shedding more DPP-4 from the cell membrane. Increased levels of soluble DPP-4 result in adipocyte insulin resistance, possibly mediated via interactions with caveolin-1 (Cav-1) expressed on the cell surface of adipocytes themselves and on adipose tissue macrophages. Increased soluble DPP-4 levels are also associated with increased hepatic insulin resistance. Reduced insulin sensitivity in adipose and hepatic tissue may lead to hyperglycaemia, further exacerbating insulin resistance. See text for further details.
In an analogous manner, DPP-4 has been linked with hepatic insulin sensitivity in several studies. Thus, in mice, hepatocyte-specific overexpression of Dpp4 is associated with hepatic insulin resistance and liver steatosis (86), whereas knockdown of Dpp4 results in improved insulin sensitivity and reduced lipid accumulation in cultured hepatocytes (95). Yet other studies have pointed toward DPP-4 acting as a hepatokine, linking the liver and adipose tissue with the development of insulin resistance, and glucose intolerance. In mice, obesity and the associated visceral adipose tissue inflammation result in insulin resistance, a process which appears to be mediated via increased synthesis and release of hepatic DPP-4, since eliminating hepatocyte Dpp4 expression suppresses inflammation and improves insulin sensitivity (82, 83). The mechanism seems to be independent of the catalytic activity, since these effects were not mimicked by systemic DPP-4 inhibition (73, 82), and it was suggested that soluble DPP-4 interacted with caveolin-1 on adipose tissue macrophages, the down-stream effects of which combined with actions of other pro-inflammatory molecules such as factor Xa, to lead to activation of the inflammatory signaling pathway (83) (Figure 3). However, it has also been reported that although deletion of hepatocyte Dpp4 expression abolished the obesity-induced rise in circulating DPP-4 and was associated with reduced levels of proinflammatory cytokines in hepatocytes and adipose tissue, levels of circulating soluble DPP-4 per se did not correlate with tissue or systemic inflammation (82). Accordingly, the role of hepatocyte DPP-4 and the precise relationship between soluble DPP-4 and inflammation is complex, and the mechanisms linking hepatocyte Dpp4 expression with inflammation remain to be clarified.
Nevertheless, taken together, these studies indicate that DPP-4 may be able to influence fat and liver biology via both direct and indirect pathways, and suggest, therefore, that metabolic control could be perturbed if DPP-4 levels change. As discussed above, DPP-4 levels are positively correlated with BMI and insulin resistance and, in some studies, also to glycaemic control, but the mechanisms regulating synthesis and release of DPP-4 are still incompletely understood. In adipose tissue, Dpp4 expression is related to adipocyte size, and both insulin and the inflammatory cytokine TNF-α increase shedding of soluble DPP-4 from the membrane (79). Weight gain is also associated with greater Dpp4 expression in the liver of mice prone to diet-induced obesity, independently of the degree of intra-hepatic fat accumulation (87). Furthermore, hepatocyte Dpp4 expression is increased following exposure to high glucose (although not to insulin) (85), with weight gain further enhancing glucose-induced transcription of hepatic Dpp4 (87). Accordingly, excess DPP-4, derived from adipocytes and/or hepatocytes, may act as a local mediator of inflammation and adipose/hepatic tissue insulin resistance, thereby forming one link between obesity and the pathogenesis of T2DM and metabolic disease (Figure 3).
Pancreatic DPP-4
As well as influencing glucose homeostasis through its more widespread systemic effects, as discussed above, there is some evidence to suggest that DPP-4 may have local actions in the endocrine pancreas. Reports that DPP-4 may be associated with glucagon in the secretory granules of the pancreatic α-cell first emerged in 1993, when positive staining in pig islets was shown using immunohistochemical and enzyme histochemical techniques (96), but the pancreatic localization of DPP-4 has also been unequivocally demonstrated more recently using molecular biological techniques (97–99). It has also been suggested that DPP-4 might be secreted into the interstitial spaces within the islets (100) and, accordingly, DPP-4 activity can be measured in the media of human islet incubations (101), but whether this is simple shedding of membrane-associated DPP-4 or regulated secretion is not known. There appear to be species differences in pancreatic Dpp4 expression; it is associated predominantly with the β-cell in rodents but with the α-cell in pig islets (102). The situation in human islets is less clear. The majority of studies have found DPP-4 to be associated primarily with the α-cell (97, 99, 102, 103), and to be present in only a subset of human β-cells (99), although one study reported that it was the β-cell which was the predominant site of expression (98). Moreover, its expression appears to be influenced by metabolic stress, with it being increased in islets from diet-induced obese mice compared to normal weight animals (104), but lower in human islets from diabetic compared to non-diabetic donors (98, 99, 104).
There is also some evidence that a local GLP-1 system may exist within the pancreas. Thus, several studies have demonstrated the presence of prohormone convertase 1/3 [PC1/3, the processing enzyme that cleaves proglucagon to generate GLP-1 (105)] and fully processed GLP-1 in rodent and human α-cells and islets in culture (106–110), and GLP-1 can be detected in the media, suggesting that it is secreted from the α-cells. Adult α-cells are not thought to produce much GLP-1, and a release of GLP-1 is barely detectable from the perfused pancreas of normal mice (111). Nevertheless, exposure to metabolic stress may be able to influence pancreatic GLP-1 expression. Thus, incubating human and rodent islets under high glucose conditions results in increased expression of PC1/3 and increased secretion of GLP-1 (101, 108–110), while exposure of α-cells to the proinflammatory cytokine, interleukin-6, also increased GLP-1 secretion (112). Moreover, the development of diabetes has been associated with greater pancreatic GLP-1 expression. Thus, the tissue content of GLP-1 is increased in islets from diabetic rodents (106, 110), and when cultured, islets obtained from human donors with T2DM or from diabetic rodents release more GLP-1 ex vivo than non-diabetic islets (98, 109, 113). It has, therefore, been suggested that metabolic stress may influence the pancreatic processing of proglucagon toward the production of GLP-1 as a compensatory response to help regulate glucose homeostasis and maintain β-cell function and survival (109, 113). In line with this, mice with an α-cell specific knock-down of PC1/3, resulting in significantly reduced islet GLP-1 content, were able to maintain normal glucose tolerance, suggesting that islet GLP-1 may not be essential and/or that it can be compensated for by intestinally derived GLP-1 under normal conditions. In contrast, when placed under metabolic stress with increased demand on the β-cell (induced by high fat feeding and a low dose of the β-cell toxin, streptozotocin), glucose tolerance did become impaired, supporting the view that paracrine GLP-1 may be required to maintain glucose homeostasis during adaptation to metabolic stress (114). On the other hand, it has also been suggested that pancreatic-derived GLP-1 may play a role even under normal circumstances. In proglucagon gene (Gcg) knockout mice, the GLP-1 receptor antagonist, exendin 9-39, led to impaired glucose tolerance when Gcg was reactivated in the pancreas, but not when intestinal Gcg was reactivated (115). This was taken to indicate that locally produced GLP-1 within the pancreas may be more important than its systemic effects for maintaining normal β-cell function. However, glucagon itself has recently been shown to be able to stimulate insulin secretion from the perfused mouse pancreas by a mechanism which appears to involve an interaction with GLP-1 receptors (since the effect was attenuated by exendin 9-39) (111), complicating assessment of the relative importance of any locally produced GLP-1.
Nevertheless, when taken together, these studies do raise the possibility that pancreatic DPP-4 may be able to influence islet function by modulating the activity of locally produced GLP-1. Accordingly, culturing human islets in the presence of a DPP-4 inhibitor resulted in increased levels of intact GLP-1 (98, 101, 104), increased insulin secretion (98, 99, 101, 104), and improved β-cell survival (99, 101). However, there is also a possibility that some of the effect may be independent of GLP-1, because DPP-4 inhibition still reduced β-cell apoptosis in human islets cultured in the presence of exendin 9-39 (99), suggesting that other mediators might be involved, but it was also suggested that an anti-inflammatory action could have played a role (99).
Collectively, the results discussed above could be interpreted to suggest that conditions likely to result in β-cell stress, such as obesity, hyperglycaemia, and inflammation, activate compensatory responses in an attempt to improve β-cell function and survival. In the α-cell, PC1/3 is up-regulated, resulting in increased processing of proglucagon to GLP-1 while DPP-4 is down-regulated in order to reduce degradation and enhance local intact GLP-1 concentrations. Whilst attractive as a theory, it should, nevertheless, be borne in mind that the majority of the studies cited above have been carried out in rodents or have examined isolated islets/α-cells under in vitro or ex vivo conditions, and whether a local pancreatic DPP-4/GLP-1 system plays an important role in normal human physiology or in the pharmacological effect of DPP-4 inhibitors is not clear.
DPP-4 Inhibitors
As mentioned above, DPP-4 inhibitors are now an established and successful class of oral anti-hyperglycaemic agents, and are firmly embedded in guidelines for treating T2DM patients without atherosclerotic cardiovascular (CV) disease (116, 117). They are all small molecules which act as reversible competitive inhibitors of the catalytic activity of DPP-4 and, when used at the appropriate therapeutic dose, are associated with at least 70% inhibition of plasma DPP-4 activity (35) (although the actual extent of inhibition in vivo is likely to be higher, due to dilution of the inhibitor in the ex vivo assay). As discussed, the anti-hyperglycaemic effects of DPP-4 inhibition are mediated via enhancing levels of endogenous DPP-4 substrates, since the inhibitors themselves do not possess any inherent glucose-lowering activity. Accordingly, the degree of DPP-4 inhibition attained in vivo is sufficient to increase circulating levels of intact (active) incretins by 2- to 4-fold and result in improved glycaemic control (118). The risk of hypoglycaemia is not increased because the glucose-lowering effects of DPP-4 inhibition are self-limiting. Thus, the enzyme cannot be inhibited by more than 100% and the actions of the incretin hormones are glucose-dependent (16), meaning that insulin secretion is only stimulated when glucose levels rise above fasting levels.
Despite the theoretical potential for multiple endogenous DPP-4 substrates to be affected and for the DPP-4 protein to have diverse actions, as discussed in the sections above, pooled safety analyses (119–123), and large cardiovascular (CV) safety outcome trials (124–127) with the individual drugs, which between them have included many thousands of patients (Table 2), have uniformly shown that DPP-4 inhibitors are well-tolerated and not generally associated with adverse effects.
TABLE 2
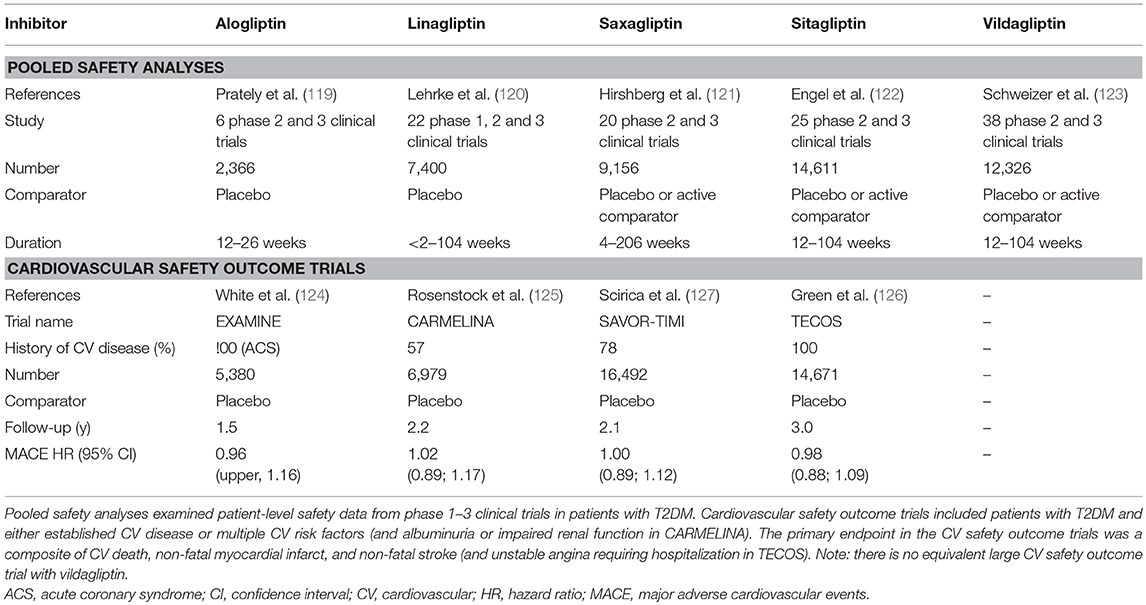
Table 2. Key published studies demonstrating the safety and tolerability of DPP-4 inhibitors.
There had been some initial questions over whether DPP-4 inhibition might compromise immune function, given the role of DPP-4/CD26 in the regulation of T-cell activity. However, neither the innate nor the adaptive immune response is affected by the DPP-4 inhibitors being used clinically (128), and reassuringly, there is no evidence for any increased risk of infections in the pooled safety analyses (119–123) or in the CV safety outcome trials (125–127), in line with the view that the catalytic activity of DPP-4 is not necessary for the co-stimulatory role of CD26 in T-cell activation. Similarly, these studies (Table 2) have not raised concern over increased cancer risk. However, in the CV safety outcome trials, a numerical imbalance in adjudicated cases of acute pancreatitis was noted (124–127, 129, 130), although the absolute risk was still very low. Thus, although the independent audit of all data carried out by regulatory authorities in the US and EU did not find evidence to support a causal relationship between incretin therapies and pancreatitis (131), it cannot be ruled out that a small increased risk of acute pancreatitis may exist in susceptible patients treated with DPP-4 inhibitors. The DPP-4 inhibitor CV safety outcome trials have unequivocally demonstrated that DPP-4 inhibition is not associated with any increased risk of major adverse CV events (MACE), with the four trials so far published (124–127) having hazard ratios (HR) for the primary outcome of between 0.96 and 1.02 (Table 2). Unexpectedly, hospitalization for heart failure was modestly increased in the SAVOR-TIMI trial with saxagliptin [3.5 vs. 2.8% for placebo, HR = 1.27 (127)]. Heart failure events were not increased in EXAMINE [alogliptin, HR = 1.07 (124)], TECOS [sitagliptin; HR = 1.00 (126)], or CARMELINA [linagliptin; HR = 0.90 (125)], even in those patients with previous heart failure (132–134), suggesting that the increased risk with saxagliptin is not mechanistically related to the inhibition of DPP-4 per se, but the cause is still unexplained. It has been suggested that it might simply be a chance finding or be related to the specific characteristics of the patients enrolled in the SAVOR-TIMI trial. However, it cannot be excluded that it could also be associated with some property of the saxagliptin molecule, unrelated to its action as a DPP-4 inhibitor. Accordingly, in vitro and ex vivo studies have shown that exposure to saxagliptin (135, 136) but not sitagliptin (136), results in altered cardiac electrophysiology and is associated with impaired cardiomyocyte contractility. The mechanism has been suggested to involve inhibition of cardiac intracellular signaling pathways (Ca2+/calmodulin-dependent protein kinase II and protein kinase C) due to saxagliptin-mediated inhibition DPP-9 (136). DPP-9 belongs to the same family as DPP-4, and it is known that saxagliptin is a less selective inhibitor, whereas sitagliptin is highly selective for DPP-4 (35) and does not alter DPP-9 activity (136). Nevertheless, although off-target inhibition of DPP-9 could be one potential explanation for the association between saxagliptin and heart failure in an experimental setting, it is uncertain whether mean exposure levels of saxagliptin reach a high enough level to affect DPP-9 activity when used at its therapeutic dose in humans.
Taken together, these pooled safety analyses (119–123) and CV outcome safety trials (124–127), together with review of published data from numerous clinical trials (39), provide reassuring evidence that the catalytic activity of the DPP-4 molecule can be modulated to enhance the beneficial metabolic effects of its substrates (such as the incretin hormones) without adversely affecting any of the protease-independent metabolic effects discussed above, or interfering with any of its other (non-metabolic) functions.
Conclusions
Once known primarily as a marker of activated T-cells and associated with immune regulation, signal transduction, and apoptosis, it is now more generally recognized that the actions of DPP-4 may be more widespread, and that it has the potential, either directly or indirectly, to influence metabolic processes. The catalytic activity of the molecule appears to be involved in at least some of these actions, and several endogenous substrates have been identified, although a role for protease-independent functions related to metabolic homeostasis has also been suggested. However, while the relative importance of enzymatic vs. non-enzymatic actions and the overall significance of DPP-4 for normal physiological regulation in humans and for the pathogenesis of metabolic disease are still largely unknown, targeting the catalytic activity has been harnessed pharmacologically, and DPP-4 inhibitors now play an important part in the therapy of T2DM.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
BMI, body mass index; DPP, dipeptidyl peptidase; DPL, DPP4-like protein; FAP, fibroblast activation protein; GIP, glucose-dependent insulinotropic polypeptide; GLP-1, glucagon-like peptide-1; QPP, quiescent cell proline dipeptidase; SDF-1α, Stromal cell-derived factor-1α T2DM, type 2 diabetes mellitus.
References
1. Mentlein R. Dipeptidyl-peptidase IV (CD26)–role in the inactivation of regulatory peptides. Regul Pept. (1999) 85:9–24. doi: 10.1016/S0167-0115(99)00089-0
2. Demuth HU, McIntosh CH, Pederson RA. Type 2 diabetes–therapy with dipeptidyl peptidase IV inhibitors. Biochim Biophys Acta (2005) 1751:33–44. doi: 10.1016/j.bbapap.2005.05.010
3. Yu DM, Yao TW, Chowdhury S, Nadvi NA, Osborne B, Church WB, et al. The dipeptidyl peptidase IV family in cancer and cell biology. FEBS J. (2010) 277:1126–44. doi: 10.1111/j.1742-4658.2009.07526.x
4. Lambeir AM, Durinx C, Scharpé S, De Meester I. Dipeptidyl-peptidase IV from bench to bedside: an update on structural properties, functions, and clinical aspects of the enzyme DPP IV. Crit Rev Clin Lab Sci. (2003) 40:209–94. doi: 10.1080/713609354
5. Ohnuma K, Dang NH, Morimoto C. Revisiting an old acquaintance: CD26 and its molecular mechanisms in T cell function. Trends Immunol. (2008) 29:295–301. doi: 10.1016/j.it.2008.02.010
6. Hühn J, Ehrlich S, Fleischer B, von Bonin A. Molecular analysis of CD26-mediated signal transduction in T cells. Immunol Lett. (2000) 72:127–32. doi: 10.1016/S0165-2478(00)00170-X
7. Vora KA, Porter G, Peng R, Cui Y, Pryor K, Eiermann G, et al. Genetic ablation or pharmacological blockade of dipeptidyl peptidase IV does not impact T cell-dependent immune responses. BMC Immunol. (2009) 10:19. doi: 10.1186/1471-2172-10-19
8. Bell GI, Santerre RF, Mullenbach GT. Hamster preproglucagon contains the sequence of glucagon and two related peptides. Nature (1983) 302:716–8. doi: 10.1038/302716a0
9. Ørskov C, Holst JJ, Knuhtsen S, Baldissera FG, Poulsen SS, Nielsen OV. Glucagon-like peptides GLP-1 and GLP-2, predicted products of the proglucagon gene, are secreted separately from pig small intestine but not pancreas. Endocrinology (1986) 119:1467–75. doi: 10.1210/endo-119-4-1467
10. Mojsov S, Weir GC, Habener JF. Insulinotropin: glucagon-like peptide I (7-37) co-encoded in the glucagon gene is a potent stimulator of insulin release in the perfused rat pancreas. J Clin Invest. (1987) 79:616–9. doi: 10.1172/JCI112855
11. Holst JJ, Ørskov C, Nielsen OV, Schwartz TW. Truncated glucagon-like peptide 1, an insulin-releasing hormone from the distal gut. FEBS Lett. (1987) 211:169–74. doi: 10.1016/0014-5793(87)81430-8
12. Orskov C, Holst JJ, Nielsen OV. Effect of truncated glucagon-like peptide-1 [proglucagon-(78-107) amide] on endocrine secretion from pig pancreas, antrum, and nonantral stomach. Endocrinology (1988) 123:2009–13. doi: 10.1210/endo-123-4-2009
13. Kreymann B, Williams G, Ghatei MA, Bloom SR. Glucagon-like peptide-1 7-36, a physiological incretin in man. Lancet (1987) 2:1300–4. doi: 10.1016/S0140-6736(87)91194-9
14. Nathan DM, Schreiber E, Fogel H, Mojsov S, Habener JF. Insulinotropic action of glucagonlike peptide-I-(7-37) in diabetic and nondiabetic subjects. Diabetes Care (1992) 15:270–6. doi: 10.2337/diacare.15.2.270
15. Gutniak M, Orskov C, Holst JJ, Ahrén B, Efendic S. Antidiabetogenic effect of glucagon-like peptide-1 (7-36)amide in normal subjects and patients with diabetes mellitus. N Engl J Med. (1992) 326:1316–22. doi: 10.1056/NEJM199205143262003
16. Nauck MA, Kleine N, Ørskov C, Holst JJ, Willms B, Creutzfeldt W. Normalization of fasting hyperglycemia by exogenous GLP-1 (7-36 amide) in type 2 diabetic patients. Diabetologia (1993) 36:741–4. doi: 10.1007/BF00401145
17. Nauck MA, Wollschläger D, Werner J, Holst JJ, Ørskov C, Creutzfeldt W, et al. Effects of subcutaneous glucagon-like peptide 1 (GLP-1 [7-36 amide]) in patients with NIDDM. Diabetologia (1996) 39:1546–53. doi: 10.1007/s001250050613
18. Mentlein R, Gallwitz B, Schmidt WE. Dipeptidyl-peptidase IV hydrolyses gastric inhibitory polypeptide, glucagon-like peptide-1(7-36)amide, peptide histidine methionine and is responsible for their degradation in human serum. Eur J Biochem. (1993) 214:829–35. doi: 10.1111/j.1432-1033.1993.tb17986.x
19. Deacon CF, Johnsen AH, Holst JJ. Degradation of glucagon-like peptide-1 by human plasma in vitro yields an N-terminally truncated peptide that is a major endogenous metabolite in vivo. J Clin Endocrinol Metab. (1995) 80:952–7.
20. Deacon CF, Nauck MA, Toft-Nielsen M, Pridal L, Willms B, Holst JJ. Both subcutaneously and intravenously administered glucagon-like peptide I are rapidly degraded from the NH2-terminus in type II diabetic patients and in healthy subjects. Diabetes (1995) 44:1126–31. doi: 10.2337/diab.44.9.1126
21. Kieffer TJ, McIntosh CH, Pederson RA. Degradation of glucose-dependent insulinotropic polypeptide and truncated glucagon-like peptide 1 in vitro and in vivo by dipeptidyl peptidase IV. Endocrinology (1995) 136:3585–96. doi: 10.1210/endo.136.8.7628397
22. Carr RD. Drug development from the bench to the pharmacy: with special reference to dipeptidyl peptidase-4 inhibitor development. Diabet Med. (2016) 33:718–22. doi: 10.1111/dme.13066
23. Schön E, Born I, Demuth HU, Faust J, Neubert K, Steinmetzer T, et al. Dipeptidyl peptidase IV in the immune system. Effects of specific enzyme inhibitors on activity of dipeptidyl peptidase IV and proliferation of human lymphocytes. Biol Chem Hoppe Seyler (1991) 372:305–11. doi: 10.1515/bchm3.1991.372.1.305
24. Deacon CF, Hughes TE, Holst JJ. Dipeptidyl peptidase IV inhibition potentiates the insulinotropic effect of glucagon-like peptide 1 in the anesthetized pig. Diabetes (1998) 47:764–9. doi: 10.2337/diabetes.47.5.764
25. Hansen L, Deacon CF, Orskov C, Holst JJ. Glucagon-like peptide-1-(7-36)amide is transformed to glucagon-like peptide-1-(9-36)amide by dipeptidyl peptidase IV in the capillaries supplying the L cells of the porcine intestine. Endocrinology (1999) 140:5356–63. doi: 10.1210/endo.140.11.7143
26. Pederson RA, White HA, Schlenzig D, Pauly RP, McIntosh CH, Demuth HU. Improved glucose tolerance in Zucker fatty rats by oral administration of the dipeptidyl peptidase IV inhibitor isoleucine thiazolidide. Diabetes (1998) 47:1253–8. doi: 10.2337/diab.47.8.1253
27. Pauly RP, Demuth HU, Rosche F, Schmidt J, White HA, Lynn F, et al. Improved glucose tolerance in rats treated with the dipeptidyl peptidase IV (CD26) inhibitor Ile-thiazolidide. Metabolism (1999) 48:385–9. doi: 10.1016/S0026-0495(99)90090-2
28. Balkan B, Kwasnik L, Miserendino R, Holst JJ, Li X. Inhibition of dipeptidyl peptidase IV with NVP-DPP728 increases plasma GLP-1 (7-36 amide) concentrations and improves oral glucose tolerance in obese Zucker rats. Diabetologia (1999) 42:1324–31. doi: 10.1007/s001250051445
29. Ahrén B, Holst JJ, Mårtensson H, Balkan B. Improved glucose tolerance and insulin secretion by inhibition of dipeptidyl peptidase IV in mice. Eur J Pharmacol. (2000) 404:239–45. doi: 10.1016/S0014-2999(00)00600-2
30. Pospisilik JA, Stafford SG, Demuth HU, Brownsey R, Parkhouse W, Finegood DT, et al. Long-term treatment with the dipeptidyl peptidase IV inhibitor P32/98 causes sustained improvements in glucose tolerance, insulin sensitivity, hyperinsulinemia, and beta-cell glucose responsiveness in VDF (fa/fa) Zucker rats. Diabetes (2002) 51:943–50. doi: 10.2337/diabetes.51.4.943
31. Reimer MK, Holst JJ, Ahrén B. Long-term inhibition of dipeptidyl peptidase IV improves glucose tolerance and preserves islet function in mice. Eur J Endocrinol. (2002) 146:717–27. doi: 10.1530/eje.0.1460717
32. Ahrén B, Simonsson E, Larsson H, Landin-Olsson M, Torgeirsson H, Jansson PA, et al. Inhibition of dipeptidyl peptidase IV improves metabolic control over a 4-week study period in type 2 diabetes. Diabetes Care (2002) 25:869–75. doi: 10.2337/diacare.25.5.869
33. Ahrén B, Landin-Olsson M, Jansson PA, Svensson M, Holmes D, Schweizer A. Inhibition of dipeptidyl peptidase-4 reduces glycemia, sustains insulin levels, and reduces glucagon levels in type 2 diabetes. J Clin Endocrinol Metab. (2004) 89:2078–84. doi: 10.1210/jc.2003-031907
34. Ahrén B, Gomis R, Standl E, Mills D, Schweizer A. Twelve- and 52-week efficacy of the dipeptidyl peptidase IV inhibitor LAF237 in metformin-treated patients with type 2 diabetes. Diabetes Care (2004) 27:2874–80. doi: 10.2337/diacare.27.12.2874
35. Deacon CF, Lebovitz HE. Comparative review of dipeptidyl peptidase-4 inhibitors and sulphonylureas. Diabetes Obes Metab. (2016) 18:333–47. doi: 10.1111/dom.12610
36. Zhu L, Tamvakopoulos C, Xie D, Dragovic J, Shen X, Fenyk-Melody JE, et al. The role of dipeptidyl peptidase IV in the cleavage of glucagon family peptides: in vivo metabolism of pituitary adenylate cyclase activating polypeptide-(1-38). J Biol Chem. (2003) 278:22418–23. doi: 10.1074/jbc.M212355200
37. Mulvihill EE, Drucker DJ. Pharmacology, physiology, and mechanisms of action of dipeptidyl peptidase-4 inhibitors. Endocr Rev. (2014) 35:992–1019. doi: 10.1210/er.2014-1035
38. Deacon CF. Peptide degradation and the role of DPP-4 inhibitors in the treatment of type 2 diabetes. Peptides (2018) 100:150–7. doi: 10.1016/j.peptides.2017.10.011
39. Scheen AJ. The safety of gliptins: updated data in 2018. Expert Opin Drug Saf. (2018) 17:387–405. doi: 10.1080/14740338.2018.1444027
40. Deacon CF, Nauck MA, Meier J, Hücking K, Holst JJ. Degradation of endogenous and exogenous gastric inhibitory polypeptide in healthy and in type 2 diabetic subjects as revealed using a new assay for the intact peptide. J Clin Endocrinol Metab. (2000) 85:3575–81.
41. Deacon CF, Danielsen P, Klarskov L, Olesen M, Holst JJ. Dipeptidyl peptidase IV inhibition reduces the degradation and clearance of GIP and potentiates its insulinotropic and antihyperglycemic effects in anesthetized pigs. Diabetes (2001) 50:1588–97. doi: 10.2337/diabetes.50.7.1588
42. Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W. Preserved incretin activity of glucagon-like peptide 1 [7-36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Invest. (1993) 91:301–7. doi: 10.1172/JCI116186
43. Hansotia T, Baggio LL, Delmeire D, Hinke SA, Yamada Y, Tsukiyama K, et al. Double incretin receptor knockout (DIRKO) mice reveal an essential role for the enteroinsular axis in transducing the glucoregulatory actions of DPP-IV inhibitors. Diabetes (2004) 53:1326–35. doi: 10.2337/diabetes.53.5.1326
44. Aulinger BA, Bedorf A, Kutscherauer G, de Heer J, Holst JJ, Göke B, et al. Defining the role of GLP-1 in the enteroinsulinar axis in type 2 diabetes using DPP-4 inhibition and GLP-1 receptor blockade. Diabetes (2014) 63:1079–92. doi: 10.2337/db13-1455
45. Nauck MA, Kind J, Köthe LD, Holst JJ, Deacon CF, Broschag M, et al. Quantification of the contribution of GLP-1 to mediating insulinotropic effects of DPP-4 inhibition with vildagliptin in healthy subjects and patients with type 2 diabetes using exendin [9-39] as a GLP-1 receptor antagonist. Diabetes (2016) 65:2440–7. doi: 10.2337/db16-0107
46. Højberg PV, Vilsbøll T, Rabøl R, Knop FK, Bache M, Krarup T, et al. Four weeks of near-normalisation of blood glucose improves the insulin response to glucagon-like peptide-1 and glucose-dependent insulinotropic polypeptide in patients with type 2 diabetes. Diabetologia (2009) 52:199–207. doi: 10.1007/s00125-008-1195-5
47. Aaboe K, Akram S, Deacon CF, Holst JJ, Madsbad S, Krarup T. Restoration of the insulinotropic effect of glucose-dependent insulinotropic polypeptide contributes to the antidiabetic effect of dipeptidyl peptidase-4 inhibitors. Diabetes Obes Metab. (2015) 17:74–81. doi: 10.1111/dom.12395
48. Xu G, Kaneto H, Laybutt DR, Duvivier-Kali VF, Trivedi N, Suzuma K, et al. Downregulation of GLP-1 and GIP receptor expression by hyperglycemia: possible contribution to impaired incretin effects in diabetes. Diabetes (2007) 56:1551–8. doi: 10.2337/db06-1033
49. Hansen KB, Vilsbøll T, Bagger JI, Holst JJ, Knop FK. Impaired incretin-induced amplification of insulin secretion after glucose homeostatic dysregulation in healthy subjects. J Clin Endocrinol Metab. (2012) 97:1363–70. doi: 10.1210/jc.2011-2594
50. Gasbjerg LS, Christensen MB, Hartmann B, Lanng AR, Sparre-Ulrich AH, Gabe MBN, et al. GIP(3-30)NH2 is an efficacious GIP receptor antagonist in humans: a randomised, double-blinded, placebo-controlled, crossover study. Diabetologia (2018) 61:413–23. doi: 10.1007/s00125-017-4447-4
51. Grandt D, Schimiczek M, Beglinger C, Layer P, Goebell H, Eysselein VE, et al. Two molecular forms of peptide YY (PYY) are abundant in human blood: characterization of a radioimmunoassay recognizing PYY 1-36 and PYY 3-36. Regul Pept. (1994) 51:151–9. doi: 10.1016/0167-0115(94)90204-6
52. Aaboe K, Knop FK, Vilsbøll T, Deacon CF, Holst JJ, Madsbad S, et al. Twelve weeks treatment with the DPP-4 inhibitor, sitagliptin, prevents degradation of peptide YY and improves glucose and non-glucose induced insulin secretion in patients with type 2 diabetes mellitus. Diabetes Obes Metab. (2010) 12:323–33. doi: 10.1111/j.1463-1326.2009.01167.x
53. Ahrén B, Larsson H. Peptide YY does not inhibit glucose-stimulated insulin secretion in humans. Eur J Endocrinol. (1996) 134:362–5. doi: 10.1530/eje.0.1340362
54. Chen CH, Stephens RL, Rogers RC. PYY and NPY: control of gastric motility via action on Y1 and Y2 receptors in the DVC. Neurogastroenterol Motil. (1997) 9:109–16. doi: 10.1046/j.1365-2982.1997.d01-26.x
55. Holst JJ, Albrechtsen NJW, Gabe MBN, Rosenkilde MM. Oxyntomodulin: actions and role in diabetes. Peptides (2018) 100:48–53. doi: 10.1016/j.peptides.2017.09.018
56. Druce MR, Minnion JS, Field BC, Patel SR, Shillito JC, Tilby M, et al. Investigation of structure-activity relationships of Oxyntomodulin (Oxm) using Oxm analogs. Endocrinology (2009) 150:1712–22. doi: 10.1210/en.2008-0828
57. Liu YL, Ford HE, Druce MR, Minnion JS, Field BC, Shillito JC, et al. Subcutaneous oxyntomodulin analogue administration reduces body weight in lean and obese rodents. Int J Obes. (2010) 34:1715–25. doi: 10.1038/ijo.2010.110
58. Bianchi E, Carrington PE, Ingallinella P, Finotto M, Santoprete A, Petrov A, et al. A PEGylated analog of the gut hormone oxyntomodulin with long-lasting antihyperglycemic, insulinotropic and anorexigenic activity. Bioorg Med Chem. (2013) 21:7064–73. doi: 10.1016/j.bmc.2013.09.016
59. Liu Z, Habener JF. Stromal cell-derived factor-1 promotes survival of pancreatic beta cells by the stabilisation of beta-catenin and activation of transcription factor 7-like 2 (TCF7L2). Diabetologia (2009) 52:1589–98. doi: 10.1007/s00125-009-1384-x
60. Liu Z, Stanojevic V, Avadhani S, Yano T, Habener JF. Stromal cell-derived factor-1 (SDF-1)/chemokine (C-X-C motif) receptor 4 (CXCR4) axis activation induces intra-islet glucagon-like peptide-1 (GLP-1) production and enhances beta cell survival. Diabetologia (2011) 54:2067–76. doi: 10.1007/s00125-011-2181-x
61. Yano T, Liu Z, Donovan J, Thomas MK, Habener JF. Stromal cell derived factor-1 (SDF-1)/CXCL12 attenuates diabetes in mice and promotes pancreatic beta-cell survival by activation of the prosurvival kinase Akt. Diabetes (2007) 56:2946–57. doi: 10.2337/db07-0291
62. Zaruba MM, Theiss HD, Vallaster M, Mehl U, Brunner S, David R, et al. Synergy between CD26/DPP-IV inhibition and G-CSF improves cardiac function after acute myocardial infarction. Cell Stem Cell (2009) 4:313–23. doi: 10.1016/j.stem.2009.02.013
63. Wang W, Choi BK, Li W, Lao Z, Lee AY, Souza SC, et al. Quantification of intact and truncated stromal cell-derived factor-1α in circulation by immunoaffinity enrichment and tandem mass spectrometry. J Am Soc Mass Spectr. (2014) 25:614–25. doi: 10.1007/s13361-013-0822-7
64. Lovshin JA, Rajasekeran H, Lytvyn Y, Lovblom LE, Khan S, Alemu R, et al. Dipeptidyl peptidase 4 inhibition stimulates distal tubular natriuresis and increases in circulating SDF-1α1-67 in patients with type 2 diabetes. Diabetes Care (2017) 40:1073–81. doi: 10.2337/dc17-0061
65. Shioda T, Kato H, Ohnishi Y, Tashiro K, Ikegawa M, Nakayama EE, et al. Anti-HIV-1 and chemotactic activities of human stromal cell-derived factor 1α (SDF-1α) and SDF-1β are abolished by CD26/dipeptidyl peptidase IV-mediated cleavage. Proc Natl Acad Sci USA. (1998) 95:6331–6. doi: 10.1073/pnas.95.11.6331
66. Fadini GP, Boscaro E, Albiero M, Menegazzo L, Frison V, de Kreutzenberg S, et al. The oral dipeptidyl peptidase-4 inhibitor sitagliptin increases circulating endothelial progenitor cells in patients with type 2 diabetes: possible role of stromal-derived factor-1alpha. Diabetes Care (2010) 33:1607–9. doi: 10.2337/dc10-0187
67. Korosi J, McIntosh CH, Pederson RA, Demuth HU, Habener JF, Gingerich R, et al. Effect of aging and diabetes on the enteroinsular axis. J Gerontol A Biol Sci Med Sci. (2001) 56:M575–9. doi: 10.1093/gerona/56.9.M575
68. Mannucci E, Pala L, Ciani S, Bardini G, Pezzatini A, Sposato I, et al. Hyperglycaemia increases dipeptidyl peptidase IV activity in diabetes mellitus. Diabetologia (2005) 48:1168–72. doi: 10.1007/s00125-005-1749-8
69. Pala L, Ciani S, Dicembrini I, Bardini G, Cresci B, Pezzatini A, et al. Relationship between GLP-1 levels and dipeptidyl peptidase-4 activity in different glucose tolerance conditions. Diabet Med. (2010) 27:691–5. doi: 10.1111/j.1464-5491.2010.03010.x
70. Meneilly GS, Demuth HU, McIntosh CH, Pederson RA. Effect of ageing and diabetes on glucose-dependent insulinotropic polypeptide and dipeptidyl peptidase IV responses to oral glucose. Diabet Med. (2000) 17:346–50. doi: 10.1046/j.1464-5491.2000.00236.x
71. McKillop AM, Duffy NA, Lindsay JR, O'Harte FP, Bell PM, Flatt PR. Decreased dipeptidyl peptidase-IV activity and glucagon-like peptide-1 (7-36) amide degradation in type 2 diabetic subjects. Diabetes Res Clin Pract. (2008) 79:79–85. doi: 10.1016/j.diabres.2007.08.001
72. Ryskjaer J, Deacon CF, Carr RD, Krarup T, Madsbad S, Holst J, et al. Plasma dipeptidyl peptidase-IV activity in patients with type-2 diabetes mellitus correlates positively with HbAlc levels, but is not acutely affected by food intake. Eur J Endocrinol. (2006) 155:485–93. doi: 10.1530/eje.1.02221
73. Fadini GP, Albiero M, Menegazzo L, de Kreutzenberg SV, Avogaro A. The increased dipeptidyl peptidase-4 activity is not counteracted by optimized glucose control in type 2 diabetes, but is lower in metformin-treated patients. Diabetes Obes Metab. (2012) 14:518–22. doi: 10.1111/j.1463-1326.2011.01550.x
74. Deacon CF, Pridal L, Klarskov L, Olesen M, Holst JJ. Glucagon-like peptide 1 undergoes differential tissue-specific metabolism in the anesthetized pig. Am J Physiol. (1996) 271:E458–64.
75. Holst JJ, Deacon CF. Glucagon-like peptide-1 mediates the therapeutic actions of DPP-IV inhibitors. Diabetologia (2005) 48:612–5. doi: 10.1007/s00125-005-1705-7
76. Pala L, Mannucci E, Pezzatini A, Ciani S, Sardi J, Raimondi L, et al. Dipeptidyl peptidase-IV expression and activity in human glomerular endothelial cells. Biochem Biophys Res Commun. (2003) 310:28–31. doi: 10.1016/j.bbrc.2003.08.111
77. Pala L, Pezzatini A, Dicembrini I, Ciani S, Gelmini S, Vannelli BG, et al. Different modulation of dipeptidyl peptidase-4 activity between microvascular and macrovascular human endothelial cells. Acta Diabetol. (2012) 49 (Suppl. 1):S59–63. doi: 10.1007/s00592-010-0195-3
78. Lugari R, Dei Cas A, Ugolotti D, Barilli AL, Camellini C, Ganzerla GC, et al. Glucagon-like peptide 1 (GLP-1) secretion and plasma dipeptidyl peptidase IV (DPP-IV) activity in morbidly obese patients undergoing biliopancreatic diversion. Horm Metab Res. (2004) 36:111–5. doi: 10.1055/s-2004-814222
79. Lamers D, Famulla S, Wronkowitz N, Hartwig S, Lehr S, Ouwens DM, et al. Dipeptidyl peptidase 4 is a novel adipokine potentially linking obesity to the metabolic syndrome. Diabetes (2011) 60:1917–25. doi: 10.2337/db10-1707
80. Kirino Y, Sei M, Kawazoe K, Minakuchi K, Sato Y. Plasma dipeptidyl peptidase 4 activity correlates with body mass index and the plasma adiponectin concentration in healthy young people. Endocr J. (2012) 59:949–53. doi: 10.1507/endocrj.EJ12-0158
81. Sell H, Blüher M, Klöting N, Schlich R, Willems M, Ruppe F, et al. Adipose dipeptidyl peptidase-4 and obesity: correlation with insulin resistance and depot-specific release from adipose tissue in vivo and in vitro. Diabetes Care (2013) 36:4083–90. doi: 10.2337/dc13-0496
82. Varin EM, Mulvihill EE, Beaudry JL, Pujadas G, Fuchs S, Tanti JF, et al. Circulating levels of soluble dipeptidyl peptidase-4 are dissociated from inflammation and induced by enzymatic DPP4 inhibition. Cell Metab. (2018) 29:320–34. doi: 10.1016/j.cmet.2018.10.001
83. Ghorpade DS, Ozcan L, Zheng Z, Nicoloro SM, Shen Y, Chen E, et al. Hepatocyte-secreted DPP4 in obesity promotes adipose inflammation and insulin resistance. Nature (2018) 555:673–7. doi: 10.1038/nature26138
84. Firneisz G, Varga T, Lengyel G, Fehér J, Ghyczy D, Wichmann B, et al. Serum dipeptidyl peptidase-4 activity in insulin resistant patients with non-alcoholic fatty liver disease: a novel liver disease biomarker. PLoS ONE (2010) 5:e12226. doi: 10.1371/journal.pone.0012226
85. Miyazaki M, Kato M, Tanaka K, Tanaka M, Kohjima M, Nakamura K, et al. Increased hepatic expression of dipeptidyl peptidase-4 in non-alcoholic fatty liver disease and its association with insulin resistance and glucose metabolism. Mol Med Rep. (2012) 5:729–33. doi: 10.3892/mmr.2011.707
86. Baumeier C, Schlüter L, Saussenthaler S, Laeger T, Rödiger M, Alaze SA, et al. Elevated hepatic DPP4 activity promotes insulin resistance and non-alcoholic fatty liver disease. Mol Metab. (2017) 6:1254–63. doi: 10.1016/j.molmet.2017.07.016
87. Baumeier C, Saussenthaler S, Kammel A, Jähnert M, Schlüter L, Hesse D, et al. Hepatic DPP4 DNA methylation associates with fatty liver. Diabetes (2017) 66:25–35. doi: 10.2337/db15-1716
88. Ahmed RH, Huri HZ, Muniandy S, Al-Hamodi Z, Al-Absi B, Alsalahi A, et al. Altered circulating concentrations of active glucagon-like peptide (GLP-1) and dipeptidyl peptidase 4 (DPP4) in obese subjects and their association with insulin resistance. Clin Biochem. (2017) 50:746–9. doi: 10.1016/j.clinbiochem.2017.03.008
89. Röhrborn D, Brückner J, Sell H, Eckel J. Reduced DPP4 activity improves insulin signaling in primary human adipocytes. Biochem Biophys Res Commun. (2016) 471:348–54. doi: 10.1016/j.bbrc.2016.02.019
90. Gorrell MD. Dipeptidyl peptidase IV and related enzymes in cell biology and liver disorders. Clin Sci. (2005) 108:277–92. doi: 10.1042/CS20040302
91. Ohnuma K, Yamochi T, Uchiyama M, Nishibashi K, Yoshikawa N, Shimizu N, et al. CD26 up-regulates expression of CD86 on antigen-presenting cells by means of caveolin-1. Proc Natl Acad Sci USA. (2004) 101:14186–91. doi: 10.1073/pnas.0405266101
92. Cohen AW, Combs TP, Scherer PE, Lisanti MP. Role of caveolin and caveolae in insulin signaling and diabetes. Am J Physiol Endocrinol Metab. (2003) 285:E1151–60. doi: 10.1152/ajpendo.00324.2003
93. Catalán V, Gómez-Ambrosi J, Rodríguez A, Silva C, Rotellar F, Gil MJ, et al. Expression of caveolin-1 in human adipose tissue is upregulated in obesity and obesity-associated type 2 diabetes mellitus and related to inflammation. Clin Endocrinol. (2008) 68:213–9. doi: 10.1111/j.1365-2265.2007.03021.x
94. Palacios-Ortega S, Varela-Guruceaga M, Martínez JA, de Miguel C, Milagro FI. Effects of high glucose on caveolin-1 and insulin signaling in 3T3-L1 adipocytes. Adipocyte (2015) 5:65–80. doi: 10.1080/21623945.2015.1122856
95. Rufinatscha K, Radlinger B, Dobner J, Folie S, Bon C, Profanter E, et al. Dipeptidyl peptidase-4 impairs insulin signaling and promotes lipid accumulation in hepatocytes. Biochem Biophys Res Commun. (2017) 485:366–71. doi: 10.1016/j.bbrc.2017.02.071
96. Poulsen MD, Hansen GH, Dabelsteen E, Høyer PE, Norén O, Sjöström H. Dipeptidyl peptidase IV is sorted to the secretory granules in pancreatic islet A-cells. J Histochem Cytochem. (1993) 41:81–8. doi: 10.1177/41.1.8093256
97. Segerstolpe Å, Palasantza A, Eliasson P, Andersson EM, Andréasson AC, Sun X, et al. Single-cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metab. (2016) 24:593–607. doi: 10.1016/j.cmet.2016.08.020
98. Zhang Y, Wu M, Htun W, Dong EW, Mauvais-Jarvis F, Fonseca VA, et al. Differential effects of linagliptin on the function of human islets isolated from non-diabetic and diabetic donors. Sci Rep. (2017) 7:7964. doi: 10.1038/s41598-017-08271-9
99. Bugliani M, Syed F, Paula FMM, Omar BA, Suleiman M, Mossuto S, et al. DPP-4 is expressed in human pancreatic beta cells and its direct inhibition improves beta cell function and survival in type 2 diabetes. Mol Cell Endocrinol. (2018) 473:186–93. doi: 10.1016/j.mce.2018.01.019
100. Grondin G, Hooper NM, LeBel D. Specific localization of membrane dipeptidase and dipeptidyl peptidase IV in secretion granules of two different pancreatic islet cells. J Histochem Cytochem. (1999) 47:489–98. doi: 10.1177/002215549904700407
101. Shah P, Ardestani A, Dharmadhikari G, Laue S, Schumann DM, Kerr-Conte J, et al. The DPP-4 inhibitor linagliptin restores beta-cell function and survival in human isolated islets through GLP-1 stabilization. J Clin Endocrinol Metab. (2013) 98:E1163–72. doi: 10.1210/jc.2013-1029
102. Liu L, Omar B, Marchetti P, Ahrén B. Dipeptidyl peptidase-4 (DPP-4): Localization and activity in human and rodent islets. Biochem Biophys Res Commun. (2014) 453:398–404. doi: 10.1016/j.bbrc.2014.09.096
103. Augstein P, Naselli G, Loudovaris T, Hawthorne WJ, Campbell P, Bandala-Sanchez E, et al. Localization of dipeptidyl peptidase-4 (CD26) to human pancreatic ducts and islet alpha cells. Diabetes Res Clin Pract. (2015) 110:291–300. doi: 10.1016/j.diabres.2015.10.010
104. Omar BA, Liehua L, Yamada Y, Seino Y, Marchetti P, Ahrén B. Dipeptidyl peptidase 4 (DPP-4) is expressed in mouse and human islets and its activity is decreased in human islets from individuals with type 2 diabetes. Diabetologia (2014) 57:1876–83. doi: 10.1007/s00125-014-3299-4
105. Rouille Y, Martin S, Steiner DF. Differential processing of proglucagon by the subtilisin-like prohormone convertases PC2 and PC3 to generate either glucagon or glucagon-like peptide. J Biol Chem. (1995) 270:26488–96. doi: 10.1074/jbc.270.44.26488
106. Nie Y, Nakashima M, Brubaker PL, Li QL, Perfetti R, Jansen E, et al. Regulation of pancreatic PC1 and PC2 associated with increased glucagon-like peptide 1 in diabetic rats. J Clin Invest. (2000) 105:955–65. doi: 10.1172/JCI7456
107. Wideman RD, Covey SD, Webb GC, Drucker DJ, Kieffer TJ. A switch from prohormone convertase (PC)-2 to PC1/3 expression in transplanted alpha-cells is accompanied by differential processing of proglucagon and improved glucose homeostasis in mice. Diabetes (2007) 56:2744–52. doi: 10.2337/db07-0563
108. Whalley NM, Pritchard LE, Smith DM, White A. Processing of proglucagon to GLP-1 in pancreatic α-cells: is this a paracrine mechanism enabling GLP-1 to act on β-cells? J Endocrinol. (2011) 211:99–106. doi: 10.1530/JOE-11-0094
109. Marchetti P, Lupi R, Bugliani M, Kirkpatrick CL, Sebastiani G, Grieco FA, et al. A local glucagon-like peptide 1 (GLP-1) system in human pancreatic islets. Diabetologia (2012) 55:3262–72. doi: 10.1007/s00125-012-2716-9
110. Sancho V, Daniele G, Lucchesi D, Lupi R, Ciccarone A, Penno G, et al. Metabolic regulation of GLP-1 and PC1/3 in pancreatic α-cell line. PLoS ONE (2017) 12:e0187836. doi: 10.1371/journal.pone.0187836
111. Svendsen B, Larsen O, Gabe MBN, Christiansen CB, Rosenkilde MM, Drucker DJ, et al. Insulin secretion depends on intra-islet glucagon signaling. Cell Rep. (2018) 25:1127–34. doi: 10.1016/j.celrep.2018.10.018
112. Ellingsgaard H, Hauselmann I, Schuler B, Habib AM, Baggio LL, Meier DT, et al. Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nat Med. (2011) 17:1481–9. doi: 10.1038/nm.2513
113. Hansen AM, Bödvarsdottir TB, Nordestgaard DN, Heller RS, Gotfredsen CF, Maedler K, et al. Upregulation of alpha cell glucagon-like peptide 1 (GLP-1) in Psammomys obesus - an adaptive response to hyperglycaemia? Diabetologia (2011) 54:1379–87. doi: 10.1007/s00125-011-2080-1
114. Traub S, Meier DT, Schulze F, Dror E, Nordmann TM, Goetz N, et al. Pancreatic α cell-derived glucagon-related peptides are required for β cell adaptation and glucose homeostasis. Cell Rep. (2017) 18:3192–3203. doi: 10.1016/j.celrep.2017.03.005
115. Chambers AP, Sorrell JE, Haller A, Roelofs K, Hutch CR, Kim KS, et al. The role of pancreatic preproglucagon in glucose homeostasis in mice. Cell Metab. (2017) 25:927–34. doi: 10.1016/j.cmet.2017.02.008
116. Davies MJ, D'Alessio DA, Fradkin J, Kernan WN, Mathieu C, Mingrone G, et al. Management of hyperglycaemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia (2018) 61:2461–98. doi: 10.1007/s00125-018-4729-5
117. Garber AJ, Abrahamson MJ, Barzilay JI, Blonde L, Bloomgarden ZT, Bush MA, et al. Consensus statement by the american association of clinical endocrinologists and american college of endocrinology on the comprehensive type 2 diabetes management algorithm - 2018 executive summary. Endocr Pract. (2018) 24:91–120. doi: 10.4158/CS-2017-0153
118. He YL, Serra D, Wang Y, Campestrini J, Riviere GJ, Deacon CF, et al. Pharmacokinetics and pharmacodynamics of vildagliptin in patients with type 2 diabetes mellitus. Clin Pharmacokinet (2007) 46:577–88. doi: 10.2165/00003088-200746070-00003
119. Pratley RE, McCall T, Fleck PR, Wilson CA, Mekki Q. Alogliptin use in elderly people: a pooled analysis from phase 2 and 3 studies. J Am Geriatr Soc. (2009) 57:2011–9. doi: 10.1111/j.1532-5415.2009.02484.x
120. Lehrke M, Marx N, Patel S, Seck T, Crowe S, Cheng K, et al. Safety and tolerability of linagliptin in patients with type 2 diabetes: a comprehensive pooled analysis of 22 placebo-controlled studies. Clin Ther. (2014) 36:1130–46. doi: 10.1016/j.clinthera.2014.06.008
121. Hirshberg B, Parker A, Edelberg H, Donovan M, Iqbal N. Safety of saxagliptin: events of special interest in 9156 patients with type 2 diabetes mellitus. Diabetes Metab Res Rev. (2014) 30:556–69. doi: 10.1002/dmrr.2502
122. Engel SS, Round E, Golm GT, Kaufman KD, Goldstein BJ. Safety and tolerability of sitagliptin in type 2 diabetes: pooled analysis of 25 clinical studies. Diabetes Ther. (2013) 4:119–45. doi: 10.1007/s13300-013-0024-0
123. Schweizer A, Dejager S, Foley JE, Kothny W. Assessing the general safety and tolerability of vildagliptin: value of pooled analyses from a large safety database versus evaluation of individual studies. Vasc Health Risk Manag. (2011) 7:49–57. doi: 10.2147/VHRM.S16925
124. White WB, Cannon CP, Heller SR, Nissen SE, Bergenstal RM, Bakris GL, et al. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N Engl J Med. (2013) 369:1327–35. doi: 10.1056/NEJMoa1305889
125. Rosenstock J, Perkovic V, Johansen OE, Cooper ME, Kahn SE, Marx N, et al. Effect of linagliptin vs placebo on major cardiovascular events in adults with type 2 diabetes and high cardiovascular and renal risk: the CARMELINA randomized clinical trial. JAMA (2018). doi: 10.1001/jama.2018.18269. [Epub ahead of print].
126. Green JB, Bethel MA, Armstrong PW, Buse JB, Engel SS, Garg J, et al. Effect of sitagliptin on cardiovascular outcomes in type 2 diabetes. N Engl J Med. (2015) 373:232–42. doi: 10.1056/NEJMoa1501352
127. Scirica BM, Bhatt DL, Braunwald E, Steg PG, Davidson J, Hirshberg B, et al. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med. (2013) 369:1317–26. doi: 10.1056/NEJMoa1307684
128. Anz D, Kruger S, Haubner S, Rapp M, Bourquin C, Endres S. The dipeptidylpeptidase-IV inhibitors sitagliptin, vildagliptin and saxagliptin do not impair innate and adaptive immune responses. Diabetes Obes Metab. (2014) 16:569–72. doi: 10.1111/dom.12246
129. Raz I, Bhatt DL, Hirshberg B, Mosenzon O, Scirica BM, Umez-Eronini A, et al. Incidence of pancreatitis and pancreatic cancer in a randomized controlled multicenter trial (SAVOR-TIMI 53) of the dipeptidyl peptidase-4 inhibitor saxagliptin. Diabetes Care (2014) 37:2435–41. doi: 10.2337/dc13-2546
130. Buse JB, Bethel MA, Green JB, Stevens SR, Lokhnygina Y, Aschner P, et al. Pancreatic safety of sitagliptin in the TECOS study. Diabetes Care (2017) 40:164–70. doi: 10.2337/dc15-2780
131. Egan AG, Blind E, Dunder K, de Graeff PA, Hummer BT, Bourcier T, et al. Pancreatic safety of incretin-based drugs–FDA and EMA assessment. N Engl J Med. (2014) 370:794–7. doi: 10.1056/NEJMp1314078
132. Zannad F, Cannon CP, Cushman WC, Bakris GL, Menon V, Perez AT, et al. Heart failure and mortality outcomes in patients with type 2 diabetes taking alogliptin versus placebo in EXAMINE: a multicentre, randomised, double-blind trial. Lancet (2015) 385:2067–76. doi: 10.1016/S0140-6736(14)62225-X
133. McGuire DK, Van de Werf F, Armstrong PW, Standl E, Koglin J, Green JB, et al. Association between sitagliptin use and heart failure hospitalization and related outcomes in type 2 diabetes mellitus: secondary analysis of a randomized clinical trial. JAMA Cardiol. (2016) 1:126–35. doi: 10.1001/jamacardio.2016.0103
134. McGuire DK, Alexander JH, Johansen OE, Perkovic V, Rosenstock J, Cooper ME, et al. Linagliptin effects on heart failure and related outcomes in individuals with type 2 diabetes mellitus at high cardiovascular and renal risk in CARMELINA. Circulation (2019) 139:351–61. doi: 10.1161/CIRCULATIONAHA.118.038352
135. Koyani CN, Kolesnik E, Wölkart G, Shrestha N, Scheruebel S, Trummer C, et al. Dipeptidyl peptidase-4 independent cardiac dysfunction links saxagliptin to heart failure. Biochem Pharmacol. (2017) 145:64–80. doi: 10.1016/j.bcp.2017.08.021
Keywords: dipeptidyl peptidase-4, glucagon-like peptide-1, incretin, peptide degradation, therapy, type 2 diabetes
Citation: Deacon CF (2019) Physiology and Pharmacology of DPP-4 in Glucose Homeostasis and the Treatment of Type 2 Diabetes. Front. Endocrinol. 10:80. doi: 10.3389/fendo.2019.00080
Received: 03 December 2018; Accepted: 30 January 2019;
Published: 15 February 2019.
Edited by:
Pierre De Meyts, de Duve Institute, BelgiumReviewed by:
Zachary Bloomgarden, Icahn School of Medicine at Mount Sinai, United StatesBurkhard Göke, Medical School Hamburg, Germany
Copyright © 2019 Deacon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carolyn F. Deacon, deacon@sund.ku.dk