Oral microbiome mediated inflammation, a potential inductor of vascular diseases: a comprehensive review
 Diego F. Gualtero*†
Diego F. Gualtero*†  Gloria Inés Lafaurie*†
Gloria Inés Lafaurie*†  Diana Marcela Buitrago
Diana Marcela Buitrago  Yormaris Castillo
Yormaris Castillo  Paula Katherine Vargas-Sanchez
Paula Katherine Vargas-Sanchez  Diana Marcela Castillo
Diana Marcela Castillo
- Universidad El Bosque, Vicerrectoría de investigaciones, Facultad de Odontología, Unidad de Investigación Básica Oral-UIBO, Bogotá, Colombia
The dysbiosis of the oral microbiome and vascular translocation of the periodontopathic microorganism to peripheral blood can cause local and systemic extra-oral inflammation. Microorganisms associated with the subgingival biofilm are readily translocated to the peripheral circulation, generating bacteremia and endotoxemia, increasing the inflammation in the vascular endothelium and resulting in endothelial dysfunction. This review aimed to demonstrate how the dysbiosis of the oral microbiome and the translocation of oral pathogen-induced inflammation to peripheral blood may be linked to cardiovascular diseases (CVDs). The dysbiosis of the oral microbiome can regulate blood pressure and activate endothelial dysfunction. Similarly, the passage of periodontal microorganisms into the peripheral circulation and their virulence factors have been associated with a vascular compartment with a great capacity to activate endothelial cells, monocytes, macrophages, and plaquettes and increase interleukin and chemokine secretion, as well as oxidative stress. This inflammatory process is related to atherosclerosis, hypertension, thrombosis, and stroke. Therefore, oral diseases could be involved in CVDs via inflammation. The preclinic and clinical evidence suggests that periodontal disease increases the proinflammatory markers associated with endothelial dysfunction. Likewise, the evidence from clinical studies of periodontal treatment in the long term evidenced the reduction of these markers and improved overall health in patients with CVDs.
1. Introduction
The oral microbiota fulfills essential homeostasis functions in the body. An increase in systemic nitrite can improve nitric oxide (NO) concentrations via oral microbiome by its capacity to reduce dietary nitrates; the NO is a vasodilatory solid and anti-inflammatory molecule that maintains vascular homeostasis (1). The dysbiosis of the oral microbiome with increased anaerobic Gram-negative microorganisms, such as Porphyromonas gingivalis, displaces commensal microorganisms with a high nitrate-reduction capacity (NRC) (2). However, P. gingivalis cannot reduce nitrate, which is significantly affected by the high concentration of nitrates (3). In addition, the dysbiosis of the subgingival microbiome induces the innate immune response, which impacts the vascular endothelium (4). Periodontitis is a multifactorial disease characterized by an inflammatory response that may lead to endothelial dysfunction (5, 6). The microbiome dysbiosis and the bacteremia and endotoxemia caused by periodontopathic microorganisms have been reported after tooth brushing and periodontal treatment in individuals with periodontitis (7, 8). These microorganisms can increase proinflammatory activation at a distance and are found in the arteries of patients suffering from periodontitis with atherosclerosis and a high risk of CVD (9–11). However, some concerns have arisen, such as what are these doing here? Do they cause or potentiate vascular lesions? How are they accomplishing this? This review discusses these and other topics to demonstrate the role of oral microorganisms and their potential to cause or worsen CVDs.
2. Periodontal disease: a chronic inflammatory disease
Periodontitis is a multifactorial chronic inflammatory mediated by a biofilm dysbiosis that generates diverse grades of tissue destruction (12). Severe periodontitis affects more than 700 million people worldwide, with a global prevalence of 10.8% (13). However, less severe forms of the disease affect 12%–55% of the population, with the prevalence varying by population (14). Periodontitis is among the most prevalent diseases worldwide with local and systemic consequences (15). Periodontitis is characterized clinically by migrating the junctional epithelium, allowing proliferation and cell migration on an altered connective tissue substrate, and inflammatory tissular status generating the depth of the gingival sulcus forming periodontal pockets. In periodontitis, the subgingival biofilm in the gingival sulcus causes tissue destruction, which expands to the periodontal ligament and alveolar bone, causing protein degradation and proteoglycan reduction (16). The 2018 world workshop diagnostic criteria are based on a multidimensional system including stages in ranges from 1 to 4 and depend on the severity of the disease and the complexity of its treatment and rehabilitation, considering clinical attachment loss (the connective tissue of the root cementum), percentage of bone loss, pocket depth and teeth lost due to periodontal disease as critical factors. It also has a grading system that provides information on the progression based on risk factors such as smoking, and diabetes used as modifiers for progression risk (17, 18).
2.1. Dysbiosis of oral biofilm in periodontitis and their effect on the cardiovascular system
The human oral cavity and gastrointestinal tract harbor the most abundant microbiota (19). The oral microbiome contains up to 750 species of microorganisms, including bacteria and other important microorganisms such as archaea, protozoa, fungi, and viruses. These can grow on the tongue, buccal mucosa, tonsils, and palate or hard surfaces such as teeth or dental prostheses. In a healthy environment, there is a balance between these species, with stable diversity and composition. However, this balance is disrupted in dysbiosis, resulting in a composition with an increase of commensals and a reduction in beneficial microorganisms (20).
It has been reported that the nitrites generated in the metabolism in the eubiotic microbiome contribute to systemic health, stimulating the circulatory system related to cardiometabolic health (21). This link occurs because hemoglobin sequentially oxidizes NO in the blood to NO2− and NO3−. This NO3− concentrated in the salivary glands is mixed with nitrate ingested from the diet and reduced to nitrite NO2− by nitrate reductase enzymes oral bacteria. Salivary nitrite can be enzymatically reduced to NO, nitrous oxide (N2O), or dinitrogen (N2) in the mouth by denitrifying bacteria. Commensals such as Actinobacteria, including Actinomyces and Rothia; Firmicutes, such as Veillonella and Streptococcus; Bacteroidetes, such as Prevotella and some Proteobacteria that include the genus Neisseria and Haemophilus are the most critical microorganisms in eubiotic oral microbiome who improved the vasodilator function of the endothelium. The dysbiosis associated with periodontopathic microorganisms can move them and play an essential role in cardiovascular risk (21, 22).
Pathobionts such as P. gingivalis, which cause dysbiosis and alter the bacterial community's relative abundances, can deregulate the inflammatory response (23–25). Subgingival microorganisms induce immune cell infiltration and inflammatory mediators in the periodontium (26, 27). Inflammatory markers can pass passively from the periodontal tissue to the peripheral circulation. Higher systemic circulating inflammatory burden of CVD risks such as interleukin (IL)-1β, IL-8, IL-6 and tumor necrosis factor-alpha (TNF-α) are directly associated with the periodontitis incipient lesions in adolescents and established periodontitis in adults (28, 29). Higher levels of circulating inflammatory markers have also been linked to patients with atherosclerosis with periodontitis (30). However, periodontal treatment reduces these biomarkers, particularly in those with CVD (31). These findings imply that periodontitis can elicit a systemic immune response that is not restricted to the localized lesion (32) (Figure 1).
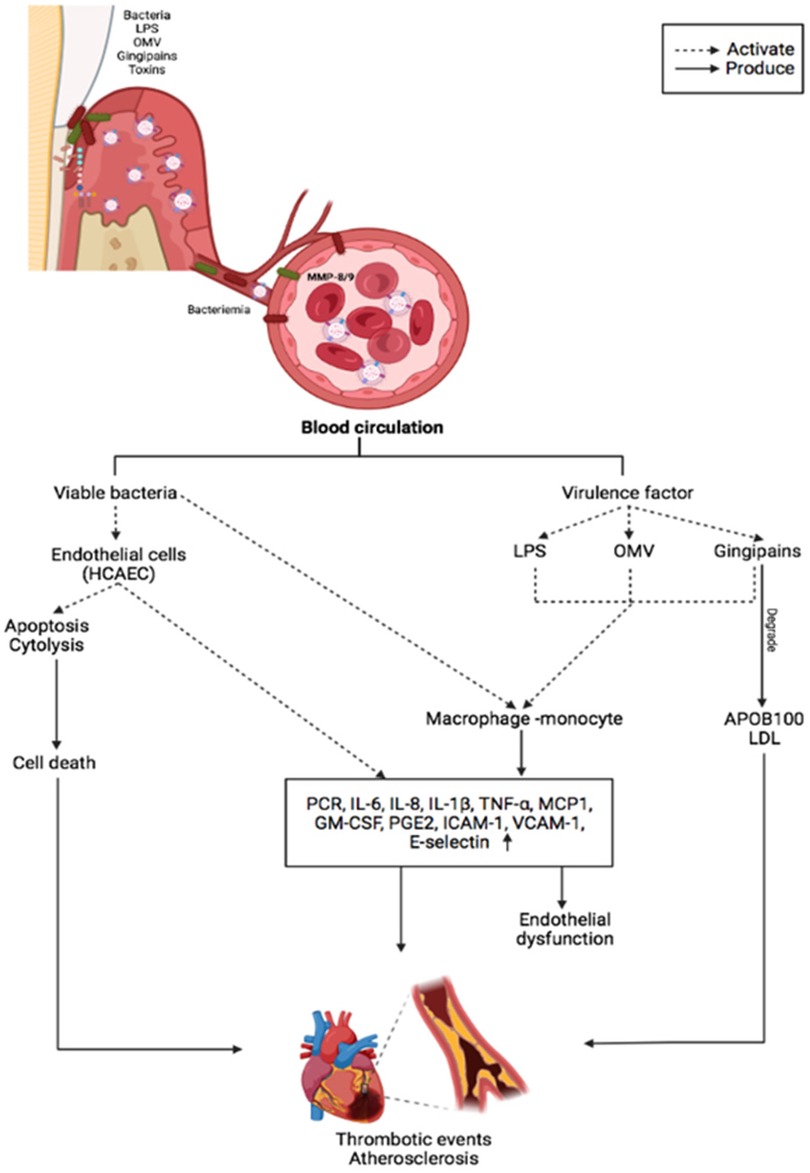
Figure 1. The main inflammatory effects of periodontopathogens and virulence factors on vascular endothelium and CVDs.
Porphyromonas species are proteolytic and degrade proteins and peptides into amino acids, which are degraded to produce short-chain fatty acids (SCFA), ammonia, sulfur compounds, and indole (22). Although SCFAs are considered metabolites that preserve endothelial function, this appears to be differential. Butyrate decreased Nlrp3 inflammasome activation in mice's carotid arterial wall after consuming a Western diet, while acetate-like propionate markedly enhanced Nlrp3 inflammasome generated an activation and carotid neointimal formation in mice in the carotid arteries (33). Due to the high metabolism of SACS in the oral microbiome, this mechanism has been proposed as another mechanism associated with increased cardiovascular risk in patients with periodontitis.
2.2. Atopobiosis of periodontopathic microorganisms and their effect on the cardiovascular system
Dysbiosis transforms the microbiome's composition into an inflammatory state, whereas atopobiosis can induce inflammation by translocating microorganisms to locations other than their usual location (34). Gut and oral dysbiosis have been linked to the pathogenesis of several immune-mediated inflammatory diseases (35, 36). The microorganisms will likely enter the bloodstream from oral niches via mechanisms that allow translocation and atopobiosis (20). Chewing, brushing, periodontal probing, scaling and root planning, and dental extractions can transfer the bacteria to capillaries and other small blood vessels, allowing bacteria to enter the systemic circulation (20, 37). P. gingivalis is the most frequent microorganism during oral bacteremia, especially in patients with periodontitis (8, 38). It is the most common microorganism in the amniotic fluid, placental tissues (39–41), brains, cerebrospinal fluid (42, 43), and vascular tissues (44, 45).
Other different routes of entry of oral organisms have been proposed, such as pathway phagocytes or dendritic cells. Many oral organisms in many tissues are related to these pathways. Periodontal pathogens such as P. gingivalis and F. nucleatum can achieve intracellular survival and disseminate to distant sites (46). P. gingivalis can invade several cell lines, including human umbilical vein endothelial cells, KB cells, and the human oral epidermoid cell line (47). P. gingivalis invasion of endothelial cells and phagocytic cells within the atheroma is also critical in atherosclerosis progression in mice (46, 48). During the invasion in endothelial cells, microvillus-like extensions are observed around bacteria, followed by the engulfment within vacuoles, using fimbriae and P. gingivalis proteases, which are essential for invasion; P. gingivalis invasion is considered an evasion mechanism of the host immune response (49).
P. gingivalis, can also colonize different oral mucosa cells, including the tongue, floor of the mouth, and buccal mucosa (50). P. gingivalis strain ATCC 33277 activates pro-survival phosphatidylinositol-3-kinase (PI3K)/protein kinase B (AKT) pathway in the primary oral epithelial cells for its effective colonization; the microorganism regulates the inflammatory response promoting host cell survival through the PI3K/Akt pathway (51). P. gingivalis is associated with developing oral cancers by upregulating IL-8 and MMPs 1 and 2 levels and increasing metalloproteinase-9 stimulated by gingipains and by the induction of the PI3K/AKT signaling along with the epithelial-mesenchymal transition (EMT) (51). F. nucleatum also invades colorectal cancer cells (CRC) using galactose sugars, l-arginine, neutralizing membrane protein antibodies, or fap2 deletion and mobilizes immune cells, increasing IL-8 and CXCL1 production in tumors to generate their progression (52). Gal-GalNAc is overexpressed in CRC and recognized by Fap2 of F. nucleatum, identifies the tumors in a Fap2-dependent manner, and uses the hematogenous to reach the colorectal tumors (53).
During bacteremia, the innate response serves as the first line of defense (54). In healthy individuals, transient bacteremia of dental origin is common and is cleared rapidly and efficiently within a few minutes to an hour (55). Because of the blood speed and bacterial size, it is practically impossible for leukocytes to recognize, trap, and destroy bacteria in the bloodstream (56). Periodontal bacteria can sequester leukocytes and erythrocytes, which bind through complement 3b (C3b) via complement receptor type 1 and spread from the oral mucosa to the vascular tissues. The erythrocytes destroy the bacteria by oxidizing their membranes, and the Kupffer cells in the liver and the splenic lymphoid tissue macrophages engulf and digest them, resulting in their clearance (57). However, erythrocytes play a fundamental role in bacterial clearance due to their abundance. Therefore, they are the only cells capable of clearing the trillions of bacteria that can enter the blood during bacteremia in minutes (56). The complement system is another mechanism that aids in bacterial clearance and is an essential component of the innate immune response and is the first line of defense against bacterial infections. Despite its lack of specificity, complement selectively recognizes foreign pathogens and damaged cells damaged by using recognition molecules from the classical and alternate pathways and lectin receptors. Massive bacteremia has been observed in knockout mice lacking C5 and C3 due to their inability to induce phagocytosis or oxidative burst (58).
Recently, the atopobiosis of oral microorganisms was assessed by sequencing bacterial DNA and comparing it to subgingival biofilm samples and coronary balloons. Microbial diversity differed significantly between the two environments. However, the bacteria translocate between periodontal pockets and coronary arteries, and 17 phylotypes were identical between atheroma and subgingival samples (59). In vascular tissues, oral bacteria invade and activate endothelial cells and promote the transmigration of leukocytes that may harbor intracellular bacteria that induce the atheroma lesion formation (20). Another systemic mechanism of periodontitis is the presence of endotoxemia (60). The release of proinflammatory cytokines via lipopolysaccharide (LPS), LPS-binding protein (LBP), CD14, and toll-like receptor (TLR) activation (61). LPS-LBP is a complex that plays a crucial role in the innate immune response to bacterial challenges in the systemic and local environment by activating proinflammatory cytokines (62).
3. Periodontopathogens and CVD risk
Studies indicate that the adverse cardiovascular effects are due to a few putative or high-risk bacteria: Aggregatibacter actinomycetemcomitans, Porphyromonas gingivalis, Tannerella forsythia, Treponema denticola or Fusobacterium nucleatum (63). P. gingivalis, and its virulence factors such as fimbriae (64, 65), LPS (66), gingipains (67), and outer membrane vesicles (OMVs) (68), A. actinomycetemcomitans with factors such as Leukotoxin (LtxA), cytolethal distending toxin (CDT), LPS and OMVs (69), and other microorganisms of dental biofilm as F. nucleatum, and Eikenella corrodens, and their virulence factors, migrate into the bloodstream and affect tissues and organs (7, 70, 71). Once in the systemic circulation, periodontal bacteria can be transported to distant sites, freely in the circulation or within circulating cells (36). Bacterial invasion can indirectly induce endothelial activation or generate endothelial dysfunction via systemic inflammation with increased acute-phase plasma proteins and proinflammatory cytokines. Furthermore, releasing bacterial products, such as OMVs, gingipains, or free soluble components, into circulation can induce proatherogenic responses in endothelial cells. Immune activation by the pathogen-derived GroEL heat shock protein (HSP) can also lead to an autoimmune response due to structural similarity between host HSP60 and GroEL (72, 73). F. nucleatum has been implicated in CVDs and is frequently detected in atherosclerotic plaques, as well as in a ruptured brain aneurysm (74). Genetic DNA from P. gingivalis and F. nucleatum in human aortic atherosclerotic lesions suggests a link between periodontal pathogens and CVDs (75).
OMVs also play a role in P. gingivalis-related systemic diseases. OMVs have been shown to activate Rho kinase in endothelial cells of umbilical veins via ERK1/2-and p38 mitogen-activated protein kinase, which may promote endothelial dysfunction and CVD (76). Preclinic experiments confirmed that P. gingivalis OMVs significantly increase vascular permeability. P. gingivalis can generate edema by altering the endothelial cell junctions such as platelet endothelial cell adhesion molecule-1 (77). The protease activity of P. gingivalis OMVs can cleave host proteins avoiding the immune response by degrading proinflammatory cytokines and disruption of the extracellular matrix, altering the tissue integrity, suggesting that these vesicles may play a vital role in the vascular damage (78). OMVs also promote vascular smooth muscle cell calcification via ERK1/2-RUNX2, inducing atherosclerosis (78, 79). In vitro, studies have demonstrated that OMVs from P. gingivalis can accelerate the foam cell formation in the murine macrophage and can induce the rupture of atherosclerotic plaque. P. gingivalis 381 also degrades fibrous caps and induce the matrix metalloproteinase (MMP)-9 activity in macrophages, which is involved in plaque disruption (80). In vitro, OMVs from P. gingivalis also induce platelet aggregation, which is essential in atherosclerotic plaque formation. In patients with periodontal disease, an increase in systemic inflammation, with elevated IL-1α and MMP-9 in plasma, causes a slight but significant decrease in cardiac function, dependent on MMP-9 (81). The proteolytic and oxidative activity of P. gingivalis also increased protein oxidation forming two apoB-100 N-terminal fragments that modified the LDL, which induce cell proliferation and could influence atherosclerosis (82). Likewise, the degradation of apoB-100 by Rgps gingipain plays a crucial role in promoting atherosclerosis by P. gingivalis infection (83) (Figure 1).
4. Endothelial dysfunction mediated by periodontopathic microorganisms
The vascular endothelium comprises monolayer endothelial cells that rest the intima formed by a matrix of proteoglycan. Endothelial cells perform various functions, including vascular tone, permeability, catabolic metabolism, SMC proliferation, angiogenesis, inflammation, white cell trafficking, fibrinolysis, thrombosis, and platelet activation (84). Therefore, the endothelial injury could alter their functionality, causing atherosclerosis (85). Furthermore, endothelial dysfunction can be caused by several factors, such as hyperlipidemia, oxidative stress, and inflammation due to bacteremia and endotoxemia (86).
Both periodontitis and CVDs are chronic inflammatory diseases. Biochemical and physiological analyses, including in vitro experiments, animal models, and clinical studies, establish a significant impact of periodontal pathogens, their virulence factors, and bacterial endotoxins on CVD mechanisms such as systemic inflammation, oxidative stress, endothelial dysfunction, foam cell formation, lipid accumulation, atherothrombosis, and vascular remodeling (6, 87). However, there is insufficient evidence to conclude that periodontitis causes CVDs. In this way, some studies have shown that periodontitis contributes to CVD through systemic bacterial exposure. For instance, the meta-analysis by Mustapha et al. demonstrated that elevated markers of systemic bacterial exposure, such as IgG to P. gingivalis, are associated with coronary heart diseases in periodontal patients (88).
In a population-based cohort study, Holtfreter et al. revealed a significant association between pocket probing depth and flow-mediated dilation of the brachial artery (6). Moreover, periodontal therapy has been shown to reduce inflammatory markers in CVDs. A previous meta-analysis demonstrated a significant weighted mean difference in CRP, proinflammatory cytokines, fibrinogen, total cholesterol, high-density lipoprotein-cholesterol, and improved endothelial function after periodontal therapy (32). In this context, we can speculate that periodontal pathogens promote endothelial dysfunction mechanisms to increase CVD risk.
Lipopolysaccharides (LPS) are biomolecules found in the outer membranes of anaerobic Gram-negative microorganisms composed of a lipidic (Lipid A) and a polysaccharide (O-antigen) region. Several studies have shown that Lipid A has heterogeneous structures; for example, some bacteria, such as Escherichia coli, have Hexa-acylated lipid A, whereas oral bacteria, such as P. gingivalis, have Penta- and Tetra-acylated lipid A (89, 90). The various structures of Lipid A from LPS are associated with multiple cell recognition and innate immune responses.
LPSs bind to TLRs in host cells, and several studies have shown that the classic structure of Hexa-acylated lipid A matches TLR-4, whereas Penta- and Tetra-acylated lipid A matches TLR-2. There are controversial studies on LPSs from P. gingivalis (LPS-Pg) and their TLRs; some studies show that LPS-Pg activates cells via TLR2, whereas others show that it activates cells via TLR4. Nonetheless, LPS-Pg heterogeneity enables recognition via Penta-acylated lipid A with TLR4 and Tetra-acylated lipid A with TLR2. LPSs from atherosclerosis-associated bacteria inhibit TLR4 and active endothelial cells via TLR2, resulting in a proinflammatory atherosclerotic response mediated by chemokines and cytokines (89, 90). Endothelial secretion of IL-8, monocyte chemoattractant protein-1 (MCP-1), IL-6, and TNF-α aids in leukocyte tracking and adhesion.
In bidimensional and three-dimensional models, human coronary artery endothelial cells (HCAECs) have low inflammatory responses to LPS-Pg (91). Nonetheless, when HCAECs were exposed to multiple doses of P. gingivalis, they produced significant amounts of MCP-1, IL-6and granulocyte-macrophage colony-stimulating factor (92). Other microorganisms, such as A. actinomycetemcomitans and E. corrodens, have a greater proinflammatory capacity than P. gingivalis. Treating HCAECs with A. actinomycetemcomitans and A.a-LPS IL-8 and IL-6 secretion increased via nuclear factor kappa beta-p65 (NF-κB p65) activation. However, this inflammatory response was inhibited by rosuvastatin due to the atheroprotective factor Kruppel-like factor 2 (93). A recent study demonstrated that A. actinomycetemcomitans infection increased transforming growth factor beta 1, IL-8, MCP-1, and IL-6 secretion in HCAECs in a dose-dependent manner in a 3D model. This proinflammatory response stimulated the THP-1 monocyte activation, adhesion, and migration (94). Similarly, E. corrodens-LPS activates HCAECs pathway TLR4, ERK, and NF-kB p65, inducing a pro-atherosclerotic endothelial response and monocyte adhesion (95). Previous evidence suggests that inflammation induced by oral pathogens and microorganisms may promote endothelial dysfunction and CVD progression (Table 1). A plausible mechanism for periodontopathogens' ability to induce endothelial dysfunction associated with CVDs and stroke is depicted in Figure 1.
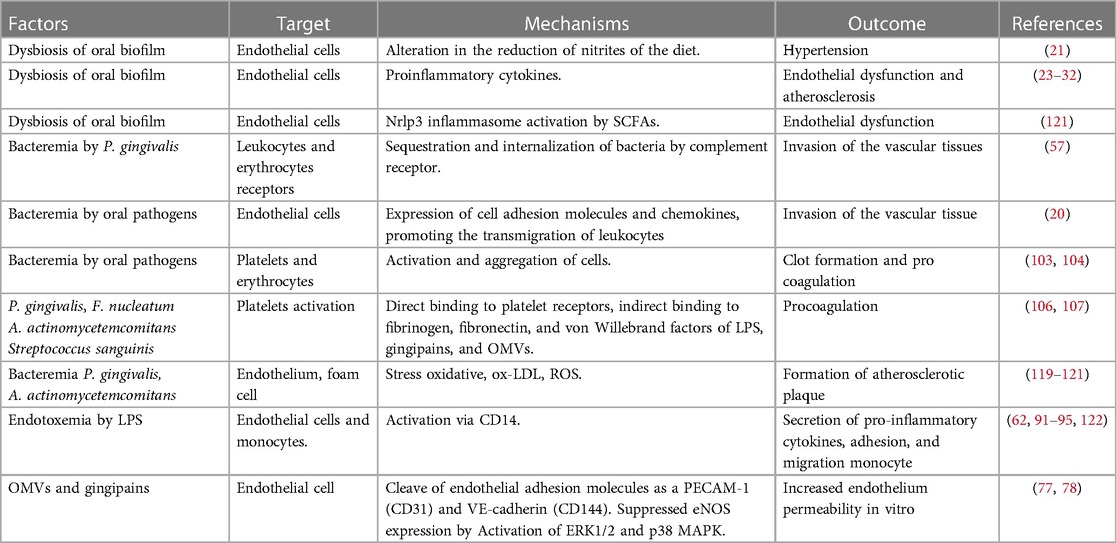
Table 1. Periodontopathogens factors related to endothelial dysfunction.
5. Macrophage–monocyte activation by periodontopathogens and cardiovascular risk
Macrophages play an active role as effectors and regulators in different phases of inflammation. Together with other cells, they are phagocytic cells that effectively modulate innate and adaptive immune responses, promoting inflammatory resolution and tissue healing (96). In periodontitis, macrophages secrete chemotactic and different cytokines such as RANTES (Regulated upon Activation, Normal T Cell Expressed and Presumably Secreted), MCP-1, macrophage inflammatory protein (MIP)-1α, MIP-3α, MIP-1β, and IL-8, which activate and migrate leukocytes, and proinflammatory cytokines such as IL-1β, TNF-α, and IL-6, which activate osteoclastogenesis processes and the degradation of periodontal tissue (97).
OMVs are essential virulence factors secreted into the external environment and serve as a generalized secretion and transport system for other virulence factors such as fimbriae, endotoxin as LPS, and enzymes, including gingipains. The role of this bacterium and its vesicles in the host immune response and tissue destruction by P. gingivalis infection has been well investigated. They have been shown in an in vitro model using the human macrophage cell line U937 the induction of proinflammatory cytokines associated with osteoclastogenesis, alveolar bone resorption, and tissue destruction (98, 99). In atherosclerosis-related endothelial dysfunction, monocytes migrate through the endothelium, differentiate into macrophages in the subendothelial tissue, and then convert to foam cells. LPSs from periodontal pathogenic bacteria induce acceleration in foam cell formation (100). LPSs from A. actinomycetemcomitans have been shown to synergistically interact.
LPSs play a role in developing atherosclerotic and CVD, including coronary artery disease, cerebrovascular disease, peripheral artery disease, and aortic atherosclerosis, increase TLR expression and respond to TLR agonists inducing significant inflammatory lesions (101). These roles and their plasticity make macrophages attractive targets for preventing and stabilizing existing atherosclerosis (102).
6. Prothrombotic state induced by periodontopathogens
Platelets play a role in various phases of the atherosclerotic process and vascular thrombosis leading to myocardial infarction. In periodontitis, low-grade systemic inflammation contributes to platelet activation, aggregation, and a procoagulant state (103, 104). Platelet activation is associated with the decrease in the release or inactivation of nitric oxide, as well as the release of platelet agonists. Platelet-derived substances, such as nitric oxide, cytokines, growth factors, chemokines, metalloproteinases, histamine, and selectins, actively participate in immune and inflammatory reactions (105, 106).
Epidemiological studies in patients with chronic periodontitis have revealed an increase in the mean platelet volume and platelet count, which has been linked to the production of cytokines such as IL-3 or IL-6, which in turn regulates megakaryocyte ploidy, producing more giant and reactive platelets (104, 105). Using in vitro studies, animal models, and clinical studies, P. gingivalis, F. nucleatum, and A. actinomycetemcomitans have been identified as the most commonly associated periodontopathogens with platelet activation and aggregation. The following mechanisms of pathogen–platelet interaction have been described: (1) direct bacterial binding to one of the platelet receptors; (2) indirect binding via other mediators such as fibrinogen, fibronectin, and von Willebrand factor (vWF); and (3) binding of bacterial products and toxins such as LPS, gingipains, and OMVs (80, 106–108).
Pathogens have been shown to express surface proteins, allowing them to bind directly to platelets. In this regard, P. gingivalis produces kazal-type (KSPI) serine proteasa and Arg-gingipains, RgpA, and RgpB, which, when bound to hemagglutinin/adhesion, stimulate platelets via Par-1 and Par-2, producing inositol trisphosphate and intracellular calcium downstream (107). Moreover, Naito et al. demonstrated that this microorganism could express the adhesin protein Hgp44, which can activate platelets by binding to glycoprotein (GP) Ib–IX–V, a vital platelet receptor essential in primary hemostasis due to its high affinity for vWF or the FcγRIIa receptor, a potent activator of hemostasis, followed by activated platelet elimination (108). Furthermore, S. sanguinis expresses highly glycosylated serine-rich protein A, which can bind to GPIbα (109), or indirectly induce platelet aggregation via the complement pathway. This interaction can aid in bacterial destruction, but it can also activate platelets inducing procoagulant factors such as the prothrombinase complex on the cell surface generating platelet–bacteria binding by complement molecules inducing an immunological mechanism rather than a hemostatic mechanism, highlighting platelets' dual role (110).
On the other hand, platelets can modulate the responses of other cells such as leukocytes and endothelial cells, via TLR2 and the GP ligand P-selectin/P-selectin (111). Platelets express TLR1, TLR2, TLR4, TLR6, TLR8, and TLR9, and the interaction of TLR2 and TLR4 with LPS from A. actinomycetemcomitans and P. gingivalis causes platelets to release cytokines such, as IL-1, TNF-α, and soluble CD40 ligand (sCD40l) (111, 112). Plasma sCD40l can predict recurrent cardiovascular events such as myocardial infarction and stroke, and arginine gingipains (RgpA and RgpB) of P. gingivalis can cause platelet activation and aggregation. Arg-ingipains enter the circulation and increase intracellular calcium levels in platelets by activating the protease-activated receptor-1 (PAR-1) and PAR-4 receptors, activate prothrombin, factor X and protein C, promoting thrombosis via thrombin release (113, 114). The vascular responses, upregulates endothelial cell adhesion molecule expression, and increases IL-1β, TNF-α, and thromboxane secretion, resulting in macrophage recruitment, aggregation, and platelet adhesion (114). Sharma et al. demonstrated that OMVs activate platelet aggregation. In contrast, fimbria is essential in bacterial adhesion to platelets via fibrinogen bridges that bind to the integrin receptor on the platelet surface. Most likely, this is what makes OMVs interact with platelet membrane receptors, which cause platelets to be stimulated and release dense and alpha granules and aggregate (80).
Platelets also link thrombosis and inflammation by producing platelet microparticles (PMPs). PMPs can release immunomodulatory factors such as RANTES, IL-1β, and CD40l. They can also modulate the activation of inflammatory cells such as neutrophils. In addition, bacterial infection with P. gingivalis induces PMP formation (112), so more studies are required to fully understand their role and association with periodontal pathogens. Studies have demonstrated that bacteria-induced platelet aggregation differs from that induced by conventional agonists. The above is recommended as a “binary” aggregation, which means that no aggregation is observed below a specific bacterial density, and aggregation is already maximal above that density (105). Therefore, it remains controversial. Several studies demonstrate that bacterial stimulation may not result in aggregation but rather in a more specific inflammatory response by activating the platelet TLR pathway (105).
We evaluated the effect of live-P. gingivalis W83 and live-A. Actinomycetemcomitans ATCC 29522 (106 CFU/kg) in an intraperitoneal infection model for six weeks in Wistar rats (ethical act No. 657), on platelet aggregation in platelet-rich plasma (PRP) using the spectrophotometric technique of aggregometry (115), against agonists adenosine diphosphate (ADP) (10 µM), collagen (10 µg/ml), arachidonic acid (AA) (150 µg/ml) and U46619 (2.10 µM thromboxane analog). The results demonstrate that after six weeks of treatment at the cumulative induction dose of live-P. gingivalis W83 and live-A. Actinomycetemcomitans on platelet aggregation in Wistar rats significantly increases platelet aggregation against agonists AA and U46619. In turn, it was shown that both periodontopathogens inhibit platelet aggregation in the presence of collagen (Table 2), which may be related to the “binary” effect of aggregation already described (105).
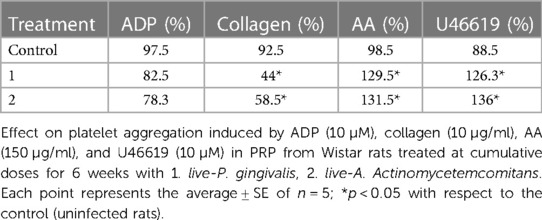
Table 2. Effect on platelet aggregation.
7. Endothelial oxidative stress
Oxidative stress and chronic inflammation are significant contributors to atherosclerosis. The oxidation of polyunsaturated fatty acids (PUFAs) by free radical lipid peroxidation (LPO) in lipoproteins to phospholipids and cholesterol esters forms a fatty streak in atherogenesis. LDL-containing PUFAs are prime targets for reactive oxygen species (ROS), resulting in non-enzymatic oxidation processes known as LPO (116–118). LPO can be induced by multiple endogenous factors such as lipoxygenases, cyclooxygenases, myeloperoxidases, NADPH oxidases, and ROS generated by the mitochondrial electron transport chain (118).
Vascular oxidative stress and inflammation can be induced by oral microorganisms such as P. gingivalis and A. actinomycetemcomitans (119). A. actinomycetemcomitans was used to assess LPO production in the mouse aorta (73). The increase in oxidized LDL (ox-LDL) levels, with an increase in NADPH expression or oxidative stress in the aorta of Aa-infected mice, suggests that A. actinomycetemcomitans plays a significant role in oxidative stress and LDL oxidation. In spontaneously hyperlipidemic mice with apolipoprotein E deficiency exposed to LPS and A. actinomycetemcomitans, ROS production depends on NOX (nicotinamide adenine dinucleotide phosphate oxidase) and myeloperoxidase activities, accelerating the formation of atherogenic plaques (118).
In P. gingivalis, ROS generation is dependent on the interaction of bacterial gingipain R with platelets and platelets with neutrophils, which promotes foam cell formation by increasing ox-LDL absorption (73). In addition, it contributes to the progression of atherosclerotic plaques by stimulating MCP-1 release in vascular endothelial cells via NOX-mediated superoxide anion production, followed by activation of multiple signaling pathways such as p38, c-Jun N-terminal kinase, and NF-κB (118). Recent studies have shown that P. gingivalis OMVs are systemic proinflammatory and prooxidant effectors of endothelial responses and that they upregulate proinflammatory cytokines such as TNF-α and IL-6. Their secretion has also been linked to uncontrolled oxidative stress via eNOS downregulation and iNOS upregulation, implying an uncoupling of NO synthase (120).
8. Association of periodontitis with CVDs
CVDs are the leading cause of death globally (121). CVDs are pathological disorders that damage the heart and blood vessels, including coronary heart disease, cerebrovascular disease, and peripheral artery disease associated with atherosclerosis (122). In 1989, Mattila et al. were the first to identify oral infection, especially periodontal disease, as an independent predictor of myocardial infarction risk. Since then, several observational studies on the association between periodontal disease and CVDs have been published (123). Thus far, several systematic reviews have demonstrated that periodontal disease has a minor association with CVDs, with odds ratios (ORs) ranging from 1.14 to 1.34 (124, 125). However, subgroup analyses revealed that exposure to infection for more than 15 years was associated with an increased OR of 1.67 (95% confidence interval (CI): 1.27–2.2) (124). Moreover, systemic bacterial exposure based on IgG antibodies against periodontal pathogens raises the risk to 1.75 (95% CI: 1.32–2.34) (88). When periodontitis or edentulism was used as a mortality indicator, there was an increased risk of all-cause mortality and CVD mortality. However, a higher risk of heart disease (RR: 2.58, 95% CI: 2.20–.03) and cerebrovascular diseases (RR: 3.11, 95% CI: 2.42–3.98) were detected (126). Likewise, patients with periodontitis and erectile dysfunction presented a higher number of cardiovascular adverse events adjusted by age and previous cardiovascular disease (127).
Due to the difficulty of conducting clinical trials to assess the impact of periodontal treatment, only a multicenter clinical trial known as the Periodontitis and Vascular Events (PAVE) was conducted as a pilot study (128). Although no differences in the number of cardiovascular events were observed between periodontally treated patients and control over 24 months, periodontal care exhibited a significant reduction in the percentage of individuals with elevated high-sensitivity CRP (hs-CRP) over 3 mg/L at 6 months (129). However, a larger sample size and a longer time frame are required to assess the effect of periodontal treatment on new cardiovascular events.
The relationship between inflammation and endothelial dysfunction has been widely demonstrated (130, 131). Several clinical studies later confirmed this hypothesis, revealing a critical epidemiological association between inflammatory markers such as CRP and acute ischemic events (132–134). CRP has been used as a universal marker to predict the risk of coronary heart disease in intermediate-risk individuals (135). The effects of hs-CRP on vascular risk are linear, with higher values above 3 mg/L predicting worse cardiovascular outcomes (135). Moreover, reducing this marker with anti-inflammatory drugs such as statins reduced the risk of coronary events in patients with high CRP (>2 mg/L) but normal cholesterol levels (136). CRP is produced in the liver in response to inflammatory molecules, especially IL-6, activated mainly by innate immune responses to infectious agents or secondary to systemic inflammatory diseases (135–137). However, hs-CRP is produced in the SMCs of the coronary artery due to endothelial dysfunction (138). Because CRP is not produced in the periodontal tissue, hs-CRP detected in the crevicular fluid is most likely of systemic origin (139). However, in patients with periodontitis, local proinflammatory cytokines and transient bacteremia overexpression may be associated with increased oxidative stress and CRP levels (140). Recently, Machado et al. showed that chronic and aggressive periodontitis is consistently associated with higher hs-CRP levels. Patients with aggressive periodontitis had hs-CRP levels more than 50% higher than patients with chronic periodontitis. Intensive nonsurgical periodontal treatment increased hs-CRP levels immediately, followed by a decrease after treatment. Non-intensive treatment reduced hs-CRP for up to 180 days. These findings indicate that periodontitis is associated with systemic inflammation using the serum hs-CRP levels as a marker (141).
Periodontitis also induces endothelial dysfunction in rats, as evidenced by reduced eNOS expression and increased iNOS and COX-2 expression (142, 143). In humans, Tonetti et al. compared patients with severe periodontitis who received intensive periodontal treatment (IPT) to a control group who received community-based treatment. Endothelial function was assessed using flow-mediated dilatation (FMD), and inflammatory, coagulation, and endothelial activation markers were assessed before and after treatment. FMD was higher in the IPT group than in the control group by the second month. There was a significant correlation between the change in FMD and improved periodontal status. However, few changes were observed in inflammatory markers 6 months after therapy (144). After 6 months of intervention, the effects of periodontal treatment on endothelial function in patients with a recent ST-segment elevation myocardial infarction were observed in individuals with severe periodontitis (145). Similarly, periodontal therapy improved endothelial function in patients with a recent myocardial infarction without causing adverse clinical events (146). Figure 2 illustrates the main inflammatory effects of periodontopathogens and virulence factors on vascular endothelium and CVDs.
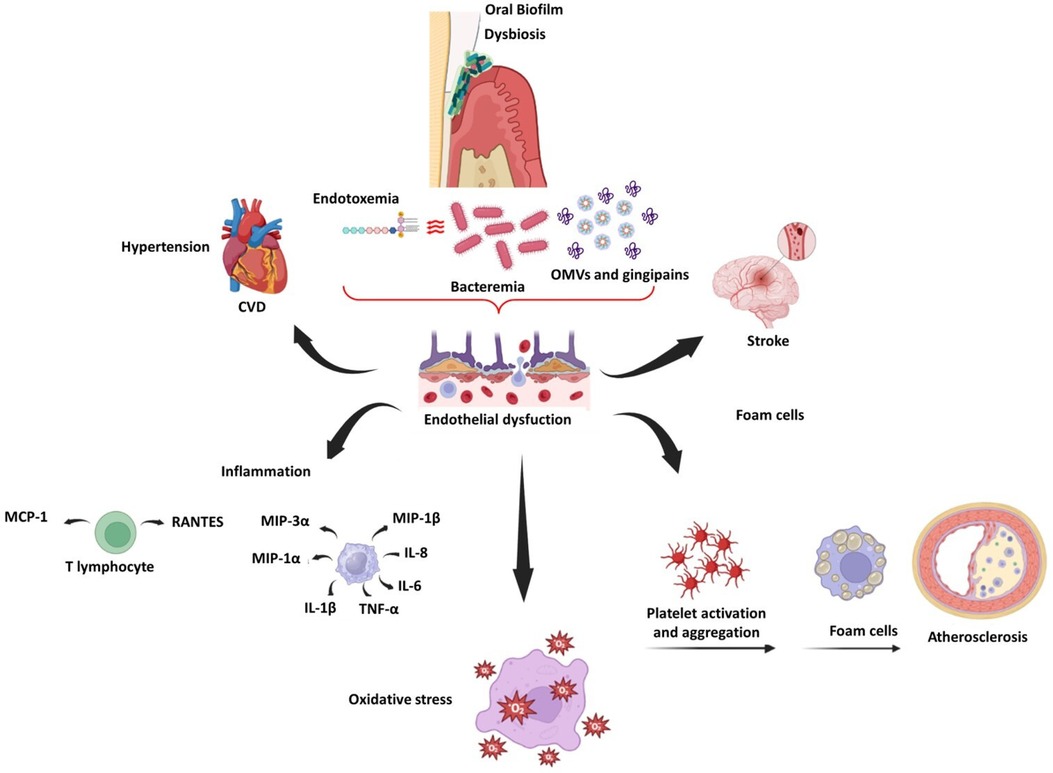
Figure 2. Schematic representation of endothelial dysfunction related to oral dysbiosis, CVDs, and stroke.
9. Association between periodontal disease and hypertension
Another effect of periodontal disease in CVDs is arterial hypertension. Periodontitis causes inflammation and oxidative stress, which causes vascular changes such as arterial stiffness, vascular dysfunction, and hypertension (147, 148). Epidemiological observational studies have supported the association between periodontal disease and hypertension (149, 150). Moreover, IPT significantly reduced the systolic blood pressure (SBP) in patients with the periodontal disease after treatment compared to those who did not receive treatment, which correlated with periodontal status improvement. Periodontal treatment also improved diastolic pressure and endothelial function (FMD), with lower levels of circulating interferon-gamma and IL-6, as well as activated and immunosenescent CD8+ T-cells, both of which have been linked to hypertension (151, 152). In 2021, an integrative study of evidence based on eight randomized controlled trials found that IPT improved SBP and diastolic blood pressure in individuals with hypertension and prehypertension, with significant reductions in CRP and improvement in endothelial function after periodontal treatment. This evidence supports the role of periodontal treatment in improving endothelial dysfunction in patients with CVDs (149).
In a multiethnic cohort study on the direct assessment of subgingival periodontal bacterial load and its relationship to BP, which restricted clinical periodontal assessment bias, a strong positive association was demonstrated between increased subgingival colonization by, P. gingivalis, T. forsythia, T. denticola, and A. actinomycetemcomitans and arterial hypertension (148). To our knowledge, this was the first study to link bacterial load to increased blood pressure. Periodontitis is associated with systemic inflammation, the mediators can affect endothelial function (92). Clinical evidence suggests that periodontitis affects systemic endothelial function, which may affect hypertension, with some reports suggesting possible direct effects of bacteremia-related P. gingivalis in mediating vascular dysfunction and immune response, resulting in elevated BP, vascular inflammation, and endothelial dysfunction (147).
As discussed above, an imbalance in NO bioavailability associated with oral microbiome dysbiosis is associated with some cardiovascular and metabolic diseases (21, 22). A representative population study using data from the third National Health and Nutrition Examination Survey (NHANES III) demonstrate a significant association of elevated BP with antibodies against Campylobacter rectus, Veillonella parvula, and Prevotella melaninogenica. C. rectus resulted in the strongest association with BP (153). In another study, in periodontitis patients, high BP was associated with higher P. intermedia, P. gingivalis, and F. nucleatum counts (154). This dysbiosis can displace the denitrifying bacteria, reduce the NO, and alter arterial vasodilatation (21). Different publications have been realized to establish the impact of mouthwashes on the elevation of BP; The use of chlorhexidine, even at lower concentrations, inhibited the nitrate activity and the Veillonella dispar counts, however, the activity of salivary nitrite production was not affected (155). Other mouthwashes, such as essential oil, povidone-iodine, and cetylpyridinium chloride, have little effect on nitrate-reducing activity (156). The use of chlorhexidine mouthwash for one-week generated changes in the microbiome of the tongue and changes in systolic pressure in normotensive individuals. Otherwise, the individuals who cleaned the tongue regularly resulted in an enrichment of nitrate-reducing bacteria on the tongue (157). However, the tongue clean may disrupt the papillary surface and favor chlorhexidine penetration, resulting in a significant alteration of bacterial community and greater SBP (157).
Regarding the local renin–angiotensin system in gingival tissue, there may be another pathogenic link between the two conditions under investigation (147), in addition to a possible variable risk of periodontitis among different renin–angiotensin system genetic polymorphisms. Moreover, Viafara et al. demonstrated that repeated exposure to live P. gingivalis or LPSs induces the release of proinflammatory cytokines and angiotensin II in HCAECs and mediators of systemic inflammation such as CRP, IL-6, and TNF-α, contributing both to endothelial dysfunction (92). This could be linked to the Th1 response induced by bacterial antigens such as P. gingivalis by increasing sensitivity to the suppressive pro-hypertensive insult evoked by low doses of angiotensin II (147). Therefore, it supports the “two-hit” hypothesis, which states that immune activation at sites of chronic inflammation exacerbates responses to low-dose angiotensin II, establishing a link between chronic immune activation and hypertension.
10. Conclusions
Periodontitis and CVDs are both inflammatory diseases caused by the systemic circulation of periodontopathogens and their virulent factors, which can cause endothelial dysfunction via ILs, cytokines, oxidative stress, monocyte and macrophage activation, plaque aggregation, and cellular proliferation. These events could be related to CVDs such as atherosclerosis and hypertension. Periodontal treatment reduces inflammatory markers related to CVDs and may reduce the risk of CVD events. Dysbiosis of the oral microbiome in saliva and tongue in periodontal patients plays a vital role in the loss of balance that regulates blood pressure mediated by NO, which induces endothelial dysfunction. A controlled preventive therapy on other cardiovascular markers could significantly contribute to managing cardiovascular patients with periodontitis. This effect should be demonstrated in the future.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
The Vicerrectoria de Investigaciones-Universidad El Bosque funded this article. PCI-2017-9568 and PCI- 2019-10705.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Cyr AR, Huckaby LV, Shiva SS, Zuckerbraun BS. Nitric oxide and endothelial dysfunction. Crit Care Clin. (2020) 36:307–21. doi: 10.1016/j.ccc.2019.12.009
2. Rosier BT, Moya-Gonzalvez EM, Corell-Escuin P, Mira A. Isolation and characterization of nitrate-reducing bacteria as potential probiotics for oral and systemic health. Front Microbiol. (2020) 11:555465. doi: 10.3389/fmicb.2020.555465
3. Rosier BT, Buetas E, Moya-Gonzalvez EM, Artacho A, Mira A. Nitrate as a potential prebiotic for the oral microbiome. Sci Rep. (2020) 10(1):12895. doi: 10.1038/s41598-020-69931-x
4. Suárez LJ, Garzón H, Arboleda S, Rodríguez A. Oral dysbiosis and autoimmunity: from local periodontal responses to an imbalanced systemic immunity. A review. Front Immunol. (2020) 11:591255. doi: 10.3389/fimmu.2020.591255
5. Kinane DF, Stathopoulou PG, Papapanou PN. Periodontal diseases. Nat Rev Dis Primers. (2017) 3:17038. doi: 10.1038/nrdp.2017.38
6. Holtfreter B, Empen K, Gläser S, Lorbeer R, Völzke H, Ewert R, et al. Periodontitis is associated with endothelial dysfunction in a general population: a cross-sectional study. PLoS One. (2013) 8:e84603. doi: 10.1371/journal.pone.0084603
7. Lafaurie GI, Mayorga-Fayad I, Torres MF, Castillo DM, Aya MR, Barón A, et al. Periodontopathic microorganisms in peripheric blood after scaling and root planing. J Clin Periodontol. (2007) 34:873–9. doi: 10.1111/j.1600-051X.2007.01125.x
8. Castillo DM, Sánchez-Beltrán MC, Castellanos JE, Sanz I, Mayorga-Fayad I, Sanz M, et al. Detection of specific periodontal microorganisms from bacteraemia samples after periodontal therapy using molecular-based diagnostics. J Clin Periodontol. (2011) 38:418–27. doi: 10.1111/j.1600-051X.2011.01717.x
9. Armingohar Z, Jørgensen JJ, Kristoffersen AK, Abesha-Belay E, Olsen I. Bacteria and bacterial DNA in atherosclerotic plaque and aneurysmal wall biopsies from patients with and without periodontitis. J Oral Microbiol. (2014) 15:6. doi: 10.3402/jom.v6.23408
10. Kozarov EV, Dorn BR, Shelburne CE, Dunn WA Jr, Progulske-Fox A. Human atherosclerotic plaque contains viable invasive Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis. Arterioscler Thromb Vasc Biol. (2005) 25:e17–8. doi: 10.1161/01.ATV.0000155018.67835.1a
11. Reyes L, Herrera D, Kozarov E, Roldá S, Progulske-Fox A. Periodontal bacterial invasion and infection: contribution to atherosclerotic pathology. J Periodontol. (2013) 84:S30–50. doi: 10.1902/jop.2013.1340012
12. Chapple ILC, Mealey BL, Van Dyke TE, Bartold PM, Dommisch H, Eickholz P, et al. Periodontal health and gingival diseases and conditions on an intact and a reduced periodontium: consensus report of workgroup 1 of the 2017 world workshop on the classification of periodontal and peri-implant diseases and conditions. J Periodontol. (2018) 89(Suppl 1):S74–84. doi: 10.1002/JPER.17-0719
13. Kassebaum NJ, Bernabé E, Dahiya M, Bhandari B, Murray CJ, Marcenes W. Global burden of severe periodontitis in 1990–2010: a systematic review and meta-regression. J Dent Res. (2014) 93:1045–53. doi: 10.1177/0022034514552491
14. Dye BA. Global periodontal disease epidemiology. Periodontol 2000. (2012) 58:10–25. doi: 10.1111/j.1600-0757.2011.00413.x
15. Tonetti MS, Jepsen S, Jin L, Otomo-Corgel J. Impact of the global burden of periodontal diseases on health, nutrition and wellbeing of mankind: a call for global action. J Clin Periodontol. (2017) 44:456–62. doi: 10.1111/jcpe.12732
16. Haapasalmi K, Mäkelä M, Oksala O, Heino J, Yamada KM, Uitto VJ, et al. Expression of epithelial adhesion proteins and integrins in chronic inflammation. Am J Pathol. (1995) 147:193–206.7541610
17. Caton JG, Armitage G, Berglundh T, Chapple ILC, Jepsen S, Kornman KS, et al. A new classification scheme for periodontal and peri-implant diseases and conditions—introduction and key changes from the 1999 classification. J Clin Periodontol. (2018) 45:S1–8. doi: 10.1111/jcpe.12935
18. Papapanou PN, Sanz M, Buduneli N, Dietrich T, Feres M, Fine DH, et al. Periodontitis: consensus report of workgroup 2 of the 2017 world workshop on the classification of periodontal and peri-implant diseases and conditions. J Periodontol. (2018) 89:S173–82. doi: 10.1002/JPER.17-0721
19. Verma D, Garg PK, Dubey AK. Insights into the human oral microbiome. Arch Microbiol. (2018) 200:525–40. doi: 10.1007/s00203-018-1505-3
20. Metsäniitty M, Hasnat S, Salo T, Salem A. Oral microbiota-A new frontier in the pathogenesis and management of head and neck cancers. Cancers (Basel). (2021) 14:46. doi: 10.3390/cancers14010046
21. Morou-Bermúdez E, Torres-Colón JE, Bermúdez NS, Patel RP, Joshipura KJ. Pathways linking oral bacteria, nitric oxide metabolism, and health. J Dent Res. (2022) 101:623–31. doi: 10.1177/00220345211064571
22. Takahashi N. Oral microbiome metabolism: from “who are they?” to “what are they doing?”. J Dent Res. (2015) 94:1628–37. doi: 10.1177/0022034515606045
23. Hajishengallis G. Periodontitis: from microbial immune subversion to systemic inflammation. Nat Rev Immunol. (2015) 15:30–44. doi: 10.1038/nri3785
24. Hajishengallis G, Krauss JL, Liang S, McIntosh ML, Lambris JD. Pathogenic microbes and community service through manipulation of innate immunity. Adv Exp Med Biol. (2012) 946:69–85. doi: 10.1007/978-1-4614-0106-3_5
25. Hajishengallis G, Diaz PI. Porphyromonas gingivalis: immune subversion activities and role in periodontal dysbiosis. Curr Oral Health Rep. (2020) 7:12–21. doi: 10.1007/s40496-020-00249-3
26. Slocum C, Kramer C, Genco CA. Immune dysregulation mediated by the oral microbiome: potential link to chronic inflammation and atherosclerosis. J Intern Med. (2016) 280:114–28. doi: 10.1111/joim.12476
27. Shaik-Dasthagirisaheb YB, Huang N, Gibson FC III. Inflammatory response to porphyromonas gingivalis partially requires interferon regulatory factor (IRF) 3. Innate Immun. (2014) 20:312–9. doi: 10.1177/1753425913492180
28. Palm E, Khalaf H, Bengtsson T. Porphyromonas gingivalis downregulates the immune response of fibroblasts. BMC Microbiol. (2013) 13:155. doi: 10.1186/1471-2180-13-155
29. Ribeiro CCC, Carmo CDS, Benatti BB, Casarin RVC, Alves CMC, et al. Systemic circulating inflammatory burden and periodontitis in adolescents. Clin Oral Investig. (2021) 25:5855–65. doi: 10.1007/s00784-021-03891-y
30. Gomes MS, Blattner TC, Sant'Ana Filho M, Grecca FS, Hugo FN, Fouad AF, et al. Can apical periodontitis modify systemic levels of inflammatory markers? A systematic review and meta-analysis. J Endod. (2013) 39:1205–17. doi: 10.1016/j.joen.2013.06.014
31. Almeida APCPSC, Fagundes NCF, Maia LC, Lima RR. Is there an association between periodontitis and atherosclerosis in adults? A systematic review. Curr Vasc Pharmacol. (2018) 16:569–82. doi: 10.2174/1570161115666170830141852
32. Teeuw WJ, Slot DE, Susanto H, Gerdes VE, Abbas F, D'Aiuto F, et al. Treatment of periodontitis improves the atherosclerotic profile: a systematic review and meta-analysis. J Clin Periodontol. (2014) 4:70–9. doi: 10.1111/jcpe.12171
33. Yuan X, Wang L, Bhat OM, Lohner H, Li PL. Differential effects of short chain fatty acids on endothelial Nlrp3 inflammasome activation and neointima formation: antioxidant action of butyrate. Redox Biol. (2018) 16:21–31. doi: 10.1016/j.redox.2018.02.007
34. Potgieter M, Bester J, Kell DB, Pretorius E. The dormant blood microbiome in chronic, inflammatory diseases. FEMS Microbiol Rev. (2015) 39:567–91. doi: 10.1093/femsre/fuv013
35. Forbes JD, Chen CY, Knox NC, Marrie RA, El-Gabalawy H, de Kievit T, et al. A comparative study of the gut microbiota in immune-mediated inflammatory diseases-does a common dysbiosis exist? Microbiome. (2018) 6:221. doi: 10.1186/s40168-018-0603-4
36. Hajishengallis G, Chavakis T. Local and systemic mechanisms linking periodontal disease and inflammatory comorbidities. Nat Rev Immunol. (2021) 21:426–40. doi: 10.1038/s41577-020-00488-6
37. Parahitiyawa NB, Jin LJ, Leung WK, Yam WC, Samaranayake LP. Microbiology of odontogenic bacteremia: beyond endocarditis. Clin Microbiol Rev. (2009) 22:46–64. doi: 10.1128/CMR.00028-08
38. Horliana AC, Chambrone L, Foz AM, Artese HP, Rabelo Mde S, Pannuti CM, et al. Dissemination of periodontal pathogens in the bloodstream after periodontal procedures: a systematic review. PLoS One. (2014) 9:e98271. doi: 10.1371/journal.pone.0098271
39. Hasegawa-Nakamura K, Tateishi F, Nakamura T, et al. The possible mechanism of preterm birth associated with periodontopathic Porphyromonas gingivalis. J Periodontal Res. (2011) 46:497–504. doi: 10.1111/j.1600-0765.2011.01366.x
40. Vanterpool SF, Been JV, Houben ML, Nikkels PG, De Krijger RR, Zimmermann LJ, et al. Porphyromonas gingivalis within placental villous mesenchyme and umbilical cord stroma is associated with adverse pregnancy outcome. PLoS One. (2016) 11(1):e0146157. doi: 10.1371/journal.pone.0146157
41. Gómez LA, De Avila J, Castillo DM, Montenegro DA, Trujillo TG, Suárez LJ, et al. Porphyromonas gingivalis placental atopobiosis and inflammatory responses in women with adverse pregnancy outcomes. Front Microbiol. (2020) 11:591626. doi: 10.3389/fmicb.2020.591626
42. Poole S, Singhrao SK, Kesavalu L, Curtis MA, Crean S. Determining the presence of periodontopathic virulence factors in short-term postmortem Alzheimer’s disease brain tissue. J Alzheimers Dis. (2013) 36:665–77. doi: 10.3233/JAD-121918
43. Dominy SS, Lynch C, Ermini F, Benedyk M, Marczyk A, Konradi A, et al. Porphyromonas gingivalis in Alzheimer’s disease brains: evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv. (2019) 5:eaau3333. doi: 10.1126/sciadv.aau3333
44. Szulc M, Kustrzycki W, Janczak D, Michalowska D, Baczynska D, Radwan-Oczko M. Presence of periodontopathic bacteria DNA in atheromatous plaques from coronary and carotid arteries. Biomed Res Int. (2015) 2015:825397. doi: 10.1155/2015/825397
45. Marcelino SL, Jr G-JE, Nakano V, Canônico LA, Nunes FD, Lotufo RF, et al. Presence of periodontopathic bacteria in coronary arteries from patients with chronic periodontitis. Anaerobe. (2010) 16:629–32. doi: 10.1016/j.anaerobe.2010.08.007
46. Schenkein HA, Papapanou PN, Genco R, Sanz M. Mechanisms underlying the association between periodontitis and atherosclerotic disease. Periodontol 2000. (2020) 83:90–106. doi: 10.1111/prd.12304
47. Dorn BR, Burks JN, Seifert KN, Progulske-Fox A. Invasion of endothelial and epithelial cells by strains of Porphyromonas gingivalis. FEMS Microbiol Lett. (2000) 187:139–44. doi: 10.1111/j.1574-6968.2000.tb09150.x
48. Amar S, Wu SC, Madan M. Is Porphyromonas gingivalis cell invasion required for atherogenesis? Pharmacotherapeutic implications. J Immunol. (2009) 182:1584–92. doi: 10.4049/jimmunol.182.3.1584
49. Deshpande RG, Khan MB, Genco CA. Invasion of aortic and heart endothelial cells by Porphyromonas gingivalis. Infect Immun. (1998) 66:5337–43. doi: 10.1128/IAI.66.11.5337-5343.1998
50. Yilmaz Ö. The chronicles of Porphyromonas gingivalis: the microbium, the human oral epithelium and their interplay. Microbiology (Reading. (2008) 154:2897–903. doi: 10.1099/mic.0.2008/021220-0
51. Yilmaz O, Jungas T, Verbeke P, Ojcius DM. Activation of the phosphatidylinositol 3-kinase/akt pathway contributes to survival of primary epithelial cells infected with the periodontal pathogen Porphyromonas gingivalis. Infect Immun. (2004) 72:3743–51. doi: 10.1128/IAI.72.7.3743-3751.2004
52. Casasanta MA, Yoo CC, Udayasuryan B, Sanders BE, Umaña A, Zhang Y, et al. Fusobacterium nucleatum host-cell binding and invasion induces IL-8 and CXCL1 secretion that drives colorectal cancer cell migration. Sci Signal. (2020) 13:eaba9157. doi: 10.1126/scisignal.aba9157
53. Abed J, Emgård JE, Zamir G, Faroja M, Almogy G, Grenov A, et al. Fap2 mediates Fusobacterium nucleatum colorectal adenocarcinoma enrichment by binding to tumor-expressed gal-GalNAc. Cell Host Microbe. (2016) 20:215–25. doi: 10.1016/j.chom.2016.07.006
54. Wong CH, Jenne CN, Petri B, Chrobok NL, Kubes P. Nucleation of platelets with blood-borne pathogens on kupffer cells precedes other innate immunity and contributes to bacterial clearance. Nat Immunol. (2013) 14:785–92. doi: 10.1038/ni.2631
55. Lockhart PB, Brennan MT, Sasser HC, Fox PC, Paster BJ, Bahrani-Mougeot FK. Bacteremia associated with toothbrushing and dental extraction. Circulation. (2008) 117:3118–25. doi: 10.1161/CIRCULATIONAHA.107.758524
56. Minasyan H. Erythrocyte and blood antibacterial defense. Eur J Microbiol Immunol (Bp). (2014) 4:138–43. doi: 10.1556/EuJMI.4.2014.2.7
57. Belstrøm D, Holmstrup P, Damgaard C, Borch TS, Skjødt MO, Bendtzen K, et al. The atherogenic bacterium Porphyromonas gingivalis evades circulating phagocytes by adhering to erythrocytes. Infect Immun. (2011) 79:1559–65. doi: 10.1128/IAI.01036-10
58. Flierl MA, Rittirsch D, Nadeau BA, Day DE, Zetoune FS, Sarma JV, et al. Functions of the complement components C3 and C5 during sepsis. FASEB J. (2008) 22:3483–90. doi: 10.1096/fj.08-110595
59. Serra e Silva Filho W, Casarin RC, Nicolela EL Jr, Passos HM, Sallum AW, Gonçalves RB. Microbial diversity similarities in periodontal pockets and atheromatous plaques of cardiovascular disease patients. PLoS One. (2014) 9:e109761. doi: 10.1371/journal.pone.0109761
60. Geerts SO, Nys M, De MP, Charpentier J, Albert A, Legrand V, et al. Systemic release of endotoxins induced by gentle mastication: association with periodontitis severity. J Periodontol. (2002) 73:73–8. doi: 10.1902/jop.2002.73.1.73
61. Marcano R, Rojo MÁ, Cordoba-Diaz D, Garrosa M. Pathological and therapeutic approach to endotoxin-secreting bacteria involved in periodontal disease. Toxins (Basel). (2021) 13:533. doi: 10.3390/toxins13080533
62. Ding PH, Jin LJ. The role of lipopolysaccharide-binding protein in innate immunity: a revisit and its relevance to oral/periodontal health. J Periodontal Res. (2014) 49:1–9. doi: 10.1111/jre.12081
63. Bale BF, Doneen AL, Vigerust DJ. High-risk periodontal pathogens contribute to the pathogenesis of atherosclerosis. Postgrad Med J. (2017) 93:215–20. doi: 10.1136/postgradmedj-2016-134279
64. Hasegawa Y, Nagano K. Porphyromonas gingivalis FimA and Mfa1 fimbriae: current insights on localization, function, biogenesis, and genotype. Jpn Dent Sci Rev. (2021) 57:190–200. doi: 10.1016/j.jdsr.2021.09.003
65. Pérez-Chaparro PJ, Lafaurie GI, Gracieux P, et al. Distribution of Porphyromonas gingivalis fimA genotypes in isolates from subgingival plaque and blood sample during bacteremia. Biomedica. (2009) 29:298–306. doi: 10.7705/biomedica.v29i2.31
66. Xu W, Zhou W, Wang H, Liang S. Roles of Porphyromonas gingivalis and its virulence factors in periodontitis. Adv Protein Chem Struct Biol. (2020) 120:45–84. doi: 10.1016/bs.apcsb.2019.12.001
67. Imamura T. The role of gingipains in the pathogenesis of periodontal disease. J Periodontol. (2003) 74:111–8. doi: 10.1902/jop.2003.74.1.111
68. Amano A, Takeuchi H, Furuta N. Outer membrane vesicles function as offensive weapons in host-parasite interactions. Microbes Infect. (2010) 12:791–8. doi: 10.1016/j.micinf.2010.05.008
69. Belibasakis GN, Maula T, Bao K, Lindholm M, Bostanci N, Oscarsson J, et al. Virulence and pathogenicity properties of Aggregatibacter actinomycetemcomitans. Pathogens. (2019) 8:222. doi: 10.3390/pathogens8040222
70. Stokowa-Sołtys K, Wojtkowiak K, Jagiełło K. Fusobacterium nucleatum—friend or foe? J Inorg Biochem. (2021) 224:111586. doi: 10.1016/j.jinorgbio.2021.111586
71. Umaña A, Sanders BE, Yoo CC, Casasanta MA, Udayasuryan B, Verbridge SS, et al. Utilizing whole Fusobacterium genomes to identify, correct, and characterize potential virulence protein families. J Bacteriol. (2019) 201:e00273–19. doi: 10.1128/JB.00273-19
72. Zhang Z, Liu D, Liu S, Zhang S, Pan Y. The role of Porphyromonas gingivalis outer membrane vesicles in periodontal disease and related systemic diseases. Front Cell Infect Microbiol. (2021) 10:585917. doi: 10.3389/fcimb.2020.585917
73. Kurita-Ochiai T, Yamamoto M. Periodontal pathogens and atherosclerosis: implications of inflammation and oxidative modification of LDL. Biomed Res Int. (2014):595981. doi: 10.1155/2014/595981
74. Han YW. Fusobacterium nucleatum: a commensal-turned pathogen. Curr Opin Microbiol. (2015) 23:141–7. doi: 10.1016/j.mib.2014.11.013
75. Chukkapalli SS, Easwaran M, Rivera-Kweh MF, Velsko IM, Ambadapadi S, Dai J, et al. Sequential colonization of periodontal pathogens in induction of periodontal disease and atherosclerosis in LDLRnull mice. Pathog Dis. (2017) 75:ftx003. doi: 10.1093/femspd/ftx003
76. Jia Y, Guo B, Yang W, Zhao Q, Jia W, Wu Y. Rho kinase mediates Porphyromonas gingivalis outer membrane vesicle-induced suppression of endothelial nitric oxide synthase through ERK1/2 and p38 MAPK. Arch Oral Biol. (2015) 60:488–95. doi: 10.1016/j.archoralbio.2014.12.009
77. Farrugia C, Stafford GP, Murdoch C. Porphyromonas gingivalis outer membrane vesicles increase vascular permeability. J Dent Res. (2020) 99:1494–501. doi: 10.1177/0022034520943187
78. Yang WW, Guo B, Jia WY, Jia Y. Porphyromonas gingivalis-derived outer membrane vesicles promote calcification of vascular smooth muscle cells through ERK1/2-RUNX2. FEBS Open Bio. (2016) 6:1310–9. doi: 10.1002/2211-5463.12151
79. Kuramitsu HK, Qi M, Kang IC, Chen W. Role for periodontal bacteria in cardiovascular diseases. Ann Periodontol. (2001) 6:41–7. doi: 10.1902/annals.2001.6.1.41
80. Sharma A, Novak EK, Sojar HT, Swank RT, Kuramitsu HK, Genco RJ. Porphyromonas gingivalis platelet aggregation activity: outer membrane vesicles are potent activators of murine platelets. Oral Microbiol Immunol. (2000) 15:393–6. doi: 10.1034/j.1399-302x.2000.150610.x
81. DeLeon-Pennell KY, de Castro Brás LE, Iyer RP, Bratton DR, Jin YF, Ripplinger CM, et al. P. gingivalis lipopolysaccharide intensifies inflammation post-myocardial infarction through matrix metalloproteinase-9. J Mol Cell Cardiol. (2014) 76:218–26. doi: 10.1016/j.yjmcc.2014.09.007
82. Bengtsson T, Karlsson H, Gunnarsson P, Skoglund C, Elison C, Leanderson P, et al. The periodontal pathogen Porphyromonas gingivalis cleaves apoB-100 and increases the expression of apoM in LDL in whole blood leading to cell proliferation. J Intern Med. (2008) 263:558–71. doi: 10.1111/j.1365-2796.2007.01917.x
83. Hashimoto M, Kadowaki T, Tsukuba T, Yamamoto K. Selective proteolysis of apolipoprotein B-100 by arg-gingipain mediates atherosclerosis progression accelerated by bacterial exposure. J Biochem. (2006) 140:713–23. doi: 10.1093/jb/mvj202
84. Félétou M. The endothelium: Part 1: Multiple functions of the endothelial cells—focus on endothelium-derived vasoactive mediators. San Rafael (CA): Morgan & Claypool Life Sciences (2011).
85. Matsubara T, Ishibashi T, Hori T, Ozaki K, Mezaki T, Tsuchida K, et al. Association between coronary endothelial dysfunction and local inflammation of atherosclerotic coronary arteries. Mol Cell Biochem. (2003) 249:67–73. doi: 10.1023/A:1024778421491
86. Gimbrone MA Jr, García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. (2016) 118:620–36. doi: 10.1161/CIRCRESAHA.115.306301
87. Chistiakov DA, Orekhov AN, Bobryshev YV. Links between atherosclerotic and periodontal disease. Exp Mol Pathol. (2016) 100:220–35. doi: 10.1016/j.yexmp.2016.01.006
88. Mustapha IZ, Debrey S, Oladubu M, Ugarte R. Markers of systemic bacterial exposure in periodontal disease and cardiovascular disease risk: a systematic review and meta-analysis. J Periodontol. (2007) 78:2289–302. doi: 10.1902/jop.2007.070140
89. Triantafilou M, Gamper FG, Lepper PM, Mouratis MA, Schumann C, Harokopakis E, et al. Lipopolysaccharides from atherosclerosis-associated bacteria antagonize TLR4, induce formation of TLR2/1/CD36 complexes in lipid rafts and trigger TLR2-induced inflammatory responses in human vascular endothelial cells. Cell Microbiol. (2007) 9:2030–9. doi: 10.1111/j.1462-5822.2007.00935.x
90. Erridge C, Spickett CM, Webb DJ. Non-enterobacterial endotoxins stimulate human coronary artery but not venous endothelial cell activation via toll-like receptor 2. Cardiovasc Res. (2007) 73:181–9. doi: 10.1016/j.cardiores.2006.11.004
91. Gualtero DF, Lafaurie GI, Fontanilla MR. Differential responses of endothelial cells on three-dimensional scaffolds to lipopolysaccharides from periodontopathogens. Mol Oral Microbiol. (2019) 34:183–93. doi: 10.1111/omi.12263
92. Viafara-García SM, Morantes SJ, Chacon-Quintero Y, Castillo DM, Lafaurie GI, Buitrago DM. Repeated Porphyromonas gingivalis W83 exposure leads to release pro-inflammatory cytokynes and angiotensin II in coronary artery endothelial cells. Sci Rep. (2019) 9:19379. doi: 10.1038/s41598-019-54259-y
93. Gualtero DF, Viafara-Garcia SM, Morantes SJ, Buitrago DM, Gonzalez OA, Lafaurie GI. Rosuvastatin inhibits interleukin (IL)-8 and IL-6 production in human coronary artery endothelial cells stimulated with Aggregatibacter actinomycetemcomitans serotype b. J Periodontol. (2017) 88:225–35. doi: 10.1902/jop.2016.160288
94. Torres MA, Gualtero DF, Lafaurie GI, Fontanilla MR. Aggregatibacter actinomycetemcomitans induces a pro-atherosclerotic response in a three-dimensional collagen scaffold model in human endothelial cells. Mol Oral Microbiol. (2021) 36:58–66. doi: 10.1111/omi.12326
95. Viafara-Garcia SM, Gualtero DF, Avila-Ceballos D, Lafaurie GI. Eikenella corrodens lipopolysaccharide stimulates the pro-atherosclerotic response in human coronary artery endothelial cells and monocyte adhesion. Eur J Oral Sci. (2018) 126:476–84. doi: 10.1111/eos.12580
96. Hasturk H, Kantarci A, Van Dyke TE. Oral inflammatory diseases and systemic inflammation: role of the macrophage. Front Immunol. (2012) 3:118. doi: 10.3389/fimmu.2012.00118
97. Kaparakis-Liaskos M, Ferrero RL. Immune modulation by bacterial outer membrane vesicles. Nat Rev Immunol. (2015) 15:375–87. doi: 10.1038/nri3837
98. Castillo Y, Castellanos JE, Lafaurie GI, Castillo DM. Porphyromonas gingivalis outer membrane vesicles modulate cytokine and chemokine production by gingipain-dependent mechanisms in human macrophages. Arch Oral Biol. (2022) 140:105453. doi: 10.1016/j.archoralbio.2022.105453
99. Nakao R, Takashiba S, Kosono S, Yoshida M, Watanabe H, Ohnishi M, et al. Effect of Porphyromonas gingivalis outer membrane vesicles on gingipain-mediated detachment of cultured oral epithelial cells and immune responses. Microbes Infect. (2014) 16:6–16. doi: 10.1016/j.micinf.2013.10.005
100. Gualtero DF, Lafaurie GI, Fontanilla MR. Two-dimensional and three-dimensional models for studying atherosclerosis pathogenesis induced by periodontopathogenic microorganisms. Mol Oral Microbiol. (2018) 33:29–37. doi: 10.1111/omi.12201
101. Barrett TJ. Macrophages in atherosclerosis regression. Arterioscler Thromb Vasc Biol. (2020) 40:20–33. doi: 10.1161/ATVBAHA.119.312802
102. Hajishengallis G, Sharma A, Russell MW, Genco RJ. Interactions of oral pathogens with toll-like receptors: possible role in atherosclerosis. Ann Periodontol. (2002) 7:72–8. doi: 10.1902/annals.2002.7.1.72
103. Loos BG, Teeuw WJ, Nicu EA. Plausible mechanisms explaining the association of periodontitis with cardiovascular diseases. In: Lynge Pedersen A, editors. Oral infections and general health (2016). p. 19–33. doi: 10.1007/978-3-319-25091-5_3
104. López R, Loos BG, Baelum V. Hematological features in adolescents with periodontitis. Clin Oral Investig. (2012) 16:1209–16. doi: 10.1007/s00784-011-0628-6
105. Hamzeh-Cognasse H, Damien P, Chabert A, Pozzetto B, Cognasse F, Garraud O. Platelets and infections—complex interactions with bacteria. Front Immunol. (2015) 6:82. doi: 10.3389/fimmu.2015.00082
106. Pham K, Feik D, Hammond BF, Rams TE, Whitaker EJ. Aggregation of human platelets by gingipain-R from Porphyromonas gingivalis cells and membrane vesicles. Platelets. (2002) 13:21–30. doi: 10.1080/09537100120104863
107. Lourbakos A, Yuan YP, Jenkins AL, Travis J, Andrade-Gordon P, Santulli R, et al. Activation of protease-activated receptors by gingipains from Porphyromonas gingivalis leads to platelet aggregation: a new trait in microbial pathogenicity. Blood. (2001) 97:3790–7. doi: 10.1182/blood.v97.12.3790
108. Naito M, Sakai E, Shi Y, Ideguchi H, Shoji M, Ohara N, et al. Porphyromonas gingivalis-induced platelet aggregation in plasma depends on Hgp44 adhesin but not rgp proteinase. Mol Microbiol. (2006) 59:152–67. doi: 10.1111/j.1365-2958.2005.04942.x
109. Plummer C, Wu H, Kerrigan SW, Meade G, Cox D, Ian Douglas CW. A serine-rich glycoprotein of Streptococcus sanguis mediates adhesion to platelets via GPIb. Br J Haematol. (2005) 129:101–9. doi: 10.1111/j.1365-2141.2005.05421.x
110. Hally K, Fauteux-Daniel S, Hamzeh-Cognasse H, Larsen P, Cognasse F. Revisiting platelets and toll-like receptors (TLRs): at the interface of vascular immunity and thrombosis. Int J Mol Sci. (2020) 21:6150. doi: 10.3390/ijms21176150
111. Cognasse F, Lafarge S, Chavarin P, Acquart S, Garraud O. Lipopolysaccharide induces sCD40l release through human platelets TLR4, but not TLR2 and TLR9. Intensive Care Med. (2007) 33:382–4. doi: 10.1007/s00134-006-0488-8
112. Assinger A, Laky M, Badrnya S, Esfandeyari A, Volf I. Periodontopathogens induce expression of CD40l on human platelets via TLR2 and TLR4. Thromb Res. (2012) 130:e73–8. doi: 10.1016/j.thromres.2012.04.017
113. Potempa J, Sroka A, Imamura T, Travis J. Gingipains, the major cysteine proteinases and virulence factors of Porphyromonas gingivalis: structure, function and assembly of multidomain protein complexes. Curr Protein Pept Sci. (2003) 4:397–407. doi: 10.2174/1389203033487036
114. McNicol A. Bacteria-induced intracellular signalling in platelets. Platelets. (2015) 26:309–16. doi: 10.3109/09537104.2015.1014470
115. Buitrago DM, Ramos G, Rincón GM. Actividad antiagregante del extracto etanólico de Solanum tuberosum en plaquetas humanas. Vitae. (2007) 14:49–54.
116. Zhong S, Li L, Shen X, Li Q, Xu W, Wang X, et al. An update on lipid oxidation and inflammation in cardiovascular diseases. Free Radic Biol Med. (2019) 144:266–78. doi: 10.1016/j.freeradbiomed.2019.03.036
117. Yin H, Xu L, Porter NA. Free radical lipid peroxidation: mechanisms and analysis. Chem Rev. (2011) 111:5944–72. doi: 10.1021/cr200084z
118. Di Pietro M, Filardo S, Falasca F, Turriziani O, Sessa R. Infectious agents in atherosclerotic cardiovascular diseases through oxidative stress. Int J Mol Sci. (2017) 18:2459. doi: 10.3390/ijms18112459
119. Jia R, Kurita-Ochiai T, Oguchi S, Yamamoto M. Periodontal pathogen accelerates lipid peroxidation and atherosclerosis. J Dent Res. (2013) 92:247–52. doi: 10.1177/0022034513475625
120. Bugueno IM, Zobairi El-Ghazouani F, Batool F, El Itawi H, Anglès-Cano E, Benkirane-Jessel N, et al. Porphyromonas gingivalis triggers the shedding of inflammatory endothelial microvesicles that act as autocrine effectors of endothelial dysfunction. Sci Rep. (2020) 10:1778. doi: 10.1038/s41598-020-58374-z
121. OMS 2. Available at: https://www.who.int/health-topics/cardiovascular-diseases#tab=tab_1
122. Arnett DK, Blumenthal RS, Albert MA, Buroker AB, Goldberger ZD, Hahn EJ, et al. 2019 ACC/AHA guideline on the primary prevention of cardiovascular disease: a report of the American college of cardiology/American heart association task force on clinical practice guidelines. Circulation. (2020) 21:141. doi: 10.1161/CIR.0000000000000678
123. Mattila KJ, Nieminen MS, Valtonen VV, Rasi VP, Kesäniemi YA, Syrjälä SL, et al. Association between dental health and acute myocardial infarction. Br Med J. (1989) 298:779–81. doi: 10.1136/bmj.298.6676.779
124. Humphrey LL, Fu R, Buckley DI, Freeman M, Helfand M. Periodontal disease and coronary heart disease incidence: a systematic review and meta-analysis. J Gen Intern Med. (2008) 23:2079–86. doi: 10.1007/s11606-008-0787-6
125. Larvin H, Kang J, Aggarwal VR, Pavitt S, Wu J. Risk of incident cardiovascular disease in people with periodontal disease: a systematic review and meta-analysis. Clin Exp Dent Res. (2021) 7:109–22. doi: 10.1002/cre2.336
126. Romandini M, Baima G, Antonoglou G, Bueno J, Figuero E, Sanz M. Periodontitis, edentulism, and risk of mortality: a systematic review with meta-analyses. J Dent Res. (2021) 100:37–49. doi: 10.1177/0022034520952401
127. Mesa F, Arrabal-Polo MA, Magan-Fernandez A, Arrabal M, Martin A, Muñoz R, et al. Patients with periodontitis and erectile dysfunction suffer a greater incidence of major adverse cardiovascular events: a prospective study in a spanish population. J Periodontol. (2022) 93:1233–42. doi: 10.1002/JPER.21-0477
128. Beck JD, Couper DJ, Falkner KL, Graham SP, Grossi SG, Gunsolley JC, et al. The periodontitis and vascular events (PAVE) pilot study: adverse events. J Periodontol. (2008) 79:90–6. doi: 10.1902/jop.2008.070223
129. Offenbacher S, Beck JD, Moss K, Mendoza L, Paquette DW, Barrow DA, et al. Results from the periodontitis and vascular events (PAVE) study: a pilot multicentered, randomized, controlled trial to study effects of periodontal therapy in a secondary prevention model of cardiovascular disease. J Periodontol. (2009) 80:190–201. doi: 10.1902/jop.2009.080007
130. Becker L, Prado K, Foppa M, Martinelli N, Aguiar C, Furian T, et al. Endothelial dysfunction assessed by brachial artery ultrasound in severe sepsis and septic shock. J Crit Care. (2012) 27:316.e9–e3. doi: 10.1016/j.jcrc.2011.08.002
131. Hänsel S, Lässig G, Pistrosch F, Passauer J. Endothelial dysfunction in young patients with long-term rheumatoid arthritis and low disease activity. Atherosclerosis. (2003) 170:177–80. doi: 10.1016/s0021-9150(03)00281-8
132. Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med. (1997) 336:973–9. doi: 10.1056/NEJM199704033361401
133. Ridker PM, Hennekens CH, Buring JE, Rifai N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med. (2000) 342:836–43. doi: 10.1056/NEJM200003233421202
134. Kaptoge S, Di Angelantonio E, Lowe G, Pepys MB, Thompson SG, Collins R, et al. C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: an individual participant meta-analysis. Lancet. (2010) 375:132–40. doi: 10.1016/S0140-6736(09)61717-7
135. Ridker PM. From C-reactive protein to interleukin-6 to interleukin-1: moving upstream to identify novel targets for atheroprotection. Circ Res. (2016) 118:145–56. doi: 10.1161/CIRCRESAHA.115.306656
136. Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM Jr, Kastelein JJ, et al. Reduction in C-reactive protein and LDL cholesterol and cardiovascular event rates after initiation of rosuvastatin: a prospective study of the JUPITER trial. Lancet. (2009) 373:1175–82. doi: 10.1016/S0140-6736(09)60447-5
137. Del Giudice M, Gangestad SW. Rethinking IL-6 and CRP: why they are more than inflammatory biomarkers, and why it matters. Brain Behav Immun. (2018) 70:61–75. doi: 10.1016/j.bbi.2018.02.013
138. Calabró P, Willerson JT, Yeh ET. Inflammatory cytokines stimulated C-reactive protein production by human coronary artery smooth muscle cells. Circulation. (2003) 108:1930–2. doi: 10.1161/01.CIR.0000096055.62724.C5
139. Megson E, Fitzsimmons T, Dharmapatni K, Bartold PM. C-reactive protein in gingival crevicular fluid may be indicative of systemic inflammation. J Clin Periodontol. (2010) 37:797–804. doi: 10.1111/j.1600-051X.2010.01603.x
140. Singer RE, Moss K, Kim SJ, Beck JD, Offenbacher S. Oxidative stress and IgG antibody modify periodontitis-CRP association. J Dent Res. (2015) 94:1698–705. doi: 10.1177/0022034515602693
141. Machado V, Botelho J, Escalda C, Hussain SB, Luthra S, Mascarenhas P, et al. Serum C-reactive protein and periodontitis: a systematic review and meta-analysis. Front Immunol. (2021) 12:706432. doi: 10.3389/fimmu.2021.706432
142. Brito LC, DalBó S, Striechen TM, Farias JM, Olchanheski LR Jr, Mendes RT, et al. Experimental periodontitis promotes transient vascular inflammation and endothelial dysfunction. Arch Oral Biol. (2013) 58:1187–98. doi: 10.1016/j.archoralbio.2013.03.009
143. Campi P, Herrera BS, de Jesus FN, Napolitano M, Teixeira SA, Maia-Dantas A, et al. Endothelial dysfunction in rats with ligature-induced periodontitis: participation of nitric oxide and cycloxygenase-2-derived products. Arch Oral Biol. (2016) 63:66–74. doi: 10.1016/j.archoralbio.2015.11.022
144. Tonetti MS, D'Aiuto F, Nibali L, Donald A, Storry C, Parkar M, et al. Treatment of periodontitis and endothelial function. N Engl J Med. (2007) 356:911–20. doi: 10.1056/NEJMoa063186
145. Gugnani N, Gugnani S. Can treatment of severe periodontitis in patients with ST-segment elevation myocardial infarction improve endothelial function? Evid Based Dent. (2021) 22:5–7. doi: 10.1038/s41432-021-0157-3
146. Lobo MG, Schmidt MM, Lopes RD, Dipp T, Feijó IP, Schmidt KES, et al. Treating periodontal disease in patients with myocardial infarction: a randomized clinical trial. Eur J Intern Med. (2020) 71:76–80. doi: 10.1016/j.ejim.2019.08.012
147. Czesnikiewicz-Guzik M, Osmenda G, Siedlinski M, Nosalski R, Pelka P, Nowakowski D, et al. Causal association between periodontitis and hypertension: evidence from Mendelian randomization and a randomized controlled trial of non-surgical periodontal therapy. Eur Heart J. (2019) 40:3459–70. doi: 10.1093/eurheartj/ehz646
148. Muñoz Aguilera E, Suvan J, Buti J, Czesnikiewicz-Guzik M, Barbosa Ribeiro A, Orlandi M, et al. Periodontitis is associated with hypertension: a systematic review and meta-analysis. Cardiovasc Res. (2020) 116:28–39. doi: 10.1093/cvr/cvz201
149. Sharma S, Sridhar S, McIntosh A, Messow CM, Aguilera EM, Del Pinto R, et al. Periodontal therapy and treatment of hypertension-alternative to the pharmacological approach. A systematic review and meta-analysis. Pharmacol Res. (2021) 166:105511. doi: 10.1016/j.phrs.2021.105511
150. Tsakos G, Sabbah W, Hingorani AD, Netuveli G, Donos N, Watt RG, et al. Is periodontal inflammation associated with raised blood pressure? Evidence from a national US survey. J Hypertens. (2010) 28:2386–93. doi: 10.1097/HJH.0b013e32833e0fe1
151. Kawabata Y, Ekuni D, Miyai H, Kataoka K, Yamane M, Mizutani S, et al. Relationship between prehypertension/hypertension and periodontal disease: a prospective cohort study. Am J Hypertens. (2016) 29:388–96. doi: 10.1093/ajh/hpv117
152. Desvarieux M, Demmer RT, Jacobs JD Jr, Rundek T, Boden-Albala B, Sacco RL, et al. Periodontal bacteria and hypertension: the oral infections and vascular disease epidemiology study (INVEST). J Hypertens. (2010) 28:1413–21. doi: 10.1097/HJH.0b013e328338cd36
153. Pietropaoli D, Del Pinto R, Ferri C, Ortu E, Monaco A. Definition of hypertension-associated oral pathogens in NHANES. J Periodontol. (2019) 90:866–76. doi: 10.1002/JPER.19-0046
154. Silveira TMD, Silva CFE, Vaucher RA, Angst PDM, Casarin M, Pola NM. Higher frequency of specific periodontopathogens in hypertensive patients. A pilot study. Braz Dent J. (2022) 33(5):64–73. doi: 10.1590/0103-6440202204914
155. Woessner M, Smoliga JM, Tarzia B, Stabler T, Van Bruggen M, Allen JD. A stepwise reduction in plasma and salivary nitrite with increasing strengths of mouthwash following a dietary nitrate load. Nitric Oxide. (2016) 1:1–7. doi: 10.1016/j.niox.2016.01.002
156. Mitsui T, Harasawa R. The effects of essential oil, povidone-iodine, and chlorhexidine mouthwash on salivary nitrate/nitrite and nitrate-reducing bacteria. J Oral Sci. (2017) 59:597–601. doi: 10.2334/josnusd.16-0593
Keywords: microbiota, dysbiosis, bacteremia, Porphyromonas gingivalis, nitric oxide, cardiovascular diseases
Citation: Gualtero DF, Lafaurie GI, Buitrago DM, Castillo Y, Vargas-Sanchez PK and Castillo DM (2023) Oral microbiome mediated inflammation, a potential inductor of vascular diseases: a comprehensive review. Front. Cardiovasc. Med. 10:1250263. doi: 10.3389/fcvm.2023.1250263
Received: 30 June 2023; Accepted: 10 August 2023;
Published: 30 August 2023.
Edited by:
Adriana Georgescu, Institute of Cellular Biology and Pathology (ICBP), RomaniaReviewed by:
Francisco Mesa, University of Granada, SpainDavid John Vigerust, Vanderbilt University, United States
© 2023 Gualtero, Lafaurie, Buitrago, Castillo, Vargas-Sanchez and Castillo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Diego F. Gualtero gualterodiego@unbosque.edu.co Gloria Inés Lafaurie lafauriegloria@unbosque.edu.co
†These authors have contributed equally to this work and share first authorship