Tribbles 3 deficiency promotes atherosclerotic fibrous cap thickening and macrophage-mediated extracellular matrix remodelling
 Laura Martinez-Campesino1
Laura Martinez-Campesino1  Klaudia Kocsy1
Klaudia Kocsy1  Jaime Cañedo1
Jaime Cañedo1  Jessica M. Johnston1
Jessica M. Johnston1  Charlotte E. Moss1
Charlotte E. Moss1  Simon A. Johnston1 Stephen Hamby2,3
Simon A. Johnston1 Stephen Hamby2,3  Alison H. Goodall2,3 Jessica Redgrave1
Alison H. Goodall2,3 Jessica Redgrave1  Sheila E. Francis1
Sheila E. Francis1  Endre Kiss-Toth1,4
Endre Kiss-Toth1,4  Heather L. Wilson1*
Heather L. Wilson1*- 1Department of Infection, Immunity and Cardiovascular Disease, Medical School, University of Sheffield, Sheffield, United Kingdom
- 2Department of Cardiovascular Sciences, University of Leicester, Leicester, United Kingdom
- 3National Institute for Healthcare Research, Leicester Biomedical Research Centre, Glenfield Hospital, Leicester, United Kingdom
- 4Biological Research Centre of the Hungarian Academy of Sciences, Szeged, Hungary
Tribbles 3 (TRIB3) modulates lipid and glucose metabolism, macrophage lipid uptake, with a gain-of-function variant associated with increased cardiovascular risk. Here we set out to examine the role of this pseudokinase in atherosclerotic plaque development. Human endarterectomy atherosclerotic tissue specimens analysed by immunofluorescence showed upregulated TRIB3 in unstable plaques and an enrichment in unstable regions of stable plaques. Atherosclerosis was induced in full body Trib3KO and Trib3WT littermate mice by injecting mPCSK9 expressing adeno-associated virus and western diet feeding for 12 weeks. Trib3KO mice showed expanded visceral adipose depot while circulatory lipid levels remained unaltered compared to wildtype mice. Trib3KO mice aortae showed a reduced plaque development and improved plaque stability, with increased fibrous cap thickness and collagen content, which was accompanied by increased macrophage content. Analysis of both mouse and human macrophages with reduced TRIB3 expression showed elongated morphology, increased actin expression and altered regulation of genes involved in extracellular matrix remodelling. In summary, TRIB3 controls plaque development and may be atherogenic in vivo. Loss of TRIB3 increases fibrous cap thickness via altered metalloproteinase expression in macrophages, thus inhibiting collagen and elastic fibre degradation, suggesting a role for TRIB3 in the formation of unstable plaques.
Introduction
Atherosclerosis is a progressive disease of the arteries where LDL particles from the circulation accumulate in the arterial intima and undergo modification by oxidation promoting endothelial dysfunction and immune cell recruitment (1, 2). As atherosclerotic lesions progress, the risk of plaque rupture and subsequent thrombosis increasingly depends on plaque composition. Fibrous cap thickness, collagen content, necrotic core size and the inflammatory microenvironment together determine plaque vulnerability, rather than plaque size per se (3). Macrophages are key effector cells in the initiation and progression of the atherosclerotic plaque; local signals influence polarisation of this immune cell type toward pro-atherogenic or at hero-protective phenotypes, thereby influencing plaque stability (4). Macrophages in the plaque engulf oxidised lipids and transform into foam cells (5). Further accumulation of foamy macrophages, inflammatory cells, and migration of vascular smooth muscle cells (VSMC) promotes atheroma development. Fibrous cap thickness can be further modulated by activated macrophages, key sources of extracellular matrix metalloproteinases in human and murine atherosclerotic plaques (6, 7).
The pseudokinase Tribbles 3 (TRIB3) has been implicated in a number of metabolic processes such as modulating lipid and glucose metabolism (8, 9), promoting insulin resistance (10–12), and inhibiting adipocyte differentiation (13–15). Human TRIB3 is ubiquitously expressed, with high levels in bone marrow, thyroid gland and peripheral blood leukocytes (16). The Q84R variant of the human TRIB3 gene has been associated with insulin resistance, altered insulin secretion and with increased cardiovascular risk (17) but the molecular mechanisms behind these associations are poorly understood. In a model of diabetic atherosclerosis using ApoE/Ldldr double knockout mice fed with high fat and high sugar diet, siRNA-depletion of Trib3 led to a reduction in insulin resistance and atherosclerotic burden (18). However, the diabetogenic diet used for the development of diabetes-accelerated atherosclerosis prevents a distinction between the systemic effects produced by promoting hyperglycaemia and diabetes from the effects associated with hyperlipidaemia and its local impact on atherosclerosis (19). In vitro, using the human THP-1 monocytic cell line, ox-LDL was found to enhance TRIB3 activity; ox-LDL treatment combined with TRIB3 over-expression promotes cholesterol accumulation and decreased production and secretion of pro- inflammatory cytokines IL-1β, MCP-1, and TNF (20).
Here we sought to examine the consequences of altered TRIB3 levels in atherosclerotic plaque development and the molecular mechanisms underpinning these, with a focus on macrophages, using a range of in vivo and in vitro human and murine models. We show TRIB3 expression is increased in unstable regions of human carotid artery plaques. Loss of TRIB3 in a full body knockout mouse model of atherosclerosis results in reduced atherosclerotic burden via increased collagen and fibrous cap thickness and increased macrophage plaque content. Human and mouse macrophages with reduced TRIB3 have altered expression of extracellular matrix metalloproteinases and actin remodelling pathways, resulting in increased actin levels, cellular size, and elongation.
Materials and methods
Human carotid plaque isolation and characterisation
Human tissue and blood samples from volunteers and patients who gave informed written consent, were collected under protocols approved by the University of Sheffield Research Ethics Committee and Sheffield Teaching Hospitals Trust Review Board (ref. STH18222 and SMBRER310) and in accordance with the Declaration of Helsinki.
Carotid plaques were removed during carotid endarterectomy and placed in 10% (v/v) neutral buffered formalin following decalcification by placing them in 0.5 M EDTA at pH8 for 7 days. Plaques were then divided and embedded into four 3 mm thick wax blocks (A, B, C, D) centred from the carotid bifurcation. 5 μm tissue sections were obtained from wax blocks using a Leica RM2245 semi-automated rotary microtome and placed on APES microscope slides. Tissue sections were stained with haematoxylin and Eosin (H&E) for histological classification. For immunofluorescence staining against TRIB3 rabbit anti-human TRIB3 ab64693 or ab50516 was used at 1:50 with NL557 anti-rabbit secondary (NL004) or Alexa 488 anti-rabbit secondary (4412S, Cell Signalling) at 1:1,000. Mouse anti-human CD68 (ab201340) was used at 1:50 with Alexa 594 anti-mouse secondary (8890S, Cell Signaling) at 1:1,000. One carotid plaque lesion was mounted per slide and imaged by tile scanning using a Leica AF6000LX inverted microscope with a 5x/0.12 dry objective. The plaque shoulder region was captured with 10x/0.3 or 20x/0.35 dry lens. Regions of interest were analysed across two sections with Fiji. The TRIB3 + area was normalised to the plaque area. The proportion of double positive stained CD68+/TRIB3+ macrophages was compared to the total CD68 + macrophages assessed separately in each fluorescent channel.
Human monocyte derived macrophage isolation and culture
Human blood samples drawn by venepuncture were mixed with 3.8% (w/v) trisodium citrate dehydrate (Na3C6O7 x H2O, Sigma). After gradient centrifugation using Ficoll-Paque PLUS (GE Healthcare), peripheral blood mononuclear cells (PBMCs) were isolated in phosphate buffer saline (PBS) containing 2 mM EDTA. RBC were lysed using an ammonium chloride solution (155 mM NH4Cl, 10 mM KHCO3, 0.1 M EDTA in H2O). Magnetic CD14 microbeads (Miltenyi Biotec) were used for the positive selection of human monocytes. Monocytes were incubated for 7 days at 37°C and 5% CO2 in RPMI 1640 medium (Gibco) containing: 10% (v/v) ultra-low endotoxin heat-inactivated foetal bovine serum (FBS) (Biowest); 1% (v/v) L-Glutamine (Lonza); 1% (v/v) penicillin-streptomycin (Gibco); and 100 nM of macrophage colony stimulated factor (M-CSF) (Peprotech). Small-interfering RNAs were transfected into human monocyte derived macrophages (HMDM) to achieve RNA silencing. The transfection reagent Viromer Green (Lipocalyx) was used following the manufacturer’s instructions. siRNA targeting TRIB3 gene (ON-TARGET plus smartpool siRNA #L- 003754-00, Dharmacon) was mixed at 12.5 nM with the transfection reagent. As a control, ON- TARGETplus smartpool control non-targeting siRNA (#D-001810-10-20, Dharmacon) was used at the same concentration.
Microarray analysis of human monocyte derived macrophages
The published Cardiogenic Consortium transcriptomic data set (21–23) was re-analysed in collaboration with Professor Alison Goodall and Dr Stephen Hamby (University of Leicester, United Kingdom) as described in Johnston et al. (24). The dataset contains microarray analysis of human monocyte derived macrophages from 596 donors (Illumina’s Human Ref-8 Sentrix Bead Chip arrays, Illumina Inc., San Diego, CA). The samples were ranked based on TRIB3 expression and top and bottom quartiles were compared, obtaining 5,782 differentially expressed genes co-expressed with TRIB3. The genes passing the cut off p-value < 0.05 and passing threshold fold change < 0.8 (directly correlated) and > 1.2 (indirectly correlated) were further analysed for a functional gene set enrichment analysis using the DAVID Functional Annotation Analysis Software1 and Metascape.org (25).
Experimental animal procedures and atherosclerosis development
All experiments were performed in accordance with UK legislation under the Animals (Scientific Procedures) Act 1986. The University of Sheffield Project Review Committee approved all animal experiments which were carried out under the UK Home Office Project Licence P5395C858. A Trib3 knock out (Trib3KO) mouse strain was developed using the gene-trap system targeting the first intron, as described previously (26). The vector contained two expression cassettes: one encoding the splice acceptor site, β-Galactosidase and neomycin; the second encoding a diagnostic marker and splice donor site. Heterozygous mice were backcrossed with C57/BL6 mice for 10 generations. New heterozygous animals were inter-crossed to obtain final Trib3KO embryos.
Mice were kept in an optimal and controlled environment to reduce stress. Mice were subjected to 12 h light/12 h dark cycle, at 22°C with 40–60% of humidity. 12-week-old Trib3KO and Trib3WT male mice [N = 6 per group, based on power calculations for number of animals from a previous similar study type (24)] were subjected to tail vein injection with an adeno-associated virus-based vector (rAAV8) carrying the mPCSK9-D377Y gene, purchased from UNC GTC Vector Core (Chapel Hill, NC). The mice received 6.1 × 1011 viral particles via a single injection. Following 7 days of recovery, the mice were fed with Western Rodent Diet (Special Diet Services, United Kingdom), with ingredients [% (w/w)]: Sucrose, 33.94; milk fat anhydrous, 20.00; casein, 19.50; maltodextrin, 10.00; corn starch, 5.00; cellulose, 5.00; corn oil, 1.00; calcium carbonate, 0.40; L-cystine, 0.30; choline bitartrate, 0.20; Cholesterol, 0.15; antioxidant, 0.01; AIN-76A-MX, 3.50; AIN-76A-VX, 1.00. Specification: Crude Fat, 21.4; Crude Protein, 17.5; Crude Fibre, 3.5; Ash, 4.1; Carbohydrate, 50.0.) The diet was fed for 12 weeks, as we described previously (24); atherosclerosis does not develop in the absence of PCSK9 or western diet (27, 28). Weekly weight measurements were performed over the duration of the diet. Mice were culled via pharmacological overdose of 0.2 ml sodium pentobarbital (200 mg/ml) applied into the peritoneal cavity followed by cervical dislocation. For mice used to assess atherosclerotic lesions or when blood was required, cervical dislocation was avoided, and cardiac puncture was performed following pentobarbital injection and prior to breath cessation. Following loss of pedal reflex, blood was collected into a heparinised syringe injected to the heart through the chest wall.
Plasma lipid profiling
Plasma was collected from the mice 3 h after fasting, separated from isolated blood by centrifugation (1,500 × g for 5 min at room temperature) and immediately stored at −80°C prior to a full lipid profile analysis: total cholesterol, low (LDL), and high (HDL) density lipoproteins, triglycerides and glucose. This was carried out in the Department of Clinical Chemistry at the Royal Hallamshire Hospital (Sheffield Teaching Hospitals) using a Roche Cobas 8000 modular analyser.
En face staining of the aorta
Quantification of atherosclerosis was performed by en face aorta and aortic root staining. Mice were euthanised and the aortic tree was perfused through the heart with PBS and then with 10% (w/v) neutral buffered formalin. Under a dissecting microscope, the aorta was dissected out from the diaphragm to the top of the aortic arch. Following careful cleaning of the adventitia and removal of surrounding material (fat and connective tissue), it was excised longitudinally and fixed in 4% (w/v) paraformaldehyde. Dissected aortas were stained with Oil Red O [ORO, 60% (v/v) in isopropanol] and secured with insect pins onto wax. Images to assess the atherosclerotic lesion area and total area of the aorta were taken with a macroscopic CCD camera and analysed by computer-assisted image analysis (NIS-Elements software, Nikon Instruments, United Kingdom).
Histological staining of the aortic root
Murine aortic sinus samples were obtained by excising the heart and transecting parallel to the atria. Following fixation in 10% formalin (v/v) buffered saline for at least 24 h, samples were serially cut (at 5 μm intervals) from the valve leaflets until the beginning of the aorta. Sections where all 3 valve leaflets were visible at different stages of the plaque development were selected. To determine lesion size and plaque features—necrotic core and fibrous cap thickness—sections were deparaffinised in xylene, rehydrated and stained with Elastic stain kit (Verhoeff Van Gieson/EVG Stain—ab150667) following manufacturer’s instructions. To determine collagen content, Picrosirius Red staining (ab246832) was used to detect fibrillar collagen networks, where the intensity threshold was set up from the mean threshold across multiple images from the same plaque. Lesion area relative to lumen area was calculated at different regions of the plaque, taken as the mean per plaque. Fibrous cap thickness was measured relative to the thickness of the plaque, taken as the mean per plaque. For immunohistochemistry, consecutive cross-sections were stained to assess smooth muscle cell (SMC) and macrophage content. SMC content was assessed using mouse anti-human smooth muscle actin (Dako, M0851) that required a mouse-on-mouse (MOM) staining kit (Vector, MP-2400). VectaFluor ready-to-use DyLight dye-labelled secondary antibody kit was used for fluorescence staining (Vector, DI-2794). For macrophage content, slides were incubated with Mac-3 (BD, 550292) and with secondary antibody Goat anti- Rat DyLight 488 (ImmunoReagents).
Bone marrow derived macrophage isolation and culture
Bone marrow was isolated from the femurs and tibias of Trib3KO and Trib3WT mice under aseptic conditions. The bone marrow was flushed using a syringe and 26-guage needle into RPMI 1640 medium + 10% (v/v) ultra-low endotoxin heat-inactivated FBS. The cell suspension was passed through a 40 μm cell strainer and centrifuged at 500 g for 5 min. The samples obtained could then be frozen after resuspension in 90% (v/v) ultra-low endotoxin heat inactivated FBS + 10% DMSO or cultured for further gene analysis.
Gene expression analysis by RT-qPCR
RNA isolation from macrophage in culture was performed using the RNeasy UCP kit (Qiagen) according to manufacturer’s instructions. cDNA was transcribed from the total RNA using the Precision nanoScriptTM 2 RT kit (Primerdesign Ltd.) according to the manufacturer’s instructions. Real time qPCR was performed using a Bio-Rad i-Cycler. The reagents used were Precision PLUS SYBR-Green master mix (Primerdesign) and specific primers (Supplementary Table 1) designed with NCBI BLAST. All assays were performed in triplicate and normalised to the expression levels of GAPDH, determined in our assays as the most suitable house-keeping gene in Trib3 deficient macrophages (Supplementary Figure 1).
Cytoskeleton staining and analysis
Bone Marrow Derived Macrophages (BMDMs) and hMDMs were fixed with 4% (v/v) paraformaldehyde (PFA) and permeabilised with 0.1% (v/v) Triton-X diluted in PBS following staining with Fluorescein Isothiocyanate Labelled Phalloidin (Sigma-Aldrich). The slides were mounted with antifade mounting medium with DAPI (Life Technology) and coverslips. All the samples were kept in the dark until imaging. Images to assess cell morphology were taken using a Leica AF6000 microscope at 10× and 20× magnification. Three fields of view per tissue per sample were captured and analysed using Image J software. The same threshold was set in all images and by using the “analyse particles” command, the above- mentioned features were selected for automated quantification analysis. Under circularity, values closer to “1” correspond to particles that have similar shapes to a perfect circle, if the value approaches zero the particle contains spindle shaped features and/or increased elongation. The aspect ratio (AR) was used to measure elongation, for this measurement the software fits an ellipse within each particle/cell and calculates the ratio within the major and minor axes, therefore an increased AR value corresponds to an increased cell elongation.
Scanning electron microscopy
BMDMs from Trib3WT and Trib3KO mice were cultured for 5 days in complete media supplemented with L929 (M-CSF rich) and seeded in coverslips to allow adhesion. After differentiation, all samples were fixed in 4% (v/v) PFA for 10 min followed by 2.5% (v/v) glutaraldehyde in 0.1 M sodium phosphate overnight. Thereafter, samples were washed with 0.1 M phosphate buffer at least twice with 10-min intervals. A secondary fixation in 2% (v/v) aqueous osmium tetroxide was performed. Samples were then dehydrated sequentially using 75, 95, and 100% (v/v) ethanol before drying using a Leica EM CPD300. When dried, samples were mounted on aluminium stubs, attached to carbon sticky tabs, and coated with 25 nm of gold in an Edwards S150B sputter coater. Images to assess cell morphology were taken using TESCAN Vega 3 LMU Scanning Electron Microscope at an acceleration voltage of 10 kV. Cell morphology was analysed using Image J software. After setting the same threshold and smoothing the background in all images, by using the “analyse particles” command, the cell surface area and elongation (as aspect ratio) features were selected for automated quantification analysis. For membrane ridge analysis, an optimised image analysis workflow based on ridge detection (29) was used to estimate ridge density in single cell SEM images. The ridge area density was calculated as ridge total cross-sectional area/cell cross-sectional area.
Statistical analysis
All statistical analysis and graphs were generated in GraphPad Prism 8 software, with the average of at least three independent replicates (individual animals, human donors or patients) with standard error of the mean shown. Differences between two or more groups were assessed using two-tailed Student’s t-test, one- or two-way ANOVA, assuming normal distribution and considering p-values < 0.05 as significant.
Results
Upregulation of tribbles 3 in unstable regions of human carotid artery plaques
Tissue sections from carotid plaques from human patients undergoing endarterectomy, stained with H&E were classified based on the Stary classification (30) to determine their stability (Figure 1A). Sequential sections from these carotid plaques were used to assess expression of TRIB3 according to disease severity and regions associated with atherosclerotic plaque instability (Figure 1A). Quantification of TRIB3 positive areas showed an increased expression in unstable plaques (Figure 1B). Staining of TRIB3 showed it was not widely expressed within the whole plaque, but was found predominated in the shoulder region, considered vulnerable sites for plaque rupture. The percentage of total TRIB3 + ve shoulder regions of stable plaques represented 44% of the total signal (Figure 1C), suggesting that TRIB3 is upregulated in unstable areas. In unstable plaques 30% of TRIB3 staining was found in this region while the remaining staining was expressed diffusely across other areas. Double staining of carotid plaques for TRIB3 and the pan-macrophage marker, CD68, demonstrated that unstable plaques show an increased proportion of macrophages with high expression of TRIB3, compared to stable plaques (Figures 1D,E). Overall, these initial results suggest that TRIB3 is upregulated in unstable plaque macrophages and is more highly expressed in the unstable (shoulder) regions of atherosclerotic lesions compared to stable regions, raising the possibility that macrophage TRIB3 may play a regulatory role in plaque stability.
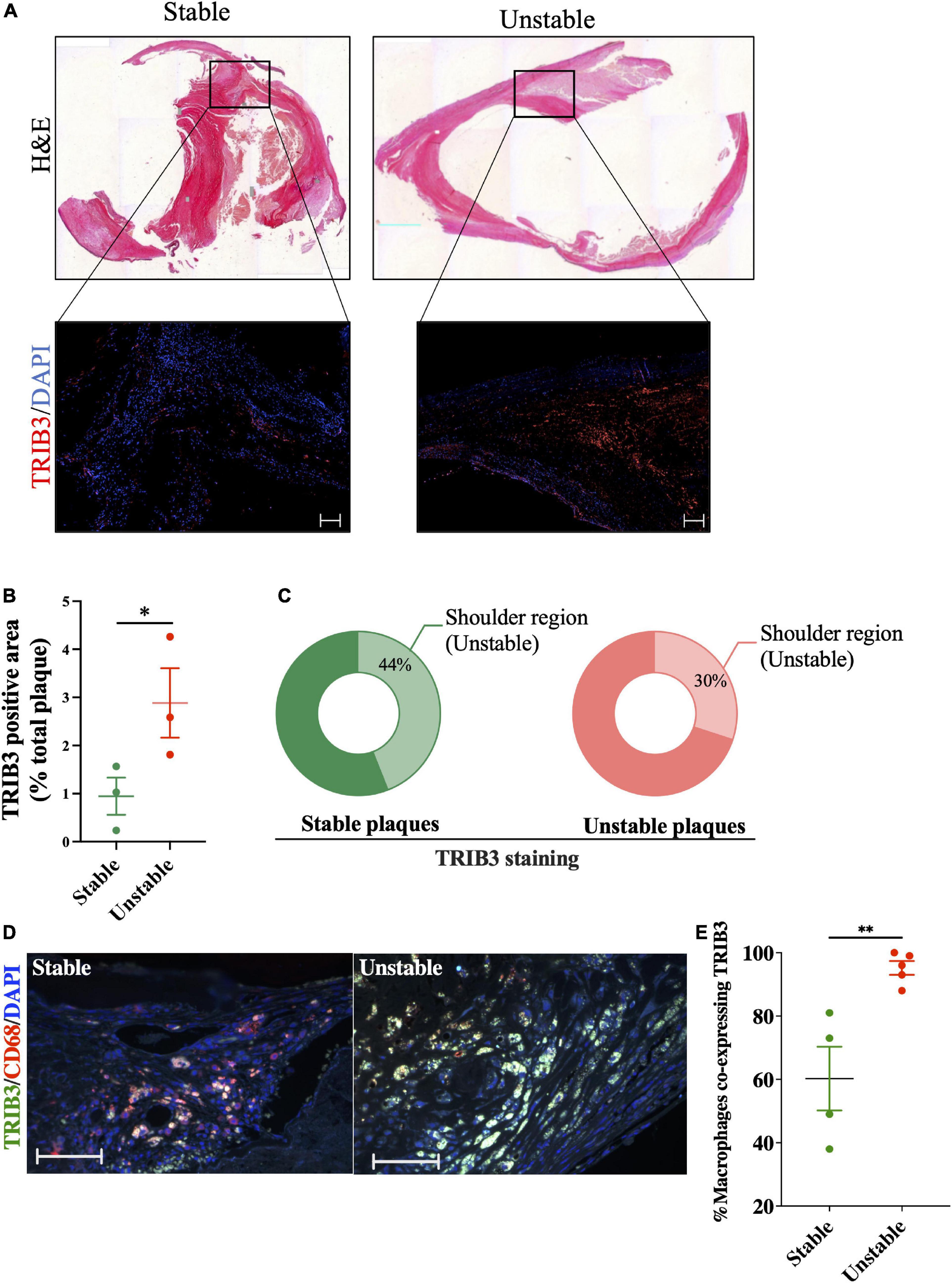
Figure 1. TRIB3 expression is increased in human unstable carotid plaques, in unstable plaque regions and in macrophages from unstable plaques, relative to stable plaques. (A) H&E staining of human carotid plaques was used to identify plaque features, classified as stable or unstable using the Stary classification guidelines. Below, representative images of the shoulder region of these plaques with TRIB3 staining (red) with DAPI (blue) as a nuclear marker (scale bars represent 100 μm). (B) Quantification of TRIB3 expression, as a percentage of the plaque area, of stable (0.944 ± 0.38) and unstable (2.88 ± 0.72) plaques. (C) Percentage of the total TRIB3 staining in the shoulder region (stable: 43.71 ± 9%; Unstable: 56 ± 9%). (D) Representative images of TRIB3 and CD68 macrophage co-staining from stable and unstable plaques (scale bars represent 100 μm). (E) Quantification of percentage CD68 + macrophages co-expressing TRIB3 in stable and unstable plaques. Data is shown as the mean ± SEM; each data point represents one donor (N = 3–5 patients). Unpaired t-test, *p < 0.05, **p < 0.01.
Atherogenic diet-fed Trib3KO mice have increased body mass and expanded visceral adipose
Since we found that TRIB3 was upregulated in vulnerable and unstable regions of human plaques we used a Trib3KO mouse model of atherosclerosis to assess the consequences of Trib3-deficiency in atherosclerosis development and in fibrous cap formation. Twelve week-old male Trib3KO and Trib3WT mice were injected with adeno-associated-virus-8, over-expressing the LDLR inhibitor PCSK9 (rAAV8-PCSK9). One week after injection, mice were fed with Western diet for 12 weeks (Figure 2A). Body weight was monitored weekly (Figure 2B) with no overall difference in weight gain between Trib3KO and Trib3WT mice, although Trib3KO started and maintained a higher average weight during the experiment (Supplementary Figures 2A,B).
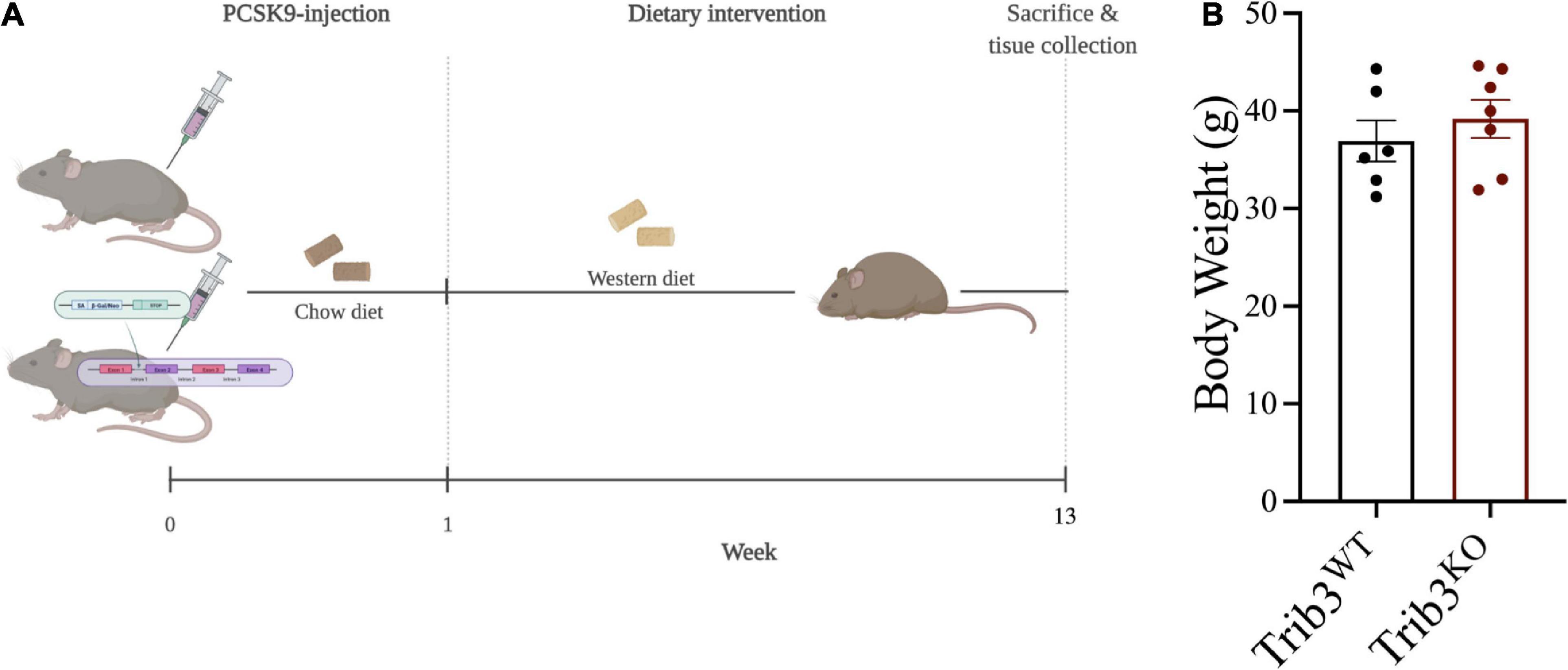
Figure 2. Atherosclerosis model and weight gain in Trib3KO mice. (A) Schematic representation of atherosclerosis development model used in Trib3WT and Trib3KO mice. (B) Body weight measurements in Trib3WT and Trib3KO mice at 13 weeks.
Analysis of the visceral adipose tissue, liver and spleen mass showed a significant increase in adipose tissue expansion in Trib3KO mice with no differences in liver or spleen weights (Supplementary Figure 2C). Despite an increased body weight and adiposity in Trib3KO mice, there were no differences in levels of circulating triglycerides, cholesterol (total, HDL, and non-HDL), and glucose (Supplementary Figure 2D).
Reduced atherosclerotic burden and increased plaque cap thickness in Trib3KO mice
Analysis of lipid deposition by Oil Red O staining of the aorta and in the aortic arch (inner curvature and carotid bifurcations) in Trib3WT and Trib3KO mice showed that mean atherosclerotic burden did not differ between genotypes (Figures 3A,B). Lesion size within the aortic roots was unaltered between Trib3WT and Trib3KO genotypes (Figure 3C). Elastin and collagen content in the aortic root lesions was assessed using Elastic van Gieson (EvG) and picosirius red staining (Figure 3D), respectively, where collagen content was increased in Trib3KO plaques (Figure 3E). Atherosclerosis development and lesion character in Trib3WT and Trib3KO mice was assessed by grading individual plaques in relation to the stage of the disease as summarised in Supplementary Table 2 and described in Otsuka et al. (31). Initial lesions with adaptive intimal thickening consisting mainly of smooth muscle cells and isolated macrophage foam cells were classified as Grade 0; Intimal xanthoma or “Fatty steak” lesions were characterised by the accumulation of macrophages within the intima and intracellular lipid accumulation and classified as Grade 1. Grades 0 and 1 are considered early manifestations of the disease and the precursors of symptomatic atherosclerosis. Grade 2 were plaques showing pathological intimal thickening, characterised by the presence of lipid pools or small necrotic cores with or without thinner fibrous caps consisting of SMC. Fibroatheroma lesions were classified as Grade 3, these plaques contained larger necrotic cores characterised by increased macrophage apoptosis and encapsulated by fibrotic tissue and collagen forming the fibrous cap.
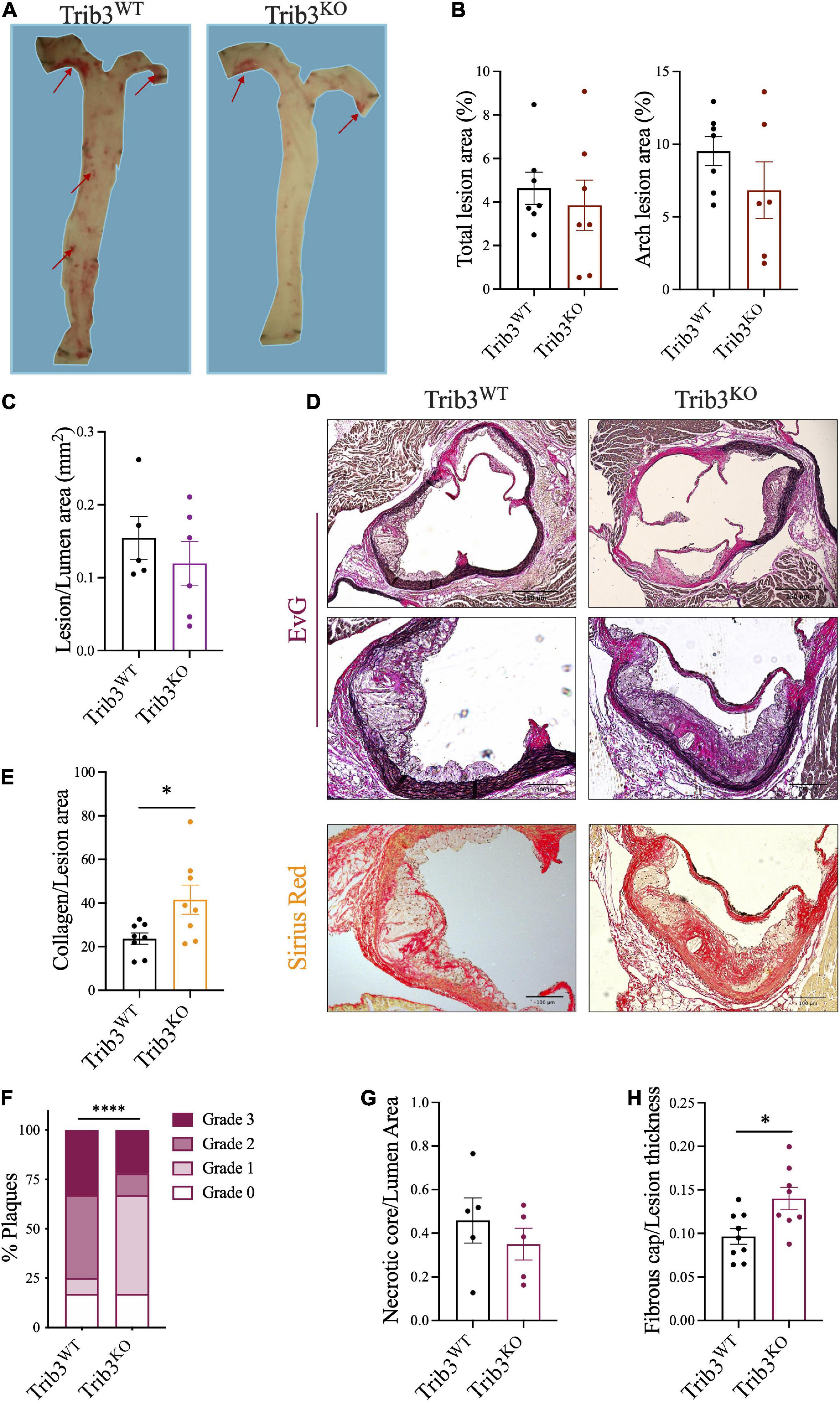
Figure 3. Atherosclerotic lesion size, disease progression and features in Trib3KO mice. (A) Representative images and quantification (B) of atherosclerotic lesions stained with ORO in the inner lumen of the aorta and in the aortic arch. (C) Total lesion quantification in the aortic root. Graphs are shown as the mean ± SEM, each data point represents a single mouse, unpaired t-test. (D) Representative images of Elastic van Gieson [elastin, original, and inset (high power image)] and picosirius red (collagen) stained cross-sections of aortic root lesions. (E) Collagen content quantification using picrosirius staining shown as mean ± SEM where each data point represents a single lesion; un-paired t-test *p < 0.05. (F) Double-blind plaque grading, made according to classification in Supplementary Table 2, presented as percentage of the total number of lesions (n = 14–17 lesions) using a Chi-square test, ****p < 0.0001. The necrotic core relative to lumen area (G) and fibrous cap relative to lumen thickness (H) quantification shown as mean ± SEM where each data point represents a single lesion where more than one lesion was assessed per individual mouse for a N = 6 mice for each group; un-paired t-test *p < 0.05. Scale bars represent 100 μm.
In a double-blinded analysis, all plaques were graded following these guidelines, revealing that Trib3KO mice present with less advanced disease with an increased content characteristic of early plaques (17% grade 0; 50% grade 1) and fewer advanced plaques (11% grade 2; 22% grade 3, Figure 3F). Wild-type littermates had a greater number of advanced lesions (42% grade 2; 33% grade 3) and fewer early plaques (17% grade 0; 8% grade 1, Figure 3F).
The association of plaque structure with plaque stability was further assessed by quantification of necrotic core area and fibrous cap thickness. No significant changes were detected in necrotic core areas (Figure 3G), however, plaques from Trib3KO mice had a significant increase in fibrous cap thickness (Figure 3H). These data, taken together with the twofold increase in collagen in plaques (Figure 3E) suggest that the absence of Trib3 does not affect atherosclerotic lesion size per se, but that it reduces the pathobiological complexity of early atherosclerotic disease associated with increased plaque stability in more advanced lesions.
Increased macrophage content in Trib3KO atherosclerotic plaques
Since Trib3 deletion increased features associated with plaque stability, mouse lesions were analysed for changes in smooth muscle cell and macrophage cell content, as major contributors to the development of stable vs. unstable plaque phenotypes. Sequential cross-sections of aortic root lesions from Trib3KO and Trib3WT mice were stained with α-SMA and MAC-3, to quantify the levels of SMC and macrophage content in the plaques, respectively (Figure 4A). Although Trib3KO plaques have increased fibrous caps, image analysis showed that smooth muscle cell content did not differ between Trib3KO and Trib3WT genotypes (Figure 4B). While an increase in macrophage content has traditionally been associated with reduced plaque stability (32) quantification of MAC3-positive staining showed a significant increase in plaque macrophages from Trib3KO mice (Figure 4C), suggesting that increased macrophage content, rather than smooth muscle cell content alone, may be driving the increase in fibrous cap thickness observed in Trib3KO lesions. To elucidate the molecular mechanisms regulating fibrous cap thickness, we next sought to analyse the consequences of Trib3 deficiency on the macrophage phenotype.
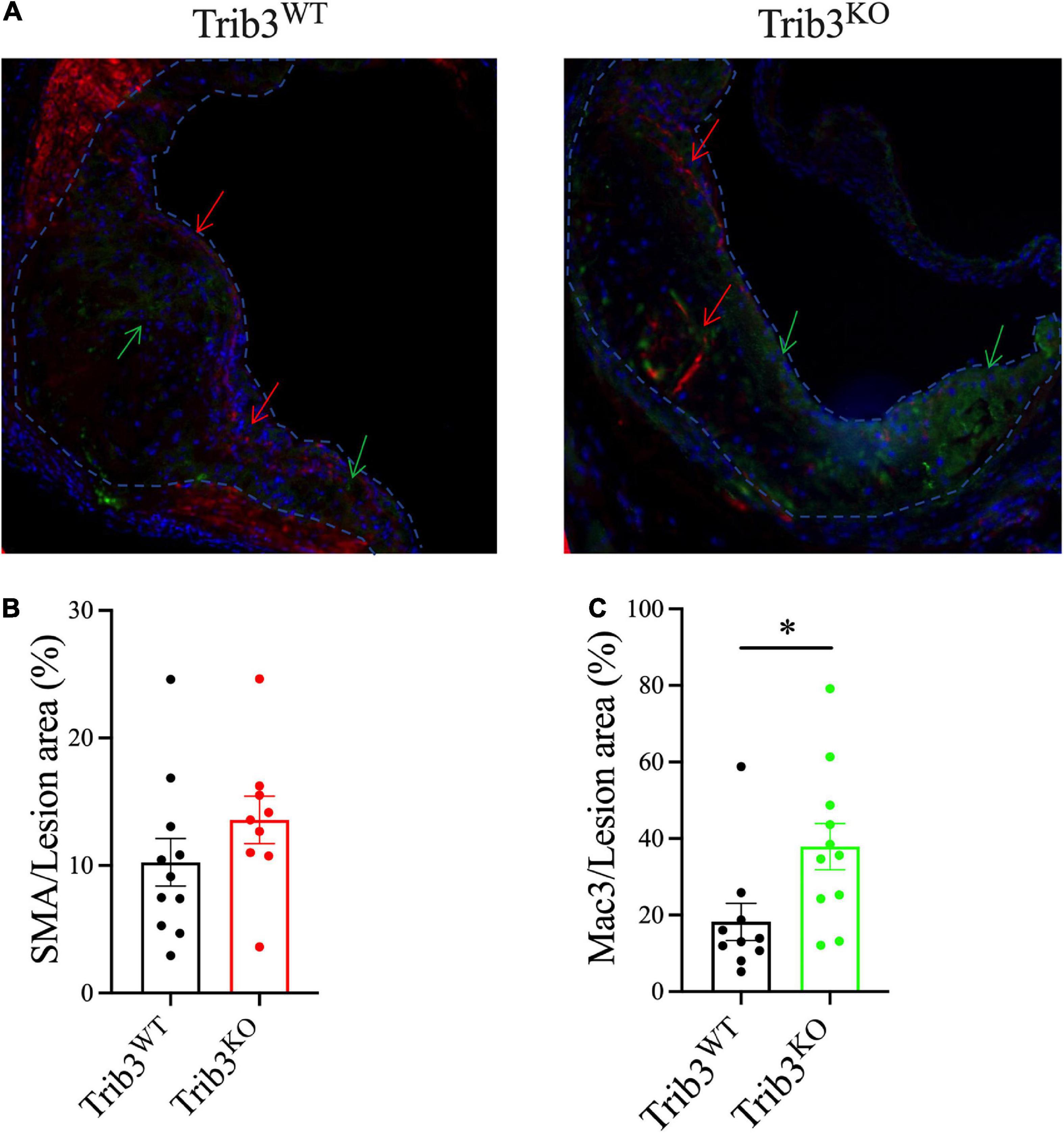
Figure 4. Genetic deletion of Trib3 has no effect on SMC content but increases macrophage presence in advanced atherosclerotic lesions with altered ECM degradative function. (A) Representative images of α-SMA (red) and MAC-3 (green) staining of aortic root lesions from Trib3KO and Trib3WT mice. DAPI (blue) was used as a marker for nuclear staining. Quantification of the SMC area (B) and macrophage area (C) immunostaining as a percentage of the total lesion area. Graphs are shown as the mean ± SEM; each data point represents a lesion (n = 9–10 lesions from N = 6 mice for each group). Unpaired t-test, *p < 0.05.
Loss of tribbles 3 dysregulates extracellular matrix remodelling pathways in human and mouse macrophages
First, we explored whether the levels of Trib3 RNA levels in macrophages are associated with an altered regulation of genes associated with increased plaque stability. To achieve this, we analysed the gene expression profile of human primary monocyte-derived macrophages (MDMs) from the Cardiogenics Consortium transcriptomic data set (21–23) of 596 donors. Samples were ranked based on TRIB3 expression then transcriptomic profiles between the upper and lower quartiles were compared. Differentially expressed genes between the samples with high and low TRIB3 levels were identified. A total of 5,782 significant genes were found to be differentially expressed between high vs. low TRIB3 expression. Genes were identified that were correlatively expressed for low TRIB3 (fold change = 0.32 from the mean); these were classified as directly correlated (fold change < 0.8) vs. inversely correlated (fold change > 1.2) to TRIB3 expression, respectively (Figure 5A and Supplementary Data).
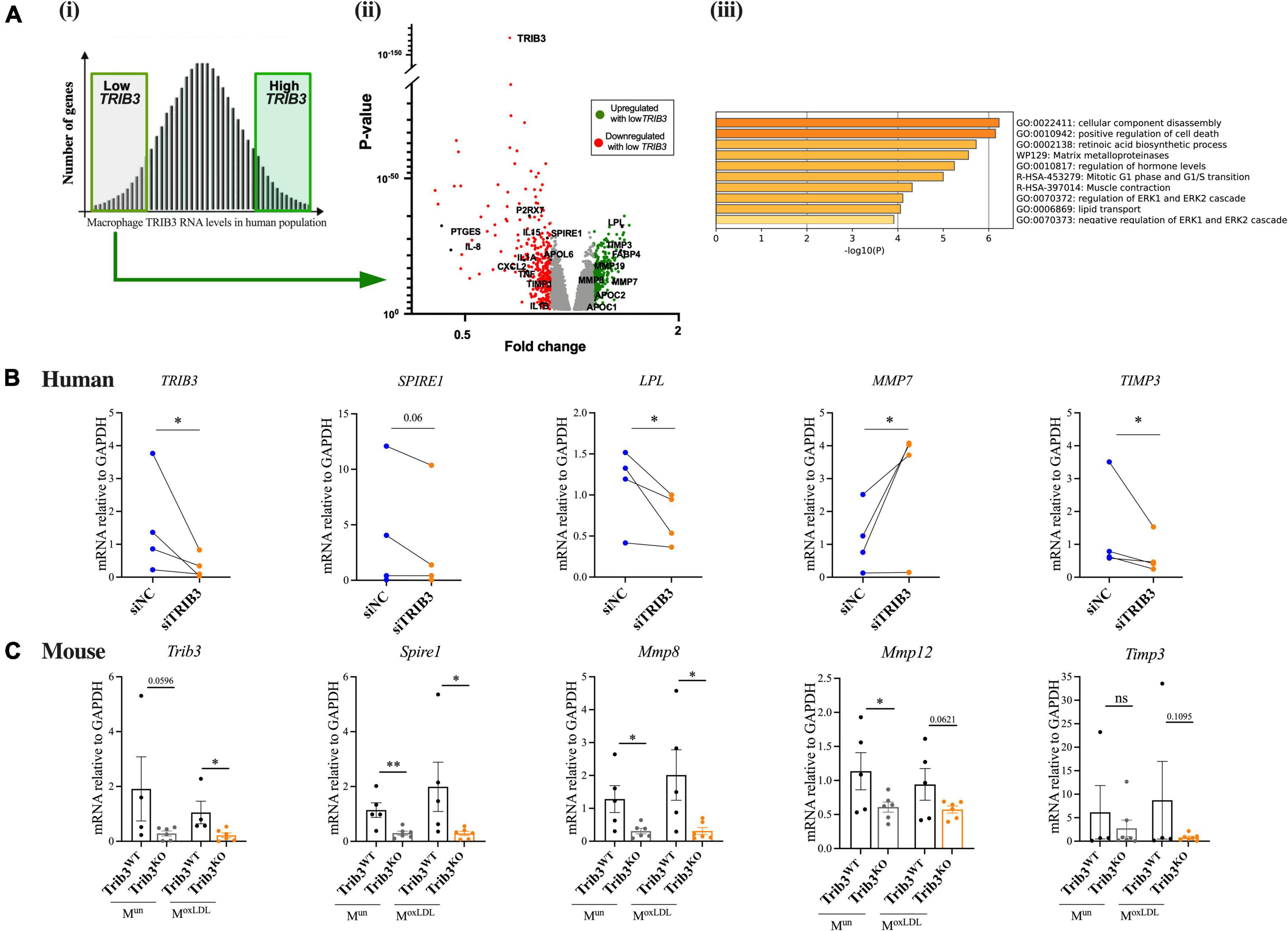
Figure 5. Loss of TRIB3 alters extracellular matrix remodelling pathways in human and mouse macrophages. (Ai) Microarray data of MDMs from 596 human donors was ranked based on TRIB3 expression and the top high and bottom low expressors were selected for further analysis. (ii) Under a low TRIB3 expression condition, the volcano plot shows the significant genes correlatively expressed with TRIB3, passing the cut off p-value < 0.05 and passing threshold fold change < 0.8 (red—downregulated, so correlatively expressed, with low TRIB3 expression; examples include P2 × 7, PTGES, IL-8, IL15, IL1A, SPIRE1, APOL6, SXSL2, TNF, TIMP1, IL1B) and > 1.2 (green—upregulated, so inversely correlated, with low TRIB3 expression; examples include LPL, TIMP3, FABP4, MMP19, MMP8, MMP7, APOC2, APOC1). (iii) Functional gene set enrichment analysis using Metascape for genes inversely correlated with low TRIB3 expression. (B) siTRIB3-knockdown mediated changes in mRNA expression levels of genes involved in lipid metabolism (LPL), cytoskeleton and ECM remodelling (SPIRE1, MMP7, and TIMP3) in human MDMs. Graphs are presented as individual connected points representing one donor, paired student’s T-test, *p < 0.05. (C) Expression of mouse Trib3 and ECM remodelling genes (Spire1, Mmp8, Mmp12, and Timp3) in Trib3WT and Trib3KO BMDMs treated or untreated with oxLDL. Graphs are shown as the mean ± SEM (N = 4–6). Unpaired t-test, *p < 0.05, **p < 0.01. GAPDH expression was used as an internal control.
Analysis of the genes in the Cardiogenics dataset that significantly correlated with low human MDM TRIB3 expression were entered into a functional gene set enrichment analysis using the DAVID Functional Annotation Analysis Software and Metascape. As summarised in Table 1 and Figure 5Aiii lower levels of TRIB3 in macrophages were significantly associated with metabolic, cardiovascular, and pharmacogenomic diseases. In the cellular component analysis, altered genes associated with TRIB3 downregulation were significantly enriched for “extracellular exosome,” “Cytosol,” “Cytoplasm,” “Plasma membrane,” “Extracellular space” and “Extracellular region.” In the analysis of biological functions, the inflammatory response was most significantly enriched (FDR = 2.2 × 106). Extracellular matrix disassembly was altered (FDR = 7.9 × 102; p = 2.0 × 104), notably with matrix regulatory genes MMP7, LPL, TIMP3, MMP8, COL22A1, MMP19, SPIRE1, and TIMP1 in the top 5% of the 5,782 genes significantly altered between high vs. low TRIB3 expression (Figure 5Aii).
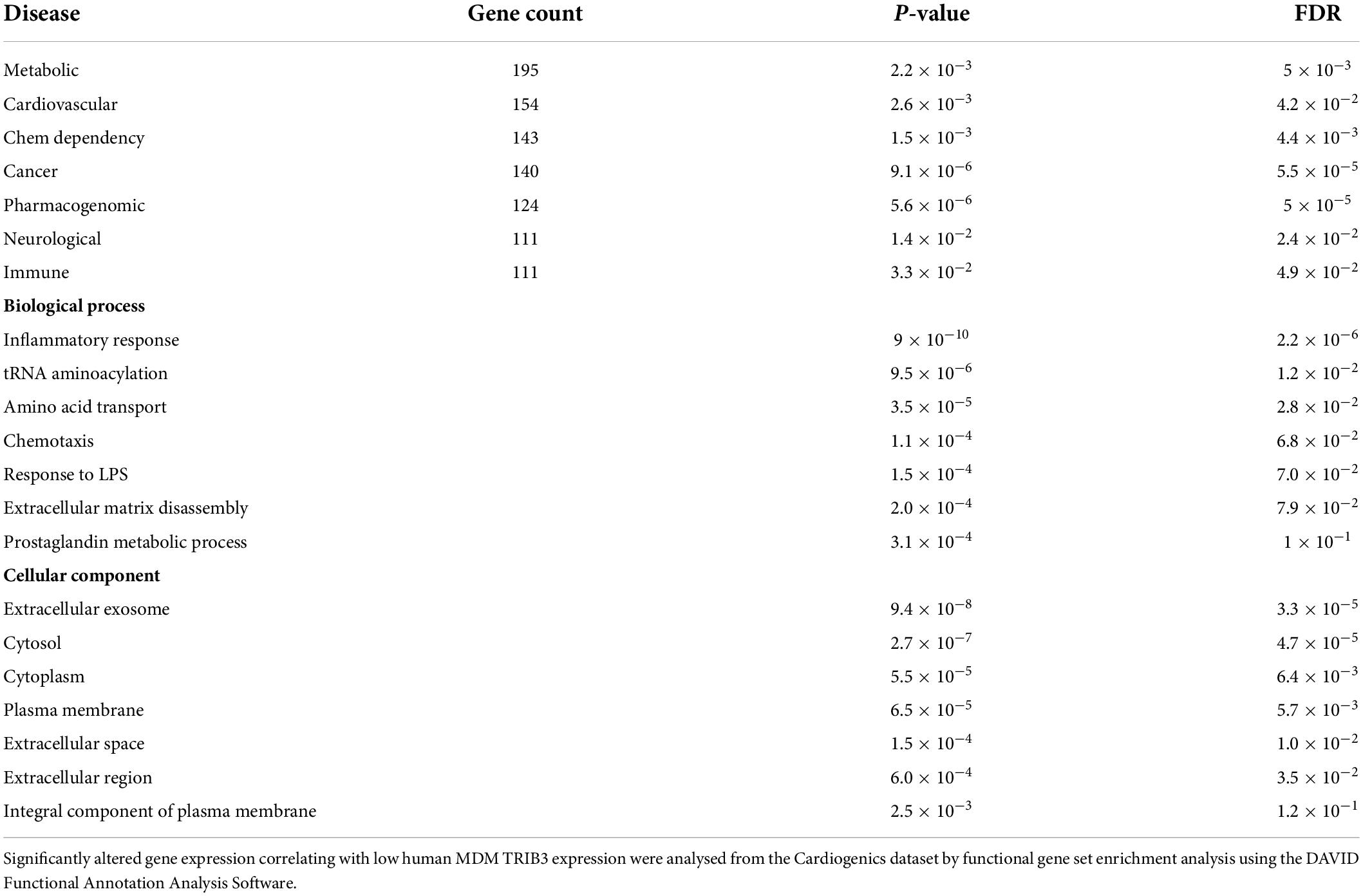
Table 1. Functional gene ontology analysis of human macrophages with low TRIB3 expression.
To substantiate whether these TRIB3-correlative transcriptomic changes in macrophages showed possible causality, expression of the most significant and altered genes associated with low TRIB3 expression in the human microarray was assessed in human MDMs transfected with TRIB3-targeting siRNA, compared to non-targeting siRNA. The efficiency of TRIB3 gene silencing was 46 ± 9%, determined by quantification of TRIB3 mRNA levels (Figure 5B). The effect of TRIB3 knockdown on expression of selected extracellular matrix remodelling genes was evaluated (Figure 5B). In some cases, changes in the opposite direction were detected in the analysis of gene expression by qRT-PCR compared to the microarray data, likely reflecting differences between TRIB3 knockdown compared to correlative, co-expression analysis of the Cardiogenics data. TRIB3 knockdown resulted in significant downregulation of LPL (lipoprotein lipase) expression, a driver of lipid metabolism and lipoprotein uptake. A significant reduction in expression of TIMP3, a metalloproteinase inhibitor, was also seen in the TRIB3 knockdowns (Figure 5B). Conversely, TRIB3 knockdown led to increased expression of matrix metalloproteinase MMP7 in contrast to the lower expression seen in the Cardiogenics dataset in relation to TRIB3 expression.
Key genes in the differentially expressed human macrophage extracellular matrix remodelling pathways identified in the Cardiogenics dataset were then compared in bone marrow derived macrophages (BMDMs) isolated from Trib3WT and Trib3KO mice. BMDMs from Trib3WT and Trib3KO mice were treated with oxLDL to mimic the plaque foam cell phenotype. Trib3 expression in these cells confirmed the genotype of the Trib3WT and Trib3KO samples (Figure 5C). Spire1, an actin nucleation factor involved in the actin cytoskeleton organisation and actin nucleation whose alteration affects cell shape and ECM remodelling, was downregulated in both unpolarised and oxLDL-treated Trib3KO macrophages. SPIRE1 expression was also compared in human MDMs where a decreased expression with TRIB3 knockdown was observed in 2 out of 3 donors tested (Figure 5C). Timp3 regulates MMPs and therefore plays a key role in extracellular matrix composition; its mRNA levels were not significantly altered upon oxLDL-treatment or Trib3 downregulation. Relative expression levels of the collagenolytic Mmp8 and elastinolytic Mmp12 metalloproteinases that degrade components of the ECM were also significantly reduced in Trib3KO unpolarised and oxLDL-treated macrophages. Taken together, these data indicate that Trib3 regulates extracellular matrix remodelling by reducing metalloproteinase-8 and −12 expression in foamy macrophages, accounting for the increased collagen and fibrous cap thickness seen in by Trib3KO atherosclerotic lesions.
Loss of tribbles 3 results in cellular elongation, increased surface area, increased actin, and altered polarisation of macrophages
Since TRIB3 knockdown or knockout in both human and mouse macrophages alters the expression of genes in extracellular matrix pathways, we assessed the functional impact of reduced TRIB3 levels on macrophage cell morphology and cytoskeleton. Human MDMs transfected with TRIB3-targeting siRNA, compared to non-targeting siRNA, were stained with F-actin-binding fluorescein isothiocyanate labelled-phalloidin. Cell size, F-actin intensity and shape descriptors (circularity and elongation) were analysed by fluorescence microscopy. This showed that TRIB3 downregulation led to an increase in cell size, measured as F-actin area per nuclei, as well as increased actin expression, measured as MFI (Figure 6A). Morphological analysis of siTRIB3 macrophages showed no significant changes in cell circularity, while a significant increase in elongation was detected compared to non-targeting siRNA control transfected cells (Figure 6A).
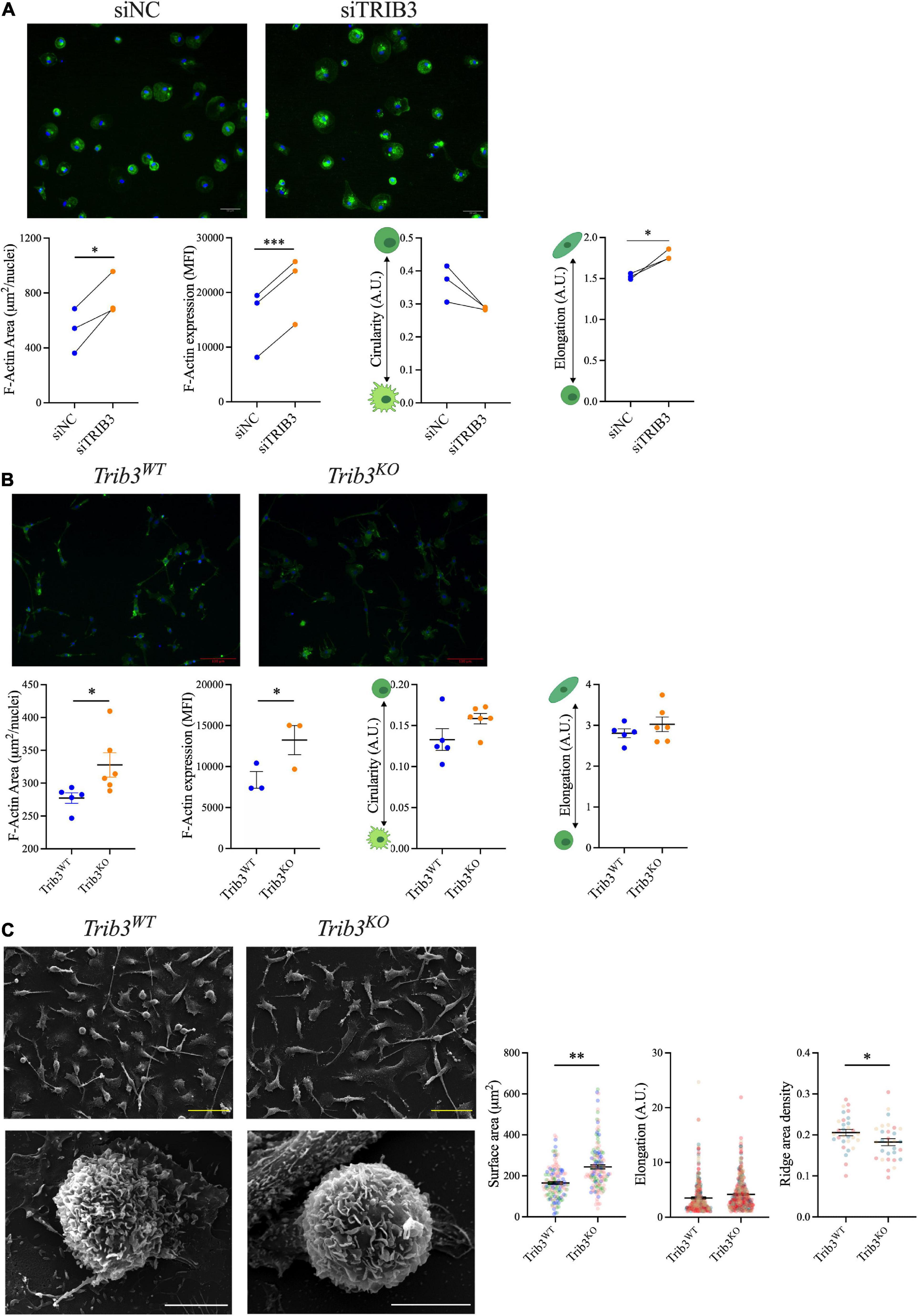
Figure 6. Increased F-actin and size of macrophages with reduced TRIB3 expression. (A) F-actin staining and quantification of human MDMs with non-targeting or siTRIB3-knockdown; representative images of phalloidin (green) and Dapi (blue) staining (upper) with quantification of cell size (F-actin area), F-actin mean fluorescence intensity and shape descriptors (circularity and elongation), shown connected points for each donor, compared by paired student’s T-test, *p < 0.05, ***p < 0.001. (B) F-actin staining and quantification of Trib3WT and Trib3KO BMDMs; representative images of phalloidin (green) and Dapi (blue) staining (upper) with quantification of cell size (F-actin area), F-actin mean fluorescence intensity and shape descriptors (circularity and elongation). Shown as Mean ± SEM where each data point is the average of all cell measurements per individual mouse (3 fields of view per mouse N = 3–6) unpaired student’s t-test, *p < 0.05 (C) Trib3WT and Trib3KO BMDMs imaged by scanning EM; quantification of cell surface area, elongation and ridge area density shown as Mean ± SEM where each data point is from an individual cell (3 fields of view per mouse N = 3, 30–60 cells for surface area; 95–148 cells for elongation; 10 cells for ridge density measurements) compared by unpaired student’s T-test, *p < 0.05, **p < 0.01. Scale bars represent 100 μm.
To assess the cellular consequences of TRIB3-dependent changes in cytoskeleton and extracellular matrix in murine macrophages, the morphology of BMDMs from Trib3WT vs. Trib3KO mice was examined (Figures 6B,C). Phalloidin staining analysis showed that Trib3KO macrophages have an increased cell size (327.9 ± 18.50 μm2) compared to Trib3WT (277.4 ± 8.058 μm2) and a significant increase in F-actin mean fluorescence intensity (MFI) staining. In line with this, increased β-actin expression was detected by qPCR in Trib3KO BMDMs compared to WT (Figure 7A).
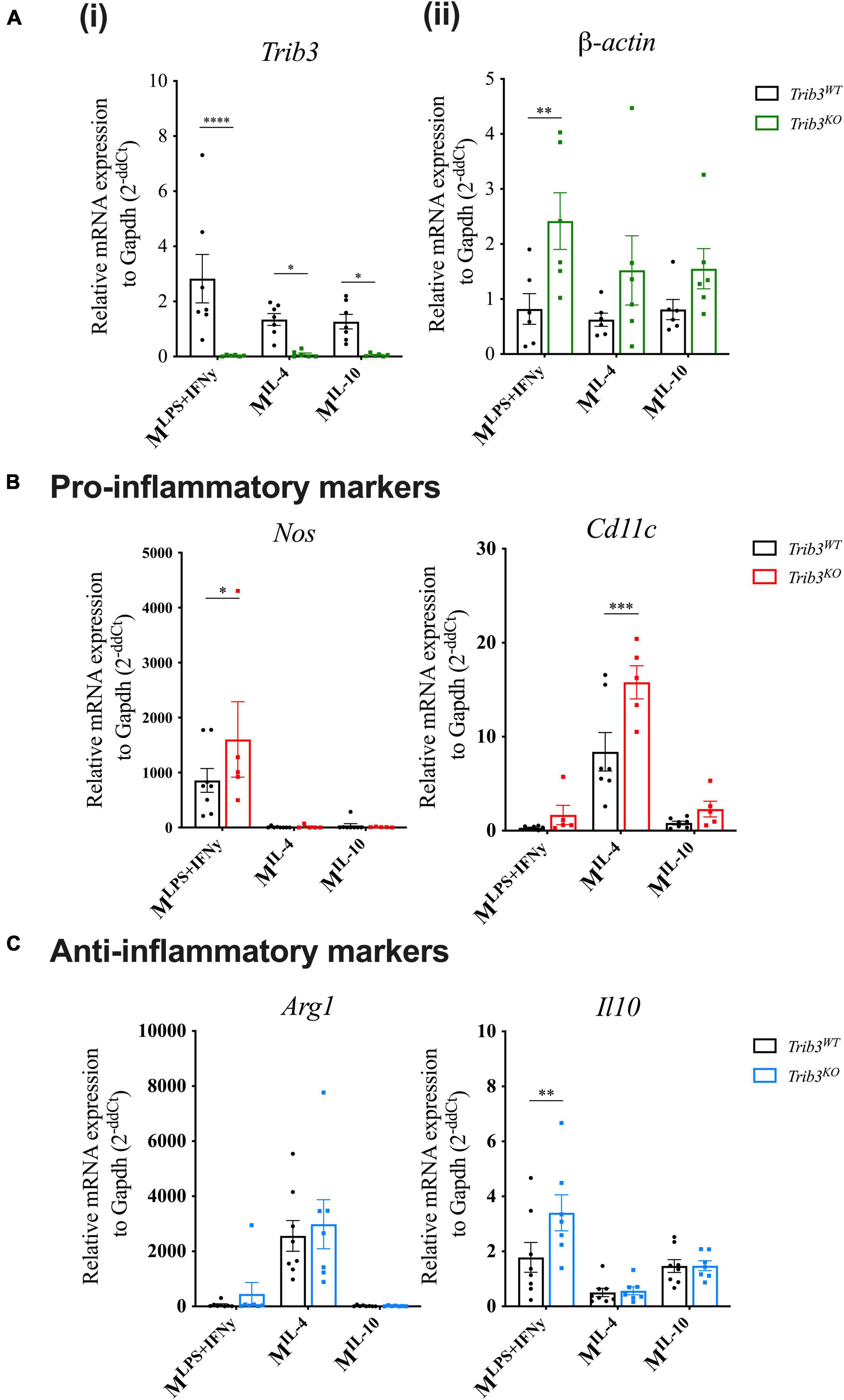
Figure 7. Trib3KO BMDMs show altered gene expression after polarisation. qPCR gene expression analysis after macrophage polarisation shows changes in the expression levels of (Ai) Trib3 to confirm the knockout genotype, (ii) β-actin for changes in the cytoskeleton, (B) and pro-inflammatory markers (Nos2 and Cd11c) and (C) anti-inflammatory markers Arg1 and Il-10. Gapdh expression was used as an internal control and normalised to unpolarised controls to give 2–ΔΔCt values. Graphs are presented as Mean ± SEM, two-way ANOVA with Fisher’s post hoc comparisons, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Changes in cellular morphology and elongation occur with macrophage polarisation. Polarised Trib3KO BMDMs showed a relative increase in inflammatory markers Nos2, when polarised with inflammatory cytokines (LPS + IFNγ), and Cd11c for polarisation with the anti-inflammatory cytokine, IL-4; while the anti-inflammatory marker Il10 was increased in Trib3KO BMDMs polarised with inflammatory cytokines (Figures 7B,C). Unlike the human MDMs, no differences in cell circularity or elongation were detected between the genotypes (Figure 6B). However, detailed examination of the morphological features of the Trib3WT and Trib3KO BMDMs using scanning electron microscopy showed that Trib3KO macrophages have increased cell size without affecting cell elongation, with a significant reduction in cell membrane ridges (Figure 6C).
Discussion
TRIB-3 modulates lipid and glucose metabolism, macrophage lipid uptake, with a gain-of-function variant associated with increased cardiovascular risk (17). In the current study, TRIB3 protein expression was increased in unstable regions of human carotid artery plaques. Using a mouse model of atherosclerosis, loss of TRIB3 resulted in increased macrophage numbers in atherosclerotic plaques and increased plaque stability features of thicker fibrous caps and an increase in collagen. Reduced or absent TRIB3 in human and mouse macrophages, respectively, led to altered expression of extracellular matrix metalloproteinases and actin remodelling pathways. This also led to an increased macrophage actin expression, increased cellular size and elongation suggesting a more reparative phenotype (33). Together these results suggest TRIB3 upregulates macrophage matrix production and reduces MMP expression, resulting in weakened fibrous caps and potentially increased plaque instability associated with acute cardiovascular disease events.
TRIB3 has been proposed as a key target to treat obesity-related diseases, as many studies have shown that its overexpression prevents the detrimental effects of diet-induced obesity by modulating lipid metabolism (9), promoting insulin resistance (10–12) and inhibiting adipocyte differentiation (13–15). In addition, in the context of inflammation and cardiovascular disease, loss of TRIB3 has been implicated in the development of diabetic atherosclerosis ameliorating insulin resistance and reducing macrophage apoptosis via modulating AKT phosphorylation (34, 35). TRIB3 upregulation has been associated with the development of a lean phenotype (9) and its over-expression in macrophages has been shown to promote cholesterol accumulation and reduced expression of pro-inflammatory cytokines (20).
Whilst previous studies have focused on the well-established roles of TRIB3 as a metabolic and AKT modulator, this is the first study to show in vivo and in vitro, using both human and murine models, that TRIB3 is mechanistically involved in modulating mechanisms associated with plaque stability. Here we have shown a clear relationship between the levels of TRIB3 and plaque stability in human carotid atherosclerotic plaques, with significantly upregulated expression in unstable compared to stable plaques. TRIB3 staining was found to be highly concentrated in macrophages within the shoulder region, considered unstable vulnerable sites of rupture where pro-inflammatory macrophages are predominantly localised (36).
Here we have shown that genetic deletion of Trib3 mediates changes in body weight supporting effects for atherosclerosis development in vivo. Along with an increased body weight, Trib3KO mice also presented an increased adiposity. There were, however, no changes in plasma lipid levels compared to WT littermates, likely to be a result of increased lipid tissue storage. The similar atherosclerotic lesion coverage in both aorta and aortic roots of Trib3WT and Trib3KO mice may reflect the similar levels of circulating lipids in both mouse genotypes. This atherosclerotic model for Trib3WT and Trib3KO mice showed no differences in blood glucose contrasting with an association of TRIB3 overexpression in human insulin resistance (11) and a diabetic mouse Trib3 siRNA knockdown, likely reflecting the difference in models and upregulation in disease. Changes in the atherosclerotic plaque morphology were noticeable visually, therefore a double blinded plaque classification analysis was carried out, revealing that Trib3KO mice had an increased number of early lesions, while Trib3WT mice had more advanced fibroatheromas. Although the lesion size was not affected by genetic deletion of Trib3, plaque character was altered with plaques from Trib3KO mice having significantly thicker fibrous caps and increased collagen content, indicating stable atherosclerotic disease.
Our results fit with other findings using a diabetic atherosclerosis model, combined with siRNA downregulated Trib3, where decreased atherosclerosis development with increased plaque stability features was observed with reduced macrophage apoptosis and increased phagocytosis suggested to prevent expansion of the necrotic core (18). Our study on whole body genetic deletion of Trib3 in a non-diabetic model of atherosclerosis also promotes mechanisms associated with increased plaque stability, by a reduction in the advancement of the plaque, with increased collagen and fibrous cap thickness, thereby delineating the impact of systemic/metabolic challenges from local changes in plaque-resident cell phenotype and function. We studied the cellular composition of the plaques since the formation of the fibrous cap is a multifactorial process with vascular smooth muscle cells (VSMC) and macrophages playing key roles in balancing the production and degradation of collagen, that ultimately determines the cap strength (37). VSMCs are the major collagen source in the artery wall; while their proliferation and migration promote plaque stabilisation, an increased production of inflammatory cytokines by activated immune cells will induce apoptosis and inhibit collagen synthesis in VSMC (38). In this regard, plaques from Trib3KO mice did not exhibit a difference in VSMC content. We cannot completely rule out a contribution of TRIB3 to VSMC phenotypes in atherosclerosis; future use of conditional Trib3-knockout mouse strains for myeloid and VSMC specific expression, will help us understand these specific cellular contributions to by Trib3 to plaque stability, VMSC phenotype and the type of collagen production, beyond the scope of this current study.
An increased macrophage content is conventionally considered a sign of plaque instability, as are inflammatory macrophage phenotypes. Mouse macrophages deficient in Trib3 showed altered expression of markers of polarisation following treatment with pro-inflammatory or anti-inflammatory cytokines where both inflammatory (“M1”) and anti-inflammatory (“M2”) markers were upregulated. Upregulation of these markers was polarisation treatment-dependent appearing more complex than simple phenotype switching. Interestingly others have found that overexpression of TRIB3 in human THP-1 macrophages has been shown to increase lipid accumulation and foam cell formation, with a reduction in inflammatory TNF secretion (20). Despite increased macrophage numbers being viewed as a sign of plaque instability, in contrast Trib3KO plaques had increased fibrous cap thickness and features of stability. Macrophages are one of the main sources of matrix metalloproteinases within the atherosclerotic plaque (39). Since we found increased fibrous cap thickness and macrophage content in Trib3KO lesions we sought to determine whether Trib3KO macrophages show an altered regulation of extracellular matrix associated with increased plaque stability.
We assessed mechanisms that could explain the role of TRIB3 in modulating gene expression and plaque macrophage phenotype using the Cardiogenics Consortium human macrophage transcriptomic dataset. When assessing causality, significant differences for macrophage TRIB3 downregulation were associated with genes involved in extracellular matrix remodelling and cell morphology regulation, with enrichment in cellular component annotations for the cytoskeleton and extracellular regions. This suggests Trib3 activation may reduce fibrous cap thickness and promote plaque instability, through macrophage-mediated extracellular matrix destruction. We assessed this in vitro in Trib3WT and Trib3KO BMDMs treated with oxLDL, to promote foam cell formation and mimic the atherosclerotic plaque microenvironment. The absence of Trib3 resulted in reduced expression of Spire1, a regulator of extracellular matrix degradation and MMP vesicular trafficking (40). In addition, Mmp8 and Mmp12 levels were reduced in Trib3KO macrophages, whose depletion in human diseased arteries and animal models of atherosclerosis results in reduced plaque size and increased collagen content, therefore improving plaque stability (41–44).
Morphological changes in macrophages mediated by TRIB3 were assessed by F-actin immunostaining. In humans and mice, TRIB3 silencing led not only to an increased cell size but also to a significant increase in cell elongation. Similarly, Trib3 deletion markedly increased macrophage cell size in unpolarised murine macrophages from the Trib3KO mice, which was associated with increased actin expression levels. The effects of Trib3 deletion on macrophage circularity and elongation were less pronounced in murine macrophages. A more detailed analysis using scanning electron microscopy of Trib3KO macrophages showed an increased cell size and a reduction of the number of cell membrane ridges in macrophages lacking Trib3 compared to WT. These changes in macrophage ridges may link to the increased phagocytosis response previously described in Trib3 knockdown macrophages (18).
In summary, our findings suggest that TRIB3 regulates macrophage cell shape, polarity, extracellular matrix remodelling and collagen, which leads to fibrous cap thinning and a reduction in features of plaque stability. TRIB3-activating therapies to promote a healthy obese phenotype may need to be balanced with the potential to cause atherosclerotic plaque instability and risk of acute cardiovascular events.
Data availability statement
The original contributions presented in this study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author/s.
Ethics statement
The studies involving human participants were reviewed and approved by the Human tissue and blood samples from volunteers and patients who gave informed written consent, were collected under protocols approved by the University of Sheffield Research Ethics Committee and Sheffield Teaching Hospitals Trust Review Board (ref. STH18222 and SMBRER310), Sheffield, United Kingdom, and in accordance with the Declaration of Helsinki. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by the University of Sheffield Project Review Committee approved all animal experiments which were carried out under the United Kingdom Home Office Project Licence P5395C858.
Author contributions
HW, EK-T, and LM-C conceived and designed the experiments. LM-C, JJ, KK, CM, and JC performed the experiments. LM-C, SF, JR, KK, AG, and SH analysed the data. LM-C and HW wrote the manuscript. JJ, KK, JC, SF, AG, and EK-T contributed to the writing of the article. All authors approved the submitted version.
Funding
LM-C, EK-T, and HW were supported by the European Marie Skłodowska Curie ITN Project TRAIN-TRIBBLES Research and Innovation Network (Grant No. 721532). EK-T was a member and supported by the UK Coronavirus Immunology Consortium (UK-CIC, MR/V028448/1). The Cardiogenic Consortium transcriptomic dataset was funded by the European Union 6th Framework Programme (LSHM-CT-2006-037593). CM was funded by a studentship to the Healthy Lifespan Institute (HELSI), University of Sheffield. KK and JR were supported/funded by the NIHR Sheffield Biomedical Research Centre (BRC)/NIHR Sheffield Clinical Research Facility (CRF).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2022.948461/full#supplementary-material
Footnotes
References
1. Doran AC, Meller N, McNamara CA. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler Thromb Vasc Biol. (2008) 28:812–9. doi: 10.1161/ATVBAHA.107.159327
2. Gimbrone MA, García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. (2016) 118:620–36. doi: 10.1161/CIRCRESAHA.115.306301
3. Falk E, Shah PK, Fuster V. Coronary plaque disruption. Circulation. (1995) 92:657–71. doi: 10.1161/01.CIR.92.3.657
4. Colin S, Chinetti-Gbaguidi G, Staels B. Macrophage phenotypes in atherosclerosis. Immunol Rev. (2014) 262:153–66. doi: 10.1111/imr.12218
5. Lewis GF, Rader DJ. New insights into the regulation of hdl metabolism and reverse cholesterol transport. Circ Res. (2005) 96:1221–32. doi: 10.1161/01.RES.0000170946.56981.5c
6. Newby AC. Metalloproteinases and vulnerable atherosclerotic plaques. Trends Cardiovasc Med. (2007) 17:253–8. doi: 10.1016/j.tcm.2007.09.001
7. Segers D, Helderman F, Cheng C, van Damme LCA, Tempel D, Boersma E, et al. Gelatinolytic activity in atherosclerotic plaques is highly localized and is associated with both macrophages and smooth muscle cells in vivo. Circulation. (2007) 115:609–16. doi: 10.1161/CIRCULATIONAHA.106.636415
8. Du K, Herzig S, Kulkarni RN, Montminy M. TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science. (2003) 300:1574–7.
9. Qi L, Heredia JE, Altarejos JY, Screaton R, Goebel N, Niessen S, et al. TRB3 links the E3 ubiquitin ligase COP1 to lipid metabolism. Science. (2006) 312:1763–6.
10. Liu J, Wu X, Franklin JL, Messina JL, Hill HS, Moellering DR, et al. Mammalian tribbles homolog 3 impairs insulin action in skeletal muscle: role in glucose-induced insulin resistance. Am J Physiol Endocrinol Metab. (2010) 298:E565–76. doi: 10.1152/ajpendo.00467.2009
11. Prudente S, Sesti G, Pandolfi A, Andreozzi F, Consoli A, Trischitta V. The mammalian tribbles homolog TRIB3, glucose homeostasis, and cardiovascular diseases. Endocr Rev. (2012) 33:526–46. doi: 10.1210/er.2011-1042
12. Weismann D, Erion DM, Ignatova-Todorava I, Nagai Y, Stark R, Hsiao JJ, et al. Knockdown of the gene encoding drosophila tribbles homologue 3 (TRIB3) improves insulin sensitivity through peroxisome proliferator-activated receptor-γ (PPAR-γ) activation in a rat model of insulin resistance. Diabetologia. (2011) 54:935–44. doi: 10.1007/s00125-010-1984-5
13. Bai XS, Lv LW, Zhou YS. [Tribbles pseudokinase 3 inhibits the adipogenic differentiation of human adipose-derived mesenchymal stem cells]. Beijing Da Xue Xue Bao Yi Xue Ban. (2020) 52:1–9.
14. Bezy O, Vernochet C, Gesta S, Farmer SR, Kahn CR. TRB3 blocks adipocyte differentiation through the inhibition of C/EBPβ transcriptional activity. Mol Cell Biol. (2007) 27:6818–31. doi: 10.1128/MCB.00375-07
15. Takahashi Y, Ohoka N, Hayashi H, Sato R. TRB3 suppresses adipocyte differentiation by negatively regulating PPARgamma transcriptional activity. J Lipid Res. (2008) 49:880–92. doi: 10.1194/jlr.M700545-JLR200
16. Stefanovska B, Andre F, Fromigue O. Tribbles pseudokinase 3 regulation and contribution to cancer. Cancers. (2021) 13:1822. doi: 10.3390/cancers13081822
17. Prudente S, Hribal ML, Flex E, Turchi F, Morini E, Cosmo SD, et al. The functional Q84r polymorphism of mammalian tribbles homolog TRB3 is associated with insulin resistance and related cardiovascular risk in caucasians from Italy. Diabetes. (2005) 54:2807–11. doi: 10.2337/diabetes.54.9.2807
18. Wang Z-H, Shang Y-Y, Zhang S, Zhong M, Wang X-P, Deng J-T, et al. Silence of TRIB3 suppresses atherosclerosis and stabilizes plaques in diabetic ApoE-/-/LDL receptor-/- mice. Diabetes. (2012) 61:463–73. doi: 10.2337/db11-0518
19. Wu KK, Huan Y. Diabetic atherosclerosis mouse models. Atherosclerosis. (2007) 191:241–9. doi: 10.1016/j.atherosclerosis.2006.08.030
20. Steverson D, Tian L, Fu Y, Zhang W, Ma E, Garvey WT. Tribbles homolog 3 promotes foam cell formation associated with decreased proinflammatory cytokine production in macrophages: evidence for reciprocal regulation of cholesterol uptake and inflammation. Metab Syndr Relat Disord. (2016) 14:7–15. doi: 10.1089/met.2015.0037
21. Heinig M, Petretto E, Wallace C, Bottolo L, Rotival M, Lu H, et al. A trans-acting locus regulates an anti-viral expression network and type 1 diabetes risk. Nature. (2010) 467:460–4. doi: 10.1038/nature09386
22. Rotival M, Zeller T, Wild PS, Maouche S, Szymczak S, Schillert A, et al. Integrating genome-wide genetic variations and monocyte expression data reveals trans-regulated gene modules in humans. PLoS Genet. (2011) 7:e1002367. doi: 10.1371/journal.pgen.1002367
23. Schunkert H, König IR, Kathiresan S, Reilly MP, Assimes TL, Holm H, et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. (2011) 43:333–8. doi: 10.1038/ng.784
24. Johnston JM, Angyal A, Bauer RC, Hamby S, Suvarna SK, Baidzajevas K, et al. Myeloid tribbles 1 induces early atherosclerosis via enhanced foam cell expansion. Sci Adv. (2019) 5:eaax9183. doi: 10.1126/sciadv.aax9183
25. Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. (2019) 10:1523. doi: 10.1038/s41467-019-09234-6
26. Salazar M, Lorente M, Garcia-Taboada E, Perez Gomez E, Davila D, Zuniga-Garcia P, et al. Loss of tribbles pseudokinase-3 promotes Akt-driven tumorigenesis via FOXO inactivation. Cell Death Differ. (2015) 22:131–44. doi: 10.1038/cdd.2014.133
27. Emini Veseli B, Perrotta P, De Meyer GRA, Roth L, Van der Donckt C, Martinet W, et al. Animal models of atherosclerosis. Eur J Pharmacol. (2017) 816:3–13. doi: 10.1016/j.ejphar.2017.05.010
28. Johnston JM, Francis SE, Kiss-Toth E. Experimental models of murine atherosclerosis: does perception match reality? Cardiovasc Res. (2018) 114:1845–7. doi: 10.1093/cvr/cvy140
29. Steger C. An unbiased detector of curvilinear structures. IEEE Trans Pattern Anal Mach Intell. (1998) 20:113–25. doi: 10.1109/34.659930
30. Stary H, Chandler A, Dinsmore R, Fuster V, Glagov S, Insull W, et al. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. a report from the committee on vascular lesions of the council on arteriosclerosis, american heart association. Circulation. (1995) 92:1355–74. doi: 10.1161/01.cir.92.5.1355
31. Otsuka F, Yasuda S, Noguchi T, Ishibashi-Ueda H. Pathology of coronary atherosclerosis and thrombosis. Cardiovasc Diagn Ther. (2016) 6:396–408. doi: 10.21037/cdt.2016.06.01
32. Chinetti-Gbaguidi G, Colin S, Staels B. Macrophage subsets in atherosclerosis. Nat Rev Cardiol. (2015) 12:10–7. doi: 10.1038/nrcardio.2014.173
33. McWhorter F, Wang T, Nguyen P, Chung T, Liu W. Modulation of macrophage phenotype by cell shape. Proc Natl Acad Sci USA. (2013) 110:17253–8. doi: 10.1073/pnas.1308887110
34. Sowers JR. Role of TRIB3 in diabetic and overnutrition-induced atherosclerosis. Diabetes. (2012) 61:265–6. doi: 10.2337/db11-1495
35. Wang W, Sun A, Lv W, Cheng J, Lv S, Liu X, et al. TRB3, up-regulated in kidneys of rats with type1 diabetes, mediates extracellular matrix accumulation in vivo and in vitro. Diabetes Res Clin Pract. (2014) 106:101–9. doi: 10.1016/j.diabres.2014.07.015
36. Stöger JL, Gijbels MJJ, Velden SVD, Manca M, Loos CMvd, Biessen EAL. Distribution of macrophage polarization markers in human atherosclerosis. Atherosclerosis. (2012) 225:461–8. doi: 10.1016/j.atherosclerosis.2012.09.013
37. Halvorsen B, Otterdal K, Dahl TB, Skjelland M, Gullestad L, Øie E, et al. Atherosclerotic plaque stability—what determines the fate of a plaque. Prog Cardiovasc Dis. (2008) 51:183–94. doi: 10.1016/j.pcad.2008.09.001
38. Libby P, Sukhova G, Lee RT, Galis ZS. Cytokines regulate vascular functions related to stability of the atherosclerotic plaque. J Cardiovasc Pharmacol. (1995) 25:S9.
39. Chang MY, Chan CK, Braun KR, Green PS, O’Brien KD, Chait A, et al. Monocyte-to-macrophage differentiation: synthesis and secretion of a complex extracellular matrix. J Biol Chem. (2012) 287:14122–35. doi: 10.1074/jbc.M111.324988
40. Lagal V, Abrivard M, Gonzalez V, Perazzi A, Popli S, Verzeroli E, et al. Spire-1 contributes to the invadosome and its associated invasive properties. J Cell Sci. (2014) 127:328–40. doi: 10.1242/jcs.130161
41. Johnson JL, George SJ, Newby AC, Jackson CL. Divergent effects of matrix metalloproteinases 3, 7, 9, and 12 on atherosclerotic plaque stability in mouse brachiocephalic arteries. Proc Natl Acad Sci USA. (2005) 102:15575–80. doi: 10.1073/pnas.0506201102
42. Laxton RC, Hu Y, Duchene J, Zhang F, Zhang Z, Leung K-Y, et al. A role of matrix metalloproteinase-8 in atherosclerosis. Circ Res. (2009) 105:921–9. doi: 10.1161/CIRCRESAHA.109.200279
43. Lenglet S, Mach F, Montecucco F. Role of matrix metalloproteinase-8 in atherosclerosis. Mediators Inflamm. (2013) 2013:e659282. doi: 10.1155/2013/659282
44. Luttun A, Lutgens E, Manderveld A, Maris K, Collen D, Carmeliet P, et al. Loss of matrix metalloproteinase-9 or matrix metalloproteinase-12 protects apolipoprotein E–deficient mice against atherosclerotic media destruction but differentially affects plaque growth. Circulation. (2004) 109:1408–14. doi: 10.1161/01.CIR.0000121728.14930.DE
Keywords: atherosclerosis, TRIB3, macrophage, fibrous, cap
Citation: Martinez-Campesino L, Kocsy K, Cañedo J, Johnston JM, Moss CE, Johnston SA, Hamby S, Goodall AH, Redgrave J, Francis SE, Kiss-Toth E and Wilson HL (2022) Tribbles 3 deficiency promotes atherosclerotic fibrous cap thickening and macrophage-mediated extracellular matrix remodelling. Front. Cardiovasc. Med. 9:948461. doi: 10.3389/fcvm.2022.948461
Received: 19 May 2022; Accepted: 02 August 2022;
Published: 26 August 2022.
Edited by:
Margreet R. De Vries, Leiden University Medical Center, NetherlandsReviewed by:
Geoff H. Werstuck, McMaster University, CanadaMenno Hoekstra, Leiden University, Netherlands
Copyright © 2022 Martinez-Campesino, Kocsy, Cañedo, Johnston, Moss, Johnston, Hamby, Goodall, Redgrave, Francis, Kiss-Toth and Wilson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Heather L. Wilson, H.L.Wilson@sheffield.ac.uk