The thromboxane receptor antagonist NTP42 promotes beneficial adaptation and preserves cardiac function in experimental models of right heart overload
 Eamon P. Mulvaney1
Eamon P. Mulvaney1  Fabiana Renzo1
Fabiana Renzo1  Rui Adão2 Emilie Dupre3 Lucia Bialesova1 Viviana Salvatore1 Helen M. Reid1 Glória Conceição2
Rui Adão2 Emilie Dupre3 Lucia Bialesova1 Viviana Salvatore1 Helen M. Reid1 Glória Conceição2  Julien Grynblat4,5 Aida Llucià-Valldeperas6,7
Julien Grynblat4,5 Aida Llucià-Valldeperas6,7  Jean-Baptiste Michel8
Jean-Baptiste Michel8  Carmen Brás-Silva2 Charles E. Laurent3,9
Carmen Brás-Silva2 Charles E. Laurent3,9  Luke S. Howard10
Luke S. Howard10  David Montani4,5,11 Marc Humbert4,5,11
David Montani4,5,11 Marc Humbert4,5,11  Anton Vonk Noordegraaf6
Anton Vonk Noordegraaf6  Frédéric Perros4,5,12,13
Frédéric Perros4,5,12,13  Pedro Mendes-Ferreira2,12
Pedro Mendes-Ferreira2,12  B. Therese Kinsella1,14*
B. Therese Kinsella1,14*- 1ATXA Therapeutics Limited, UCD Conway Institute of Biomolecular and Biomedical Research, University College Dublin, Dublin, Ireland
- 2Department of Surgery and Physiology, Cardiovascular R&D Centre—UnIC@RISE, Faculty of Medicine of the University of Porto, Porto, Portugal
- 3IPS Therapeutique Inc., Sherbrooke, QC, Canada
- 4School of Medicine, Université Paris-Saclay, Le Kremlin-Bicêtre, France
- 5INSERM UMR_S 999, Pulmonary Hypertension: Pathophysiology and Novel Therapies, Hôpital Marie Lannelongue, Le Plessis-Robinson, France
- 6PHEniX Laboratory, Department of Pulmonary Medicine, Amsterdam UMC (Location VUMC), Amsterdam Cardiovascular Sciences, Vrije Universiteit Amsterdam, Amsterdam, Netherlands
- 7Amsterdam Cardiovascular Sciences, Pulmonary Hypertension and Thrombosis, Amsterdam, Netherlands
- 8INSERM UMR_S 1116, Université de Lorraine, Vandoeuvre-lès-Nancy, France
- 9ToxiPharm Laboratories Inc., Ste-Catherine-de-Hatley, QC, Canada
- 10Imperial College London, National Heart and Lung Institute, London, United Kingdom
- 11AP-HP, Dept of Respiratory and Intensive Care Medicine, Pulmonary Hypertension National Referral Centre, Hôpital Bicêtre, Le Kremlin-Bicêtre, France
- 12Paris-Porto Pulmonary Hypertension Collaborative Laboratory (3PH), INSERM UMR_S 999, Université Paris-Saclay, Le Kremlin-Bicêtre, France
- 13INSERM, INRAE, CarMeN Laboratory and Centre de Recherche en Nutrition Humaine Rhône-Alpes (CRNH-RA), Claude Bernard University Lyon 1, University of Lyon, Lyon, France
- 14UCD School of Biomolecular and Biomedical Research, UCD Conway Institute of Biomolecular and Biomedical Research, University College Dublin, Dublin, Ireland
Background: Pulmonary arterial hypertension (PAH) is a progressive disease characterized by increased pulmonary artery pressure leading to right ventricular (RV) failure. While current PAH therapies improve patient outlook, they show limited benefit in attenuating RV dysfunction. Recent investigations demonstrated that the thromboxane (TX) A2 receptor (TP) antagonist NTP42 attenuates experimental PAH across key hemodynamic parameters in the lungs and heart. This study aimed to validate the efficacy of NTP42:KVA4, a novel oral formulation of NTP42 in clinical development, in preclinical models of PAH while also, critically, investigating its direct effects on RV dysfunction.
Methods: The effects of NTP42:KVA4 were evaluated in the monocrotaline (MCT) and pulmonary artery banding (PAB) models of PAH and RV dysfunction, respectively, and when compared with leading standard-of-care (SOC) PAH drugs. In addition, the expression of the TP, the target for NTP42, was investigated in cardiac tissue from several other related disease models, and from subjects with PAH and dilated cardiomyopathy (DCM).
Results: In the MCT-PAH model, NTP42:KVA4 alleviated disease-induced changes in cardiopulmonary hemodynamics, pulmonary vascular remodeling, inflammation, and fibrosis, to a similar or greater extent than the PAH SOCs tested. In the PAB model, NTP42:KVA4 improved RV geometries and contractility, normalized RV stiffness, and significantly increased RV ejection fraction. In both models, NTP42:KVA4 promoted beneficial RV adaptation, decreasing cellular hypertrophy, and increasing vascularization. Notably, elevated expression of the TP target was observed both in RV tissue from these and related disease models, and in clinical RV specimens of PAH and DCM.
Conclusion: This study shows that, through antagonism of TP signaling, NTP42:KVA4 attenuates experimental PAH pathophysiology, not only alleviating pulmonary pathologies but also reducing RV remodeling, promoting beneficial hypertrophy, and improving cardiac function. The findings suggest a direct cardioprotective effect for NTP42:KVA4, and its potential to be a disease-modifying therapy in PAH and other cardiac conditions.
Introduction
Pulmonary arterial hypertension (PAH) is a rare yet devastating disease with progressively debilitating symptoms and high mortality. The underlying etiology of PAH is characterized by excessive vasoconstriction and remodeling of the pulmonary vasculature leading to increased pulmonary vascular resistance (PVR). However, the ultimate determinant of survival in PAH patients is the response of the right ventricle (RV) (1, 2).
Upon elevated afterload due to increased PVR, the RV in PAH patients initially responds through compensatory mechanisms termed adaptive hypertrophy. These physiological responses aim to preserve systolic and diastolic right heart function and are characterized by an increased RV wall thickness facilitated by hypertrophic remodeling, increased angiogenesis, altered sarcomere organization and increased intrinsic cardiomyocyte contractility (1, 3, 4). However, in most PAH patients, these adaptive mechanisms are either insufficient or become exhausted, and RV hypertrophy ultimately transitions to pathological maladaptive mechanisms. Maladaptive remodeling of the RV is characterized by a transition to a more eccentric pattern of hypertrophy and a progressive RV dilation resulting in a leftward septal shift impacting left ventricular (LV) function (5, 6). In addition, the consequent elevation in RV wall tension results in an increased metabolic demand and a simultaneous decrease in myocardial perfusion capacity, leading to decreased RV contractility despite progressive increases in afterload (7). Furthermore, RV diastolic function is compromised through increased stiffness primarily due to cardiomyocyte hypertrophy and fibrosis. In PAH patients, all these factors contribute to progressive right heart dysfunction, ultimately resulting in heart failure.
While considerable advances have been made in the clinical management of PAH, patient mortality remains high. Current PAH standard-of-care (SOC) therapies include the phosphodiesterase type-5 inhibitors (PDE5is), endothelin receptor antagonists (ERAs), prostacyclin analogs (PCAs) or prostacyclin receptor agonists (PRAs) and soluble guanylate cyclase (sGC) stimulators, with various other pipeline compounds in clinical development. The key focus of the SOCs and those in clinical development is to reduce PVR, either by alleviating excessive pulmonary vasoconstriction or enhancing pulmonary vasodilation, and/or reduce pulmonary vascular remodeling. However, while it is recognized that RV function is the main determinant of prognosis in PAH, current SOC therapies have limited cardiac-specific effects (8, 9). Furthermore, in PAH there is a paradox where RV function continues to deteriorate and have poorer survival outcomes despite reductions in PVR observed using current PAH SOC therapies (10). Consequently, there has been a shift in the clinical thinking from solely considering the effects of PAH therapies on PVR to instead investigating their potential for directly addressing the effects on the RV.
The thromboxane (TX) A2 receptor, or TP, primarily mediates signaling of the prostanoid TXA2 and of the free-radical derived isoprostane 8-iso-prostaglandin F2α (8-iso-PGF2α), as well as other endogenous ligands, regulating processes including platelet aggregation, and constriction and proliferation of vascular and pulmonary smooth muscle. TP-mediated signaling also mediates potent pro-inflammatory, pro-mitogenic, and pro-fibrotic effects, and levels of TXA2, 8-iso-PGF2α, and TP expression are elevated in many cardiovascular and pulmonary diseases, inflammatory disorders and in certain cancers (11, 12). Multiple studies have shown the importance of TP signaling in the development and progression of PAH (13–19). TXA2 mimetics induce ventricular arrhythmia, and TP signaling contributes to cardiac hypertrophy and fibrosis in multiple animal models (19–26). TP expression has been demonstrated to be specifically elevated in certain pathological cardiac conditions, and both TP receptor occupancy and expression is elevated in the RV of PAH patients compared to non-diseased subjects (18, 19). In addition, the TXA2/TP signaling axis contributes to cardiac hypertrophy in multiple animal models of systemic hypertension (24, 25). While activation of the TP is profibrotic in multiple systems, including within the heart, TP antagonism with CPI211 (Ifetroban) decreased RV fibrosis and improved cardiac function in a pulmonary artery banding (PAB) model of RV pressure overload (19). Furthermore, TP antagonism improved cardiac output, increased ejection fraction while decreasing cardiac fibrosis and transforming growth factor (TGF)−β signaling in mouse models of Duchenne muscular dystrophy (DMD) (26). Taken together, these studies suggest a broader pattern of deleterious consequences of TP activation on the heart.
The TP antagonist NTP42 is currently in clinical development for PAH and other cardiopulmonary indications. Previous efficacy evaluations demonstrated that NTP42 attenuates preclinical PAH in both the monocrotaline (MCT)-and Sugen/Hypoxia (SuHx)-induced animal models of PAH (27, 28). As a drug specifically developed as an oral formulation for clinical use, NTP42:KVA4 was recently evaluated in a randomized, placebo-controlled first-in-human Phase I clinical trial (NCT04919863) in 79 healthy male volunteers where it was confirmed as safe, well-tolerated, with good pharmacokinetic and pharmacodynamic profiles following single and repeat oral dosing. To specifically assess its potential to impart direct cardioprotective effects on the RV, the aim of this study was to validate the efficacy of NTP42, delivered as the orally formulated NTP42:KVA4, in the MCT-PAH model while also exploring its effect in the pulmonary artery banding (PAB) preclinical model of RV dysfunction and pressure overload. Furthermore, in this study, we also examined expression levels of the TP, the target receptor for NTP42, in RV tissues from several highly relevant disease models as well as in clinical specimens from subjects with PAH and dilated cardiomyopathy (DCM), where the data further supports the hypothesis that the TP is a bona fide target for treatment of PAH and certain other cardiac dysfunctions.
Materials and methods
Animal models
All experiments were carried out in accordance with US NIH guidelines. Male Sprague-Dawley rats (Charles River Laboratories) were used in all models. MCT-PAH was induced using a single injection of 60 mg/kg MCT, where twice-daily oral treatment with placebo, NTP42:KVA4 (1 mg/kg), the PDE5i Sildenafil (50 mg/kg), the ERA Macitentan (30 mg/kg), the PRA Selexipag (1 mg/kg), or the sGC stimulator Riociguat (5 mg/kg) was commenced on Day 7 post-MCT and continued to Day 28 (Supplementary material). The PAB model used surgical banding of the pulmonary artery, where twice-daily oral treatment with placebo, NTP42:KVA4 (1 mg/kg), or the sGC stimulator Riociguat (5 mg/kg) was started on Day 2 post-PAB and continued to Day 27 (Supplementary material).
Human tissues
Human tissues, following autopsy, were obtained from the Institute of Cardiometabolism and Nutrition BioCollection (Paris, France), detailed in Supplementary Table 2. Protocols to obtain human biospecimens conformed with the recommendations of the Declaration of Helsinki.
A detailed description of the Materials and methods is presented in Supplementary material. Detailed descriptions of all materials and methods used in this study, including chemicals, animals and surgical procedures, tissue harvesting, preparation and histological staining and analysis, isolated cardiomyocyte force transduction experiments, quantitative real-time PCR, Western blotting, and statistical analyses are presented in Supplementary material. Statistical methods are also summarized in each figure legend, where values are expressed as mean ± standard error of the mean (SEM) and number of replicates (n). Throughout, P-values < 0.05 were considered to indicate significant differences.
Results
NTP42:KVA4 improves right ventricular adaptation in the monocrotaline- pulmonary arterial hypertension model
We previously reported that NTP42 alleviates pulmonary pathologies and cardiopulmonary hemodynamics in both the MCT- and SuHx-induced PAH animal models (27, 28). In those studies, NTP42 was administered orally as the active pharmaceutical ingredient (API) following its dissolution in an organic-based drug vehicle unsuited and not approved for use in man. Hence, for use in the clinical setting, NTP42 has since been uniquely formulated with the widely used pharmaceutical polymer Kollidon® VA 64 yielding the investigational medicinal product (IMP) referred to as NTP42:KVA4. Herein, it was first necessary to confirm or validate the oral efficacy of NTP42 administered as NTP42:KVA4 in preclinical PAH. Thereafter, the study specifically aimed to investigate the preclinical efficacy of NTP42:KVA4 in the PAB model of RV pressure overload to assess the potential of NTP42 to impart direct cardiac benefits.
Hence, efficacy of NTP42:KVA4 was first evaluated in a delayed interventional MCT-PAH model in rodents, where disease was allowed to develop for 7 days prior to initiating treatment, where efficacy was also compared with drugs from each of the four clinical SOC PAH therapies.
MCT led to increased RV systolic pressure (RVSP) and RV hypertrophy, as measured by Fulton’s Index (Figures 1A,B). Quantification of cardiomyocyte size, measured as cross-sectional area at the cellular level, and vascularization demonstrated that this RV hypertrophy was typical of maladaptive processes. Specifically, RV cardiomyocytes from MCT-treated animals were significantly enlarged, while RV capillary density was significantly reduced which, combined, resulted in a decrease in the RV Adaptation Index, the ratio of vascularization to cardiomyocyte size (Figures 1C–E,G). Consistent with the myocardial disorganization that occurs in abnormal hypertrophic processes, Masson’s trichrome staining also revealed pronounced fibrosis in the RV of MCT-treated animals (Figures 1F,H). Treatment with NTP42:KVA4 significantly alleviated the MCT-induced increases in RVSP and Fulton’s Index parameters and alleviated the MCT-induced increase in cardiomyocyte size (Figures 1A–C). While NTP42:KVA4 did not significantly affect RV vascularization in this PAH model per se, it did result in an improvement in the RV Adaptation Index (Figures 1D–E,G). Moreover, NTP42:KVA4 significantly attenuated MCT-induced RV fibrosis (Figures 1F,H). In line with previous investigations,(27) treatment with NTP42:KVA4 also significantly alleviated the MCT-induced increase in mean pulmonary arterial pressure (mPAP), and significantly attenuated pulmonary pathologies, including vessel occlusion and muscularization, CD68+ macrophage infiltration, perivascular fibrosis, and edema (Supplementary Figure 1).
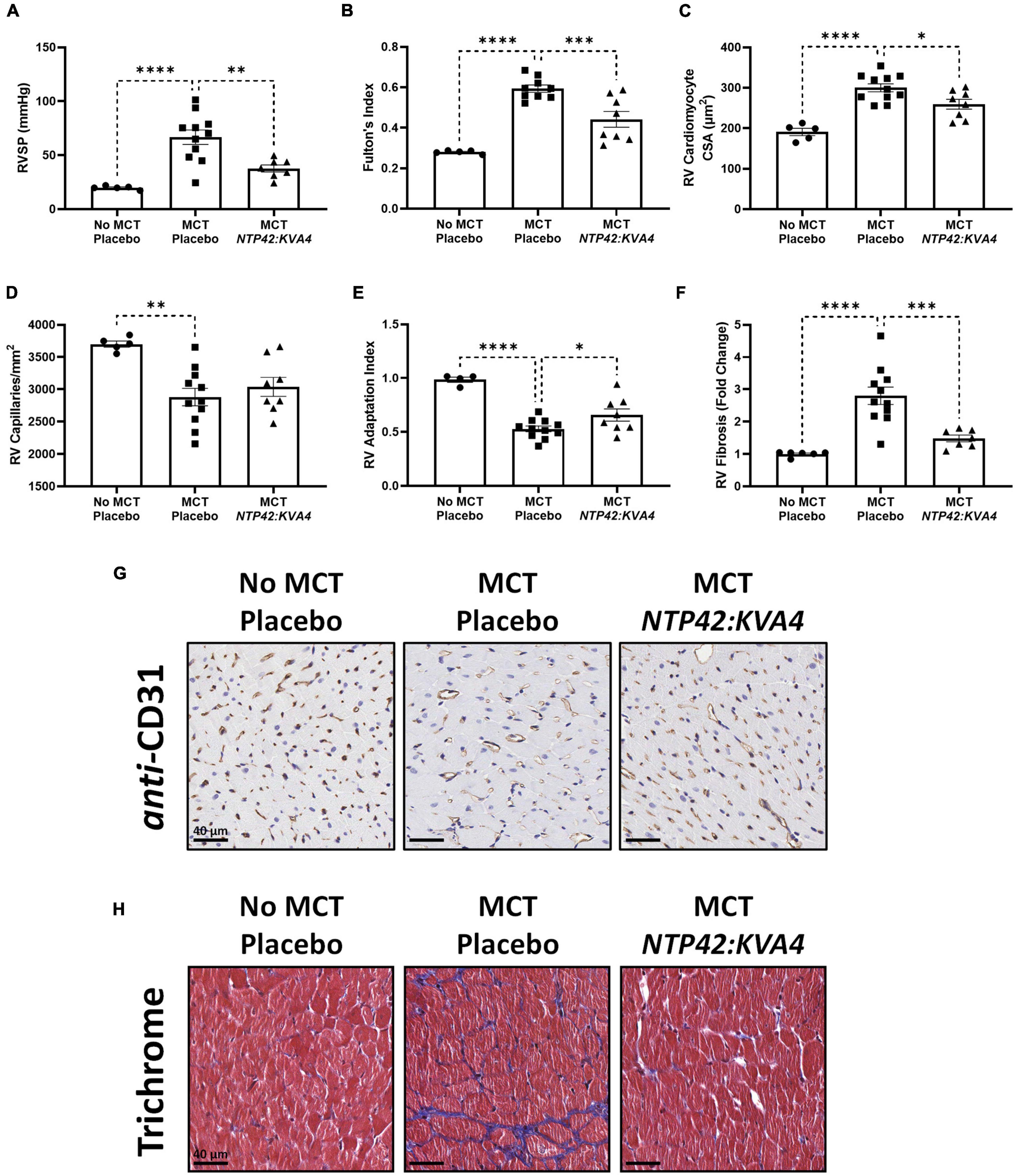
Figure 1. Effect of NTP42:KVA4 treatment on RV hypertrophy and pathology in the MCT-PAH model. Normal control rats (“No MCT—Placebo”) and rats injected with MCT (60 mg/kg) were treated twice daily orally (PO BID) with either drug placebo (“MCT—Placebo”) or NTP42:KVA4 (1 mg/kg PO BID), starting from Day 7 following administration of MCT. (A–F) show: (A) RVSP in the “No MCT—Placebo,” “MCT—Placebo,” and NTP42:KVA4 groups [n = 5, 11, 7], (B) Fulton’s Index [n = 5, 9, and 8, respectively]; (C) RV cardiomyocyte size [n = 5, 11, 8]; (D) RV vascularization [n = 5, 11, 8]; (E) RV adaptation index [n = 6, 11, 8], and (F) RV fibrosis [n = 5, 11, 7]. Data presented are the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. “MCT—Placebo,” according to one-way analysis of variance (ANOVA) with Holm-Šídák correction applied for multiple comparisons. (G,H) Show representative photomicrographs, selected from a random animal from each treatment group, of: (G) Anti-CD31-stained RV tissue captured at 200 × magnification (scale bars represent 40 μm), and (H) Masson’s trichrome-stained RV tissue captured at 200 × magnification (scale bars, 40 μm). Note that while Supplementary Table 1 provides details on numbers of animals enrolled into the studies reported herein and those that survived through to terminal surgery, the numbers (n) given in the square brackets in all figure legends refer to the number of input data used for the individual experimental parameter following removal of any justifiable outliers identified using the method of Interquartile Range with Tukey fences.
Treatment with the PAH SOCs Sildenafil, Macitentan and Riociguat, but not with Selexipag, also resulted in decreased mPAP, RVSP and Fulton’s Index (Supplementary Table 4). In contrast to NTP42:KVA4, none of these SOCs significantly reduced cardiomyocyte size (Supplementary Table 4). Sildenafil alone resulted in increased RV vascularization while Sildenafil, Macitentan, and Riociguat, but not Selexipag, significantly increased the RV Adaptation Index and decreased the levels of RV fibrosis (Supplementary Table 4). While the PAH SOCs decreased vessel occlusion and muscularization as well as CD68+ macrophage infiltration (Supplementary Table 4), they did not significantly reduce perivascular fibrosis or edema (Supplementary Table 4). Notably, while neither NTP42:KVA4, Sildenafil or Macitentan affected mean systemic arterial pressure (mAP) or heart rate (HR) per se, animals treated with Riociguat or Selexipag displayed either increased mAP or decreased HR, respectively (Supplementary Table 4).
NTP42:KVA4 attenuates right ventricular structural changes and dysfunction in the pulmonary artery banding model
Following validation of the efficacy of the formulated NTP42:KVA4 in the MCT-PAH model, the potential for a direct benefit of TP antagonism on cardiac adaptation and function was investigated using the PAB model of RV pressure overload. Notably, while RV hypertrophy and dysfunction are features of the MCT-PAH model, and findings of cardiac benefits for therapeutic agents in this model are indeed valuable, the MCT-PAH model has two important limitations in this regard (29). Firstly, as the RV and the pulmonary vasculature are functionally coupled, direct cardiac-specific effects for an interventional therapy cannot be readily distinguished from afterload reductions due any benefits of the therapy on the pulmonary vasculature, i.e., its pulmonary-specific effects. In addition, the toxic MCT alkaloid itself may have direct effects on the RV, inducing confounding pathologies including myocarditis and arrythmias (30). The use of the PAB model herein to induce a chronic pressure load on the RV aimed to circumvent these limitations. Treatment with NTP42:KVA4 (1 mg/kg PO BID) or Riociguat (5 mg/kg PO, BID) was initiated 2 days after PAB surgery, where Riociguat was chosen as an appropriate comparator compound as, in previous preclinical investigations, it has been shown to prevent deterioration of RV function induced by PAB (31). In addition, Riociguat is the only PAH SOC which has demonstrated potential clinical uses in heart failure settings, (32) where other SOC compounds have demonstrated conflicting findings, often with adverse outcomes (33, 34). Pre-treatment echocardiogram (ECHO) showed robust and comparable pulmonary arterial (PA) pressure gradients across the randomized PAB animal groups (Supplementary Figures 2A,B). At study termination, animals subjected to PAB showed comparable bodyweight and no differences in bodyweight gain over the course of the study was observed (Supplementary Figures 2C,D).
In ECHO assessments, PAB resulted in increased RV free wall thickness (RVFWT) and RV dilation as evidenced by increased RV end-diastolic dimension (RVEDD) and RV end-diastolic area (RVEDA) (Figures 2A–C). In addition, the right atrial area (RAA) was enlarged (Figure 2D). Treatment with NTP42:KVA4 improved RV geometries and attenuated RV dilation, where both RVEDD and RVEDA were reduced (Figures 2B,C). Notably, the attenuated RV dilation observed following NTP42:KVA4 treatment was not paralleled by compromised RV hypertrophy, where RVFWT was unchanged (Figure 2A). In addition, NTP42:KVA4 alleviated PAB-induced RAA enlargement (Figure 2D). Comparable benefits on right heart geometry were not observed following treatment with Riociguat (Supplementary Table 5).
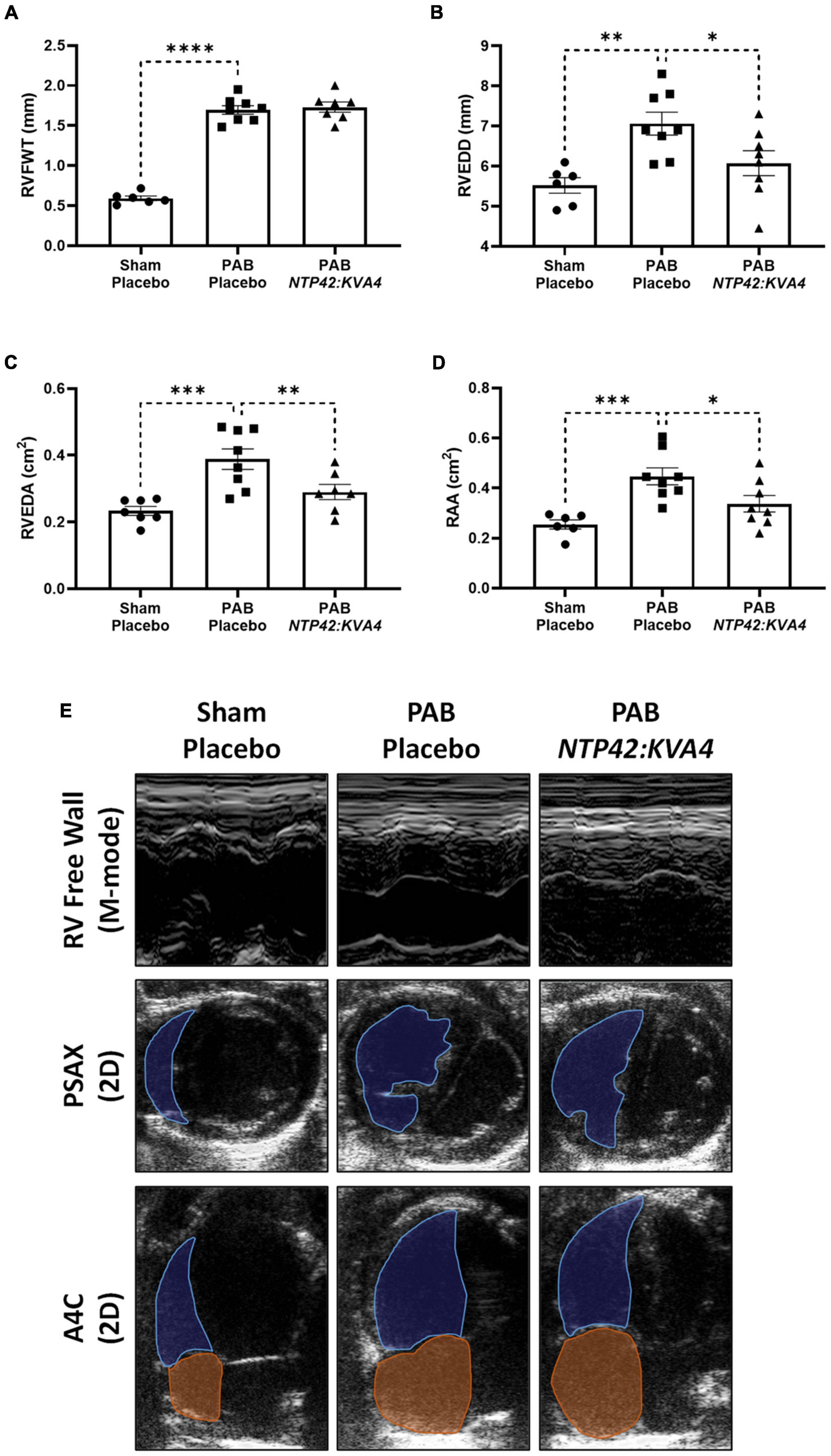
Figure 2. Effect of NTP42:KVA4 treatment on right heart dimensions and geometry in the PAB model. Normal control rats (“Sham—Placebo”) and rats subjected to pulmonary arterial banding (PAB) were treated with either drug placebo (“PAB—Placebo”) or NTP42:KVA4 (1 mg/kg PO BID), starting from Day 2 following PAB. (A–D) Show ECHO-derived measurements of: (A) RVFWT in the “Sham—Placebo,” “PAB—Placebo,” and NTP42:KVA4 groups [n = 6, 8, and 7, respectively]; (B) RVEDD [n = 6, 8, 8]; (C) RVEDA [n = 7, 8, 7], and (D) RA area (RAA) [n = 6, 8, 7]. Data presented are the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. “PAB—Placebo,” according to one-way ANOVA with Holm-Šídák correction. (E) Shows representative ECHO images from M-mode recordings and from 2D parasternal short-axis (PSAX) and apical four-chamber (A4C) views selected from a random animal from each treatment group and where blue and orange shading and lines delineate the RV and RA, respectively.
Detailed pressure-volume (PV) loop analyses showed that PAB animals demonstrated profound RV overload with marked signs of RV dysfunction (Figure 3). In PAB animals, HR was decreased with unchanged mAP (Figures 3A,B), and RV end-systolic pressure (ESP) was fourfold higher than in Sham animals (Figure 3C). RV filling pressure (end diastolic pressure, EDP) was also increased in PAB animals (Figure 3D), and RV dilation was observed when considering both end-systolic and end-diastolic volumes (ESV and EDV; Figures 3E,F). While cardiac output (CO) was reduced upon PAB (–18%, P = 0.1998, Figure 3G), the RV ejection fraction (RV EF) was significantly compromised relative to the Sham control (Figure 3H). End-systolic elastance (Ees) was increased in PAB animals (Figure 3I), and significant diastolic dysfunction was evident in this group, as demonstrated by increased end-diastolic elastance (Eed) (Figure 3J).
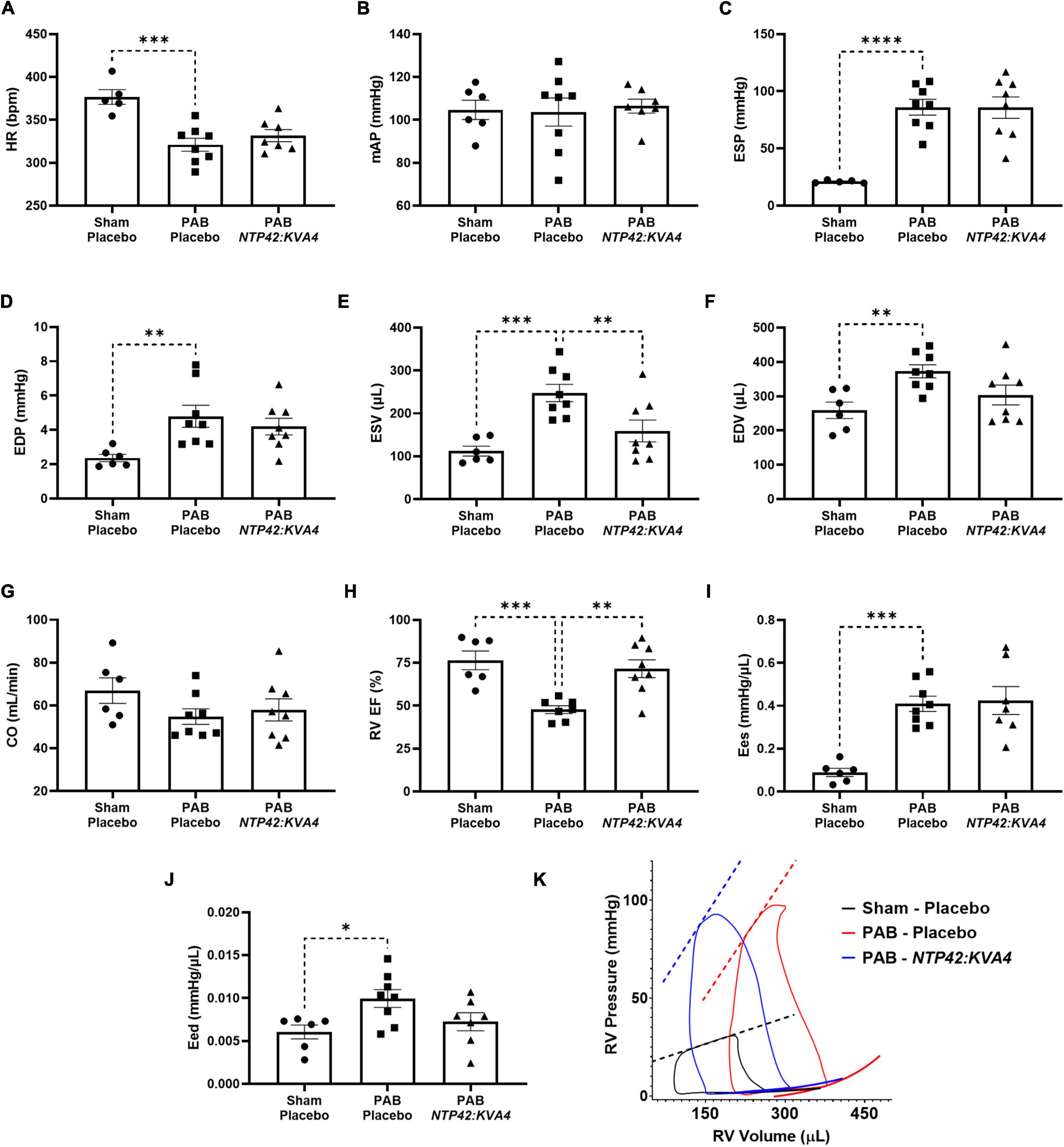
Figure 3. Effect of NTP42:KVA4 treatment on RV pressure, volume and function in the PAB model. Hemodynamic measurements of: (A) HR in the “Sham—Placebo,” “PAB—Placebo,” and NTP42:KVA4 groups [n = 5, 8, and 7, respectively]; (B) mAP [n = 6, 8, 7]; (C) RV ESP [n = 5, 8, 8]; (D) RV EDP [n = 6, 8, 8]; (E) RV ESV [n = 6, 8, 8]; (F) RV EDV [n = 6, 8, 8]; (G) CO [n = 6, 8, 8]; (H) RV EF [n = 6, 7, 8]; (I) RV Ees [n = 6, 8, 7], and (J) RV Eed [n = 6, 8, 7]. Data presented are the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. “PAB—Placebo,” according to one-way ANOVA with Holm-Šídák correction. (K) shows representative RV PV loops from PAB study animals. The linear end-systolic and exponential end-diastolic PV relationships within each group are displayed as thick dashed or solid lines, respectively. Maximum/minimum PV points on the displayed representative loops, and the PV relationships plotted thereon, were adjusted to correspond approximately with the average values determined within treatment group.
In this model, treatment with NTP42:KVA4 significantly improved RV function. In line with ECHO data (Figure 2), NTP42:KVA4 markedly reduced RV dilation, where both ESV and EDV were reduced relative to the PAB control (Figures 3E,F). Most notably, NTP42:KVA4 significantly improved RV EF relative to PAB control (Figure 3H), resulting in near-normalized values compared with Sham animals. In addition, NTP42:KVA4 trended toward an improvement (27%, P = 0.0641) in diastolic function, as measured by Eed (Figure 3J).
While treatment with Riociguat led to improvements in measures of RV dilation, and increased RV EF, albeit to a lesser extent than NTP42:KVA4, Riociguat treatment trended toward a further worsening in Eed (24%, P = 0.2908; Supplementary Table 5).
NTP42:KVA4 promotes an adaptive pattern of right ventricular hypertrophy and reduces expression of genes associated with cardiac dysfunction in the pulmonary artery banding model
While NTP42:KVA4 treatment did not lead to reductions in the gross RV wall enlargement induced by PAB (Fulton’s Index, Figure 4A), it significantly decreased cardiomyocyte size relative to PAB controls (Figures 4B,I), consistent with findings from the MCT-PAH model (Figure 1C). In addition, in this PAB model, NTP42:KVA4 treatment significantly increased RV vascularization (Figures 4C,I). Considering both factors, NTP42:KVA4 treatment resulted in a trend toward improvement in the RV Adaptation Index (P = 0.0797; Figure 4D). Furthermore, expression analysis of genes associated with RV hypertrophy showed that levels of atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) were significantly increased upon PAB, relative to Sham levels (Figures 4E,F). Treatment with NTP42:KVA4 significantly reduced ANP levels (Figure 4E) and trended toward reduced BNP levels (Figure 4F).
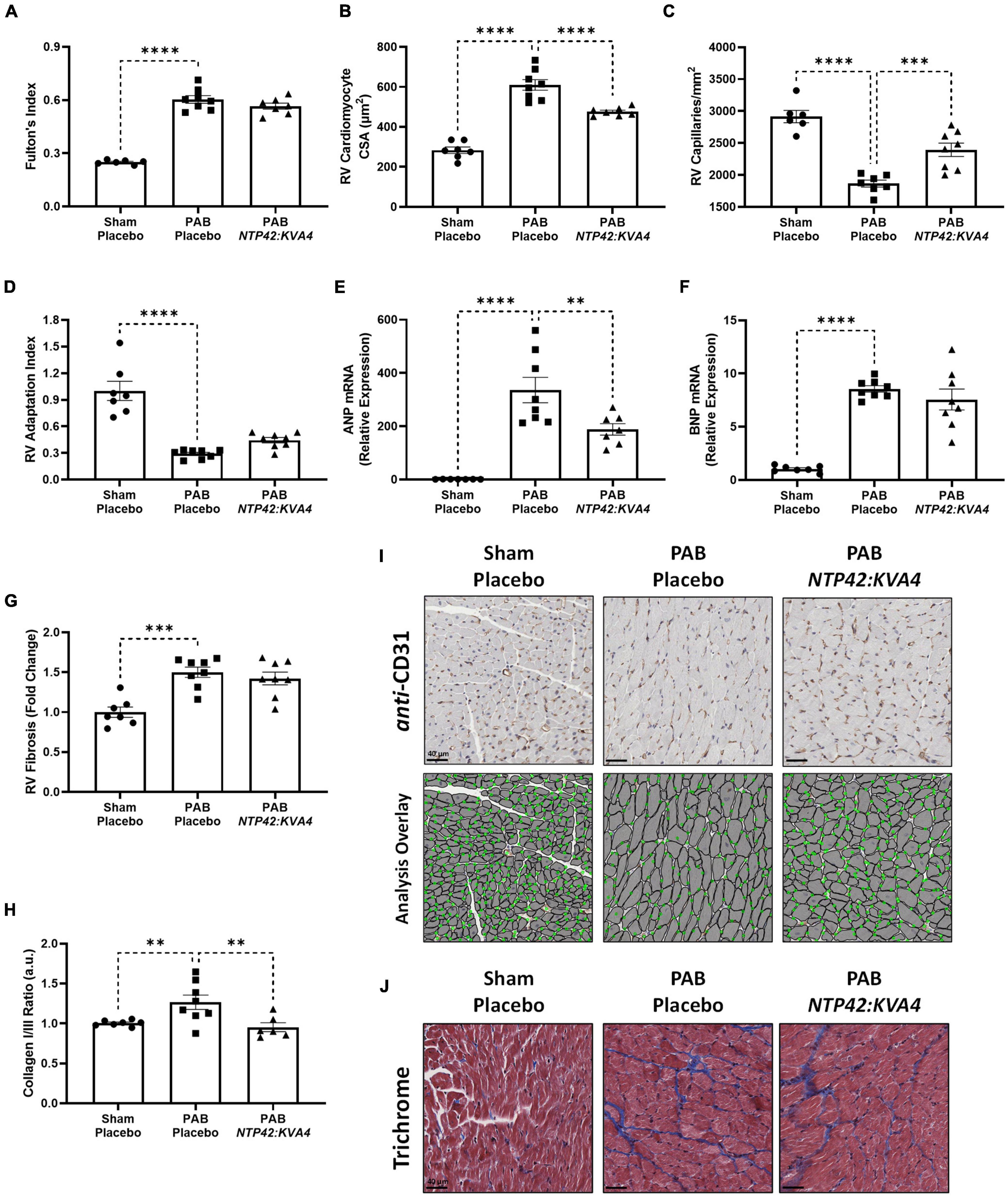
Figure 4. Effect of NTP42:KVA4 treatment on RV hypertrophy and pathology in the PAB model. (A–H) show: (A) Fulton’s Index in the “Sham—Placebo,” “PAB—Placebo,” and NTP42:KVA4 groups [n = 6, 8, and 7, respectively]; (B) RV cardiomyocyte size [n = 7, 8, 7]; (C) RV vascularization [n = 6, 7, 8]; (D) RV adaptation index [n = 7, 8, 8], (E) RV ANP mRNA expression [n = 7, 8, 7], (F) RV BNP mRNA expression [n = 7, 8, 8], (G) RV fibrosis [n = 7, 8, 8], and (H) RV collagen type I/III ratio, calculated from the RV collagen type I and III mRNA expression for each animal, and expressing the ratio in arbitrary units (a.u.) [n = 7, 8, 6]. Data presented are the mean ± SEM. **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. “PAB—Placebo,” according to one-way ANOVA with Holm-Šídák correction. (I,J) show representative photomicrographs, selected from a random animal from each treatment group, of: (I) (Upper panel): Anti-CD31-stained RV tissue captured at 400 × magnification (scale bars represent 40 μm), (Lower panel): Image analysis overlay showing annotated cardiomyocyte cross-sectional area (gray outline) and CD31+ vessels (green dots), and (J) Masson’s trichrome-stained RV tissue captured at 400 × magnification (scale bars, 40 μm).
Notably, RV fibrosis, while increased upon PAB (Figures 4G,J), was less pronounced than that observed in the MCT-PAH model (Figure 1G), and reductions in this gross level of fibrosis were not observed with NTP42:KVA4 in the PAB model. Notably, fibrosis-mediated myocardial stiffness is influenced by the predominant collagen isoform, where an increased ratio of the stiff type I isoform relative to the elastic type III isoform is linked with more severe RV dysfunction (35). Herein, analysis of collagen isoform expression levels demonstrated a marked increase in the collagen I/III ratio upon PAB (Figure 4H). Treatment with NTP42:KVA4 significantly reduced the collagen I/III ratio (Figure 4H), indicative of predominant expression of the more flexible collagen III isoform.
In this model, total heart weight was significantly increased in all PAB groups (Supplementary Figure 3A). Consistent with ECHO assessments showing RA chamber enlargement (Figure 2D), measurements of the RA wall weight demonstrated that substantial RA remodeling occurred in response to PAB, and where NTP42:KVA4 treatment led to significant reductions in this index (Supplementary Figure 3B). Notably, while PAB did not induce changes in left ventricular (LV) cardiomyocyte size or vascularization, an increased level of LV fibrosis was appreciable which was somewhat reduced by NTP42:KVA4 treatment (–28%, P = 0.1386, Supplementary Figure 3C–G).
In these analyses, the PAH SOC Riociguat did not lead to significant improvements in Fulton’s Index, cardiomyocyte size, RV vascularization, ANP mRNA expression levels, or RV fibrosis, and an improved pattern of adaptive hypertrophy was not observed upon Riociguat treatment (Supplementary Table 5). However, like NTP42:KVA4, Riociguat led to a reduction in the collagen I/III ratio (Supplementary Table 5).
NTP42:KVA4 results in normalized cardiomyocyte passive tension and reduces proteolytic degradation of calcium-handling proteins in the pulmonary artery banding model
While PAB-induced right heart pressure overload resulted in significant structural remodeling and systolic and diastolic dysfunction, intrinsic changes in cardiomyocyte tension development were also observed in this model. Passive tension (PT) was significantly increased across all sarcomere lengths in PAB animals, indicative of increased cardiomyocyte stiffness (Figure 5A). Consistent with the improvement in diastolic function observed for NTP42:KVA4 (Figure 3J), PT was significantly attenuated in cardiomyocytes isolated from NTP42:KVA4-treated animals (Figure 5A) and was indistinguishable from the profile of Sham control animals. Regarding systolic function, cardiomyocytes isolated from PAB control animals showed somewhat increased maximum active tension (AT) compared with Sham animals (Figure 5B), while relative AT profiles were similar between Sham, PAB and NTP42:KVA4 groups (Figure 5C). Similar benefits on intrinsic cardiomyocyte function were not observed following Riociguat treatment in this model and both PT and AT were increased in comparison with the PAB control (Supplementary Table 5).
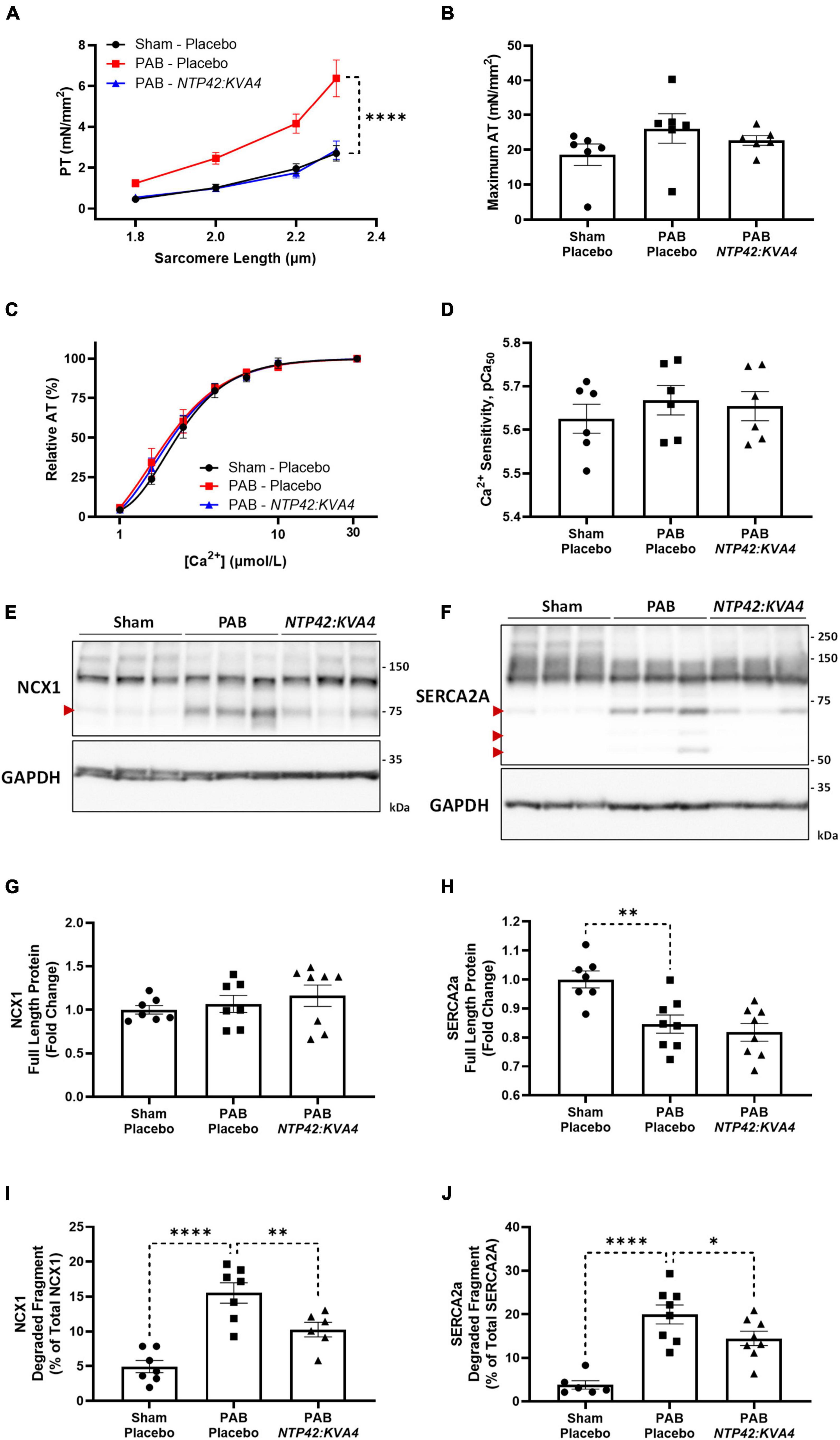
Figure 5. Effect of NTP42:KVA4 treatment on isolated cardiomyocyte force transduction and calcium-handling protein expression in the PAB model. (A–D) show: (A) Steady-state PT measured at increasing sarcomere lengths (1.8–2.3 μm) from cardiomyocytes isolated from animals in the “Sham—Placebo,” “PAB—Placebo,” and NTP42:KVA4 groups [n = 6, 6, and 6, respectively]; (B) Maximum AT development at [Ca2+] 31.6 μmol/L [n = 6, 6, 6]; (C) Relative AT development in response to increasing submaximal free Ca2+ concentration ([Ca2+], 1–31.6 μmol/L) [n = 6, 6, 6], where calcium response curves were fitted using non-linear regression, and (D) Calcium sensitivity (pCa50) determined from individual regression analyses [n = 6, 6, 6]. (E,F) Show western blot protein expression of: (E) NCX1 and (F) SERCA2a, where representative RV lysates are displayed from 3 random animals from each treatment group and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression levels were used as loading control. The relative positions of the molecular size markers (kDa) are indicated to the right of the panels and the positions of the observed degraded fragments of NCX1 and SERCA2a are marked with red arrowheads to the left of the panels. (G–J) Show mean relative RV expression levels of (G) NCX1 full-length protein (120 kDa) [n = 7, 7, 8]; (H) SERCA2a full-length protein (110 kDa) [n = 7, 8, 8]; (I) NCX1 75 kDa degraded fragment, expressed as a percentage of total NCX1 [n = 7, 7, 6], and (J) SERCA2a 70 kDa degraded fragment, expressed as a percentage of total SERCA2a [n = 6, 8, 8]. Data presented are the mean ± SEM, and where in (A–D), results are presented from 6 animals per group, with an average of 5 independent cardiomyocytes (technical replicates) analyzed per animal. *P < 0.05, **P < 0.01, ****P < 0.0001 vs. “PAB—Placebo,” according to two-way ANOVA with Holm-Šídák correction (A,C) or one-way ANOVA with Holm-Šídák correction (B,D,G–J).
Notably, despite the altered profiles of contraction, no changes in calcium sensitivity were observed between the groups (Figure 5D). Besides altered calcium sensitivity, a further mechanism contributing to cardiomyocyte dysfunction involves decreased capacity for diastolic calcium clearance (36). For efficient cardiomyocyte relaxation, cytosolic calcium levels must promptly drop following contraction, where this is facilitated either by its efflux from the cell by Na+/Ca2+ exchanger 1 (NCX1) or sequestration into internal cellular stores by sarco/endoplasmic reticulum Ca2+-ATPase 2a (SERCA2a) (37). Alterations in the expression of these calcium-handling proteins contribute to cardiomyocyte dysfunction during pressure-induced hypertrophy and cardiac failure (38–40), and decreases in NCX1 and SERCA2a have been observed in the RV from PAH patients (36). Furthermore, degradation and inactivation of NCX1 and SERCA2a, mediated by the calcium-activated protease calpain, occurs in multiple animal models of heart failure (41–44). Herein, while expression of intact full-length NCX1 was unchanged, a significant decrease in SERCA2a protein was observed in PAB animals (Figures 5E–H). Moreover, elevated degradation of both NCX1 and SERCA2a were observed following PAB (Figures 5E,F,I,J). Specifically, increased expression of a single degradation fragment of NCX-1 and up to three SERCA2a degradation fragments were observed (Figures 5E,F), where these inactive fragments have been previously described (41, 44). Treatment with NTP42:KVA4, but not Riociguat, attenuated calcium-handling protein degradation, with significantly reduced NCX1 and SERCA2a degradation fragments evident (Figures 5E,F,I,J).
Thromboxane receptor expression is elevated in the right ventricle in experimental models and in human pulmonary arterial hypertension and other right ventricular conditions
While signaling through the TP is implicated in pathological cardiac conditions, few studies have examined TP expression levels in RVs of subjects with PAH. Thus, as a rationale for the therapeutic potential and utility of TP antagonists per se, expression of the TP was examined in RV tissues from experimental PAH and cardiac disease models, as well as in clinical specimens of PAH and dilated cardiomyopathy (DCM).
While low levels of TP expression were noted in the RV myocardium in No MCT and Sham animals, increased TP expression occurred in all diseased groups (Figures 6A–D). In addition, genomic analysis confirmed elevated TP expression levels in the PAB model (Figure 6E). Administration of NTP42:KVA4 in both MCT-PAH and PAB models led to a non-significant trend toward reduction in TP expression (P = 0.2091 and P = 0.0920, respectively; Figures 6A–D). As no significant effects on TP expression were observed following treatment with PAH SOCs in either model (Supplementary Tables 4, 5), a potential effect on TP expression following specific receptor engagement with NTP42:KVA4 is notable and may indicate a mechanism whereby TP antagonism may lead to beneficial RV effects.
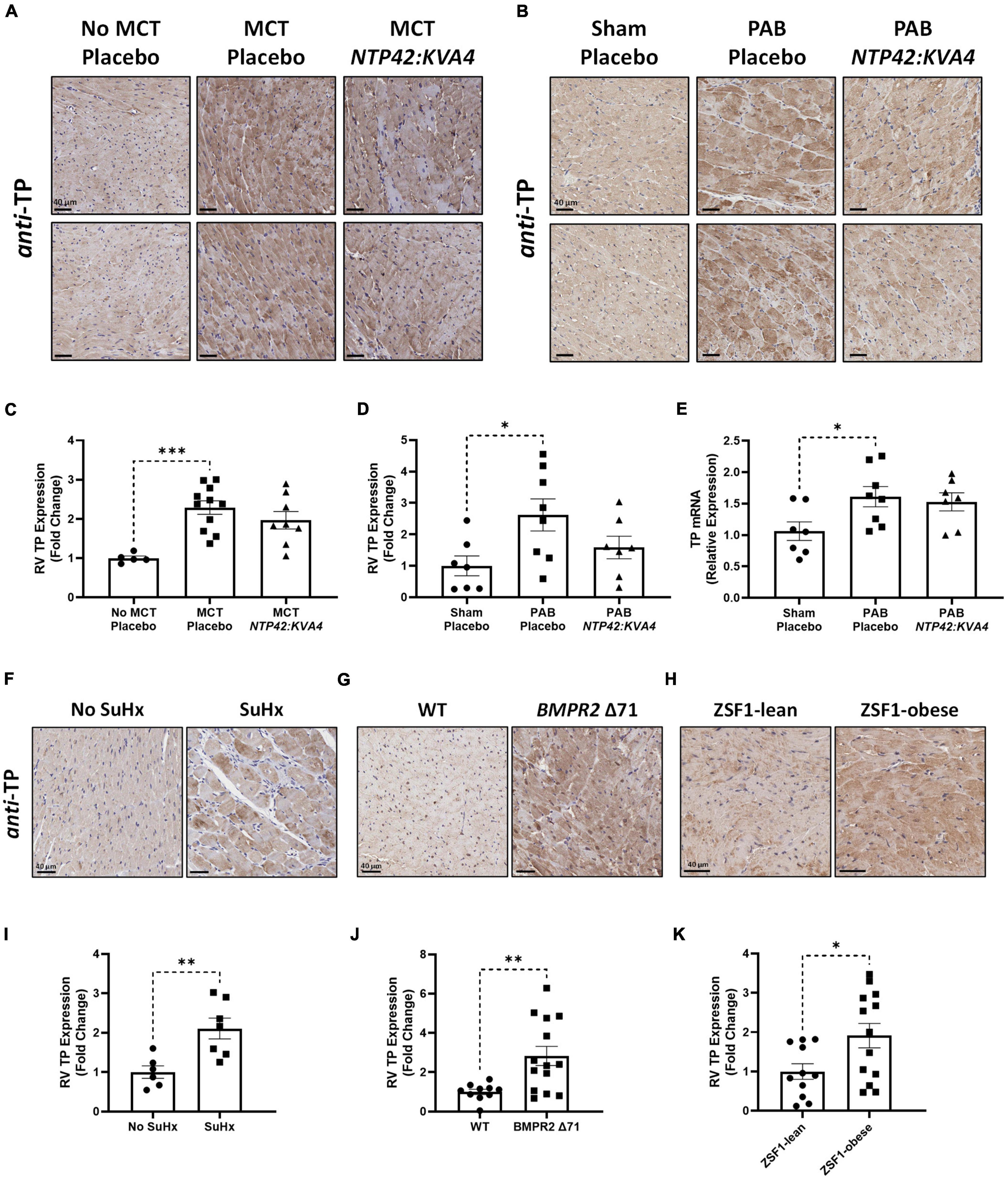
Figure 6. RV expression of the TP in preclinical models of PAH and RV dysfunction. (A,B) Show two representative photomicrographs, selected from a random animal from each treatment group, of anti-TP-stained RV tissue from the: (A) MCT-PAH model, and (B) PAB model, where images were captured at 400 × magnification (scale bars represent 40 μm). (C) Shows relative immunohistochemical (IHC) expression of the TP in the MCT-PAH model in RV tissue from animals of the “No MCT—Placebo,” “MCT—Placebo,” and NTP42:KVA4 groups [n = 5, 11, and 8, respectively]. (D–E) Show: (D) relative TP IHC expression in the PAB model in RV tissue from animals of the “Sham—Placebo,” “PAB—Placebo,” and NTP42:KVA4 groups [n = 7, 8, and 7, respectively], and (E) RV TP mRNA expression [n = 7, 8, 7]. (F–H) Show representative photomicrographs, selected from a random animal from each treatment group, of anti-TP-stained RV tissue from: (F) Control groups of a previously described SuHx-PAH model (28), namely “No SuHx” and “SuHx,” where normal control rats (“No SuHx”) and rats treated with SuHx (“SuHx”) were treated PO BID with vehicle upon removal from hypoxia and continuing in normoxia until Day 49 (i.e., 4 weeks duration); (G) BMPR2 Δ 71 rats or their WT counterparts, and (H) ZSF1-obese rats or their lean counterparts (ZSF1-lean). (I–K) show relative expression levels of the TP in RV tissue from: (I) “No SuHx” and “SuHx” animals [n = 6 and 7, respectively]; (J) WT or BMPR2 Δ71 animals [n = 14 and 11, respectively], and (K) ZSF1-lean or ZSF1-obese animals [n = 13 and 11, respectively]. Data presented are the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 vs. the respective disease control group in each case according to one-way ANOVA with Holm-Šídák correction (A–C) or unpaired Student’s t-tests (I–K).
Further evidence for increased TP expression in the RV in PAH and, potentially in other cardiac dysfunctions, was found in RV specimens from both the Sugen5416/Hypoxia (SuHx)-induced PAH model (Figures 6F,I), and notably from rat strains harboring a mutation in the BMPR2 gene, the primary genetic cause of heritable PAH in humans (Figures 6G,J). Furthermore, TP expression was also increased in RVs from obese ZSF1 rats, a recognized model of heart failure with preserved ejection fraction (HFpEF) which also manifests RV dysfunction (Figures 6H,K).
In clinical specimens from healthy human donors (Figure 7A), TP expression was observed at a low level throughout the myocardium, with stronger expression evident in perinuclear regions, consistent with previous reports (19). In diseased tissue, TP expression was augmented in the enlarged cardiomyocytes of RV samples from PAH patients (Figure 7B), with increased expression also observed in RV samples from DCM (Figure 7C), a primary cardiomyopathy which results in ventricular dilation and functional impairment. Quantitative analysis confirmed these elevated TP expression levels in PAH & DCM cases, relative to healthy donors (Figure 7D). Notably, while increased TP expression occurred in RVs in experimental models and in clinical PAH samples, no significant changes were observed in the matching LV tissues (Supplementary Figure 4).
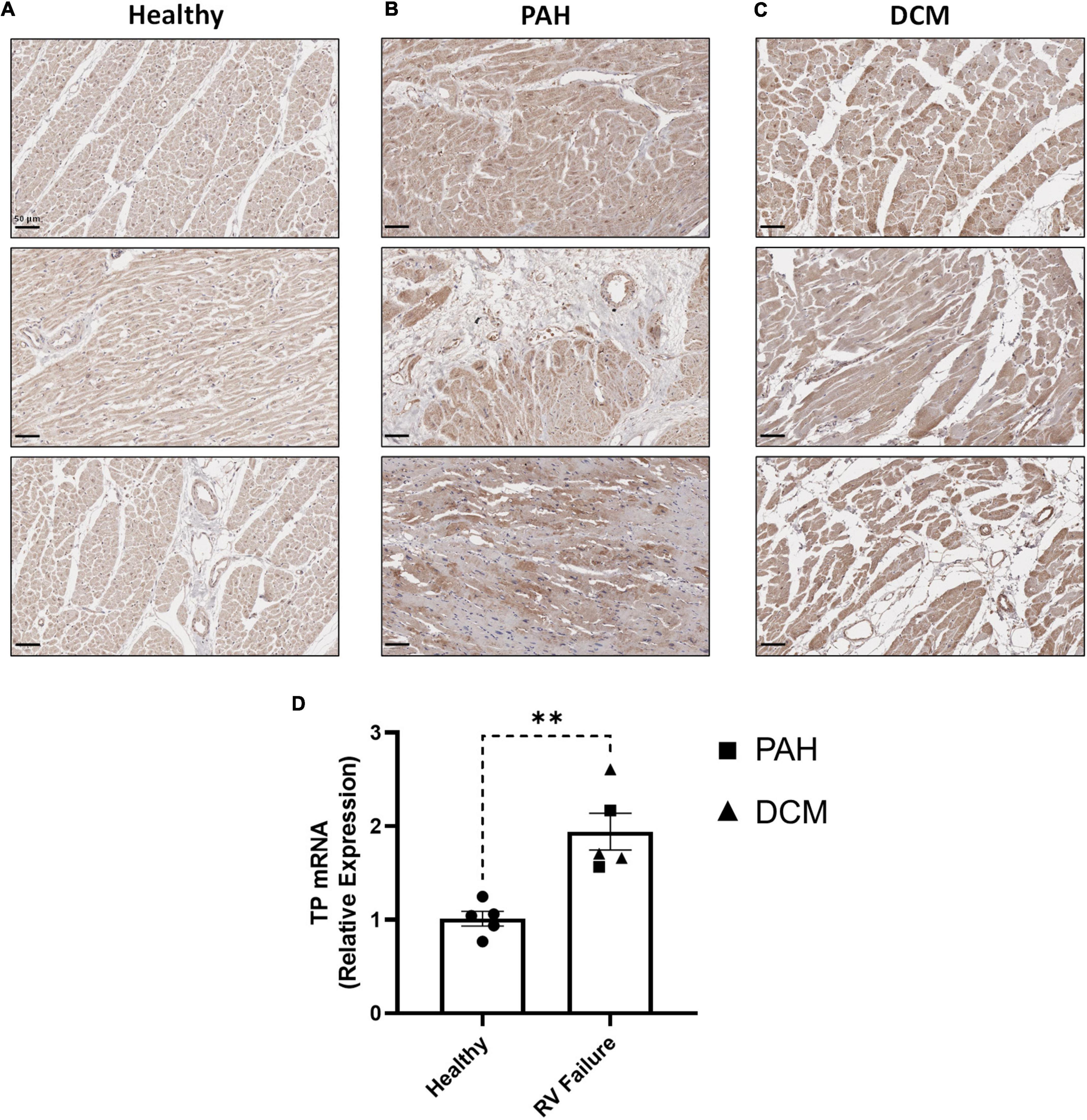
Figure 7. RV expression of the TP in human PAH and other RV pathologies. (A–C) show three representative photomicrographs of anti-TP-stained human RV tissue obtained from: (A) Healthy donors; (B) PAH patients, and (C) DCM patients, where all images were captured at 150 × magnification (scale bars represent 50 μm). (D) Shows relative RV TP mRNA expression levels in Healthy (n = 5) or RV Failure patients (n = 5, data for PAH and DCM combined). Data presented are the mean ± SEM. **P < 0.01 according to unpaired Student’s t-test.
Discussion
Right heart function is widely viewed as the most important determinant of clinical outcome in PAH and indeed in various other forms of pulmonary hypertension (PH) (45). Insufficient or aberrant RV adaptation, the development of RV dysfunction, and the progression to right heart failure in PAH involves complex pathological mechanisms and a precise understanding of the causes underlying these mechanisms remains to be fully elucidated (46). Unfortunately, no currently available PAH SOC therapy directly targets right heart adaptation and function.
While PAH SOCs demonstrate robust pulmonary vasodilatory enhancing effects and lead to meaningful reductions in PVR, they show limited evidence of directly targeting right heart adaptation and function. In PAH, there is an unmet need to not only alleviate pulmonary pathology and PVR, but to also directly target mechanisms underlying RV dysfunction to enhance patient quality-of-life and improve survival. Current PAH SOCs have variable effects on RV function in both experimental and clinical settings (47). In preclinical models, the prostacyclin analog Iloprost improved RV contractility, and a recent trial in PAH patients demonstrated that Iloprost increases contractility and RV-PA coupling (48). However, large-scale clinical trials of Epoprostenol in heart failure patients demonstrated an association with increased mortality (49, 50). In preclinical models, endothelin receptor blockade worsens cardiomyocyte contractility (51). Clinical trials of endothelin receptor antagonists in patients with heart failure have never fully reported, making it impossible to assess efficacy or, at worst, suggestive of unfavorable effects (52–54). In PAH patients, while acute treatment with the phosphodiesterase (PDE)5 inhibitor Sildenafil improved RV diastolic function (55), recent trials in non-PAH heart failure patients with Sildenafil or the soluble guanylate cyclase (sGC) stimulator Riociguat failed to meet their primary clinical endpoints (56, 57).
Early studies targeting the TXA2/TP pathway in preclinical PAH demonstrated conflicting results (58, 59). However, through recent evaluations in both the MCT- and SuHx-induced PAH models, we have demonstrated that the TP antagonist NTP42 attenuates multiple features of experimental PAH (27, 28). In this study, the efficacy of the NTP42:KVA4, a novel oral formulation of NTP42 specifically developed for clinical use and recently validated in a Phase I clinical trial (NCT04919863) in alleviating pulmonary pathologies in the MCT-PAH model was confirmed to be in line with previous findings using the NTP42 API. Moreover, this MCT-PAH study demonstrated that NTP42:KVA4 attenuates RV hypertrophy and promotes a more beneficial pattern of RV adaptation. Thereafter, we aimed to investigate the potential for NTP42:KVA4 to directly target the compromised RV using the PAB model of right heart pressure overload. With an absence of confounding pulmonary pathologies, the PAB model allows for a direct assessment of therapeutic intervention on RV structure and function (60). In this PAB model, NTP42:KVA4 reduced RV dilation as observed from ECHO and PV loop indices and also alleviated RA enlargement. While not affecting gross RV hypertrophy per se as evidenced by unchanged wall thickness and Fulton’s Index, NTP42:KVA4 treatment also resulted in a more adaptive pattern of hypertrophy, with significantly decreased cardiomyocyte size and a significant increase in capillary density in RV tissue. In individually isolated cardiomyocytes, the PAB-induced increase in passive tension development, a key indicator of cell stiffness, was attenuated by NTP42:KVA4 being indistinguishable from that of healthy control animals, and decreased degradation of the Ca2+-handling proteins NCX1 and SERCA2a. Moreover, due to the overall improved RV geometry, decreased RV dilation, and attenuated profiles of intrinsic diastolic and systolic cardiomyocyte tension development, treatment with NTP42:KVA4 resulted in significantly improved RV function in PAB animals. Most notably, NTP42:KVA4 markedly improved RV EF, near normalizing this key parameter relative to control Sham animals.
Together, the findings from these two independent preclinical models demonstrate that NTP42:KVA4 not only alleviates pulmonary pathologies akin to those observed in clinical PAH, but also may act as a direct cardioprotective agent in settings of right heart pressure overload. Throughout these preclinical models, the efficacy seen with NTP42:KVA4 was similar or indeed greater than the PAH SOCs used herein (Supplementary Tables 4, 5). Furthermore, in translating the preclinical efficacy findings generated in these rodent PAH models to that predicted to clinically occur in man, it is important to also note that the API NTP42 was rationally designed and selected using the human and not the rodent TP drug target (61, 62). Thus, due to key evolutionary differences in the TP in primates vs. lower species (63), NTP42 is a highly potent antagonist of both TPα and TPβ isoforms of the human TP (61, 62), inhibiting TXA2 mimetic U46619-induced calcium mobilization in cell lines stably over-expressing the human TP, and U46619-induced aggregation of human platelets, with IC50 values of 8.86 and 10.6 nM, respectively (27). However, in similar studies in the rat, NTP42 is substantially less potent inhibiting U46619-induced calcium mobilization by the rat TP and aggregation of rat platelets ex vivo with IC50 values of 1.91 and 3.2 μM, respectively (Supplementary Figure 5). Thus, based on its relative IC50 for the TP in rats and humans, NTP42 will be substantially more efficacious (250–300-fold) in man than in rat. Consistent with this proposition, in the recent Phase I clinical trial of the IMP NTP42:KVA4 in healthy subjects (NCT04919863), the IC50 of NTP42 for inhibition of U46619-induced aggregation of human platelets ex vivo was confirmed to be 9.9 nM. Extending this translation of preclinical to predicted clinical data for the PAH SOCs, based on their relative IC50 in rats and humans, both Sildenafil (IC50 in rat and man, approx. 3 nM) (64, 65) and Macitentan (IC50 in rat and man, 1 nM) (66, 67) are expected to be equally efficacious in both species. In contrast to this, based on its EC50 of 170 and 4 nM in rat and man (67, 68), the prostacyclin receptor agonist Selexipag is predicted to be 42.5-fold more efficacious in man than in rat and, therefore, is likely to elicit a better outcome clinically than observed in this or other preclinical studies. Indeed, this may account for the poor efficacy observed for Selexipag on key parameters such as mPAP, RVSP and Fulton’s Index (Supplementary Table 4). With regard to Riociguat, based on its relative potency (EC50 in rat and man, 30 and 80 nM, respectively) (69, 70), it is predicted to be 2.6-fold more efficacious in rat than in man, likely generating a poorer clinical outcome than it does in the preclinical studies carried out in the rat. In addition to such key species-dependent differences in target specificity, it was also notable there were fewer systemic effects apparent for NTP42:KVA4 relative to the PAH SOCs. Specifically, while Selexipag and Riociguat led to changes in HR and mAP, respectively, and treatment with all the PAH SOCs tested led to increased liver weight indices, similar indicators of potential off-target or toxicological effects were not observed with NTP42:KVA4 (Supplementary Tables 4, 5). In addition, while demonstrating effects in the compromised RV, PV loop analysis showed that NTP42:KVA4 did not affect LV parameters (data not shown). As an important regulatory-compliant safety parameter required by both the European and US EMA and FDA agencies before proceeding to FIH Phase I clinical trials, the in vivo effect of NTP42:KVA4 on the cardiovascular system was also investigated in conscious telemetered dogs, where no inotropic or chronotropic effects were observed at doses up to 450 mg/kg NTP42:KVA4 PO (Supplementary Figure 6).
In previous studies investigating the role of the TXA2/TP pathway on RV dysfunction, TP antagonism was protective against mild RV pressure overload in a mouse PAB model, where pressures and cardiac output were improved (19). TP antagonism also attenuated PAB-induced increases in end-diastolic calcium levels and improved cardiac repolarization and reduced ECG abnormalities through restoration of the gap junction protein connexin 43 (71–73). Targeting of TXA2/TP pathway has been previously investigated in PAH clinical therapy. Terbogrel, a dual TP antagonist and TXA2 synthase (TXAS) inhibitor was evaluated in a Phase II clinical trial, but this study was prematurely terminated during enrolment due to the development of acute leg pain in trial participants (74). As subsequently reported, this leg pain occurred due to Terbogrel’s inhibition of TXAS which, while blocking TXA2 generation, resulted in a shift toward synthesis of prostacyclin, a potent pain inducer. In contrast to Terbogrel, NTP42 is a highly selective TP antagonist, which does not inhibit TXAS and therefore, as also confirmed in the recent Phase I clinical trial even at high doses, will not induce leg pain (27). In addition, the specificity of NTP42 for the TP has been previously reported, with no agonist or antagonist activity at the 7 other prostanoid receptors, namely the prostaglandin (PG) D2 (DP1), PGE2 (EP1, EP2, EP3, EP4), PGF2α (FP) and PGI2/prostacyclin (IP) receptors, with no agonist activity at the TP itself (27).
As depicted in the model in Figure 8, there are many putative mechanisms by which signaling via the TXA2/TP pathway may elicit detrimental effects within the myocardium, where the findings from this study provide important mechanistic insights into this and how NTP42 alleviates this dysfunction. Consistent with a large body of data, including from this laboratory (63), signaling through the TP, TXA2 induces profound increases in intracellular calcium (Ca2+) in many cell types. Specifically, in cardiomyocytes, basal and peak Ca2+ concentrations as well as width of Ca2+ transients are increased following treatment with the TXA2 mimetic U46619, and prolonged stimulation results in irregular Ca2+ oscillations and a marked increase in cytosolic-free Ca2+ concentrations (20). Direct injections of U46619 also induce ventricular arrhythmia in rabbits, where this effect occurs through a mechanism independent of reductions in coronary blood flow or activation of the autonomic nervous system (22). As a G protein-coupled receptor, the TP primarily couples to Gq, resulting in phospholipase (PL)Cβ activation and liberation of inositol trisphosphate (IP3) and diacylglycerol (DAG) from phosphatidylinositol 4,5-bisphosphate (PIP2) cellular stores (63). IP3 release leads to rapid mobilization of intracellular Ca2+ from the sarco/endoplasmic reticulum (SR) via activation of ligand-gated IP3 receptors (IP3Rs). In this regard, treatment of isolated cardiomyocytes with U46619 increased intracellular Ca2+ in a dose-dependent manner, where these increases were blocked by the TP antagonist SQ29548 or inhibitors of the IP3 pathway (23). IP3-mobilized Ca2+, along with the depolarization phase of the action potential and extracellular Ca2+ influx via L-type Ca2+ channels (LTCCs), triggers a larger Ca2+ release from the SR via ryanodine receptors (RyRs), leading to cardiac contraction. A further mechanism through which the TP may lead to increased intracellular Ca2+ is following protein kinase (PK) C activation due to second-messenger DAG liberation from PIP2. It has been shown by multiple groups that TP stimulation in vascular smooth muscle cells leads to dysregulation of resting membrane potential and subsequent activation of LTCCs (75–78). Mechanistically, Cogolludo et al. demonstrated that U46619 treatment of pulmonary artery smooth muscle cells directly inhibits voltage-gated K+ channels via a PKCζ-mediated mechanism, leading to subsequent depolarization, LTCC-mediated increase in intracellular Ca2+, and vasoconstriction (76). Chronically elevated or dysregulated intracellular Ca2+ cycling plays a central role in hypertrophic signaling in cardiomyocytes (79). In particular, the distinct Ca2+ duty cycle produced following IP3-mediated Ca2+ release has been shown to activate pro-hypertrophic pathways, including those involving nuclear factor of activated T cells (NFAT) transcriptional mechanisms (80–82). Notably in the context of TP-mediated signaling, Ca2+ overload, aberrant Ca2+ cycling, or PKC-mediated phosphorylation, activates the cysteine protease calpain (83, 84). In turn, activated calpain proteolytically degrades myofibrillar proteins including myosin and titin, as well as Ca2+-handling proteins including NCX1 and SERCA2a (85–87). Herein, this study shows decreased degradation of both NCX1 and SERCA2a following NTP42:KVA4 treatment in the PAB model. We propose that this is one possible mechanism where NTP42 antagonism of TP signaling may be involved in preventing progression to cardiac dysfunction and heart failure. Besides this, there are other mechanisms which may drive cardiac dysfunction and account for the observed benefits of NTP42, including potentially involving free-radical mechanisms and/or contractile machinery modifications (35, 36). Levels of 8-iso-PGF2α, a non- enzymatic-, free-radical- derived product of arachidonic acid, are increased in line with heart failure severity and associated with increased ventricular dilation (88). Notably, the TP also mediates the actions of 8-iso-PGF2α, where uniquely TP antagonism is predicted to have the additional benefit of blocking this important pathological mediator of oxidative injury. Furthermore, in the context of the myofibril machinery, as a key determinant of myocardial passive stiffness, the distensibility of titin is heavily regulated by phosphorylation (89). In contrast to PKA/PKG-mediated effects, PKC phosphorylation of titin is widely known to increase cardiomyocyte stiffness (90–92). Mechanistically, as a Gq-coupled receptor directly linked to PKC activation (63), a plausible working hypothesis of how TP antagonism by NTP42 may lead to decreased stiffness is by reducing PKC-mediated titin phosphorylation. While beyond the scope of this study, further mechanistic investigations are warranted to explore these proposed TXA2/TP-mediated mechanisms.
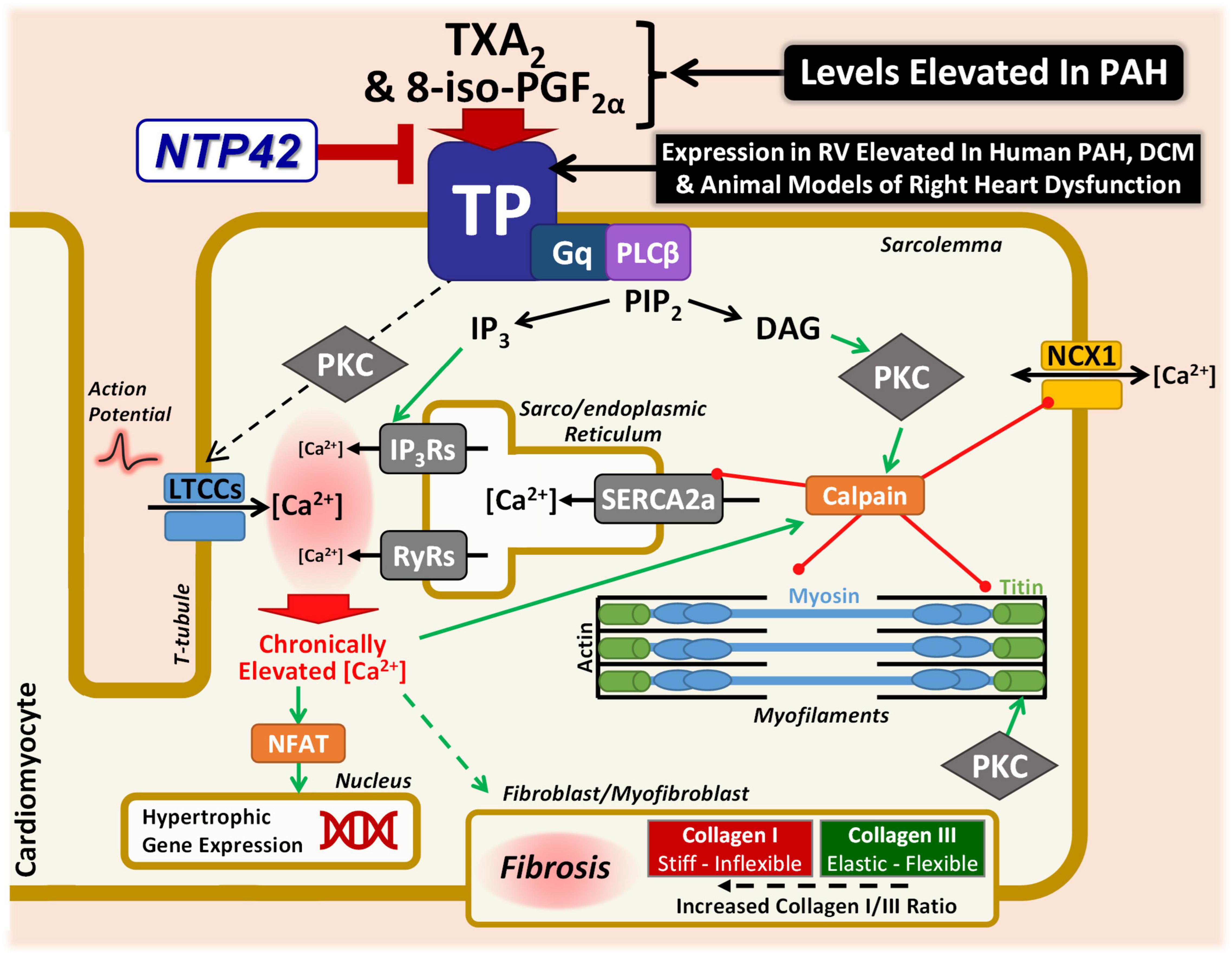
Figure 8. Specific antagonism of TP signaling may alleviate key mechanisms involved in cardiac dysfunction in PAH and other cardiac conditions. Schematic representation focusing on the potential mechanisms through which the TXA2/TP signaling axis may lead to cardiac dysfunction in PAH and other cardiac conditions. Signaling through the Gq-coupled TP leads to PLCβ activation, liberating IP3 and DAG from PIP2. IP3 release mobilizes Ca2+ from the sarco/endoplasmic reticulum via IP3R activation. IP3-mobilized Ca2+, along with extracellular Ca2+ influx via LTCCs, triggers a larger Ca2+ release via RyRs. LTCC-mediated Ca2+ influx may also occur via a TP-mediated PKC mechanism. Chronically elevated Ca2+ leads to hypertrophic signaling via NFAT. Ca2+ overload also activates the protease calpain, which degrades myofibrillar proteins including myosin and titin, as well as Ca2+-handling proteins including NCX1 and SERCA2a. This study has demonstrated decreased degradation of both NCX1 and SERCA2a following NTP42:KVA4 treatment. Furthermore, PKC-mediated phosphorylation of titin leads to increased stiffness, and it is hypothesized that the decreased cardiomyocyte stiffness observed herein following TP antagonism may be as a result of reduced PKC-mediated titin phosphorylation. In addition, TP antagonism may also act on the cardiac fibroblast/myofibroblast, decreasing cardiomyocyte stiffness by resulting in a decreased Collagen I/III ratio and/or net RV fibrosis levels, as demonstrated in this study. Finally, not only are levels of the TP ligands TXA2 and 8-iso-PGF2α increased in PAH and in other cardiopulmonary conditions, but this study has demonstrated elevated expression of the TP in the myocardium in clinical PAH and dilated cardiomyopathy (DCM), as well as in multiple independent experimental models of PAH and/or cardiac dysfunction. Both TXA2 and 8-iso-PGF2α would be predicted to further compound the contribution of the TXA2/TP pathway to detrimental effects within the diseased myocardium.
Several limitations with the current study are acknowledged. While anesthesia may lead to depressed cardiac function and systemic hemodynamic effects, the anesthetics isoflurane or sevoflurane used in our studies are known to have only mild effects on cardiac function in rodents (46, 93). Furthermore, as anesthetic regimens were used identically in all animal groups, we would not contend that this affected the study findings. Regarding the pathologies seen in the MCT-PAH and PAB models, it is accepted that these develop in a short time frame and that these preclinical studies may not completely recapitulate the changes that develop progressively over many years in the human condition. However, it is important to acknowledge that our data shows that increased expression of the TP occurred in RVs of several independent and relevant preclinical models, including in the MCT- and SuHx-PAH models, in the PAB model of RV overload, in the ZSF-1 model of heart failure and spontaneously in the BMPR Δ71 rodents without intervention. Importantly, increased expression of the TP was also found in clinical RV specimens from subjects with PAH as well as from DCM subjects. While historically most clinical attention in the study of DCM has been LV function and morphology, recent advances in cardiac imaging show that RV involvement is common in DCM, and the presence of RV dysfunction is in fact a major negative prognostic determinant in DCM morbidity and mortality (94). The observations reported herein for the potential involvement of the TP and the TXA2 pathway in DCM pathogenesis or progression warrant further investigation in relevant preclinical models. The studies reported herein are in line with the current recommendations that, where possible, the effects of an intervention be tested in multiple animal models (27–29). With regard to the choice of preclinical models, and the specific timing of intervention used herein, treatment with NTP42:KVA4 or PAH SOC was commenced in as delayed a schedule as possible while still permitting sufficient animal survival for analyses through complementary modalities, including invasive assessments of pressure-volume relationships. While the findings from the early interventional MCT-PAH approach reported herein add substantially to previous reports from a preventative MCT-PAH model (27) it is acknowledged that the therapeutic effects of NTP42:KVA4 could be further investigated using conditions viewed as more reminiscent of a reversal approach, such as by further delaying treatment in an MCT-PAH or PAB model. Finally, regarding the findings from the PAB model, while NTP42:KVA4 treatment resulted in normalization of isolated cardiomyocyte passive tension, statistical significance for the functional consequence of this improvement in diastolic stiffness, such as in Eed, was not achieved (P = 0.091, Figure 3J). While trending toward benefit, a possible explanation for this disparity is that while Eed measurement corresponds with the stiffness of the heart in vivo, tension measurements are taken ex vivo from individual isolated cardiomyocytes. While this isolation procedure preserves the structural and functional properties of the myofibrillar apparatus, these cells present with sarcolemmal damage and loss of intracellular organelles (95). These isolated cardiomyocytes also lack supporting extracellular matrix, fibrotic deposition, or cell-cell interactions that may contribute to diastolic stiffness in vivo (95). In addition, while assessments of NCX-1 expression showed unchanged levels of intact functional protein following PAB, the finding of increased degraded protein likely indicates that net expression of NCX-1 may be increased upon PAB, presumably as a compensatory or adaptative mechanism to cope with elevated Ca2+ levels and aberrant Ca2+ cycling in the compromised cardiomyocyte. Increased NCX-1 degradation in this setting would hence be predicted to have a negative effect on this adaptive response. However, and as demonstrated in this study, treatment with NTP42:KVA4 led to significantly decreased levels of NCX-1 degradation and this benefit on NCX-1 turnover is hypothesized to be a component of the mechanism of TP antagonism in this model.
In conclusion, these preclinical studies provide evidence that, through antagonism of TP signaling, NTP42, administered orally as the clinical formulation NTP42:KVA4, may attenuate PAH pathophysiology by not only alleviating pulmonary pathologies but also by reducing RV remodeling and promoting beneficial hypertrophy, resulting in improved cardiac function. These findings in experimental models point to a cardioprotective effect for NTP42:KVA4 as a component of its therapeutic potential not only in PAH, but possibly in other RV dysfunctions. Finally, expanding on the growing evidence for the role for the TP in PAH and the potential for antagonism of the TP as a therapeutic strategy in its clinical management, such as with NTP42:KVA4, this current study also demonstrated elevated expression of the TP in RV tissue from human PAH and other cardiomyopathy patients, validating the potential of this largely ignored target ripe for pharmaceutical intervention.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
Protocols for the collection of human biomaterials used in this study were reviewed and approved by the Institute of Cardiometabolism and Nutrition BioCollection (Paris, France). All patients/participants, or their surrogates, provided written informed consent for the use of these biomaterials for research purposes. Animal studies were reviewed and approved by the Institutional Animal Care and Use Committee of IPS Therapeutique (Sherbrooke, QC, Canada), or by the Ethical Committee of the University of Porto (Porto, Portugal), certified by the Portuguese National Authority for Animal Health.
Author contributions
EM, FR, RA, ED, LB, VS, HR, GC, JG, AL-V, J-BM, CB-S, CL, LH, DM, MH, AV, FP, PM-F, and BK: substantial contributions to the conception or experimental design of the work or the acquisition, analysis, and interpretation of data for the work. EM, LH, DM, MH, AV, FP, PM-F, and BK: drafting the manuscript or revising it critically for important intellectual content. All authors have read and approved the final manuscript.
Funding
This work was supported by the ATXA Therapeutics Limited from Enterprise Ireland’s Commercialization Fund Program (Grant Nos. CFTD/2009/0122 and CF/2012/2608) and from the European Commission Horizon 2020 SME Instrument (Grant No. 822258). In addition, researchers at University of Porto were supported by funding from the Portuguese Foundation for Science and Technology (FCT) (Grant No. UID/IC/00051/2013 [COMPETE_2020, POCI]). These funding sources supported ATXA Therapeutics’ research and development program, and the individual funding bodies did not play a role in the study design, data collection, data analysis, data interpretation or in writing the manuscript. CB-S, PM-F, RA, and GC were supported by the Portuguese Foundation for Science and Technology (FCT), under the scope of the Cardiovascular R&D Centre—UnIC (Grant Nos. UIDB/00051/2020 and UIDP/00051/2020) and projects IMPAcT (Grant Nos. PTDC/MED-FSL/31719/2017; POCI-01–0145-FEDER-031719).
Acknowledgments
We thank Frances S. de Man for her careful reading and discussion of the manuscript and Audrey Chausson and Sara Cipriani for their assistance in the provision of human clinical tissue samples.
Conflict of interest
EM, FR, LB, VS, HR, and BK were employees of ATXA Therapeutics Limited at the time of conducting the work reported in this manuscript. BK was also the founder and a member of the board of directors of ATXA Therapeutics Limited. Studies performed at the University of Porto or at IPS Therapeutique Inc./ToxiPharm Laboratories Inc. were sponsored by ATXA Therapeutics Ltd. LH reported grants from Janssen, honoraria for speaking from Janssen and MSD, supported for attending congresses from Janssen, and participates on the advisory board or steering committee for Janssen, Gossamer Bio, MSD, and Acceleron. DM reported grants from Acceleron, Janssen, and Merck, consulting fees from Acceleron and speakers’ honoraria from Bayer, Janssen, and Merck. MH reported grants from Acceleron, AOP Orphan, Janssen, Merck, and Shou Ti, consulting fees from Acceleron, Aerovate, Altavant, AOP Orphan, Bayer, Chiesi, Ferrer, Janssen, Merck, MorphogenIX, and United Therapeutics, speakers’ honoraria from Janssen and Merck, and participates on the advisory board for Janssen, Altavant, Merck, United Therapeutics, and Acceleron.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2022.1063967/full#supplementary-material
References
1. Ryan J, Archer S. The right ventricle in pulmonary arterial hypertension: disorders of metabolism, angiogenesis and adrenergic signaling in right ventricular failure. Circ Res. (2014) 115:176–88. doi: 10.1161/CIRCRESAHA.113.301129
2. Vonk Noordegraaf A, Galie N. The role of the right ventricle in pulmonary arterial hypertension. Eur Respir Rev. (2011) 20:243–53. doi: 10.1183/09059180.00006511
3. Voelkel N, Quaife R, Leinwand L, Barst R, McGoon M, Meldrum D, et al. Right ventricular function and failure: report of a national heart, lung, and blood institute working group on cellular and molecular mechanisms of right heart failure. Circulation. (2006) 114:1883–91. doi: 10.1161/CIRCULATIONAHA.106.632208
4. Vonk-Noordegraaf A, Haddad F, Chin K, Forfia P, Kawut S, Lumens J, et al. Right heart adaptation to pulmonary arterial hypertension: physiology and pathobiology. J Am Coll Cardiol. (2013) 62(Suppl. 25):D22–33. doi: 10.1016/j.jacc.2013.10.027
5. Ren X, Johns R, Gao W. Express: right heart in pulmonary hypertension: from adaptation to failure. Pulm Circ. (2019) 9:2045894019845611. doi: 10.1177/2045894019845611
6. Sanz J, Sanchez-Quintana D, Bossone E, Bogaard H, Naeije R. Anatomy, function, and dysfunction of the right ventricle: jacc state-of-the-art review. J Am Coll Cardiol. (2019) 73:1463–82. doi: 10.1016/j.jacc.2018.12.076
7. Frump A, Bonnet S, de Jesus Perez V, Lahm T. Emerging role of angiogenesis in adaptive and maladaptive right ventricular remodeling in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. (2018) 314:L443–60. doi: 10.1152/ajplung.00374.2017
8. Handoko M, de Man F, Allaart C, Paulus W, Westerhof N, Vonk-Noordegraaf A. Perspectives on novel therapeutic strategies for right heart failure in pulmonary arterial hypertension: lessons from the left heart. Eur Respir Rev. (2010) 19:72–82. doi: 10.1183/09059180.00007109
9. Tello K, Seeger W, Naeije R, Vanderpool R, Ghofrani H, Richter M, et al. Right heart failure in pulmonary hypertension: diagnosis and new perspectives on vascular and direct right ventricular treatment. Br J Pharmacol. (2021) 178:90–107. doi: 10.1111/bph.14866
10. van de Veerdonk M, Kind T, Marcus J, Mauritz G, Heymans M, Bogaard H, et al. Progressive right ventricular dysfunction in patients with pulmonary arterial hypertension responding to therapy. J Am Coll Cardiol. (2011) 58:2511–9. doi: 10.1016/j.jacc.2011.06.068
11. Ekambaram P, Lambiv W, Cazzolli R, Ashton A, Honn K. The thromboxane synthase and receptor signaling pathway in cancer: an emerging paradigm in cancer progression and metastasis. Cancer Metastasis Rev. (2011) 30:397–408. doi: 10.1007/s10555-011-9297-9
12. Mulvaney E, Shilling C, Eivers S, Perry A, Bjartell A, Kay E, et al. Expression of the tpalpha and tpbeta isoforms of the thromboxane prostanoid receptor (Tp) in prostate cancer: clinical significance and diagnostic potential. Oncotarget. (2016) 7:73171–87. doi: 10.18632/oncotarget.12256
13. Fuse S, Kamiya T. Plasma thromboxane B2 concentration in pulmonary hypertension associated with congenital heart disease. Circulation. (1994) 90:2952–5.
14. Bui K, Hammerman C, Hirschl R, Snedecor S, Cheng K, Chan L, et al. Plasma prostanoids in neonatal extracorporeal membrane oxygenation. Influence of meconium aspiration. J Thorac Cardiovasc Surg. (1991) 101:612–7.
15. Dobyns E, Wescott J, Kennaugh J, Ross M, Stenmark K. Eicosanoids decrease with successful extracorporeal membrane oxygenation therapy in neonatal pulmonary hypertension. Am J Respir Crit Care Med. (1994) 149(4 Pt 1):873–80. doi: 10.1164/ajrccm.149.4.8143049
16. Christman B, McPherson C, Newman J, King G, Bernard G, Groves B, et al. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N Engl J Med. (1992) 327:70–5. doi: 10.1056/NEJM199207093270202
17. Al-Naamani N, Palevsky H, Lederer D, Horn E, Mathai S, Roberts K, et al. Prognostic significance of biomarkers in pulmonary arterial hypertension. Ann Am Thorac Soc. (2016) 13:25–30. doi: 10.1513/AnnalsATS.201508-543OC
18. Katugampola S, Davenport A. Thromboxane receptor density is increased in human cardiovascular disease with evidence for inhibition at therapeutic concentrations by the at(1) receptor antagonist losartan. Br J Pharmacol. (2001) 134:1385–92. doi: 10.1038/sj.bjp.0704416
19. West J, Voss B, Pavliv L, de Caestecker M, Hemnes A, Carrier E. Antagonism of the thromboxane-prostanoid receptor is cardioprotective against right ventricular pressure overload. Pulm Circ. (2016) 6:211–23. doi: 10.1086/686140
20. Hoffmann P, Heinroth-Hoffmann I, Toraason M. Alterations by a thromboxane A2 analog (U46619) of calcium dynamics in isolated rat cardiomyocytes. J Pharmacol Exp Ther. (1993) 264:336–44.
21. Nakamura F, Minshall R, Le Breton G, Rabito S. Thromboxane A2 mediates the stimulation of inositol 1,4,5-trisphosphate production and intracellular calcium mobilization by bradykinin in neonatal rat ventricular cardiomyocytes. Hypertension. (1996) 28:444–9. doi: 10.1161/01.hyp.28.3.444
22. Wacker M, Best S, Kosloski L, Stachura C, Smoot R, Porter C, et al. Thromboxane A2-induced arrhythmias in the anesthetized rabbit. Am J Physiol Heart Circ Physiol. (2006) 290:H1353–61. doi: 10.1152/ajpheart.00930.2005
23. Wacker M, Kosloski L, Gilbert W, Touchberry C, Moore D, Kelly J, et al. Inhibition of thromboxane A2-induced arrhythmias and intracellular calcium changes in cardiac myocytes by blockade of the inositol trisphosphate pathway. J Pharmacol Exp Ther. (2009) 331:917–24. doi: 10.1124/jpet.109.157677
24. Francois H, Athirakul K, Mao L, Rockman H, Coffman T. Role for thromboxane receptors in angiotensin-ii-induced hypertension. Hypertension. (2004) 43:364–9. doi: 10.1161/01.HYP.0000112225.27560.24
25. Francois H, Makhanova N, Ruiz P, Ellison J, Mao L, Rockman H, et al. A role for the thromboxane receptor in l-name hypertension. Am J Physiol Renal Physiol. (2008) 295:F1096–102. doi: 10.1152/ajprenal.00369.2007
26. West J, Galindo C, Kim K, Shin J, Atkinson J, Macias-Perez I, et al. Antagonism of the thromboxane-prostanoid receptor as a potential therapy for cardiomyopathy of muscular dystrophy. J Am Heart Assoc. (2019) 8:e011902. doi: 10.1161/JAHA.118.011902
27. Mulvaney E, Reid H, Bialesova L, Bouchard A, Salvail D, Kinsella B. Ntp42, a novel antagonist of the thromboxane receptor, attenuates experimentally induced pulmonary arterial hypertension. BMC Pulm Med. (2020) 20:85. doi: 10.1186/s12890-020-1113-2
28. Mulvaney E, Reid H, Bialesova L, Mendes-Ferreira P, Adao R, Bras-Silva C, et al. Efficacy of the thromboxane receptor antagonist Ntp42 Alone, or in combination with sildenafil, in the sugen/hypoxia-induced model of pulmonary arterial hypertension. Eur J Pharmacol. (2020) 889:173658. doi: 10.1016/j.ejphar.2020.173658
29. Boucherat O, Agrawal V, Lawrie A, Bonnet S. The Latest in animal models of pulmonary hypertension and right ventricular failure. Circ Res. (2022) 130:1466–86. doi: 10.1161/CIRCRESAHA.121.319971
30. Gomez-Arroyo J, Farkas L, Alhussaini A, Farkas D, Kraskauskas D, Voelkel N, et al. The monocrotaline model of pulmonary hypertension in perspective. Am J Physiol Lung Cell Mol Physiol. (2012) 302:L363–9. doi: 10.1152/ajplung.00212.2011
31. Rai N, Veeroju S, Schymura Y, Janssen W, Wietelmann A, Kojonazarov B, et al. Effect of riociguat and sildenafil on right heart remodeling and function in pressure overload induced model of pulmonary arterial banding. Biomed Res Int. (2018) 2018:3293584. doi: 10.1155/2018/3293584
32. Dachs T, Duca F, Rettl R, Binder-Rodriguez C, Dalos D, Ligios L, et al. Riociguat in pulmonary hypertension and heart failure with preserved ejection fraction: the haemodynamic trial. Eur Heart J. (2022) 43:3402–13. doi: 10.1093/eurheartj/ehac389
33. Guay C, Morin-Thibault L, Bonnet S, Lacasse Y, Lambert C, Lega J, et al. Pulmonary hypertension-targeted therapies in heart failure: a systematic review and meta-analysis. PLoS One. (2018) 13:e0204610. doi: 10.1371/journal.pone.0204610
34. Kido K, Coons J. Efficacy and safety of the use of pulmonary arterial hypertension pharmacotherapy in patients with pulmonary hypertension secondary to left heart disease: a systematic review. Pharmacotherapy. (2019) 39:929–45. doi: 10.1002/phar.2314
35. Rain S, Andersen S, Najafi A, Gammelgaard Schultz J, da Silva Goncalves Bos D, Handoko M, et al. Right ventricular myocardial stiffness in experimental pulmonary arterial hypertension: relative contribution of fibrosis and myofibril stiffness. Circ Heart Fail. (2016) 9:e002636. doi: 10.1161/CIRCHEARTFAILURE.115.002636
36. Rain S, Bos Dda S, Handoko M, Westerhof N, Stienen G, Ottenheijm C, et al. Protein changes contributing to right ventricular cardiomyocyte diastolic dysfunction in pulmonary arterial hypertension. J Am Heart Assoc. (2014) 3:e000716. doi: 10.1161/JAHA.113.000716
37. Lou Q, Janardhan A, Efimov I. Remodeling of calcium handling in human heart failure. Adv Exp Med Biol. (2012) 740:1145–74. doi: 10.1007/978-94-007-2888-2_52
38. Moon M, Aziz A, Lee A, Moon C, Okada S, Kanter E, et al. Differential calcium handling in two canine models of right ventricular pressure overload. J Surg Res. (2012) 178:554–62. doi: 10.1016/j.jss.2012.04.066
39. Muller O, Lange M, Rattunde H, Lorenzen H, Muller M, Frey N, et al. Transgenic rat hearts overexpressing serca2a show improved contractility under baseline conditions and pressure overload. Cardiovasc Res. (2003) 59:380–9. doi: 10.1016/s0008-6363(03)00429-2
40. Sande J, Sjaastad I, Hoen I, Bokenes J, Tonnessen T, Holt E, et al. Reduced level of serine(16) phosphorylated phospholamban in the failing rat myocardium: a major contributor to reduced serca2 activity. Cardiovasc Res. (2002) 53:382–91. doi: 10.1016/s0008-6363(01)00489-8
41. Wanichawan P, Hafver T, Hodne K, Aronsen J, Lunde I, Dalhus B, et al. Molecular basis of calpain cleavage and inactivation of the sodium-calcium exchanger 1 in heart failure. J Biol Chem. (2014) 289:33984–98. doi: 10.1074/jbc.M114.602581
42. Bidwell P, Liu G, Nagarajan N, Lam C, Haghighi K, Gardner G, et al. Hax-1 regulates serca2a oxidation and degradation. J Mol Cell Cardiol. (2018) 114:220–33. doi: 10.1016/j.yjmcc.2017.11.014
43. French J, Quindry J, Falk D, Staib J, Lee Y, Wang K, et al. Ischemia-reperfusion-induced calpain activation and serca2a degradation are attenuated by exercise training and calpain inhibition. Am J Physiol Heart Circ Physiol. (2006) 290:H128–36. doi: 10.1152/ajpheart.00739.2005
44. Roczkowsky A, Chan B, Lee T, Mahmud Z, Hartley B, Julien O, et al. Myocardial Mmp-2 contributes to serca2a proteolysis during cardiac ischaemia-reperfusion injury. Cardiovasc Res. (2020) 116:1021–31. doi: 10.1093/cvr/cvz207
45. Lahm T, Douglas I, Archer S, Bogaard H, Chesler N, Haddad F, et al. Assessment of right ventricular function in the research setting: knowledge gaps and pathways forward. an official american thoracic society research statement. Am J Respir Crit Care Med. (2018) 198:e15–43. doi: 10.1164/rccm.201806-1160ST
46. Prisco S, Thenappan T, Prins K. Treatment targets for right ventricular dysfunction in pulmonary arterial hypertension. JACC Basic Transl Sci. (2020) 5:1244–60. doi: 10.1016/j.jacbts.2020.07.011
47. Gomez-Arroyo J, Sandoval J, Simon M, Dominguez-Cano E, Voelkel N, Bogaard H. Treatment for pulmonary arterial hypertension-associated right ventricular dysfunction. Ann Am Thorac Soc. (2014) 11:1101–15. doi: 10.1513/AnnalsATS.201312-425FR
48. Tello K, Kremer N, Richter M, Gall H, Muenks J, Ghofrani A, et al. Inhaled iloprost improves right ventricular load-independent contractility in pulmonary hypertension. Am J Respir Crit Care Med. (2022) 206:111–4. doi: 10.1164/rccm.202201-0095LE
49. Sueta C, Gheorghiade M, Adams K Jr., Bourge R, Murali S, Uretsky B, et al. Safety and efficacy of epoprostenol in patients with severe congestive heart failure. epoprostenol multicenter research group. Am J Cardiol. (1995) 75:34A–43A. doi: 10.1016/s0002-9149(99)80381-6
50. Califf R, Adams K, McKenna W, Gheorghiade M, Uretsky B, McNulty S, et al. A randomized controlled trial of epoprostenol therapy for severe congestive heart failure: the flolan international randomized survival trial (First). Am Heart J. (1997) 134:44–54. doi: 10.1016/s0002-8703(97)70105-4
51. Nagendran J, Sutendra G, Paterson I, Champion H, Webster L, Chiu B, et al. Endothelin axis is upregulated in human and rat right ventricular hypertrophy. Circ Res. (2013) 112:347–54. doi: 10.1161/CIRCRESAHA.111.300448
52. Kaluski E, Cotter G, Leitman M, Milo-Cotter O, Krakover R, Kobrin I, et al. Clinical and hemodynamic effects of bosentan dose optimization in symptomatic heart failure patients with severe systolic dysfunction, associated with secondary pulmonary hypertension–a multi-center randomized study. Cardiology. (2008) 109:273–80. doi: 10.1159/000107791
53. Kalra P, Moon J, Coats A. Do results of the enable (endothelin antagonist bosentan for lowering cardiac events in heart failure) study spell the end for non-selective endothelin antagonism in heart failure? Int J Cardiol. (2002) 85:195–7. doi: 10.1016/s0167-5273(02)00182-1
54. Anand I, McMurray J, Cohn J, Konstam M, Notter T, Quitzau K, et al. Long-term effects of darusentan on left-ventricular remodelling and clinical outcomes in the endothelina receptor antagonist trial in heart failure (earth): randomised, double-blind, placebo-controlled trial. Lancet. (2004) 364:347–54. doi: 10.1016/S0140-6736(04)16723-8
55. Gan C, Holverda S, Marcus J, Paulus W, Marques K, Bronzwaer J, et al. Right ventricular diastolic dysfunction and the acute effects of sildenafil in pulmonary hypertension patients. Chest. (2007) 132:11–7. doi: 10.1378/chest.06-1263
56. Redfield M, Chen H, Borlaug B, Semigran M, Lee K, Lewis G, et al. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA. (2013) 309:1268–77. doi: 10.1001/jama.2013.2024
57. Bonderman D, Ghio S, Felix S, Ghofrani H, Michelakis E, Mitrovic V, et al. Riociguat for patients with pulmonary hypertension caused by systolic left ventricular dysfunction: a phase iib double-blind, randomized, placebo-controlled, dose-ranging hemodynamic study. Circulation. (2013) 128:502–11. doi: 10.1161/CIRCULATIONAHA.113.001458
58. Langleben D, Carvalho A, Reid L. The platelet thromboxane inhibitor, dazmegrel, does not reduce monocrotaline-induced pulmonary hypertension. Am Rev Respir Dis. (1986) 133:789–91.
59. Nagata T, Uehara Y, Hara K, Igarashi K, Hazama H, Hisada T, et al. Thromboxane inhibition and monocrotaline-induced pulmonary hypertension in rats. Respirology. (1997) 2:283–9. doi: 10.1111/j.1440-1843.1997.tb00090.x
60. Akazawa Y, Okumura K, Ishii R, Slorach C, Hui W, Ide H, et al. Pulmonary artery banding is a relevant model to study the right ventricular remodeling and dysfunction that occurs in pulmonary arterial hypertension. J Appl Physiol. (2020) 129:238–46. doi: 10.1152/japplphysiol.00148.2020
61. Kinsella B, Reid H. Thromboxane Receptor Antagonists. US PTO (Patent No. 10,357,504, Fully Granted July 23, 2019). Alexandria, VA: United States Patent and Trademark Office (US PTO) (2019).
62. Kinsella B, Reid H. Thromboxane Receptor Antagonists. US PTO (Patent No. 9,932,304, Fully Granted April 3, 2018). Alexandria, VA: United States Patent and Trademark Office (US PTO) (2018).
63. Kinsella B. Thromboxane A2 signalling in humans: a ‘Tail’ of two receptors. Biochem Soc Trans. (2001) 29(Pt 6):641–54.
64. Pauvert O, Lugnier C, Keravis T, Marthan R, Rousseau E, Savineau J. Effect of sildenafil on cyclic nucleotide phosphodiesterase activity, vascular tone and calcium signaling in rat pulmonary artery. Br J Pharmacol. (2003) 139:513–22. doi: 10.1038/sj.bjp.0705277
65. Wallis R. The pharmacology of sildenafil, a novel and selective inhibitor of phosphodiesterase (Pde) type 5. Nihon Yakurigaku Zasshi. (1999) 114(Suppl. 1):22–6. doi: 10.1254/fpj.114.supplement_22
66. European Medicines Agency. Assessment Report for Opsumit (International Non-Proprietary Name: Macitentan). (2013). Available online at: https://www.ema.europa.eu/en/documents/assessment-report/opsumit-epar-public-assessment-report_en.pdf (accessed January 26, 2022).
67. Iglarz M, Binkert C, Morrison K, Fischli W, Gatfield J, Treiber A, et al. Pharmacology of Macitentan, an Orally Active Tissue-Targeting Dual Endothelin Receptor Antagonist. J Pharmacol Exp Ther. (2008) 327:736–45. doi: 10.1124/jpet.108.142976
68. European Medicines Agency. Assessment Report for Uptravi (International Non-Proprietary Name: Selexipag). (2016). Available online at: https://www.ema.europa.eu/en/documents/assessment-report/uptravi-epar-public-assessment-report_en.pdf (accessed January 26, 2022).
69. European Medicines Agency. Assessment Report for Adempas (International Non-Proprietary Name: Riociguat). (2014). Available online at: https://www.ema.europa.eu/en/documents/assessment-report/adempas-epar-public-assessment-report_en.pdf (accessed January 26, 2022).
70. Follmann M, Ackerstaff J, Redlich G, Wunder F, Lang D, Kern A, et al. Discovery of the soluble guanylate cyclase stimulator vericiguat (bay 1021189) for the treatment of chronic heart failure. J Med Chem. (2017) 60:5146–61. doi: 10.1021/acs.jmedchem.7b00449
71. West J, Kim K, Suzuki T, Moore C, Knollmann B, Carrier E. Abstract 824: thromboxane/prostanoid receptor activation increases calpain-mediated proteolysis and alters calcium handling and fibrosis following right ventricular pressure overload. Circulation Res. (2019) 125(Suppl. 1):A824–A. doi: 10.1161/res.125.suppl_1.824
72. Carrier E, Kim K, Shay S, Moore C, Knollmann B, West J. Abstract 443: blockade of the thromboxane/prostanoid receptor prevents Ecg abnormalities in Rv pressure overload. Circulation Res. (2020) 127(Suppl. 1):A443–A. doi: 10.1161/res.127.suppl_1.443
73. Carrier E, Kim K, Noll N, Macias-Perez I, Merryman W, Knollmann B, et al. Abstract 261: activation of the thromboxane/prostanoid receptor contributes to elevated end-diastolic calcium in cardiomyocytes and cardiac fibrosis following right ventricular pressure overload. Circulation Res. (2018) 123(Suppl. 1):A261–A. doi: 10.1161/res.123.suppl_1.261
74. Langleben D, Christman B, Barst R, Dias V, Galie N, Higenbottam T, et al. Effects of the thromboxane synthetase inhibitor and receptor antagonist terbogrel in patients with primary pulmonary hypertension. Am Heart J. (2002) 143:E4. doi: 10.1067/mhj.2002.121806
75. Grann M, Comerma-Steffensen S, Arcanjo D, Simonsen U. Mechanisms involved in thromboxane A2 -induced vasoconstriction of rat intracavernous small penile arteries. Basic Clin Pharmacol Toxicol. (2016) 119(Suppl. 3):86–95. doi: 10.1111/bcpt.12544
76. Cogolludo A, Moreno L, Bosca L, Tamargo J, Perez-Vizcaino F. Thromboxane A2-induced inhibition of voltage-gated K+ channels and pulmonary vasoconstriction: role of protein kinase czeta. Circ Res. (2003) 93:656–63. doi: 10.1161/01.RES.0000095245.97945.FE
77. Ozen G, Aljesri K, Celik Z, Turkyilmaz G, Turkyilmaz S, Teskin O, et al. Mechanism of thromboxane receptor-induced vasoconstriction in human saphenous vein. Prostaglandins Other Lipid Mediat. (2020) 151:106476. doi: 10.1016/j.prostaglandins.2020.106476
78. Tosun M, Paul R, Rapoport R. Role of extracellular Ca++ Influx Via L-Type and Non-L-Type Ca++ channels in thromboxane A2 RECEPTOR-MEDIATED CONTRACTION IN RAT AORTA. J Pharmacol Exp Ther. (1998) 284:921–8.
79. Hunt H, Tilunaite A, Bass G, Soeller C, Roderick H, Rajagopal V, et al. Ca(2+) release Via Ip3 receptors shapes the cardiac Ca(2+) transient for hypertrophic signaling. Biophys J. (2020) 119:1178–92. doi: 10.1016/j.bpj.2020.08.001
80. Rinne A, Blatter L. Activation of Nfatc1 is directly mediated by ip3 in adult cardiac myocytes. Am J Physiol Heart Circ Physiol. (2010) 299:H1701–7. doi: 10.1152/ajpheart.00470.2010
81. Nakayama H, Bodi I, Maillet M, DeSantiago J, Domeier T, Mikoshiba K, et al. The Ip3 receptor regulates cardiac hypertrophy in response to select stimuli. Circ Res. (2010) 107: 659–66. doi: 10.1161/CIRCRESAHA.110.220038
82. Higazi DR, Fearnley CJ, Drawnel FM, Talasila A, Corps EM, Ritter O, et al. Endothelin-1-stimulated insp3-induced ca2+ release is a nexus for hypertrophic signaling in cardiac myocytes. Mol Cell. (2009) 33:472-82. doi: 10.1016/j.molcel.2009.02.005
83. Yamaguchi O, Taneike M, Otsu K. Cooperation between proteolytic systems in cardiomyocyte recycling. Cardiovasc Res. (2012) 96:46–52. doi: 10.1093/cvr/cvs236
84. Quiniou C, Sennlaub F, Beauchamp M, Checchin D, Lahaie I, Brault S, et al. Dominant role for calpain in thromboxane-induced neuromicrovascular endothelial cytotoxicity. J Pharmacol Exp Ther. (2006) 316:618–27. doi: 10.1124/jpet.105.093898
85. Barta J, Toth A, Edes I, Vaszily M, Papp J, Varro A, et al. Calpain-1-sensitive myofibrillar proteins of the human myocardium. Mol Cell Biochem. (2005) 278:1–8. doi: 10.1007/s11010-005-1370-7
86. Huang J, Forsberg N. Role of calpain in skeletal-muscle protein degradation. Proc Natl Acad Sci U.S.A. (1998) 95:12100–5. doi: 10.1073/pnas.95.21.12100
87. Whipple G, Koohmaraie M. Degradation of myofibrillar proteins by extractable lysosomal enzymes and M-calpain, and the effects of zinc chloride. J Anim Sci. (1991) 69:4449–60. doi: 10.2527/1991.69114449x
88. Mallat Z, Philip I, Lebret M, Chatel D, Maclouf J, Tedgui A. Elevated levels of 8-Iso-prostaglandin F2alpha in pericardial fluid of patients with heart failure: a potential role for in vivo oxidant stress in ventricular dilatation and progression to heart failure. Circulation. (1998) 97:1536–9. doi: 10.1161/01.cir.97.16.1536
89. Linke W. Sense and Stretchability: the role of titin and titin-associated proteins in myocardial stress-sensing and mechanical dysfunction. Cardiovasc Res. (2008) 77:637–48. doi: 10.1016/j.cardiores.2007.03.029
90. Hidalgo C, Hudson B, Bogomolovas J, Zhu Y, Anderson B, Greaser M, et al. Pkc phosphorylation of Titin’s pevk element: a novel and conserved pathway for modulating myocardial stiffness. Circ Res. (2009) 105:631–8. doi: 10.1161/CIRCRESAHA.109.198465
91. Hudson B, Hidalgo C, Saripalli C, Granzier H. Hyperphosphorylation of mouse cardiac titin contributes to transverse aortic constriction-induced diastolic dysfunction. Circ Res. (2011) 109:858–66. doi: 10.1161/CIRCRESAHA.111.246819
92. Yamasaki R, Wu Y, McNabb M, Greaser M, Labeit S, Granzier H. Protein kinase a phosphorylates Titin’s cardiac-specific N2b domain and reduces passive tension in rat cardiac myocytes. Circ Res. (2002) 90:1181–8. doi: 10.1161/01.res.0000021115.24712.99
93. Janssen B, De Celle T, Debets J, Brouns A, Callahan M, Smith T. Effects of anesthetics on systemic hemodynamics in mice. Am J Physiol Heart Circ Physiol. (2004) 287:H1618–24. doi: 10.1152/ajpheart.01192.2003
94. Manca P, Nuzzi V, Cannata A, Castrichini M, Bromage D, De Luca A, et al. The right ventricular involvement in dilated cardiomyopathy: prevalence and prognostic implications of the often-neglected child. Heart Fail Rev. (2022) 27:1795–805. doi: 10.1007/s10741-022-10229-7
Keywords: pulmonary arterial hypertension (PAH), thromboxane receptor, NTP42, right ventricle (RV), heart failure
Citation: Mulvaney EP, Renzo F, Adão R, Dupre E, Bialesova L, Salvatore V, Reid HM, Conceição G, Grynblat J, Llucià-Valldeperas A, Michel J-B, Brás-Silva C, Laurent CE, Howard LS, Montani D, Humbert M, Vonk Noordegraaf A, Perros F, Mendes-Ferreira P and Kinsella BT (2022) The thromboxane receptor antagonist NTP42 promotes beneficial adaptation and preserves cardiac function in experimental models of right heart overload. Front. Cardiovasc. Med. 9:1063967. doi: 10.3389/fcvm.2022.1063967
Received: 07 October 2022; Accepted: 22 November 2022;
Published: 14 December 2022.
Edited by:
Serafino Fazio, Federico II University Hospital, ItalyReviewed by:
Alessandra Cuomo, Federico II University Hospital, ItalyErica J. Carrier, Vanderbilt University Medical Center, United States
Copyright © 2022 Mulvaney, Renzo, Adão, Dupre, Bialesova, Salvatore, Reid, Conceição, Grynblat, Llucià-Valldeperas, Michel, Brás-Silva, Laurent, Howard, Montani, Humbert, Vonk Noordegraaf, Perros, Mendes-Ferreira and Kinsella. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: B. Therese Kinsella, therese.kinsella@atxatherapeutics.com