Identification of Rare Variants in Right Ventricular Outflow Tract Obstruction Congenital Heart Disease by Whole-Exome Sequencing
 Yue Zhou1† Kai Bai1† Yu Wang1,2 Zhuo Meng1 Shuang Zhou1 Shiwei Jiang1
Yue Zhou1† Kai Bai1† Yu Wang1,2 Zhuo Meng1 Shuang Zhou1 Shiwei Jiang1  Hualin Wang1 Jian Wang1 Mei Yang1 Qingjie Wang1*
Hualin Wang1 Jian Wang1 Mei Yang1 Qingjie Wang1*  Kun Sun1*
Kun Sun1*  Sun Chen1*
Sun Chen1*- 1Department of Pediatric Cardiology, Xinhua Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, China
- 2Department of Pediatric Cardiology, The Second Affiliated Hospital & Yuying Children's Hospital of Wenzhou Medical University, Wenzhou, China
Background: Pulmonary atresia (PA) is a kind of congenital heart disease characterized by right ventricular outflow tract obstruction. It is divided into PA with intact ventricular septum (PA/IVS) whose favorable form is pulmonary valvular stenosis (PS), and PA with ventricular septal defect (PA/VSD) whose favorable form is tetralogy of Fallot (TOF). Due to limitations in genetics etiology, whole-exome sequencing (WES) was utilized to identify new variants associated with the diseases.
Methods: The data from PS-PA/IVS (n = 74), TOF-PA/VSD (n = 100), and 100 controls were obtained. The common sites between PS and PA/IVS, PA/VSD and TOF, were compared. The novel rare damage variants, and candidate genes were identified by gene-based burden analysis. Finally, the enrichment analysis of differential genes was conducted between case and control groups.
Results: Seventeen rare damage variants located in seven genes were predicted to be associated with the PS through burden analysis. Enrichment analysis identified that the Wnt and cadherin signaling pathways were relevant to PS-PA/IVS.
Conclusion: This study put forth seven candidate genes (APC, PPP1R12A, PCK2, SOS2, TNR, MED13, and TIAM1), resulting in PS-PA/IVS. The Wnt and cadherin signaling pathways were identified to be related to PS-PA/IVS by enrichment analysis. This study provides new evidence for exploring the genetic mechanism of PS-PA/IVS.
Introduction
Congenital heart disease (CHD) is the most common congenital disorder that affects about 1% liveborn infants (1). The incidence of CHD among live birth in China is ~0.9% (2). Pulmonary atresia (PA), a rare malformation of complex cyanotic CHD characterized by right ventricular outflow tract obstruction (RVOTO), accounts for about 1.3–3.4% of all heart abnormalities (3). The definition of PA is that there is no direct communication between the ventricle and the pulmonary blood flow. PA is traditionally divided into two groups: PA with intact ventricular septum (PA/IVS) and PA with ventricular septal defect (PA/VSD) (4). The characteristics of PA/IVS are membranous or muscular atresia of the RVOT without communication between the ventricle and pulmonary vessel (5). The pulmonary valvular stenosis (PS) might be a favorable form of PA/IVS. It is also a cyanotic CHD and comprises 8–12% of all CHDs (6, 7). PA/VSD is another group of PA and is considered as the extreme form of tetralogy of Fallot (TOF) (8). TOF is a heart defects syndrome with heart malformations of TOF are VSD, right ventricular hypertrophy, variable obstruction of the right ventricular outflow tract, and overriding aorta (9, 10). Patients born with these diseases might need to relieve RVOTO by intervention or surgery (11, 12). Although these diseases belong to or are similar to the PA family, they differ in structural abnormalities, hemodynamics, intervention strategies, and prognosis, implying varied pathogenesis (3).
Single-gene disorders and chromosomal anomalies could be the genetic etiology of CHD (13). Approximately 25% of CHDs could be explained based on genetic mutations (14). CHD patients with existing family history or suspected with other birth abnormalities, intellectual disability/developmental delay may undergo the clinical genetics evaluation; however, such patients constitute a small proportion. The majority of CHDs are isolated, sporadic, and non-syndromic cases (15). Due to the limitations in genetics etiology, current studies have utilized whole-exome sequencing (WES) to identify variants associated with the disease. Currently, known genes associated with PA/IVS include HNRNPC, DANH10, GAJ1, and GDF1 (3, 16), while the genes related to PA/VSD are GAJ5, MTHFR, MYH6, DANH1, PPP4C, FLT4, RICTOR, and FGF22 (16, 17). For sporadic TOF, GATA4, NKX2.5, JAG1, FOXC2, and TBX1 are correlated with its genetic etiology (18). A previous study reported a plausible gene-disease association between TOF/PS and CDC42BPA and FGD5 (19). Another study found that the pathogenesis of PS and PA/IVS might be related to the increased percentage of homozygous TT genotypes of MTHFR (20). However, only a few studies identified the rare variants of PS. The genetic etiology of PS and the differences in the genetic pattern between PS and PA are yet to be elucidated. In this study, we compared the genetic etiology of the four diseases according to the WES. In addition, we analyzed the potentially pathogenic genes of PS and PA/IVS.
Materials and Methods
Study Population
The flow chart of the study was illustrated in Figure 1. The cohort comprised 174 sporadic patients diagnosed with CHD by echocardiography and 100 healthy controls. The cohort comprised Chinese Han individuals aged 0–14-year-old with no family history of CHD. The CHD cases were further divided into four groups: PS (n = 42), PA/IVS (n =32), TOF (n =40), and PA/VSD (n =60) (Supplementary Table 1). The patients were selected according to the inclusion criteria that included patients with neither other CHDs nor identified syndromic or chromosomal disorders. The study was approved by the Ethics Committee of Xinhua Hospital affiliated with Shanghai Jiao Tong University (XHEC-C-2019-083), and conducted in accordance with the Declaration of Helsinki. Informed consent was obtained from all participants' parents.
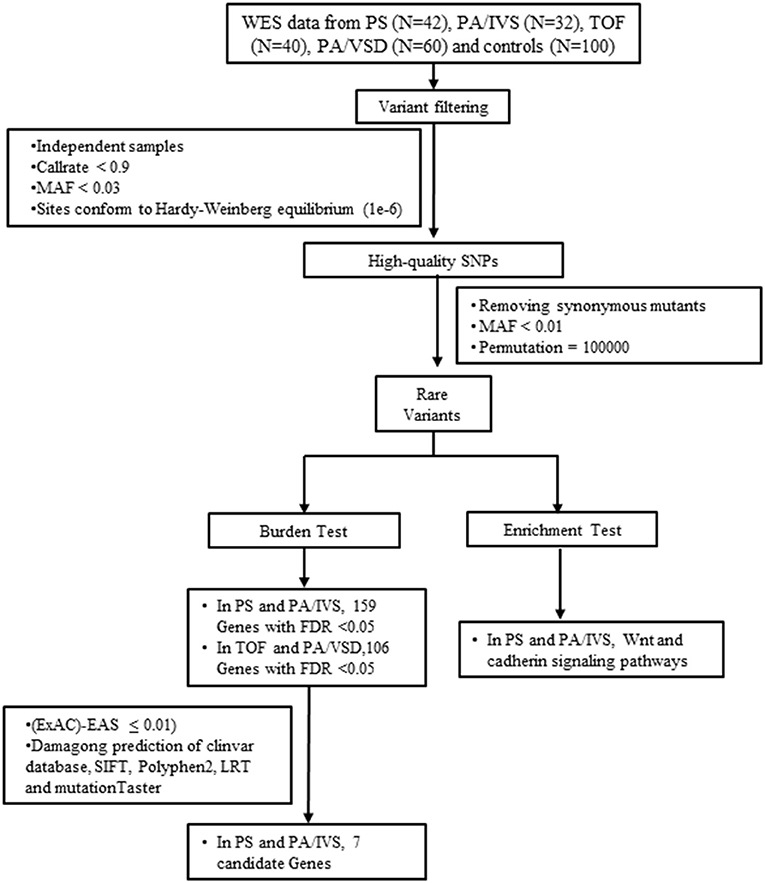
Figure 1. The flow chart showed the different steps taken during whole-exome sequencing analysis. After variant calling, annotation and screening, variants were filtered by gene-based burden analysis. Candidate genes were collected by gene expression analysis. FDR, false discovery rate; MAF, minor allele frequency; PA/IVS, pulmonary atresia with intact ventricular septum; PA/VSD, pulmonary atresia with ventricular septal defect; PS, Pulmonary valvular stenosis; SNP, single nucleotide polymorphism; TOF, tetralogy of Fallot; WES, whole-exome sequencing.
WES
Peripheral blood DNA was extracted using the QIAampTM DNA and Blood Mini Kit (Qiagen, Germany) following the standard instructions. The DNA concentration and quality were assessed by measuring the 260/280 optical density value on a NanoDrop 2000c spectrophotometer (Thermo Fisher Scientific, USA). WES was performed using the Agilent Sure Select Target Enrichment kit (V6 58 Mbp; Agilent Technologies, USA) for sequence capture and Illumina HiSeq2500 for sequencing (Illumina, USA).
Reads Mapping, Variant Calling, and Annotation
Sequencing reads were mapped to the Genome Reference Consortium Human Genome Build 37 (GRCh37). Single nucleotide variant (SNV) and Insert/Delete (Indel) results were processed simultaneously by Genome Analysis Toolkit (GATK) HiplotypeCaller. Subsequently, ANNOVAR software was employed to complete the mutation annotation, including the population control databases (ExAC, genomAD, 1000 Genomes Projects, etc.) and mutation prediction programs (SIFT, Polyphen-2, and MutationTaster, etc.).
Single Nucleotide Polymorphisms Screening
Original data should be controlled to remove the sites with low detection rate, low allele frequency, and not in agreement with the Hardy–Weinberg equilibrium. The details are as follows:
(1) Duplicate samples and related samples were removed.
(2) Callrate ≥ 0.9.
(3) Minor allele frequency (MAF) ≥ 0.03.
(4) Remove sites that do not conform to Hardy-Weinberg equilibrium (1e-6).
Burden Analysis
This study used the “T1” criterion: if a variation site (SNV or Indels) had a minor allele frequency (MAF) <0.01, then the variation site was regarded as a rare variant site (21).
After removing synonymous mutations, the non-synonymous mutations in rare mutations were divided into five groups: (1) non-synonymous rare mutations (non-synonymous sets); (2) deleterious rare mutations labeled as “deleterious” by Polyphen-2 humdiv (deleterious sets); (3) these rare mutations labeled as nonsense, Indel frameshift, and splice site (disruptive sets); (4) deleterious rare mutations labeled as “deleterious” in at least one of the five protein prediction algorithms, including LRT Score, MutationTaster, Polyphen-2 humdiv, Polyphen-2 humvar, and SIFT (Broad deleterious sets); (5) rare mutations labeled “deleterious” by five protein prediction algorithms (Strict deleterious sets) (21).
In this study, a variant of the Combined Multivariate Collapsing test, which grouped the count of alleles of SNVs in cases and controls, was performed. To assign a statistical significance, we permuted the phenotype labels 100,000 times. Association analysis was performed using PLINK/SEQ. Frequency-weighted test (FW) and variable threshold test (VT) methods were used to calculate the correlation between rare mutations in each gene region and the diseases (21).
Rare Variants Validation
Furthermore, these candidate variants were confirmed by Sanger sequencing. The primers for PCR amplification were designed using Primer premier5; the sequences were listed in Supplementary Table 2.
Results
Overview of SNPs
The obtained data were filtered by total sample variation screening, callrate (missing rate) test, singleton (defined as a variant present in only one sample) test, heterozygosity test, and Ti/Tv ratio test. Based on the above quality control methods, one control sample was removed due to many singletons, which indicated the low quality of DNA. Another control sample was removed because of high heterozygosity (Supplementary Figure 1).
We compared the common and different SNPs between PS and PA/IVS, TOF and PA/VSD. After quality control, 18549, 21326, 16594, and 20230 SNPs and Indels were identified in PS, PA/IVS, PA/VSD, and TOF, respectively. Next, we counted and compared the localization of selected variants, variants type, the function of exonic variants, and SNV classes of four diseases (Figures 2A–D). The most selected variants were in introns, followed by exons. Most of the exonic variants were SNVs, and the number of Inserts (INSs) and Deletes (DELs) was small. Regarding the exonic function, the number of synonymous and non-synonymous SNVs were significantly larger than the others (non-frameshift INS/DEL, stoploss, stopgain, frameshift INS/DEL). In these SNVs, T>C was the most common class. The number of common sites between PS and PA/IVS was 15644, while those between PA/VSD and TOF were 14854 (Figure 2E). A previous study speculated that PA/IVS was the extremely severe form of PS, and PA/VSD, another group of PA, was the extreme form of TOF (6–8). Because of the limited sample size of each disease, we combined PS and PA/IVS as one group PS-PA/IVS, and PA/VSD and TOF as another group TOF-PA/VSD for further analysis.
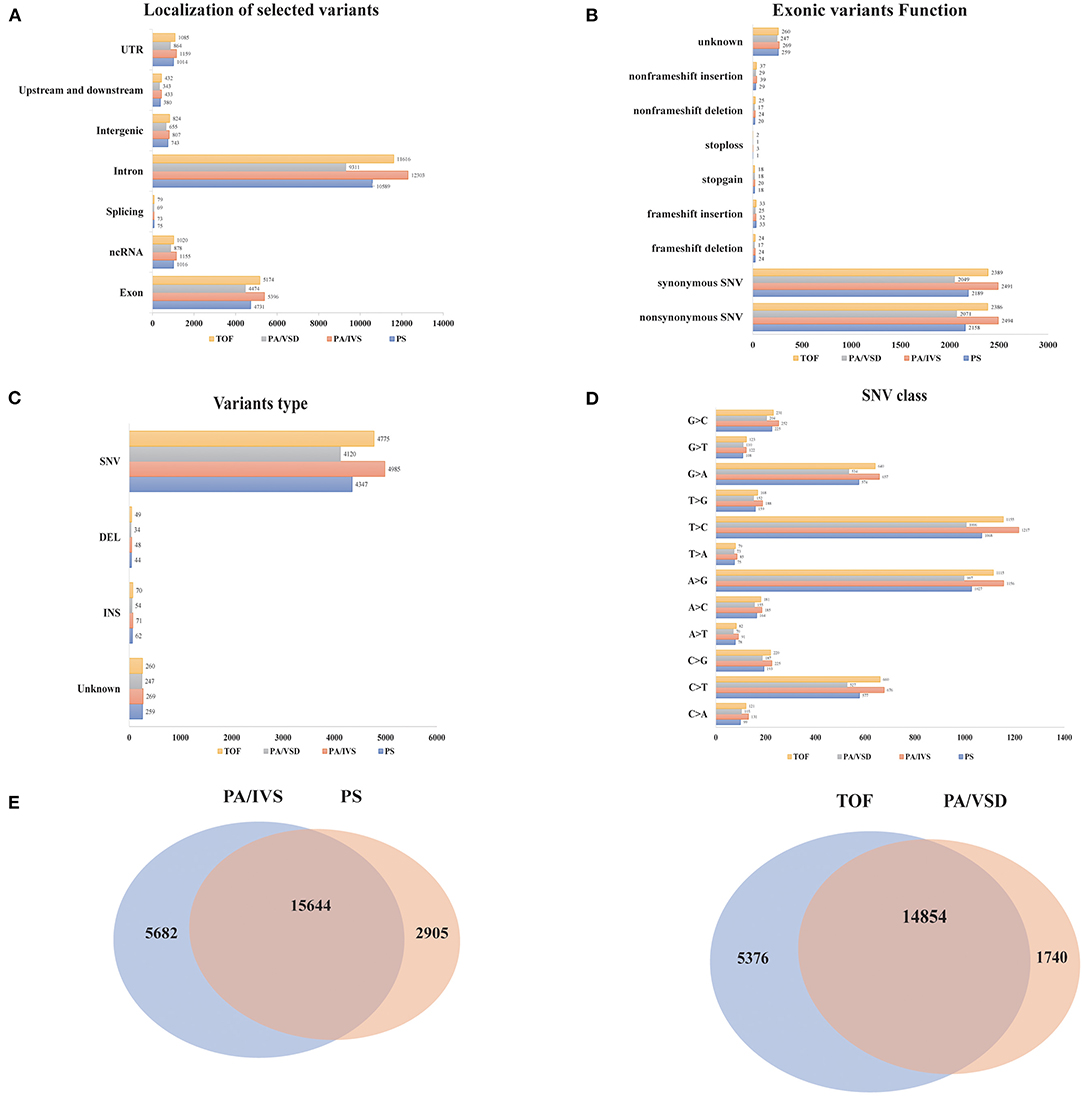
Figure 2. The comparisons of the variants among four disease groups. The localization of selected variants (A), variants type (B), the function of exonic variants (C), and SNV classes (D) of four diseases. (E) The comparisons of the variants between PA/IVS and PS, TOF and PA/VSD.
Rare Variants Analysis
We conducted a principal component analysis (PCA) between these two groups with control and found that the distribution of disease and control samples was consistent (Supplementary Figures 2A,B).
Burden Test
The definition of rare variants was that the MAF of variants was <1% or <0.5% (22). In order to conduct the correlation analysis on the rare mutants, after setting MAF <0.01, we conducted the burden test of rare variants undergoing selection and classification. To enrich the harmful alleles, we considered three groups of variants (non-synonymous, deleterious and disruptive sets) (21). Given the threshold of p = 0.01, 21, and 10 genes with potential pathogenicity of PS-PA/IVS were found in the non-synonymous and deleterious sets, respectively; 20 and 9 genes with potential pathogenicity of TOF-PA/VSD were found in the non-synonymous and deleterious sets, respectively; no gene in two diseases was found in disruptive sets. Since only a few studies have assessed the rare variants of PS-PA/IVS, we mainly analyzed the genes with potential pathogenicity of the two diseases. A total of 26 genes with potential disease pathogenicity were identified (Table 1). 218 unique non-synonymous or splice-site SNVs or Indel frameshifts with MAF <0.01 were identified among these genes. Also, 26 alternate allele counts in cases and 19 alternate allele counts in controls were observed.
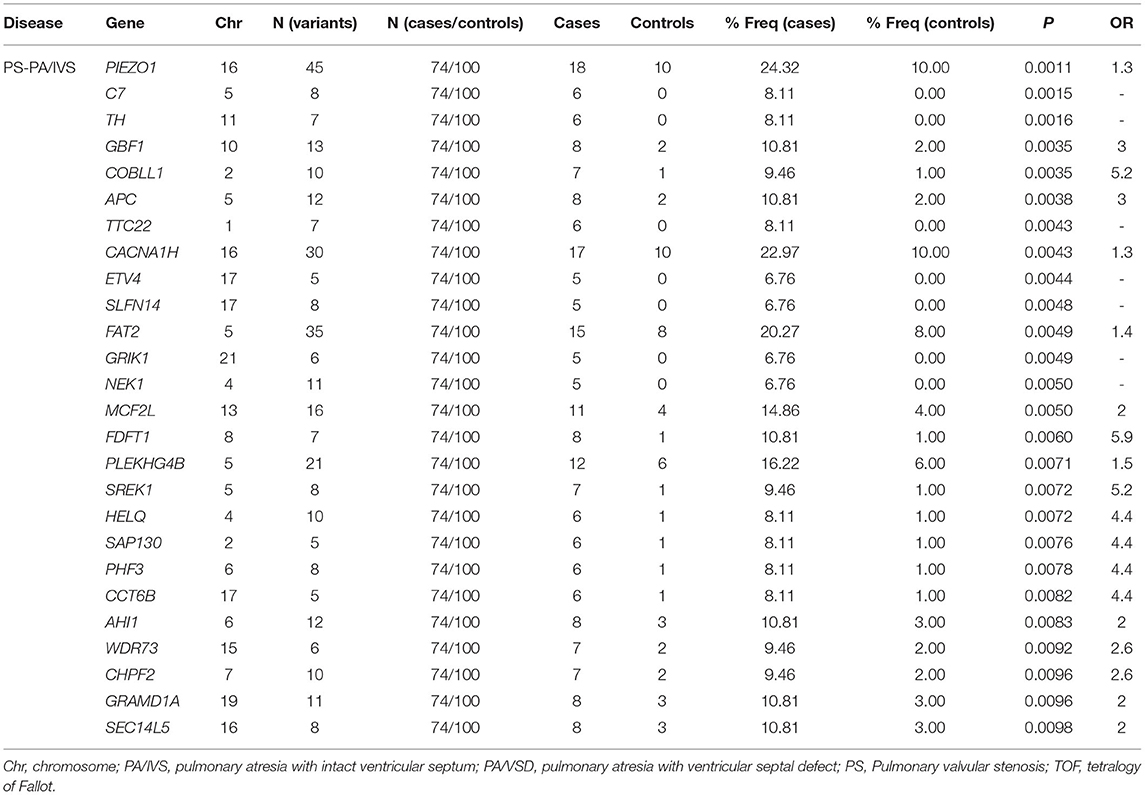
Table 1. A list of genes with the potential pathogenicity of disease were identified by burden test.
The variant sets based on multiple protein prediction algorithms might yield strong association signals. Therefore, in exploratory analyses, we investigated two additional variant sets (broad deleterious and strict deleterious sets) (21). The threshold was set to p = 0.05 for these two sets, following which, we identified 159 genes of PS-PA/IVS and 106 genes of TOF-PA/VSD. Next, we searched and summarized the known genes related to the PS phenotypes (Figure 3A; http://phenolyzer.wglab.org/). After excluding the genes related to the syndromes, only those associated with the PS phenotype remained (Figure 3B). Then, we predicted the association between genes tested in this study and disease phenotype (Figure 3C) and identified two seed genes, SOS2 and DOCK6. A seed gene meant that this gene was directly correlated to the input term based on the existing databases. SOS2 was a known gene related to the PS phenotype, while DOCK6 was associated with Adams–Oliver syndrome, whose clinical performance might exist PS phenotype. Next, we listed the first 25 genes in Figure 3D. Except for the two seed genes, the remaining genes, such as CDKN1B, TIAM1, APC, and PPP1R12A, were also predicted to be relevant to disease phenotypes by gene-gene linkage (Figure 3D).
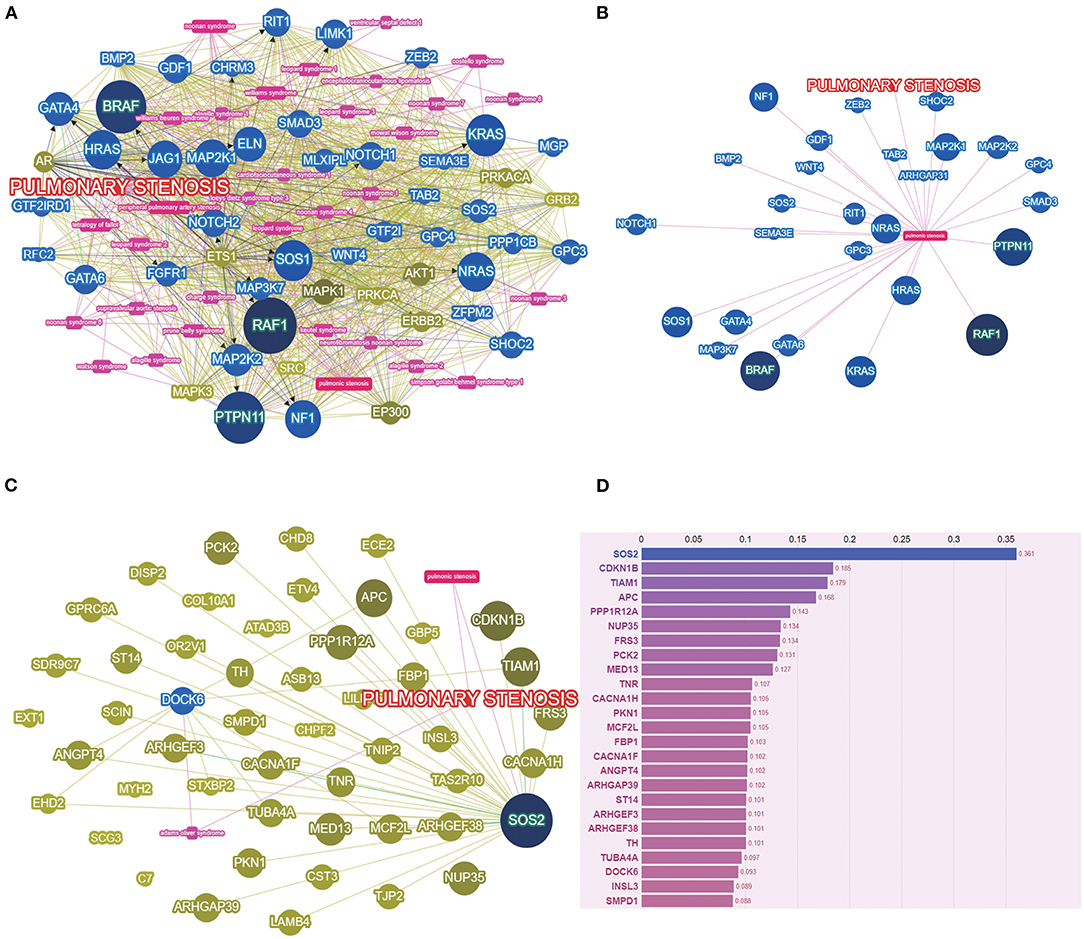
Figure 3. The PS-associated genes were identified by gene-based burden analysis. (A) The known genes related to the PS phenotypes were searched and summarized. (B) Genes associated with PS phenotypes were shown after excluding the genes related to the syndromes. (C) The association between genes tested and disease phenotype was predicted. (D) The score of genes tested was listed by Phenolyzer (http://phenolyzer.wglab.org/).
Enrichment Analysis
To further analyze the function of differential genes between case and control groups, we conducted the enrichment analysis of genes in strict deleterious set. The enrichment analysis mainly included disease, ontology, and pathway enrichment (Figure 4). Regarding disease enrichment, these genes were involved in a variety of systemic diseases, among which cardiovascular diseases mainly included hypertrophic cardiomyopathy and hypertension. In addition, pathway enrichment found Wnt and cadherin signaling pathways. The Wnt signaling pathway was a highly conserved regulatory pathway during embryonic development. When dysregulated, it could result in congenital malformations, including aortic valve stenosis (23, 24). The cadherin signaling pathway also played a role during aortic valve maturation (25). However, whether these pathways affect PS or PA/IVS was yet to be explored. The enrichment analysis might be limited by fewer pathogenic genes associated with PS and PA/IVS.
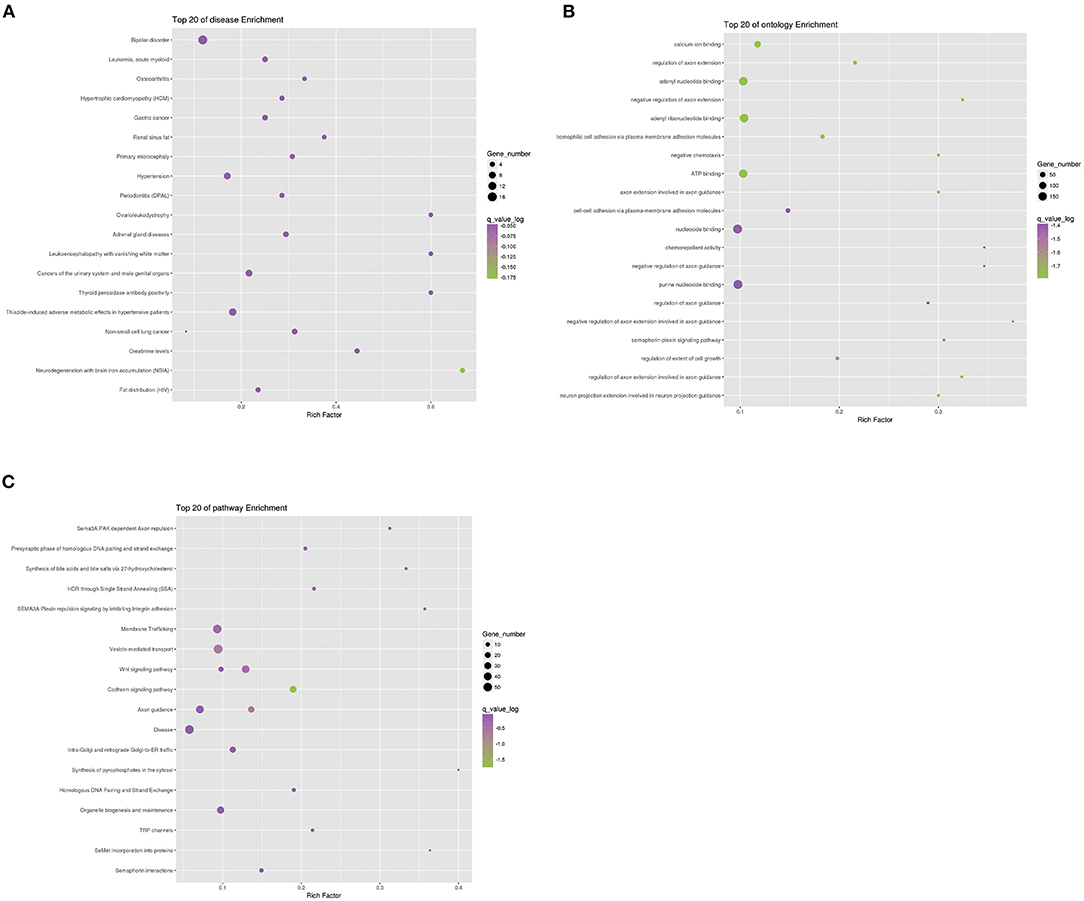
Figure 4. Enrichment analysis of differential genes between PS-PA/IVS and control groups. Top 20 of disease (A), ontology (B), and pathway (C) enrichment were listed.
Rare Variants Validation
Among the above 159 selected genes of PS-PA/IVS, we selected the first ten genes for further analysis. After being predicted to affect the protein function according to ClinVar database, SIFT, Polyphen-2, LRT, and MutationTaster prediction, 19 SNPs located in seven genes, SOS2, TIAM1, APC, PPP1R12A, PCK2, MED13, and TNR were left (Table 2; Supplementary Table 3). Thus, seven genes with 19 SNPs were validated. Next, according to the position of SNPs, we designed primers to validate the mutations by Sanger sequencing. We found that only two SNPs were not validated successfully (APC chr5:112170769 and PPP1R12A chr12:80203601) (Figure 5). However, whether these genes were related to phenotypes needs to be substantiated further by clinical studies.
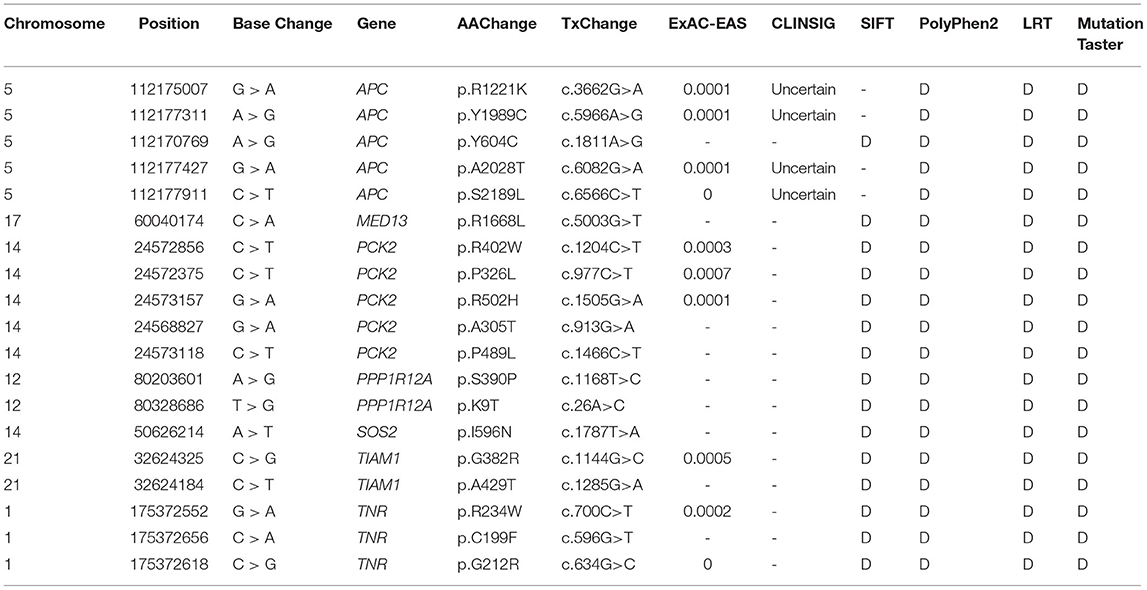
Table 2. Rare variant of seven candidate genes associated with PS-PA/IVS.
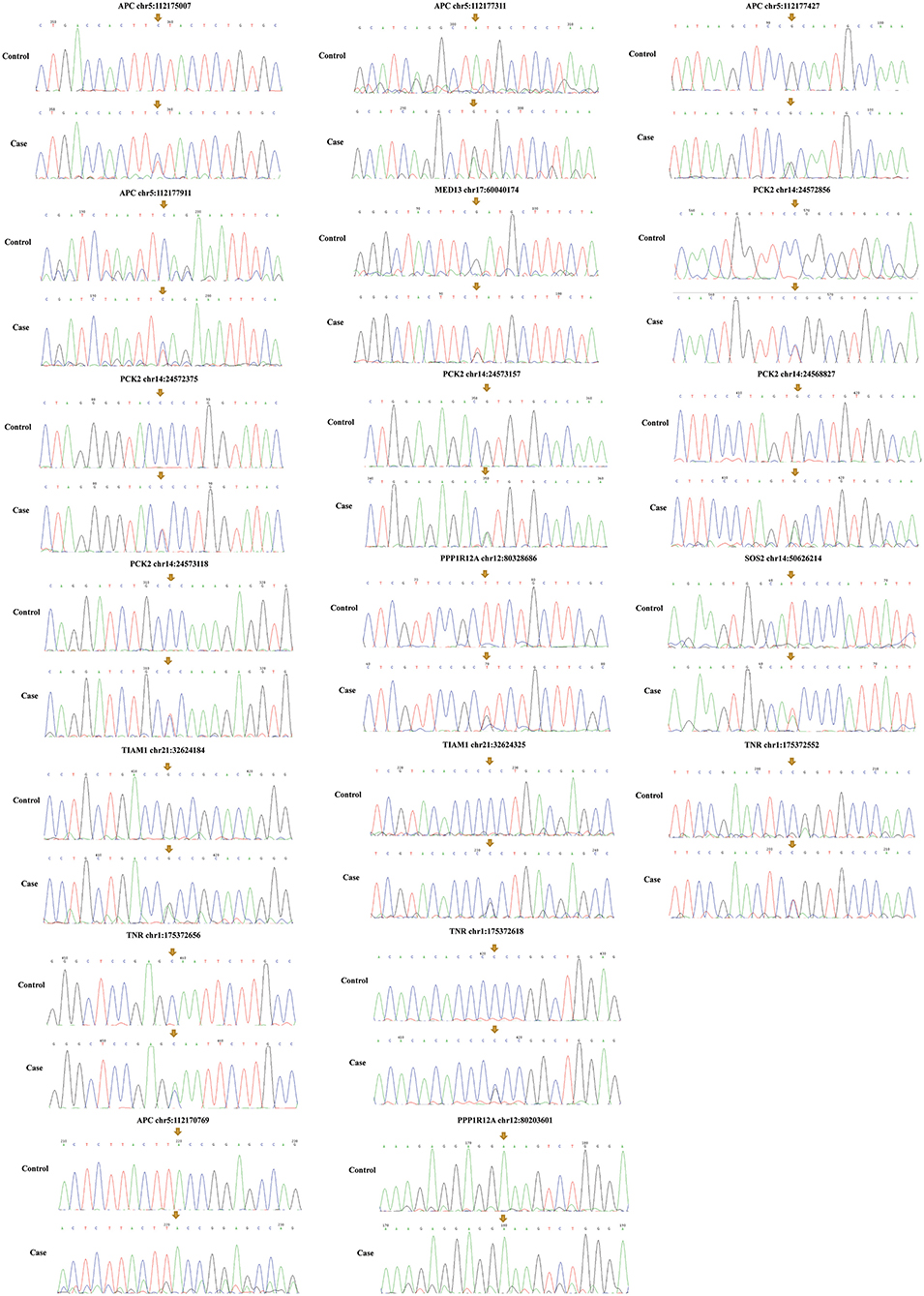
Figure 5. Mutation validation of seven genes with 19 SNPs. Only two SNPs (APC chr5:112170769 and PPP1R12A chr12:80203601) were not validated successfully. The yellow arrows represent the sites of SNPs.
Discussion
After negative results in chromosomal microarray and karyotype testing, WES was a common method to diagnose the underlying genetic cause in 25–35% of children with an unexplained presumed genetic disease (such as a birth defect) (26). As it is the most common birth defect, the pathogenesis for CHD needs to be elucidated. Hitherto, only a few studies have specifically focused on PS or PA/IVS. In order to clarify the genetic cause underlying PS-PA/IVS, we adopted WES to identify the damaging variants and potential pathogenic genes in 74 PS-PA/IVS cases and 100 healthy controls. Further burden test and phenotype analysis identified seven candidate genes, including SOS2, TIAM1, APC, PPP1R12A, PCK2, MED13, and TNR. Enrichment analysis identified the Wnt and cadherin signaling pathways linked to PS. Due to the little-known genetic cause of PS-PA/IVS, the disease enrichment analysis enriched a variety of systemic diseases. However, additional basic and clinical studies are required to confirm whether these genes were pathogenic in PS-PA/IVS.
In order to identify the rare variants associated with PS-PA/IVS, we conducted the burden test and identified seven genes that might be related to the disease. Based on the analysis of a phenolyzer, SOS2 was the seed gene of PS. A previous study found that SOS2 was homologous to SOS1, the second gene frequently related to Noonan syndrome (27). It is the second most common syndromic cause of CHD, and its most common phenotype is PS (28). Noonan syndrome was caused by functional dysregulation in the ERK signal transduction pathway. A recent study demonstrated that SOS2 mutation causes Noonan syndrome (29). However, in the current study, we found that the SOS2 mutation might be a genetic cause of non-syndromic PS.
Except for the seed gene SOS2, the other six genes associated with PS were predicted and found to be involved in three pathways. The first pathway was Wnt signaling pathway and mainly included APC and PPP1R12A in this study. APC, a Wnt signaling pathway regulator, plays a critical role in cellular processes, including signal transduction and cell adhesion. Mutant mice and cells studies showed that APC inhibits the Wnt signaling pathway (30). Moreover, the signaling pathway has been proven to be critical for cardiac development. Canonical Wnt signaling pathway was associated with cardiac valve formation. The mutated APC protein would give rise to an excessive endocardial layer fused with the atrioventricular outflow tract and markedly expand the endocardial cushions through overexpressing β-catenin during EMT (31). Another gene related to the Wnt pathway was PPP1R12A, which was identified as a putative cofactor of NKX2.5 (32). It encoded a regulatory subunit of myosin phosphatase and was related to significant roles in several cellular processes, such as gene expression regulation, cell cycle, and embryonic development (33). The knockdown of PPP1R12A enhanced cardiomyogenesis and rescued Wnt3a-mediated inhibition in mice embryonic stem cells (32).
The second pathway was the thyroid hormone (TH) signaling pathway, including only MED13. MED13 was the major target of microRNA-208, which was specific to cardiac muscle, and modulated the activity of the thyroid hormone receptor related to cardiac hypertrophy. In cyanotic CHD children, adenosine-to-inosine RNA editing in MED13 was significantly higher than in acyanotic patients (34), thereby implying that MED13 might be involved in cyanotic CHD.
The last pathway was the PI3K-AKT pathway, including TIAM1, PCK2, and TNR. The triplication of the TIAM1-Knj6 region was necessary to cause Down's syndrome-associated heart defects in mice (35). TIAM1 was related to the non-syndromic mitral valve prolapse (36). A previous study found that TIAM1 activates the PI3K pathway when hypertrophy is induced in neonatal rat cardiomyocytes (37). PCK2 encoded the mitochondrial phosphoenolpyruvate carboxykinase. Increasing PCK2 promotes hypertrophic growth in mice while knocking down the gene attenuates norepinephrine-induced cardiac hypertrophy (38). A recent study found that PCK2 regulates the osteogenic capacity of mesenchymal stem cells through the AMPK pathway (39). However, the biological role of PCK2 is not yet clarified. The Tenascin family modulates the cellular response to growth factors and cell adhesion. TNR is an extracellular matrix protein specific to the central nervous system (40). Strikingly, TNR and CHD have not been investigated previously. Nonetheless, TNR is involved in focal adhesion and PI3K-AKT signaling pathway (41, 42). All these genes were predicted to correlate with the phenotype of PS-PA/IVS; however, clinical and basic studies are still lacking.
Limitations
Firstly, due to the small sample size, establishing a validation cohort to confirm the candidate genes was difficult. Secondly, because of the small sample size of each disease group, we combined PS and PA/IVS into one group for further rare variants analysis. Hence, fewer genes differed between the two groups. Lastly, the genetic background of patients was limited by the lack of parental samples.
Conclusion
In conclusion, an effective analytical bioinformatics method allowed us to identify rare damage variants. Thus, this study put forth seven candidate genes (APC, PPP1R12A, PCK2, SOS2, TNR, MED13, and TIAM1), resulting in PS-PA/IVS, and which have not been reported in either animals or humans. Enrichment analysis identified the Wnt and cadherin signaling pathways linked to PS. This study provides new evidence for exploring the genetic mechanism of PS-PA/IVS. However, additional studies are required to verify these genes.
Data Availability Statement
The datasets presented in this article are not readily available due to ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
The protocol was approved by the Ethical Committee of Xinhua Hospital (XHEC-C-2019-083). Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
SC, KS, and QW contributed equally to the study and conceived and designed the study. YZ, KB, and YW prepared an analytical plan, analyzed data, and drafted the initial manuscript. ZM, SZ, and SJ were involved in sample collection. HW, JW, and MY collaborated in the revision and interpretation of the data and results. All authors reviewed and revised the manuscript, approved the final manuscript as submitted, and agreed to be accountable for all aspects of the work.
Funding
This work was supported by the Key Program for International S&T Cooperation Projects of China (81720108003), the National Key R&D Program of China (2018YFC1002400 and 2018YFC1002403), and the National Natural Science Foundation of China (81800281).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We express our gratitude to all subjects who participated in this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2021.811156/full#supplementary-material
References
1. Zaidi S, Brueckner Brueckner M, Genetics and genomics of congenital heart disease. Circ Res. (2017) 120:923–40. doi: 10.1161/CIRCRESAHA.116.309140
2. Zhao Z, Zhan Y, Chen W, Ma X. Sheng W, Huang G Functional analysis of rare variants of GATA4 identified in Chinese patients with congenital heart defect. Genesis. (2019) 57:e23333. doi: 10.1002/dvg.23333
3. He X, Zhang X, Jing H, Zhang X, Gao M, Chen H, et al. Rare copy number variations might not be involved in the molecular pathogenesis of PA-IVS in an unselected chinese cohort. Pediatr Cardiol. (2019) 40:762–7. doi: 10.1007/s00246-019-02062-x
4. Montanaro C, Merola A, Kempny A, Alvarez-Alvarez B, Alonso-Gonzalez R, Swan L, et al. The outcome of adults born with pulmonary atresia: high morbidity and mortality irrespective of repair. Int J Cardiol. (2019) 280:61–6. doi: 10.1016/j.ijcard.2018.11.011
5. Gorla SR, Singh AP Pulmonary Atresia With Intact Ventricular Septum. Treasure Island, FL: StatPearls (2020).
6. Kwiatkowski DM, Hanley FL, Krawczeski CD. Right ventricular outflow tract obstruction: pulmonary atresia with intact ventricular septum, pulmonary stenosis, and ebstein's malformation. Pediatr Crit Care Med. (2016) 17:S323–329. doi: 10.1097/PCC.0000000000000818
7. Anderson K, Cnota J, James J, Miller EM, Parrott A, Pilipenko V, et al. Prevalence of Noonan spectrum disorders in a pediatric population with valvar pulmonary stenosis. Congenit Heart Dis. (2019) 14:264–73. doi: 10.1111/chd.12721
8. Soquet J, Barron DJ, d'Udekem Y. A review of the management of pulmonary atresia, ventricular septal defect, and major aortopulmonary collateral arteries. Ann Thorac Surg. (2019) 108:601–12. doi: 10.1016/j.athoracsur.2019.01.046
9. Guo T, Repetto GM, McDonald McGinn DM, Chung JH, Nomaru H, Campbell CL, et al. Genome-wide association study to find modifiers for tetralogy of fallot in the 22q11.2 deletion syndrome identifies variants in the GPR98 locus on 5q14.3. Circ Cardiovasc Genet. (2017) 10:e001690. doi: 10.1161/CIRCGENETICS.116.001690
10. Page DJ, Miossec MJ, Williams SG, Monaghan RM, Fotiou E, Cordell HJ, et al. Whole exome sequencing reveals the major genetic contributors to nonsyndromic tetralogy of fallot. Circ Res. (2019) 124:553–63. doi: 10.1161/CIRCRESAHA.118.313250
11. Bokma JP, Winter MM, Oosterhof T, Vliegen HW, van Dijk AP, Pieper PG, et al. pulmonary valve replacement after repair of pulmonary stenosis compared with tetralogy of fallot. J Am Coll Cardiol. (2016) 67:1123–4. doi: 10.1016/j.jacc.2015.12.032
12. Baumgartner H, Hung J, Bermejo J, Chambers JB, Evangelista A, Griffin BP, et al. Echocardiographic assessment of valve stenosis: EAE/ASE recommendations for clinical practice. J Am Soc Echocardiogr. (2009) 22:1–23. doi: 10.1016/j.echo.2008.11.029
13. Hu P, Qiao F, Wang Y, Meng L, Ji X, Luo C, et al. Clinical application of targeted next-generation sequencing in fetuses with congenital heart defect. Ultrasound Obstet Gynecol. (2018) 52:205–11. doi: 10.1002/uog.19042
14. Grunert M, Dorn C, Cui H, Dunkel I, Schulz K, Schoenhals S, et al. Comparative DNA methylation and gene expression analysis identifies novel genes for structural congenital heart diseases. Cardiovasc Res. (2016) 112:464–77. doi: 10.1093/cvr/cvw195
15. LaHaye S, Corsmeier D, Basu M, Bowman JL, Fitzgerald-Butt S, Zender G, et al. Utilization of whole exome sequencing to identify causative mutations in familial congenital heart disease. Circ Cardiovasc Genet. (2016) 9:320–9. doi: 10.1161/CIRCGENETICS.115.001324
16. Shi X, Zhang L, Bai K, Xie H, Shi T, Zhang R, et al. Identification of rare variants in novel candidate genes in pulmonary atresia patients by next generation sequencing. Comput Struct Biotechnol J. (2020) 18:381–92. doi: 10.1016/j.csbj.2020.01.011
17. Xie H, Hong N, Zhang E, Li F. Sun K, Yu Y Identification of rare copy number variants associated with pulmonary atresia with ventricular septal defect. Front Genet. (2019) 10:15. doi: 10.3389/fgene.2019.00015
18. Morgenthau A, Frishman WH. Genetic origins of tetralogy of fallot. Cardiol Rev. (2018) 26:86–92. doi: 10.1097/CRD.0000000000000170
19. Reuter MS, Chaturvedi RR, Liston E, Manshaei R, Aul RB, Bowdin S, et al. The cardiac genome clinic: implementing genome sequencing in pediatric heart disease. Genet Med. (2020) 22:1015–24. doi: 10.1038/s41436-020-0757-x
20. Lee CN, Su YN, Cheng WF, Lin MT, Wang JK, Wu MH, et al. Association of the C677T methylenetetrahydrofolate reductase mutation with congenital heart diseases. Acta Obstet Gynecol Scand. (2005) 84:1134–40. doi: 10.1111/j.0001-6349.2005.00611.x
21. Do R, Stitziel NO, Won HH, Jorgensen AB, Duga S, Angelica Merlini P, et al. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature. (2015) 518:102–6. doi: 10.1038/nature13917
22. Nicolae DL. Association tests for rare variants. Annu Rev Genomics Hum Genet. (2016) 17:117–30. doi: 10.1146/annurev-genom-083115-022609
23. Griffin JN, Del Viso F, Duncan AR, Robson A, Hwang W, Kulkarni S, et al. RAPGEF5 regulates nuclear translocation of beta-catenin. Dev Cell. (2018) 44:248–260 e244 doi: 10.1016/j.devcel.2017.12.001
24. Khan K, Yu B, Kiwan C, Shalal Y, Filimon S, Cipro M, et al. The Role of Wnt/beta-catenin pathway mediators in aortic valve stenosis. Front Cell Dev Biol. (2020) 8:862. doi: 10.3389/fcell.2020.00862
25. Bowen CJ, Zhou J. Sung DC, Butcher JT Cadherin-11 coordinates cellular migration and extracellular matrix remodeling during aortic valve maturation. Dev Biol. (2015) 407:145–57. doi: 10.1016/j.ydbio.2015.07.012
26. Petrovski S, Aggarwal V, Giordano JL, Stosic M, Wou K, Bier L, et al. Whole-exome sequencing in the evaluation of fetal structural anomalies: a prospective cohort study. Lancet. (2019) 393:758–67. doi: 10.1016/S0140-6736(18)32042-7
27. Yamamoto GL, Aguena M, Gos M, Hung C, Pilch J, Fahiminiya S, et al. Rare variants in SOS2 and LZTR1 are associated with Noonan syndrome. J Med Genet. (2015) 52:413–21. doi: 10.1136/jmedgenet-2015-103018
28. Roberts AE, Allanson JE. Tartaglia M, Gelb BD Noonan syndrome. Lancet. (2013) 381:333–42. doi: 10.1016/S0140-6736(12)61023-X
29. Cordeddu V, Yin JC, Gunnarsson C, Virtanen C, Drunat S, Lepri F, et al. Activating mutations affecting the dbl homology domain of SOS2 cause noonan syndrome. Hum Mutat. (2015) 36:1080–7. doi: 10.1002/humu.22834
30. Li Z, Li W. Song L, Zhu W Cilia, adenomatous polyposis coli and associated diseases. Oncogene. (2012) 31:1475–83. doi: 10.1038/onc.2011.351
31. Joziasse IC, van de Smagt JJ, Smith K, Bakkers J, Sieswerda GJ, Mulder BJ, et al. Genes in congenital heart disease: atrioventricular valve formation. Basic Res Cardiol. (2008) 103:216–27. doi: 10.1007/s00395-008-0713-4
32. Ryan T, Shelton M, Lambert JP, Malecova B, Boisvenue S, Ruel M, et al. Myosin phosphatase modulates the cardiac cell fate by regulating the subcellular localization of Nkx25 in a Wnt/Rho-associated protein kinase-dependent pathway. Circ Res. (2013) 112:257–66. doi: 10.1161/CIRCRESAHA.112.275818
33. Niclass T, Le Guyader G, Beneteau C, Joubert M, Pizzuti A, Giuffrida MG. 12q21 deletion syndrome: Narrowing the critical region down to 16 Mb including SYT1 and PPP1R12A. Am J Med Genet A. (2020) 182:2133–8. doi: 10.1002/ajmg.a.61734
34. Borik S, Simon AJ, Nevo-Caspi Y, Mishali D, Amariglio N, Rechavi G, et al. Increased RNA editing in children with cyanotic congenital heart disease. Intensive Care Med. (2011) 37:1664–71. doi: 10.1007/s00134-011-2296-z
35. Liu C, Morishima M, Jiang X, Yu T, Meng K, Ray D, et al. Engineered chromosome-based genetic mapping establishes a 37 Mb critical genomic region for Down syndrome-associated heart defects in mice. Hum Genet. (2014) 133:743–53. doi: 10.1007/s00439-013-1407-z
36. Fulmer D, Toomer KA, Glover J, Guo L, Moore K, Moore R, et al. Desert hedgehog-primary cilia cross talk shapes mitral valve tissue by organizing smooth muscle actin. Dev Biol. (2020) 463:26–38. doi: 10.1016/j.ydbio.2020.03.003
37. Vettel C, Wittig K, Vogt A, Wuertz CM, El-Armouche A, Lutz S, et al. A novel player in cellular hypertrophy: Gibetagamma/PI3K-dependent activation of the RacGEF TIAM-1 is required for alpha(1)-adrenoceptor induced hypertrophy in neonatal rat cardiomyocytes. J Mol Cell Cardiol. (2012) 53:165–75. doi: 10.1016/j.yjmcc.2012.04.015
38. Ma H, Yu S, Liu X, Zhang Y, Fakadej T, Liu Z, et al. Lin28a Regulates pathological cardiac hypertrophic growth through Pck2-mediated enhancement of anabolic synthesis. Circulation. (2019) 139:1725–40. doi: 10.1161/CIRCULATIONAHA.118.037803
39. Li Z, Liu X, Zhu Y, Du Y, Liu X, Lv L, et al. Mitochondrial phosphoenolpyruvate carboxykinase regulates osteogenic differentiation by modulating AMPK/ULK1-dependent autophagy. Stem Cells. (2019) 37:1542–55. doi: 10.1002/stem.3091
40. Chiovaro F, Chiquet-Ehrismann R, Chiquet M. Transcriptional regulation of tenascin genes. Cell Adh Migr. (2015) 9:34–47. doi: 10.1080/19336918.2015.1008333
41. Probstmeier R, Michels M, Franz T. Chan BM, Pesheva P Tenascin-R interferes with integrin-dependent oligodendrocyte precursor cell adhesion by a ganglioside-mediated signalling mechanism. Eur J Neurosci. (1999) 11:2474–88. doi: 10.1046/j.1460-9568.1999.00670.x
Keywords: whole-exome sequencing, congenital heart disease, right ventricular outflow tract obstruction, pulmonary valvular stenosis, pulmonary atresia with intact ventricular septum, single nucleotide polymorphism
Citation: Zhou Y, Bai K, Wang Y, Meng Z, Zhou S, Jiang S, Wang H, Wang J, Yang M, Wang Q, Sun K and Chen S (2022) Identification of Rare Variants in Right Ventricular Outflow Tract Obstruction Congenital Heart Disease by Whole-Exome Sequencing. Front. Cardiovasc. Med. 8:811156. doi: 10.3389/fcvm.2021.811156
Received: 08 November 2021; Accepted: 30 December 2021;
Published: 24 January 2022.
Edited by:
Xuejun Yuan, Max Planck Institute for Heart and Lung Research, GermanyReviewed by:
Christoph Preuss, Jackson Laboratory, United StatesPatrick G. Burgon, Qatar University, Qatar
Copyright © 2022 Zhou, Bai, Wang, Meng, Zhou, Jiang, Wang, Wang, Yang, Wang, Sun and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qingjie Wang, wangqingjie@xinhuamed.com.cn; Kun Sun, sunkun@xinhuamed.com.cn; Sun Chen, chensun@xinhuamed.com.cn
†These authors have contributed equally to this work and share first authorship