Transcription Factor KLF14 and Metabolic Syndrome
 Qianyi Yang1*
Qianyi Yang1*  Mete Civelek1,2*
Mete Civelek1,2*- 1Center for Public Health Genomics, University of Virginia, Charlottesville, VA, United States
- 2Department of Biomedical Engineering, University of Virginia, Charlottesville, VA, United States
Metabolic syndrome (MetSyn) is a combination of metabolic abnormalities that lead to the development of cardiovascular disease (CVD) and Type 2 Diabetes (T2D). Although various criteria for defining MetSyn exist, common abnormalities include abdominal obesity, elevated serum triglyceride, insulin resistance, and blood glucose, decreased high-density lipoprotein cholesterol (HDL-C), and hypertension. MetSyn prevalence has been increasing with the rise of obesity worldwide, with significantly higher prevalence in women compared with men and in Hispanics compared with Whites. Affected individuals are at a higher risk of developing T2D (5-fold) and CVD (2-fold). Heritability estimates for individual components of MetSyn vary between 40 and 70%, suggesting a strong contribution of an individual's genetic makeup to disease pathology. The advent of next-generation sequencing technologies has enabled large-scale genome-wide association studies (GWAS) into the genetics underlying MetSyn pathogenesis. Several such studies have implicated the transcription factor KLF14, a member of the Krüpple-like factor family (KLF), in the development of metabolic diseases, including obesity, insulin resistance, and T2D. How KLF14 regulates these metabolic traits and increases the risk of developing T2D, atherosclerosis, and liver dysfunction is still unknown. There have been some debate and controversial results with regards to its expression profile and functionality in various tissues, and a systematic review of current knowledge on KLF14 is lacking. Here, we summarize the research progress made in understanding the function of KLF14 and describe common attributes of its biochemical, physiological, and pathophysiological roles. We also discuss the current challenges in understanding the role of KLF14 in metabolism and provide suggestions for future directions.
Introduction
GWAS identified several genetic variants near the KLF14 gene on chromosome 7 to be associated with a multitude of metabolic pathologies, including insulin resistance, T2D (1–3), and coronary artery disease (CAD) (4). The associations were stronger in females than in males (Table 1). While the initial GWAS was performed in people of European ancestry, many of the results have now been replicated in different ancestries (8–11). Expression quantitative trait locus studies linked the GWAS-associated genetic variants to the abundance of KLF14 transcript in adipose tissue (1, 12, 13). KLF14 is a single exon imprinted gene, and only the allele inherited from the mother is expressed. As a transcription factor, the targets of KLF14 are largely unknown. In addition to genetic variants, studies showed environmental factors, such as diet, affected KLF14 expression in various metabolic tissues. In this review, we outline the current state of knowledge about KLF14 biology, and we provide suggestions for future studies to delineate its role in metabolism.
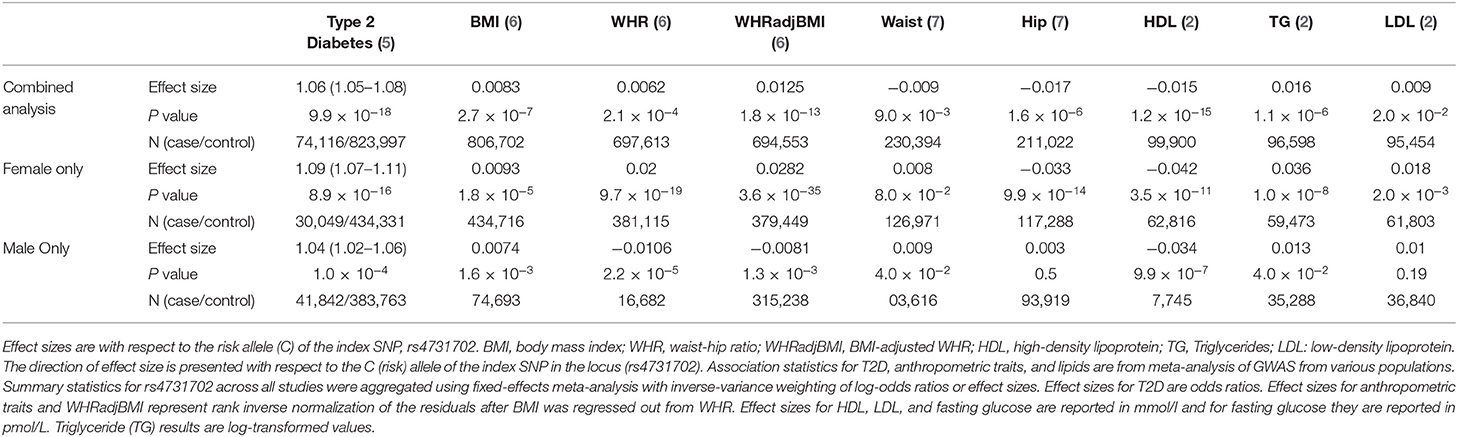
Table 1. Sex-specific and pleiotropic effects of the genetic variants at the KLF14 locus with cardiometabolic phenotypes.
Krüpple-Like Family of Transcription Factors
KLF14 is a member of the Krüpple-Like family of transcription factors which are responsible for modulation of gene expression in mammals (14, 15). There are 18 KLF proteins which are grouped into three distinct families [reviewed in (14)]. Group 1 KLFs primarily play a role in transcriptional repression while members of Group 2 function as activators of transcription. KLF14 belongs to Group 3 KLFs which typically play repressive roles by binding to DNA with Sin3A, another transcriptional co-repressor protein. However, KLF14 has been shown to activate transcription. KLF proteins contain conserved C2H2-type zinc finger domains in their C-termini which bind to GC-rich sites in the regulatory regions of target genes [reviewed in (14)]. In the absence of experiments that show direct binding of KLFs to the enhancers and promoters of target genes, it has been possible to predict common transcriptional targets using the structural similarities among KLFs (16). This strategy is especially important for KLF14 for which an antibody that can be used for immunoprecipitation for sequencing is not available.
The N-terminus regions of KLF proteins possess unique repetitive sequences that allow for binding with other protein partners, including co-activators, co-repressors, and histone-modifying enzymes (17). Sequence similarities in the amino-terminal domains lead to a correlation between structure and function. KLF14 has three C2H2-type zinc finger structural motifs at the C-terminus with the first and second domains comprising 25 amino acid residues and a shorter third domain containing 23 amino acid residues (18, 19). These three zinc finger domains can each recognize three DNA base pairs thereby contacting the gene regulatory region at three sites (20). There is evidence that the zinc finger domains have positively charged amino acids that may aid in nuclear localization. Several studies have examined the DNA binding site preference for KLFs and it appears that KLF proteins interact with similar GC-rich sequences or 5′-CACCC-3′ of the gene promoter regions (21, 22). KLF14 exerts its repressive activity on transcription by interacting with Sin3A, which acts as a transcriptional hub (23). Sin3A and Sin3B are large proteins that belong to a family of histone deacetylases that generally repress transcription by tethering to DNA. These large multi-domain Sin3 proteins integrate the function of transcriptional factors, such as KLF14, with chromatin-modifying activity.
The expression patterns of KLFs vary drastically in different tissues [reviewed in (14, 24)]; many members are widely expressed, including KLF6 (25), KLF7 (26), KLF9 (27), KLF10 (28), KLF11 (29), KLF13 (30) and KLF15 (31), whereas some exhibit tissue-specific expression. For example, KLF1 is mostly expressed in red blood cells and megakaryocytes (32, 33), KLF2 is largely present in white adipose tissue (34, 35), and KLF4 and KLF5 are predominant in the blood vessels, and white adipose tissue (36–39).
KLF14 is widely expressed in various tissues with higher expression in females compared to males (Figure 1). Given its wide-spread expression pattern, its role in various pathologies has been explored. Below we summarize the current state of knowledge of KLF14's role in various diseases.
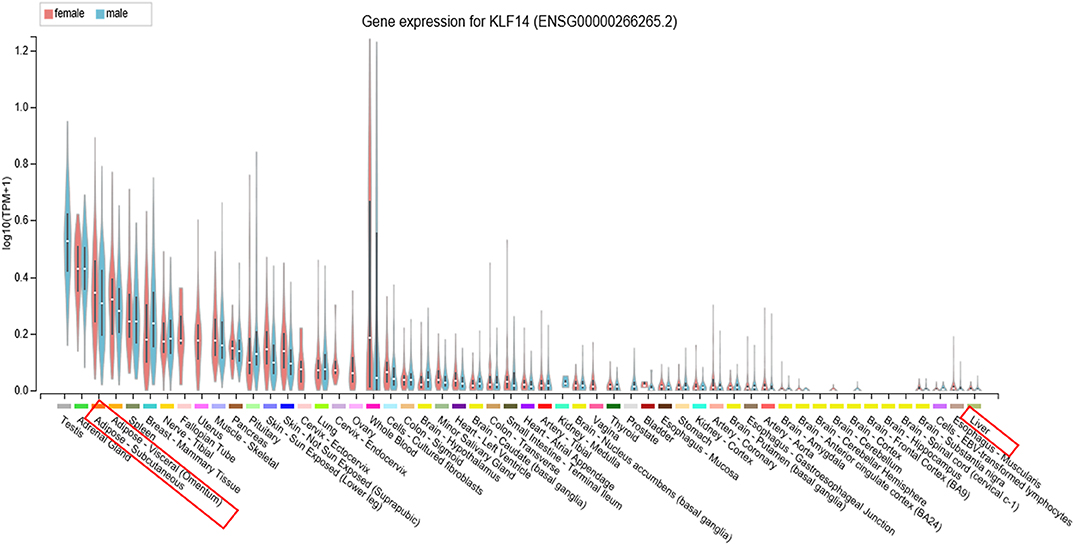
Figure 1. KLF14 expression in human tissues. Expression of KLF14 in 54 tissues or cell types from several hundred male and female donors was measured using RNA sequencing as part of the Genotype-Tissue Expression project. The results show widespread expression of KLF14 in various tissues, variation in the abundance of KLF14 in each human donor, and generally higher expression level in females compared to males. Image obtained from the GTEx Portal (40).
Human Genetic Evidence for the Role of KLF14 in Disease
The genetic variants associated with T2D and other metabolic phenotypes map to a region of 3–48 kb upstream of KLF14. GWAS-associated single nucleotide polymorphisms (SNPs) are also associated with the KLF14 expression level in adipose tissue (1, 12, 13). In other words, the GWAS signal for metabolic disorders and the expression quantitative trait locus for KLF14 are co-localized. Further, the same genetic variants are also associated with nearly 400 genes in trans. This is one of the largest trans-eQTL hotspots known in the human genome (41). GWAS of gene expression performed in the Multiple Tissue Human Expression Resource (MuTHER) and the Metabolic Syndrome in Men (METSIM) cohorts identified KLF14 as a master regulator of gene expression in subcutaneous adipose tissue (12, 41, 42). Interestingly, despite the nearly ubiquitous expression of KLF14 in many tissues and variation in its expression among humans (Figure 1) (43), the genetic variants regulate gene expression only in adipose tissue (1). This strongly suggests that disease-associated variants act in adipose tissue to increase the disease risk; however, given that adipose tissue contains a multitude of cell types, it is still not clear which cells are ultimately responsible for KLF14's effect on disease risk. KLF14 is an imprinted gene and only the allele inherited from the mother is expressed (44); only the maternally inherited SNPs have significant associations with adipose tissue expression of KLF14 (1). Small et al. have been able to fine-map the GWAS locus by using functional regulatory features from the Encyclopedia of DNA elements (ENCODE) (45) and the Roadmap Epigenomics Projects (46). They used chromatin-state maps generated by these projects to identify a ~1.6-kb enhancer region which is ~5 kb upstream of the KLF14 transcription start site in adipose tissue (1). Overall, three distinct lines of evidence predict that the transcription factor KLF14 is the causal gene in this GWAS locus and it functions in the adipose tissue: 1. association of KLF14 expression in adipose tissue with T2D-associated SNPs but not in other tissues, 2. association of KLF14 expression with maternal, but not paternal, alleles, and 3. the presence of an adipose-specific enhancer near the KLF14 transcription start site in the locus associated with T2D.
In more than 70 thousand whole genome sequences from a diverse human population, only five potential loss-of-function (LoF) variants have been identified with very low frequency suggesting coding changes in KLF14 are under purifying selection (https://gnomad.broadinstitute.org/). However, to date, no LoF variants in KLF14 have been shown to cause a rare disease, suggesting that regulatory variants that affect KLF14 expression are responsible for the GWAS findings.
The Role of KLF14 in Cardiometabolic Disorders
T2D is the prevalent form of diabetes affecting ~10% of the US population. It is characterized by high blood sugar, and insulin resistance resulting in dysfunction of several organs. The expression pattern of most KLF members has been shown to be altered during disease progression with multiple lines of evidence suggesting that KLF14 is a key metabolic transcriptional regulator (42).
Insulin is a hormone produced in the pancreas that helps maintain the blood glucose homeostasis (47–49). Several members of the KLF family members, including KLF14, have confirmed roles in the regulation of insulin sensitivity [reviewed in (50)]. Yang et al. used high-fat diet (HFD)-fed mice and leptin receptor-deficient (db/db) mice that have impaired insulin sensitivity and showed that there is significant down-regulation of Klf14 expression in skeletal muscle (51). At the molecular level, KLF14 activates PI3K/Akt signaling pathway to increase insulin sensitivity in the livers of HFD-fed mice and the db/db obese mice (51).
It has also been shown that KLF14 mediates lipid signaling (52). In cultured human endothelial cells, fibroblast growth factor 2 (FGF2) stimulation leads to the up-regulation of KLF14, which further induces sphingosine kinase 1 (SK1) expression (52). Mechanistically, KLF14 forms a complex with the coactivator p300 and then binds to a GC-rich region 633 bp upstream of the SK1 transcription start site (52) and activates the expression of SK1 (52). It has been shown that SK1 regulates adiposity by catalyzing the formation of lipid second messengers which leads to insulin resistance (53). Inhibition of SK1 resulted in accumulation of the lipid second messenger ceramides in liver cancer cells (54), while elevated muscle ceramides have been associated with muscle insulin resistance in obese humans (55). As such, some of the KLF14-mediated metabolic phenotypes may be attributed to regulation of lipid signaling via dysregulation of SK1 (50, 53). Below we summarize the studies that elucidated the role of KLF14 in metabolically active organs.
The Role of KLF14 in Adipose Tissue
Excessive fat accumulation occurs when caloric intake is higher than energy expenditure. In response to the change in energy status, adipose tissue goes through rapid and dynamic remodeling by changing the number and/or size of adipocytes (56). Adipose tissues expand themselves to accommodate potential excess calories. In obesity, subcutaneous adipose tissue fails to expand appropriately in response to the excess energy intake, which leads to fat overflow and fat deposition in other ectopic sites, such as skeletal muscle, liver, and visceral fat depots. The consequence of this excess ectopic fat accumulation is insulin resistance (57–59). This shift in body fat distribution from subcutaneous to visceral is strongly associated with metabolic diseases (60–64). Adipocytes can be enlarged by excessive lipid deposition or generated from precursors through adipogenesis. Several members of KLFs play a role in energy homeostasis by regulating lipid and glucose metabolism, and many of them have been implicated in adipogenesis, including promoting [KLF4 (65, 66), KLF5 (37, 67, 68), KLF6 (69, 70), KLF8 (71), KLF9 (72), KLF13 (73), and KLF15 (74–77)] or inhibiting [KLF2 (78, 79), KLF3 (80), KLF7 (81, 82), KLF14 (1) and KLF16 (83)] adipocyte differentiation [reviewed in (84, 85)]. However, the role of KLF14 in hyperplasia is not known.
KLF14 appears to impact both pre-adipocyte differentiation to mature adipocytes and the function of mature adipocytes (1). To investigate the role of KLF14 in adipogenesis, Small et al. isolated primary pre-adipocytes from human abdominal subcutaneous fat tissue and assayed KLF14 levels over a 14-day differentiation time course. Cells from males and females had distinct patterns of expression throughout the differentiation (1). While KLF14 expression did not change in male cells, it decreased and then increased as female adipocyte precursors matured into adipocytes. Interestingly, KLF14 levels were higher in female subcutaneous adipocytes compared to male adipocytes throughout the time course (1). This was consistent with higher KLF14 level in the adipose tissue of female donors compared to male donors both in the deCODE Icelandic cohort and the Genotype-Tissue Expression (GTEx) datasets (Figure 1) (1, 43). Lowering KLF14 expression in cells from females prevented maturation as measured by the expression of adipogenesis markers and lipid accumulation suggesting a role of KLF14 in adipocyte differentiation (1). H&E staining of subcutaneous adipose tissue biopsies showed that females who were homozygous for the T2D risk allele had adipocytes with larger cell surface area, suggesting that lower KLF14 expression is associated with adipocyte dysfunction (1). These studies collectively suggested a role of KLF14 in both adipogenesis and mature adipocyte size and function. However, a recent study that used machine learning to quantify adipocyte size from hundreds of histology images of adipose tissue did not find an association with adipocyte size at the KLF14 locus (86).
In a recent study, Iwaya et al. analyzed the DNA methylation status of the Klf14 promoter in adipose tissue to investigate if there was a correlation between aging and state of obesity, both of which are risk factors of T2D (87). Their study showed that, the expression of Klf14 was reduced in adipose tissue of aged wild type mice and HFD-fed mice and consistent with this change in expression, Klf14 promoter region was hypermethylated in adipose tissue in response to aging and HFD. Consistent with reduced Klf14 expression, downstream target genes, many of which were associated with metabolic traits (88, 89), were also downregulated (87). In a study by Bacos et al. KLF14 hypermethylation was also associated with aging, insulin secretion and T2D (90). Taken together, these studies suggest that KLF14 loss-of-function via epigenetic regulation may trigger the onset of metabolic diseases. However, Argmann et al. showed that mice harboring whole-body germ-line deletion of Klf14 were resilient to HFD-induced insulin resistance (91). Their results were inconsistent with the previously reported role of KLF14 in regulating HDL-C levels in the liver (92, 93). This is possibly due to the utilization of different mouse lines (Table 2). It is also likely that KLF14 function is redundant in mice and can be substituted with other KLFs. It is also possible that human and mouse KLF14 have different functions.
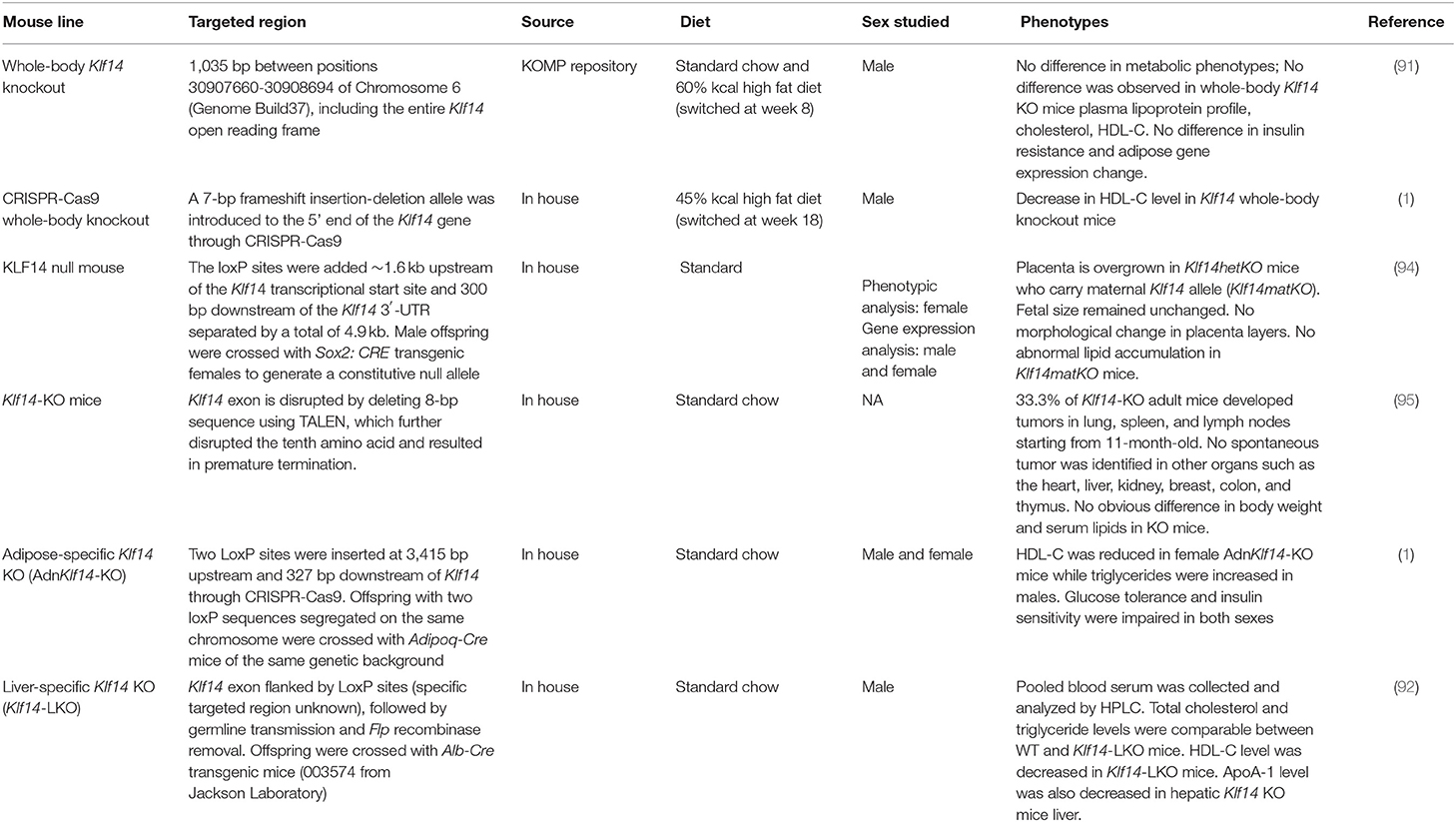
Table 2. Summary of published KLF14 mouse lines.
The Role of KLF14 in Liver Tissue
T2D-associated variants identified in GWAS also have strong associations with serum HDL-C and triglyceride levels (96, 97) (Table 1), specifically, T2D risk alleles at KLF14 are associated with decreased HDL-C (1). However, the role of KLF14 in regulating cholesterol metabolism is not known. The liver is a specialized tissue comprising of mostly hepatocytes that regulate cholesterol and glucose metabolism. To maintain blood glucose levels, the liver provides glucose during low insulin and high glucagon conditions by both gluconeogenesis and glycogen breakdown (98). Insulin and glucagon are two hormones that work in concert to keep the blood glucose level balanced. When food is ingested insulin levels rise and glucagon levels dip; the liver removes excess glucose by breaking it down into pyruvate or using it for synthesizing glycogen (99).
It has been shown that hepatic KLF14 regulates glucose production in fasted and re-fed mice as well as diet-induced and genetically obese mice (93). Several lines of evidence suggest that increased KLF14 expression facilitates gluconeogenesis (93). Fasted animals had higher Klf14 expression compared to ad libitum fed animals, and the expression levels returned to baseline levels after refeeding, suggesting a role of Klf14 in gluconeogenesis (93). In the same study, Wang et al. also demonstrated that a high level of Klf14 in wildtype C57BL/6J mice resulted in higher hepatic glucose production, high serum insulin levels and impaired glucose and insulin tolerance (93). shRNA-mediated silencing of Klf14 in obese animals reduced blood glucose levels in fasting mice and increased glucose tolerance. Klf14 overexpression in primary hepatocytes increased the transcript and protein levels of peroxisome proliferator-activated receptor-gamma coactivator 1-alpha (PGC-1α) a protein that stimulates glucose production. At the same time, Klf14 knockdown reduced PGC-1α, thereby constraining glucose production (93). Taken together, these results suggested that KLF14 modulates hepatic glucose metabolism by regulating the activity of PGC-1α.
The liver regulates whole-body energy homeostasis in response to insulin stimulation (100). It has been suggested that higher Klf14 expression protects the liver from insulin sensitivity. When Klf14 is overexpressed, there is increased glucose uptake concomitant with Akt kinase activation in mouse hepatoma cells (51). Overexpression of Klf14 leads to increased transcription activation of PGC-1α, the histone deacetylase Sirtuin 1 and hepatocyte nuclear transcription factor 4α, all of which are insulin signaling-associated factors and play a role in glucose production in the liver (50, 51, 93, 101).
Guo et al. studied the role of KLF14 in regulating lipid metabolism in the liver and atherosclerosis using KLF14 overexpression and genetic inactivation in the livers of atherosclerosis mouse models (92). In contrast to Wang et al. they found that the KLF14 expression was reduced in the livers of HFD-fed and leptin-deficient (ob/ob) obese mice (92). Overexpression of KLF14 by injecting adenovirus encoding KLF14 via the tail vein increased HDL-C levels without affecting the levels of triglycerides, total cholesterol (TC), low-density lipoprotein cholesterol (LDL-C), and fasting blood glucose (92). Albumin Cre-mediated hepatic deletion of Klf14 resulted in reduced HDL-C levels. Through promoter-bashing experiments and ChIP analysis, the authors showed that KLF14 directly binds to the promoter of apolipoprotein A-1 (ApoA1), which encodes the major protein component of HDL particles, and regulates its expression (92). Modulating KLF14 levels in hepatocytes in vitro or in mouse livers in vivo resulted in changes in the efflux capacity of HDL-C in agreement with KLF14's role in regulating ApoA1 expression (92). The authors also identified perhexiline from a drug screen, as a KLF14 activator, and used this drug to treat ApoE-deficient mice, which have very high total cholesterol levels and develop atherosclerosis. Administration of perhexiline increased hepatic KLF14 expression and HDL-C plasma levels which has a protective effect against atherosclerosis in ApoE-deficient mice (92). Lipids are transported in the blood serum as lipoproteins. HDL and LDL are the primary carriers of blood cholesterol. However, many laboratory animal models carry most cholesterol in HDL, while humans transport majority of cholesterol in LDL (102, 103). It is remarkable that there is consistency in the effect of the manipulation of KLF14 expression on HDL levels in humans and mice.
Wei et al. demonstrated that ApoE-deficient mice exhibited increased expression of KLF14 in their aortas compared to wild type controls when fed HFD or standard chow diet, which indicates a role for KLF14 in the vessel wall (104). Contrary to Guo et al. silencing KLF14 expression reduced circulating levels of inflammatory cytokines in HFD-fed ApoE-null mice (104). The authors noted decreased atherosclerotic lesions in the same ApoE-null mice likely brought upon by suppression of the M2 inflammatory phenotype of macrophages due to the inhibition of MAPK signaling, particularly extracellular signal-regulated kinase 1/2 and p38. These studies demonstrated a direct role of KLF14 in regulating atherosclerotic plaque and lesion formation independent of its risk factors (104).
It is not clear why there are discrepancies among the three studies which observed different effects of high-fat feeding and obesity on liver Klf14 levels, fasting glucose, and the formation of atherosclerotic plaque. The conflicting expression profiles of KLF14 reported in these studies can be attributed to the differences in the expression patterns of KLF14 in different tissues. In addition, external factors, such as handling and stress, housing conditions, or methodological differences in measuring mRNA and protein levels may contribute to the observed discrepancy. In previous studies, we and others could not detect the expression of Klf14 mRNA in human or mouse livers (1, 91, 105). The Genotype-Tissue Expression study, in which mRNA from 226 human livers were sequenced, also did not detect KLF14 expression (Figure 1) (40). Our meta-analysis showed that risk allele is associated with decreased expression of adipose tissue KLF14 and decreased HDL-C levels and risk of developing T2D (Table 1), suggesting that higher levels of KLF14 may serve to protect against the development of adverse metabolic conditions. The association of genetic variants with KLF14 expression is only observed in adipose tissue suggesting that, instead of the liver, the metabolic effects of the risk variants are in the adipose tissue, which was not addressed in the aforementioned studies.
The Role of KLF14 in Endothelial Cells
Endothelial cells (ECs), which line blood vessels, play significant roles in angiogenesis and the regulation of vascular function (106, 107). Hu et al. investigated the function of KLF14 in ECs. They showed that KLF14 is downregulated in mouse aortic ECs under acute and chronic inflammatory conditions (108). Overexpression of KLF14 suppressed inflammation in human ECs by inhibiting the pro-inflammatory response mediated by interleukin-1β (IL-1β) and tumor necrosis factor α (TNFα). Klf14 knockout mice also exhibited increased expression of adhesion molecules in primary ECs when stimulated by IL-1 β (108). These results suggest that KLF14 protects ECs against inflammatory stresses. Mechanistically, KLF14 was shown to inhibit the expression of p65 subunit of the Nuclear Factor Associated Activator of Kappa B-Cells (NF-κB) signaling pathway, resulting in markedly reduced leukocyte adhesion to activated ECs (108). In addition, the KLF14 activator perhexiline was observed to induce KLF14 expression in ECs which in turn prevented leukocyte adhesion in mice (108). These results demonstrate that KLF14 inhibits the macrophage-mediated inflammatory response in ECs by transcriptionally inhibiting the NF-κB signaling pathway (108).
The Role of KLF14 in Cancer and Placenta Development
The link between KLF proteins and regulation of cell cycle processes such as proliferation, differentiation, as well as cell adhesion and migration (15), and maintenance of pluripotency of stem cells (16) have been well established. More recently, several studies showed that KLFs also play important roles in cancer (14, 109) [reviewed in (15)]. Stacey et al. reported a new genomic locus associated with increased susceptibility to basal cell carcinoma (BCC) (110). This locus is 167 kb upstream of KLF14 and is distinct from the T2D susceptibility locus (110). The authors showed that increased BCC incidence is coupled to the paternal allele, despite the observation that the maternal allele is responsible for KLF14 expression (110). Fan et al. reported that disruption of Klf14 gene in mice causes mitotic defects ranging from centrosome over-amplification, missegregation to chromosome aneuploidy, all of which contribute to the pathogenesis of tumors (95). Specifically, KLF14 negatively regulates Polo-like Kinase 4 (Plk4), a protein which when overexpressed leads to centrosome overamplification (111, 112). KLF14 downregulation is inversely correlated with Plk4 upregulation in many cancers; these findings were affirmed by data analysis from cancer microarray database Oncomine (www.oncomine.org) (95). The authors also demonstrated that it is essential to maintain KLF14 activity within a normal range for cells to keep their genomic integrity. Deletion of KLF14 using transcription activator-like effector nucleases (TALEN) caused genomic instability in mice, such as chromosomal aneuploidy and chromosome missegregation, whereas KLF14 overexpression induced a host of mitotic defects that proceed with initiation of apoptotic programs and end with cell death (95). Consistent with a negative regulatory role of KLF14 in relation to Plk4, overexpression of Plk4 lead to chromosomal instability in gastric cancer whereas Plk4-null mouse embryos had a remarkable increase of mitotic cells (95). It is known that mitotic catastrophe is induced as a cellular mechanism to avoid genome instability (113) and as such, it is plausible that cells undergoing stress enhance expression of KLF14 subsequently initiating mitotic catastrophe to circumvent genomic instability in dividing cells. They proposed that KLF14 plays a tumor suppressor role, since Klf14 transcription is significantly downregulated in multiple types of cancers (95). As such, KLF14 presents as a new biomarker or potential target for cancer therapies.
Parker-Katiraee et al. identified KLF14 as a maternally imprinted gene with near-ubiquitous expression in mice and human (105). The authors observed greater KLF14 expression levels in embryonic tissues than in adult tissues, indicating a role in embryogenesis (105), which is consistent with the finding that several imprinted genes have been observed to regulate growth and embryonic development [reviewed in (114, 115)]. It has been proposed that maternally expressed genes are responsible for increasing the mothers' survival rate by suppressing fetal growth, whereas paternally expressed genes augment fetal growth and development to guarantee the viability of their genetic offspring (116). This would implicate KLF14 to play a repressive role in fetal and/or placental development and growth. In addition to adipose tissue, KLF14 is also highly expressed in the placenta (43). A recent study by Koppes et al. in mice showed that offsprings from Klf14-deficient mothers showed an overgrowth of the placenta in late gestation, however, fetal weight remained unchanged (94). RNA sequencing of placentas from Klf14-deficient and wild type mice identified 21 differentially expressed genes such as Col26a1 (94). Consistent with KLF14's implicated role as a transcriptional repressor (1, 117), the loss of Klf14 activated many placental genes. However, none of the significantly differentially expressed genes overlapped with previously reported adipose tissue KLF14 trans-regulated target genes nor with previously reported liver tissue targets in Klf14-deficient mice (1, 52, 92, 93), possibly due to distinct transcriptional regulation profiles in different tissues.
Mouse Models of KLF14
Several mouse studies showed conflicting results, for example, for the role of liver Klf14 in atherosclerosis. One major factor that may be causing the discrepancies in these studies is the utilization of distinct mouse lines and experimental designs, which are different in many aspects, including targeted regions, targeting approaches, tests performed, and phenotypes observed. We provide a summary of the different Klf14 mouse lines which have been reported in the literature (Table 2). Thus far, there are four Klf14 whole-body knockout mice generated by four different groups. Argmann et al. obtained Klf14tm1.1(KOMP)Vlcg mouse sperm from the Knockout Mouse Project (KOMP) repository (91). In these mice, 1035 bp between positions 30907660-30908694 on chromosome 6 (Genome Build 37), which includes the entire Klf14 open reading frame, was deleted through homologous recombination (91). The authors reported no significant differences in metabolic phenotypes comparing these knockout animals to their wild type littermate controls. In contrast to previous reports, no changes in HDL-C, cholesterol and ApoA-I plasma levels were observed in Klf14-deficient mice. However, the authors did not study female and male mice separately, a consequential omission given that in humans, KLF14 plays sexual-dimorphic roles. The second whole-body knockout mouse was generated by the Karpe Lab by inserting a 7 bp frameshift sequence using CRISPR-Cas9 at the beginning of the Klf14 gene to mitigate the disruption of neighboring regulatory regions (1). They reported that serum HDL-C level was reduced following the disruption of Klf14 in these mice when fed with HFD. The third Klf14-null mouse line was reported recently by Koppes et al. (94). This mouse line was generated by flanking the Klf14 gene with two loxP sites, separated by a total of 4.9 kb. This strategy targets a much bigger region than other whole-body knockout mouse lines. It was not clear if the neighboring gene expression was analyzed to ensure adjacent regulatory elements were not disrupted. This group reported no abnormal lipid accumulation in the placentas of Klf14-deficient mice. Lastly, Fan et al. generated a Klf14-KO mouse line by deleting an 8 bp sequence using TALEN, which resulted in the disruption of the tenth amino acid and premature termination (95). These Klf14-KO mice did not display abnormal changes in their body weight or serum lipids. Instead, they developed spontaneous tumors at various locations. However, spontaneous tumorigenesis was not reported in other Klf14 whole-body knockout mice. Chen and Musunuru groups generated two tissue-specific Klf14 knockout mouse lines: a liver-specific knockout (Klf14-LKO) (92) and an adipose-specific knockout (AdnKlf14-KO) (1). Both lines were generated by inserting two LoxP sites flanking the Klf14 gene, followed by crossing with mice harboring liver (Alb)- or adipose (Adipoq)-specific Cre transgenes. AdnKlf14-KO mice showed decreased HDL-C in females and increased triglycerides in males with both sexes exhibiting impaired glucose tolerance and insulin sensitivity (1). Klf14-LKO mice showed decreased HDL-C levels but no change in TC and triglyceride levels in male mice (92). Female mice were not studied. While mouse models are useful to establish causality, they do not capture the subtle differences in KLF14 abundance among individuals as observed in human studies. Further, developmental compensation by related KLFs cannot be neglected as a possibility for the lack of an observed metabolic phenotype in some studies or conflicting results in others.
Transcriptional Targets of KLF14
Direct transcriptional targets of KLF14 are largely unknown. Several studies identified individual transcriptional targets of KLF14 using multiple approaches. Sarmento et al. showed that KLF14 regulated regulatory T-cell differentiation via chromatin remodeling at the FOXP3 Treg-specific demethylation region based on ChIP analysis suggesting that FOXP3 is a potential target of KLF14 (118). Truty et al. showed that KLF14 exerts repressive pressure on the TGFβRII promoter by binding to GC-rich binding motif and by competing with Sp1 using ChIP assays, site-directed mutagenesis, and electromobility shift (119). They further observed that the N-terminus region of KLF14 binds with Sin3A to form a co-repressor complex that represses the TGFβRII promoter, which is consistent with the fact that KLF14 employs the chromatin-modifying capability of Sin3A to repress transcription (119). Guo et al. showed that ApoA-I is decreased in Klf14 liver-specific knockout mouse model, indicating KLF14-mediated transcriptional regulation of ApoA-I (92). They demonstrated that the human APOA1 promoter region contains two KLF14 binding sites (CACCC box). ChIP assay showed that KLF14 binds to regions containing the CACCC box, demonstrating that APOA1 is a functional KLF14 transcriptional target. De Assuncao et al. first demonstrated the transcriptional activation role of KLF14 by identifying Sphingosine kinase 1 (SK1) as its transcriptional target (52). SK1 mRNA levels were observed to decrease following KLF14 siRNA treatment in endothelial cells. Specifically, using ChIP and promoter binding assays, the group demonstrated that KLF14 binds to the promoter region of SK1 and represses its transcription (52). Wei et al. have shown that adenovirus-mediated KLF14 knockdown is significantly associated with the increased level of MAPK proteins including ERK1/2 and p38, stress-induced signaling pathways that are associated with the transcription of some proinflammatory factors and pathogenesis of atherosclerosis (104). However, whether or not P38 MAPK or ERK1/2 are transcriptional targets of KLF14 still require further validation.
Recent advances in human genetic and bioinformatic approaches have been employed to identify genetics variants that are associated with KLF14 in adipose tissue. The same genetic variants are also associated with nearly 400 genes in trans. The promoter regions of the trans-regulated genes were enriched for the KLF14 binding motif (1), although, there is conflicting evidence about the exact binding motif of KLF14 (120, 121). Many of these trans-genes have been demonstrated to regulate MetSyn. For example, SLC2A4 impairs glucose tolerance (122); STARD10 impairs glucose-stimulated insulin secretion (123, 124); and the IDE regulates the degradation of insulin (125). Many of the putative target genes were also linked to a range of metabolic characteristics, such as waist-to-hip ratio, body mass index (BMI), cholesterol, insulin, and blood glucose levels. These putative transcription targets are waiting for further experimental validation.
Conclusions and Future Studies
It has been nearly 20 years since Klf14 was first described and cloned (19). In the intervening years, its role in metabolic disorders and cancer has been established through a series of human genetics and mouse studies (Figure 2). However, conflicting results in mouse models show the need for additional carefully performed studies to elucidate its role in human disease.
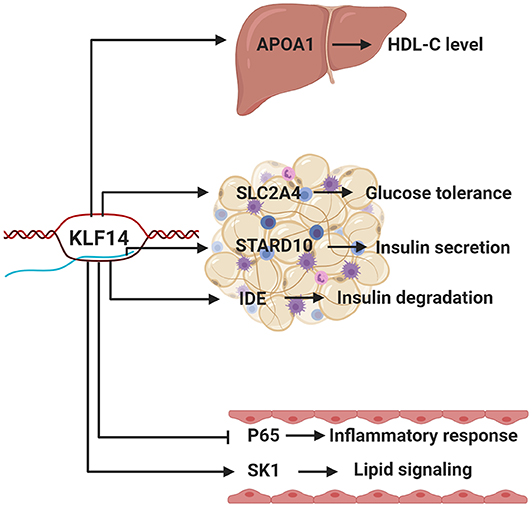
Figure 2. The function of KLF14 in metabolic tissues. Putative KLF14 transcription targets in different metabolically active tissues/organs, as well as their potential roles, are shown. Human genetics studies show that risk allele is associated with lower KLF14 expression in adipose tissue. Schematic depicts the KLF14 risk allele.
The genetic variants of the KLF14 locus in the human genome are significantly associated with a multitude of MetSyn-related traits and disorders. However, the causal variant(s) that is responsible for metabolic disorders, increased T2D/CAD risk, and changes in KLF14 expression remains elusive. These variants appear to regulate KLF14 expression only in adipose tissue and the cause of this tissue specificity is not known. Trans-ethnic fine-mapping studies, massively parallel reporter assays, or creating cell lines with different alleles on an isogenic background may help to identify the causal variant(s) in the locus (126). However, it will be more difficult to identify the tissue-specific effect of the causal variants since adipose tissue consists of a multitude of cells.
It is also crucial to identify the direct KLF14 targets to understand how KLF14 functions. A systematic approach identifying the direct KLF14 targets is lacking. Approximately half of the nearly 400 genes identified in the Small et al. study (1) had KLF14 binding sites in their promoter/enhancer regions, but having a binding site is not enough evidence to show that the transcription factor actually binds to that sequence to regulate target gene expression. The currently preferred method for identifying targets of transcriptional factors is to reduce or induce their expression using shRNA or overexpression vectors, followed by the identification of differentially expressed genes. A subset of genes with a consensus binding sequence near the transcription start sites is then defined as transcriptional targets. However, in this approach, the effects of prolonged modulation of transcription factor expression are dominated by secondary (and beyond) effects and it is impossible to discern primary transcriptional targets. Furthermore, proximal transcription factor binding alone is not sufficient to modulate gene expression (117). To avoid these complications, novel approaches that allow the identification of primary KLF14 targets by determining the impact of rapid KLF14 depletion on nascent RNA transcription should be employed (127–129).
Finally, the sex-biased effects of KLF14 on metabolic phenotypes are intriguing, especially since these effects do not appear to be hormonally-driven (1). Multiple studies show that KLF14 expression is higher in females, but the effect of the genetic variants on KLF14 is similar in males and females (1, 12). There is also a great deal of overlap between the adipose tissue genes whose expression is associated with the KLF14 locus in trans in males and females (1, 12). However, this is based on a study that identified the overlap in trans-regulated genes in two distinct studies. A more careful approach would be to calculate the association of all the adipose tissue-expressed genes in males and females recruited from the same population and whose gene expression was measured with the same method. GTEx study provides an opportunity for such a study to be performed (43). It is also possible that KLF14 shows an effect based on a threshold of abundance and since females have more KLF14 expression, the metabolic effects are more pronounced.
In summary, based on human genetics findings, inducing KLF14 expression in adipose tissue would be predicted to be metabolically beneficial, at least in females. However, greater insights into the mechanistic pathways that engender KLF14-mediated effects on adipose tissue are warranted to comprehensively evaluate its potential as a therapeutic target. It has been shown that perhexiline, a small drug that is approved for clinical use in many countries except the United States, to treat angina and heart failure, can effectively induce KLF14 expression in the liver while reducing atherosclerosis in ApoE-null mice (92). However, chronic use of perhexiline is associated with hepatotoxicity and neurotoxicity, which is why it has been removed from the markets of several countries. To this end, the derivatives of perhexiline could be potential substitutes (130). Another alternative to avoid unwanted side effects would be to deliver the drug directly to adipose tissue. Xue et al. have developed nanoparticles that can deliver anti-obesity drugs directly to adipose tissue (131). This approach can be optimized to deliver KLF14 agonists to improve adipose tissue function in metabolic disorders.
Author Contributions
QY and MC planned and wrote the manuscript.
Funding
This work was supported by R01 DK118287 from the National Institute of Diabetes and Digestive and Kidney Diseases and 1-19-IBS-105 from the American Diabetes Association.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank members of the Civelek Lab for feedback on the manuscript.
References
1. Small KS, Todorčević M, Civelek M, Moustafa JSE-S, Wang X, Simon MM, et al. Regulatory variants at KLF14 influence type 2 diabetes risk via a female-specific effect on adipocyte size and body composition. Nat Genet. (2018) 50:572–80. doi: 10.1038/s41588-018-0088-x
2. Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. (2010) 466:707–13. doi: 10.1038/nature09270
3. Voight BF, Scott LJ, Steinthorsdottir V, Morris AP, Dina C, Welch RP, et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet. (2010) 42:579–89. doi: 10.1038/ng.609
4. Chen X, Li S, Yang Y, Yang X, Liu Y, Liu Y, et al. Genome-wide association study validation identifies novel loci for atherosclerotic cardiovascular disease. J Thromb Haemost. (2012) 10:1508–14. doi: 10.1111/j.1538-7836.2012.04815.x
5. Mahajan A, Taliun D, Thurner M, Robertson NR, Torres JM, Rayner NW, et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet. (2018) 50:1505–13. doi: 10.1038/s41588-018-0241-6
6. Pulit SL, Stoneman C, Morris AP, Wood AR, Glastonbury CA, Tyrrell J, et al. Meta-analysis of genome-wide association studies for body fat distribution in 694 649 individuals of European ancestry. Hum Mol Genet. (2019) 28:166–74. doi: 10.1093/hmg/ddy327
7. Shungin D, Winkler TW, Croteau-Chonka DC, Ferreira T, Locke AE, Mägi R, et al. New genetic loci link adipose and insulin biology to body fat distribution. Nature. (2015) 518:187–96. doi: 10.1038/nature14132
8. Chen G, Bentley A, Adeyemo A, Shriner D, Zhou J, Doumatey A, et al. Genome-wide association study identifies novel loci association with fasting insulin and insulin resistance in African Americans. Hum Mol Genet. (2012) 21:4530–6. doi: 10.1093/hmg/dds282
9. Huang P, Yin R-X, Huang K-K, Zeng X-N, Guo T, Lin Q-Z, et al. Association of the KLF14 rs4731702 SNP and serum lipid levels in the Guangxi Mulao and Han populations. BioMed Res Int. (2013) 2013:231515. doi: 10.1155/2013/231515
10. Ohshige T, Iwata M, Omori S, Tanaka Y, Hirose H, Kaku K, et al. Association of new loci identified in European genome-wide association studies with susceptibility to type 2 diabetes in the Japanese. PLoS ONE. (2011) 6:e26911. doi: 10.1371/journal.pone.0026911
11. Rees SD, Hydrie MZI, Shera AS, Kumar S, O'Hare JP, Barnett AH, et al. Replication of 13 genome-wide association (GWA)-validated risk variants for type 2 diabetes in Pakistani populations. Diabetologia. (2011) 54:1368–74. doi: 10.1007/s00125-011-2063-2
12. Civelek M, Wu Y, Pan C, Raulerson CK, Ko A, He A, et al. Genetic regulation of adipose gene expression and cardio-metabolic traits. Am J Hum Genet. (2017) 100:428–43. doi: 10.1016/j.ajhg.2017.01.027
13. Small KS, Hedman AK, Grundberg E, Nica AC, Thorleifsson G, Kong A, et al. Identification of an imprinted master trans regulator at the KLF14 locus related to multiple metabolic phenotypes. Nat Genet. (2011) 43:561–4. doi: 10.1038/ng.833
14. McConnell BB, Yang VW. Mammalian krüppel-like factors in health and diseases. Physiol Rev. (2010) 90:1337–81. doi: 10.1152/physrev.00058.2009
15. Tetreault M-P, Yang Y, Katz JP. Krüppel-like factors in cancer. Nat Rev Cancer. (2013) 13:701–13. doi: 10.1038/nrc3582
16. Jiang J, Chan Y-S, Loh Y-H, Cai J, Tong G-Q, Lim C-A, et al. A core Klf circuitry regulates self-renewal of embryonic stem cells. Nat Cell Biol. (2008) 10:353–60. doi: 10.1038/ncb1698
17. Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, et al. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. (2008) 36:W465–9. doi: 10.1093/nar/gkn180
18. Krishna SS, Majumdar I, Grishin NV. Structural classification of zinc fingers survey and summary. Nucleic Acids Res. (2003) 31:532–50. doi: 10.1093/nar/gkg161
19. Scohy S, Gabant P, van Reeth T, Hertveldt V, Drèze PL, van Vooren P, et al. Identification of KLF13 and KLF14 (SP6), novel members of the SP/XKLF transcription factor family. Genomics. (2000) 70:93–101. doi: 10.1006/geno.2000.6362
20. Nagai R, Friedman SL, Kasuga M (editors). The Biology of Krüppel-Like Factors. Japan: Springer (2009). doi: 10.1007/978-4-431-87775-2
21. Pandya AY, Talley LI, Frost AR, Fitzgerald TJ, Trivedi V, Chakravarthy M, et al. Nuclear localization of KLF4 is associated with an aggressive phenotype in early-stage breast cancer. Clin Cancer Res. (2004) 10:2709–19. doi: 10.1158/1078-0432.CCR-03-0484
22. Quadrini KJ, Bieker JJ. Krüppel-like zinc fingers bind to nuclear import proteins and are required for efficient nuclear localization of erythroid krüppel-like factor. J Biol Chem. (2002) 277:32243–52. doi: 10.1074/jbc.M205677200
23. Knoepfler PS, Eisenman RN. Sin meets NuRD and other tails of repression. Cell. (1999) 99:447–50. doi: 10.1016/S0092-8674(00)81531-7
24. Pearson R, Fleetwood J, Eaton S, Crossley M, Bao S. Krüppel-like transcription factors: a functional family. Int J Biochem Cell Biol. (2008) 40:1996–2001. doi: 10.1016/j.biocel.2007.07.018
25. Matsumoto N, Kubo A, Liu H, Akita K, Laub F, Ramirez F, et al. Developmental regulation of yolk sac hematopoiesis by Kruppel-like factor 6. Blood. (2006) 107:1357–65. doi: 10.1182/blood-2005-05-1916
26. Laub F, Lei L, Sumiyoshi H, Kajimura D, Dragomir C, Smaldone S, et al. Transcription factor KLF7 is important for neuronal morphogenesis in selected regions of the nervous system. Mol Cell Biol. (2005) 25:5699–711. doi: 10.1128/MCB.25.13.5699-5711.2005
27. Morita M, Kobayashi A, Yamashita T, Shimanuki T, Nakajima O, Takahashi S, et al. Functional analysis of basic transcription element binding protein by gene targeting technology. Mol Cell Biol. (2003) 23:2489–500. doi: 10.1128/MCB.23.7.2489-2500.2003
28. Subramaniam M, Gorny G, Johnsen SA, Monroe DG, Evans GL, Fraser DG, et al. TIEG1 null mouse-derived osteoblasts are defective in mineralization and in support of osteoclast differentiation in vitro. Mol Cell Biol. (2005) 25:1191–9. doi: 10.1128/MCB.25.3.1191-1199.2005
29. Song C-Z, Gavriilidis G, Asano H, Stamatoyannopoulos G. Functional study of transcription factor KLF11 by targeted gene inactivation. Blood Cells Mol Dis. (2005) 34:53–9. doi: 10.1016/j.bcmd.2004.08.027
30. Zhou M, McPherson L, Feng D, Song A, Dong C, Lyu S-C, et al. Krüppel-like transcription factor 13 regulates T lymphocyte survival in vivo. J Immunol. (2007) 178:5496–504. doi: 10.4049/jimmunol.178.9.5496
31. Fisch S, Gray S, Heymans S, Haldar SM, Wang B, Pfister O, et al. Kruppel-like factor 15 is a regulator of cardiomyocyte hypertrophy. Proc Natl Acad Sci USA. (2007) 104:7074–9. doi: 10.1073/pnas.0701981104
32. Perkins AC, Sharpe AH, Orkin SH. Lethal beta-thalassaemia in mice lacking the erythroid CACCC-transcription factor EKLF. Nature. (1995) 375:318–22. doi: 10.1038/375318a0
33. Tallack MR, Magor GW, Dartigues B, Sun L, Huang S, Fittock JM, et al. Novel roles for KLF1 in erythropoiesis revealed by mRNA-seq. Genome Res. (2012) 22:2385–98. doi: 10.1101/gr.135707.111
34. Kuo CT, Veselits ML, Barton KP, Lu MM, Clendenin C, Leiden JM. The LKLF transcription factor is required for normal tunica media formation and blood vessel stabilization during murine embryogenesis. Genes Dev. (1997) 11:2996–3006. doi: 10.1101/gad.11.22.2996
35. Wani MA, Means RT, Lingrel JB. Loss of LKLF function results in embryonic lethality in mice. Transgenic Res. (1998) 7:229–38. doi: 10.1023/A:1008809809843
36. Katz JP, Perreault N, Goldstein BG, Lee CS, Labosky PA, Yang VW, et al. The zinc-finger transcription factor Klf4 is required for terminal differentiation of goblet cells in the colon. Development. (2002) 129:2619–28.
37. Oishi Y, Manabe I, Tobe K, Tsushima K, Shindo T, Fujiu K, et al. Krüppel-like transcription factor KLF5 is a key regulator of adipocyte differentiation. Cell Metab. (2005) 1:27–39. doi: 10.1016/j.cmet.2004.11.005
38. Segre JA, Bauer C, Fuchs E. Klf4 is a transcription factor required for establishing the barrier function of the skin. Nat Genet. (1999) 22:356–60. doi: 10.1038/11926
39. Shindo T, Manabe I, Fukushima Y, Tobe K, Aizawa K, Miyamoto S, et al. Krüppel-like zinc-finger transcription factor KLF5/BTEB2 is a target for angiotensin II signaling and an essential regulator of cardiovascular remodeling. Nat Med. (2002) 8:856–63. doi: 10.1038/nm738
40. GTEx Consortium Laboratory Data Analysis &Coordinating Center (LDACC)—Analysis Working Group Statistical Methods groups—Analysis Working Group Enhancing GTEx (eGTEx) groups NIH Common Fund, et al. Genetic effects on gene expression across human tissues. Nature. (2017) 550:204–13. doi: 10.1038/nature24277
41. Civelek M, Lusis AJ. Systems genetics approaches to understand complex traits. Nat Rev Genet. (2014) 15:34–48. doi: 10.1038/nrg3575
42. Civelek M, Lusis AJ. Conducting the metabolic syndrome orchestra. Nat Genet. (2011) 43:506–8. doi: 10.1038/ng.842
43. GTEx Consortium. The genotype-tissue expression (GTEx) project. Nat Genet. (2013) 45:580–5. doi: 10.1038/ng.2653
44. Kong A, Steinthorsdottir V, Masson G, Thorleifsson G, Sulem P, Besenbacher S, et al. Parental origin of sequence variants associated with complex diseases. Nature. (2009) 462:868–74. doi: 10.1038/nature08625
45. Consortium TEP. A user's guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol. (2011) 9:e1001046. doi: 10.1371/journal.pbio.1001046
46. Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, et al. The NIH roadmap epigenomics mapping consortium. Nat Biotechnol. (2010) 28:1045–8. doi: 10.1038/nbt1010-1045
47. Best CH, Haist RE. The effect of insulin administration on the insulin content of the pancreas1. J Physiol. (1941) 100:142–6. doi: 10.1113/jphysiol.1941.sp003930
48. Bonner-Weir S. Life and death of the pancreatic β cells. Trends Endocrinol Metab. (2000) 11:375–8. doi: 10.1016/S1043-2760(00)00305-2
49. Leibiger IB, Leibiger B, Berggren P-O. Insulin signaling in the pancreatic β-Cell. Ann Rev Nutr. (2008) 28:233–51. doi: 10.1146/annurev.nutr.28.061807.155530
50. Pollak NM, Hoffman M, Goldberg IJ, Drosatos K. Krüppel-like factors: crippling and uncrippling metabolic pathways. JACC Basic Transl Sci. (2018) 3:132–56. doi: 10.1016/j.jacbts.2017.09.001
51. Yang M, Ren Y, Lin Z, Tang C, Jia Y, Lai Y, et al. Krüppel-like factor 14 increases insulin sensitivity through activation of PI3K/Akt signal pathway. Cell Signal. (2015) 27:2201–8. doi: 10.1016/j.cellsig.2015.07.019
52. de Assuncao TM, Lomberk G, Cao S, Yaqoob U, Mathison A, Simonetto DA, et al. New role for kruppel-like factor 14 as a transcriptional activator involved in the generation of signaling lipids. J Biol Chem. (2014) 289:15798–809. doi: 10.1074/jbc.M113.544346
53. Wang J, Badeanlou L, Bielawski J, Ciaraldi TP, Samad F. Sphingosine kinase 1 regulates adipose proinflammatory responses and insulin resistance. Am J Physiol Endocrinol Metab. (2014) 306:E756–68. doi: 10.1152/ajpendo.00549.2013
54. Chatzakos V, Rundlöf AK, Ahmed D, de Verdier PJ, Flygare J. Inhibition of sphingosine kinase 1 enhances cytotoxicity, ceramide levels and ROS formation in liver cancer cells treated with selenite. Biochem Pharmacol. (2012) 84:712–21. doi: 10.1016/j.bcp.2012.06.009
55. Amati F, Dubé JJ, Alvarez-Carnero E, Edreira MM, Chomentowski P, Coen PM, et al. Skeletal muscle triglycerides, diacylglycerols, and ceramides in insulin resistance: another paradox in endurance-trained athletes? Diabetes. (2011) 60:2588–97. doi: 10.2337/db10-1221
56. Choe SS, Huh JY, Hwang IJ, Kim JI, Kim JB. Adipose tissue remodeling: its role in energy metabolism and metabolic disorders. Front Endocrinol. (2016) 7:30. doi: 10.3389/fendo.2016.00030
57. Goossens GH. The role of adipose tissue dysfunction in the pathogenesis of obesity-related insulin resistance. Physiol Behav. (2008) 94:206–18. doi: 10.1016/j.physbeh.2007.10.010
58. Goossens GH. The metabolic phenotype in obesity: fat mass, body fat distribution, and adipose tissue function. Obes Facts. (2017) 10:207–15. doi: 10.1159/000471488
59. Rosen ED, Spiegelman BM. What we talk about when we talk about fat. Cell. (2014) 156:20–44. doi: 10.1016/j.cell.2013.12.012
60. Björntorp P. Metabolic implications of body fat distribution. Diabetes Care. (1991) 14:1132–43. doi: 10.2337/diacare.14.12.1132
61. Després J-P. Body fat distribution and risk of cardiovascular disease. Circulation. (2012) 126:1301–13. doi: 10.1161/CIRCULATIONAHA.111.067264
62. Goodpaster BH, Krishnaswami S, Harris TB, Katsiaras A, Kritchevsky SB, Simonsick EM, et al. Obesity, regional body fat distribution, and the metabolic syndrome in older men and women. Arch Intern Med. (2005) 165:777–83. doi: 10.1001/archinte.165.7.777
63. Jensen MD. Role of body fat distribution and the metabolic complications of obesity. None. (2008) 93:s57–63. doi: 10.1210/jc.2008-1585
64. Kwon H, Kim D, Kim JS. Body fat distribution and the risk of incident metabolic syndrome: a longitudinal cohort study. Sci Rep. (2017) 7:10955. doi: 10.1038/s41598-017-09723-y
65. Birsoy K, Chen Z, Friedman J. Transcriptional regulation of adipogenesis by KLF4. Cell Metabol. (2008) 7:339–47. doi: 10.1016/j.cmet.2008.02.001
66. Chen Z, Torrens JI, Anand A, Spiegelman BM, Friedman JM. Krox20 stimulates adipogenesis via C/EBPbeta-dependent and -independent mechanisms. Cell Metab. (2005) 1:93–106. doi: 10.1016/j.cmet.2004.12.009
67. Lee DS, Choi H, Han BS, Kim WK, Lee SC, Oh K-J, et al. c-Jun regulates adipocyte differentiation via the KLF15-mediated mode. Biochem Biophys Res Commun. (2016) 469:552–8. doi: 10.1016/j.bbrc.2015.12.035
68. Oishi Y, Manabe I, Tobe K, Ohsugi M, Kubota T, Fujiu K, et al. SUMOylation of Krüppel-like transcription factor 5 acts as a molecular switch in transcriptional programs of lipid metabolism involving PPAR-δ. Nat Med. (2008) 14:656–66. doi: 10.1038/nm1756
69. Li D, Yea S, Li S, Chen Z, Narla G, Banck M, et al. Krüppel-like factor-6 promotes preadipocyte differentiation through histone deacetylase 3-dependent repression of DLK1. J Biol Chem. (2005) 280:26941–52. doi: 10.1074/jbc.M500463200
70. Smas CM, Sul HS. Pref-1, a protein containing EGF-like repeats, inhibits adipocyte differentiation. Cell. (1993) 73:725–34. doi: 10.1016/0092-8674(93)90252-L
71. Lee H, Kim HJ, Lee YJ, Lee M-Y, Choi H, Lee H, et al. Krüppel-like factor KLF8 plays a critical role in adipocyte differentiation. PLoS ONE. (2012) 7:e52474. doi: 10.1371/journal.pone.0052474
72. Pei H, Yao Y, Yang Y, Liao K, Wu J-R. Krüppel-like factor KLF9 regulates PPARγ transactivation at the middle stage of adipogenesis. Cell Death Differ. (2011) 18:315–327. doi: 10.1038/cdd.2010.100
73. Jiang S, Wei H, Song T, Yang Y, Zhang F, Zhou Y, et al. KLF13 promotes porcine adipocyte differentiation through PPARγ activation. Cell Biosci. (2015) 5:28. doi: 10.1186/s13578-015-0016-z
74. Asada M, Rauch A, Shimizu H, Maruyama H, Miyaki S, Shibamori M, et al. DNA binding - dependent glucocorticoid receptor activity promotes adipogenesis via krüppel-like factor 15 gene expression. Lab Invest. (2011) 91:203–15. doi: 10.1038/labinvest.2010.170
75. Gray S, Feinberg MW, Hull S, Kuo CT, Watanabe M, Sen-Banerjee S, et al. The Krüppel-like factor KLF15 regulates the insulin-sensitive glucose transporter GLUT4. J Biol Chem. (2002) 277:34322–28. doi: 10.1074/jbc.M201304200
76. Mori T, Sakaue H, Iguchi H, Gomi H, Okada Y, Takashima Y, et al. Role of krüppel-like factor 15 (KLF15) in transcriptional regulation of adipogenesis. J Biol Chem. (2005) 280:12867–75. doi: 10.1074/jbc.M410515200
77. Uchida S, Tanaka Y, Ito H, Saitoh-Ohara F, Inazawa J, Yokoyama KK, et al. Transcriptional regulation of the CLC-K1 promoter by myc-associated zinc finger protein and kidney-enriched Krüppel-like factor, a novel zinc finger repressor. Mol Cell Biol. (2000) 20:7319–31. doi: 10.1128/MCB.20.19.7319-7331.2000
78. Banerjee SS, Feinberg MW, Watanabe M, Gray S, Haspel RL, Denkinger DJ, et al. The Krüppel-like factor KLF2 inhibits peroxisome proliferator-activated receptor-γ expression and adipogenesis. J Biol Chem. (2003) 278:2581–4. doi: 10.1074/jbc.M210859200
79. Lee H, Kang R, Kim YS, Chung S-I, Yoon Y. Platycodin D inhibits adipogenesis of 3T3-L1 cells by modulating Kruppel-like factor 2 and peroxisome proliferator-activated receptor gamma. Phytother Res. (2010) 24(Suppl. 2):S161–7. doi: 10.1002/ptr.3054
80. Sue N, Jack BHA, Eaton SA, Pearson RCM, Funnell APW, Turner J, et al. Targeted disruption of the basic krüppel-like factor gene (Klf3) reveals a role in adipogenesis. Mol Cell Biol. (2008) 28:3967–78. doi: 10.1128/MCB.01942-07
81. Kawamura Y, Tanaka Y, Kawamori R, Maeda S. Overexpression of Kruppel-like factor 7 regulates adipocytokine gene expressions in human adipocytes and inhibits glucose-induced insulin secretion in pancreatic beta-cell line. Mol Endocrinol. (2006) 20:844–56. doi: 10.1210/me.2005-0138
82. Zhang Z, Wang H, Sun Y, Li H, Wang N. Klf7 modulates the differentiation and proliferation of chicken preadipocyte. Acta Biochim Biophys Sin. (2013) 45:280–8. doi: 10.1093/abbs/gmt010
83. Jang M-K, Lee S, Jung MH. RNA-seq analysis reveals a negative role of KLF16 in adipogenesis. PLoS ONE. (2016) 11:e0162238. doi: 10.1371/journal.pone.0162238
84. Wei S, Zhang L, Zhou X, Du M, Jiang Z, Hausman GJ, et al. Emerging roles of zinc finger proteins in regulating adipogenesis. Cell Mol Life Sci. (2013) 70:4569–84. doi: 10.1007/s00018-013-1395-0
85. Wu Z, Wang S. Role of kruppel-like transcription factors in adipogenesis. Dev Biol. (2013) 373:235–43. doi: 10.1016/j.ydbio.2012.10.031
86. Machine Learning Based Histology Phenotyping to Investigate Epidemiologic and Genetic Basis of Adipocyte Morphology and Cardiometabolic Traits. bioRxiv. Available online at: https://www.biorxiv.org/content/10.1101/680637v1 (accessed September 29, 2019).
87. Iwaya C, Kitajima H, Yamamoto K, Maeda Y, Sonoda N, Shibata H, et al. DNA methylation of the Klf14 gene region in whole blood cells provides prediction for the chronic inflammation in the adipose tissue. Biochem Biophys Res Commun. (2018) 497:908–15. doi: 10.1016/j.bbrc.2017.12.104
88. Franks PW, McCarthy MI. Exposing the exposures responsible for type 2 diabetes and obesity. Science. (2016) 354:69–73. doi: 10.1126/science.aaf5094
89. Guewo Fokeng M, Atogho Tiedeu B. The Kruppel-like factor 14 (KLF14), master gene of multiple metabolic phenotypes: putative trans-regulator network. Transl Biomed. (2016) 7:67. doi: 10.21767/2172-0479.100067
90. Bacos K, Gillberg L, Volkov P, Olsson AH, Hansen T, Pedersen O, et al. Blood-based biomarkers of age-associated epigenetic changes in human islets associate with insulin secretion and diabetes. Nat Commun. (2016) 7:11089. doi: 10.1038/ncomms11089
91. Argmann CA, Violante S, Dodatko T, Amaro MP, Hagen J, Gillespie VL, et al. Germline deletion of Krüppel-like factor 14 does not increase risk of diet induced metabolic syndrome in male C57BL/6 mice. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:3277–3285. doi: 10.1016/j.bbadis.2017.09.021
92. Guo Y, Fan Y, Zhang J, Lomberk GA, Zhou Z, Sun L, et al. Perhexiline activates KLF14 and reduces atherosclerosis by modulating ApoA-I production. J Clin Invest. (2015) 125:3819–30. doi: 10.1172/JCI79048
93. Wang L, Tong X, Gu F, Zhang L, Chen W, Cheng X, et al. The KLF14 transcription factor regulates hepatic gluconeogenesis in mice. J Biol Chem. (2017) 292:21631–42. doi: 10.1074/jbc.RA117.000184
94. Koppes E, Shaffer B, Sadovsky E, Himes K, Barak Y, Sadovsky Y, et al. Klf14 is an imprinted transcription factor that regulates placental growth. Placenta. (2019) 88:61–7. doi: 10.1016/j.placenta.2019.09.013
95. Fan G, Sun L, Shan P, Zhang X, Huan J, Zhang X, et al. Loss of KLF14 triggers centrosome amplification and tumorigenesis. Nat Commun. (2015) 6:8450. doi: 10.1038/ncomms9450
96. Liu DJ, Peloso GM, Yu H, Butterworth AS, Wang X, Mahajan A, et al. Exome-wide association study of plasma lipids in >300,000 individuals. Nat Genet. (2017) 49:1758–66. doi: 10.1038/ng.3977
97. Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, et al. Discovery and refinement of loci associated with lipid levels. Nat Genet. (2013) 45:1274–83. doi: 10.1038/ng.2797
98. Rui L. Energy metabolism in the liver. Compr Physiol. (2014) 4:177–97. doi: 10.1002/cphy.c130024
99. Rodgers JT, Puigserver P. Fasting-dependent glucose and lipid metabolic response through hepatic sirtuin 1. PNAS. (2007) 104:12861–6. doi: 10.1073/pnas.0702509104
100. Leclercq IA, Da Silva Morais A, Schroyen B, Van Hul N, Geerts A. Insulin resistance in hepatocytes and sinusoidal liver cells: mechanisms and consequences. J Hepatol. (2007) 47:142–56. doi: 10.1016/j.jhep.2007.04.002
101. Rodgers JT, Lerin C, Gerhart-Hines Z, Puigserver P. Metabolic adaptations through the PGC-1α and SIRT1 pathways. FEBS Lett. (2008) 582:46–53. doi: 10.1016/j.febslet.2007.11.034
102. Bergen WG, Mersmann HJ. Comparative aspects of lipid metabolism: impact on contemporary research and use of animal models. J Nutr. (2005) 135:2499–502. doi: 10.1093/jn/135.11.2499
103. Vitić J, Stevanović J. Comparative studies of the serum lipoproteins and lipids in some domestic, laboratory and wild animals. Comp Biochem Physiol B. (1993) 106:223–9. doi: 10.1016/0305-0491(93)90030-9
104. Wei X, Yang R, Wang C, Jian X, Li L, Liu H, et al. A novel role for the Krüppel-like factor 14 on macrophage inflammatory response and atherosclerosis development. Cardiovasc. Pathol. (2017) 27:1–8. doi: 10.1016/j.carpath.2016.11.003
105. Parker-Katiraee L, Carson AR, Yamada T, Arnaud P, Feil R, Abu-Amero SN, et al. Identification of the imprinted KLF14 transcription factor undergoing human-specific accelerated evolution. PLoS Genet. (2007) 3:e65. doi: 10.1371/journal.pgen.0030065
106. Sattler S, Kennedy-Lydon T (editors). The Immunology of Cardiovascular Homeostasis and Pathology. Springer International Publishing (2017). doi: 10.1007/978-3-319-57613-8
107. Stevens T, Brough GH, Moore TM, Babal P, Thompson WJ. Endothelial cells. In S. Uhlig, A. E. Taylor, editors. Methods in Pulmonary Research. Basel: Birkhäuser, 403–26. doi: 10.1007/978-3-0348-8855-4_16
108. Hu W, Lu H, Zhang J, Fan Y, Chang Z, Liang W, et al. Krüppel-like factor 14, a coronary artery disease associated transcription factor, inhibits endothelial inflammation via NF-κB signaling pathway. Atherosclerosis. (2018) 278:39–48. doi: 10.1016/j.atherosclerosis.2018.09.018
109. Luo X-H, Liu J-Z, Wang B, Men Q-L, Ju Y-Q, Yin F-Y, et al. KLF14 potentiates oxidative adaptation via modulating HO-1 signaling in castrate-resistant prostate cancer. Endocr Relat Cancer. (2019) 26:181–95. doi: 10.1530/ERC-18-0383
110. Stacey SN, Sulem P, Masson G, Gudjonsson SA, Thorleifsson G, Jakobsdottir M, et al. New common variants affecting susceptibility to basal cell carcinoma. Nat Genet. (2009) 41:909–14. doi: 10.1038/ng.412
111. Habedanck R, Stierhof Y-D, Wilkinson CJ, Nigg EA. The polo kinase Plk4 functions in centriole duplication. Nat Cell Biol. (2005) 7:1140–6. doi: 10.1038/ncb1320
112. Rosario CO, Ko MA, Haffani YZ, Gladdy RA, Paderova J, Pollett A, et al. Plk4 is required for cytokinesis and maintenance of chromosomal stability. Proc Natl Acad Sci USA. (2010) 107:6888–93. doi: 10.1073/pnas.0910941107
113. Vitale I, Galluzzi L, Castedo M, Kroemer G. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol. (2011) 12:385–92. doi: 10.1038/nrm3115
114. Piedrahita JA. The role of imprinted genes in fetal growth abnormalities. Birth Defects Res A Clin Mol Teratol. (2011) 91:682–92. doi: 10.1002/bdra.20795
115. Reik W, Walter J. Genomic imprinting: parental influence on the genome. Nat Rev Genet. (2001) 2:21–32. doi: 10.1038/35047554
116. Moore T, Haig D. Genomic imprinting in mammalian development: a parental tug-of-war. Trends Genet. (1991) 7:45–9. doi: 10.1016/0168-9525(91)90040-W
117. Lomberk G, Urrutia R. The family feud: turning off Sp1 by Sp1-like KLF proteins. Biochem J. (2005) 392(Pt 1):1–11. doi: 10.1042/BJ20051234
118. Sarmento OF, Svingen PA, Xiong Y, Xavier RJ, McGovern D, Smyrk TC, et al. A novel role for Kruppel-like factor 14 (KLF14) in T-regulatory cell differentiation. Cell Mol Gastroenterol Hepatol. (2014) 1:188–202.e4. doi: 10.1016/j.jcmgh.2014.12.007
119. Truty MJ, Lomberk G, Fernandez-Zapico ME, Urrutia R. Silencing of the transforming growth factor-β (TGFβ) receptor II by Krüppel-like factor 14 underscores the importance of a negative feedback mechanism in TGFβ signaling. J Biol Chem. (2009) 284:6291–300. doi: 10.1074/jbc.M807791200
120. Jolma A, Yan J, Whitington T, Toivonen J, Nitta KR, Rastas P, et al. DNA-binding specificities of human transcription factors. Cell. (2013) 152:327–39. doi: 10.1016/j.cell.2012.12.009
121. Najafabadi HS, Mnaimneh S, Schmitges FW, Garton M, Lam KN, Yang A, et al. C2H2 zinc finger proteins greatly expand the human regulatory lexicon. Nat Biotechnol. (2015) 33:555–62. doi: 10.1038/nbt.3128
122. Nakata M, Nagasaka S, Kusaka I, Matsuoka H, Ishibashi S, Yada T. Effects of statins on the adipocyte maturation and expression of glucose transporter 4 (SLC2A4): implications in glycaemic control. Diabetologia. (2006) 49:1881–92. doi: 10.1007/s00125-006-0269-5
123. Carrat GR, Hu M, Nguyen-Tu M-S, Chabosseau P, Gaulton KJ, van de Bunt M, et al. Decreased STARD10 expression is associated with defective insulin secretion in humans and mice. Am J Hum Genet. (2017) 100:238–56. doi: 10.1016/j.ajhg.2017.01.011
124. Hu M, Gadue P, Rutter GA. Characterization of a type 2 diabetes–associated islet-specific enhancer cluster in STARD10 by genome editing of endoC-ßH1 cells. Diabetes. (2018) 67:1708–P. doi: 10.2337/db18-1708-P
125. Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, et al. Insulin-degrading enzyme regulates the levels of insulin, amyloid β-protein, and the β-amyloid precursor protein intracellular domain in vivo. PNAS. (2003) 100:4162–7. doi: 10.1073/pnas.0230450100
126. Jennie L, Kiran M. From genotype to phenotype. Circulation. (2018) 11:e001946. doi: 10.1161/CIRCGEN.117.001946
127. Holland AJ, Fachinetti D, Han JS, Cleveland DW. Inducible, reversible system for the rapid and complete degradation of proteins in mammalian cells. Proc Natl Acad Sci USA. (2012) 109:E3350–7. doi: 10.1073/pnas.1216880109
128. Nishimura K, Fukagawa T, Takisawa H, Kakimoto T, Kanemaki M. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat Methods. (2009) 6:917–22. doi: 10.1038/nmeth.1401
129. Sathyan KM, McKenna BD, Anderson WD, Duarte FM, Core L, Guertin MJ. An improved auxin-inducible degron system preserves native protein levels and enables rapid and specific protein depletion. Genes Dev. (2019) 33:1441–55. doi: 10.1101/gad.328237.119
130. Marciniak G, Decolin D, Leclerc G, Decker N, Schwartz J. Synthesis and pharmacological properties of soft drug derivatives related to perhexiline. J Med Chem. (1988) 31:2289–96. doi: 10.1021/jm00120a007
Keywords: transcription factor, cardiometabolic diseases, human genetics, mouse models, transcriptional targets, sexual dimorphism
Citation: Yang Q and Civelek M (2020) Transcription Factor KLF14 and Metabolic Syndrome. Front. Cardiovasc. Med. 7:91. doi: 10.3389/fcvm.2020.00091
Received: 16 March 2020; Accepted: 29 April 2020;
Published: 27 May 2020.
Edited by:
Xia Yang, University of California, Los Angeles, United StatesReviewed by:
Ville-Petteri Makinen, South Australian Health and Medical Research Institute (SAHMRI), AustraliaMarcus M. Seldin, University of California, Irvine, United States
Copyright © 2020 Yang and Civelek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qianyi Yang, qy5sy@virginia.edu; Mete Civelek, mete@virginia.edu