COVID-19 alters human microbiomes: a meta-analysis
 Rine Christopher Reuben
Rine Christopher Reuben Rémy Beugnon
Rémy Beugnon Stephanie D. Jurburg
Stephanie D. Jurburg- 1German Centre of Integrative Biodiversity Research (iDiv) Halle-Jena-Leipzig, Leipzig, Germany
- 2Institute of Biology, Leipzig University, Leipzig, Germany
- 3Leipzig Institute for Meteorology, Universität Leipzig, Leipzig, Germany
- 4CEFE, Université de Montpellier, CNRS, EPHE, IRD, Montpellier, France
- 5Department of Environmental Microbiology, Helmholtz Centre for Environmental Research - UFZ, Leipzig, Germany
Introduction: Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) has infected a substantial portion of the world’s population, and novel consequences of COVID-19 on the human body are continuously being uncovered. The human microbiome plays an essential role in host health and well-being, and multiple studies targeting specific populations have reported altered microbiomes in patients infected with SARS-CoV-2. Given the global scale and massive incidence of COVID on the global population, determining whether the effects of COVID-19 on the human microbiome are consistent and generalizable across populations is essential.
Methods: We performed a synthesis of human microbiome responses to COVID-19. We collected 16S rRNA gene amplicon sequence data from 11 studies sampling the oral and nasopharyngeal or gut microbiome of COVID-19-infected and uninfected subjects. Our synthesis included 1,159 respiratory (oral and nasopharyngeal) microbiome samples and 267 gut microbiome samples from patients in 11 cities across four countries.
Results: Our reanalyses revealed communitywide alterations in the respiratory and gut microbiomes across human populations. We found significant overall reductions in the gut microbial diversity of COVID-19-infected patients, but not in the respiratory microbiome. Furthermore, we found more consistent community shifts in the gut microbiomes of infected patients than in the respiratory microbiomes, although the microbiomes in both sites exhibited higher host-to-host variation in infected patients. In respiratory microbiomes, COVID-19 infection resulted in an increase in the relative abundance of potentially pathogenic bacteria, including Mycoplasma.
Discussion: Our findings shed light on the impact of COVID-19 on the human-associated microbiome across populations, and highlight the need for further research into the relationship between long-term effects of COVID-19 and altered microbiota.
Introduction
The emergence and rapid spread of the novel beta-coronavirus, severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), caused severe and unprecedented public health and socioeconomic challenges globally (Waters et al., 2022). Primarily, COVID-19 presents as a multifaceted and multi-organ infection with variable severity. Symptoms range from acute respiratory distress syndrome to pneumonia and include non-specific flu-like symptoms, gastrointestinal symptoms, myocardial dysfunction, multiple organ failure, and death (Kumar Singh et al., 2019; Baud et al., 2020; Onder et al., 2020). In most cases, SARS-CoV-2-infected persons are either asymptomatic or show mild symptoms. However, approximately 5% of those infected, usually the elderly and/or individuals with comorbidities, develop a severe form of the disease, resulting in intensive medical care and death (Grasselli et al., 2020; Wiersinga et al., 2020). As of April 2023, there have been 762,791,152 confirmed COVID-19 cases with 6,897,025 mortalities worldwide (WHO Coronavirus (COVID-19) Dashboard).
Among the many long-term effects associated with COVID-19 infection, numerous studies have reported altered microbiota in COVID-19 patients. The human microbiome plays a vital role in host health (Kumpitsch et al., 2019) and has been suggested to act as an additional organ (Baquero and Nombela, 2012). These microbial communities maintain host homeostasis through complex and essential interactions, which result in improved immunomodulation, metabolism, organ functions, mucosal barrier integrity, and structural protection against intruding pathogens (Jandhyala et al., 2015; Kumpitsch et al., 2019). Specific microbial communities are associated with different human tissues (Human Microbiome Project Consortium, 2012; Pflughoeft and Versalovic, 2012).
Perturbations such as COVID-19 can result in microbiome dysbiosis in human microbiomes, in which the composition and diversity of beneficial and/or commensal microorganisms are altered, promoting the growth or opportunistic pathogens (Hoque et al., 2021b; Ren et al., 2021; Sun et al., 2022). In particular, the observation of increased host-to-host variability in the microbiomes associated with unhealthy hosts has been dubbed the Anna Karenina Principle (AKP), derived from the opening line of Tolstoy’s Anna Karenina: “All happy families are all alike; each unhappy family is unhappy in its own way”.
Different human diseases including obesity, psoriasis, arthritis, inflammatory bowel disease (IBD), influenza, HBV, and HIV have been reported to significantly alter human microbiomes (Ling et al., 2015; Lu et al., 2017; Gilbert et al., 2018; Gonçalves et al., 2019; Yun et al., 2019; Sencio et al., 2021). Similarly, several reports have demonstrated changes in the microbiomes (intestinal, nasopharyngeal, and oral) of COVID-19 patients during active infection and convalescent state, and these are usually characterized by the depletion of beneficial commensal microbes and a higher abundance of opportunistic pathogens (Zuo et al., 2020; Zuo et al., 2020; Hoque et al., 2021a; Hoque et al., 2021b; Jochems et al., 2021; Ren et al., 2021; Xu et al., 2021; Yeoh et al., 2021). The composition and diversity of the gut, nasal, or oral microbiome of COVID-19 patients are now widely believed to be predictive of COVID-19 prognosis, progression, and severity (Mathieu et al., 2018; Wypych et al., 2019; Chen et al., 2020; He et al., 2020; Hoque et al., 2021b). Moreover, the abundance of specific microbes within human microbiomes are now identified as biomarkers to distinguish COVID-19-infected individuals from healthy persons (Gou et al., 2020; Zuo et al., 2020; Tian et al., 2021).
Extensive interactions exist between the host immune system and microbiome. These interactions seem to induce immune responses to diseases, and in turn, the immune system affects the composition and diversity of the microbiome (Round and Mazmanian, 2009; Mangalmurti and Hunter, 2020; Attaway et al., 2021; Sun et al., 2022). COVID-19 has been reported to induce aberrant immune responses that not only elevate inflammatory markers including tumor necrosis factor-α, interleukin (IL)-10, and C-reactive protein, but also affect gut microbiome composition with a decreased population of beneficial bacteria especially bifidobacteria, Eubacterium rectale, and Faecalibacterium prausnitzii (Mangalmurti and Hunter, 2020; Attaway et al., 2021; Yeoh et al., 2021). Understanding microbiome changes across multiple microbiome compartments can shed light on the level and mechanisms of microbiome perturbation/dysbiosis associated with COVID-19, and may in turn aid the development of effective strategies for COVID-19 diagnosis, long-term management, and prevention.
All humans are susceptible to SARS-CoV-2 infection. Nevertheless, the human microbiome slightly differs across age, ethnicity, sex, race, and even geography (Human Microbiome Project Consortium, 2012; Brooks et al., 2018). Consequently, determining the effects of COVID-19 infections on the human microbiome requires assessing changes in the microbiomes of a wide range of patients across geographic regions. To this end, we performed a synthesis of human microbiome responses to COVID-19. We collected 16S rRNA gene amplicon sequence data from 11 studies sampling the oral and nasopharyngeal, or gut microbiome of COVID-19-infected and uninfected subjects. Our synthesis included 1,159 respiratory (oral and nasopharyngeal) microbiome samples and 267 gut microbiome samples and spanned four countries. We hypothesized that (1) because of stronger immune responses, infected patients would have a lower microbiome richness across microbiome compartments; (2) the microbiomes of infected patients would be more variable from host to host than that of healthy individuals, in line with the AKP; (3) as SARS-COV-2 is primarily a respiratory disease, the oral and nasopharyngeal microbiomes would be more strongly affected than the gut microbiomes; and (4) COVID-19 infection would result in consistent shifts in microbiome composition in both compartments.
Materials and methods
Literature search, selection criteria, and data extraction
This study was conducted through a robust literature search and selection using the RepOrting standards for Systematic Evidence Syntheses (ROSES) guidelines (Haddaway et al., 2018) (Figure 1). In January 2022, we performed a keyword search on the Web of Science database (www.webofscience.com) to identify and select relevant published articles. We used the terms “COVID-19” OR “SARS-CoV-2” OR “severe acute respiratory syndrome coronavirus 2” OR “coronavirus disease 2019” OR “nCoV” OR “novel coronavirus” AND “microbiome” OR “microbiota” OR “microflora” or “flora’’ OR “biome”. Furthermore, additional studies were included from other sources including PubMed and Google Scholar. We included published articles that performed 16S rRNA gene or transcript amplicon sequencing from gut/stool, nasopharyngeal, and oral samples collected from COVID-19+ individuals. We only included articles that were published in the English language. The authors independently reviewed the titles and abstracts of all the selected studies. Other COVID-19-related publications outside the scope of this study, as well as related commentaries, editorials, reviews, systematic reviews, and meta-analyses, were excluded.
Figure 1 ROSES flowchart illustrating the systematic search, identification, screening, and final selection of articles.
For each selected study, we extracted information about the NCBI accession numbers to the 16S rRNA gene/transcript sequences, author names, patient characteristics, sample types, sample size, the 16S rRNA gene region sequenced, and the sequencing platform used. Studies were grouped based on the systems examined: gut microbiome, or oral, nasopharyngeal, and upper respiratory tract (URT) microbiomes (heretofore oral/URT).
Bioinformatics and statistical analyses
All sequences were downloaded from NCBI as.fastq files and sequence data were processed using R (v 4.1) software (Bunn, 2008) and the dada2 (Callahan et al., 2016) package. Following preliminary assessments of quality, we downloaded data for 1,588 samples from 11 studies. First, forward reads from each study were inspected to determine the optimal processing parameters using the plotQualityProfile function (detailed for each study in Supplementary Table 1), and trimmed to 100 base pairs with the filterAndTrim function, with maxEE = 2 and truncQ = 2. Reads were assigned a taxonomy using SILVA V.132 (Quast et al., 2013). The proportion of reads lost at each processing step for each study is shown in Supplementary Figure S1. According to available metadata, technical controls (e.g., blanks, mock communities) were removed prior to further processing.
Statistical analyses were performed with the phyloseq (McMurdie and Holmes, 2013), vegan (Oksanen et al., 2022), Maaslin2 (Mallick et al., 2021), and lmerTest (Kuznetsova et al., 2017) packages. Prior to analyses, all samples were standardized to 2,000 reads per sample using the rarefy_even_depth function, which led to a loss of 63 samples. To assess the effects of COVID-19 infection on different compartments of microbial diversity (i.e., rare and dominant), we calculated Hill numbers (richness, effective Shannon diversity, and inverse Simpson diversity, or q = 0, q = 1, and q = 2, respectively; Chao et al., 2014). Richness is more heavily affected by the diversity of rare taxa, while inverse Simpson diversity is more affected by the diversity of dominant taxa. To determine the contribution of COVID-19 infection to oral/URT and gut microbiomes, we performed a distance-based variance partitioning analysis using the varpart function of vegan, with Bray–Curtis dissimilarities. The Bray–Curtis dissimilarities between samples taken from the same region among COVID-19-infected and uninfected patients were used to measure microbiome variance. Unless otherwise noted, diversity measures are presented as mean ± standard deviation.
To test the effect of COVID-19 infection on microbiome diversity (i.e., Hill numbers, H1) and variability (i.e., Bray–Curtis dissimilarities, H2), we used linear mixed effect models, with the study as a random effect, and COVID-19 infection as a fixed effect using the lmer function from the lmerTest package (Kuznetsova et al., 2017). To compare the effect of SARS-CoV-2 infection on gut and oral/URT microbiomes (H3), we used linear mixed effect models, with the study as a random effect, and the interaction between COVID-19 status and the sampled region (i.e., gut vs. oral/URT) as a fixed effect. In addition, a contrast analysis was performed using the emmeans function from the emmeans package (Lenth et al., 2023) to quantify the effect of COVID-19 infection within sampled regions. Model assumptions and performances were tested using the performance package (Lüdecke et al., 2021); all model outputs and performances are found in the Supplementary Materials.
To identify bacterial genera that were consistently under- or overrepresented in SARS-CoV-2-infected patients, we used microbiome-oriented linear models (MaAsLin2 package; Mallick et al., 2021) for gut and oral/URT samples separately, with the study and sample type as random effects, SARS-CoV-2 infection as a fixed effect, and a prevalence threshold of 0.2.
Results
In total, we collected 1,426 high-quality, processed samples from the USA (Chicago, Jackson, Nashville, New York City, Philadelphia, and San Diego), Spain (Alicante), France (Paris), and China (Guangdong, Shanghai, and Wuhan) (Supplementary Figure S2), represented by 29,491 amplicon sequence variants, or ASVs (Supplementary Table 1). Of the 11 studies surveyed, 8 included COVID-19-infected patients and controls (Engen et al., 2021; Merenstein et al., 2021; Minich et al., 2021; Newsome et al., 2021; Shilts et al., 2021; Smith et al., 2021; Ventero et al., 2021; Wu et al., 2021), and 2 of these (Newsome et al., 2021; Wu et al., 2021) also included samples of recovered patients. Three of the studies only sampled infected patients (Shilts et al., 2021; Xu et al., 2021; Ventero et al., 2022), and one sampled infected and recovered patients (Tian et al., 2021).
On average, gut microbiome samples were more diverse (136 ± 54 ASVs) than oral/URT samples (80 ± 79 ASVs). Within studies, COVID-19 infection caused minor, but consistent decreases in gut microbial richness (Hill q = 0, estimate ± SE = 22.46 ± 6.22, p < 0.001), but not in oral/URT richness (p = 0.55, Figure 2; Supplementary Figure S3). This decrease was also significant for q = 1 and q = 2 (q = 1: 8.20 ± 2.50, p = 0.001; q = 2: 3.71 ± 1.38, p = 0.008, Figure 2), highlighting richness losses in the dominant portion of the community.
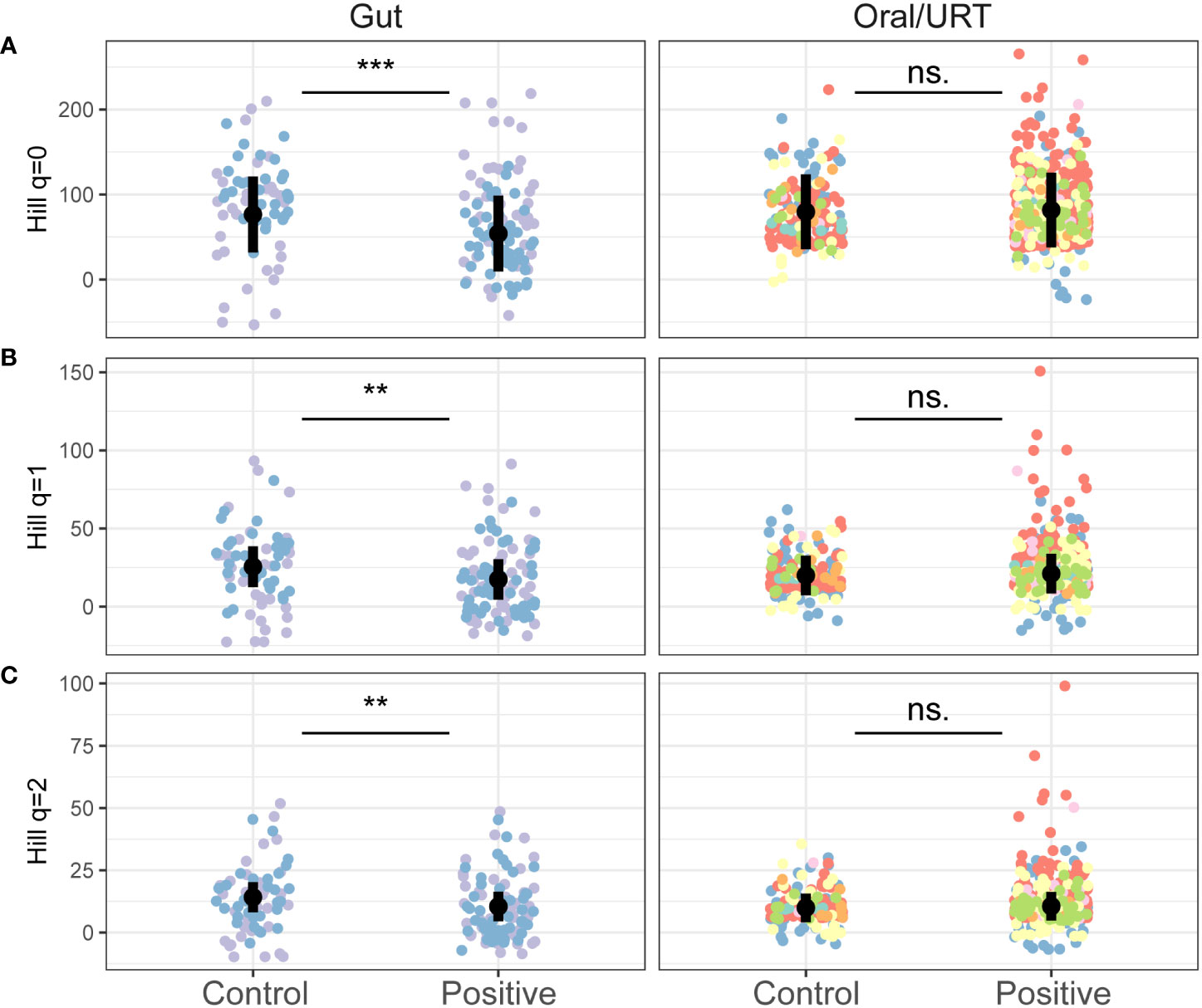
Figure 2 The effect of COVID-19 infection on the alpha diversity of the gut and oral/URT microbiomes. Hill richness (A: q = 0), effective Shannon (B: q = 1) and Inverse Simpson (C: q = 2) diversity indices were calculated to assess the impact of increasingly dominant portions of the community. Points are colored by studies, and the average across studies is shown with a black point. Significant differences between infected and non-infected patients across studies are indicated with asterisks where significant (***p < 0.001; ** p < 0.01) and with “n.s.” otherwise.
SARS-CoV-2 infection led to changes in the microbiome composition, significantly explaining 2% of the variation in the microbial community, although these changes were study-dependent (p < 0.001, Supplementary Figure S4). Notably, of the 14.9% of the variance in community composition explained by each study, 7.3% could be ascribed to the participant’s country of origin (Supplementary Figure S4). In line with the AKP, patients infected with COVID-19 had a higher host-to-host variance in both the oral/URT and gut microbiome than uninfected patients (an increase of 0.06 ± 0.003 in Bray–Curtis dissimilarity relative to uninfected patients, p < 0.001, Figure 3).
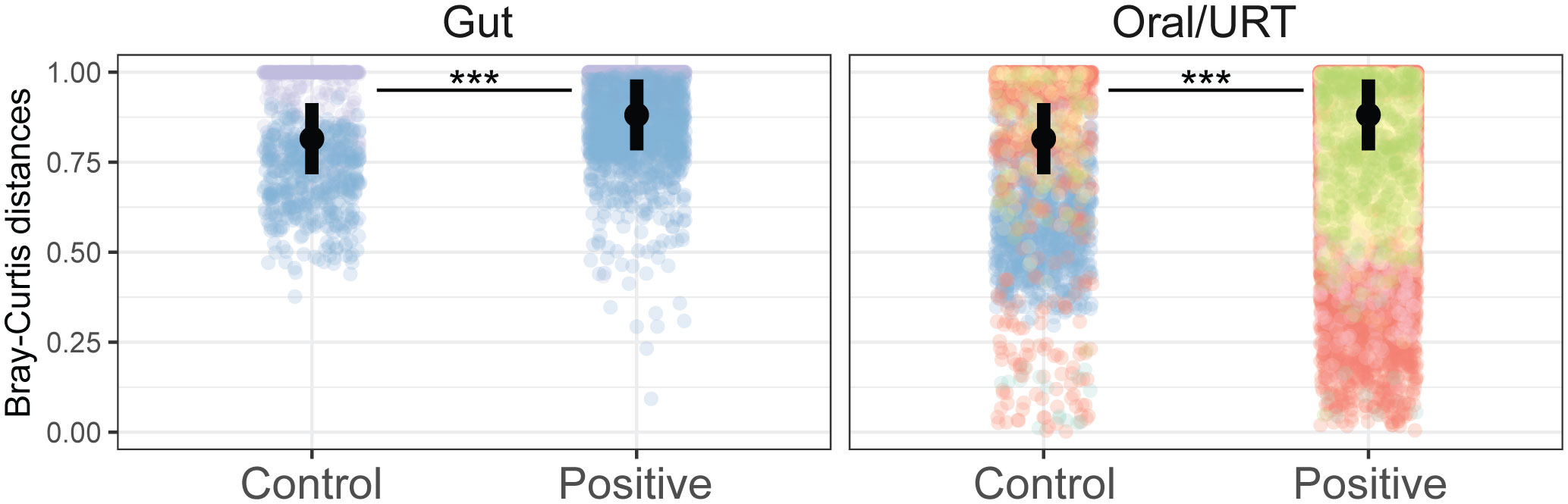
Figure 3 COVID-19 increases host-to-host variability across microbiomes. Community distances were measured as the Bray–Curtis distance between individuals in the same study, from the same body site, and with the same infection status. Points are colored by studies, and the average across studies is shown with a black point. Significant differences between infected and non-infected patients across studies are indicated with asterisks where significant (***p < 0.001).
In general, the gut microbiomes of infected and non-infected patients had more consistent shifts across studies for the gut than for the oral/URT microbiomes. We identified 51 dominant genera (38.3% ± 22.2% of the community) in the gut microbiome, whose relative abundances consistently and significantly differed between infected and non-infected patients across studies (Figure 4). These taxa belonged predominantly to Firmicutes, with the most consistent decreases in Fusicatenibacter, Lachnospiraceae NK4A316 group, Lachnoclostridum, Blautia, and Roseburia and the most consistent increases in Finegoldia, Porphyromonas, Anaerococcus, and Peptoniphilus for COVID-19-infected patients, relative to uninfected patients. In contrast, we only found 16 genera (23.8% ± 25.9% of the community on average) that consistently differed between the oral/URT microbiome of COVID-19-infected and uninfected patients (Figure 5). Of these, Enterococcus, Pseudomonas, unclassified Enterobacteriaceae, and Solobacterium were lower in uninfected patients, whereas Prevotella, Mycoplasma, Veillonella, Cutibacterium, Atopobium, and Megasphaera were consistently and significantly more abundant in COVID-19-infected patients.
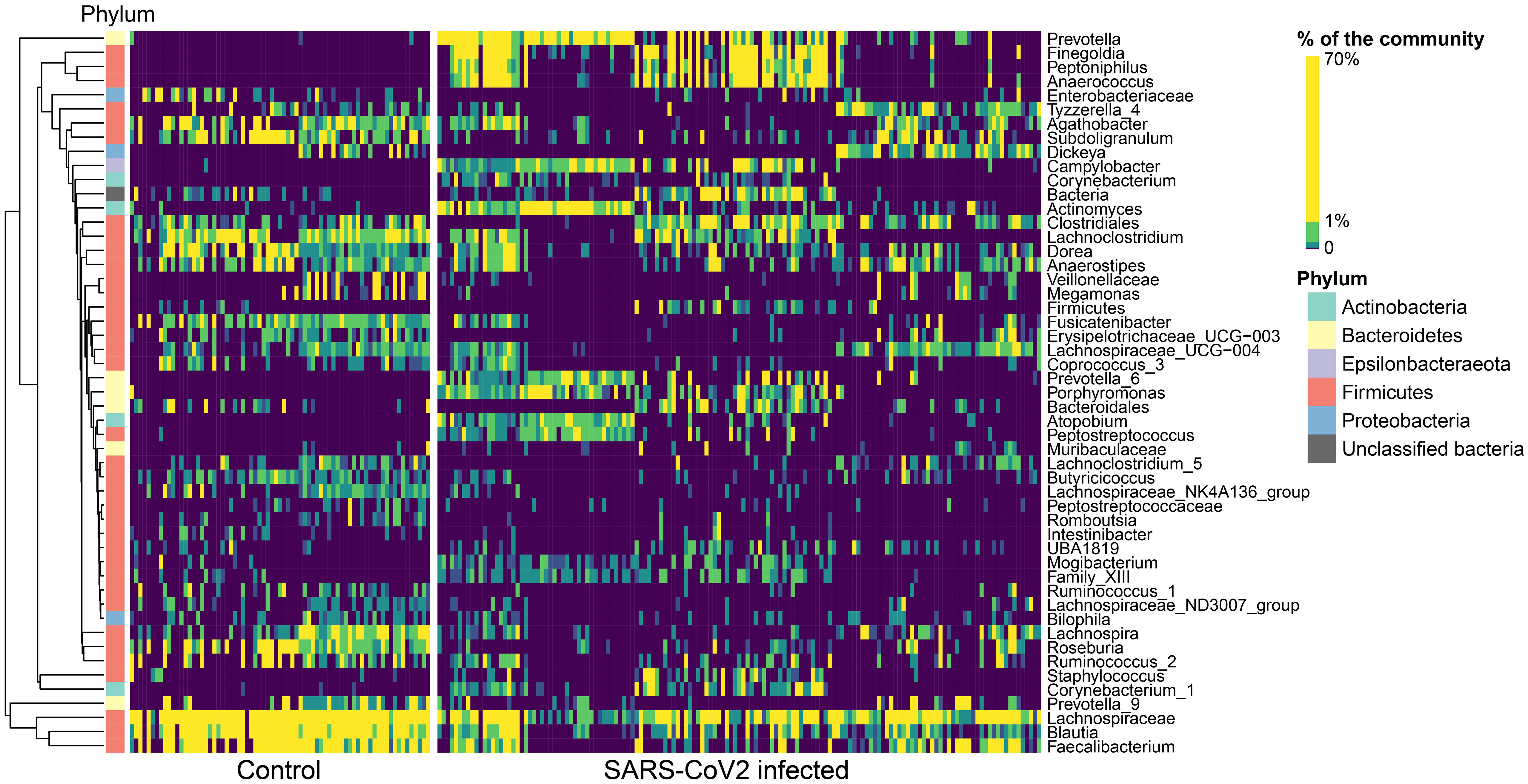
Figure 4 Altered relative abundances of bacterial genera in the gut microbiome of SARS-CoV-2-infected individuals. Bacterial genera that exhibit significantly different (p < 0.01) relative abundances between SARS-CoV- 2-infected and uninfected individuals in the gut microbiome were selected using the MaAsLin2 approach, which included random effects for sample type and study. Only significantly different genera are displayed, relative abundances are colored by quantiles, and genera are grouped according to Ward’s clustering method. Phylum membership is displayed on the left bar. These 51 genera make up 38.3% ± 22.2% of the community, on average across all samples.
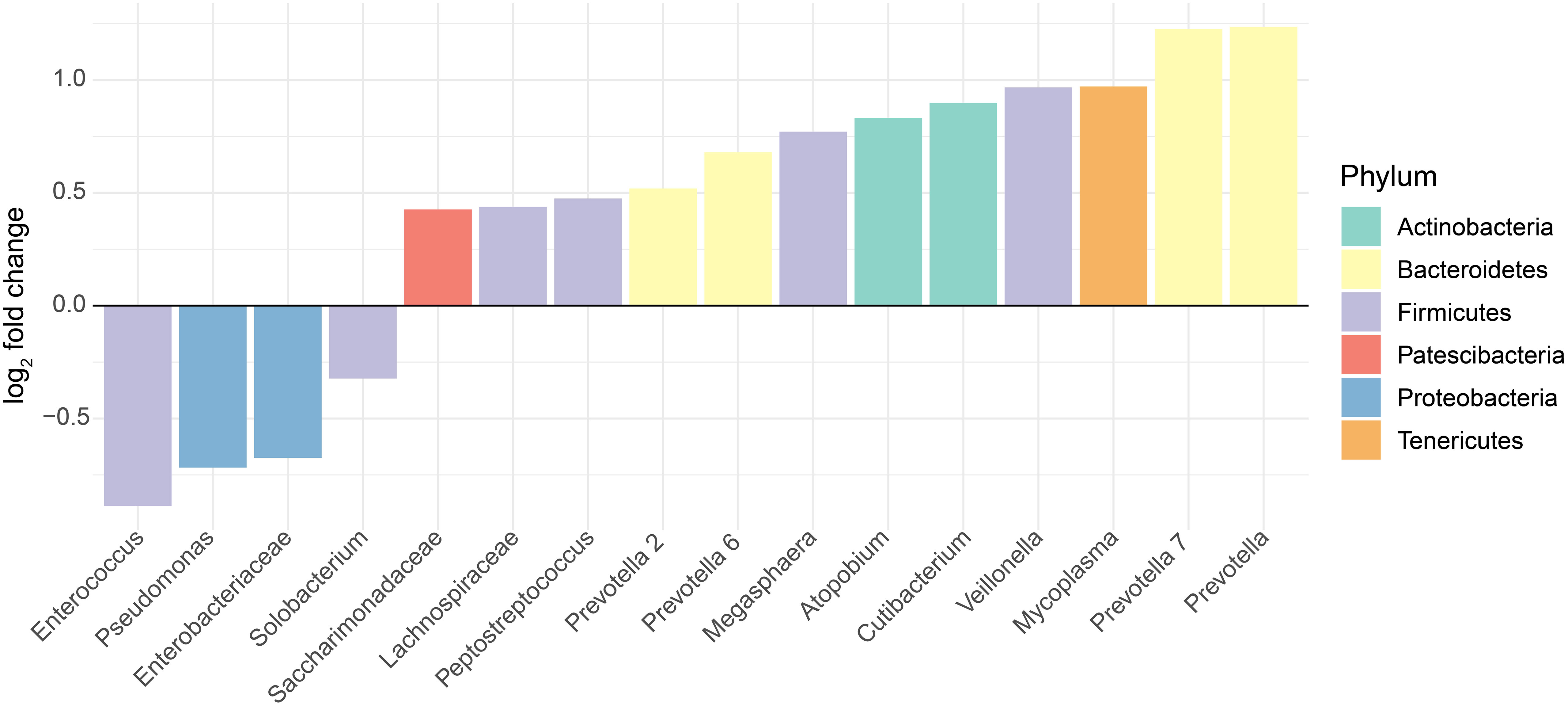
Figure 5 Altered relative abundances of bacterial genera in the oral and URT microbiome of SARS-CoV-2-infected individuals. Bacterial genera that exhibit significantly different (p < 0.01) relative abundances between SARS-CoV-2-infected and uninfected individuals in the oral or URT microbiome were selected using the MaAsLin2 approach, which included random effects for sample type and study. The log2 fold changes in taxon abundances for infected patients relative to non-infected patients are displayed. Only genera with significantly different (p < 0.01) abundances between these two groups are displayed, and in total, they represent 23.8% ± 25.9% of the whole community, on average across all samples.
Discussion
SARS-CoV-2 invades the human body mainly through the angiotensin-converting enzyme 2 (ACE2) and cofactor transmembrane serine protease 2 (TMPRSS2) receptors in the epithelial cells of the nasopharyngeal tract, and then gradually moves to initiate infection in the lungs, which gradually results in gastrointestinal involvement as well as affects other organs including the heart, kidneys, pancreas, eyes, and skin (Gavriatopoulou et al., 2020; Hoque et al., 2021a; Rahman et al., 2021; Rahman et al., 2021). Interestingly, high levels of both ACE2 and TMPRSS2 receptors are naturally expressed in multiple organs of the human respiratory and gastrointestinal tracts (Perlot & Penninger, 2013; Roncon et al., 2020; Xiao et al., 2020), thus enabling SARS-CoV-2 to circulate and induce severe inflammation, immune imbalance, and microbiome dysbiosis within these systems (Hoffmann et al., 2020; Villapol, 2020). Moreover, recent insights into coronavirus biology and SARS-CoV-2-human interactions attribute COVID-19 pathophysiology to aberrant and aggressive immune responses in SARS-CoV-2 clearance (Hoque et al., 2021b; Yeoh et al., 2021). COVID-19 infection can therefore result in a wide variety of responses in the human-associated microbiome, which may be further modulated by the host’s environment, such as diet and exposure to pollutants, which is population-specific. By simultaneously reanalyzing microbiome data from various microbiome compartments in infected and uninfected patients across the world, we sought to identify consistent, COVID-19 infection-specific changes in the human microbiome.
Consistent with our hypotheses, we found that SARS-CoV-2-infected individuals had a lower microbial diversity in the gut, but not in the upper respiratory tract. The reduction of gut microbial diversity in COVID-19 has been similarly reported (Zuo et al., 2020; Mazzarelli et al., 2021; Tian et al., 2021), regardless of antibiotic use (Zuo et al., 2020; Mazzarelli et al., 2021) and even several weeks after viral clearance (Zuo et al., 2020; Tian et al., 2021; Xu et al., 2021). The gut microbiome is relatively stable, and a major predictor of normal gut functioning, immunomodulation, and overall host health (Rooks and Garrett, 2016; Tian et al., 2021; Xu et al., 2021). Our study highlights SARS-CoV-2’s ability to disrupt human gut microbiome eubiosis through the depletion of gut microbial diversity, which may contribute to disease severity and opportunistic infections. The consistent decrease in diversity found across Hill numbers for the gut microbiome richness highlights that richness loss occurs in dominant taxa. This may have major implications on the composition and diversity of the gut microbiome in COVID-19 infection resulting in dysbiosis, impaired immune functioning, pro-inflammatory conditions, etc.
Our study did not control for COVID-19 patients’ medication use (e.g., antibiotics and antivirals), age, genetic background, sex, or diet, which may also affect the gut microbial diversity and further confound COVID-19-associated gut microbial signatures. However, the consistent results we recorded across studies after controlling for study–study particularities suggest that the decrease in gut microbial diversity is indeed due to SARS-CoV-2 infection. Intriguingly, the intestinal ACE2, which is the receptor of SARS-CoV-2, plays a vital role in maintaining the gut microbiome eubiosis (Hamming et al., 2004; Hashimoto et al., 2012; Tian et al., 2021), and the SARS-CoV-2 infection may downregulate the expression and availability of ACE2, which could disrupt gut homeostasis, adversely impacting microbial diversity. Our inability to detect a significant effect on the microbial richness of the oral/URT microbiome may be attributed to the oral/URT microbiome being more dynamic, resilient, and transient than the gut microbiome due to frequent bidirectional air and mucus movement as well as its regular exposure to the environment (Huffnagle et al., 2017). Recent studies have found inconsistent effects of COVID-19 on the microbial diversity of the URT (De Maio et al., 2020; Mostafa et al., 2020; Braun et al., 2021; Miao et al., 2021; Yamamoto et al., 2021; Zhang et al., 2021; Rafiqul Islam et al., 2022; Uehara et al., 2022), and others have proposed that SARS-CoV-2 has weak effects on the URT microbiome (De Maio et al., 2020; Braun et al., 2021; Yamamoto et al., 2021) akin to acute respiratory virus infections in humans (Ramos-Sevillano et al., 2019; De Maio et al., 2020; Yamamoto et al., 2021).
Higher variability is generally associated with lower stability and predictability. In accordance with previous studies (Ma, 2020; Altabtbaei et al., 2021), we also found that host-to-host gut and URT microbiome variability was greater in SARS-CoV-2-infected patients than in non-infected controls, in line with the AKP (Zaneveld et al., 2017) and with a previous synthesis, which found that most human-associated diseases result in a higher microbiome variability across patients (Ma, 2020).
Notably, our study shows that COVID-19 infection resulted in an overall loss of beneficial bacteria, and worryingly, a consistent increase in pathogenic bacteria, particularly in the oral/URT microbiome. The increased relative abundances and colonization of opportunistic pathogens including Mycoplasma, Prevotella, Peptostreptococcus, Veillonella, Cutibacterium, and Saccharibacteria in the oral/URT recorded in our study may be associated with the early-stage SARS-CoV-2-induced inflammation, the loss of beneficial bacteria, and the increased exposure and receptiveness to allochthonous and indigenous microorganisms (Man et al., 2017; Dubourg et al., 2019).
In the gut microbiome, increased relative abundances of pathogenic bacteria including Campylobacter, Corynebacterium, Staphylococcus, Clostridium, Peptostreptococcus, Prevotella, Anaerococcus, Actinomyces, Porphyromonas, and Bacteroides were recorded in SARS-CoV-2 infection. Increasingly, emerging reports posit that alterations in the gut microbiome may facilitate blooms of both pathogenic and previously rare bacteria, which can further aggravate overall gut inflammation (Mazzarelli et al., 2021; Tian et al., 2021; Xu et al., 2021). The presence and increased abundance of common oral/URT commensals and pathogens in the gut (e.g., Corynebacterium, Peptostreptococcus, Porphyromonas, Prevotella, and Staphylococcus) may suggest a possible translocation of these organisms from the oral/URT to the gut. Previously, inflammation, disruption, and increased permeability of membrane mucosa were associated with COVID-19 (Cao and Li, 2020). Increased permeability of membrane mucosa facilitates the translocation of some oral/URT microbes as well as enriched opportunistic pathogens to the gut (Man et al., 2017; Cao and Li, 2020; Giron et al., 2021).
We also detected the loss of beneficial microbes Fusicatenibacter, Lachnospiraceae NK4A316 group, Lachnoclostridium, Blautia, and Roseburia in the gut. These beneficial bacteria often enhance and maintain the integrity and function of mucosal barriers, metabolism, and immunomodulation, and protect against pathogen invasion through several mechanisms including the secretion of short-chain fatty acids (SCFA) and antimicrobial peptides (Gallo and Hooper, 2012; Abt & Pamer, 2014; Zhang et al., 2015). In line with our findings, SARS-CoV-2-associated microbiome perturbations were previously associated with a decline in SCFA (Lv et al., 2021; Sokol et al., 2021), thus promoting a systemic pro-inflammatory condition (Qin et al., 2015; Esquivel-Elizondo et al., 2017) and the severity of pulmonary viral infections such as COVID-19 (Chemudupati et al., 2020; Friedland and Haribabu, 2020; Tang et al., 2020). In the same vein, Lv and colleagues reported a pathogen-regulated feedback loop between the decline in SCFA production and SARS-CoV-2 infection (Lv et al., 2021).
Whether increased abundances of gut and oral/URT pathogenic and pro-inflammatory bacteria in SARS-CoV-2 infection actually play an active part in COVID-19 or mainly thrive opportunistically, exploiting the depletion of commensal bacteria, remains unknown. Nevertheless, our findings demonstrate that oral/URT and gut microbiomes are systematically perturbed by COVID-19, resulting in a lower microbial diversity, loss of beneficial microbes, and increased presence of pathogenic bacteria, which could trigger prolonged pro-inflammatory reactions, immunological changes, and secondary bacterial infections that could account for chronic COVID-19-associated symptoms, as well as prolonged sequelae. Understanding the dynamics of COVID-19-associated microbiome alterations may help identify microbiome-based strategies with potential applications in COVID-19 management and treatment. Furthermore, our findings highlight that non-invasive organ and/or system-based microbiome profiling may serve not only for COVID-19 diagnosis and prognosis but also, for the identification of individuals at risk of secondary infections, chronic disease, and/or degenerative inflammatory symptoms, including Kawasaki-like disease (KLD) and multisystem inflammation, as is the case with children and young adults (Akca et al., 2020; Cheung et al., 2020; Sokolovsky et al., 2021; Elouardi et al., 2022).
Finally, human microbiome composition and diversity are highly heterogeneous and largely driven by biogeographies, environments, ethnicity, and socioeconomic status (Amato et al., 2021; Yamamoto et al., 2021; Yeoh et al., 2021). While our study design highlights the importance of sampling across human populations to understand a disease, our study lacks samples from the Southern Hemisphere, in line with recent reports indicating very limited public human microbiome data from the Global South (Abdill et al., 2022). A global representation of data in human microbiome studies is critical to understanding global drivers and patterns of disease (in this case, COVID-19) to provide sustainable interventions to all populations without bias.
Data availability statement
INSDC accession numbers for each dataset reused in this study are included in Supplementary Table 1. Code used for data analysis is available in https://github.com/drcarrot/COVID, and detailed model descriptions are included in the Supplementary Material.
Author contributions
RR conceptualized the study, carried out the literature search, reviewed and selected relevant literature, and wrote the article. RB performed the statistical analyses. SJ conceptualized the study, extracted and assembled the 16S rRNA data and metadata, organized the presentation of the results, and created all figures. All authors contributed substantially to subsequent revisions. All authors approved the submitted version.
Acknowledgments
RR was funded by the Alexander von Humboldt Foundation, Germany. RB acknowledges support by the Saxon State Ministry for Science, Culture and Tourism (SMWK), Germany – [3-7304/35/6-2021/48880]. SJ acknowledges support of sDiv, the Synthesis Center of the German Centre for Integrative Biodiversity Research (iDiv) Halle-Jena-Leipzig, funded by the German Research Foundation (DFG– FZT 118, 202548816). We acknowledge support by the iDiv Open Science Publication Fund as well as the Open Access Publishing Fund of Leipzig University, supported by the German Research Foundation within the program Open Access Publication Funding.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2023.1211348/full#supplementary-material
References
Abdill, R. J., Adamowicz, E. M., Blekhman, R. (2022). Public human microbiome data are dominated by highly developed countries. PLoS Biol. 20(2), e3001536. doi: 10.1371/journal.pbio.3001536
Abt, M. C., Pamer, E. G. (2014). Commensal bacteria mediated defenses against pathogens. Curr. Opin. Immunol. 29, 16–22. doi: 10.1016/j.coi.2014.03.003
Akca, U. K., Kesici, S., Ozsurekci, Y., Aykan, H. H., Batu, E. D., Atalay, E., et al. (2020). Kawasaki-like disease in children with COVID-19. Rheumatol. Int. 40(12), 2105–2115. doi: 10.1007/s00296-020-04701-6
Altabtbaei, K., Maney, P., Ganesan, S. M., Dabdoub, S. M., Nagaraja, H. N., Kumar, P. S. (2021). Anna Karenina and the subgingival microbiome associated with periodontitis. Microbiome 9 (1), 97. doi: 10.1186/s40168-021-01056-3
Amato, K. R., Arrieta, M.-C., Azad, M. B., Bailey, M. T., Broussard, J. L., Bruggeling, C. E., et al. (2021). The human gut microbiome and health inequities. Proc. Natl. Acad. Sci. U.S.A. 118 (25), 1–10. doi: 10.1073/pnas.2017947118
Attaway, A. H., Scheraga, R. G., Bhimraj, A., Biehl, M., Hatipoğlu, U. (2021). Severe Covid-19 pneumonia: pathogenesis and clinical management. BMJ (Clinical Res. Ed.) 372, n436. doi: 10.1136/bmj.n436
Baquero, F., Nombela, C. (2012). The microbiome as a human organ. Clin. Microbiol. Infection 18 Suppl 4, 2–4. doi: 10.1111/j.1469-0691.2012.03916.x
Baud, D., Qi, X., Nielsen-Saines, K., Musso, D., Pomar, L., Favre, G. (2020). Real estimates of mortality following COVID-19 infection. Lancet Infect. Dis. 20 (7), 773. doi: 10.1016/S1473-3099(20)30195-X
Braun, T., Halevi, S., Hadar, R., Efroni, G., Glick Saar, E., Keller, N., et al. (2021). SARS-CoV-2 does not have a strong effect on the nasopharyngeal microbial composition. Sci. Rep. 11 (1), 8922. doi: 10.1038/s41598-021-88536-6
Brooks, A. W., Priya, S., Blekhman, R., Bordenstein, S. R. (2018). Gut microbiota diversity across ethnicities in the United States. PLoS Biol. 16 (12), e2006842. doi: 10.1371/journal.pbio.2006842
Bunn, A. G. (2008). A dendrochronology program library in R (dplR). Dendrochronologia 26 (2), 115–124. doi: 10.1016/j.dendro.2008.01.002
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., Holmes, S. P. (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13 (7), 581–583. doi: 10.1038/nmeth.3869
Cao, W., Li, T. (2020). COVID-19: towards understanding of pathogenesis. Cell Res. 30 (5), 367–369. doi: 10.1038/s41422-020-0327-4
Chao, A., Chiu, C.-H., Jost, L. (2014). Unifying species diversity, phylogenetic diversity, functional diversity, and related similarity and differentiation measures through hill numbers. Annu. Rev. Ecol. Evol. Syst. 45 (1), 297–324. doi: 10.1146/annurev-ecolsys-120213-091540
Chemudupati, M., Kenney, A. D., Smith, A. C., Fillinger, R. J., Zhang, L., Zani, A., et al. (2020). Butyrate reprograms expression of specific interferon-stimulated genes. J. Virol. 94 (16), 1–13. doi: 10.1128/JVI.00326-20
Chen, Y., Liu, Q., Guo, D. (2020). Emerging coronaviruses: genome structure, replication, and pathogenesis. J. Med. Virol. 92 (4), 418–423. doi: 10.1002/jmv.25681
Cheung, E. W., Zachariah, P., Gorelik, M., Boneparth, A., Kernie, S. G., Orange, J. S., et al. (2020). Multisystem inflammatory syndrome related to COVID-19 in previously healthy children and adolescents in New York City. J. Am. Med. Assoc. 324 (3), 294–296. doi: 10.1001/jama.2020.10374
De Maio, F., Posteraro, B., Ponziani, F. R., Cattani, P., Gasbarrini, A., Sanguinetti, M. (2020). Nasopharyngeal microbiota profiling of SARS-CoV-2 infected patients. Biol. Procedures Online 22, 18. doi: 10.1186/s12575-020-00131-7
Dubourg, G., Edouard, S., Raoult, D. (2019). Relationship between nasopharyngeal microbiota and patient’s susceptibility to viral infection. Expert Rev. Anti-Infective Ther. 17 (6), 437–447. doi: 10.1080/14787210.2019.1621168
Elouardi, Y., Rebahi, H., Zarrouki, Y., Ziadi, A., Younous, S., Samkaoui, M. A. (2022). COVID-19 associated Kawasaki-like multisystem inflammatory syndrome in an adult. Rev. Espanola Anestesiologia Y Reanimacion 69 (1), 43–47. doi: 10.1016/j.redare.2020.11.009
Engen, P. A., Naqib, A., Jennings, C., Green, S. J., Landay, A., Keshavarzian, A., et al. (2021). Nasopharyngeal microbiota in SARS-CoV-2 positive and negative patients. Biol. Procedures Online 23 (1), 10. doi: 10.1186/s12575-021-00148-6
Esquivel-Elizondo, S., Ilhan, Z. E., Garcia-Peña, E. I., Krajmalnik-Brown, R. (2017). Insights into Butyrate Production in a Controlled Fermentation System via Gene Predictions. MSystems 2 (4), 1–13. doi: 10.1128/mSystems.00051-17
Friedland, R. P., Haribabu, B. (2020). The role for the metagenome in the pathogenesis of COVID-19. EBioMedicine 61, 103019. doi: 10.1016/j.ebiom.2020.103019
Gallo, R. L., Hooper, L. V. (2012). Epithelial antimicrobial defence of the skin and intestine. Nat. Rev. Immunol. 12 (7), 503–516. doi: 10.1038/nri3228
Gavriatopoulou, M., Korompoki, E., Fotiou, D., Ntanasis-Stathopoulos, I., Psaltopoulou, T., Kastritis, E., et al. (2020). Organ-specific manifestations of COVID-19 infection. Clin. Exp. Med. 20 (4), 493–506. doi: 10.1007/s10238-020-00648-x
Gilbert, J. A., Blaser, M. J., Caporaso, J. G., Jansson, J. K., Lynch, S. V., Knight, R. (2018). Current understanding of the human microbiome. Nat. Med. 24 (4), 392–400. doi: 10.1038/nm.4517
Giron, L. B., Dweep, H., Yin, X., Wang, H., Damra, M., Goldman, A. R., et al. (2021). Plasma markers of disrupted gut permeability in severe COVID-19 patients. Front. Immunol. 12. doi: 10.3389/fimmu.2021.686240
Gonçalves, L. S., Ferreira, D., de, C., Heng, N. C. K., Vidal, F., Santos, H. F., et al. (2019). Oral bacteriome of HIV-1-infected children from Rio de Janeiro, Brazil: Next-generation DNA sequencing analysis. J. Clin. Periodontol. 46 (12), 1192–1204. doi: 10.1111/jcpe.13176
Gou, W., Fu, Y., Yue, L., Chen, G., Cai, X., Shuai, M., et al. (2020). Gut microbiota may underlie the predisposition of healthy individuals to COVID-19. MedRxiv. 1–44. doi: 10.1101/2020.04.22.20076091
Grasselli, G., Zangrillo, A., Zanella, A., Antonelli, M., Cabrini, L., Castelli, A., et al. (2020). Baseline characteristics and outcomes of 1591 patients infected with SARS-CoV-2 admitted to ICUs of the Lombardy region, Italy. J. Am. Med. Assoc. 323 (16), 1574–1581. doi: 10.1001/jama.2020.5394
Haddaway, N., Macura, B., Whaley, P., Pullin, A. (2018). ROSES Flow Diagram for Systematic Reviews. Version 1.0. Figshare. doi: 10.6084/m9.figshare.5897389.v3
Hamming, I., Timens, W., Bulthuis, M. L. C., Lely, A. T., Navis, G. J., van Goor, H. (2004). Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 203 (2), 631–637. doi: 10.1002/path.1570
Hashimoto, T., Perlot, T., Rehman, A., Trichereau, J., Ishiguro, H., Paolino, M., et al. (2012). ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature 487 (7408), 477–481. doi: 10.1038/nature11228
He, X., Lau, E. H. Y., Wu, P., Deng, X., Wang, J., Hao, X., et al. (2020). Temporal dynamics in viral shedding and transmissibility of COVID-19. Nat. Med. 26 (5), 672–675. doi: 10.1038/s41591-020-0869-5
Hoffmann, M., Kleine-Weber, H., Schroeder, S., Krüger, N., Herrler, T., Erichsen, S., et al. (2020). SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181 (2), 271–280.e8. doi: 10.1016/j.cell.2020.02.052
Hoque, M. N., Akter, S., Mishu, I. D., Islam, M. R., Rahman, M. S., Akhter, M., et al. (2021a). Microbial co-infections in COVID-19: Associated microbiota and underlying mechanisms of pathogenesis. Microbial Pathogenesis 156, 104941. doi: 10.1016/j.micpath.2021.104941
Hoque, M. N., Sarkar, M. M. H., Rahman, M. S., Akter, S., Banu, T. A., Goswami, B., et al. (2021b). SARS-CoV-2 infection reduces human nasopharyngeal commensal microbiome with inclusion of pathobionts. Sci. Rep. 11 (1), 24042. doi: 10.1038/s41598-021-03245-4
Huffnagle, G. B., Dickson, R. P., Lukacs, N. W. (2017). The respiratory tract microbiome and lung inflammation: a two-way street. Mucosal Immunol. 10 (2), 299–306. doi: 10.1038/mi.2016.108
Human Microbiome Project Consortium (2012). Structure, function and diversity of the healthy human microbiome. Nature 486 (7402), 207–214. doi: 10.1038/nature11234
Jandhyala, S. M., Talukdar, R., Subramanyam, C., Vuyyuru, H., Sasikala, M., Nageshwar Reddy, D. (2015). Role of the normal gut microbiota. World J. Gastroenterol. 21 (29), 8787–8803. doi: 10.3748/wjg.v21.i29.8787
Jochems, S. P., Ferreira, D. M., Smits, H. H. (2021). Microbiota and compartment matter in the COVID-19 response. Nat. Immunol. 22 (11), 1350–1352. doi: 10.1038/s41590-021-01041-w
Kumar Singh, A., Cabral, C., Kumar, R., Ganguly, R., Kumar Rana, H., Gupta, A., et al. (2019). Beneficial effects of dietary polyphenols on gut microbiota and strategies to improve delivery efficiency. Nutrients 11 (9), 2216. doi: 10.3390/nu11092216
Kumpitsch, C., Koskinen, K., Schöpf, V., Moissl-Eichinger, C. (2019). The microbiome of the upper respiratory tract in health and disease. BMC Biol. 17 (1), 87. doi: 10.1186/s12915-019-0703-z
Kuznetsova, A., Brockhoff, P. B., Christensen, R. H. B. (2017). lmertest package: tests in linear mixed effects models. J. Stat. Software 82 (13), 1–26. doi: 10.18637/jss.v082.i13
Lenth, R. V., Bolker, B., Buerkner, P., Giné-Vázquez, I., Herve, M., Jung, M., et al. (2023). Emmeans: estimated marginal means, aka least-squares means. Version 1.8.7. Available at: https://github.com/rvlenth/emmeans.
Ling, Z., Liu, X., Cheng, Y., Jiang, X., Jiang, H., Wang, Y., et al. (2015). Decreased diversity of the oral microbiota of patients with hepatitis B virus-induced chronic liver disease: A pilot project. Sci. Rep. 5, 17098. doi: 10.1038/srep17098
Lu, H.-F., Li, A., Zhang, T., Ren, Z.-G., He, K.-X., Zhang, H., et al. (2017). Disordered oropharyngeal microbial communities in H7N9 patients with or without secondary bacterial lung infection. Emerg. Microbes Infect. 6 (12), e112. doi: 10.1038/emi.2017.101
Lüdecke, D., Ben-Shachar, M., Patil, I., Waggoner, P., Makowski, D. (2021). performance: an R package for assessment, comparison and testing of statistical models. J. Open Source Software 6 (60), 3139. doi: 10.21105/joss.03139
Lv, L., Gu, S., Jiang, H., Yan, R., Chen, Y., Chen, Y., et al. (2021). Gut mycobiota alterations in patients with COVID-19 and H1N1 infections and their associations with clinical features. Commun. Biol. 4 (1), 480. doi: 10.1038/s42003-021-02036-x
Ma, Z. S. (2020). Testing the anna karenina principle in human microbiome-associated diseases. IScience 23 (4), 101007. doi: 10.1016/j.isci.2020.101007
Mallick, H., Rahnavard, A., McIver, L. J., Ma, S., Zhang, Y., Nguyen, L. H., et al. (2021). Multivariable association discovery in population-scale meta-omics studies. PLoS Comput. Biol. 17 (11), e1009442. doi: 10.1371/journal.pcbi.1009442
Man, W. H., de Steenhuijsen Piters, W. A. A., Bogaert, D. (2017). The microbiota of the respiratory tract: gatekeeper to respiratory health. Nat. Rev. Microbiol. 15 (5), 259–270. doi: 10.1038/nrmicro.2017.14
Mangalmurti, N., Hunter, C. A. (2020). Cytokine storms: understanding COVID-19. Immunity 53 (1), 19–25. doi: 10.1016/j.immuni.2020.06.017
Mathieu, E., Escribano-Vazquez, U., Descamps, D., Cherbuy, C., Langella, P., Riffault, S., et al. (2018). Paradigms of lung microbiota functions in health and disease, particularly, in asthma. Front. Physiol. 9. doi: 10.3389/fphys.2018.01168
Mazzarelli, A., Giancola, M. L., Farina, A., Marchioni, L., Rueca, M., Gruber, C. E. M., et al. (2021). 16S rRNA gene sequencing of rectal swab in patients affected by COVID-19. PLoS One 16 (2), e0247041. doi: 10.1371/journal.pone.0247041
McMurdie, P. J., Holmes, S. (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8 (4), e61217. doi: 10.1371/journal.pone.0061217
Merenstein, C., Liang, G., Whiteside, S. A., Cobián-Güemes, A. G., Merlino, M. S., Taylor, L. J., et al. (2021). Signatures of COVID-19 severity and immune response in the respiratory tract microbiome. MBio 12 (4), e0177721. doi: 10.1128/mBio.01777-21
Miao, Q., Ma, Y., Ling, Y., Jin, W., Su, Y., Wang, Q., et al. (2021). Evaluation of superinfection, antimicrobial usage, and airway microbiome with metagenomic sequencing in COVID-19 patients: A cohort study in Shanghai. J. Microbiol. Immunol. Infect. = Wei Mian Yu Gan Ran Za Zhi 54 (5), 808–815. doi: 10.1016/j.jmii.2021.03.015
Minich, J. J., Ali, F., Marotz, C., Belda-Ferre, P., Chiang, L., Shaffer, J. P., et al. (2021). Feasibility of using alternative swabs and storage solutions for paired SARS-CoV-2 detection and microbiome analysis in the hospital environment. Microbiome 9 (1), 25. doi: 10.1186/s40168-020-00960-4
Mostafa, H. H., Fissel, J. A., Fanelli, B., Bergman, Y., Gniazdowski, V., Dadlani, M., et al. (2020). Metagenomic next-generation sequencing of nasopharyngeal specimens collected from confirmed and suspect COVID-19 patients. MBio 11 (6), 1–13. doi: 10.1128/mBio.01969-20
Newsome, R. C., Gauthier, J., Hernandez, M. C., Abraham, G. E., Robinson, T. O., Williams, H. B., et al. (2021). The gut microbiome of COVID-19 recovered patients returns to uninfected status in a minority-dominated United States cohort. Gut Microbes 13 (1), 1–15. doi: 10.1080/19490976.2021.1926840
Oksanen, J., Simpson, G. L., Blanchet, F. G., Kindt, R., Legendre, R., et al. (2022). Ordination methods, diversity analysis and other functions for community and vegetation ecologists, Version 2.6-4. Available at: https://github.com/vegandevs/vegan.
Onder, G., Rezza, G., Brusaferro, S. (2020). Case-fatality rate and characteristics of patients dying in relation to COVID-19 in Italy. J. Am. Med. Assoc. 323 (18), 1775–1776. doi: 10.1001/jama.2020.4683
Perlot, T., Penninger, J. M. (2013). ACE2 - from the renin-angiotensin system to gut microbiota and malnutrition. Microbes Infect. 15 (13), 866–873. doi: 10.1016/j.micinf.2013.08.003
Pflughoeft, K. J., Versalovic, J. (2012). Human microbiome in health and disease. Annu. Rev. Pathol. 7, 99–122. doi: 10.1146/annurev-pathol-011811-132421
Qin, N., Zheng, B., Yao, J., Guo, L., Zuo, J., Wu, L., et al. (2015). Influence of H7N9 virus infection and associated treatment on human gut microbiota. Sci. Rep. 5, 14771. doi: 10.1038/srep14771
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41 (Database issue), D590–D596. doi: 10.1093/nar/gks1219
Rafiqul Islam, S. M., Foysal, M. J., Hoque, M. N., Mehedi, H. M. H., Rob, M. A., Salauddin, A., et al. (2022). Dysbiosis of oral and gut microbiomes in SARS-CoV-2 infected patients in Bangladesh: elucidating the role of opportunistic gut microbes. Front. Med. 9. doi: 10.3389/fmed.2022.821777
Rahman, M.S., Hoque, M. N., Islam, M. R., Islam, I., Mishu, I. D., Rahaman, M. M., et al. (2021). Mutational insights into the envelope protein of SARS-CoV-2. Gene Rep. 22, 100997. doi: 10.1016/j.genrep.2020.100997
Rahman, M. S., Islam, M. R., Hoque, M. N., Alam, A. S. M. R. U., Akther, M., Puspo, J. A., et al. (2021). Comprehensive annotations of the mutational spectra of SARS-CoV-2 spike protein: a fast and accurate pipeline. Transbound. Emerg. Dis. 68 (3), 1625–1638. doi: 10.1111/tbed.13834
Ramos-Sevillano, E., Wade, W. G., Mann, A., Gilbert, A., Lambkin-Williams, R., Killingley, B., et al. (2019). The effect of influenza virus on the human oropharyngeal microbiome. Clin. Infect. Dis. 68 (12), 1993–2002. doi: 10.1093/cid/ciy821
Ren, Z., Wang, H., Cui, G., Lu, H., Wang, L., Luo, H., et al. (2021). Alterations in the human oral and gut microbiomes and lipidomics in COVID-19. Gut 70 (7), 1253–1265. doi: 10.1136/gutjnl-2020-323826
Roncon, L., Zuin, M., Rigatelli, G., Zuliani, G. (2020). Diabetic patients with COVID-19 infection are at higher risk of ICU admission and poor short-term outcome. J. Clin. Virol. 127, 104354. doi: 10.1016/j.jcv.2020.104354
Rooks, M. G., Garrett, W. S. (2016). Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 16 (6), 341–352. doi: 10.1038/nri.2016.42
Round, J. L., Mazmanian, S. K. (2009). The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 9 (5), 313–323. doi: 10.1038/nri2515
Sencio, V., Machado, M. G., Trottein, F. (2021). The lung-gut axis during viral respiratory infections: the impact of gut dysbiosis on secondary disease outcomes. Mucosal Immunol. 14 (2), 296–304. doi: 10.1038/s41385-020-00361-8
Shilts, M. H., Rosas-Salazar, C., Strickland, B. A., Kimura, K. S., Asad, M., Sehanobish, E., et al. (2021). Severe COVID-19 is associated with an altered upper respiratory tract microbiome. Front. Cell. Infect. Microbiol. 11. doi: 10.3389/fcimb.2021.781968
Smith, N., Goncalves, P., Charbit, B., Grzelak, L., Beretta, M., Planchais, C., et al. (2021). Distinct systemic and mucosal immune responses during acute SARS-CoV-2 infection. Nat. Immunol. 22 (11), 1428–1439. doi: 10.1038/s41590-021-01028-7
Sokol, H., Contreras, V., Maisonnasse, P., Desmons, A., Delache, B., Sencio, V., et al. (2021). SARS-CoV-2 infection in nonhuman primates alters the composition and functional activity of the gut microbiota. Gut Microbes 13 (1), 1–19. doi: 10.1080/19490976.2021.1893113
Sokolovsky, S., Soni, P., Hoffman, T., Kahn, P., Scheers-Masters, J. (2021). COVID-19 associated Kawasaki-like multisystem inflammatory disease in an adult. Am. J. Emergency Med. 39, 253.e1–253.e2. doi: 10.1016/j.ajem.2020.06.053
Sun, Z., Song, Z.-G., Liu, C., Tan, S., Lin, S., Zhu, J., et al. (2022). Gut microbiome alterations and gut barrier dysfunction are associated with host immune homeostasis in COVID-19 patients. BMC Med. 20 (1), 24. doi: 10.1186/s12916-021-02212-0
Tang, Y., Liu, J., Zhang, D., Xu, Z., Ji, J., Wen, C. (2020). Cytokine storm in COVID-19: the current evidence and treatment strategies. Front. Immunol. 11. doi: 10.3389/fimmu.2020.01708
Tian, Y., Sun, K.-Y., Meng, T.-Q., Ye, Z., Guo, S.-M., Li, Z.-M., et al. (2021). Gut microbiota may not be fully restored in recovered COVID-19 patients after 3-month recovery. Front. Nutr. 8. doi: 10.3389/fnut.2021.638825
Uehara, O., Abiko, Y., Nagasawa, T., Morikawa, T., Hiraki, D., Harada, F., et al. (2022). Alterations in the oral microbiome of individuals with a healthy oral environment following COVID-19 vaccination. BMC Oral. Health 22 (1), 50. doi: 10.1186/s12903-022-02093-6
Ventero, M. P., Cuadrat, R. R. C., Vidal, I., Andrade, B. G. N., Molina-Pardines, C., Haro-Moreno, J. M., et al. (2021). Nasopharyngeal microbial communities of patients infected with SARS-CoV-2 that developed COVID-19. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.637430
Ventero, M. P., Moreno-Perez, O., Molina-Pardines, C., Paytuví-Gallart, A., Boix, V., Escribano, I., et al. (2022). Nasopharyngeal Microbiota as an early severity biomarker in COVID-19 hospitalised patients. J. Infect. 84 (3), 329–336. doi: 10.1016/j.jinf.2021.12.030
Villapol, S. (2020). Gastrointestinal symptoms associated with COVID-19: impact on the gut microbiome. Trans. Res. 226, 57–69. doi: 10.1016/j.trsl.2020.08.004
Waters, M. D., Dhawan, A., Marrs, T., Anderson, D., Warren, S., Hughes, C. L. (Eds.). (2022). The coronavirus pandemic and the future: virology, epidemiology, translational toxicology and therapeutics (Vol. 2). Royal Society of Chemistry, p. 604–624. doi: 10.1039/9781839166839
WHO Coronavirus (COVID-19) Dashboard | WHO Coronavirus (COVID-19) Dashboard With Vaccination Data. (n.d.). Available at: https://covid19.who.int/ (Retrieved April 28, 2022).
Wiersinga, W. J., Rhodes, A., Cheng, A. C., Peacock, S. J., Prescott, H. C. (2020). Pathophysiology, transmission, diagnosis, and treatment of coronavirus disease 2019 (COVID-19): A review. J. Am. Med. Assoc. 324 (8), 782–793. doi: 10.1001/jama.2020.12839
Wu, Y., Cheng, X., Jiang, G., Tang, H., Ming, S., Tang, L., et al. (2021). Altered oral and gut microbiota and its association with SARS-CoV-2 viral load in COVID-19 patients during hospitalization. NPJ Biofilms Microbiomes 7 (1), 61. doi: 10.1038/s41522-021-00232-5
Wypych, T. P., Wickramasinghe, L. C., Marsland, B. J. (2019). The influence of the microbiome on respiratory health. Nat. Immunol. 20 (10), 1279–1290. doi: 10.1038/s41590-019-0451-9
Xiao, F., Tang, M., Zheng, X., Liu, Y., Li, X., Shan, H. (2020). Evidence for gastrointestinal infection of SARS-CoV-2. Gastroenterology 158 (6), 1831–1833.e3. doi: 10.1053/j.gastro.2020.02.055
Xu, R., Lu, R., Zhang, T., Wu, Q., Cai, W., Han, X., et al. (2021). Temporal association between human upper respiratory and gut bacterial microbiomes during the course of COVID-19 in adults. Commun. Biol. 4 (1), 240. doi: 10.1038/s42003-021-01796-w
Yamamoto, S., Saito, M., Tamura, A., Prawisuda, D., Mizutani, T., Yotsuyanagi, H. (2021). The human microbiome and COVID-19: A systematic review. PloS One 16 (6), e0253293. doi: 10.1371/journal.pone.0253293
Yeoh, Y. K., Zuo, T., Lui, G. C.-Y., Zhang, F., Liu, Q., Li, A. Y., et al. (2021). Gut microbiota composition reflects disease severity and dysfunctional immune responses in patients with COVID-19. Gut 70 (4), 698–706. doi: 10.1136/gutjnl-2020-323020
Yun, Y., Chang, Y., Kim, H.-N., Ryu, S., Kwon, M.-J., Cho, Y. K., et al. (2019). Alterations of the gut microbiome in chronic hepatitis B virus infection associated with alanine aminotransferase level. J. Clin. Med. 8 (2), 173. doi: 10.3390/jcm8020173
Zaneveld, J. R., McMinds, R., Vega Thurber, R. (2017). Stress and stability: applying the Anna Karenina principle to animal microbiomes. Nat. Microbiol. 2, 17121. doi: 10.1038/nmicrobiol.2017.121
Zhang, H., Ai, J.-W., Yang, W., Zhou, X., He, F., Xie, S., et al. (2021). Metatranscriptomic characterization of coronavirus disease 2019 identified a host transcriptional classifier associated with immune signaling. Clin. Infect. Dis. 73 (3), 376–385. doi: 10.1093/cid/ciaa663
Zhang, Y.-J., Li, S., Gan, R.-Y., Zhou, T., Xu, D.-P., Li, H.-B. (2015). Impacts of gut bacteria on human health and diseases. Int. J. Mol. Sci. 16 (4), 7493–7519. doi: 10.3390/ijms16047493
Zuo, T., Liu, Q., Zhang, F., Lui, G. C., Tso, E. Y., Yeoh, Y. K., et al. (2020). Depicting SARS-CoV-2 faecal viral activity in association with gut microbiota composition in patients with COVID-19. Gut 70 (2), 276–284. doi: 10.1136/gutjnl-2020-322294
Keywords: COVID-19, gut microbiome, host health, SARS-C0V-2, infection, human microbiome
Citation: Reuben RC, Beugnon R and Jurburg SD (2023) COVID-19 alters human microbiomes: a meta-analysis. Front. Cell. Infect. Microbiol. 13:1211348. doi: 10.3389/fcimb.2023.1211348
Received: 24 April 2023; Accepted: 23 June 2023;
Published: 02 August 2023.
Edited by:
Piyush Baindara, University of Missouri, United StatesReviewed by:
Projoyita Samanta, All India Institute of Medical Sciences, IndiaPhoolwanti Rani, San Diego Biomedical Research Institute, United States
Dinata Roy, Mizoram University, India
Copyright © 2023 Reuben, Beugnon and Jurburg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rine Christopher Reuben, reubenrine@yahoo.com; Stephanie D. Jurburg, s.d.jurburg@gmail.com