Transformation of (allo)securinine to (allo)norsecurinine via a molecular editing strategy
 Seoyoung Kim
Seoyoung Kim Hee-Seung Lee
Hee-Seung Lee Sunkyu Han
Sunkyu Han- Department of Chemistry, Korea Advanced Institute of Science & Technology (KAIST), Daejeon, Republic of Korea
Securinega alkaloids have intrigued chemists since the isolation of securinine in 1956. This family of natural products comprises a securinane subfamily with a piperidine substructure and norsecurinane alkaloids featuring a pyrrolidine core. From a biosynthetic perspective, the piperidine moiety in securinane alkaloids derives from lysine, whereas the pyrrolidine moiety in norsecurinane natural products originates from ornithine, marking an early biogenetic divergence. Herein, we introduce a single-atom deletion strategy that enables the late-stage conversion of securinane to norsecurinane alkaloids. Notably, for the first time, this method enabled the transformation of piperidine-based (allo)securinine into pyrrolidine-based (allo)norsecurinine. Straightforward access to norsecurinine from securinine, which can be readily extracted from the plant Flueggea suffruticosa, abundant across the Korean peninsula, holds promise for synthetic studies of norsecurinine-based oligomeric securinega alkaloids.
Introduction
Securinega alkaloids, composed of over 100 members, have fascinated the synthetic community owing to their structural diversity (Wehlauch and Gademann, 2017; Kang et al., 2023) and potent bioactivities (Hou et al., 2023) since the isolation of securinine (2) in 1956 (Muraveva and Bankovskii, 1956). Securinane alkaloids, exemplified by allosecurinine (1) and securinine (2), are characterized by an azabicyclo[3.2.1]octane core (B/C rings), fused with a piperidine (A) and butenolide (D) rings (Scheme 1A). Norsecurinane alkaloids such as allonorsecurinine (3) (Alibés et al., 2004; Antien et al., 2019; Hetzler et al., 2022) and norsecurinine (4) share a structural resemblance with securinanes. However, they feature a pyrrolidine moiety in place of the piperidine-based A ring. It is established that the piperidine (A) ring of the securinane alkaloids is derived from 1-piperideine (5), the biosynthetic downstream derivative of lysine (6) (Marek Golebiewski et al., 1976). On the other hand, the pyrrolidine (A) ring of the norsecurinane alkaloids is envisioned to have originated from 1-pyrroline (7), the biosynthetic derivative of ornithine (8). Hence, the size of the A ring is determined at the early stage of the biogenesis.
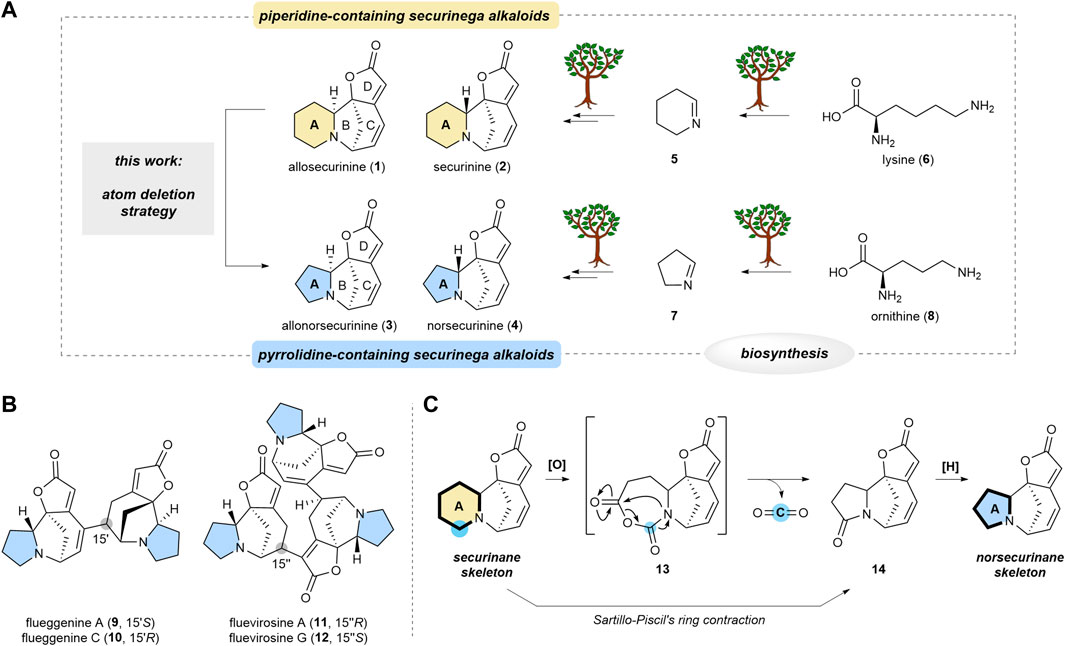
SCHEME 1. Transformation of securinane framework to norsecurinane framework via molecular editing strategy. (A) Parallel biosynthesis of (allo)securinine and (allo)norsecurinine and the direct transformation of the former natural products to the latter natural product (this work). (B) Representative high-order securinega alkaloids with the pyrrolidine core. (C) Overview of our atom deletion strategy that is based on Sartillo-Piscil’s ring contraction method.
Notably, various dimeric and oligomeric high-order securinega alkaloids such as flueggenines A (9), C (10), and fluevirosines A (11), G (12) have been isolated (Scheme 1B) (Jeon et al., 2017; Kang et al., 2023). In 2017, we reported the total synthesis of flueggenine C (10) via an accelerated Rauhut–Currier (RC) reaction (Jeon and Han, 2017). Subsequently, we also succeeded in the total synthesis of flueggenines D and I via a Stille cross-coupling and stereoselective conjugate reduction dimerization strategy (Jeon et al., 2020). Notably, the primary constituent among the monomeric units forming high-order securinega alkaloids is norsecurinine (4). In pursuit of a synthetic program centered on direct RC-based conjugations among norsecurinines (4) (Han, 2023; Park et al., 2023), we sought a solution for obtaining a substantial quantity of norsecurinine (4). In consideration of this goal, we conceived a semisynthetic method to derive (allo)norsecurinine (3 and 4) from (allo)securinine (1 and 2), natural substances abundantly found in Flueggea suffruticosa, a widely distributed plant in the Korean peninsula.
Single-atom skeletal editing has emerged as a powerful approach to efficiently diversify the molecular framework (Jurczyk et al., 2022). This molecular editing strategy has also been applied in natural product synthesis (Hui et al., 2022). We envisioned that the securinane skeleton could be transformed into the norsecurinane skeleton via a single-atom deletion logic (Ha, 2023). Herein, we describe a synthetic protocol that enables a single carbon deletion of the piperidine (A) (Srivastava and Ha, 2023) ring of securinanes to yield pyrrolidine-based norsecurinanes via the intermediacy of N-carboxyanhydride intermediate 13 and lactam 14 (Scheme 1C) based on Sartillo-Piscil’s ring contraction method (Romero-Ibañez et al., 2019).
Results and discussion
Our synthesis commenced with regioselective double oxidation of C5 and C6 carbons of allosecurinine (1) to produce lactam 20 in 60% yield as a 4:1 diastereomeric mixture (Scheme 2). In 2016, Sartillo-Piscil and coworkers reported a double sp3 C−H oxidation of cyclic amines to α-alkoxyamine lactams (Osorio-Nieto et al., 2016; Romero-Ibañez et al., 2023). In the instance of allosecurinine (1), the oxoammonium cation of TEMPO (15) selectively abstracted the C6 hydridic α-hydrogen from the amine moiety due to its greater steric accessibility consistent with Sartillo-Piscil’s reports (Osorio-Nieto et al., 2016; Romero-Ibañez et al., 2023). This led to the formation of the iminium ion intermediate 16, which subsequently underwent tautomerization, resulting in the formation of enamine 17. Subsequently, enamine 17 reacted with the oxoammonium cation of TEMPO (15) to produce iminium ion 18, which is consequently trapped by the chlorine dioxide anion to afford intermediate 19 (Osorio-Nieto et al., 2016). The extrusion of hypochlorous acid from intermediate 19 resulted in lactam 20. The structure of the major diastereomer of 20 was unambiguously confirmed by a single crystal X-ray diffraction (SCXD) analysis. To our delight, when the TEMPO adduct 20 was allowed to react with 3 equiv of m-CPBA, the ring contraction product 14 was obtained in 54% yield (Romero-Ibañez et al., 2019). The structure of lactam 14 was confirmed by a single crystal X-ray diffraction (SCXD) analysis. Chemoselective reduction of the lactam moiety of 14 in the presence of the lactone group was achieved by a synthetic sequence involving the conversion of the lactam moiety to the thiolactam group (96% yield), the methylation of the resulting thiolactam moiety, and the sodium cyanoborohydride-mediated reduction of the consequent thioimidate ester salt (55% yield for the conversion of 21 to 3).
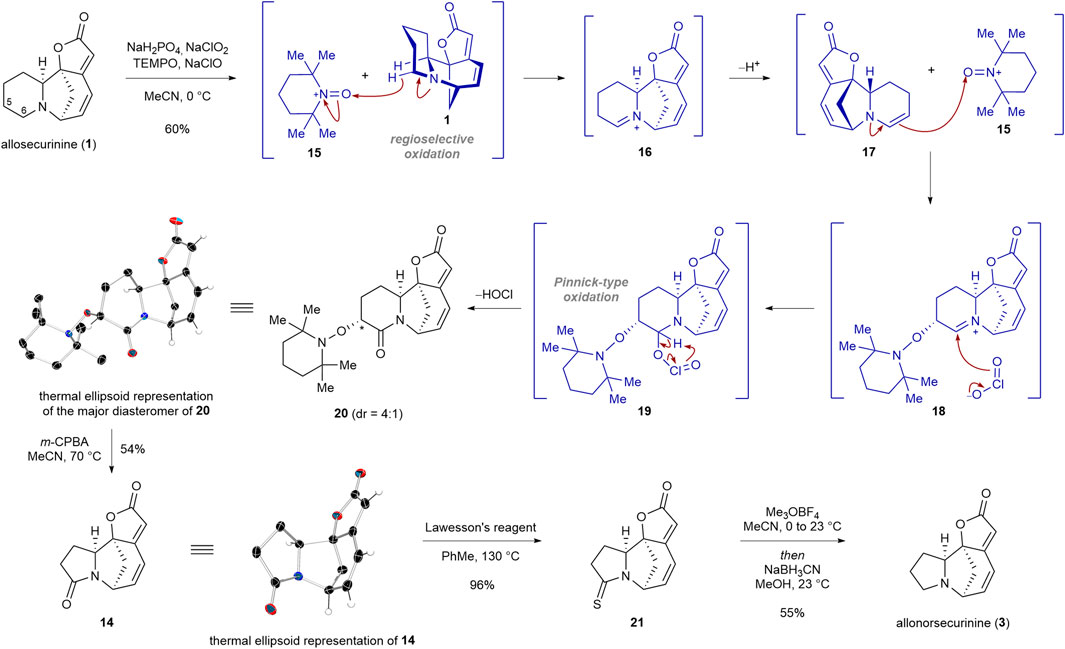
SCHEME 2. Transformation of allosecurinine (1) to allonorsecurinine (3) via an atom deletion strategy.
The plausible reaction mechanism for converting TEMPO adduct 20 to lactam 14 is depicted in Scheme 3 (Romero-Ibañez et al., 2019). The m-CPBA-mediated oxidation of TEMPO adduct 20 would result in the formation of N-oxide intermediate 23. Extrusion of TEMPOH from 23 would afford α-ketolactam intermediate 24. Ketone 24 would then undergo a regioselective Baeyer–Villiger oxidation via intermediate 25 to produce N-carboxyanhydride intermediate 13. Transannular 1,2-addition of the nitrogen nucleophile in 13 would yield pentacyclic intermediate 26, which subsequently undergoes a carbon dioxide extrusion via intermediate 27 to forge lactam 14.
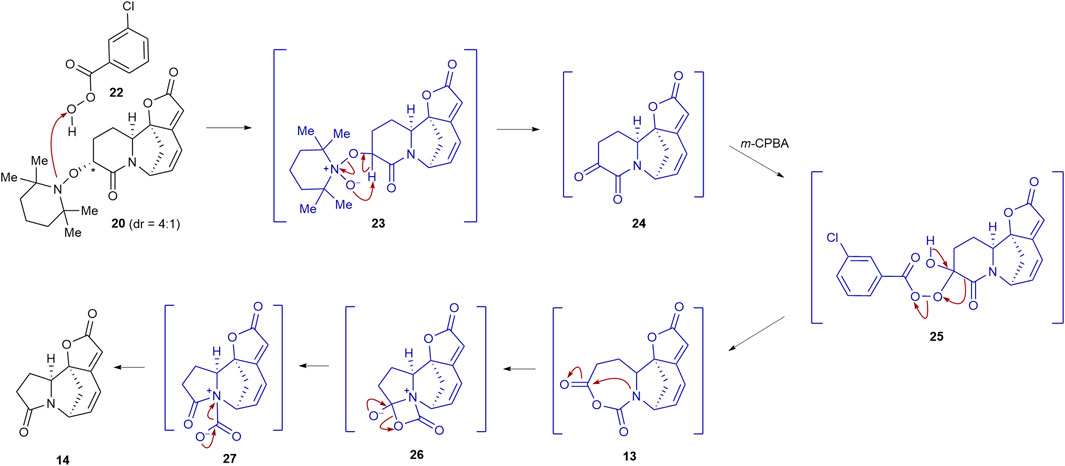
SCHEME 3. Plausible reaction mechanism for the formation of 14 from 20 in the presence of m-CPBA.
After establishing a synthetic protocol for the “methylene deletion” of allosecurinine (1), we set out to transform securinine (2) to norsecurinine (4) in an analogous manner. In the event, treatment of securinine (2) with monosodium phosphate, sodium chlorite, TEMPO, sodium hypochlorite in acetonitrile resulted in TEMPO adduct 28 in 85% yield as a 2.5:1 mixture of diastereomers (Scheme 4). Subsequent m-CPBA-mediated ring contraction (51% yield) and the thionylation of the resulting lactam with Lawesson’s reagent (91%) afforded thiolactam 29. Interestingly, when thiolactam 29 was allowed to react with Meerwein’s salt and subsequently with sodium triacetoxyborohydride, thioaminal product 30 was obtained in 60% yield. This was in stark contrast to the aforementioned reduction of thioamide 21 to the amine product under analogous reduction conditions. 2D-NMR analysis of compound 30 revealed that the proton at C5 is axially positioned. Hence, we reasoned that the stereoelectronic misalignment of the N1-lone pair p-orbital and the C5–S σ* orbital hindered the formation of the iminium ion intermediate. To our pleasure, we discovered that exposure of thioaminal product 30 to more forcing conditions (TFA and triethylsilane in refluxing toluene) afforded norsecurinine (4) in 97% yield.
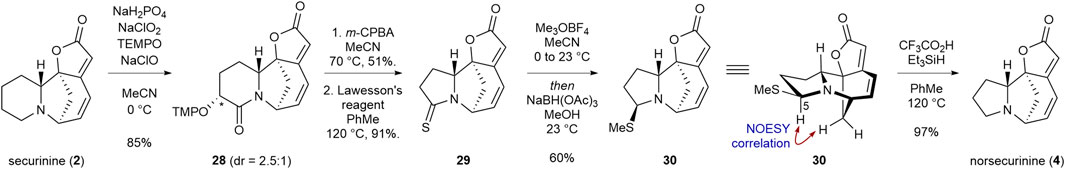
SCHEME 4. Transformation of securinine (2) to norsecurinine (4).
Conclusion
In summary, a molecular editing protocol for the transformation of (allo)securinine to (allo)norsecurinine was established. The key to our success was the employment of the oxidative ring contraction method pioneered by the Sartillo-Piscil group (Romero-Ibañez et al., 2019). Chemoselective reduction of the lactam moiety over the lactone moiety was possible via selective conversion of the amide group to the thioamide group and its subsequent chemoselective reduction. Our findings enabled a straightforward transformation of securinine into norsecurinine, leveraging the abundant supply of securinine derived from the F. suffruticosa plant commonly found across the Korean peninsula. Norsecurinine, a pyrrolidine-based compound, serves as the monomeric unit within the high-order securinega alkaloids. Hence, our discovery stands poised to enable the synthesis of more complex securinega alkaloids. Those studies are currently ongoing and will be the subject of forthcoming reports.
Materials and methods
General information
All reactions were performed in oven-dried or flame-dried round-bottomed flasks and vials. Unless otherwise noted, the flasks were fitted with rubber septa and reactions were conducted under a positive pressure of argon, and vials were tightly sealed with plastic septa, Teflon tape, and parafilm. Stainless steel syringes were used to transfer air- and moisture-sensitive liquids. Flash column chromatography was performed as described by Still et al. using silica gel (60-Å pore size, 40–63 μm, 4%–6% H2O content, Merck) (Still et al., 1978). Analytical thin–layer chromatography (TLC) was performed using glass plates pre-coated with 0.25 mm silica gel impregnated with a fluorescent indicator (254 nm). Thin layer chromatography plates were visualized by exposure to ultraviolet light, and/or a basic aqueous potassium permanganate (KMnO4).
Unless otherwise stated, all commercial reagents and solvents were used without additional purification.
1H and 13C nuclear magnetic resonance spectra were recorded with Bruker Avance Ⅲ HD (400 MHz), Bruker Avance NEO (500 MHz), Agilent DD-2 (600 MHz) and calibrated by using the residual undeuterated chloroform (δH = 7.24 ppm) and CDCl3 (δC = 77.23 ppm) as internal references. Data are reported in the following manners: chemical shift in ppm [multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, p = quintet, m = multiplet, app = apparent, br = broad), coupling constant(s) in Hertz, integration]. The NMR solvent CDCl3 was taken from a stock containing anhydrous K2CO3 to remove residual DCl. High resolution mass spectra were obtained from KAIST Analysis Center for Research (Daejeon) by using ESI method. Specific rotation
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author. Single crystal X-ray diffraction analysis data of compounds 14 (deposition number: 2314385) and 20 (deposition number: 2314384) were deposited in the Cambridge Crystallographic Data Center (CCDC).
Author contributions
SK: Conceptualization, Investigation, Writing–original draft, Writing–review and editing. H-SL: Funding acquisition, Supervision, Writing–review and editing. SH: Conceptualization, Funding acquisition, Investigation, Supervision, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Research Foundation of Korea (NRF-2021R1A2C2011203). Furthermore, we acknowledge support by the National Research Foundation of Korea (NRF-2018R1A5A1025208). This research was also supported by the KAIST UP project. We finally thank the support by the KAIST Grand Challenge 30 project.
Acknowledgments
We thank Dongwook Kim for the assistance with the single crystal X-ray diffraction analyses of compounds 14 and 20.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2024.1355636/full#supplementary-material
References
Alibés, R., Ballbé, M., Busqué, F., de March, P., Elias, L., Figueredo, M., et al. (2004). A new general access to either type of securinega alkaloids: synthesis of securinine and (–)-Allonorsecurinine. Org. Lett. 6, 1813–1816. doi:10.1021/ol049455+
Antien, K., Lacambra, A., Cossío, F. P., Massip, S., Deffieux, D., Pouységu, L., et al. (2019). Bio-inspired total synthesis of twelve securinega alkaloids: structural reassignments of (+)-Virosine B and (–)-Episecurinol A. Chem. Eur. J. 25, 11574–11580. doi:10.1002/chem.201903122
Ha, H.-J. (2023). Recent advances in synthesizing and utilizing nitrogen-containing heterocycles. Front. Chem. 11, 1279418. doi:10.3389/fchem.2023.1279418
Han, S. (2023). “K-synthesis”: recent advancements in natural product synthesis enabled by unique methods and strategies development in Korea. Bull. Korean Chem. Soc. 44, 172–201. doi:10.1002/bkcs.12654
Hetzler, B. E., Trauner, D., and Lawrence, A. L. (2022). Natural product anticipation through synthesis. Nat. Rev. Chem. 6, 170–181. doi:10.1038/s41570-021-00345-7
Hou, W., Huang, H., Wu, X.-Q., and Lan, J.-X. (2023). Bioactivities and mechanism of action of securinega alkaloids derivatives reported prior to 2022. Biomed. Pharmacother. 158, 114190. doi:10.1016/j.biopha.2022.114190
Hui, C., Wang, Z., Wang, S., and Xu, C. (2022). Molecular editing in natural product synthesis. Org. Chem. Front. 9, 1451–1457. doi:10.1039/D2QO00043A
Jeon, S., and Han, S. (2017). An accelerated intermolecular rauhut–currier reaction enables the total synthesis of (–)-flueggenine C. J. Am. Chem. Soc. 139, 6302–6305. doi:10.1021/jacs.7b02751
Jeon, S., Lee, J., Park, S., and Han, S. (2020). Total synthesis of dimeric Securinega alkaloids (–)-flueggenines D and I. Chem. Sci. 11, 10934–10938. doi:10.1039/d0sc03057k
Jeon, S., Park, J., and Han, S. (2017). Syntheses of dimeric securinega alkaloids. Synlett 28, 2353–2359. doi:10.1055/s-0036-1590864
Jurczyk, J., Woo, J., Kim, S. F., Dherange, B. D., Sarpong, R., and Levin, M. D. (2022). Single-atom logic for heterocycle editing. Nat. Synth. 1, 352–364. doi:10.1038/s44160-022-00052-1
Kang, G., Park, S., and Han, S. (2023). Synthesis of high-order and high-oxidation state securinega alkaloids. Acc. Chem. Res. 56, 140–156. doi:10.1021/acs.accounts.2c00719
Marek Golebiewski, W., Horsewood, P., and Spenser, I. D. (1976). Biosynthesis of securinine: the mode of incorporation of lysine. J. Chem. Soc. Chem. Comm. 24, 217–218. doi:10.1039/C39760000217
Muraveva, V. I., and Bankovskii, A. I. (1956). Chemical study of alkaloids of Securinega suffruticosa. Dokl. Akad. Nauk. SSSR. 110, 998–1000.
Osorio-Nieto, U., Chamorro-Arenas, D., Quintero, L., Höpfl, H., and Sartillo-Piscil, F. (2016). Transition metal-free selective double sp3 C-H oxidation of cyclic amines to 3-alkoxyamine lactams. J. Org. Chem. 81, 8625–8632. doi:10.1021/acs.joc.6b01566
Park, S., Kim, D., Yang, W., and Han, S. (2023). Total synthesis of 4α-hydroxyallosecurinine and securingine F, securinega alkaloids with a C4-hydroxy handle for biofunctional derivatizations. Synlett 34. doi:10.1055/a-2047-9680
Romero-Ibañez, J., Cruz-Gregorio, S., Sandoval-Lira, J., Hernández-Pérez, J. M., Quintero, L., and Sartillo-Piscil, F. (2019). Transition-metal-free deconstructive lactamization of piperidines. Angew. Chem. Int. Ed. 58, 8867–8871. doi:10.1002/anie.201903880
Romero-Ibañez, J., Sandoval-Lira, J., Cruz-Gregorio, S., Hernández-Pérez, J. M., Quintero, L., and Sartillo-Piscil, F. (2023). On the reaction mechanism of the selective C(sp3)–H functionalization of N-benzylpiperidines mediated by TEMPO oxoammonium cation. Synthesis 55, 3662–3669. doi:10.1055/a-2053-9558
Srivastava, N., and Ha, H.-J. (2023). Regioselective ring opening of aziridine for synthesizing azaheterocycle. Front. Chem. 11, 1280633. doi:10.3389/fchem.2023.1280633
Still, W. C., Kahn, M., and Mitra, A. (1978). Rapid chromatographic technique for preparative separations with moderate resolution. J. Org. Chem. 43, 2923–2925. doi:10.1021/jo00408a041
Keywords: securinega alkaloids, ring contraction, molecular editing, natural products, atom deletion strategy, heterocycle
Citation: Kim S, Lee H-S and Han S (2024) Transformation of (allo)securinine to (allo)norsecurinine via a molecular editing strategy. Front. Chem. 12:1355636. doi: 10.3389/fchem.2024.1355636
Received: 14 December 2023; Accepted: 08 January 2024;
Published: 22 January 2024.
Edited by:
Hyun-Joon Ha, Hankuk University of Foreign Studies, Republic of KoreaReviewed by:
Prakash Sekar, Syganture Discovery Ltd., United KingdomJung Min Joo, Kyung Hee University, Republic of Korea
Cheon-Gyu Cho, Hanyang University, Republic of Korea
Sanghee Kim, Seoul National University, Republic of Korea
Copyright © 2024 Kim, Lee and Han. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sunkyu Han, sunkyu.han@kaist.ac.kr