Perspectives on current models of Friedreich’s ataxia
 Simge Kelekçi
Simge Kelekçi Abdullah Burak Yıldız
Abdullah Burak Yıldız Kenan Sevinç
Kenan Sevinç Deniz Uğurlu Çimen
Deniz Uğurlu Çimen  Tamer Önder
Tamer Önder- School of Medicine, Koc University, Istanbul, Turkey
Friedreich’s ataxia (FRDA, OMIM#229300) is the most common hereditary ataxia, resulting from the reduction of frataxin protein levels due to the expansion of GAA repeats in the first intron of the FXN gene. Why the triplet repeat expansion causes a decrease in Frataxin protein levels is not entirely known. Generation of effective FRDA disease models is crucial for answering questions regarding the pathophysiology of this disease. There have been considerable efforts to generate in vitro and in vivo models of FRDA. In this perspective article, we highlight studies conducted using FRDA animal models, patient-derived materials, and particularly induced pluripotent stem cell (iPSC)-derived models. We discuss the current challenges in using FRDA animal models and patient-derived cells. Additionally, we provide a brief overview of how iPSC-based models of FRDA were used to investigate the main pathways involved in disease progression and to screen for potential therapeutic agents for FRDA. The specific focus of this perspective article is to discuss the outlook and the remaining challenges in the context of FRDA iPSC-based models.
Introduction
The prevalence of FRDA is 1 in 50,000 people, and the median age of onset is 10–15 years (Indelicato et al., 2020). Wheelchair use is generally necessary for around 15 years, and the average lifespan of FRDA patients is reported to be 36 years (Delatycki and Bidichandani, 2019). Approximately 96% of FRDA patients carry homozygous GAA triplet repeat expansion in the first intron of the frataxin (FXN) gene. The remaining 4% carry a heterozygous phenotype for GAA repeats with missense mutations in one allele and an expanded allele in the other (Al-Mahdawi et al., 2018). Normally, there are 5–33 GAA repeats in the first intron of the FXN gene; however, FRDA patients may have up to 1300 GAA repeats. Individuals with longer repeats show symptoms earlier and with increased severity (Delatycki et al., 2000; Delatycki and Bidichandani, 2019).
Friedreich’s ataxia is an autosomal recessive disorder with neurological and non-neurological manifestations. Neurological symptoms include progressive ataxia of gait and limbs, increased muscle tone, decrease in or loss of position sense and tendon reflexes, difficulty swallowing, and dysarthria (Filla et al., 1990; Parkinson et al., 2013). Additionally, neuroinflammation and upregulation of glial activation in the cerebellum and brainstem are observed (Apolloni et al., 2022; Khan et al., 2022). Non-neurological symptoms include hypertrophic cardiomyopathy, glucose intolerance, and diabetes mellitus (Campuzano et al., 1996; Gottesfeld, 2019). Foot deformity and scoliosis are the early signs of this disease in some cases. In others, cardiomyopathy is the first clinical symptom (Pandolfo, 2009). Diabetes mellitus usually develops in the later stage (De Michele et al., 1996).
GAA repeat expansions lead to a reduction in frataxin protein levels to 5–35% (Gellera et al., 2007; Abruzzo et al., 2013; Bürk, 2017; Vannocci et al., 2018). Why GAA repeat expansion leads to a reduction in frataxin protein levels and the role of frataxin in FRDA pathologies remain unclear. Likewise, the molecular pathology of cardiomyopathy in FRDA is not entirely elucidated, even though the most common cause of death is cardiomyopathy (Crombie et al., 2017). There are neither effective therapeutics for this disease nor any drugs that can slow its progression (Strawser et al., 2014). Therefore, modeling approaches are crucial. However, the selection of suitable models is an important step. The selected model should appropriately reflect FRDA pathophysiology.
Animal models have been nicely covered (Perdomini et al., 2013), whereas recent reviews about the molecular pathways involved in FRDA progression have been discussed in detail (Gottesfeld, 2019). In this perspective article, we focused on the challenges of FRDA in vivo (both non-complex and complex organisms) and patient-derived models and discussed iPSC-based models more comprehensively.
In vivo models of FRDA include yeast, nematode worm, fruit fly, and mice (Babcock et al., 1997; Radisky et al., 1999; Puccio et al., 2001; Al-Mahdawi et al., 2004; Vázquez-Manrique et al., 2006; Virmouni et al., 2014; Monnier et al., 2018), whereas FRDA in vitro models include patient-derived cells such as immortalized lymphoblastoid cells, primary fibroblasts (Li et al., 2016; Agro and Diaz-Nido, 2020; Misiorek et al., 2020; Johnson et al., 2021), FRDA-derived iPSCs (Angulo et al., 2021; Kelekci et al., 2021), and iPSC-based models such as neurons, cardiomyocytes, and beta cells (Crombie et al., 2016; Schreiber et al., 2019) (Figure 1). All these FRDA models need to imitate the symptoms of FRDA patients. For instance, they should be exhibiting a progression of sensory ataxia and/or cardiomyopathy due to reduced frataxin protein level, ideally as a result of GAA expansions in the first intron of the FXN gene (Perdomini et al., 2013).
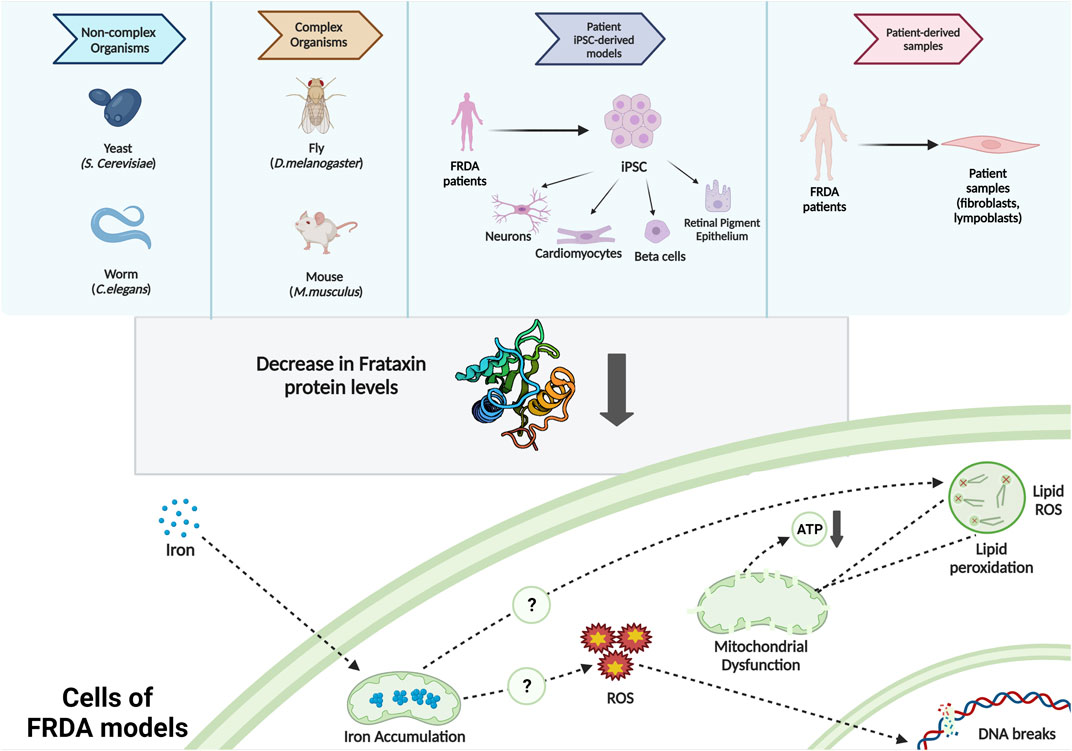
FIGURE 1. Commonly used FRDA models and the intracellular molecular phenotypes observed.
Challenges of Friedreich’s ataxia in vivo, and patient-derived models
The genetic basis of FRDA makes it challenging to study in vivo. In mice, knockout of the FXN gene is embryonically lethal (at E 6.5) (Cossee, 2000). Therefore, conditional knockout FRDA mouse models were generated. Expression of Cre recombinase was induced under MCK (muscle creatine kinase, a gene expressed in the heart cells) and NSE (neuron-specific enolase, a gene expressed in the neurons) promoters (Puccio et al., 2001; Puccio, 2009). Along with developing cardiomyopathy, MCK mice exhibited ISC enzyme deficiencies and iron accumulation at the early stages (Puccio, 2009; Simon et al., 2004). NSE mice developed progressive ataxia with a loss of proprioception (Simon et al., 2004; Perdomini et al., 2013). Additionally, a reversible frataxin knockdown mouse model was developed, and it had cardiac conduction defects, degeneration of dorsal root ganglia, and early mortality (Chandran et al., 2017). Although conditional models reproduce most of the characteristic features of FRDA, they cannot accurately model the effects of genetic background. Moreover, conditional models lead to a complete loss of frataxin, but in FRDA patients, partial frataxin deficiency is observed (Puccio, 2009; Perdomini et al., 2013).
GAA insertion models were also developed. Y4R and YG8R were generated by inserting expanded (GAA)90 and (GAA)190 alleles, respectively. Both express only human frataxin (Al-Mahdawi et al., 2004; Al-Mahdawi et al., 2006; Perdomini et al., 2013). The frataxin protein level was 57% in YG8R, which exhibited mild progressive motor coordination deficits (Al-Mahdawi et al., 2006; Clark et al., 2007; Martelli et al., 2012). Later, YG8sR mice were generated from YG8R. YG8sR mice have a single copy of the FXN transgene and a single (GAA)120 repeat expansion mutation. They exhibited behavioral deficits along with decreased expression of FXN, and reduced aconitase activity in brain regions (Anjomani Virmouni et al., 2015).
However, the short life span of in vivo models leads to incomplete development of pathological hallmarks. Also, although there is physiological accordance between humans and rodents, there are genomic differences that result in profound implications in disease modeling (Dawson et al., 2018). Therefore, further studies on FRDA animal models to overcome these challenges and thereby represent the disease phenotype better and faster are needed.
In 1999, human FRDA primary fibroblasts were used to understand whether this cell type is a reasonable model to study GAA expansions and to test potential therapeutic drugs. The fibroblasts had reduced levels of frataxin mRNA and were more sensitive to ROS-inducers (A. Wong, 1999). This study led FRDA fibroblasts and lymphoblastoid cells to be used in understanding FRDA phenotypes and investigating the effect of potential therapeutic agents. Although primary fibroblasts and lymphoblastoid cells are easily accessible, they are not the main cell types affected in the course of FRDA and they may not represent the phenotypes of FRDA adequately (Perdomini et al., 2013). Georges et al. (2019) showed that resveratrol, an antioxidant drug, and nicotinamide, vitamin B3, did not induce a significant increase in frataxin protein levels in FRDA iPSC-based neurons, although both drugs were shown to increase frataxin mRNA levels in fibroblasts and lymphoblastoid cells of FRDA patients. Additionally, one has to keep in mind that there are limited sources to work with fibroblasts because they do not grow indefinitely. Therefore, in this perspective article, we did not focus on studies where these patient-derived cells were used.
To summarize, model organisms and patient-derived cells can be used to model FRDA, but due to the technical and biological limitations, there is a need for more human FRDA-like models such as FRDA iPSC-based cells or organoids.
Generation of patient-specific induced pluripotent stem cells
The discovery that exogenous expression of OCT4, SOX2, KLF4, and c-MYC can reprogram mammalian somatic cells back to an embryonic-like state (Takahashi and Yamanaka, 2006) enabled scientists to generate disease-specific pluripotent stem cell lines (Soldner and Jaenisch, 2018). As iPSCs can be autologous, they can replace embryonic stem cells in the field of clinical applications of cellular replacement therapies and screening for potential pharmaceuticals (Onder and Daley, 2012). The first iPSC-based disease model was familial amyotrophic lateral sclerosis (ALS) (Dimos et al., 2008). Since then, iPSC-based disease models have been developed to study a diverse set of neurological diseases, such as spinal muscular atrophy (SMA) (Ebert et al., 2009), Rett syndrome (Marchetto et al., 2010), and Friedreich’s ataxia (Ku et al., 2010).
Impact of Friedreich’s ataxia-derived induced pluripotent stem cells and induced pluripotent stem cell-based models on Friedreich’s ataxia research
In 2010, iPSCs were generated from fibroblasts of FRDA patients for the first time. FXN downregulation observed in fibroblasts was found to be retained in the iPSCs (Ku et al., 2010). Since then, several additional FRDA patient-derived iPSCs were generated to study the pathophysiology of this disease (Liu et al., 2011; Bolotta et al., 2019; Schreiber et al., 2019; Dionisi et al., 2020; Mazzara et al., 2020; Angulo et al., 2021; Kelekci et al., 2021).
Modeling GAA repeat instability with induced pluripotent stem cells
When the first FRDA-derived iPSCs were generated, genomic instability during reprogramming and culturing of iPSCs was observed (Ku et al., 2010; Sharma, 2002). The reason for GAA repeat instability in iPSCs was due to elevated expression levels of mismatch repair enzymes (MSH2, MSH3, and MSH6) (Jintang Du et al., 2012). Defects in replication fork progression and stalling in the 3′–5′ direction were also observed during the replication of FXN (Gerhardt et al., 2016). Interestingly, treatment of FRDA fibroblasts with certain epigenetic drugs such as sodium butyrate (NaB), an HDAC class I inhibitor, and Parnate, a LSD1 inhibitor, led to decreased repeat instability during reprogramming. These compounds increased FXN gene expressions (Polak et al., 2016).
Recently, DNA methylation patterns on GAA repeats were studied. FRDA-specific differentially methylated regions (DMRs) were found to be closer to GAA repeats in FRDA iPSC-based models. Interestingly, the prevalence of FXN genes that lack FRDA-specific hypomethylation (unmethylated epialleles) was found to be a predictor of FXN expression and age of onset of FRDA (Rodden et al., 2021). However, the reason for the absence of unmethylated epialleles in GAA repeats near the FXN gene and the effect of methylation on GAA expansion remains to be elucidated.
Considering all these, further studies are necessary to understand the mechanisms behind the repeat expansions and repeat instability observed in FRDA iPSCs. Specifically, the effect of mismatch repair enzymes, the role of methylation in the repeats, and the reason for replication fork stalling should be further investigated in the future.
Modeling of Friedreich’s ataxia phenotypes using induced pluripotent stem cell-based models
Decrease in frataxin expression causes iron accumulation, impaired iron–sulfur cluster biogenesis, increased oxidative stress, mitochondrial dysfunctioning (Calap-Quintana et al., 2018; Gonzalez-Cabo & Palau, 2013; Santos et al., 2010), and ferroptosis (Cotticelli et al., 2019; La Rosa et al., 2020; Turchi et al., 2020; Terzi et al., 2021). These phenotypes were also recapitulated in iPSC-based in vitro models of FRDA (Schreiber et al., 2019). Here, we highlight recent studies utilizing FRDA iPSC-based models (Table 1).

TABLE 1. FRDA iPSC-based models and main findings observed.
FRDA iPSC-based neurons exhibited reduced mitochondrial transcripts and altered expression levels in transcripts involved in ECM organization and focal adhesion (Lai et al., 2019). Primary proprioceptive neurons differentiated from FRDA iPSCs had reduced expression of proprioceptor-specific markers and exhibited shorter survival in vitro (Dionisi et al., 2020).
FRDA iPSC-derived cardiomyocytes (FRDA iPSC-Cms) showed decreased levels of frataxin and mitochondrial potential and impaired functioning of mitochondria (Hick et al., 2013). Lee et al. (2014) exposed FRDA iPSC-Cms to iron-overloading conditions, and this resulted in hypertrophic cardiac stress responses, iron accumulation, and an increase in ROS formation. FRDA iPSC-Cms had calcium handling deficiencies (Crombie et al., 2017). They also exhibited lipid droplet accumulation (J. Li et al., 2019).
FRDA iPSCs were differentiated into endothelial cells, as well. FRDA iPSC-based endothelial cells (iPSC-ECs) exhibited decreased proliferative activity along with an increase in the expression of cellular senescence markers. Forced FXN expression in FRDA iPSC-ECs did not rescue this phenotype, showing that this senescence phenotype is irreversible (Culley et al., 2021).
FRDA iPSC-based β cells were generated in 2019, and those cells had lower FXN gene expression. However, FRDA iPSC-based β cells had functional mitochondria and functional secretion of glucose-induced insulin (Igoillo-Esteve et al., 2020).
Therapeutic solutions for Friedreich’s ataxia with the help of induced pluripotent stem cell-based technologies
Heterochromatin formation around the FXN gene is the major reason for the downregulation of the FXN gene in FRDA (Chutake et al., 2014; Kumari et al., 2011). Histone deacetylation, DNA methylation, and heterochromatin mark (H3K9me3 and H3K27me3) were detected around the FXN gene (Al-Mahdawi et al., 2008). Therefore, epigenetic regulators such as HDAC inhibitors have been studied using FRDA iPSC-based in vitro models (Zhang et al., 2019). HDAC inhibitor treatment of FRDA iPSC-based neurons resulted in a significant increase in FXN levels along with a decrease in oxidative stress response (Codazzi et al., 2016). More recently, large-scale chemical screens on FRDA iPSC-based neuronal progenitor cells containing a FXN reporter system revealed HDAC inhibitors as the only compounds that could upregulate FXN levels (Schreiber et al., 2022).
Recent advances in genome editing enabled the correction of expanded alleles in FRDA. Three-fold higher expression of frataxin in both mRNA and protein levels was observed in FRDA iPSC-based neurons after zinc finger nuclease (ZFN)-mediated excision of expanded GAA repeat regions (Li et al., 2015). In addition, an increase in aconitase activity and ATP levels was observed (Li et al., 2015). Similarly, GAA expanded repeats were excised using ZFNs in FRDA iPSC-based cardiomyocytes, which resulted in the upregulation of frataxin expression, a decrease in lipid droplet accumulation, and a decrease in the expression of cardiac hypertrophy-related genes (J. Li et al., 2019). Recently, dorsal root ganglia organoid-derived sensory neurons were generated from FRDA iPSCs. When the entire FXN intron 1 was excised using CRISPR-Cas9, cellular and molecular deficits observed in these organoids were rescued. This effect was not observed when only the expanded regions were removed in those organoids (Mazzara et al., 2020). All of these studies show that nuclease-mediated removal systems can be used to rescue FRDA phenotypes in the future.
Recently, repeat-targeted nucleic acids (L. Li et al., 2016; Shen et al., 2019; Shen et al., 2020) and synthetic transcription elongation factors (Syn-TEFs) (Erwin et al., 2017) were used to elevate FXN expression in FRDA iPSC-based models. In addition, particle-mediated delivery of frataxin-encoding plasmid DNA has been used to increase FXN levels in FRDA iPSC-based sensory neurons (Czuba-Wojnilowicz et al., 2020). Oligonucleotides targeting 5′ and/or 3’ untranslated regions of the FXN transcript increased FXN mRNA and protein levels in FRDA iPSC-based neuronal progenitor cells (Belbellaa et al., 2020). Taken together, these studies show that overexpression of FXN can have therapeutic potential, but further optimization is required.
Discussion and future perspectives on Friedreich’s ataxia models
FRDA is a rare neurodegenerative disorder, mediated by triplet repeat expansion which results in the downregulation of frataxin. In the recent years, there have been considerable efforts to find a promising drug for FRDA. Therapeutic approaches to augment frataxin production or modulate mitochondrial malfunctioning, increase ROS production, and Nrf2 activation have been used in phase II clinical trials (Rodden & Lynch, 2021). Additionally, there are many studies addressing potentially promising therapeutic approaches for FRDA pathophysiology. For instance, smoothened agonist (SAG) treatment in FXN knockdown human astrocytes resulted in increased neuron viability, neurite length, and synapse formation when these cells were injected into the mice brain (Vicente-Acosta et al., 2022). Additionally, Cur@SF, which are nanospheres containing curcumin in silk fibroins, reduced the levels of ROS in FRDA patient-derived fibroblasts and reduced FRDA phenotypes in YG8R mice models (Xu et al., 2022). Lastly, after eight single FDA-approved drugs were tested on FRDA fibroblasts, dimethyl fumarate and resveratrol treatments were shown to increase FXN mRNA levels. The combination of these two drugs also increased rotarod performance in FRDA mice models (Abeti et al., 2022). However, all these promising drugs should be further tested in FRDA iPSC-derived neurons and cardiomyocytes as these cells are the main cell types affected in FRDA.
In this perspective article, we have discussed recent models of FRDA and their contributions to identify molecular mechanisms in FRDA development and develop potential therapeutics through drug screening and gene therapy studies. Animal models are composed of nematode worms, fruit flies, and mice. Many of these models were successful in recapitulating the FRDA phenotypes in vivo. However, the general concern about the animal models of FRDA is the difficulty in creating such models that are able to exhibit all FRDA-related symptoms and the cellular phenotypes due to reduced FXN levels resulting from GAA repeat expansion. In vitro models include primary cells, genetically modified cell lines (Perdomini et al., 2013), iPSC-based cells (Schreiber et al., 2019), and, recently, organoids (Mazzara et al., 2020). Consideration of models using primary patient-derived fibroblasts and lymphoblastoid cells remains to be controversial because these cells are disease-irrelevant. Differentiated FRDA-derived iPSC-based models exhibit gene expression profiles that are significantly different from the isogenic iPSC-based models created and show similar phenotypes observed in patients’ cells (Lai et al., 2019). Therefore, FRDA iPSC-based models can be considered promising models for future FRDA studies. However, FRDA-derived iPSC in vitro models have certain limitations as well. First is the repeat instability observed during the reprogramming and culturing duration of FRDA-derived iPSCs. This limitation may potentially be overcome by using therapeutic agents that may prevent this instability. Another challenge is the lack of paired isogenic lines. In the case of FRDA-derived iPSCs, isogenic derivatives can be created simply via the excision of repeats. Lastly, it may not be possible to study tissue and organ level phenotypes observed in FRDA patients via 2D differentiated cells. iPSC-based organoids provide an alternative in this regard. Nevertheless, thanks to FRDA iPSC-based models, the reasons behind GAA repeat expansion and mechanisms of disease-relevant phenotypes were studied thoroughly and efficient therapies using CRISPR/Cas9 to increase FXN expression were investigated over the past decades. However, there are still many open questions that need to be addressed and challenges to overcome.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material; further inquiries can be directed to the corresponding authors.
Author contributions
Contributed substantially to the conception or design of the work: SK and AY. Drafted the manuscript: SK, AY, and KS. Revision of the manuscript for important intellectual content: SK, AY, KS, DÇ, and TÖ. All authors approved the final version of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abeti, R., Jasoliya, M., Al-Mahdawi, S., Pook, M., Gonzalez-Robles, C., Hui, C. K., et al. (2022). A drug combination rescues frataxin-dependent neural and cardiac pathophysiology in FA models. Front. Mol. Biosci. 9, 830650. doi:10.3389/fmolb.2022.830650
Abruzzo, P. M., Marini, M., Bolotta, A., Malisardi, G., Manfredini, S., Ghezzo, A., et al. (2013). Frataxin mRNA isoforms in FRDA patients and normal subjects: effect of tocotrienol supplementation. Biomed. Res. Int., 276808. doi:10.1155/2013/276808
Agro, M., and Diaz-Nido, J. (2020). Effect of mitochondrial and cytosolic FXN isoform expression on mitochondrial dynamics and metabolism. Int. J. Mol. Sci. 21 (21), E8251. doi:10.3390/ijms21218251
Al-Mahdawi, S., Ging, H., Bayot, A., Cavalcanti, F., La Cognata, V., Cavallaro, S., et al. (2018). Large interruptions of GAA repeat expansion mutations in friedreich ataxia are very rare. Front. Cell. Neurosci. 12, 443. doi:10.3389/fncel.2018.00443
Al-Mahdawi, S., Pinto, R. M., Ruddle, P., Carroll, C., Webster, Z., Pook, M., et al. (2004). GAA repeat instability in friedreich ataxia YAC transgenic mice. Genomics 84, 301–310. doi:10.1016/j.ygeno.2004.04.003
Al-Mahdawi, S., Pinto, R. M., Varshney, D., Lawrence, L., Lowrie, M. B., Hughes, S., et al. (2006). GAA repeat expansion mutation mouse models of friedreich ataxia exhibit oxidative stress leading to progressive neuronal and cardiac pathology. Genomics 88 (5), 580–590. doi:10.1016/j.ygeno.2006.06.015
Al-Mahdawi, S., Pinto, R. M., Ismail, O., Varshney, D., Lymperi, S., Sandi, C., et al. (2008). The Friedreich ataxia GAA repeat expansion mutation induces comparable epigenetic changes in human and transgenic mouse brain and heart tissues. Hum. Mol. Genet. 17 (5), 735–746. doi:10.1093/hmg/ddm346
Angulo, M. B., Yang, J., Argenziano, M. A., Bertalovitz, A. C., Beidokhti, M. N., McDonald, T. V., et al. (2021). Generation of a Friedreich's ataxia patient-derived iPSC line USFi001-A. Stem Cell Res. 54, 102399. doi:10.1016/j.scr.2021.102399
Anjomani Virmouni, S., Ezzatizadeh, V., Sandi, C., Sandi, M., Al-Mahdawi, S., Chutake, Y., et al. (2015). A novel GAA-repeat-expansion-based mouse model of Friedreich's ataxia. Dis. Model. Mech. 8 (3), 225–235. doi:10.1242/dmm.018952
Apolloni, S., Milani, M., and D'Ambrosi, N. (2022). Neuroinflammation in friedreich's ataxia. Int. J. Mol. Sci. 23 (11), 6297. doi:10.3390/ijms23116297
Babcock, M., De Silva, D., Oaks, R., Davis-Kaplan, S., Jiralerspong, S., Montermini, L., et al. (1997). Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of Frataxin. Science 276, 1709–1712. doi:10.1126/science.276.5319.1709
Belbellaa, B., Reutenauer, L., Messaddeq, N., Monassier, L., and Puccio, H. (2020). High levels of frataxin overexpression lead to mitochondrial and cardiac toxicity in mouse models. Mol. Ther. Methods Clin. Dev. 19, 120–138. doi:10.1016/j.omtm.2020.08.018
Bolotta, A., Abruzzo, P. M., Baldassarro, V. A., Ghezzo, A., Scotlandi, K., Marini, M., et al. (2019). New insights into the hepcidin-ferroportin Axis and iron homeostasis in iPSC-derived cardiomyocytes from friedreich’s ataxia patient. Oxid. Med. Cell. Longev., 7623023. doi:10.1155/2019/7623023
Bürk, K. (2017). Friedreich Ataxia: current status and future prospects. Cerebellum Ataxias 4, 4. doi:10.1186/s40673-017-0062-x
Campuzano, V., Montermini, L., Moltò, M. D., Pianese, L., Cossée, M., Cavalcanti, F., et al. (1996). Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271, 1423–1427. doi:10.1126/science.271.5254.1423
Chandran, V., Gao, K., Swarup, V., Versano, R., Dong, H., Jordan, M. C., et al. (2017). Inducible and reversible phenotypes in a novel mouse model of friedreich's ataxia. Elife 6, e30054. doi:10.7554/eLife.30054
Chutake, Y. K., Lam, C., Costello, W. N., Anderson, M., and Bidichandani, S. I. (2014). Epigenetic promoter silencing in Friedreich ataxia is dependent on repeat length. Ann. Neurol. 76 (4), 522–528. doi:10.1002/ana.24249
Clark, R. M., De Biase, I., Malykhina, A. P., Al-Mahdawi, S., Pook, M., Bidichandani, S. I., et al. (2007). The GAA triplet-repeat is unstable in the context of the human FXN locus and displays age-dependent expansions in cerebellum and DRG in a transgenic mouse model. Hum. Genet. 120 (5), 633–640. doi:10.1007/s00439-006-0249-3
Codazzi, F., Hu, A., Rai, M., Donatello, S., Salerno Scarzella, F., Mangiameli, E., et al. (2016). Friedreich ataxia-induced pluripotent stem cell-derived neurons show a cellular phenotype that is corrected by a benzamide HDAC inhibitor. Hum. Mol. Genet. 25 (22), 4847–4855. doi:10.1093/hmg/ddw308
Cossee, M., Puccio, H., GAnsmuller, A., Koutnikova, H., Dierich, A., LeMeurM., , et al. (2000). Inactivation of the Friedreich ataxia mouse gene leads to early embryonic lethality without iron accumulation. Hum. Mol. Genet. 9, 1219–1226. doi:10.1093/hmg/9.8.1219
Cotticelli, M. G., Xia, S., Lin, D., Lee, T., Terrab, L., Wipf, P., et al. (2019). Ferroptosis as a novel therapeutic target for friedreich's ataxia. J. Pharmacol. Exp. Ther. 369 (1), 47–54. doi:10.1124/jpet.118.252759
Crombie, D. E., Curl, C. L., Raaijmakers, A. J. A., Sivakumaran, P., Kulkarni, T., Wong, R. C. B., et al. (2017). Friedreich’s ataxia induced pluripotent stem cell-derived cardiomyocytes display electrophysiological abnormalities and calcium handling deficiency. Aging 9, 1440–1452. doi:10.18632/aging.101247
Crombie, D. E., Curl, C. L., Raaijmakers, A. J., Sivakumaran, P., Kulkarni, T., Wong, R. C., et al. (2017). Friedreich's ataxia induced pluripotent stem cell-derived cardiomyocytes display electrophysiological abnormalities and calcium handling deficiency. Aging (Albany NY) 9 (5), 1440–1452. doi:10.18632/aging.101247
Crombie, D. E., Pera, M. F., Delatycki, M. B., and Pebay, A. (2016). Using human pluripotent stem cells to study friedreich ataxia cardiomyopathy. Int. J. Cardiol. 212, 37–43. doi:10.1016/j.ijcard.2016.03.040
Culley, M. K., Zhao, J., Tai, Y. Y., Tang, Y., Perk, D., Negi, V., et al. (2021). Frataxin deficiency promotes endothelial senescence in pulmonary hypertension. J. Clin. Invest. 131 (11), 136459. doi:10.1172/JCI136459
Czuba-Wojnilowicz, E., Viventi, S., Howden, S. E., Maksour, S., Hulme, A. E., Cortez-Jugo, C., et al. (2020). Particle-mediated delivery of frataxin plasmid to a human sensory neuronal model of Friedreich's ataxia. Biomater. Sci. 8 (9), 2398–2403. doi:10.1039/c9bm01757g
Dawson, T. M., Golde, T. E., and Lagier-Tourenne, C. (2018). Animal models of neurodegenerative diseases. Nat. Neurosci. 21, 1370–1379. doi:10.1038/s41593-018-0236-8
De Michele, G., DiMaio, L., Filla, A., Majello, M., Cocozza, S., Cavalcanti, F., et al. (1996). Childhood onset of friedreich ataxia: a clinical and genetic study of 36 cases. Neuropediatrics 27 (1), 3–7. doi:10.1055/s-2007-973740
Delatycki, M. B., and Bidichandani, S. I. (2019). “Friedreich ataxia- pathogenesis and implications for therapies,” in Neurobiology of disease. doi:10.1016/j.nbd.2019.104606
Delatycki, M. B., Williamson, R., and Forrest, S. M. (2000). Friedreich ataxia: an overview. J. Med. Genet. 37 (1), 1–8. doi:10.1136/jmg.37.1.1
Dimos, J. T., Rodolfa, K. T., Niakan, K. K., Weisenthal, L. M., Mitsumoto, H., Chung, W., et al. (2008). Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science 321 (5893), 1218–1221. doi:10.1126/science.1158799
Dionisi, C., Rai, M., Chazalon, M., Schiffmann, S. N., and Pandolfo, M. (2020). Primary proprioceptive neurons from human induced pluripotent stem cells: a cell model for afferent ataxias. Sci. Rep. 10 (1), 7752. doi:10.1038/s41598-020-64831-6
Du, J., Campau, E., Soragni, E., Ku, S., Puckett, J. W., Dervan, P. B., et al. (2012). Role of mismatch repair enzymes in GAA·TTC triplet-repeat expansion in friedreich ataxia induced pluripotent stem cells. J. Biol. Chem. 287, 29861–29872. doi:10.1074/jbc.M112.391961
Ebert, A. D., Yu, J., Rose, F. F., Mattis, V. B., Lorson, C. L., Thomson, J. A., et al. (2009). Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 457 (7227), 277–280. doi:10.1038/nature07677
Erwin, G. S., Grieshop, M. P., Ali, A., Qi, J., Lawlor, M., Kumar, D., et al. (2017). Synthetic transcription elongation factors license transcription across repressive chromatin. Science 358 (6370), 1617–1622. doi:10.1126/science.aan6414
Filla, A., DeMichele, G., Caruso, G., Marconi, R., and Campanella, G. (1990). Genetic data and natural history of friedreich's disease: a study of 80 Italian patients. J. Neurol. 237 (6), 345–351. doi:10.1007/bf00315657
Gellera, C., Castellotti, B., Mariotti, C., Mineri, R., Seveso, V., DiDonato, S., et al. (2007). Frataxin gene point mutations in italian friedreich ataxia patients. Neurogenetics 8, 289–299. doi:10.1007/s10048-007-0101-5
Georges, P., Boza-Moran, M. G., Gide, J., Pêche, G. A., Forêt, B., Bayot, A., et al. (2019). Induced pluripotent stem cells-derived neurons from patients with friedreich ataxia exhibit differential sensitivity to resveratrol and nicotinamide. Sci. Rep. 9, 14568. doi:10.1038/s41598-019-49870-y
Gerhardt, J., Bhalla, A. D., Butler, J. S., Puckett, J. W., Dervan, P. B., Rosenwaks, Z., et al. (2016). Stalled DNA replication forks at the endogenous GAA repeats drive repeat expansion in friedreich’s ataxia cells. Cell Rep. 16, 1218–1227. doi:10.1016/j.celrep.2016.06.075
Gonzalez-Cabo, P., and Palau, F. (2013). Mitochondrial pathophysiology in friedreich's ataxia. J. Neurochem. 126, 53–64. doi:10.1111/jnc.12303
Gottesfeld, J. M. (2019). Molecular mechanisms and therapeutics for the GAA·TTC expansion disease friedreich ataxia. Neurotherapeutics 16, 1032–1049. doi:10.1007/s13311-019-00764-x
Hick, A., Wattenhofer-Donze, M., Chintawar, S., Tropel, P., Simard, J. P., Vaucamps, N., et al. (2013). Neurons and cardiomyocytes derived from induced pluripotent stem cells as a model for mitochondrial defects in friedreich's ataxia. Dis. Model. Mech. 6 (3), 608–621. doi:10.1242/dmm.010900
Igoillo-Esteve, M., Gurgul-Convey, E., Hu, A., Romagueira Bichara Dos Santos, L., Abdulkarim, B., Chintawar, S., et al. (2015). Unveiling a common mechanism of apoptosis in beta-cells and neurons in friedreich's ataxia. Hum. Mol. Genet. 24 (8), 2274–2286. doi:10.1093/hmg/ddu745
Igoillo-Esteve, M., Oliveira, A. F., Cosentino, C., Fantuzzi, F., Demarez, C., Toivonen, S., et al. (2020). Exenatide induces frataxin expression and improves mitochondrial function in friedreich ataxia. JCI Insight 5 (2), 134221. doi:10.1172/jci.insight.134221
Indelicato, E., Nachbauer, W., Eigentler, A., Amprosi, M., Matteucci Gothe, R., Giunti, P., et al. (2020). Onset features and time to diagnosis in friedreich's Ataxia. Orphanet J. Rare Dis. 15 (1), 198. doi:10.1186/s13023-020-01475-9
Johnson, J., Mercado-Ayon, E., Clark, E., Lynch, D., and Lin, H. (2021). Drp1-dependent peptide reverse mitochondrial fragmentation, a homeostatic response in friedreich ataxia. Pharmacol. Res. Perspect. 9 (3), e00755. doi:10.1002/prp2.755
Kelekci, S., Ugurlu-Cimen, D., Demir, A. B., Ozcimen, B., Burak Yildiz, A., Batuhan Karakus, M., et al. (2021). Generation of transgene-free iPSC lines from three patients with friedreich's ataxia (FRDA) carrying GAA triplet expansions in the first intron of FXN gene. Stem Cell Res. 54, 102438. doi:10.1016/j.scr.2021.102438
Khan, W., Corben, L. A., Bilal, H., Vivash, L., Delatycki, M. B., Egan, G. F., et al. (2022). Neuroinflammation in the cerebellum and brainstem in friedreich ataxia: An [18F]-FEMPA PET study. Mov. Disord. 37 (1), 218–224. doi:10.1002/mds.28825
Ku, S., Soragni, E., Campau, E., Thomas, E. A., Altun, G., Laurent, L. C., et al. (2010). Friedreich’s ataxia induced pluripotent stem cells model intergenerational GAATTC triplet repeat instability. Cell Stem Cell 7, 631–637. doi:10.1016/j.stem.2010.09.014
Kumari, D., Biacsi, R. E., and Usdin, K. (2011). Repeat expansion affects both transcription initiation and elongation in friedreich ataxia cells. J. Biol. Chem. 286 (6), 4209–4215. doi:10.1074/jbc.M110.194035
La Rosa, P., Petrillo, S., Fiorenza, M. T., Bertini, E. S., and Piemonte, F. (2020). Ferroptosis in friedreich's ataxia: a metal-induced neurodegenerative disease. Biomolecules 10 (11), E1551. doi:10.3390/biom10111551
Lai, J. I., Nachun, D., Petrosyan, L., Throesch, B., Campau, E., Gao, F., et al. (2019). Transcriptional profiling of isogenic friedreich ataxia neurons and effect of an HDAC inhibitor on disease signatures. J. Biol. Chem. 294, 1846–1859. doi:10.1074/jbc.RA118.006515
Lee, Y. K., Ho, P. W., Schick, R., Lau, Y. M., Lai, W. H., Zhou, T., et al. (2014). Modeling of Friedreich ataxia-related iron overloading cardiomyopathy using patient-specific-induced pluripotent stem cells. Pflugers Arch. 466 (9), 1831–1844. doi:10.1007/s00424-013-1414-x
Li, J., Rozwadowska, N., Clark, A., Fil, D., Napierala, J. S., Napierala, M., et al. (2019). Excision of the expanded GAA repeats corrects cardiomyopathy phenotypes of iPSC-derived friedreich's ataxia cardiomyocytes. Stem Cell Res. 40, 101529. doi:10.1016/j.scr.2019.101529
Li, Y., Polak, U., Clark, A. D., Bhalla, A. D., Chen, Y. Y., Li, J., et al. (2016). Establishment and maintenance of primary fibroblast repositories for rare diseases-friedreich's ataxia example. Biopreserv. Biobank. 14 (4), 324–329. doi:10.1089/bio.2015.0117
Liu, J., Verma, P. J., Evans-Galea, M. V., Delatycki, M. B., Michalska, A., Leung, J., et al. (2011). Generation of induced pluripotent stem cell lines from friedreich ataxia patients. Stem Cell Rev. Rep. 7, 703–713. doi:10.1007/s12015-010-9210-x
Li, Y., Polak, U., Bhalla, A. D., Rozwadowska, N., Butler, J. S., Lynch, D. R., et al. (2015). Excision of Expanded GAA Repeats Alleviates the Molecular Phenotype of Friedreich’s Ataxia. Mol. Ther. 23 (6), 1055–1065. doi:10.1038/mt.2015.41
Marchetto, M. C., Carromeu, C., Acab, A., Yu, D., Yeo, G. W., Mu, Y., et al. (2010). A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 143 (4), 527–539. doi:10.1016/j.cell.2010.10.016
Martelli, A., Napierala, M., and Puccio, H. (2012). Understanding the genetic and molecular pathogenesis of friedreich's ataxia through animal and cellular models. Dis. Model. Mech. 5 (2), 165–176. doi:10.1242/dmm.008706
Mazzara, P. G., Muggeo, S., Luoni, M., Massimino, L., Zaghi, M., Valverde, P. T., et al. (2020). Frataxin gene editing rescues friedreich's ataxia pathology in dorsal root ganglia organoid-derived sensory neurons. Nat. Commun. 11 (1), 4178. doi:10.1038/s41467-020-17954-3
Misiorek, J. O., Schreiber, A. M., Urbanek-Trzeciak, M. O., Jazurek-Ciesiolka, M., Hauser, L. A., Lynch, D. R., et al. (2020). A comprehensive transcriptome analysis identifies FXN and BDNF as novel targets of miRNAs in friedreich's ataxia patients. Mol. Neurobiol. 57 (6), 2639–2653. doi:10.1007/s12035-020-01899-1
Monnier, V., Llorens, J. V., and Navarro, J. A. (2018). Impact of drosophila models in the study and treatment of friedreich’s ataxia. Int. J. Mol. Sci. 19, E1989. doi:10.3390/ijms19071989
Onder, T. T., and Daley, G. Q. (2012). New lessons learned from disease modeling with induced pluripotent stem cells. Curr. Opin. Genet. Dev. 22 (5), 500-508. doi:10.1016/j.gde.2012.05.005
Pandolfo, M. (2009). Friedreich ataxia: the clinical picture. J. Neurol. 256, 3–8. doi:10.1007/s00415-009-1002-3
Parkinson, M. H., Boesch, S., Nachbauer, W., Mariotti, C., and Giunti, P. (2013). Clinical features of friedreich's ataxia: classical and atypical phenotypes. J. Neurochem. 126, 103–117. doi:10.1111/jnc.12317
Perdomini, M., Hick, A., Puccio, H., and Pook, M. A. (2013). Animal and cellular models of friedreich ataxia. J. Neurochem. 126, 65–79. doi:10.1111/jnc.12219
Polak, U., Li, Y., Butler, J. S., and Napierala, M. (2016). Alleviating GAA repeat induced transcriptional silencing of the friedreich’s ataxia gene during somatic cell reprogramming. Stem Cells Dev. 25, 1788–1800. doi:10.1089/scd.2016.0147
Puccio, H. (2009). Multicellular models of friedreich ataxia. J. Neurol. 256 Suppl 1, 18–24. doi:10.1007/s00415-009-1004-1
Puccio, H., Simon, D., Cossée, M., Criqui-Filipe, P., Tiziano, F., Melki, J., et al. (2001). Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat. Genet. 27, 181–186. doi:10.1038/84818
Radisky, D. C., Babcock, M. C., and Kaplan, J. (1999). The yeast frataxin homologue mediates mitochondrial iron efflux: evidence for a mitochondrial iron cycle. J. Biol. Chem. 274, 4497–4499. doi:10.1074/jbc.274.8.4497
Rodden, L. N., Chutake, Y. K., Gilliam, K., Lam, C., Soragni, E., Hauser, L., et al. (2021). Methylated and unmethylated epialleles support variegated epigenetic silencing in Friedreich ataxia. Hum. Mol. Genet. 29 (23), 3818–3829. doi:10.1093/hmg/ddaa267
Rodden, L. N., and Lynch, D. R. (2021). Designing phase II clinical trials in friedreich ataxia. Expert Opin. Emerg. Drugs 26 (4), 415–423. doi:10.1080/14728214.2021.1998452
Santos, R., Lefevre, S., Sliwa, D., Seguin, A., Camadro, J. M., Lesuisse, E., et al. (2010). Friedreich ataxia: molecular mechanisms, redox considerations, and therapeutic opportunities. Antioxid. Redox Signal. 13 (5), 651–690. doi:10.1089/ars.2009.3015
Schreiber, A. M., Li, Y., Chen, Y., Napierala, J. S., and Napierala, M. (2022). Selected histone deacetylase inhibitors reverse the frataxin transcriptional defect in a novel friedreich’s ataxia induced pluripotent stem cell-derived neuronal reporter system. Front. Neurosci. 16, 836476. doi:10.3389/fnins.2022.836476
Schreiber, A. M., Misiorek, J. O., Napierala, J. S., and Napierala, M. (2019). Progress in understanding friedreich's ataxia using human induced pluripotent stem cells. Expert Opin. Orphan Drugs 7 (2), 81–90. doi:10.1080/21678707.2019.1562334
Shan, B., Xu, C., Zhang, Y., Xu, T., Gottesfeld, J. M., Yates, J. R., et al. (2014). Quantitative proteomic analysis identifies targets and pathways of a 2-aminobenzamide HDAC inhibitor in Friedreich's ataxia patient iPSC-derived neural stem cells. J. Proteome Res. 13 (11), 4558–4566. doi:10.1021/pr500514r
Sharma, R., Bhatti, S., Gomez, M., Clark, R. M., Murray, C., Ashizawa, T., et al. (2002). The GAA triplet-repeat sequence in Friedreich ataxia shows a high level of somatic instability in vivo, with a significant predilection for large contractions. Hum. Mol. Genet. 11, 2175–2187. doi:10.1093/hmg/11.18.2175
Shen, X., Beasley, S., Putman, J. N., Li, Y., Prakash, T. P., Rigo, F., et al. (2019). Efficient electroporation of neuronal cells using synthetic oligonucleotides: identifying duplex RNA and antisense oligonucleotide activators of human frataxin expression. RNA 25 (9), 1118–1129. doi:10.1261/rna.071290.119
Simon, D., Seznec, H., Gansmuller, A., Carelle, N., Weber, P., Metzger, D., et al. (2004). Friedreich ataxia mouse models with progressive cerebellar and sensory ataxia reveal autophagic neurodegeneration in dorsal root ganglia. J. Neurosci. 24 (8), 1987–1995. doi:10.1523/JNEUROSCI.4549-03.2004
Soldner, F., and Jaenisch, R. (2018). Stem cells, genome editing, and the path to translational medicine. Cell 175, 615–632. doi:10.1016/j.cell.2018.09.010
Strawser, C. J., Schadt, K. A., and Lynch, D. R. (2014). Therapeutic approaches for the treatment of Friedreich’s ataxia. Expert Rev. Neurother. 14, 949–957. doi:10.1586/14737175.2014.939173
Takahashi, K., and Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. doi:10.1016/j.cell.2006.07.024
Terzi, E. M., Sviderskiy, V. O., Alvarez, S. W., Whiten, G. C., and Possemato, R. (2021). Iron-sulfur cluster deficiency can be sensed by IRP2 and regulates iron homeostasis and sensitivity to ferroptosis independent of IRP1 and FBXL5. Sci. Adv. 7 (22), eabg4302. doi:10.1126/sciadv.abg4302
Turchi, R., Faraonio, R., Lettieri-Barbato, D., and Aquilano, K. (2020). An overview of the ferroptosis hallmarks in friedreich's ataxia. Biomolecules 10 (11), E1489. doi:10.3390/biom10111489
Vannocci, T., Manzano, R. N., Beccalli, O., Bettegazzi, B., Grohovaz, F., Cinque, G., et al. (2018). Adding a temporal dimension to the study of Friedreich’s ataxia: the effect of frataxin overexpression in a human cell model. Dis. Model. Mech. 11, dmm032706. doi:10.1242/dmm.032706
Vázquez-Manrique, R. P., González‐Cabo, P., Ros, S., Aziz, H., Baylis, H. A., Palau, F., et al. (2006). Reduction of Caenorhabditis elegans frataxin increases sensitivity to oxidative stress, reduces lifespan, and causes lethality in a mitochondrial complex II mutant. FASEB J. 20, 172–174. doi:10.1096/fj.05-4212fje
Vicente-Acosta, A., Gimenez-Cassina, A., Diaz-Nido, J., and Loria, F. (2022). The smoothened agonist SAG reduces mitochondrial dysfunction and neurotoxicity of frataxin-deficient astrocytes. J. Neuroinflammation 19 (1), 93. doi:10.1186/s12974-022-02442-w
Virmouni, S. A., Sandi, C., Al-Mahdawi, S., and Pook, M. A. (2014). Cellular, molecular and functional characterisation of YAC transgenic mouse models of friedreich ataxia. PLoS ONE 9, e107416. doi:10.1371/journal.pone.0107416
Wong, A., Yang, J., Cavadini, P., Gellera, C., Lonnerdal, B., Taroni, B., et al. (1999). The Friedreich’s ataxia mutation confers cellular sensitivity to oxidant stress which is rescued by chelators of iron and calcium and inhibitors of apoptosis. Hum. Mol. Genet. 8 (3), 425–430. doi:10.1093/hmg/8.3.425
Wong, A. O., Wong, G., Shen, M., Chow, M. Z., Tse, W. W., Gurung, B., et al. (2019)., Correlation between frataxin expression and contractility revealed by in vitro friedreich's ataxia cardiac tissue models engineered from human pluripotent stem cellsStem Cell Res. Ther.1. 10. PMCID, 203PMC6615274. PMID: 31286988. doi:10.1186/s13287-019-1305-y
Xu, L., Sun, Z., Xing, Z., Liu, Y., Zhao, H., Tang, Z., et al. (2022). Cur@SF NPs alleviate friedreich's ataxia in a mouse model through synergistic iron chelation and antioxidation. J. Nanobiotechnology 20 (1), 118. doi:10.1186/s12951-022-01333-9
Keywords: iPSC (induced pluripotent stem cell), disease model cell, triplet repeat disease, frataxin, ataxia
Citation: Kelekçi S, Yıldız AB, Sevinç K, Çimen DU and Önder T (2022) Perspectives on current models of Friedreich’s ataxia. Front. Cell Dev. Biol. 10:958398. doi: 10.3389/fcell.2022.958398
Received: 31 May 2022; Accepted: 13 July 2022;
Published: 11 August 2022.
Edited by:
Yavuz Oktay, Dokuz Eylul University, TurkeyReviewed by:
Gunes Ozhan, Dokuz Eylül University, TurkeyJiajia Yang, University of South Florida, United States
Copyright © 2022 Kelekçi, Yıldız, Sevinç, Çimen and Önder. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Simge Kelekçi, skelekci18@ku.edu.tr, simge.kelekci@dkfz.de; Tamer Önder, tonder@ku.edu.tr