To Join or Not to Join: Decision Points Along the Pathway to Double-Strand Break Repair vs. Chromosome End Protection
 Stephanie M. Ackerson
Stephanie M. Ackerson  Jason A. Stewart
Jason A. Stewart- Department of Biological Sciences, University of South Carolina, Columbia, SC, United States
The regulation of DNA double-strand breaks (DSBs) and telomeres are diametrically opposed in the cell. DSBs are considered one of the most deleterious forms of DNA damage and must be quickly recognized and repaired. Telomeres, on the other hand, are specialized, stable DNA ends that must be protected from recognition as DSBs to inhibit unwanted chromosome fusions. Decisions to join DNA ends, or not, are therefore critical to genome stability. Yet, the processing of telomeres and DSBs share many commonalities. Accordingly, key decision points are used to shift DNA ends toward DSB repair vs. end protection. Additionally, DSBs can be repaired by two major pathways, namely homologous recombination (HR) and non-homologous end joining (NHEJ). The choice of which repair pathway is employed is also dictated by a series of decision points that shift the break toward HR or NHEJ. In this review, we will focus on these decision points and the mechanisms that dictate end protection vs. DSB repair and DSB repair choice.
Introduction
DNA double strand breaks (DSBs) originate from exposure to both external DNA damaging agents, such as genotoxic chemicals and ionizing radiation (IR), and endogenous sources, such replication fork collapse, reactive oxygen species and chromosome fusions (Symington and Gautier, 2011; Ceccaldi et al., 2016). DSBs can be beneficial or detrimental depending on the context. On the one hand, programmed DSBs can be beneficial to promote genome and antibody diversity in meiosis and V(D)J recombination, respectively. However, DSBs caused by DNA damage are almost always detrimental and result in deletions, translocations, and chromosome fusions, which leads to senescence, apoptosis or oncogenesis (Phillips and McKinnon, 2007; Bohgaki et al., 2010; Bunting and Nussenzweig, 2013; Rulten and Caldecott, 2013; Ghosh et al., 2018; Seol et al., 2018). To prevent such outcomes, cells activate a DNA damage response (DDR), which is predominantly mediated by the phosphatidylinositol 3-kinase-related kinase (PIKK) family members, DNA-dependent protein kinase (DNA-PK), ataxia-telangiectasia mutated (ATM), and ATM and RAD3-related (ATR) (Blackford and Jackson, 2017). These kinases signal to downstream cell cycle checkpoints and localize repair machinery to the break (Blackford and Jackson, 2017). DSBs are repaired by two major pathways, namely homologous recombination (HR) and non-homologous end joining (NHEJ) (Figure 1).
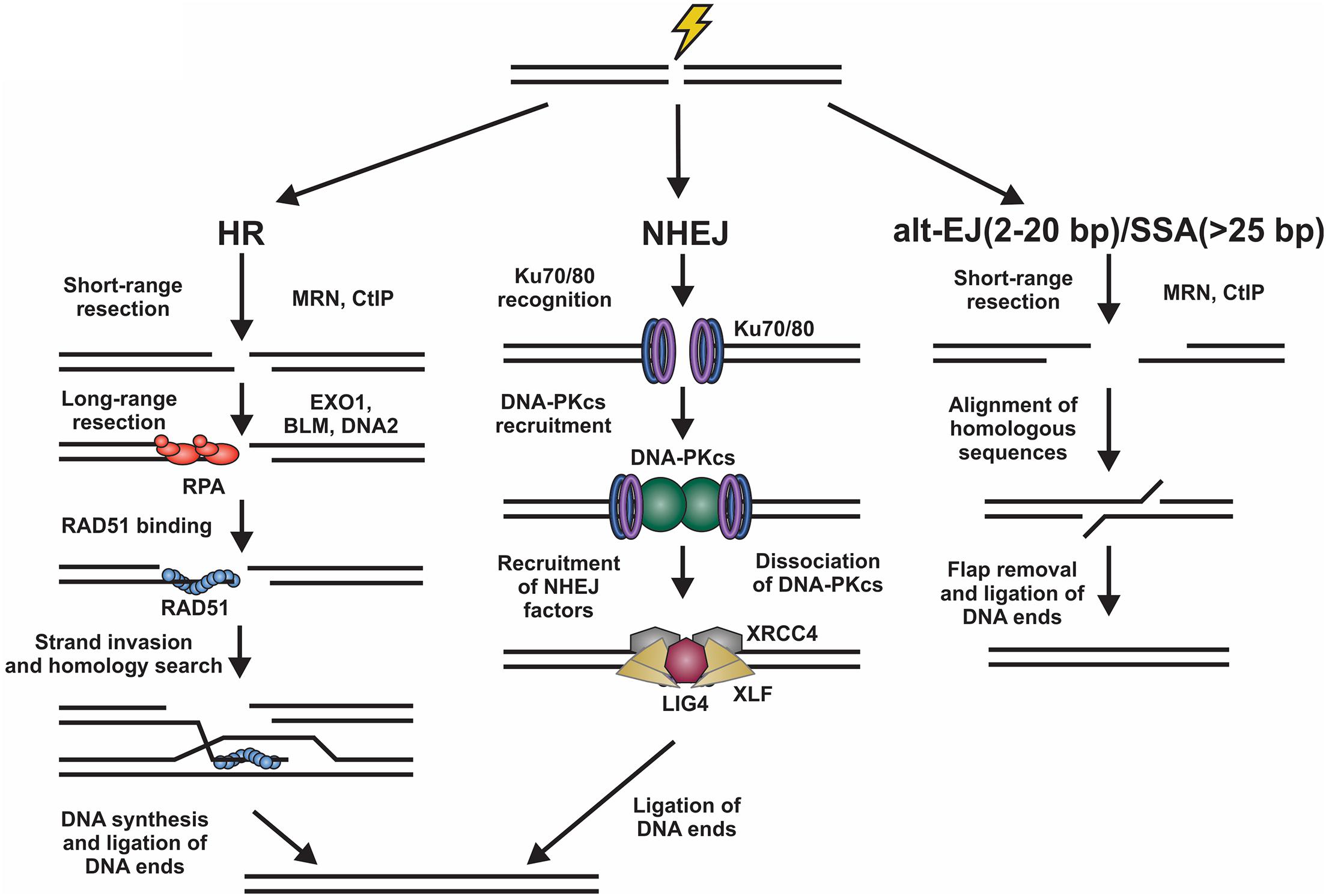
Figure 1. Overview of DSB repair pathways. Left, homologous recombination (HR) involves resection of the DNA ends by various nucleases. The ssDNA generated is then bound by RPA. Next, there is an exchange of RPA for RAD51, which facilitates the homology search and repair of the DSB. Center, for non-homologous end joining (NHEJ), the DNA ends are bound by Ku70/80 heterodimers promoting the binding of DNA-PKcs. This creates a binding platform for XRCC4, XLF, and LIG4, which facilitate ligation of the DNA ends. Right, DSBs can also be repaired by alternative-end joining (alt-EJ) or single strand annealing (SSA) pathways. These involve the use of short homologous sequences that are exposed by resection of the break. Following alignment, the DNA flaps are removed and the DNA ligated. Alt-EJ uses around 2 to 20 base pairs (bp) of homology and SSA > 25 bp to align sequences.
Homologous recombination is highly accurate and typically occurs in S/G2 phases of the cell cycle when a replicated sister chromatid is present (Burma et al., 2006; Sullivan and Bernstein, 2018). To initiate HR, the DNA ends are resected to generate long 3′ single-stranded (ss)DNA overhangs, which pair with homologous sequences. These templates are then used for DNA synthesis and repair of the break (Symington and Gautier, 2011). This process is mostly error-free, can repair protein-blocked ends and is facilitated by RAD51, a recombinase with ATPase activity which initiates strand invasion and DNA synthesis (Mehta and Haber, 2014). NHEJ, on the other hand, is fast, selective for two-ended DSBs, and often mutagenic (Ranjha et al., 2018; Stinson et al., 2020). Although NHEJ is active in all phases of the cell cycle, it occurs most frequently in G1 phase and repairs about 80% of IR-induced DSBs, making it the predominant repair pathway in mammalian cells (Burma et al., 2006; Beucher et al., 2009). To initiate NHEJ, the Ku70/80 heterodimer (hereafter referred to as Ku) and the DNA-PK catalytic subunit (DNA-PKcs) are recruited to damage sites to generate the DNA-PK holoenzyme (Gell and Jackson, 1999; Singleton et al., 1999; Jette and Lees-Miller, 2015). DNA-PK bridges the DNA ends creating a long-range synapse (Graham et al., 2016; Chen et al., 2021). Additional proteins X-Ray Repair Cross Complementing 4 (XRCC4), XRCC4-like factor (XLF) and DNA ligase 4 (LIG4) are recruited to align and ligate the DNA ends (Blackford and Jackson, 2017). To complicate matters, DSBs can also be repaired by alternative-end joining (alt-EJ; also known as DNA polymerase θ-mediated end joining) and single-strand annealing (SSA) pathways (Figure 1; McVey and Lee, 2008; Frit et al., 2014; Iliakis et al., 2015; Sallmyr and Tomkinson, 2018; Seol et al., 2018). Both pathways require some resection and utilize short regions of homology to pair the DNA ends together (Seol et al., 2018).
While DSBs must be quickly recognized and repaired to preserve genome stability, the natural chromosome ends, known as telomeres, must be protected from the DDR to prevent genome instability in the form of chromosome fusions and degradation. Telomeric DNA ranges in length from a few hundred base pairs in yeast to tens of kilobases in mammals (Blackburn, 1991; Greider, 1991). In humans, telomeres consist of short tandem 5′-TTAGGG-3′ repeats on the G-rich strand and complimentary 5′-CCCTAA-3′ repeats on the C-rich strand (Figure 2A). The G-rich strand also contains a 3′ ssDNA region referred to as the G-overhang (Makarov et al., 1997; McElligott and Wellinger, 1997). In mammals, telomeres are protected by the shelterin complex, comprised of telomere repeat-binding factors 1 and 2 (TRF1 and TRF2), repressor activator protein 1 (RAP1), TRF1-interacting nuclear factor 2 (TIN2), telomere protection protein 1 (TPP1) and protection of telomeres 1 (POT1) (Figure 2B; de Lange, 2018). Shelterin components have been identified in most eukaryotes, however, the number of known components can vary or shelterin subunits may be missing entirely, such as in Saccharomyces cerevisiae (de Lange, 2001, 2005). The duplex DNA is bound by TRF1 and TRF2/RAP1 whereas the G-overhang region is protected by POT1, which complexes with TPP1. Unlike humans, which contain a single POT1 gene, mice have two separate POT1 genes, POT1a and POT1b. These genes are proposed to have arisen from a duplication event (Hockemeyer et al., 2006). While clearly orthologous, POT1a and POT1b have evolved to provide slightly different activities in telomere protection (Hockemeyer et al., 2006; Wu et al., 2006; Palm et al., 2009; Kibe et al., 2010). POT1a has been shown to repress the DDR while POT1b controls 5′-end resection (Hockemeyer et al., 2006; Wu et al., 2006; Kibe et al., 2010). TPP1 interacts with TIN2 to bridge the double-stranded and single-stranded bound portions of shelterin. As described in more detail below, shelterin plays a critical role in telomere end protection and preventing the recognition of telomeres as DNA damage.
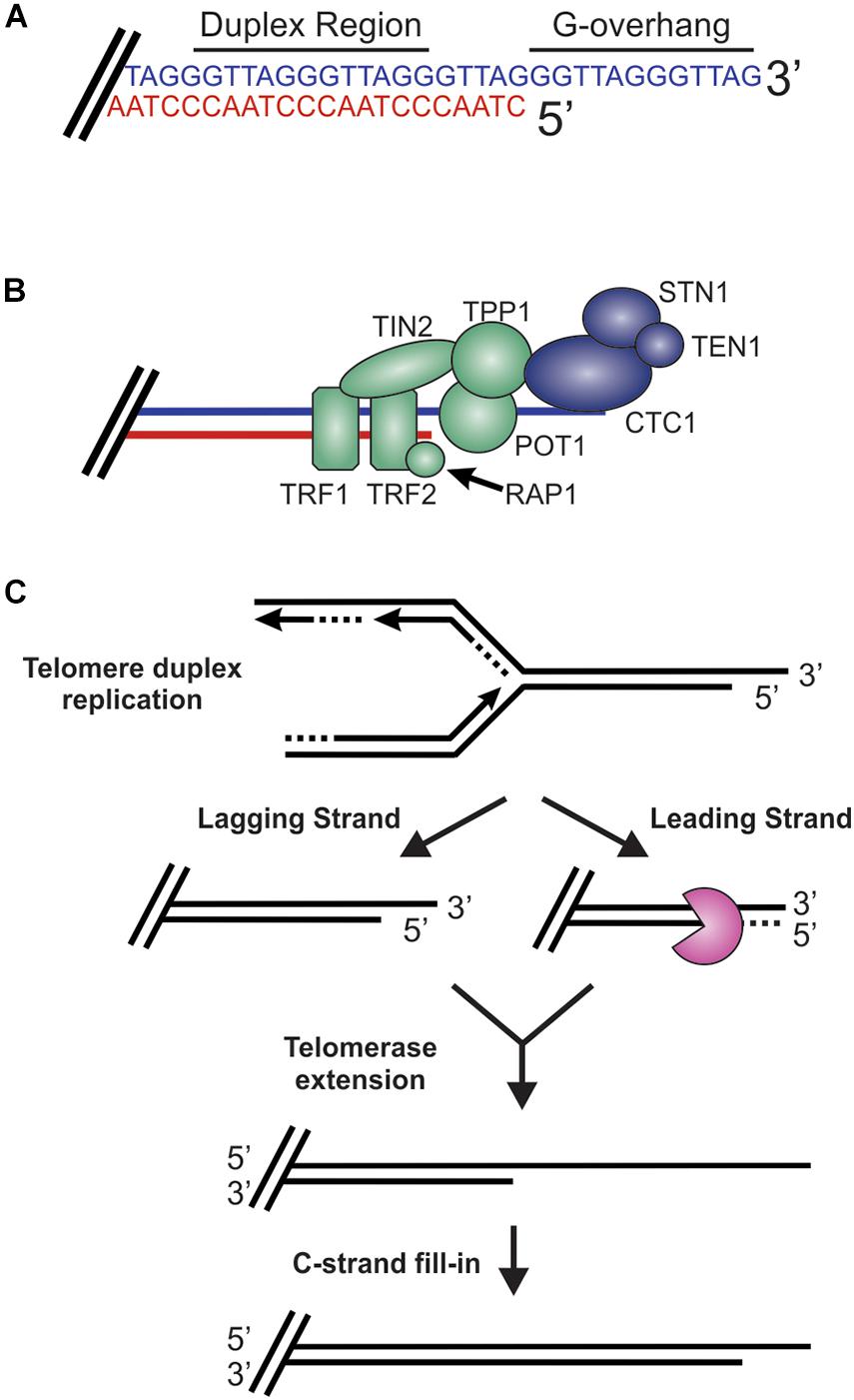
Figure 2. Overview of telomeres. (A) Telomeres consist of a repetitive DNA sequence that forms a duplex DNA region and a 3′ G-rich ssDNA overhang (C-strand, red: G-strand, blue). (B) Telomeres are bound by the shelterin complex (TRF1-TRF2-RAP1-TIN2-TPP1-POT1) and the CST complex (CTC1-STN1-TEN1), which aid in telomere maintenance. (C) Steps in telomere replication. First, the telomere duplex is replicated resulting in either a blunt end (leading strand replication) or an overhang (lagging strand replication). The leading strand end is then processed to generate a G-overhang. Telomerase then extends the G-overhangs followed by C-strand fill-in to convert most of the ssDNA to duplex DNA, leaving a short G-overhang.
During S-phase, telomeres are replicated in three distinct steps (Figure 2C; Stewart et al., 2012). First, the duplex DNA is replicated by the conventional replication machinery. While replication on the leading strand is presumed to reach the chromosome terminus, the lagging strand machinery is unable to fully replicate the ends, a phenomenon known as the end-replication problem (Watson, 1972; Olovnikov, 1973). To overcome this, telomeres are extended by telomerase, which is recruited and stimulated by TPP1/POT1. Recent work suggests that TIN2 also mediates telomerase recruitment and functions with TPP1/POT1 to stimulate telomerase processivity (Frank et al., 2015; Pike et al., 2019). Prior to extension, telomeres are resected to create a binding site for telomerase. After extension, telomerase is then dissociated from the telomere by CTC1-STN1-TEN1 (CST), a replication protein A (RPA)-like ssDNA binding protein, to prevent extensive G-overhang elongation (Stewart et al., 2018; Lim and Cech, 2021). Both CST and RPA are heterotrimeric proteins that contain multiple ssDNA binding folds and recruit proteins to the DNA to perform various activities (Chen and Wold, 2014; Lim et al., 2020). Work in yeast also suggest that the Pif1 helicase may function to remove telomerase (Boule et al., 2005). CST is then proposed to stimulate DNA polymerase α-primase (pol α) to convert most of the G-overhang to duplex DNA (Giraud-Panis et al., 2010). The remaining short G-overhang can form a lariat structure called a telomere loop (t-loop) (Griffith et al., 1999). This structure is thought to protect the DNA terminus and restrict further access by telomerase, as discussed in more detail below. Telomeres can also be extended by a telomerase-independent mechanism that relies on recombination, a pathway known as alternative lengthening of telomeres (ALT) (Cesare and Reddel, 2010).
Together, telomerase and ALT make up the telomere maintenance pathways, which can coexist in vivo (Perrem et al., 2001). However, telomerase is the predominant elongation pathway under normal conditions. In humans, telomerase expression is typically restricted to germline and stem cells with most somatic cells having a finite number of cellular divisions. Once telomeres become critically short, cells lose the ability to divide, a state known as replicative senescence (Munoz-Espin and Serrano, 2014). Telomere shortening is associated with normal aging, and premature shortening is associated with a number of premature aging-related diseases (Armanios and Blackburn, 2012). A hallmark of cancer is replicative immortality; thus, pre-cancerous cells must maintain telomere length to prevent senescence (Hanahan and Weinberg, 2011). More specifically, 85 to 90% of human tumors re-express or upregulate telomerase while 10 to 15% maintain telomeres through ALT (Shay and Wright, 2019).
DSBs and telomeres resemble each other in many ways. Both are terminal DNA ends with a ssDNA overhang or blunt end. When such substrates arise in cells, decisions on whether or not to repair the DNA must be made. These decisions are often critical to maintaining genome stability with incorrect decisions potentially leading to cell death or chromosome instability. Accordingly, each decision point is highly regulated to ensure the proper repair pathway is engaged, or, in the case of chromosome ends, prevented. Many of these decision points are reversible, allowing a way back should the incorrect decision be made, or downstream factors are not available. However, the initial pathway choice often dictates the mechanism of repair. In this review, we will focus on these decision points and how they are regulated in mammals. Since HR-mediated DSB repair requires the most processing, it will be used as the focal point on which to frame decisions that direct pathway choice, including the mechanisms that shift repair toward HR vs. NHEJ and those protecting telomeres from “repair.”
Mechanisms Regulating DSB Repair and End Protection
Based on current understanding, there are at least four key decision points required for HR-mediated DSB repair (Figure 3). First, the DNA ends are recognized and bound by the MRE11-RAD50-NBS1 (MRN) complex to initiate DNA repair and recruit the repair machinery. Second, the break is subjected to short-range resection by MRN. Third, long-range resection occurs to generate an overhang that is bound by RPA. Finally, RPA is replaced by RAD51, which formally initiates the homology search and HR-mediated repair. At each of these decision points, it is crucial to recognize whether the DNA end is a bona fide DSB vs. a chromosome terminus as well as whether a homologous sister chromatid is present. This will dictate how the DNA ends are processed and what factors are recruited. Below, we will broadly discuss each of these decision points and the factors regulating the choice to join or not join the DNA ends. For more detailed assessments of individual decisions points, we refer readers to several recent reviews (de Lange, 2018; Pannunzio et al., 2018; Wright et al., 2018; Krenning et al., 2019; Wu, 2019; Blackford and Stucki, 2020; Ensminger and Lobrich, 2020; Vitor et al., 2020; Yue et al., 2020; Panigrahi and Glover, 2021).
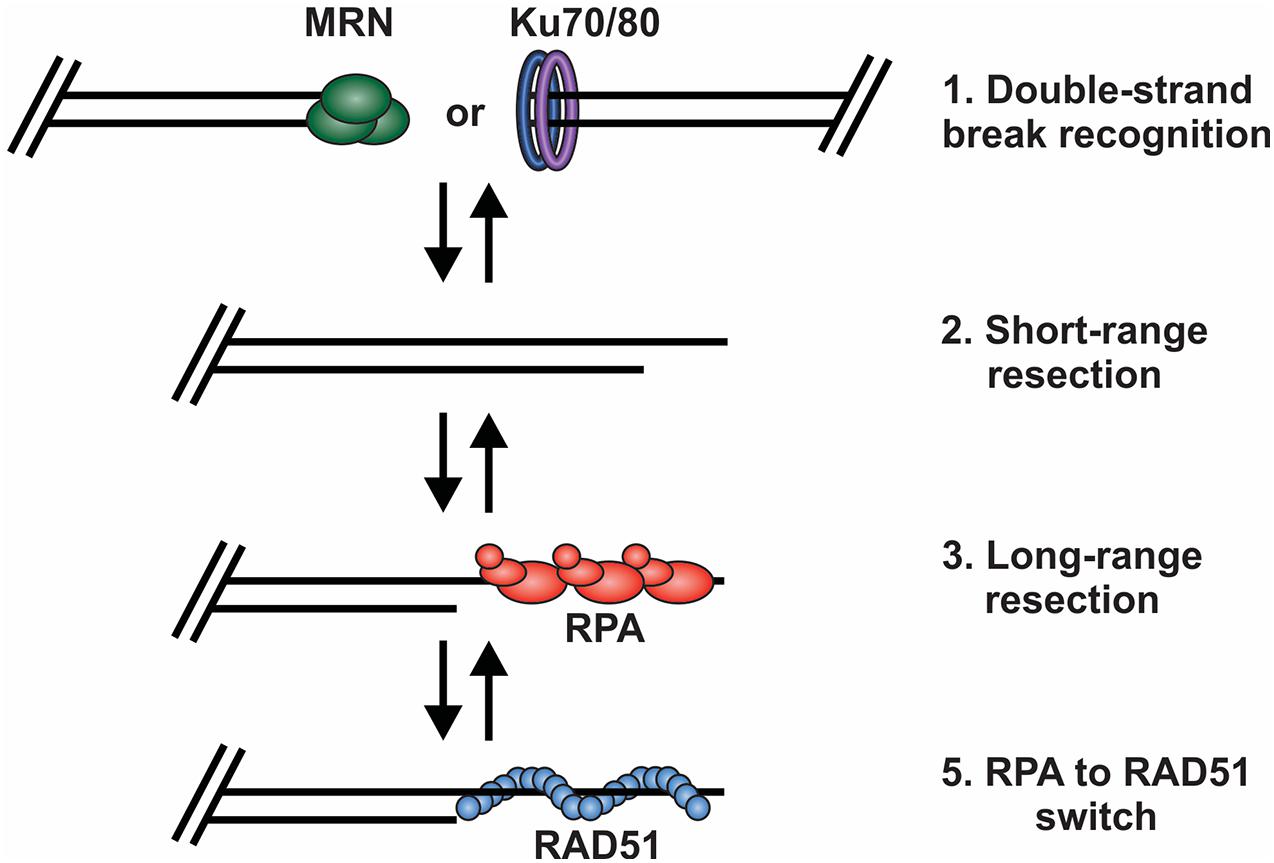
Figure 3. Key decision points in the repair of DSBs by HR. (1) The DNA ends are bound by either MRN or the Ku70/80 heterodimer. Binding and retention of MRN will shift repair toward HR and the binding of Ku shifts repair toward to NHEJ. (2) Short range resection by MRN. (3) Long-range resection of the DNA and RPA binding. (4) RPA is exchanged for RAD51, which facilitates strand invasion, DNA synthesis and HR repair.
DSB Recognition
In response to DSBs, the lesion must first be recognized by DNA damage sensors. Ku binding is traditionally associated with repair by NHEJ whereas MRN is associated with HR-mediated repair. While still not completely understood, recent work, in both yeast and mammals, suggest that the recruitment and binding of these sensors is context dependent and not mutually exclusive (Langerak et al., 2011; Ingram et al., 2019; Chen et al., 2021). Instead, it is the subsequent steps that determine the displacement of these factors to promote HR, NHEJ or end protection. Recent biochemical and single molecule studies even suggest that Ku binding may be required for MRN-dependent resection (Deshpande et al., 2020). Much of this groundbreaking work has been performed in the model organism S. cerevisiae and then subsequently verified in mammals and other organisms. A major distinction between budding yeast and mammals is that NBS1 is not conserved in yeast. Instead, the S. cerevisiae complex is composed of Mre11, Rad50 and Xrs2 (MRX) with Xrs2 being the functional homolog of NBS1 (Rupnik et al., 2010; Tisi et al., 2020). For simplicity, we will use the designation MRN unless referring to studies exclusively performed in S. cerevisiae.
HR
MRN is one of the first responders to a DSB and, thus, it is key to instigating downstream steps in the repair process (Lisby et al., 2004). While recognition of the break by MRN is still under investigation, in vitro single molecule studies suggest that MRN uses facilitated 1D diffusion to search along nucleosome-bound DNA for DSBs (Myler et al., 2017). MRN can function at both unblocked and blocked DNA ends to promote resection of the DSB (Figure 4). At blocked ends, MRN can remove Ku as well as other protein-DNA adducts to access the break. MRN also promotes the recruitment and stimulation of ATM at the DSB, which in turn promotes H2AX phosphorylation on S139 (γH2AX) (Carney et al., 1998; Lee and Paull, 2004, 2005). Mediator of DNA damage checkpoint 1 (MDC1) is then recruited by γH2AX around the break and acts as a bridge between ATM and γH2AX to create a positive feedback loop (Burma et al., 2001; Kolas et al., 2007). Further expansion of γH2AX leads to the recruitment of additional downstream repair factors, the initiation of cell cycle arrest and resection of the DNA (Stewart et al., 2003; Stucki et al., 2005; Lou et al., 2006).
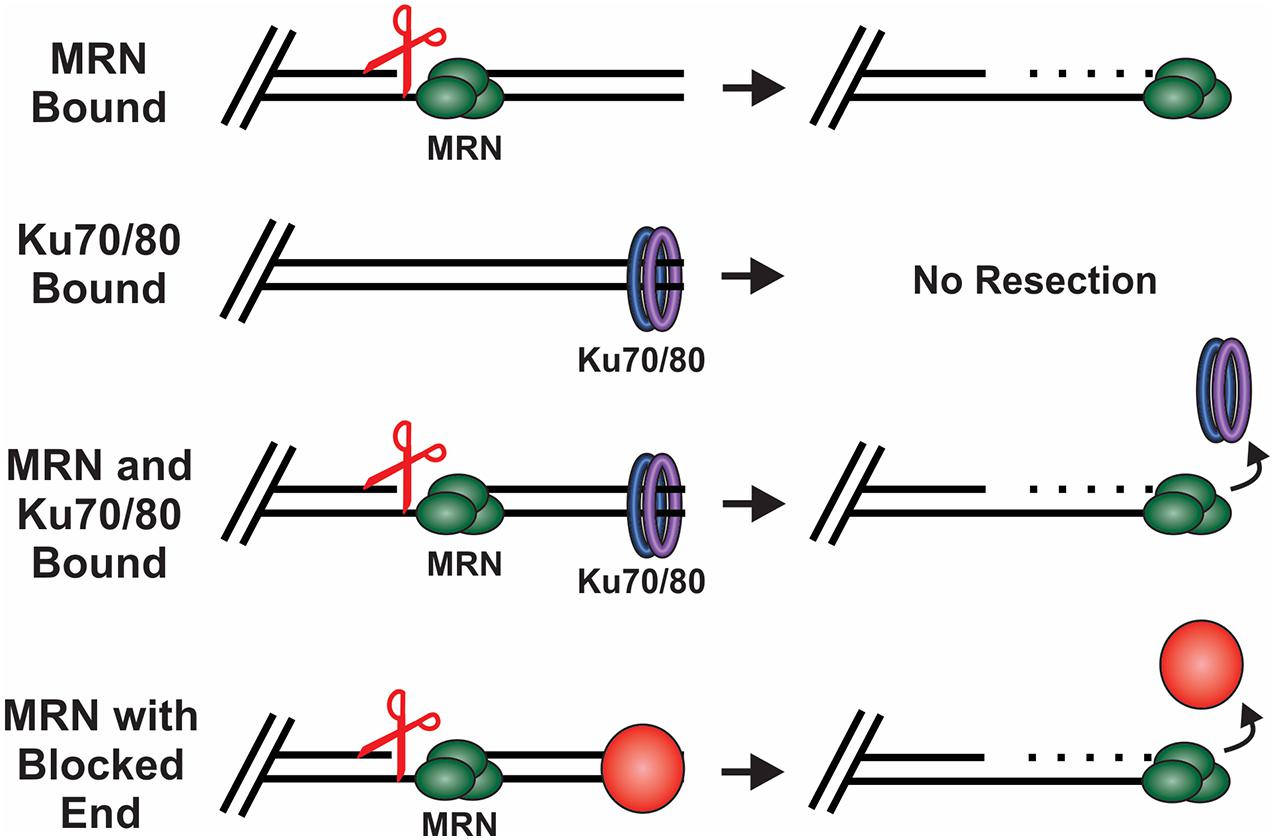
Figure 4. How DSB ends are processed under different conditions. Scissors indicate MRN endonuclease activity. MRN binding to unblocked ends leads to resection of the DNA. When Ku binds in the absence of MRN, no resection occurs. However, when both Ku and MRN are bound, stimulation of the MRN nuclease activity promotes resection and the removal of Ku. MRN can similarly function at ends blocked by a protein adduct, damaged bases or DNA secondary structures.
NHEJ
Like MRN, Ku is a first responder at DSBs and provides a docking site for DNA-PKcs (Yaneva et al., 1997). Unlike MRN, which can bind internally, Ku requires a free DNA end for binding and cannot associate with most blocked ends (Blier et al., 1993; Myler et al., 2017). Accordingly, when NHEJ is the preferred pathway, such as in G1, blocked ends must be freed to allow Ku binding (Mirman and de Lange, 2020). How these blocks are removed is still under investigation, but several nucleases, including tyrosyl-DNA phosphodiesterase 1 and 2 (TDP1/2) and Artemis, can remove hairpins, damaged bases or protein-DNA adducts (Menon and Povirk, 2016; Pannunzio et al., 2018; Meek, 2020). DNA blocks can also be removed through the stimulation of Artemis by DNA-PKcs (Gerodimos et al., 2017). Interestingly, in the event of a nucleosome blocked end, an in vitro study found that Ku can displace histone H1 from the DNA, however, it does not displace the nucleosome (Roberts and Ramsden, 2007). This could expose DNA ends to allow Ku binding, but more work is needed to uncover other possible roles of Ku in unblocking these DNA ends.
Whether Ku remains bound at the break appears to be one of the most critical steps in preventing resection and shifting repair outcomes toward NHEJ vs. HR. For Ku removal, several pathways can be employed to promote HR or, in the case of telomeres, end protection. These include the dissociation of Ku through short-range resection (discussed in more detail below), phosphorylation of Ku by DNA-PKcs, ubiquitination of Ku by RNF8 or RNF138 and blockage of DNA-PKcs autophosphorylation (Feng and Chen, 2012; Ismail et al., 2015; Lee et al., 2016). Autophosphorylation of DNA-PKcs is promoted through its interaction with the TIP60 histone acetyltransferase, which stimulate the activity of DNA-PKcs and recruitment of the ligation machinery (Ding et al., 2003). To block pro-NHEJ activity during S-phase, breast cancer gene 1 (BRCA1) directly blocks DNA-PKcs autophosphorylation (Davis et al., 2014). SUMOylation of TIP60 has also been proposed to inhibition autophosphorylation and facilitate a switch toward HR (Gao et al., 2020). Additionally, when homologous sequences are available during S and G2 phases, DNA-PKcs autophosphorylation favors the binding of MRN and other HR factors (Ding et al., 2003; Zhou and Paull, 2013; Symington, 2016). MRN can stimulate resection in the presence of Ku and DNA-PKcs through the recruitment of exonuclease 1 (EXO1) (Zhou and Paull, 2013). This allows the recruitment of EXO1 to DNA ends to promote HR rather than NHEJ, through mechanisms that are still poorly understood.
Telomeres
Although both Ku and MRN have been positively implicated in telomere maintenance (Wang et al., 2009; Lamarche et al., 2010), the exclusion of Ku and MRN from telomeres is one mechanism used to prevent the misrepair of chromosome ends. Mounting evidence indicates that t-loops serve a major role in blocking Ku and MRN access to chromosome ends (Figure 5). Recent analysis using super-resolution microscopy have helped define the mechanism of t-loop formation (Doksani et al., 2013; de Lange, 2018; Van Ly et al., 2018). This process is mediated by TRF2 and involves invasion by the G-overhang into duplex DNA to create a large lariat structure (Griffith et al., 1999; Doksani et al., 2013). While t-loops prevent initial recognition of telomeres as DSBs, these elegant structures must also be protected from the HR machinery. At the base of the t-loop, the DNA is presumed to form a Holliday junction (HJ)-like structure that can be cleaved by HJ resolvases, leading to telomere loss. Again, TRF2 is involved in preventing t-loop cleavage through inhibition of the Werner syndrome (WRN) helicase (Nora et al., 2010). This prevents WRN strand displacement of HJs with telomeric repeats (Nora et al., 2010). RAP1 has also been implicated in t-loop protection although some of the reports are conflicting (Bae and Baumann, 2007; Cesare et al., 2008; Benarroch-Popivker et al., 2016).
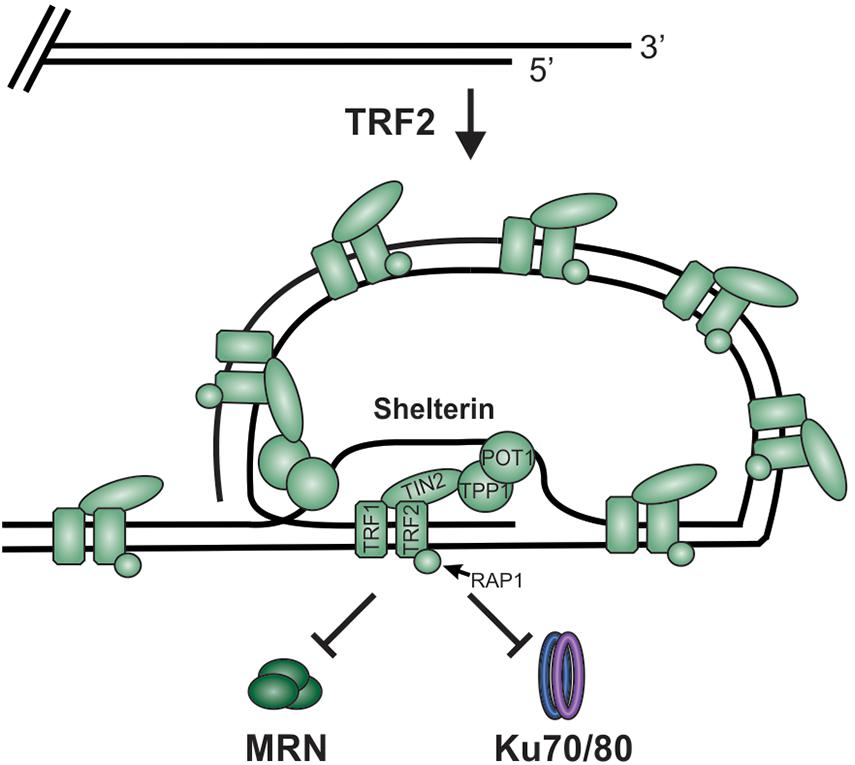
Figure 5. TRF2 facilitates telomere loop (t-loop) formation by promoting invasion of the G-overhang into the duplex region to form a displacement loop (D-loop). This t-loop combined with shelterin blocks MRN and Ku from accessing the chromosome ends and initiating DSB repair.
TRF2 deletion in mice results in removal of the 3′-ssDNA overhang by MRE11 and the loss of t-loops, promoting the “repair” of dysfunctional telomeres and chromosomal fusions (Attwooll et al., 2009; Deng et al., 2009; Cesare et al., 2013). When TRF2 is absent, ATM is phosphorylated in an MRN-dependent manner. However, the nuclease activity of MRE11 is dispensable, suggesting that MRN association alone is sufficient to recruit ATM to telomeres. Additionally, upon inhibition of TRF2, excision repair cross-complementation group 1 (ERCC1) and ERCC4 (also known as XPF) target the telomeric overhang for degradation (Zhu et al., 2003). This indicates a previously unexpected role of ERCC1/ERCC4 in the “repair” of unprotected telomeres through NHEJ. Interestingly, during G2, unprotected telomeres lacking a G-overhang are bound by Ku, which leads to chromosome fusions, although the DDR and formation of end-to-end fusions is functionally distinct from NHEJ (Zhu et al., 2003). In this case, RNF8 promotes the accumulation of ubiquitinated H2A, which in turn, recruits p53-binding protein 1 (53BP1), ATM and REV7 to promote DNA ligase IV-dependent NHEJ (Smogorzewska et al., 2002; Peuscher and Jacobs, 2011; Boersma et al., 2015).
Short-Range Resection
Once a DSB is recognized by MRN, resection can be initiated to promote HR. This occurs in a two-step process with short-range resection by MRN followed by long-range resection to allow RPA-binding (Shibata et al., 2014). Once resection initiates, HR is enabled. This was previously seen as a point of no return. However, recent studies suggest that this decision point may be more flexible than initially imagined. Furthermore, resection is an essential step in telomere elongation so, under these conditions, resection needs to be achieved without engaging HR. In this section, the mechanisms regulating short-range resection will be discussed.
HR
Biochemical and single-molecule studies have significantly contributed to our understating of both short- and long-range resection in recent years. Combined with the many genetic studies performed in both yeast and mammals, the following model of short-range resection has emerged (Figure 6). First, MRE11 forms a nick in the dsDNA 20 to 40 nt from the break (Anand et al., 2016). Interaction between CtBP interacting protein (CtIP) (Sae2 in budding yeast) and MRN is essential for short-range resection (Huertas et al., 2008; Huertas and Jackson, 2009). MRE11 is a 3′-to-5′ exonuclease with weak endonuclease activity. Since resection creates a 3′-ssDNA overhang, it remained unclear for many years how resection was achieved through MRN. However, CtIP was discovered to stimulate the endonuclease activity of MRE11, allowing it to nick the dsDNA and create a template for MRN endonuclease activity (Anand et al., 2016). CtIP localization to DSBs is regulated by cyclin dependent kinase (CDK), BRCA1 and ATM (Yu and Chen, 2004; Yun and Hiom, 2009; Wang et al., 2013). In addition to regulating CtIP, CDKs also regulates other key factors involved in resection, checkpoint activation and downstream steps in the recombination process, making CDKs a vital player in pathway choice (Ferretti et al., 2013; Zhao et al., 2017). Since CDKs are cell cycle regulated, this helps prevent HR outside of S and G2 phase, which can have disastrous consequences.
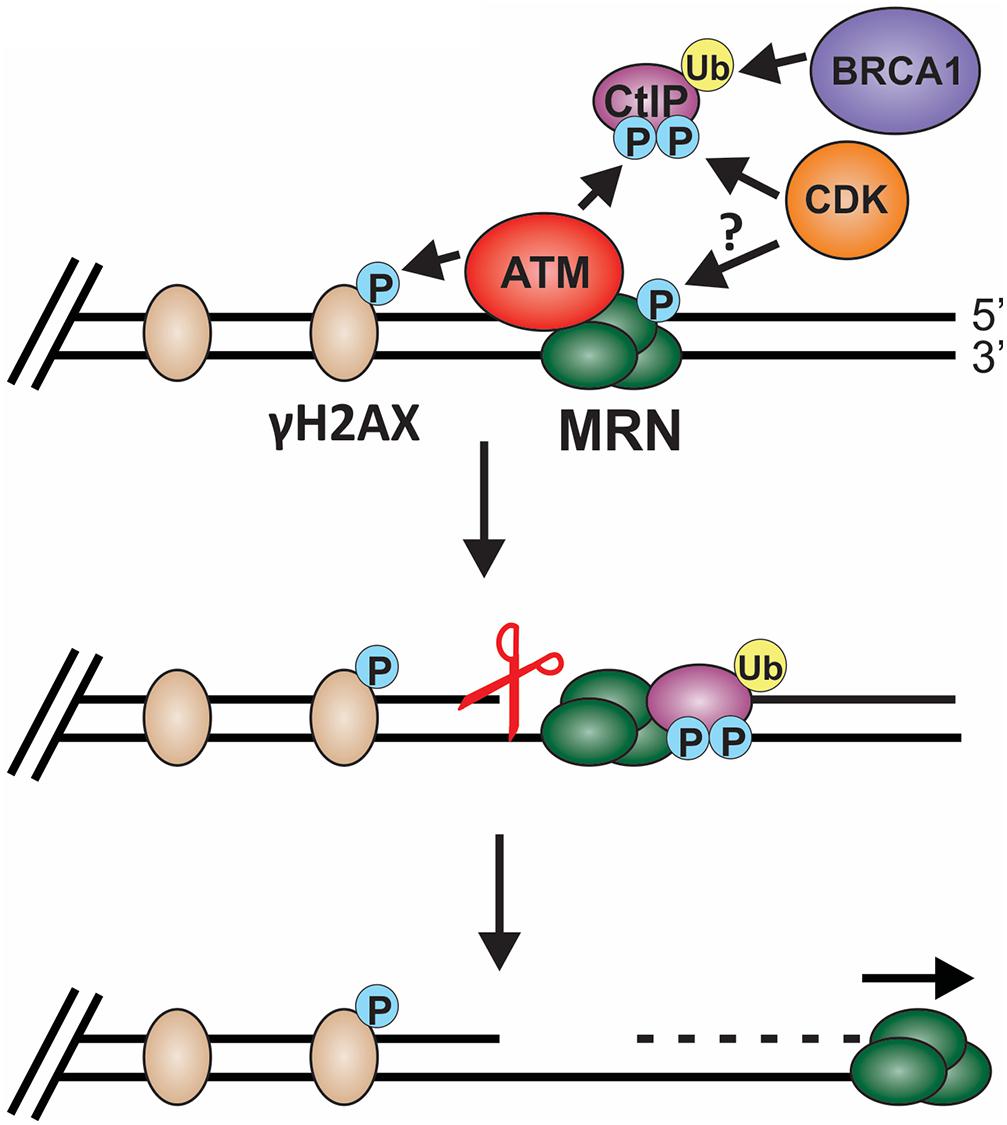
Figure 6. Model of short-range resection. Short-range resection by MRN is stimulated by CtIP. Localization of CtIP to the chromatin is dependent on phosphorylation of CtIP by CDK and ATM and ubiquitination by BRCA1. CDK has also been proposed to phosphorylate MRX in budding yeast in a cell cycle-dependent manner, suggesting that CDK may regulate MRN in mammals. Phosphorylation of H2AX by ATM also promotes short-range resection and the recruitment of DSB machinery. Upon localization with MRN, CtIP stimulates both MRN endonuclease (indicated by the scissors) and 3′-to-5′ exonuclease activity to facilitate short-range resection. (P: phosphorylation; Ub: ubiquitination).
Stimulation of MRE11 nuclease activity may also be regulated by cell cycle and apoptosis regulator protein 2 (CCAR2) (Lopez-Saavedra et al., 2016). Work by López-Saavedra et al. suggests that MRN and CtIP are found in an inactive complex with CCAR2 and, upon DNA damage, phosphorylation of CtIP disrupts the CCAR2-CtIP-MRN complex to promote MRN nuclease activity. A complex of BRCA1 and BRCA1-associated RING domain-1 (BARD1) as well as exonuclease 3′-5′ domain-containing protein 2 (EXD2) also stimulate MRN-dependent nuclease activity and resection (Wang et al., 2007; Coleman and Greenberg, 2011; Broderick et al., 2016; Nieminuszczy et al., 2016). Recently, the lysine specific histone demethylase, PHF2 (KDM7C/JHDM1E), was shown to regulate CtIP and BRCA1 mRNA levels, suggesting an additional layer of regulation (Alonso-de Vega et al., 2020).
Once the nick is formed, MRE11 engages its 3′-to-5′ exonuclease activity to resect back toward the break (Shibata et al., 2014). This activity is also stimulated by CtIP, which is recruited by NBS1 (Wang et al., 2013; Anand et al., 2019). Additionally, the RAD50 subunit of MRN moderates MRE11 nuclease activity, indicating that MRN complex formation is critical to support short-range resection (Cannavo et al., 2019). The innerworkings of MRE11, RAD50 and NBS1 complex formation and activity has been well studied and reviewed extensively elsewhere (Lafrance-Vanasse et al., 2015; Reginato and Cejka, 2020; Qiu and Huang, 2021). Short-range resection by MRN is proposed to remove obstacles, such as Ku and other DNA adducts, freeing the DNA ends for repair by HR, alt-EJ or SSA (Figure 4; Chanut et al., 2016). In yeast, MRX works not only on blunt ends but also on chemically modified DNA, short overhangs and DNA containing secondary structures (Cejka, 2015).
NHEJ
Ku binding at DSBs serves as the major mechanism to block end resection (Rothkamm et al., 2003; Zhou and Paull, 2013; Ceccaldi et al., 2016; Graham et al., 2016; Mladenov et al., 2019). Accordingly, the decision to proceed toward NHEJ requires the prevention of MRN-dependent resection so that Ku remains bound. This can be achieved in several different ways. First, ATM limits resection by phosphorylating Ubiquilin 4 (UBQLN4), a proteasomal shuttle factor, which leads to MRE11 degradation (Jachimowicz et al., 2019). Thus, ATM plays seemingly contradictory roles in short-range resection. On the one hand, it promotes MRN-CtIP-BRCA1 recruitment to DSBs, while on the other, it promotes MRE11 degradation. Future work is needed to fully understand the role of ATM in DSB repair, but context (e.g., cell cycle stage, location of the break, etc.) and the localization of other factors likely contribute to the use of ATM in NHEJ vs. HR. Another proposed mechanism to inhibit Ku removal is preventing MRN localization to the break site. While such a mechanism has not been directly demonstrated, cell cycle dependent phosphorylation of MRE11 by CDK1 was observed in S. cerevisiae and could serve to prevent MRE11 localization to DSBs (Simoneau et al., 2014). Once bound, MRN requires CtIP and other pro-resection factors to stimulate MRE11 nuclease activities. Thus, regulation of these factors can prevent resection and Ku removal. One strategy used to limit MRN nuclease activity is keeping CtIP levels low in G1 (Yu and Baer, 2000). Additionally, CtIP localization to DSBs is regulated by CDKs (CDK1 in yeast and CDK2 in mammals), as mentioned above. CDK-dependent phosphorylation of CtIP precipitates BRCA1 ubiquitination and CtIP localization to DSBs (Figure 6; Yun and Hiom, 2009; Wang et al., 2013). These modifications are mainly restricted to S/G2, limiting CtIP interaction with MRN. Interestingly, a recent report also found that long-term ATR kinase inhibition or conditional topoisomerase II binding protein 1 (TopBP1) degradation affects E2F-dependent transcription of BRCA1, CtIP and Bloom syndrome protein (BLM) (Dibitetto et al., 2020). Loss of these pro-resection factors results in impaired end resection and a shift toward toxic NHEJ, suggesting that translational regulation of pro-resection factors could also serve to regulate resection.
While NHEJ relies on the suppression of end resection for repair, in situations where NHEJ is impaired, alt-EJ and SSA works to join DSBs in a resection-dependent and Ku-independent manner (Figure 1; Ceccaldi et al., 2016). Generally, in these pathways, the DSB is first resected to expose ssDNA that varies in length depending on the pathway. In both pathways, MRN and CtIP initiate resection (Bennardo et al., 2008; Dinkelmann et al., 2009; Xie et al., 2009; Yun and Hiom, 2009; Zhang and Jasin, 2011; Truong et al., 2013; Biehs et al., 2017). If this initial end resection occurs in G1, it inhibits NHEJ and drives repair toward alt-EJ or SSA (Xiong et al., 2015; Bakr et al., 2016). Unlike HR, alt-EJ and SSA can repair DSBs during G1 of the cell cycle because a homologous sister chromatid is not required (Xiong et al., 2015; Sallmyr and Tomkinson, 2018). Alt-EJ is characterized by limited resection and short 2 to 20 nt regions of complementary sequences, while resection in SSA is longer and generally requires sequences of > 25 nt (Sallmyr and Tomkinson, 2018). After resection, the DNA ends are used to align short homologous sequences for repair. Non-homologous 3′ tails are then removed, gaps filled and the ends are ligated to complete repair (Figure 1).
Telomeres
During replication and telomere extension, telomeres are susceptible to recognition as DSBs and degradation. This is because the t-loop must be resolved to access the chromosome end (Figure 7). Furthermore, proteins involved in resection, including CtIP, EXO1 and DNA2, are involved in telomere duplex replication and could potentially act on chromosome ends (Lin et al., 2013; Stroik et al., 2019, 2020). On the leading strand, replication can theoretically proceed to the end of the chromosome to create a blunt end. This model is supported by studies in yeast, plants and mammals, where blunt-ends or very short (1 to 2 nt) overhangs have been identified after duplex replication (Chow et al., 2012; Valuchova et al., 2017). On the lagging strand, the RNA primer of the last Okazaki fragment must be removed, leading to a loss of at least 10 to 12 nt of DNA each replication cycle. However, analysis of G-overhangs suggests that the final Okazaki fragment actually initiates 70 to 100 nt from the chromosome terminus (Chow et al., 2012). Accordingly, the leading strand sister chromatid resembles a blunt-end or “clean” one-sided break whereas the lagging strand sister chromatid contains a significant ssDNA overhang, comparable to a resected DSB.
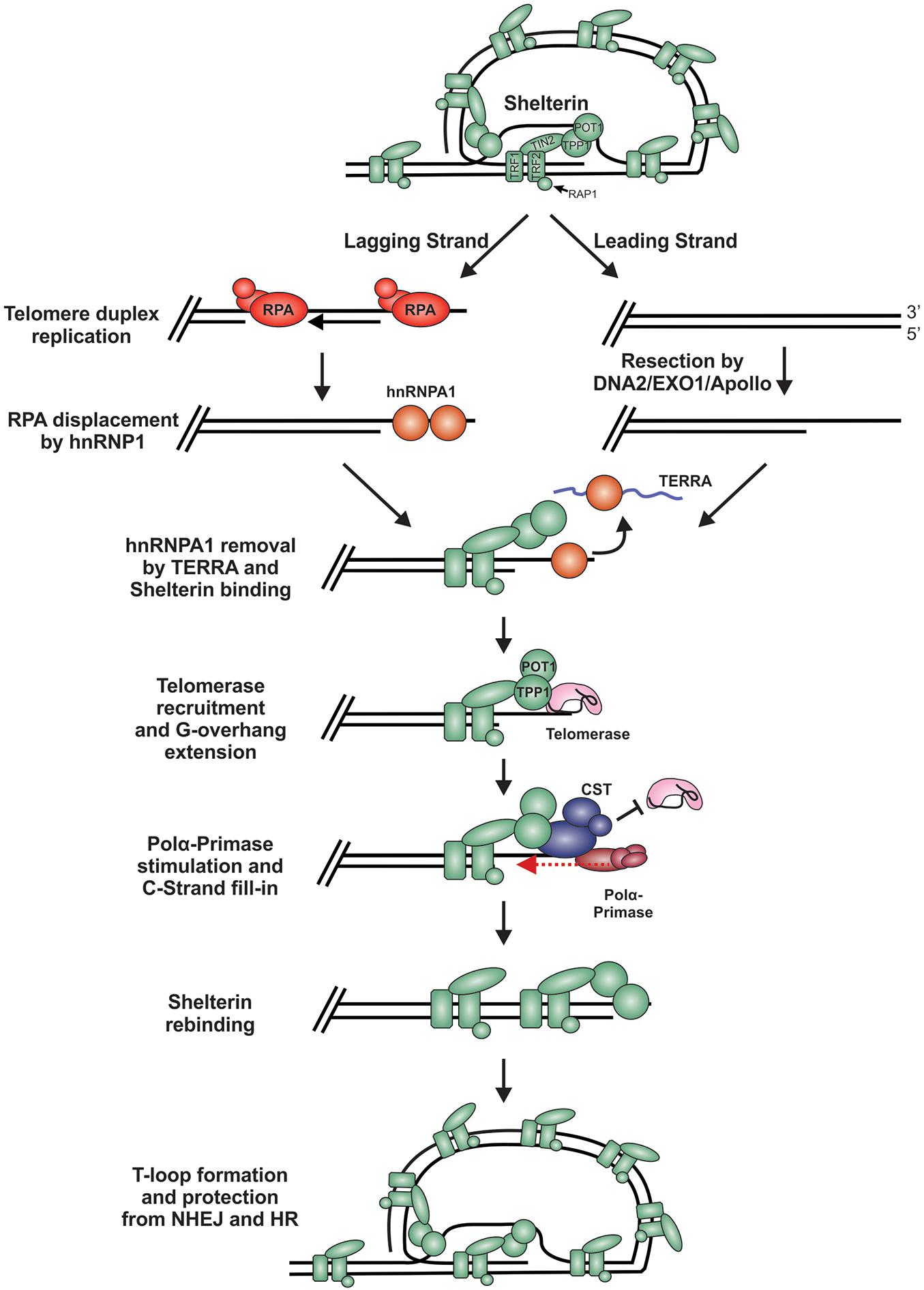
Figure 7. Model of telomere replication and end protection. During telomere replication, t-loops are resolved exposing chromosome ends to potential repair mechanisms. On the lagging strand, regions of ssDNA are bound by RPA as part of the normal replication process. To prevent stable RPA binding, an RPA-to-POT1 switch is facilitated by hnRNPA1. TERRA then removes hnRNPA1 to allow POT1 binding. On the leading strand, blunt telomere ends are resected by DNA2, EXO1 and/or Apollo to generate a G-overhang. (RPA may also bind to these ends and require removal). Telomerase is localized to the telomeres through its interaction with TPP1 to extend the G-overhang. To prevent G-overhang hyperextension, CST is localized to telomeres by TPP1 and inhibits telomerase activity. Additionally, CST promotes C-strand fill-in by stimulating pol α. After telomere processing, t-loops are reformed to protect the chromosome end from DSB repair.
In telomerase positive cells, the 3′-ssDNA overhang is extended in a highly controlled process to prevent potentially catastrophic events that may arise from telomere under- or over-extension. For extension by telomerase, a ssDNA binding site is needed to allow telomerase RNA component (TERC) to base pair, creating a primer for reverse transcription. While the lagging strand has a ready-made overhang, the leading strand requires processing. Studies in mice suggest that Apollo and EXO1 facilitate resection of the telomeric C-strand (van Overbeek and de Lange, 2006; Lam et al., 2010; Wu et al., 2010, 2012; Chow et al., 2012). In yeast, Dna2 has also been implicated in end processing, although it has been difficult to separate its role in duplex replication vs. end resection (Tomita et al., 2004; Budd and Campbell, 2013; Markiewicz-Potoczny et al., 2018). Currently, how Apollo, EXO1 and/or DNA2 are recruited for telomere end resection and whether the ends are immediately bound and protected by shelterin, following duplex replication, remains unclear. However, Apollo recruitment requires interaction with TRF2 and is subsequently blocked by POT1b (van Overbeek and de Lange, 2006; Wu et al., 2012). Therefore, shelterin, or at least shelterin subunits, are likely to bind immediately after passage of the replication fork, which could prevent recognition of the ends as DSBs.
T-loops are thought to serve as a major deterrent to HR and NHEJ. How t-loops are resolved to allow replication remains largely unknown but recent work by the Boulton group identified key steps in the assembly and disassembly process (Sarek et al., 2019). Outside of S-phase, t-loop formation is maintained by phosphorylation of TRF2 by CDK. During S-phase, TRF2 is transiently dephosphorylated by protein phosphatase 6 regulator subunit 3 (PP6R3), allowing t-loop disassembly and the completion of DNA replication. This t-loop disassembly appears to be very transient and another study suggests that t-loops may be present during S-phase (Timashev and De Lange, 2020). Thus, defining how this switch is regulated, and specifically how the DDR is inhibited during this period, warrants further investigation.
Long-Range Resection
Short-range resection leads to additional processing to generate a long ssDNA overhang that becomes a substrate for RPA binding and the promotion of HR. The switch from short- to long-range resection, like other steps, is highly regulated and often described as a point of no return. However, as described in more detail below, mechanisms do exist to shift repair back to NHEJ, making this decision point reversible under certain conditions. Nevertheless, long-range resection and RPA binding are one of the most critical points in the pathway to joining DNA ends through HR vs. NHEJ. Moreover, telomeric G-overhangs must be protected from RPA binding and unwanted HR.
HR
The goal of long-range resection is to create a stretch of ssDNA long enough for significant RPA binding. RPA is then exchanged for RAD51, which facilitates the homology search. Critical to the initiation of long-range resection is the binding of BRCA1 and blockage of 53BP1 (Daley and Sung, 2014). Generation of the 3′-overhang by MRN likely leads to initial RPA binding and ATR recruitment. One model suggests an ATM to ATR switch in which ATM initiates resection and triggers ATR activation to regulate later steps in HR (Cuadrado et al., 2006; Shiotani and Zou, 2009). It is proposed that ATR drives HR by facilitating the stabilization of BRCA1 through TopBP1. This counteracts 53BP1 recruitment (Liu et al., 2017). BRCA1 and BARD1 then form a complex, which stabilizes BRCA1, to facilitate resection and recruit the partner and localizer of BRCA2 (PALB2)-BRCA2 complex for RAD51 loading (Joukov et al., 2006; Sy et al., 2009; Zhang et al., 2009b; Orthwein et al., 2015). The BRCA1-BARD1 complex also acts as an E3 ubiquitin ligase to prevent 53BP1 localization at DSBs during S phase (Chapman et al., 2012; Kakarougkas et al., 2013). While BRCA1 is not intrinsically required for resection, it is critical to overcome the 53BP1-mediated block and activation of DSB processing by facilitating CtIP phosphorylation (Cao et al., 2009; Bunting et al., 2010, 2012; Peterson et al., 2013; Nacson et al., 2018).
Long-range resection is primarily executed by two distinct pathways (Figure 8A; Nimonkar et al., 2011). The first involves EXO1, a 5′-to-3′ exonuclease (Tomimatsu et al., 2012; Myler et al., 2016). In this pathway, EXO1 enters at the 5′ site and generates an overhang several kilobases in length. In vitro biochemical assays have determined that EXO1 alone can perform end resection, although the process is slow (Soniat et al., 2019). Based on genetic and in vitro studies, MRN and the BLM helicase (Sgs1 in budding yeast) are proposed to recruit EXO1 and stimulate end resection (Mohaghegh et al., 2001; Nimonkar et al., 2011; Soniat et al., 2019). A second mechanism involves the joint effort of DNA2 and BLM. DNA2 is a structure-specific endonuclease that also possesses weak ATP-dependent helicase activity (Zheng et al., 2020). Since DNA2 does not possess exonuclease activity, the DNA is displaced into a flap structure for cleavage. Owing to the weak helicase activity of DNA2, current thinking, backed, by in vitro single-molecule studies, posits that BLM is required to create a 5′-DNA flap for cleavage (Mimitou and Symington, 2008; Nimonkar et al., 2011). Why two separate pathways exist is still unclear. However, recent biochemical reconstitution studies suggest that each may be tailored to deal with specific obstacles at or nearby the break site, such as ribonucleotides and DNA damage (Daley et al., 2020).
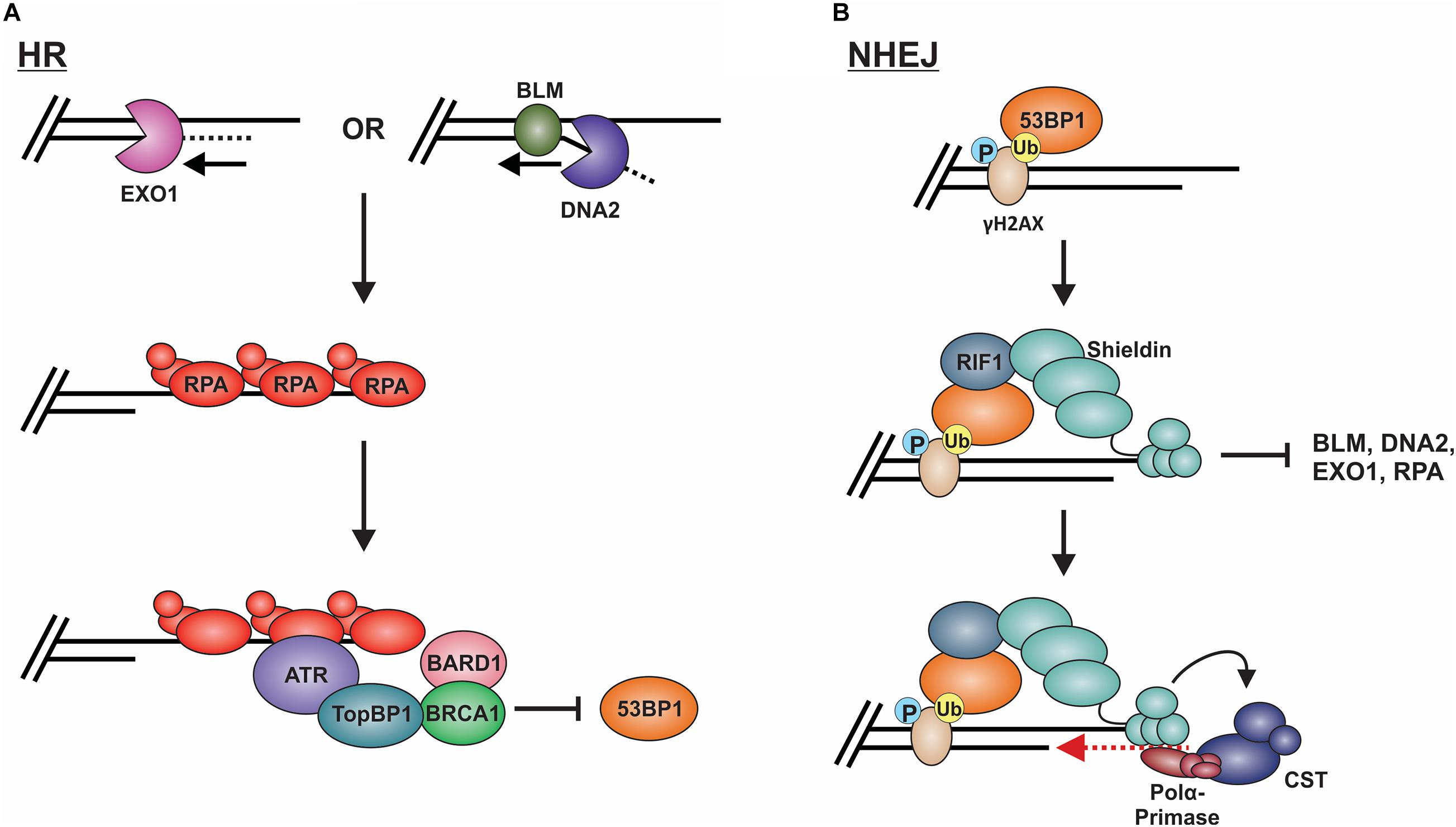
Figure 8. Models of long-range resection and how long-range resection is inhibited/reversed. (A) Long-range resection is mediated by EXO1 or BLM/DNA2. The resulting ssDNA is bound by RPA, which serves as a platform for ATR binding. To inhibit NHEJ, TopBP1 bridges interactions between ATR and the BRCA1-BARD1 complex, blocking 53BP1. (B) 53BP1 is localized to DSBs through its interaction with specific histone marks. 53BP1 then interacts with RIF1, which localizes the shieldin complex. Shieldin inhibits long-range resection by preventing access to the resected DNA and reverses resection through its interaction with CST-pol α, which can mediate fill-in of the resected DNA. This promotes repair of the break by NHEJ. (P: phosphorylation; Ub: ubiquitination).
CtIP and RPA have also been implicated as key stimulators of end resection. CDK phosphorylation of CtIP Thr847 is needed for effective ssDNA generation and RPA recruitment during long-range resection (Huertas and Jackson, 2009). In vitro, RPA stimulates BLM helicase activity at the nick created by MRN. However, phosphorylation of RPA70 inhibits DNA resection mediated by the BLM/EXO1 and BLM/DNA2 in vitro (Soniat et al., 2019; Qin et al., 2020). The dual roles of RPA in long range resection may explain how resection of the DNA is prevented while still promoting enough resection for HR.
NHEJ
To prevent long-range resection and promote NHEJ, genetic studies suggest that 53BP1 must outcompete BRCA1 for binding to MRN-generated overhangs (Setiaputra and Durocher, 2019). 53BP1 is localized to DSBs through its interaction with specific histone marks, including di-methylated histone H4 lysine 20 and ubiquitinated histone H2A lysine 15 (Figure 8B; Sanders et al., 2004; Botuyan et al., 2006; Kim et al., 2006; Yang et al., 2008; Hsiao and Mizzen, 2013). Upon localization to the DSB, 53BP1 works with various factors to prevent or reverse DSB resection. Pax2 transactivation domain interaction protein (PTIP) and Rap1-interacting protein 1 (RIF1) are two main factors downstream of 53BP1 which impair end resection at DSBs (Sfeir and de Lange, 2012; Zimmermann et al., 2013). PTIP acts by blocking DNA2 activity and interacts with the endonuclease Artemis to promote NHEJ (Wang et al., 2014; Callen et al., 2020). On the other hand, Rif1 limits the accumulation of BRCA1-BARD1 at DNA damage sites, preventing CtIP recruitment (Zimmermann et al., 2013).
The shieldin complex (SHLD1 [RINN3], SHLD2 [RINN2/FAM35A], SHLD3 [RINN1] and REV7 [MAD2L2/MAD2B]) also prevents and/or reverses end resection (Figure 8B; Setiaputra and Durocher, 2019). Shieldin was only recently discovered so many questions remain about how it functions in DSB repair. However, based on several mechanistic studies, a model has emerged in which shieldin both limits end resection and promotes the conversion of MRN-generated ssDNA back to duplex DNA. SHLD2 contains three predicted OB-folds that are proposed to bind to MRN-generated ssDNA overhangs, thus preventing long-range resection and RPA loading (Callen et al., 2020). To reverse end resection, shieldin recruits pol α and its stimulatory factor, CST (Mirman et al., 2018). CST/pol α then convert the ssDNA back to duplex DNA (Mirman et al., 2018; Noordermeer et al., 2018). How shieldin and CST/pol α are recruited to DSBs and under what circumstances is still not well understood. Nevertheless, recent studies suggest that TRIP13 and p31comet promote HR by inactivating REV7, which could provide another reversal point back to HR (Clairmont et al., 2020; Sarangi et al., 2020). Another open question is whether 53BP1-shieldin-CST-pol α can act at DNA ends that escape initial 53BP1 binding and have already been resected. Such a mechanism could prevent the more disastrous effects of using alt-EJ or SSA rather than NHEJ in G1 (Ceccaldi et al., 2016). Shieldin can also act independently of 53BP1 to inhibit DSB resection, although the mechanism is still unclear (Ghezraoui et al., 2018). Future work on this newly discovered complex will undoubtedly uncover novel insight into this critical decision point.
Telomeres
Two key factors, POT1 and CST, are critical to prevent the recognition of telomeres as HR intermediates. In mammals, POT1 prevents RPA from binding the G-overhang, which in turn suppresses ATR activation and unwanted HR (Wu et al., 2006; Kratz and de Lange, 2018). Interestingly, studies in the protozoa Leishmania amazonensis and Trypanosoma cruzi, which appear to lack homologs of POT1 or CST, found that RPA-1 is involved in end protection, suggesting that under certain situations RPA can adapt telomere protection capabilities (Pavani et al., 2014, 2018; Fernandes et al., 2020). Since RPA has a higher binding affinity and is ∼70-fold more abundant, POT1 cannot outcompete RPA in vitro (Flynn et al., 2011; Hein et al., 2015). However, in vivo POT1 is tethered to shelterin through TPP1, giving POT1 a competitive advantage over RPA. Loss of TPP1 or the disruption of POT1/TPP1 and TIN2/TPP1 interaction results in telomeric RPA (Hockemeyer et al., 2007; Barrientos et al., 2008; Gong and de Lange, 2010; Kratz and de Lange, 2018). Additionally, POT1/TPP1 can protect uncapped telomeres following extensive resection, caused by the absence of TRF2 (Deng et al., 2009). During replication, however, shelterin is displaced and RPA binds to telomeres as part of the normal replication process (Figure 7). RPA must then be displaced for allow POT1 binding. Yet, POT1/TPP1 is unable to displace RPA from telomeric DNA in vitro. Instead, an elegant mechanism was uncovered in which RPA is displaced by heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1) (Flynn et al., 2011; Figure 7). This RPA-to-POT1 switch is regulated in a cell cycle-dependent manner by expression of telomeric repeat-containing RNA (TERRA), a telomeric non-coding RNA.
Following replication, telomeres are extended in telomerase-positive cells. Under homeostatic conditions, telomerase adds ∼10 telomeric hexanucleotide repeats to each chromosome end and is then dissociated to prevent excessive G-overhang lengthening (Zhao et al., 2009, 2011). Termination of telomerase activity is primarily mediated by CST (Chen et al., 2012). The PIF1 helicase can also remove telomerase, although it is unclear whether this function is solely used to prevent de novo telomere addition at DSBs or can also promote telomerase dissociation from telomeres under certain conditions (Boule et al., 2005; Zhang et al., 2006; Churikov and Geli, 2017). Removal of telomerase is critical for end protection, as hyper-extension of G-overhangs can result in telomeric RPA due to the exhaustion of available POT1 (Feng et al., 2017, 2018). While still not completely understood, mammalian CST is thought to localize to telomeres through interactions with TPP1/POT1 (Wan et al., 2009; Chen et al., 2012). TPP1/POT1 also recruits telomerase so a switch likely occurs where telomerase is replaced by CST. A similar mechanism of overhang processing has been extensively studied in S. cerevisiae. Since shelterin is not present in S. cerevisiae, Cdc13 recruits telomerase for telomere extension and Stn1-Ten1 for C-strand fill-in, in place of TPP1/POT1 (Tseng et al., 2006; Li et al., 2009; Shen et al., 2014; Gopalakrishnan et al., 2017). This process is regulated by a series of post-translational modifications. In humans, phosphorylation of TPP1 by NIMA related kinase 6 (NEK6) is proposed to facilitate telomerase recruitment but much of the mechanism remains unknown (Zhang et al., 2013; Hirai et al., 2016). Furthermore, how CST is recruited to telomeres, whether its recruitment displaces telomerase from TPP1 and the role of CST DNA binding activity in this process remain open questions.
In addition to inhibiting telomerase, CST promotes C-strand fill-in by stimulating pol α (Stewart et al., 2018). CST regulated fill-in does not appear to be as critical as its ability to modulate telomerase inhibition for end protection. Deletion of human TEN1 resulted in defective C-strand fill-in but CTC1-STN1 were still able to inhibit telomerase. This led to only a minor increase in G-overhang elongation and the absence of RPA binding, suggesting that under these conditions POT1 levels were sufficient to block RPA (Feng et al., 2018). Similar results were obtained with knockdown of CST subunits (Surovtseva et al., 2009; Wang et al., 2012; Kasbek et al., 2013). Interestingly, analysis of G-overhangs across the cell cycle in STN1 knockdown cells showed that elongated G-overhangs were reset to near wild-type levels upon entry into the next G1 (Wang et al., 2012). The mechanism behind this reset is unknown but may be due to low levels of STN1 that promote fill-in of the overhang in G2 or M phase. It is also possible that backup mechanisms exist to rescue lingering, elongated G-overhangs and prevent potential RPA binding.
RPA-to-RAD51 Exchange
Following long-range resection, RPA is exchanged for RAD51 to promote the homology search for HR repair. The homologous template is then used to fill-in the missing sequence for error-free repair of the DSB. The switch to RAD51 is generally thought to be an irreversible step toward HR over NHEJ. However, there is new evidence that even at this late stage, mechanisms may be in place to prevent HR under specific situations. At telomeres, the best way to prevent HR is preventing RPA binding, but what happens to G-overhangs that become stably bound by RPA? Below, we will discuss regulation of the RPA-to-RAD51 switch at DSBs and telomeres as well as how ALT is used to extend telomeres but prevent end joining.
HR
After resection, ssDNA is quickly bound by RPA. For HR-mediated repair, RPA is replaced with RAD51 to form a RAD51-ssDNA nucleoprotein filament, also known as the presynaptic complex (Lee et al., 2015; Qi et al., 2015). In vitro single-molecule studies of RPA and RAD51 exchange indicated that although RPA binds to ssDNA with a higher affinity than RAD51, high concentrations of RAD51 can undergo facilitated exchange following ATP hydrolysis by RAD51 (Ma et al., 2017). However, in vivo RAD51 mediator proteins facilitate the binding, elongating, and stabilization of RAD51 onto ssDNA. BRCA2 is primarily responsible for delivering RAD51 monomers (Wong et al., 1997; Pellegrini et al., 2002; Esashi et al., 2007; Carreira et al., 2009; Jensen et al., 2010). BRC repeats in BRCA2 act as a scaffold to bind RAD51, a process facilitated by BRCA1 and PALB2. PALB2 bridges the interaction between BRCA1 and BRCA2 and localizes RAD51 to the RPA-ssDNA, where RAD51 is exchanged for RPA (Scully et al., 1997; Xia et al., 2006; Sy et al., 2009; Zhang et al., 2009a, b; Zong et al., 2019). After RAD51 filament formation, RAD51 paralogs aid in stabilization and elongation of the filament (Thacker, 1999; Suwaki et al., 2011; Sullivan and Bernstein, 2018). While filament formation is essential for HR, the exchange of RAD51 for RPA can be detrimental at other sites of RPA-ssDNA in the cell, such as replication forks. To prevent these untimely exchanges, RPA1-related ssDNA binding protein, X-linked (RADX) was found to antagonize RAD51-ssDNA filament formation, inhibiting RPA displacement as well as promoting the disassembly of existing RAD51 filaments at stalled replication forks (Zhang et al., 2020). Once formed, RAD51 filaments search for homologous regions in the sister chromatid to instigate repair, as reviewed in detail elsewhere (Haber, 2018; Bonilla et al., 2020).
Although most studies have focused on 53BP1 function prior to DSB resection, recent data in budding yeast raises the possibility that 53BP1 functions after long-range resection and RPA binding. The 53BP1 homolog Rad9 was shown to promote D-loop extension by limiting Sgs1 (BLM in mammals) and Mph1 (FANCM in mammals) helicase activity, suggesting a role in HR sub-pathway choice after DSB end resection (Ferrari et al., 2020). The proposed model is that after a DSB, Rad9 limits hyper-loading of RPA, Rad51, and Rad52. This limits Sgs1 and Mph1 from strand rejection to facilitate long-lived D-loops, thus favoring repair through sub-pathways that require stable D-loops such as break induced replication (BIR) or long tract gene conversion. If strand rejection occurs, then SSA is favored. This unprecedented role of Rad9 in controlling the fate of HR contradicts the commonly thought of role of 53BP1 in DSB repair, where it acts as a pre-resection block to HR in eukaryotes. Whether 53BP1 functions in the same manner in humans is still unclear, but it could have paradigm-shifting implications for 53BP1 activity at later steps in DSB repair, if true.
Telomeres
In the majority of cancers, telomeres are maintained in a telomerase-dependent manner, however, 10 to 15% of human cancers maintain their telomeres through the use of ALT (Bryan et al., 1997). ALT is a homology-based mechanism to lengthen telomeres (Dunham et al., 2000; Hoang and O’Sullivan, 2020; Recagni et al., 2020; Sommer and Royle, 2020; Zhang and Zou, 2020). The mechanisms of ALT are still under investigation, but recent evidence points to ALT using a BIR-like mechanism, which requires the exchange of RPA to induce recombination (Dilley et al., 2016). ALT is characterized by ALT-associated promyelocytic leukemia (PML) bodies (APBs), which consist of telomeric DNA, PML, and proteins involved in DNA repair, recombination and replication (Zhang and Zou, 2020). It is still not fully understood how APBs are assembled and how they promote ALT. However, a model has been proposed where BLM is critical for APB formation and telomeric DNA synthesis (Stavropoulos et al., 2002; Zhang et al., 2019). Additionally, MRN is localized to telomeres during S and G2 phases through its interaction with TRF2 and is required for ALT (Zhu et al., 2000; Jiang et al., 2005; Zhong et al., 2007). Within the APBs, the telomere is lengthened through RAD51- or RAD52-dependent BIR-like pathways. In both cases, a homology search is utilized to initiate BIR. In the RAD51-dependent pathway, RAD51 and HOP2-MND1 are recruited, and then RAD51 mediates homology searches and subsequent DNA polymerization (Cho et al., 2014). In RAD52-dependent BIR, BLM and DNA2 are proposed to resect the telomere and then replication factor C (RFC) mediates proliferating cell nuclear antigen (PCNA) loading (Dilley et al., 2016). PCNA recruits pol δ through interaction with POLD3 stimulating pol δ activity and DNA synthesis (Dilley et al., 2016; Roumelioti et al., 2016). Thus, for ALT, the engagement of BIR-related factors vs. those required for HR-mediated repair appears to underlie telomere extension while preventing chromosome fusions.
As described above, preventing stable binding and localization of HR factors is the major mechanism used to prevent unwanted “repair” at telomeres, but what happens when RPA remains stably bound to G-overhangs? Loss of POT1 or CST results in telomeric RPA foci but surprisingly only a minor increase in chromosome fusions, particularly in comparison to TRF2 loss (Denchi and de Lange, 2007; Feng et al., 2017). Once bound by RPA, telomeres, in essence, resemble resected RPA-bound DSB intermediates. Even in the absence of POT1, other shelterin subunits are present and likely play a role in maintaining end protection. Yet, how these RPA-bound telomeres remain protected from HR is not entirely clear. It is likely that the presence of 53BP1 contributes to protection from HR. 53BP1 foci have been observed at telomeres in both cells lacking POT1 (Hockemeyer et al., 2007; Gong and de Lange, 2010) and cells expressing a CTC1 G503R mutant construct, which is unable to localize to telomeres (Chen et al., 2013; Gu and Chang, 2013; Takai et al., 2016). In both cases, hyper-extended G-overhangs are generated. Under such conditions, 53BP1 may continue to block HR, while TRF2 is used to block NHEJ (Figure 9). In addition, deletion of CTC1 decreases TopBP1 and CHK1 phosphorylation, despite RPA-binding and ATR activation at telomeres (Ackerson et al., 2020). The loss of CHK1 signaling and 53BP1 localization could be sufficient to block the RPA-to-RAD51 exchange and, thus, HR-mediated telomere fusions at RPA-bound telomeres. However, additional studies are needed to determine the fate of RPA-bound telomeres. Unraveling such mechanism(s) will undoubtedly provide novel insights into chromosome end protection and HR-mediated repair.
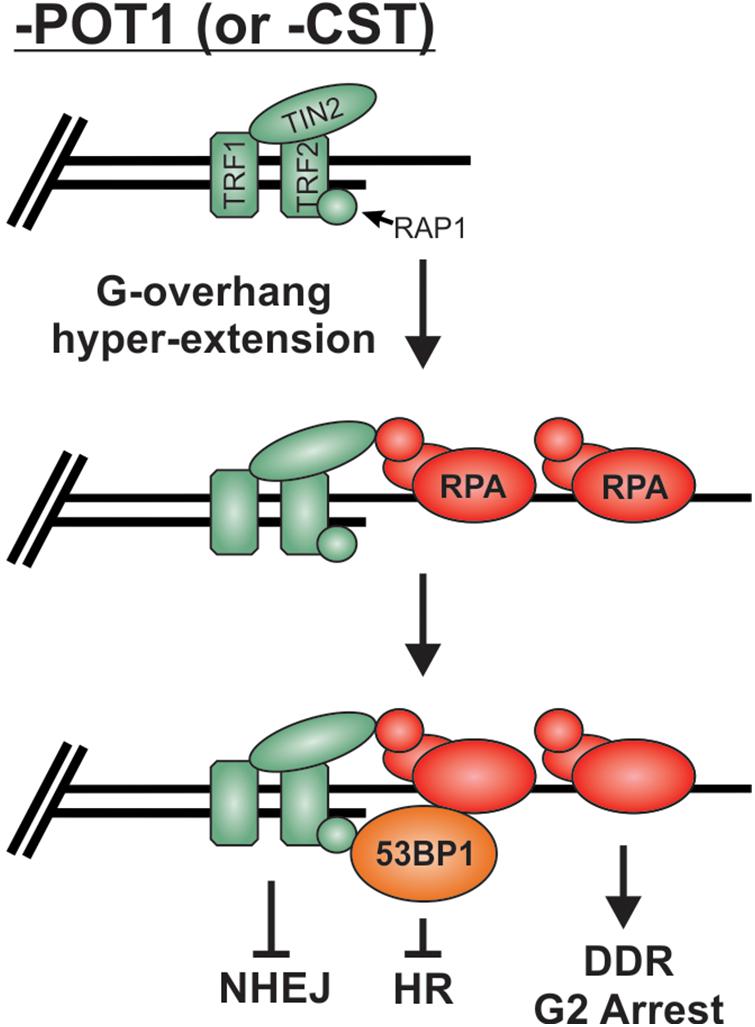
Figure 9. Potential mechanism for the protection of RPA-bound telomeres. Loss of POT1 or CST results in hyperextension of G-overhangs and telomeric RPA but very few fusions events. We propose that the combination of TRF2 and 53BP1 prevent fusions by blocking NHEJ and HR mediated repair, respectively.
Conclusion
While our understanding of DSB recognition and repair has progressed by leaps and bounds in recent years, important questions remain unanswered. Two key topics that still need to be fully elucidated are the context and timing of the key decision points described in this review. To date, much of our current understanding has centered on how specific factors interact with each other. To grasp the larger picture, we must now understand the temporal and contextual organization of these processes. This includes addressing how and when t-loops are formed, when shelterin disassembly/reassembly occurs and elucidating the mechanisms that protect exposed ends. Furthermore, understanding DSB pathway choice will require addressing questions such as the timing of Ku vs. MRN binding at DSBs, whether MRN is removed in situations where a homologous sequence is unavailable and how stimulation of MRN nuclease activities are temporally regulated. Such advances will pave the way for a more mechanistic understanding of these complex processes. Finally, both dysfunctional DSB repair and telomeres are linked to cancer and aging-related diseases. Therefore, defining the decision points that dictate whether to join or not to join the DNA ends will enlighten how these diseases arise and uncover vulnerabilities that might be exploited for therapeutic purposes.
Author Contributions
SA designed the graphics. All authors wrote and edited the manuscript, contributed to the article and approved the submitted version.
Funding
This work was supported by startup funds from the University of South Carolina. CR was supported by the NIH grant R25 GM076277. PS was supported in part by a SPARC grant from the University of South Carolina.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Drs. Eric Hendrickson, Alan Waldman, and Mike Wyatt for their feedback and critical reading of the manuscript.
References
Ackerson, S. M., Gable, C. I., and Stewart, J. A. (2020). Human CTC1 promotes TopBP1 stability and CHK1 phosphorylation in response to telomere dysfunction and global replication stress. Cell Cycle 19, 3491–3507. doi: 10.1080/15384101.2020.1849979
Alonso-de Vega, I., Paz-Cabrera, M. C., Rother, M. B., Wiegant, W. W., Checa-Rodriguez, C., and Hernandez-Fernaud, J. R. (2020). PHF2 regulates homology-directed DNA repair by controlling the resection of DNA double strand breaks. Nucleic Acids Res. 48, 4915–4927. doi: 10.1093/nar/gkaa196
Anand, R., Jasrotia, A., Bundschuh, D., Howard, S. M., Ranjha, L., and Stucki, M. (2019). NBS1 promotes the endonuclease activity of the MRE11-RAD50 complex by sensing CtIP phosphorylation. EMBO J. 38:e101005.
Anand, R., Ranjha, L., Cannavo, E., and Cejka, P. (2016). Phosphorylated CtIP functions as a Co-factor of the MRE11-RAD50-NBS1 Endonuclease in DNA end resection. Mol. Cell 64, 940–950. doi: 10.1016/j.molcel.2016.10.017
Attwooll, C. L., Akpinar, M., and Petrini, J. H. (2009). The mre11 complex and the response to dysfunctional telomeres. Mol. Cell. Biol. 29, 5540–5551. doi: 10.1128/mcb.00479-09
Bae, N. S., and Baumann, P. (2007). A RAP1/TRF2 complex inhibits nonhomologous end-joining at human telomeric DNA ends. Mol. Cell. 26, 323–334. doi: 10.1016/j.molcel.2007.03.023
Bakr, A., Kocher, S., Volquardsen, J., Petersen, C., Borgmann, K., and Dikomey, E. (2016). Impaired 53BP1/RIF1 DSB mediated end-protection stimulates CtIP-dependent end resection and switches the repair to PARP1-dependent end joining in G1. Oncotarget 7, 57679–57693. doi: 10.18632/oncotarget.11023
Barrientos, K. S., Kendellen, M. F., Freibaum, B. D., Armbruster, B. N., Etheridge, K. T., and Counter, C. M. (2008). Distinct functions of POT1 at telomeres. Mol. Cell. Biol. 28, 5251–5264. doi: 10.1128/mcb.00048-08
Benarroch-Popivker, D., Pisano, S., Mendez-Bermudez, A., Lototska, L., Kaur, P., and Bauwens, S. (2016). TRF2-mediated control of telomere DNA topology as a mechanism for chromosome-end protection. Mol. Cell 61, 274–286. doi: 10.1016/j.molcel.2015.12.009
Bennardo, N., Cheng, A., Huang, N., and Stark, J. M. (2008). Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 4:e1000110. doi: 10.1371/journal.pgen.1000110
Beucher, A., Birraux, J., Tchouandong, L., Barton, O., Shibata, A., and Conrad, S. (2009). ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J. 28, 3413–3427. doi: 10.1038/emboj.2009.276
Biehs, R., Steinlage, M., Barton, O., Juhasz, S., Kunzel, J., and Spies, J. (2017). DNA double-strand break resection occurs during non-homologous end joining in G1 but is distinct from resection during homologous recombination. Mol. Cell. 65, 671–684 e675.
Blackburn, E. H. (1991). Structure and function of telomeres. Nature 350, 569–573. doi: 10.1038/350569a0
Blackford, A. N., and Jackson, S. P. (2017). ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol. Cell 66, 801–817. doi: 10.1016/j.molcel.2017.05.015
Blackford, A. N., and Stucki, M. (2020). How cells respond to DNA breaks in mitosis. Trends Biochem. Sci. 45, 321–331. doi: 10.1016/j.tibs.2019.12.010
Blier, P. R., Griffith, A. J., Craft, J., and Hardin, J. A. (1993). Binding of Ku protein to DNA. measurement of affinity for ends and demonstration of binding to nicks. J. Biol. Chem. 268, 7594–7601. doi: 10.1016/s0021-9258(18)53216-6
Boersma, V., Moatti, N., Segura-Bayona, S., Peuscher, M. H., van der Torre, J., and Wevers, B. A. (2015). MAD2L2 controls DNA repair at telomeres and DNA breaks by inhibiting 5’ end resection. Nature 521, 537–540. doi: 10.1038/nature14216
Bohgaki, T., Bohgaki, M., and Hakem, R. (2010). DNA double-strand break signaling and human disorders. Genome Integr. 1:15. doi: 10.1186/2041-9414-1-15
Bonilla, B., Hengel, S. R., Grundy, M. K., and Bernstein, K. A. (2020). RAD51 gene family structure and function. Annu. Rev. Genet. 54, 25–46. doi: 10.1146/annurev-genet-021920-092410
Botuyan, M. V., Lee, J., Ward, I. M., Kim, J. E., Thompson, J. R., and Chen, J. (2006). Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell 127, 1361–1373. doi: 10.1016/j.cell.2006.10.043
Boule, J. B., Vega, L. R., and Zakian, V. A. (2005). The yeast Pif1p helicase removes telomerase from telomeric DNA. Nature 438, 57–61. doi: 10.1038/nature04091
Broderick, R., Nieminuszczy, J., Baddock, H. T., Deshpande, R., Gileadi, O., and Paull, T. T. (2016). EXD2 promotes homologous recombination by facilitating DNA end resection. Nat. Cell Biol. 18, 271–280. doi: 10.1038/ncb3303
Bryan, T. M., Englezou, A., Dalla-Pozza, L., Dunham, M. A., and Reddel, R. R. (1997). Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat. Med. 3, 1271–1274. doi: 10.1038/nm1197-1271
Budd, M. E., and Campbell, J. L. (2013). Dna2 is involved in CA strand resection and nascent lagging strand completion at native yeast telomeres. J. Biol. Chem. 288, 29414–29429. doi: 10.1074/jbc.m113.472456
Bunting, S. F., and Nussenzweig, A. (2013). End-joining, translocations and cancer. Nat. Rev. Cancer 13, 443–454. doi: 10.1038/nrc3537
Bunting, S. F., Callen, E., Kozak, M. L., Kim, J. M., Wong, N., and Lopez-Contreras, A. J. (2012). BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Mol. Cell 46, 125–135. doi: 10.1016/j.molcel.2012.02.015
Bunting, S. F., Callen, E., Wong, N., Chen, H. T., Polato, F., and Gunn, A. (2010). 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 141, 243–254. doi: 10.1016/j.cell.2010.03.012
Burma, S., Chen, B. P., and Chen, D. J. (2006). Role of non-homologous end joining (NHEJ) in maintaining genomic integrity. DNA Repair 5, 1042–1048. doi: 10.1016/j.dnarep.2006.05.026
Burma, S., Chen, B. P., Murphy, M., Kurimasa, A., and Chen, D. J. (2001). ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 276, 42462–42467. doi: 10.1074/jbc.c100466200
Callen, E., Zong, D., Wu, W., Wong, N., Stanlie, A., and Ishikawa, M. (2020). 53BP1 enforces distinct pre- and post-resection blocks on homologous recombination. Mol. Cell 77, 26–38 e27.
Cannavo, E., Reginato, G., and Cejka, P. (2019). Stepwise 5’ DNA end-specific resection of DNA breaks by the Mre11-Rad50-Xrs2 and Sae2 nuclease ensemble. Proc. Natl. Acad. Sci. U. S. A. 116, 5505–5513. doi: 10.1073/pnas.1820157116
Cao, L., Xu, X., Bunting, S. F., Liu, J., Wang, R. H., and Cao, L. L. (2009). A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Mol. Cell 35, 534–541. doi: 10.1016/j.molcel.2009.06.037
Carney, J. P., Maser, R. S., Olivares, H., Davis, E. M., Le Beau, M., Yates, J. R., et al. (1998). The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell 93, 477–486. doi: 10.1016/s0092-8674(00)81175-7
Carreira, A., Hilario, J., Amitani, I., Baskin, R. J., Shivji, M. K., and Venkitaraman, A. R. (2009). The BRC repeats of BRCA2 modulate the DNA-binding selectivity of RAD51. Cell 136, 1032–1043. doi: 10.1016/j.cell.2009.02.019
Ceccaldi, R., Rondinelli, B., and D’Andrea, A. D. (2016). Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 26, 52–64. doi: 10.1016/j.tcb.2015.07.009
Cejka, P. (2015). DNA end resection: nucleases team up with the right partners to initiate homologous recombination. J. Biol. Chem. 290, 22931–22938. doi: 10.1074/jbc.r115.675942
Cesare, A. J., and Reddel, R. R. (2010). Alternative lengthening of telomeres: models, mechanisms and implications. Nat. Rev. Genet. 11, 319–330. doi: 10.1038/nrg2763
Cesare, A. J., Groff-Vindman, C., Compton, S. A., McEachern, M. J., and Griffith, J. D. (2008). Telomere loops and homologous recombination-dependent telomeric circles in a Kluyveromyces lactis telomere mutant strain. Mol. Cell. Biol. 28, 20–29. doi: 10.1128/mcb.01122-07
Cesare, A. J., Hayashi, M. T., Crabbe, L., and Karlseder, J. (2013). The telomere deprotection response is functionally distinct from the genomic DNA damage response. Mol. Cell 51, 141–155. doi: 10.1016/j.molcel.2013.06.006
Chanut, P., Britton, S., Coates, J., Jackson, S. P., and Calsou, P. (2016). Coordinated nuclease activities counteract Ku at single-ended DNA double-strand breaks. Nat. Commun. 7:12889.
Chapman, J. R., Sossick, A. J., Boulton, S. J., and Jackson, S. P. (2012). BRCA1-associated exclusion of 53BP1 from DNA damage sites underlies temporal control of DNA repair. J. Cell Sci. 125(Pt 15), 3529–3534.
Chen, L. Y., Majerska, J., and Lingner, J. (2013). Molecular basis of telomere syndrome caused by CTC1 mutations. Genes Dev. 27, 2099–2108. doi: 10.1101/gad.222893.113
Chen, L. Y., Redon, S., and Lingner, J. (2012). The human CST complex is a terminator of telomerase activity. Nature 488, 540–544. doi: 10.1038/nature11269
Chen, R., and Wold, M. S. (2014). Replication protein a: single-stranded DNA’s first responder: dynamic DNA-interactions allow replication protein a to direct single-strand DNA intermediates into different pathways for synthesis or repair. Bioessays 36, 1156–1161. doi: 10.1002/bies.201400107
Chen, S., Lee, L., Naila, T., Fishbain, S., Wang, A., and Tomkinson, A. E. (2021). Structural basis of long-range to short-range synaptic transition in NHEJ. Nature 593, 294–298. doi: 10.1038/s41586-021-03458-7
Cho, N. W., Dilley, R. L., Lampson, M. A., and Greenberg, R. A. (2014). Interchromosomal homology searches drive directional ALT telomere movement and synapsis. Cell 159, 108–121. doi: 10.1016/j.cell.2014.08.030
Chow, T. T., Zhao, Y., Mak, S. S., Shay, J. W., and Wright, W. E. (2012). Early and late steps in telomere overhang processing in normal human cells: the position of the final RNA primer drives telomere shortening. Genes Dev. 26, 1167–1178. doi: 10.1101/gad.187211.112
Churikov, D., and Geli, V. (2017). De novo telomere addition at chromosome breaks: dangerous liaisons. J. Cell Biol. 216, 2243–2245. doi: 10.1083/jcb.201705156
Clairmont, C. S., Sarangi, P., Ponnienselvan, K., Galli, L. D., Csete, I., and Moreau, L. (2020). TRIP13 regulates DNA repair pathway choice through REV7 conformational change. Nat. Cell Biol. 22, 87–96. doi: 10.1038/s41556-019-0442-y
Coleman, K. A., and Greenberg, R. A. (2011). The BRCA1-RAP80 complex regulates DNA repair mechanism utilization by restricting end resection. J. Biol. Chem. 286, 13669–13680. doi: 10.1074/jbc.m110.213728
Cuadrado, M., Martinez-Pastor, B., Murga, M., Toledo, L. I., Gutierrez-Martinez, P., and Lopez, E. (2006). ATM regulates ATR chromatin loading in response to DNA double-strand breaks. J. Exp. Med. 203, 297–303. doi: 10.1084/jem.20051923
Daley, J. M., and Sung, P. (2014). 53BP1, BRCA1, and the choice between recombination and end joining at DNA double-strand breaks. Mol. Cell. Biol. 34, 1380–1388. doi: 10.1128/mcb.01639-13
Daley, J. M., Tomimatsu, N., Hooks, G., Wang, W., Miller, A. S., and Xue, X. (2020). Specificity of end resection pathways for double-strand break regions containing ribonucleotides and base lesions. Nat. Commun. 11:3088.
Davis, A. J., Chi, L., So, S., Lee, K. J., Mori, E., and Fattah, K. (2014). BRCA1 modulates the autophosphorylation status of DNA-PKcs in S phase of the cell cycle. Nucleic Acids Res. 42, 11487–11501. doi: 10.1093/nar/gku824
de Lange, T. (2001). Cell biology. Telomere capping–one strand fits all. Science 292, 1075–1076. doi: 10.1126/science.1061032
de Lange, T. (2005). Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 19, 2100–2110. doi: 10.1101/gad.1346005
de Lange, T. (2018). Shelterin-mediated telomere protection. Annu. Rev. Genet. 52, 223–247. doi: 10.1146/annurev-genet-032918-021921
Denchi, E. L., and de Lange, T. (2007). Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature 448, 1068–1071. doi: 10.1038/nature06065
Deng, Y., Guo, X., Ferguson, D. O., and Chang, S. (2009). Multiple roles for MRE11 at uncapped telomeres. Nature 460, 914–918. doi: 10.1038/nature08196
Deshpande, R. A., Myler, L. R., Soniat, M. M., Makharashvili, N., Lee, L., and Lees-Miller, S. P. (2020). DNA-dependent protein kinase promotes DNA end processing by MRN and CtIP. Sci. Adv. 6:eaay0922. doi: 10.1126/sciadv.aay0922
Dibitetto, D., Sims, J. R., Ascencao, C. F. R., Feng, K., Kim, D., and Oberly, S. (2020). Intrinsic ATR signaling shapes DNA end resection and suppresses toxic DNA-PKcs signaling. NAR Cancer 2:zcaa006.
Dilley, R. L., Verma, P., Cho, N. W., Winters, H. D., Wondisford, A. R., and Greenberg, R. A. (2016). Break-induced telomere synthesis underlies alternative telomere maintenance. Nature 539, 54–58. doi: 10.1038/nature20099
Ding, Q., Reddy, Y. V., Wang, W., Woods, T., Douglas, P., and Ramsden, D. A. (2003). Autophosphorylation of the catalytic subunit of the DNA-dependent protein kinase is required for efficient end processing during DNA double-strand break repair. Mol. Cell. Biol. 23, 5836–5848. doi: 10.1128/mcb.23.16.5836-5848.2003
Dinkelmann, M., Spehalski, E., Stoneham, T., Buis, J., Wu, Y., and Sekiguchi, J. M. (2009). Multiple functions of MRN in end-joining pathways during isotype class switching. Nat. Struct. Mol. Biol. 16, 808–813. doi: 10.1038/nsmb.1639
Doksani, Y., Wu, J. Y., de Lange, T., and Zhuang, X. (2013). Super-resolution fluorescence imaging of telomeres reveals TRF2-dependent T-loop formation. Cell 155, 345–356. doi: 10.1016/j.cell.2013.09.048
Dunham, M. A., Neumann, A. A., Fasching, C. L., and Reddel, R. R. (2000). Telomere maintenance by recombination in human cells. Nat. Genet. 26, 447–450. doi: 10.1038/82586
Ensminger, M., and Lobrich, M. (2020). One end to rule them all: non-homologous end-joining and homologous recombination at DNA double-strand breaks. Br. J. Radiol. 93:20191054. doi: 10.1259/bjr.20191054
Esashi, F., Galkin, V. E., Yu, X., Egelman, E. H., and West, S. C. (2007). Stabilization of RAD51 nucleoprotein filaments by the C-terminal region of BRCA2. Nat. Struct. Mol. Biol. 14, 468–474. doi: 10.1038/nsmb1245
Feng, L., and Chen, J. (2012). The E3 ligase RNF8 regulates KU80 removal and NHEJ repair. Nat. Struct. Mol. Biol. 19, 201–206. doi: 10.1038/nsmb.2211
Feng, X., Hsu, S. J., Bhattacharjee, A., Wang, Y., Diao, J., and Price, C. M. (2018). CTC1-STN1 terminates telomerase while STN1-TEN1 enables C-strand synthesis during telomere replication in colon cancer cells. Nat. Commun. 9:2827.
Feng, X., Hsu, S. J., Kasbek, C., Chaiken, M., and Price, C. M. (2017). CTC1-mediated C-strand fill-in is an essential step in telomere length maintenance. Nucleic Acids Res. 45, 4281–4293. doi: 10.1093/nar/gkx125
Fernandes, C. A. H., Morea, E. G. O., Dos Santos, G. A., da Silva, V. L., Vieira, M. R., and Viviescas, M. A. (2020). A multi-approach analysis highlights the relevance of RPA-1 as a telomere end-binding protein (TEBP) in Leishmania amazonensis. Biochim. Biophys. Acta Gen. Subj. 1864:129607. doi: 10.1016/j.bbagen.2020.129607
Ferrari, M., Rawal, C. C., Lodovichi, S., Vietri, M. Y., and Pellicioli, A. (2020). Rad9/53BP1 promotes DNA repair via crossover recombination by limiting the Sgs1 and Mph1 helicases. Nat. Commun. 11:3181.
Ferretti, L. P., Lafranchi, L., and Sartori, A. A. (2013). Controlling DNA-end resection: a new task for CDKs. Front. Genet. 4:99.
Flynn, R. L., Centore, R. C., O’Sullivan, R. J., Rai, R., Tse, A., and Songyang, Z. (2011). TERRA and hnRNPA1 orchestrate an RPA-to-POT1 switch on telomeric single-stranded DNA. Nature 471, 532–536. doi: 10.1038/nature09772
Frank, A. K., Tran, D. C., Qu, R. W., Stohr, B. A., Segal, D. J., and Xu, L. (2015). The Shelterin TIN2 subunit mediates recruitment of telomerase to telomeres. PLoS Genet. 11:e1005410. doi: 10.1371/journal.pgen.1005410
Frit, P., Barboule, N., Yuan, Y., Gomez, D., and Calsou, P. (2014). Alternative end-joining pathway(s): bricolage at DNA breaks. DNA Repair 17, 81–97. doi: 10.1016/j.dnarep.2014.02.007
Gao, S. S., Guan, H., Yan, S., Hu, S., Song, M., and Guo, Z. P. (2020). TIP60 K430 SUMOylation attenuates its interaction with DNA-PKcs in S-phase cells: facilitating homologous recombination and emerging target for cancer therapy. Sci. Adv. 6:eaba7822. doi: 10.1126/sciadv.aba7822
Gell, D., and Jackson, S. P. (1999). Mapping of protein-protein interactions within the DNA-dependent protein kinase complex. Nucleic Acids Res. 27, 3494–3502. doi: 10.1093/nar/27.17.3494
Gerodimos, C. A., Chang, H. H. Y., Watanabe, G., and Lieber, M. R. (2017). Effects of DNA end configuration on XRCC4-DNA ligase IV and its stimulation of artemis activity. J. Biol. Chem. 292, 13914–13924. doi: 10.1074/jbc.m117.798850
Ghezraoui, H., Oliveira, C., Becker, J. R., Bilham, K., Moralli, D., and Anzilotti, C. (2018). 53BP1 cooperation with the REV7-shieldin complex underpins DNA structure-specific NHEJ. Nature 560, 122–127. doi: 10.1038/s41586-018-0362-1
Ghosh, R., Das, D., and Franco, S. (2018). The role for the DSB response pathway in regulating chromosome translocations. Adv. Exp. Med. Biol. 1044, 65–87. doi: 10.1007/978-981-13-0593-1_6
Giraud-Panis, M. J., Teixeira, M. T., Geli, V., and Gilson, E. (2010). CST meets shelterin to keep telomeres in check. Mol. Cell 39, 665–676. doi: 10.1016/j.molcel.2010.08.024
Gong, Y., and de Lange, T. (2010). A Shld1-controlled POT1a provides support for repression of ATR signaling at telomeres through RPA exclusion. Mol. Cell 40, 377–387. doi: 10.1016/j.molcel.2010.10.016
Gopalakrishnan, V., Tan, C. R., and Li, S. (2017). Sequential phosphorylation of CST subunits by different cyclin-Cdk1 complexes orchestrate telomere replication. Cell Cycle 16, 1271–1287. doi: 10.1080/15384101.2017.1312235
Graham, T. G., Walter, J. C., and Loparo, J. J. (2016). Two-stage synapsis of DNA ends during non-homologous end joining. Mol. Cell 61, 850–858. doi: 10.1016/j.molcel.2016.02.010
Griffith, J. D., Comeau, L., Rosenfield, S., Stansel, R. M., Bianchi, A., and Moss, H. (1999). Mammalian telomeres end in a large duplex loop. Cell 97, 503–514. doi: 10.1016/s0092-8674(00)80760-6
Gu, P., and Chang, S. (2013). Functional characterization of human CTC1 mutations reveals novel mechanisms responsible for the pathogenesis of the telomere disease Coats plus. Aging Cell 12, 1100–1109. doi: 10.1111/acel.12139
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. doi: 10.1016/j.cell.2011.02.013
Hein, M. Y., Hubner, N. C., Poser, I., Cox, J., Nagaraj, N., and Toyoda, Y. (2015). A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 163, 712–723. doi: 10.1016/j.cell.2015.09.053
Hirai, Y., Tamura, M., Otani, J., and Ishikawa, F. (2016). NEK6-mediated phosphorylation of human TPP1 regulates telomere length through telomerase recruitment. Genes Cells 21, 874–889. doi: 10.1111/gtc.12391
Hoang, S. M., and O’Sullivan, R. J. (2020). Alternative lengthening of telomeres: building bridges to connect chromosome ends. Trends Cancer 6, 247–260. doi: 10.1016/j.trecan.2019.12.009
Hockemeyer, D., Daniels, J. P., Takai, H., and de Lange, T. (2006). Recent expansion of the telomeric complex in rodents: two distinct POT1 proteins protect mouse telomeres. Cell 126, 63–77. doi: 10.1016/j.cell.2006.04.044
Hockemeyer, D., Palm, W., Else, T., Daniels, J. P., Takai, K. K., and Ye, J. Z. (2007). Telomere protection by mammalian Pot1 requires interaction with Tpp1. Nat. Struct. Mol. Biol. 14, 754–761. doi: 10.1038/nsmb1270
Hsiao, K. Y., and Mizzen, C. A. (2013). Histone H4 deacetylation facilitates 53BP1 DNA damage signaling and double-strand break repair. J. Mol. Cell Biol. 5, 157–165. doi: 10.1093/jmcb/mjs066
Huertas, P., and Jackson, S. P. (2009). Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J. Biol. Chem. 284, 9558–9565. doi: 10.1074/jbc.m808906200
Huertas, P., Cortes-Ledesma, F., Sartori, A. A., Aguilera, A., and Jackson, S. P. (2008). CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature 455, 689–692. doi: 10.1038/nature07215
Iliakis, G., Murmann, T., and Soni, A. (2015). Alternative end-joining repair pathways are the ultimate backup for abrogated classical non-homologous end-joining and homologous recombination repair: implications for the formation of chromosome translocations. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 793, 166–175. doi: 10.1016/j.mrgentox.2015.07.001
Ingram, S. P., Warmenhoven, J. W., Henthorn, N. T., Smith, E. A. K., Chadwick, A. L., and Burnet, N. G. (2019). Mechanistic modelling supports entwined rather than exclusively competitive DNA double-strand break repair pathway. Sci. Rep. 9:6359.
Ismail, I. H., Gagne, J. P., Genois, M. M., Strickfaden, H., McDonald, D., and Xu, Z. (2015). The RNF138 E3 ligase displaces Ku to promote DNA end resection and regulate DNA repair pathway choice. Nat. Cell Biol. 17, 1446–1457. doi: 10.1038/ncb3259
Jachimowicz, R. D., Beleggia, F., Isensee, J., Velpula, B. B., Goergens, J., and Bustos, M. A. (2019). UBQLN4 represses homologous recombination and is overexpressed in aggressive tumors. Cell 176, 505–519 e522.
Jensen, R. B., Carreira, A., and Kowalczykowski, S. C. (2010). Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 467, 678–683. doi: 10.1038/nature09399
Jette, N., and Lees-Miller, S. P. (2015). The DNA-dependent protein kinase: a multifunctional protein kinase with roles in DNA double strand break repair and mitosis. Prog. Biophys. Mol. Biol. 117, 194–205. doi: 10.1016/j.pbiomolbio.2014.12.003
Jiang, W. Q., Zhong, Z. H., Henson, J. D., Neumann, A. A., Chang, A. C., and Reddel, R. R. (2005). Suppression of alternative lengthening of telomeres by Sp100-mediated sequestration of the MRE11/RAD50/NBS1 complex. Mol. Cell. Biol. 25, 2708–2721. doi: 10.1128/mcb.25.7.2708-2721.2005
Joukov, V., Groen, A. C., Prokhorova, T., Gerson, R., White, E., and Rodriguez, A. (2006). The BRCA1/BARD1 heterodimer modulates ran-dependent mitotic spindle assembly. Cell 127, 539–552. doi: 10.1016/j.cell.2006.08.053
Kakarougkas, A., Ismail, A., Katsuki, Y., Freire, R., Shibata, A., and Jeggo, P. A. (2013). Co-operation of BRCA1 and POH1 relieves the barriers posed by 53BP1 and RAP80 to resection. Nucleic Acids Res. 41, 10298–10311. doi: 10.1093/nar/gkt802
Kasbek, C., Wang, F., and Price, C. M. (2013). Human TEN1 maintains telomere integrity and functions in genome-wide replication restart. J. Biol. Chem. 288, 30139–30150. doi: 10.1074/jbc.m113.493478
Kibe, T., Osawa, G. A., Keegan, C. E., and de Lange, T. (2010). Telomere protection by TPP1 is mediated by POT1a and POT1b. Mol. Cell. Biol. 30, 1059–1066. doi: 10.1128/mcb.01498-09
Kim, J., Daniel, J., Espejo, A., Lake, A., Krishna, M., and Xia, L. (2006). Tudor, MBT and chromo domains gauge the degree of lysine methylation. EMBO Rep. 7, 397–403. doi: 10.1038/sj.embor.7400625
Kolas, N. K., Chapman, J. R., Nakada, S., Ylanko, J., Chahwan, R., and Sweeney, F. D. (2007). Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science 318, 1637–1640. doi: 10.1126/science.1150034
Kratz, K., and de Lange, T. (2018). Protection of telomeres 1 proteins POT1a and POT1b can repress ATR signaling by RPA exclusion, but binding to CST limits ATR repression by POT1b. J. Biol. Chem. 293, 14384–14392. doi: 10.1074/jbc.ra118.004598
Krenning, L., van den Berg, J., and Medema, R. H. (2019). Life or death after a break: what determines the choice? Mol. Cell 76, 346–358. doi: 10.1016/j.molcel.2019.08.023
Lafrance-Vanasse, J., Williams, G. J., and Tainer, J. A. (2015). Envisioning the dynamics and flexibility of Mre11-Rad50-Nbs1 complex to decipher its roles in DNA replication and repair. Prog. Biophys. Mol. Biol. 117, 182–193. doi: 10.1016/j.pbiomolbio.2014.12.004
Lam, Y. C., Akhter, S., Gu, P., Ye, J., Poulet, A., and Giraud-Panis, M. J. (2010). SNMIB/Apollo protects leading-strand telomeres against NHEJ-mediated repair. EMBO J. 29, 2230–2241. doi: 10.1038/emboj.2010.58
Lamarche, B. J., Orazio, N. I., and Weitzman, M. D. (2010). The MRN complex in double-strand break repair and telomere maintenance. FEBS Lett. 584, 3682–3695. doi: 10.1016/j.febslet.2010.07.029
Langerak, P., Mejia-Ramirez, E., Limbo, O., and Russell, P. (2011). Release of Ku and MRN from DNA ends by Mre11 nuclease activity and Ctp1 is required for homologous recombination repair of double-strand breaks. PLoS Genet. 7:e1002271. doi: 10.1371/journal.pgen.1002271
Lee, J. H., and Paull, T. T. (2004). Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science 304, 93–96. doi: 10.1126/science.1091496
Lee, J. H., and Paull, T. T. (2005). ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 308, 551–554. doi: 10.1126/science.1108297
Lee, J. Y., Terakawa, T., Qi, Z., Steinfeld, J. B., Redding, S., and Kwon, Y. (2015). DNA RECOMBINATION. base triplet stepping by the Rad51/RecA family of recombinases. Science 349, 977–981. doi: 10.1126/science.aab2666
Lee, K. J., Saha, J., Sun, J., Fattah, K. R., Wang, S. C., and Jakob, B. (2016). Phosphorylation of Ku dictates DNA double-strand break (DSB) repair pathway choice in S phase. Nucleic Acids Res. 44, 1732–1745. doi: 10.1093/nar/gkv1499
Li, S., Makovets, S., Matsuguchi, T., Blethrow, J. D., Shokat, K. M., and Blackburn, E. H. (2009). Cdk1-dependent phosphorylation of Cdc13 coordinates telomere elongation during cell-cycle progression. Cell 136, 50–61. doi: 10.1016/j.cell.2008.11.027
Lim, C. J., and Cech, T. R. (2021). Shaping human telomeres: from shelterin and CST complexes to telomeric chromatin organization. Nat. Rev. Mol. Cell Biol. 22, 283–298. doi: 10.1038/s41580-021-00328-y
Lim, C. J., Barbour, A. T., Zaug, A. J., Goodrich, K. J., McKay, A. E., and Wuttke, D. S. (2020). The structure of human CST reveals a decameric assembly bound to telomeric DNA. Science 368, 1081–1085. doi: 10.1126/science.aaz9649
Lin, W., Sampathi, S., Dai, H., Liu, C., Zhou, M., and Hu, J. (2013). Mammalian DNA2 helicase/nuclease cleaves G-quadruplex DNA and is required for telomere integrity. EMBO J. 32, 1425–1439. doi: 10.1038/emboj.2013.88
Lisby, M., Barlow, J. H., Burgess, R. C., and Rothstein, R. (2004). Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell 118, 699–713. doi: 10.1016/s1568-7864(02)00076-9
Liu, Y., Cussiol, J. R., Dibitetto, D., Sims, J. R., Twayana, S., and Weiss, R. S. (2017). TOPBP1(Dpb11) plays a conserved role in homologous recombination DNA repair through the coordinated recruitment of 53BP1(Rad9). J. Cell Biol. 216, 623–639. doi: 10.1083/jcb.201607031
Lopez-Saavedra, A., Gomez-Cabello, D., Dominguez-Sanchez, M. S., Mejias-Navarro, F., Fernandez-Avila, M. J., and Dinant, C. (2016). A genome-wide screening uncovers the role of CCAR2 as an antagonist of DNA end resection. Nat. Commun. 7:12364.
Lou, Z., Minter-Dykhouse, K., Franco, S., Gostissa, M., Rivera, M. A., and Celeste, A. (2006). MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol. Cell 21, 187–200. doi: 10.1016/j.molcel.2005.11.025
Ma, C. J., Gibb, B., Kwon, Y., Sung, P., and Greene, E. C. (2017). Protein dynamics of human RPA and RAD51 on ssDNA during assembly and disassembly of the RAD51 filament. Nucleic Acids Res. 45, 749–761. doi: 10.1093/nar/gkw1125
Makarov, V. L., Hirose, Y., and Langmore, J. P. (1997). Long G tails at both ends of human chromosomes suggest a C strand degradation mechanism for telomere shortening. Cell 88, 657–666. doi: 10.1016/s0092-8674(00)81908-x
Markiewicz-Potoczny, M., Lisby, M., and Lydall, D. (2018). A critical role for Dna2 at unwound telomeres. Genetics 209, 129–141. doi: 10.1534/genetics.118.300809
McElligott, R., and Wellinger, R. J. (1997). The terminal DNA structure of mammalian chromosomes. EMBO J. 16, 3705–3714. doi: 10.1093/emboj/16.12.3705
McVey, M., and Lee, S. E. (2008). MMEJ repair of double-strand breaks (director’s cut): deleted sequences and alternative endings. Trends Genet. 24, 529–538. doi: 10.1016/j.tig.2008.08.007
Meek, K. (2020). Activation of DNA-PK by hairpinned DNA ends reveals a stepwise mechanism of kinase activation. Nucleic Acids Res. 48, 9098–9108. doi: 10.1093/nar/gkaa614
Mehta, A., and Haber, J. E. (2014). Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 6:a016428. doi: 10.1101/cshperspect.a016428
Menon, V., and Povirk, L. F. (2016). End-processing nucleases and phosphodiesterases: an elite supporting cast for the non-homologous end joining pathway of DNA double-strand break repair. DNA Repair 43, 57–68. doi: 10.1016/j.dnarep.2016.05.011
Mimitou, E. P., and Symington, L. S. (2008). Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 455, 770–774.
Mirman, Z., and de Lange, T. (2020). 53BP1: a DSB escort. Genes Dev. 34, 7–23. doi: 10.1101/gad.333237.119
Mirman, Z., Lottersberger, F., Takai, H., Kibe, T., Gong, Y., and Takai, K. (2018). 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polalpha-dependent fill-in. Nature 560, 112–116. doi: 10.1038/s41586-018-0324-7
Mladenov, E., Fan, X., Dueva, R., Soni, A., and Iliakis, G. (2019). Radiation-dose-dependent functional synergisms between ATM, ATR and DNA-PKcs in checkpoint control and resection in G2-phase. Sci. Rep. 9:8255.
Mohaghegh, P., Karow, J. K., Brosh, R. M. Jr., Bohr, V. A., and Hickson, I. D. (2001). The Bloom’s and Werner’s syndrome proteins are DNA structure-specific helicases. Nucleic Acids Res. 29, 2843–2849. doi: 10.1093/nar/29.13.2843
Munoz-Espin, D., and Serrano, M. (2014). Cellular senescence: from physiology to pathology. Nat. Rev. Mol. Cell Biol. 15, 482–496. doi: 10.1038/nrm3823
Myler, L. R., Gallardo, I. F., Soniat, M. M., Deshpande, R. A., Gonzalez, X. B., and Kim, Y. (2017). Single-molecule imaging reveals how Mre11-Rad50-Nbs1 initiates DNA break repair. Mol. Cell. 67, 891–898 e894.
Myler, L. R., Gallardo, I. F., Zhou, Y., Gong, F., Yang, S. H., and Wold, M. S. (2016). Single-molecule imaging reveals the mechanism of Exo1 regulation by single-stranded DNA binding proteins. Proc. Natl. Acad. Sci. U. S. A. 113, E1170–E1179.
Nacson, J., Krais, J. J., Bernhardy, A. J., Clausen, E., Feng, W., and Wang, Y. (2018). BRCA1 mutation-specific responses to 53BP1 loss-induced homologous recombination and PARP inhibitor resistance. Cell Rep. 25:1384. doi: 10.1016/j.celrep.2018.10.009
Nieminuszczy, J., Broderick, R., and Niedzwiedz, W. (2016). EXD2 - a new player joins the DSB resection team. Cell Cycle 15, 1519–1520. doi: 10.1080/15384101.2016.1161997
Nimonkar, A. V., Genschel, J., Kinoshita, E., Polaczek, P., Campbell, J. L., and Wyman, C. (2011). BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 25, 350–362. doi: 10.1101/gad.2003811
Noordermeer, S. M., Adam, S., Setiaputra, D., Barazas, M., Pettitt, S. J., and Ling, A. K. (2018). The shieldin complex mediates 53BP1-dependent DNA repair. Nature 560, 117–121.
Nora, G. J., Buncher, N. A., and Opresko, P. L. (2010). Telomeric protein TRF2 protects holliday junctions with telomeric arms from displacement by the Werner syndrome helicase. Nucleic Acids Res. 38, 3984–3998. doi: 10.1093/nar/gkq144
Olovnikov, A. M. (1973). A theory of marginotomy. the incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J. Theor. Biol. 41, 181–190.
Orthwein, A., Noordermeer, S. M., Wilson, M. D., Landry, S., Enchev, R. I., and Sherker, A. (2015). A mechanism for the suppression of homologous recombination in G1 cells. Nature 528, 422–426. doi: 10.1038/nature16142
Palm, W., Hockemeyer, D., Kibe, T., and de Lange, T. (2009). Functional dissection of human and mouse POT1 proteins. Mol. Cell Biol. 29, 471–482. doi: 10.1128/mcb.01352-08
Panigrahi, R., and Glover, J. N. M. (2021). Structural insights into DNA double-strand break signaling. Biochem. J. 478, 135–156. doi: 10.1042/bcj20200066
Pannunzio, N. R., Watanabe, G., and Lieber, M. R. (2018). Nonhomologous DNA end-joining for repair of DNA double-strand breaks. J. Biol. Chem. 293, 10512–10523. doi: 10.1074/jbc.tm117.000374
Pavani, R. S., Fernandes, C., Perez, A. M., Vasconcelos, E. J., Siqueira-Neto, J. L., and Fontes, M. R. (2014). RPA-1 from Leishmania amazonensis (LaRPA-1) structurally differs from other eukaryote RPA-1 and interacts with telomeric DNA via its N-terminal OB-fold domain. FEBS Lett. 588, 4740–4748. doi: 10.1016/j.febslet.2014.11.005
Pavani, R. S., Vitarelli, M. O., Fernandes, C. A. H., Mattioli, F. F., Morone, M., and Menezes, M. C. (2018). Replication Protein A-1 Has a preference for the telomeric G-rich sequence in Trypanosoma cruzi. J. Eukaryot. Microbiol. 65, 345–356. doi: 10.1111/jeu.12478
Pellegrini, L., Yu, D. S., Lo, T., Anand, S., Lee, M., and Blundell, T. L. (2002). Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature 420, 287–293. doi: 10.1038/nature01230
Perrem, K., Colgin, L. M., Neumann, A. A., Yeager, T. R., and Reddel, R. R. (2001). Coexistence of alternative lengthening of telomeres and telomerase in hTERT-transfected GM847 cells. Mol. Cell. Biol. 21, 3862–3875. doi: 10.1128/mcb.21.12.3862-3875.2001
Peterson, S. E., Li, Y., Wu-Baer, F., Chait, B. T., Baer, R., and Yan, H. (2013). Activation of DSB processing requires phosphorylation of CtIP by ATR. Mol. Cell 49, 657–667. doi: 10.1016/j.molcel.2012.11.020
Peuscher, M. H., and Jacobs, J. J. (2011). DNA-damage response and repair activities at uncapped telomeres depend on RNF8. Nat. Cell Biol. 13, 1139–1145. doi: 10.1038/ncb2326
Phillips, E. R., and McKinnon, P. J. (2007). DNA double-strand break repair and development. Oncogene 26, 7799–7808.
Pike, A. M., Strong, M. A., Ouyang, J. P. T., and Greider, C. W. (2019). TIN2 Functions with TPP1/POT1 To stimulate telomerase processivity. Mol. Cell. Biol. 39:e00593-18.
Qi, Z., Redding, S., Lee, J. Y., Gibb, B., Kwon, Y., and Niu, H. (2015). DNA sequence alignment by microhomology sampling during homologous recombination. Cell 160, 856–869. doi: 10.1016/j.cell.2015.01.029
Qin, Z., Bi, L., Hou, X. M., Zhang, S., Zhang, X., and Lu, Y. (2020). Human RPA activates BLM’s bidirectional DNA unwinding from a nick. Elife 9:e54098.
Qiu, S., and Huang, J. (2021). MRN complex is an essential effector of DNA damage repair. J. Zhejiang Univ. Sci. B 22, 31–37. doi: 10.1631/jzus.b2000289
Ranjha, L., Howard, S. M., and Cejka, P. (2018). Main steps in DNA double-strand break repair: an introduction to homologous recombination and related processes. Chromosoma 127, 187–214. doi: 10.1007/s00412-017-0658-1
Recagni, M., Bidzinska, J., Zaffaroni, N., and Folini, M. (2020). The role of alternative lengthening of telomeres mechanism in cancer: translational and therapeutic implications. Cancers 12:949. doi: 10.3390/cancers12040949
Reginato, G., and Cejka, P. (2020). The MRE11 complex: a versatile toolkit for the repair of broken DNA. DNA Repair 9:102869. doi: 10.1016/j.dnarep.2020.102869
Roberts, S. A., and Ramsden, D. A. (2007). Loading of the nonhomologous end joining factor, Ku, on protein-occluded DNA ends. J. Biol. Chem. 282, 10605–10613. doi: 10.1074/jbc.m611125200
Rothkamm, K., Kruger, I., Thompson, L. H., and Lobrich, M. (2003). Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol. Cell. Biol. 23, 5706–5715. doi: 10.1128/mcb.23.16.5706-5715.2003
Roumelioti, F. M., Sotiriou, S. K., Katsini, V., Chiourea, M., Halazonetis, T. D., and Gagos, S. (2016). Alternative lengthening of human telomeres is a conservative DNA replication process with features of break-induced replication. EMBO Rep. 17, 1731–1737. doi: 10.15252/embr.201643169
Rulten, S. L., and Caldecott, K. W. (2013). DNA strand break repair and neurodegeneration. DNA Repair 12, 558–567. doi: 10.1016/j.dnarep.2013.04.008
Rupnik, A., Lowndes, N. F., and Grenon, M. (2010). MRN and the race to the break. Chromosoma 119, 115–135. doi: 10.1007/s00412-009-0242-4
Sallmyr, A., and Tomkinson, A. E. (2018). Repair of DNA double-strand breaks by mammalian alternative end-joining pathways. J. Biol. Chem. 293, 10536–10546. doi: 10.1074/jbc.tm117.000375
Sanders, S. L., Portoso, M., Mata, J., Bahler, J., Allshire, R. C., and Kouzarides, T. (2004). Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell 119, 603–614. doi: 10.1016/j.cell.2004.11.009
Sarangi, P., Clairmont, C. S., Galli, L. D., Moreau, L. A., and D’Andrea, A. D. (2020). p31(comet) promotes homologous recombination by inactivating REV7 through the TRIP13 ATPase. Proc. Natl. Acad. Sci. U. S. A. 117, 26795–26803. doi: 10.1073/pnas.2008830117
Sarek, G., Kotsantis, P., Ruis, P., Van Ly, D., Margalef, P., and Borel, V. (2019). CDK phosphorylation of TRF2 controls t-loop dynamics during the cell cycle. Nature 575, 523–527. doi: 10.1038/s41586-019-1744-8
Scully, R., Chen, J., Plug, A., Xiao, Y., Weaver, D., and Feunteun, J. (1997). Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell 88, 265–275. doi: 10.1016/s0092-8674(00)81847-4
Seol, J. H., Shim, E. Y., and Lee, S. E. (2018). Microhomology-mediated end joining: good, bad and ugly. Mutat. Res. 809, 81–87. doi: 10.1016/j.mrfmmm.2017.07.002
Sfeir, A., and de Lange, T. (2012). Removal of shelterin reveals the telomere end-protection problem. Science 336, 593–597. doi: 10.1126/science.1218498
Shay, J. W., and Wright, W. E. (2019). Telomeres and telomerase: three decades of progress. Nat. Rev. Genet. 20, 299–309. doi: 10.1038/s41576-019-0099-1
Shen, Z. J., Hsu, P. H., Su, Y. T., Yang, C. W., Kao, L., and Tseng, S. F. (2014). PP2A and aurora differentially modify Cdc13 to promote telomerase release from telomeres at G2/M phase. Nat. Commun. 5:5312.
Shibata, A., Moiani, D., Arvai, A. S., Perry, J., Harding, S. M., and Genois, M. M. (2014). DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol. Cell 53, 7–18.
Shiotani, B., and Zou, L. (2009). Single-stranded DNA orchestrates an ATM-to-ATR switch at DNA breaks. Mol. Cell 33, 547–558. doi: 10.1016/j.molcel.2009.01.024
Simoneau, A., Robellet, X., Ladouceur, A. M., and D’Amours, D. (2014). Cdk1-dependent regulation of the Mre11 complex couples DNA repair pathways to cell cycle progression. Cell Cycle 13, 1078–1090. doi: 10.4161/cc.27946
Singleton, B. K., Torres-Arzayus, M. I., Rottinghaus, S. T., Taccioli, G. E., and Jeggo, P. A. (1999). The C terminus of Ku80 activates the DNA-dependent protein kinase catalytic subunit. Mol. Cell. Biol. 19, 3267–3277. doi: 10.1128/mcb.19.5.3267
Smogorzewska, A., Karlseder, J., Holtgreve-Grez, H., Jauch, A., and de Lange, T. (2002). DNA ligase IV-dependent NHEJ of deprotected mammalian telomeres in G1 and G2. Curr. Biol. 12, 1635–1644. doi: 10.1016/s0960-9822(02)01179-x
Sommer, A., and Royle, N. J. (2020). ALT: a multi-faceted Phenomenon. Genes 11:133. doi: 10.3390/genes11020133
Soniat, M. M., Myler, L. R., Kuo, H. C., Paull, T. T., and Finkelstein, I. J. (2019). RPA phosphorylation inhibits DNA resection. Mol. Cell 75, 145–153 e145.
Stavropoulos, D. J., Bradshaw, P. S., Li, X., Pasic, I., Truong, K., and Ikura, M. (2002). The bloom syndrome helicase BLM interacts with TRF2 in ALT cells and promotes telomeric DNA synthesis. Hum. Mol. Genet. 11, 3135–3144. doi: 10.1093/hmg/11.25.3135
Stewart, G. S., Wang, B., Bignell, C. R., Taylor, A. M., and Elledge, S. J. (2003). MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature 421, 961–966. doi: 10.1038/nature01446
Stewart, J. A., Chaiken, M. F., Wang, F., and Price, C. M. (2012). Maintaining the end: roles of telomere proteins in end-protection, telomere replication and length regulation. Mutat. Res. 730, 12–19. doi: 10.1016/j.mrfmmm.2011.08.011
Stewart, J. A., Wang, Y., Ackerson, S. M., and Schuck, P. L. (2018). Emerging roles of CST in maintaining genome stability and human disease. Front. Biosci. 23:1564–1586. doi: 10.2741/4661
Stinson, B. M., Moreno, A. T., Walter, J. C., and Loparo, J. J. (2020). A mechanism to minimize errors during non-homologous end joining. Mol. Cell 77, 1080–1091 e1088.
Stroik, S., Kurtz, K., and Hendrickson, E. A. (2019). CtIP is essential for telomere replication. Nucleic Acids Res. 47, 8927–8940. doi: 10.1093/nar/gkz652
Stroik, S., Kurtz, K., Lin, K., Karachenets, S., Myers, C. L., and Bielinsky, A. K. (2020). EXO1 resection at G-quadruplex structures facilitates resolution and replication. Nucleic Acids Res. 48, 4960–4975. doi: 10.1093/nar/gkaa199
Stucki, M., Clapperton, J. A., Mohammad, D., Yaffe, M. B., Smerdon, S. J., and Jackson, S. P. (2005). MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 123, 1213–1226. doi: 10.1016/j.cell.2005.09.038
Sullivan, M. R., and Bernstein, K. A. (2018). RAD-ical New Insights into RAD51 regulation. Genes 9:629. doi: 10.3390/genes9120629
Surovtseva, Y. V., Churikov, D., Boltz, K. A., Song, X., Lamb, J. C., and Warrington, R. (2009). Conserved telomere maintenance component 1 interacts with STN1 and maintains chromosome ends in higher eukaryotes. Mol. Cell 36, 207–218. doi: 10.1016/j.molcel.2009.09.017
Suwaki, N., Klare, K., and Tarsounas, M. (2011). RAD51 paralogs: roles in DNA damage signalling, recombinational repair and tumorigenesis. Semin. Cell Dev. Biol. 22, 898–905. doi: 10.1016/j.semcdb.2011.07.019
Sy, S. M., Huen, M. S., and Chen, J. (2009). PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc. Natl. Acad. Sci. U. S. A. 106, 7155–7160. doi: 10.1073/pnas.0811159106
Symington, L. S. (2016). Mechanism and regulation of DNA end resection in eukaryotes. Crit. Rev. Biochem. Mol. Biol. 51, 195–212. doi: 10.3109/10409238.2016.1172552
Symington, L. S., and Gautier, J. (2011). Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 45, 247–271. doi: 10.1146/annurev-genet-110410-132435
Takai, H., Jenkinson, E., Kabir, S., Babul-Hirji, R., Najm-Tehrani, N., and Chitayat, D. A. (2016). A POT1 mutation implicates defective telomere end fill-in and telomere truncations in Coats plus. Genes Dev. 30, 812–826. doi: 10.1101/gad.276873.115
Thacker, J. (1999). A surfeit of RAD51-like genes? Trends Genet. 15, 166–168. doi: 10.1016/s0168-9525(99)01733-3
Timashev, L. A., and De Lange, T. (2020). Characterization of t-loop formation by TRF2. Nucleus 11, 164–177. doi: 10.1080/19491034.2020.1783782
Tisi, R., Vertemara, J., Zampella, G., and Longhese, M. P. (2020). Functional and structural insights into the MRX/MRN complex, a key player in recognition and repair of DNA double-strand breaks. Comput. Struct. Biotechnol. J. 18, 1137–1152. doi: 10.1016/j.csbj.2020.05.013
Tomimatsu, N., Mukherjee, B., Deland, K., Kurimasa, A., Bolderson, E., and Khanna, K. K. (2012). Exo1 plays a major role in DNA end resection in humans and influences double-strand break repair and damage signaling decisions. DNA Repair 11, 441–448. doi: 10.1016/j.dnarep.2012.01.006
Tomita, K., Kibe, T., Kang, H. Y., Seo, Y. S., Uritani, M., and Ushimaru, T. (2004). Fission yeast Dna2 is required for generation of the telomeric single-strand overhang. Mol. Cell. Biol. 24, 9557–9567. doi: 10.1128/mcb.24.21.9557-9567.2004
Truong, L. N., Li, Y., Shi, L. Z., Hwang, P. Y., He, J., and Wang, H. (2013). Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc. Natl. Acad. Sci. U. S. A. 110, 7720–7725. doi: 10.1073/pnas.1213431110
Tseng, S. F., Lin, J. J., and Teng, S. C. (2006). The telomerase-recruitment domain of the telomere binding protein Cdc13 is regulated by Mec1p/Tel1p-dependent phosphorylation. Nucleic Acids Res. 34, 6327–6336. doi: 10.1093/nar/gkl786
Valuchova, S., Fulnecek, J., Prokop, Z., Stolt-Bergner, P., Janouskova, E., and Hofr, C. (2017). Protection of arabidopsis blunt-ended telomeres is mediated by a physical association with the Ku heterodimer. Plant Cell 29, 1533–1545. doi: 10.1105/tpc.17.00064
Van Ly, D., Low, R. R. J., Frolich, S., Bartolec, T. K., Kafer, G. R., and Pickett, H. A. (2018). Telomere loop dynamics in chromosome end protection. Mol. Cell 71, 510–525 e516.
van Overbeek, M., and de Lange, T. (2006). Apollo, an Artemis-related nuclease, interacts with TRF2 and protects human telomeres in S phase. Curr. Biol. 16, 1295–1302. doi: 10.1016/j.cub.2006.05.022
Vitor, A. C., Huertas, P., Legube, G., and de Almeida, S. F. (2020). Studying DNA double-strand break repair: an ever-growing toolbox. Front. Mol. Biosci. 7:24.
Wan, M., Qin, J., Songyang, Z., and Liu, D. (2009). OB fold-containing protein 1 (OBFC1), a human homolog of yeast Stn1, associates with TPP1 and is implicated in telomere length regulation. J. Biol. Chem. 284, 26725–26731. doi: 10.1074/jbc.m109.021105
Wang, B., Matsuoka, S., Ballif, B. A., Zhang, D., Smogorzewska, A., and Gygi, S. P. (2007). Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science 316, 1194–1198. doi: 10.1126/science.1139476
Wang, F., Stewart, J. A., Kasbek, C., Zhao, Y., Wright, W. E., and Price, C. M. (2012). Human CST has independent functions during telomere duplex replication and C-strand fill-in. Cell Rep. 2, 1096–1103. doi: 10.1016/j.celrep.2012.10.007
Wang, H., Shi, L. Z., Wong, C. C., Han, X., Hwang, P. Y., and Truong, L. N. (2013). The interaction of CtIP and Nbs1 connects CDK and ATM to regulate HR-mediated double-strand break repair. PLoS Genet. 9:e1003277. doi: 10.1371/journal.pgen.1003277
Wang, J., Aroumougame, A., Lobrich, M., Li, Y., Chen, D., and Chen, J. (2014). PTIP associates with Artemis to dictate DNA repair pathway choice. Genes Dev. 28, 2693–2698. doi: 10.1101/gad.252478.114
Wang, Y., Ghosh, G., and Hendrickson, E. A. (2009). Ku86 represses lethal telomere deletion events in human somatic cells. Proc. Natl. Acad. Sci. U. S. A. 106, 12430–12435. doi: 10.1073/pnas.0903362106
Watson, J. D. (1972). Origin of concatemeric T7 DNA. Nat. New Biol. 239, 197–201. doi: 10.1038/newbio239197a0
Wong, A. K., Pero, R., Ormonde, P. A., Tavtigian, S. V., and Bartel, P. L. (1997). RAD51 interacts with the evolutionarily conserved BRC motifs in the human breast cancer susceptibility gene brca2. J. Biol. Chem. 272, 31941–31944. doi: 10.1074/jbc.272.51.31941
Wright, W. D., Shah, S. S., and Heyer, W. D. (2018). Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 293, 10524–10535.
Wu, L., Multani, A. S., He, H., Cosme-Blanco, W., Deng, Y., and Deng, J. M. (2006). Pot1 deficiency initiates DNA damage checkpoint activation and aberrant homologous recombination at telomeres. Cell 126, 49–62. doi: 10.1016/j.cell.2006.05.037
Wu, P., Takai, H., and de Lange, T. (2012). Telomeric 3’ overhangs derive from resection by Exo1 and Apollo and fill-in by POT1b-associated CST. Cell 150, 39–52. doi: 10.1016/j.cell.2012.05.026
Wu, P., van Overbeek, M., Rooney, S., and de Lange, T. (2010). Apollo contributes to G overhang maintenance and protects leading-end telomeres. Mol. Cell 39, 606–617. doi: 10.1016/j.molcel.2010.06.031
Wu, Q. (2019). Structural mechanism of DNA-end synapsis in the non-homologous end joining pathway for repairing double-strand breaks: bridge over troubled ends. Biochem. Soc. Trans. 47, 1609–1619. doi: 10.1042/bst20180518
Xia, B., Sheng, Q., Nakanishi, K., Ohashi, A., Wu, J., and Christ, N. (2006). Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol. Cell 22, 719–729. doi: 10.1016/j.molcel.2006.05.022
Xie, A., Kwok, A., and Scully, R. (2009). Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat. Struct. Mol. Biol. 16, 814–818. doi: 10.1038/nsmb.1640
Xiong, X., Du, Z., Wang, Y., Feng, Z., Fan, P., and Yan, C. (2015). 53BP1 promotes microhomology-mediated end-joining in G1-phase cells. Nucleic Acids Res. 43, 1659–1670. doi: 10.1093/nar/gku1406
Yaneva, M., Kowalewski, T., and Lieber, M. R. (1997). Interaction of DNA-dependent protein kinase with DNA and with Ku: biochemical and atomic-force microscopy studies. EMBO J. 16, 5098–5112. doi: 10.1093/emboj/16.16.5098
Yang, H., Pesavento, J. J., Starnes, T. W., Cryderman, D. E., Wallrath, L. L., and Kelleher, N. L. (2008). Preferential dimethylation of histone H4 lysine 20 by Suv4-20. J. Biol. Chem. 283, 12085–12092. doi: 10.1074/jbc.m707974200
Yu, X., and Baer, R. (2000). Nuclear localization and cell cycle-specific expression of CtIP, a protein that associates with the BRCA1 tumor suppressor. J. Biol. Chem. 275, 18541–18549. doi: 10.1074/jbc.m909494199
Yu, X., and Chen, J. (2004). DNA damage-induced cell cycle checkpoint control requires CtIP, a phosphorylation-dependent binding partner of BRCA1 C-terminal domains. Mol. Cell. Biol. 24, 9478–9486. doi: 10.1128/mcb.24.21.9478-9486.2004
Yue, X., Bai, C., Xie, D., Ma, T., and Zhou, P. K. (2020). DNA-PKcs: a multi-faceted player in DNA damage response. Front. Genet. 11:607428.
Yun, M. H., and Hiom, K. (2009). CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature 459, 460–463. doi: 10.1038/nature07955
Zhang, D. H., Zhou, B., Huang, Y., Xu, L. X., and Zhou, J. Q. (2006). The human Pif1 helicase, a potential Escherichia coli RecD homologue, inhibits telomerase activity. Nucleic Acids Res. 34, 1393–1404. doi: 10.1093/nar/gkl029
Zhang, F., Fan, Q., Ren, K., and Andreassen, P. R. (2009a). PALB2 functionally connects the breast cancer susceptibility proteins BRCA1 and BRCA2. Mol. Cancer Res. 7, 1110–1118. doi: 10.1158/1541-7786.mcr-09-0123
Zhang, F., Ma, J., Wu, J., Ye, L., Cai, H., and Xia, B. (2009b). PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr. Biol. 19, 524–529. doi: 10.1016/j.cub.2009.02.018
Zhang, H., Schaub, J. M., and Finkelstein, I. J. (2020). RADX condenses single-stranded DNA to antagonize RAD51 loading. Nucleic Acids Res. 48, 7834–7843. doi: 10.1093/nar/gkaa559
Zhang, J. M., and Zou, L. (2020). Alternative lengthening of telomeres: from molecular mechanisms to therapeutic outlooks. Cell Biosci. 10:30.
Zhang, J. M., Yadav, T., Ouyang, J., Lan, L., and Zou, L. (2019). Alternative lengthening of telomeres through two distinct break-induced replication pathways. Cell Rep. 26, 955–968 e953.
Zhang, Y., and Jasin, M. (2011). An essential role for CtIP in chromosomal translocation formation through an alternative end-joining pathway. Nat. Struct. Mol. Biol. 18, 80–84. doi: 10.1038/nsmb.1940
Zhang, Y., Chen, L. Y., Han, X., Xie, W., Kim, H., and Yang, D. (2013). Phosphorylation of TPP1 regulates cell cycle-dependent telomerase recruitment. Proc. Natl. Acad. Sci. U. S. A. 110, 5457–5462. doi: 10.1073/pnas.1217733110
Zhao, X., Wei, C., Li, J., Xing, P., Li, J., and Zheng, S. (2017). Cell cycle-dependent control of homologous recombination. Acta Biochim. Biophys. Sin. 49, 655–668. doi: 10.1093/abbs/gmx055
Zhao, Y., Abreu, E., Kim, J., Stadler, G., Eskiocak, U., and Terns, M. P. (2011). Processive and distributive extension of human telomeres by telomerase under homeostatic and nonequilibrium conditions. Mol. Cell 42, 297–307. doi: 10.1016/j.molcel.2011.03.020
Zhao, Y., Sfeir, A. J., Zou, Y., Buseman, C. M., Chow, T. T., and Shay, J. W. (2009). Telomere extension occurs at most chromosome ends and is uncoupled from fill-in in human cancer cells. Cell 138, 463–475. doi: 10.1016/j.cell.2009.05.026
Zheng, L., Meng, Y., Campbell, J. L., and Shen, B. (2020). Multiple roles of DNA2 nuclease/helicase in DNA metabolism, genome stability and human diseases. Nucleic Acids Res. 48, 16–35. doi: 10.1093/nar/gkz1101
Zhong, Z. H., Jiang, W. Q., Cesare, A. J., Neumann, A. A., Wadhwa, R., and Reddel, R. R. (2007). Disruption of telomere maintenance by depletion of the MRE11/RAD50/NBS1 complex in cells that use alternative lengthening of telomeres. J. Biol. Chem. 282, 29314–29322. doi: 10.1074/jbc.m701413200
Zhou, Y., and Paull, T. T. (2013). DNA-dependent protein kinase regulates DNA end resection in concert with Mre11-Rad50-Nbs1 (MRN) and ataxia telangiectasia-mutated (ATM). J. Biol. Chem. 288, 37112–37125. doi: 10.1074/jbc.m113.514398
Zhu, X. D., Kuster, B., Mann, M., Petrini, J. H., and de Lange, T. (2000). Cell-cycle-regulated association of RAD50/MRE11/NBS1 with TRF2 and human telomeres. Nat. Genet. 25, 347–352. doi: 10.1038/77139
Zhu, X. D., Niedernhofer, L., Kuster, B., Mann, M., Hoeijmakers, J. H., and de Lange, T. (2003). ERCC1/XPF removes the 3’ overhang from uncapped telomeres and represses formation of telomeric DNA-containing double minute chromosomes. Mol. Cell 12, 1489–1498. doi: 10.1016/s1097-2765(03)00478-7
Zimmermann, M., Lottersberger, F., Buonomo, S. B., Sfeir, A., and de Lange, T. (2013). 53BP1 regulates DSB repair using Rif1 to control 5’ end resection. Science 339, 700–704. doi: 10.1126/science.1231573
Keywords: telomeres, double-strand break, DNA repair, homologous recombination, non-homologous end joining
Citation: Ackerson SM, Romney C, Schuck PL and Stewart JA (2021) To Join or Not to Join: Decision Points Along the Pathway to Double-Strand Break Repair vs. Chromosome End Protection. Front. Cell Dev. Biol. 9:708763. doi: 10.3389/fcell.2021.708763
Received: 12 May 2021; Accepted: 17 June 2021;
Published: 12 July 2021.
Edited by:
Marcelo Santos Da Silva, São Paulo State University, BrazilReviewed by:
Jennifer Ann Black, University of Glasgow, United KingdomYlli Doksani, IFOM - The FIRC Institute of Molecular Oncology, Italy
Copyright © 2021 Ackerson, Romney, Schuck and Stewart. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jason A. Stewart, jason.stewart@sc.edu
†Present address: Carlan Romney, Department of Biology, Fisk University, Nashville, TN, United States