Branebrutinib (BMS-986195), a Bruton’s Tyrosine Kinase Inhibitor, Resensitizes P-Glycoprotein-Overexpressing Multidrug-Resistant Cancer Cells to Chemotherapeutic Agents
 Chung-Pu Wu1,2,3*
Chung-Pu Wu1,2,3*  Megumi Murakami4 Yu-Shan Wu5
Megumi Murakami4 Yu-Shan Wu5  Ya-Chen Chi1 Sung-Han Hsiao1 Yang-Hui Huang1,2 Tai-Ho Hung3,6
Ya-Chen Chi1 Sung-Han Hsiao1 Yang-Hui Huang1,2 Tai-Ho Hung3,6  Suresh V. Ambudkar4
Suresh V. Ambudkar4- 1Graduate Institute of Biomedical Sciences, College of Medicine, Chang Gung University, Taoyuan City, Taiwan
- 2Department of Physiology and Pharmacology, College of Medicine, Chang Gung University, Taoyuan City, Taiwan
- 3Department of Obstetrics and Gynecology, Taipei Chang Gung Memorial Hospital, Taipei, Taiwan
- 4Laboratory of Cell Biology, Center for Cancer Research, National Cancer Institute, NIH, Bethesda, MD, United States
- 5Department of Chemistry, Tunghai University, Taichung, Taiwan
- 6Department of Medicine, College of Medicine, Chang Gung University, Taoyuan City, Taiwan
The overexpression of P-glycoprotein (P-gp/ABCB1), an ATP-binding cassette (ABC) drug transporter, often contributes to the development of multidrug resistance (MDR) in cancer cells. P-gp mediates the ATP hydrolysis-dependent efflux of a wide range of chemotherapeutic agents out of cancer cells, thereby reducing the intracellular drug accumulation and decreasing the chemosensitivity of these multidrug-resistant cancer cells. Studies with tyrosine kinase inhibitors (TKIs) in P-gp-overexpressing cells have shown that certain TKIs could reverse MDR mediated by P-gp, while some TKIs are transported by P-gp. In the present work, we explored the prospect of repositioning branebrutinib (BMS-986195), a highly selective inhibitor of Bruton’s tyrosine kinase (BTK), to resensitize P-gp-overexpressing multidrug-resistant cancer cells to chemotherapeutic agents. Our results demonstrated that branebrutinib is capable of reversing P-gp-mediated MDR at sub-toxic concentrations, most likely by directly inhibiting the drug transport function of P-gp. Our findings were supported by the result of branebrutinib stimulating the ATPase activity of P-gp in a concentration-dependent manner and the in silico study of branebrutinib binding to the substrate-binding pocket of P-gp. In addition, we found that branebrutinib is equally cytotoxic to drug-sensitive parental cell lines and the respective P-gp-overexpressing multidrug-resistant variants, suggesting that it is unlikely that the overexpression of P-gp in cancer cells plays a significant role in reduced susceptibility or resistance to branebrutinib. In summary, we discovered an additional pharmacological action of branebrutinib against the activity of P-gp, which should be investigated further in future drug combination studies.
Introduction
P-glycoprotein (P-gp or ABCB1) is the most well-characterized member of the human ATP-binding cassette (ABC) transporter family that has been linked to the development of multidrug resistance (MDR) in cancer (Gottesman and Ambudkar, 2001; Robey et al., 2018). P-gp uses energy derived from ATP hydrolysis to actively efflux structurally unrelated chemotherapeutic drugs out of cancer cells and reduces the intracellular accumulation of these drugs. Some of the most well-known P-gp substrate drugs include Vinca alkaloids, paclitaxel, colchicine, and anthracyclines (Ambudkar et al., 1999; Gottesman et al., 2002). Consequently, the overexpression of P-gp in cancer cells frequently contributes to reduced chemosensitivity, treatment failure, and recurrence in cancer patients (Szakacs et al., 2006; Wu et al., 2011; Robey et al., 2018). In particular, the link between the high expression of P-gp and poor clinical outcome has been reported in patients with metastatic breast cancer (MBC) (Kovalev et al., 2013) and blood cancers (Ross, 2000; Robey et al., 2018) such as chronic lymphocytic leukemia (CLL) (Matthews et al., 2006), chronic myeloid leukemia (CML) (Maia et al., 2018), and multiple myeloma (MM) (Schwarzenbach, 2002; Tsubaki et al., 2012). Therefore, the discovery and development of P-gp modulators for clinical use is of great significance.
For years, the advancement of P-gp inhibitors has not been successful, which is frequently due to unforeseen toxicities and adverse drug-drug interactions (Dong et al., 2020). For example, tariquidar (XR9576) was developed as a selective and potent inhibitor of P-gp (Mistry et al., 2001), capable of increasing P-gp substrate drug accumulation in drug-resistant tumors (Agrawal et al., 2003). Unfortunately, unexpected toxicity caused two phase III clinical trials of vinorelbine combined with tariquidar (ClinicalTrials.gov Identifier: NCT00042315) and paclitaxel/carboplatin combined with tariquidar (NCT00042302) as first-line therapy in non-small cell lung cancer (NSCLC) to terminate prematurely. To date, the US Food and Drug Administration (FDA) has not approve any agent for the treatment of multidrug-resistant cancers.
As an alternative to developing novel inhibitors, the drug repositioning approach has been exploited by various research groups to utilize tyrosine kinase inhibitors (TKIs) against P-gp-mediated MDR (Beretta et al., 2017). As the result, some well-known TKIs such as osimertinib (Hsiao et al., 2016) and midostaurin (Hsiao et al., 2019), were identified as drug candidates for resensitizing P-gp-overexpressing cancer cells to chemotherapeutic agents (Wu and Fu, 2018). Branebrutinib (BMS-986195) is an orally available, highly selective inhibitor of Bruton’s tyrosine kinase (BTK) (Watterson et al., 2019; Zheng et al., 2019). The safety, tolerability and pharmacokinetics of branebrutinib in healthy participants (Catlett et al., 2020), both as a single drug or in combination with other therapeutic agents, have been studied in several clinical trials (NCT03245515, NCT02705989, NCT03262740, NCT03131973). Currently, branebrutinib is being evaluated as monotherapy in clinical trials in patients with moderate to severe psoriasis (NCT02931838), or active systemic Lupus Erythematosus or Primary Sjögren’s Syndrome (NCT04186871).
In the present study, we found that branebrutinib could inhibit P-gp-mediated drug transport and consequently resensitize P-gp-overexpressing multidrug-resistant cancer cells to apoptosis and cytotoxicity induced by P-gp substrate drugs. Moreover, we observed that P-gp-overexpressing cell lines do not confer significant resistance to branebrutinib as compared to their respective parental cell lines. In summary, our study revealed an additional pharmacological action of branebrutinib, which could potentially be utilized in combination therapies against multidrug-resistant cancers and warrant further studies.
Materials and Methods
Chemicals
All culture media and supplements were obtained from Gibco, Invitrogen (Carlsbad, CA, United States). Tools Cell Counting (CCK-8) kit was purchased from Biotools Co., Ltd. (Taipei, Taiwan). Annexin V FITC-Apoptosis Detection Kit was obtained from BD Pharmingen (San Diego, CA, United States). Primary and secondary antibodies were purchased from Abcam (Cambridge, MA, United States). Branebrutinib was purchased from Selleckchem (Houston, TX, United States). Tariquidar (XR9576) and all other chemicals were purchased from Sigma (St. Louis, MO, United States) unless stated otherwise.
Cell Lines
Human embryonic kidney 293 cells (HEK293) and P-gp-transfected HEK293 cells (MDR19-HEK293); the mouse NIH3T3 and P-gp -transfected NIH3T3-G185 fibroblast cells; the human epidermal cancer cell line KB-3-1 and its P-gp-overexpressing variant KB-V-1 were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM). The human ovarian cancer cell line OVCAR-8 and its P-gp-overexpressing variant NCI-ADR-RES; the human myelogenous leukemia K562 and the P-gp-expressing K562/i-S9 cell lines were maintained in Roswell Park Memorial Institute (RPMI-1640) medium (Mechetner et al., 1997). KB-V-1 cells were maintained in the presence of 1 μg/mL of vinblastine (Shen et al., 1986), NIH3T3-G185 cells were maintained in the presence of 60 ng/mL colchicine (Currier et al., 1992), whereas the HEK293 transfectants were maintained in the presence of 2 mg/mL of G418 (Wu et al., 2007). All cells were cultured at 37°C in 5% CO2 humidified air and grown in media supplemented with 10% FCS, L-glutamine and 100 units/mL of penicillin and streptomycin. Cells were maintained in a drug-free medium for 7 days before assay.
Cytotoxicity Assay
Cytotoxicity was measured with MTT and Cell Counting Kit-8 (CCK-8) assays. Cultured cells were seeded in 96-well flat-bottom plates and allowed to attach for 24 h before incubated in increasing concentrations of a single drug or drug combination for an additional 72 h. Cytotoxicity (IC50 value) of each drug regimen was calculated using fitted concentration-response curve from at least three independent experiments. The extent of drug resistance was presented as a resistance-factor (RF) value, whereas the chemosensitizing effect was presented as a fold-reversal (FR) value as previously described (Wu et al., 2007; Dai et al., 2008).
Apoptosis Assay
The concurrent annexin V–FITC and propidium iodide (PI) staining method was used to determine the effect of branebrutinib on colchicine-induced apoptosis in cancer cells as previously described (Anderson et al., 2003). Briefly, cells were treated with either DMSO, branebrutinib, a known apoptosis inducer colchicine, or the combination of colchicine and branebrutinib for 48 h. Cells were subsequently stained with 1.25 μg/mL of annexin V–FITC and 0.1 mg/mL of PI for 15 min at room temperature, and analyzed using the FACSCalibur flow cytometer equipped with CellQuest software (Becton-Dickinson Biosciences, San Jose, CA, United States) as previously described (Hsiao et al., 2016).
Flow Cytometry
The intracellular accumulation of the fluorescent P-gp substrate drug calcein (Hollo et al., 1994) was determined in the presence of DMSO (control), branebrutinib, or verapamil using the FACSCalibur flow cytometer, and analyzed using the CellQuest or FlowJo software (Tree Star, Inc., Ashland, OR, United States) software according to the method described by Gribar et al. (2000) and as previously described (Robey et al., 2004; Wu et al., 2013).
Immunoblotting
Cells were treated with either DMSO (control) or branebrutinib (1–10 μM) for 72 h before being harvested and subjected to SDS-polyacrylamide electrophoresis and immunoblotting as described previously (Wu et al., 2007). Primary antibodies C219 (1:3,000 dilution), anti-alpha tubulin (1:100,000 dilution) (Abcam, Cambridge, MA, United States) and the secondary horseradish peroxidase-conjugated goat anti-mouse IgG (1:100,000 dilution) were used to detect P-gp and the positive loading control tubulin, respectively. The enhanced chemiluminescence (ECL) kit was used for signal detection (Merck Millipore, Billerica, MA, United States).
ATPase Assay
ATPase activity of P-gp in total membranes prepared from High-Five insect cells (Invitrogen, Carlsbad, CA, United States) infected with recombinant baculovirus carrying the MDR1 gene was measured by endpoint inorganic phosphate (Pi) assay [33] and recorded as vanadate (Vi)-sensitive ATPase activity as previously described (Ambudkar, 1998; Wu et al., 2019). GraphPad Prism software (GraphPad Software, La Jolla, CA, United States) was used to calculate the EC50 values based on fitted concentration-response curves obtained from three independent experiments as previously described (Wu et al., 2019).
In silico Analysis of Docking of Branebrutinib in the Drug-Binding Pocket of P-gp
The cryo-EM structure of P-gp was obtained from the Protein Data Bank (PDB:6QEX) and the protein was prepared by addition of hydrogen atoms and partial charges based on CHARMM force field at pH of 7.4 using Accelrys Discovery Studio 4.0 (Alam et al., 2019). Branebrutinib structure was optimized and docking was performed by the CDOCKER module of the same software. The conformation with the lowest CDOCKER Interaction Energy was selected and the respective interaction energy was calculated.
Quantification and Statistical Analysis
Data are presented as mean ± standard deviation (SD) from at least three independent experiments unless stated otherwise. Statistical analysis was performed using KaleidaGraph software (Synergy Software, Reading, PA, United States). Two-tailed Student’s t-test was performed to analyze the difference between mean values of experimental and control or improvement in fit and labeled with asterisks as “statistically significant” if the probability, p, was less than 0.05.
Results
Branebrutinib Resensitizes P-gp-Overexpressing Multidrug-Resistant Cells to Cytotoxic Therapeutic Agents
We investigated the potential chemosensitizing effect of branebrutinib on P-gp-mediated resistance to known P-gp drug substrates (Kartner et al., 1983) such as vincristine, paclitaxel and colchicine in P-gp-overexpressing multidrug-resistant human epidermal KB-V-1 cancer cells, human ovarian NCI-ADR-RES cancer cells, and P-gp-transfected MDR19-HEK293 cells. We discovered that without significantly affecting the respective drug-sensitive parental cells (Figure 1, left panels), branebrutinib significantly reversed P-gp-mediated resistance to vincristine in KB-V-1 (Figure 1A, right panel), NCI-ADR-RES (Figure 1B, right panel), and MDR19-HEK293 (Figure 1C, right panel) cells in a concentration-dependent manner. Moreover, we found that P-gp-mediated resistance to paclitaxel (Figures 1D–F) and colchicine (Figures 1G–I) in these P-gp-overexpressing multidrug-resistant cells was reversed by branebrutinib in the same manner. The respective IC50 values and the extent of chemosensitization by branebrutinib in these cell lines, represented by the fold-reversal (FR) values (Dai et al., 2008), are summarized in Tables 1, 2. The FR value was calculated by dividing the IC50 value of a P-gp drug substrate by the IC50 value of the same drug substrate in the presence of branebrutinib or the P-gp reference inhibitor verapamil (Dai et al., 2008). Our data show that branebrutinib restores the chemosensitivity of P-gp-overexpressing cells to chemotherapeutic drugs at sub-toxic concentrations.
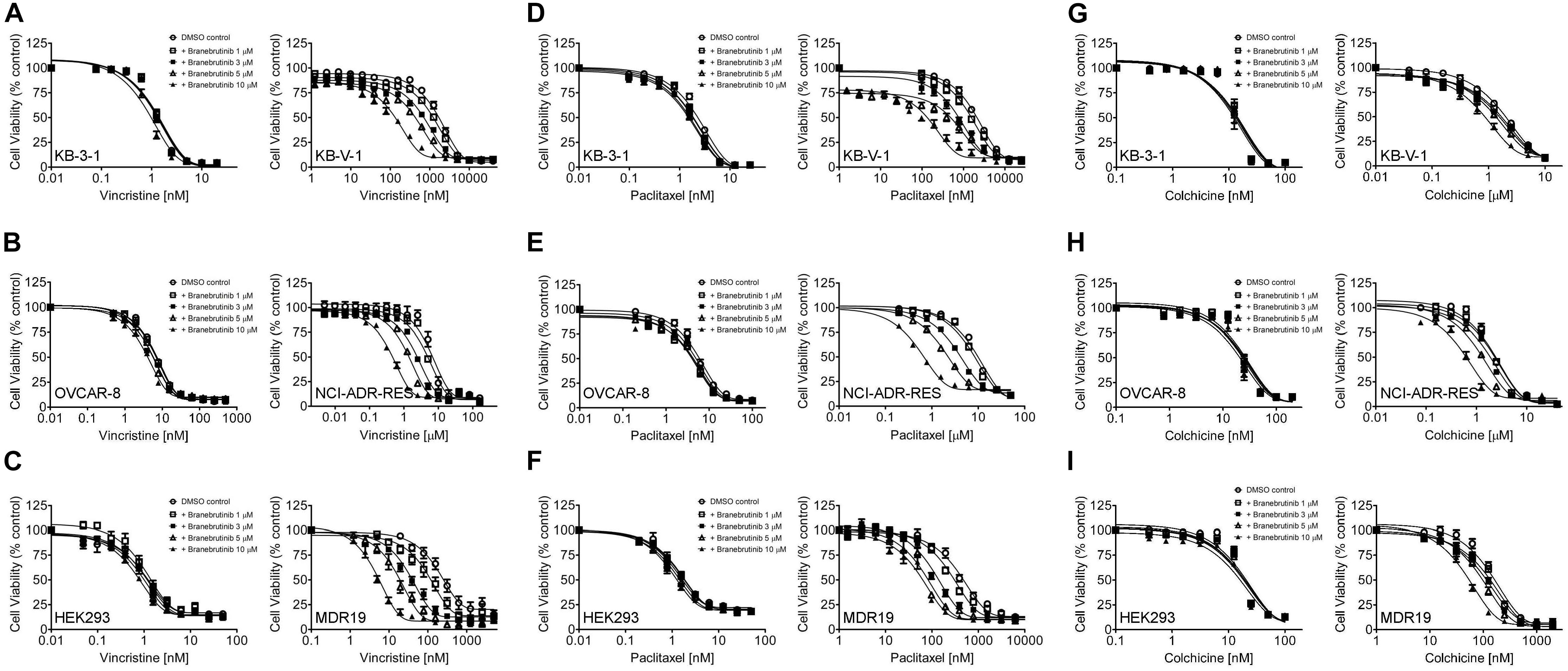
Figure 1. Branebrutinib reverses multidrug resistance (MDR) in P-glycoprotein (P-gp) -overexpressing cells. The effect of branebrutinib on P-gp-mediated resistance to vincristine (A–C), paclitaxel (D–F) and colchicine (G–I) was tested in drug-sensitive parental human KB-3-1 epidermal cancer cell line (A,D,G-left panel) and its P-gp-overexpressing multidrug-resistant KB-V-1 subline (A,D,G-right panel); drug-sensitive parental human OVCAR-8 ovarian cancer cell line (B,E,H-left panel) and its P-gp-overexpressing multidrug-resistant NCI-ADR-RES subline (B,E,H-right panel); as well as parental HEK293 cells (C,F,I-left panel) and MDR19-HEK293 cells, which are HEK293 cells transfected with human P-gp (C,F,I-right panel). Cells were treated with increasing concentration of vincristine, paclitaxel or colchicine in the presence of DMSO (open circles) or branebrutinib at 1 μM (open squares), 3 μM (filled squares), 5 μM (open triangles), or at 10 μM for 72 h before analysis as described in Materials and methods. Points, mean values from at least three independent experiments; bars; S.E.M.
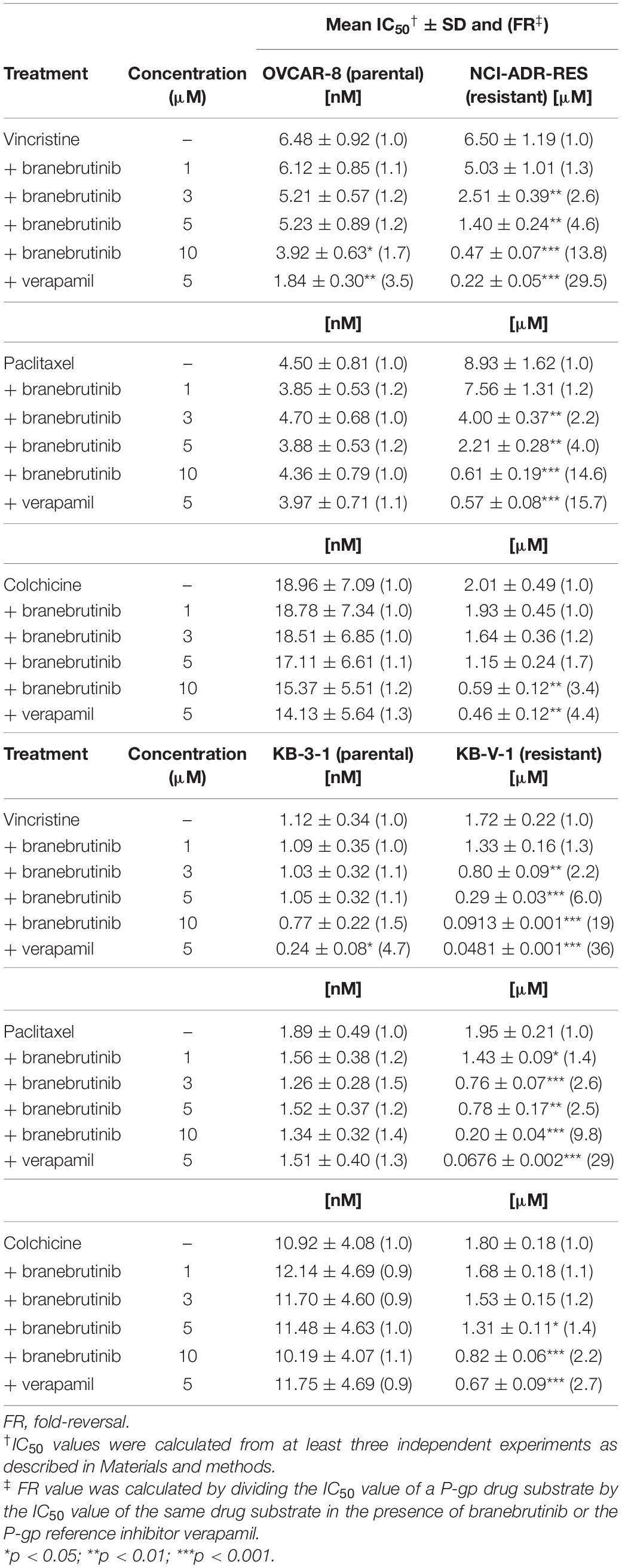
Table 1. Chemosensitizing effect of branebrutinib on drug-selected P-glycoprotein (P-gp)-overexpressing human cancer cell lines.
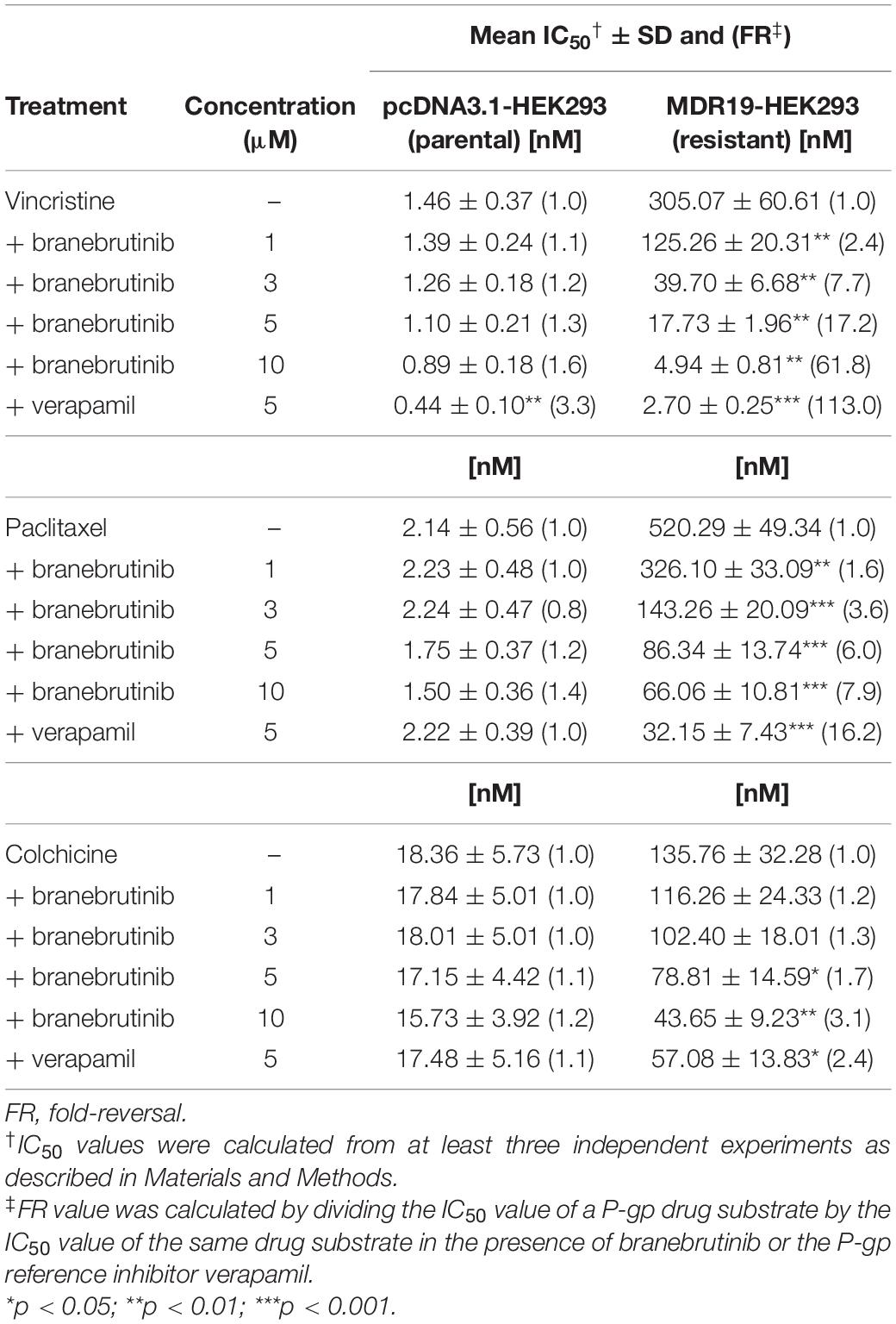
Table 2. Chemosensitizing effect of branebrutinib on HEK293 cells transfected with human P-glycoprotein (P-gp).
Branebrutinib Antagonizes the Drug Efflux Function of P-gp
Previous studies have demonstrated that a common way for a modulator to resensitize P-gp-overexpressing cells to P-gp drug substrates is by directly inhibiting the drug efflux function of P-gp (Hsiao et al., 2016, 2018, 2019; Wu et al., 2019, 2020b). To this end, we examined the effect of branebrutinib on the drug transport function of P-gp by performing a short-term fluorescent drug efflux assay in NCI-ADR-RES (Figure 2A) and KB-V-1 (Figure 2B) cancer cells, as well as in MDR9-HEK293 (Figure 2C) cells. Cells were incubated with a P-gp substrate calcein-AM (Hollo et al., 1994) in the presence of DMSO (solid line), or 20 μM of branebrutinib (filled solid line) or 20 μM of verapamil (dotted line), and the intracellular accumulation of calcein, a fluorescent product of calcein-AM, was monitored for 10 min as described in Materials and methods. We discovered that without affecting the accumulation of calcein in drug-sensitive parental cells (Figure 2, left panels), branebrutinib significantly increased the intracellular accumulation of fluorescent calcein in P-gp-overexpressing NCI-ADR-RES, KB-V-1, and MDR9-HEK293 cells (Figure 2, right panels).
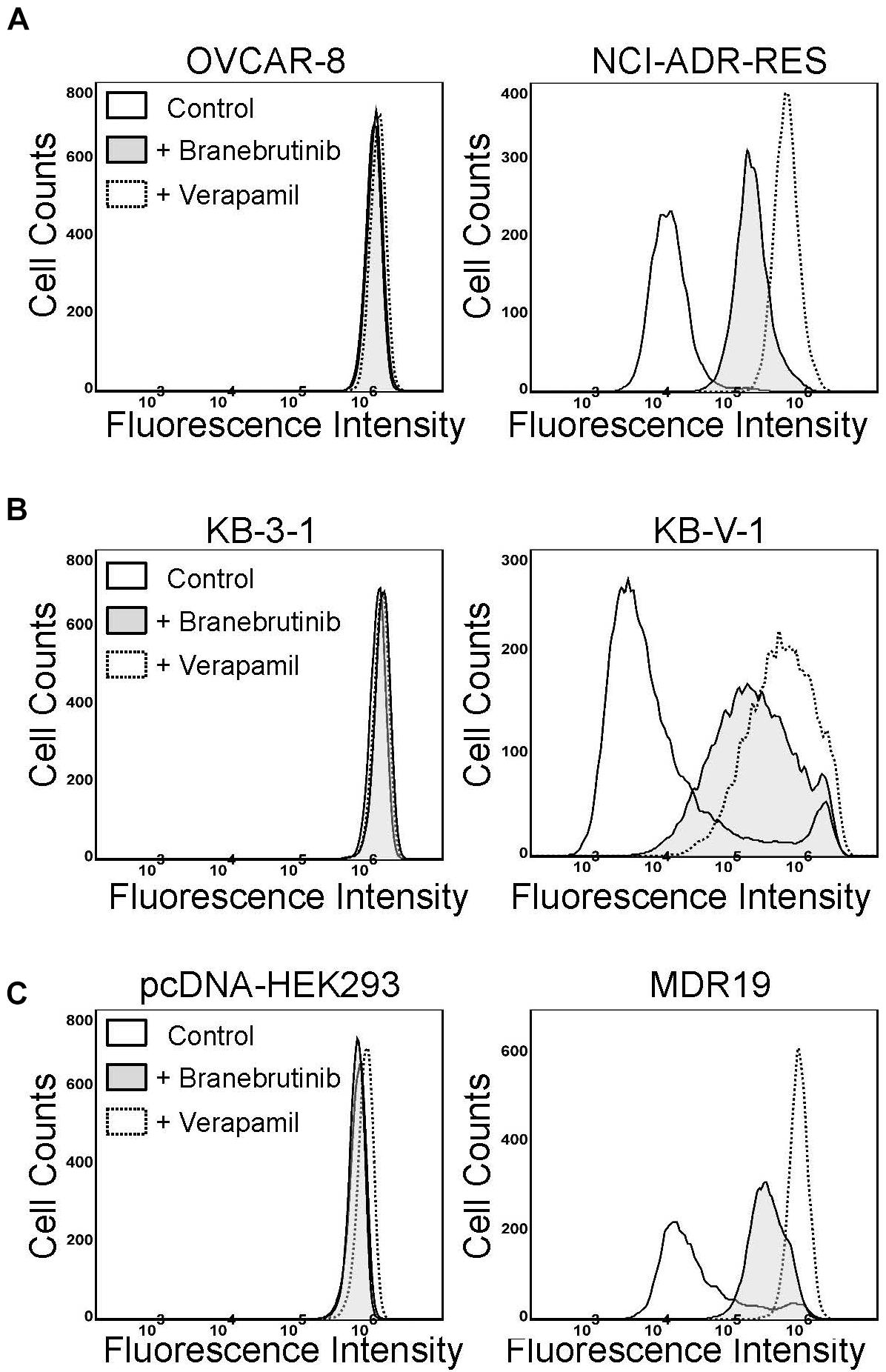
Figure 2. Branebrutinib inhibits P-glycoprotein (P-gp)-mediated drug efflux. The effect of branebrutinib on the intracellular accumulation of calcein, a fluorescent product of a known P-gp substrate calcein-AM, was determined in (A) OVCAR-8 (left panel) and NCI-ADR-RES cancer cells (right panel); (B) KB-3-1 (left panel) and KB-V-1 (right panel); and (C) HEK293 cells (left panel) and P-gp-transfected MDR19-HEK293 cells (right panel). Cells were treated with DMSO (control, solid lines), 20 μM of branebrutinib (filled solid lines) or 20 μM of a reference inhibitor verapamil (dotted lines) as a positive control. The fluorescence signal was analyzed by flow cytometry as described previously (Wu et al., 2007). Representative histograms of at least three independent experiments are shown.
Next, studies have also reported that a drug-induced, transient down-regulation of a drug transporter is another mechanism to resensitize multidrug-resistant cancer cells to anticancer drugs (Cuestas et al., 2012; Natarajan et al., 2013). For that reason, we examined the protein expression of P-gp by immunoblotting with specific antibodies after treating P-gp-overexpressing NCI-ADR-RES and KB-V-1 cancer cells with branebrutinib (1–10 μM) for 72 h as described in Materials and methods. Our results show that the expression of P-gp at protein level in NCI-ADR-RES (Figure 3A) and KB-V-1 (Figure 3B) cancer cells was not significantly altered by branebrutinib over a period of 72 h, suggesting that branebrutinib reverses P-gp-mediated MDR in these cancer cell lines (Table 1) by blocking the drug efflux function of P-gp.
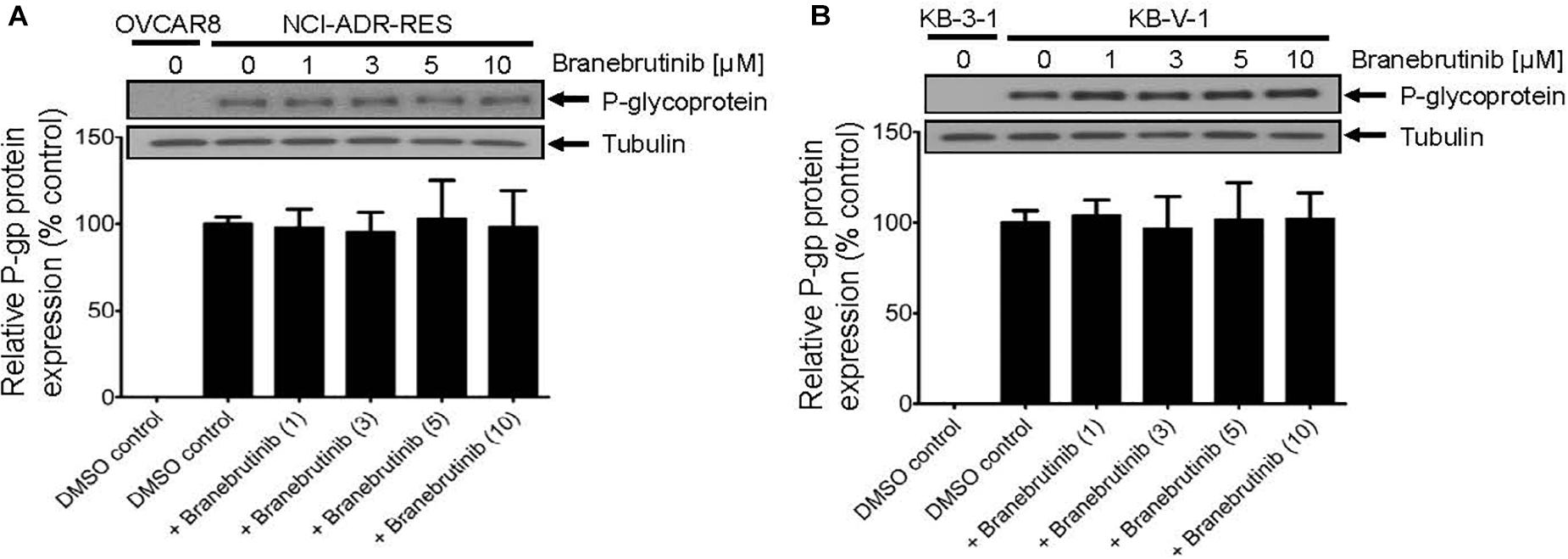
Figure 3. Branebrutinib has no significant effect on the P-glycoprotein (P-gp) protein levels after 72 h exposure. The P-gp-overexpressing (A) NCI-ADR-RES and (B) KB-V-1 cancer cells were treated with DMSO (vehicle control) or branebrutinib at 1, 3, 5, or 10 μM for 72 h and processed for immunoblotting with indicated antibodies as described in Materials and methods. Representative Western blots (upper panel) and the corresponding quantification (lower panel) of P-gp are shown. α-Tubulin was used as an internal loading control. Values are presented as mean ± SD. Calculated from at least three independent experiments.
Branebrutinib Enhances Colchicine-Induced Apoptosis in P-gp-Overexpressing Cancer Cells
To exclude the potential growth retardation effect of branebrutinib on P-gp-overexpressing multidrug-resistant cancer cells, we tested the effect of branebrutinib on drug-induced apoptosis in P-gp-overexpressing cancer cells. In addition to being a known drug substrate of P-gp (Kartner et al., 1983), colchicine is also a known inducer of apoptosis (Riordan and Ling, 1979). OVCAR-8 and NCI-ADR-RES cancer cells were treated with 0.5 μM of colchicine in the presence of DMSO (control) or 20 μM of branebrutinib for 48 h and proceed as described in Materials and methods. As expected, we found that colchicine induced the level of apoptosis in drug-sensitive parental OVCAR-8 cancer cells, from approximately 3% basal level to 82% of total apoptosis (Figure 4A, upper panels). In contrast, due to the activity of P-gp, the apoptosis-inducing effect of colchicine in P-gp-overexpressing NCI-ADR-RES cancer cells was significantly less (Figure 4A, lower panels). More importantly, we found that branebrutinib by itself does not induce substantial apoptosis in either cell line, however, it significantly enhanced colchicine-induced apoptosis in NCI-ADR-RES cancer cells, from 10 to 60% of early and late apoptosis (Figure 4B).
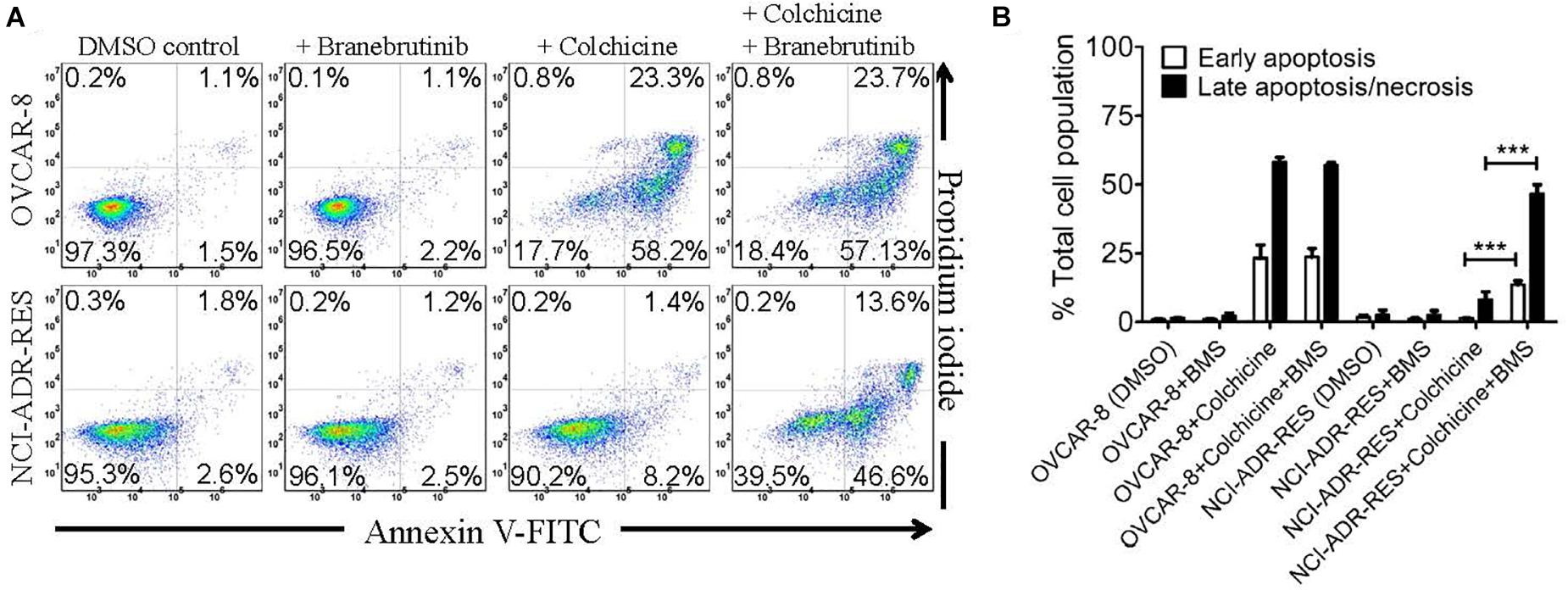
Figure 4. Branebrutinib enhances colchicine-induced apoptosis in P-glycoprotein (P-gp)-overexpressing cancer cells. Drug-sensitive OVCAR-8 and the P-gp-overexpressing multidrug-resistant NCI-ADR-RES cancer cells were treated with DMSO (control), branebrutinib at 20 μM (+BMS), colchicine at 500 nM (+ colchicine), or a combination of colchicine and branebrutinib (+colchicine + BMS) for 48 h, processed and analyzed by flow cytometry as described in Materials and methods. Representative flow cytometric dot plots are shown (A) and the corresponding quantification (B) are presented as mean ± SD. Calculated from at least three independent experiments. ***p < 0.001, versus the same treatment in the absence of branebrutinib.
Branebrutinib Stimulates ATPase Activity of P-gp
Knowing that the transport activity of P-gp is coupled to ATP hydrolysis (Ambudkar et al., 1999, 2003), the effect of branebrutinib on its ATP hydrolysis was examined to gain additional biochemical information on the interactions between branebrutinib and P-gp. As shown in Figure 5, branebrutinib stimulated Vi-sensitive ATPase activity of P-gp in a concentration-dependent manner, with maximum stimulation of almost 85% higher than the basal level of 45.6 ± 4.3 nmole Pi/min/mg protein, and the half maximal effective concentration (EC50) value (concentration for branebrutinib to obtain 50% of maximum stimulation of P-gp ATPase activity) of approximately 4.5 μM. Furthermore, to determine whether branebrutinib and verapamil interact at the same substrate site of P-gp, we examined the effect of branebrutinib on verapamil-stimulated ATPase activity of P-gp. We found that branebrutinib had no significant effect on the ATPase activity stimulated by 5 μM of verapamil (Supplementary Figure 1). These results suggested that branebrutinib does not interact at the same P-gp substrate binding site as verapamil.
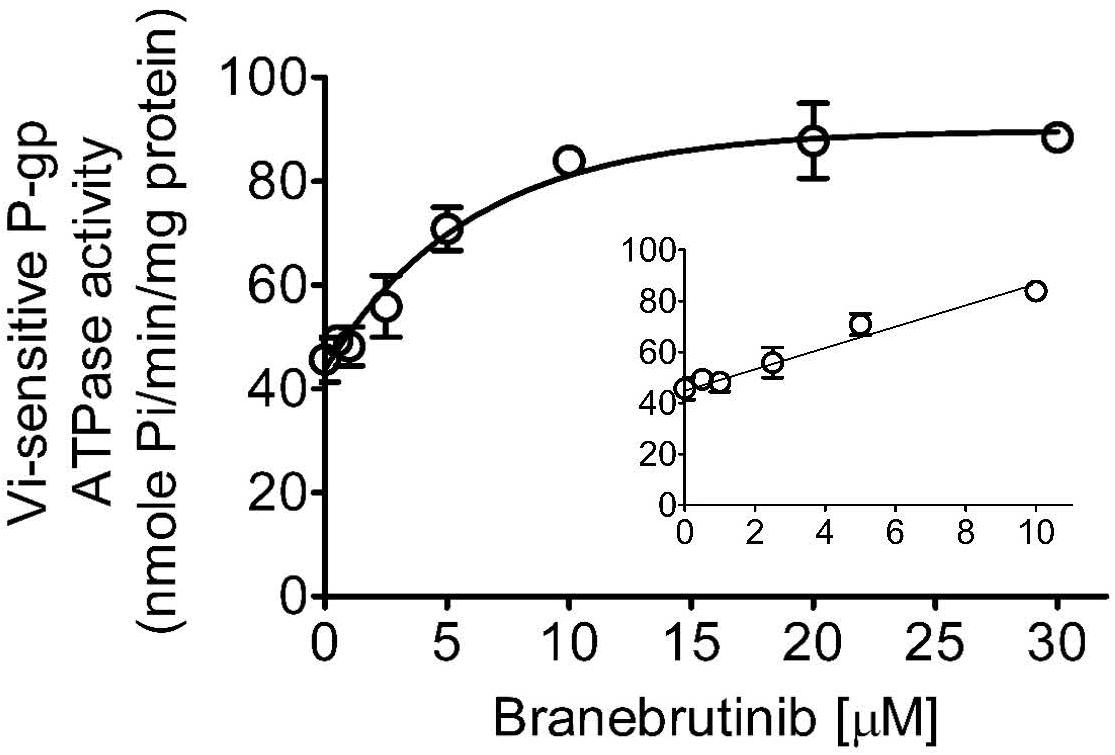
Figure 5. Branebrutinib stimulates the ATPase activity of P-glycoprotein (P-gp). The effect of 0–30 μM of branebrutinib (0–10 μM, inset) on P-gp-mediated ATP hydrolysis was measured in the membrane vesicles prepared from High-Five insect cells overexpressing human P-gp and recorded as vanadate (Vi)-sensitive ATPase activity as previously described (Wu et al., 2013). Points, mean from at least three independent experiments; bars, SD.
Docking Analysis of Branebrutinib Binding to the Drug-Binding Pocket of P-gp
Branebrutinib was docked into the human P-gp model based on cryo-EM structure (pdb.6QEX) and the binding was predicted to take place in the drug-binding cavity in the transmembrane region. The best binding conformation was selected with the binding energy calculated to be −52.83 kcal/mol. The residues from transmembrane helix (TMH) 1, 5, 6, 7, 11, and 12 were found to interact with branebrutinib. Most amino acids in the binding site interact with branebrutinib via hydrophobic interactions. The residues Met68, Met69, Phe72, Phe336, and Tyr953 of TMH 1, 6, and 11, respectively were predicted to interact with the propynyl, the residue Leu339 of TMH 6 with piperidine ring, and the indole moiety was predicted to interact with Met986 of TMH12 of P-gp. Hydrogen bonds were also found between Gln990 of TMH 12 and Gln725 of TMH7 with the amide moiety on 7-indole position of branebrutinib (Figure 6).
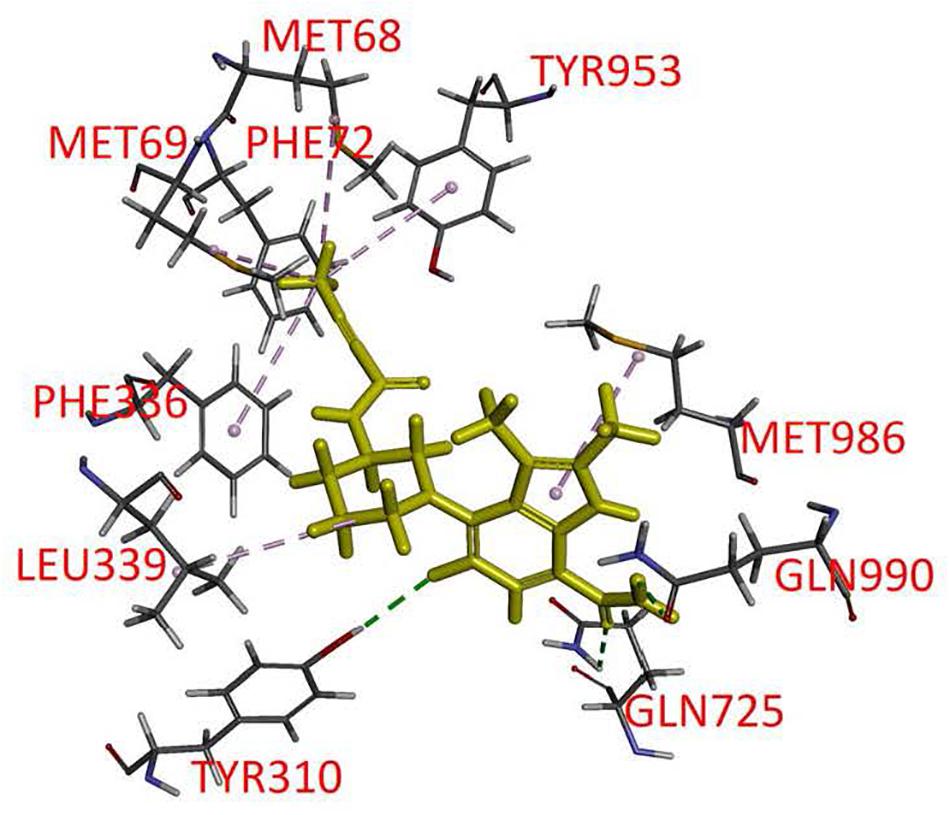
Figure 6. Docking of branebrutinib in the drug-binding pocket of P-glycoprotein (P-gp). Binding modes of branebrutinib with the protein structure of P-gp (PDB: 6QEX) was predicted by Accelrys Discovery Studio 4.0 software as described in Materials and methods. Branebrutinib is shown as a molecular model with highlighted yellow color and the atoms for interacting amino acid residues were colored as oxygen-red, nitrogen-blue, hydrogen-light gray and carbon- gray. Proposed interactions are presented as dotted lines.
P-gp-Overexpressing Cells Are Not Resistant to Branebrutinib
P-glycoprotein is known to mediate the transport of many TKIs (Eechoute et al., 2011; Tang et al., 2011, 2012; Robey et al., 2018; van Hoppe et al., 2019) and confer resistance to some of these TKIs (Mahon et al., 2003, 2008; Hiwase et al., 2008), including the BTK inhibitor ibrutinib (van Hoppe et al., 2018). To this end, we examined whether P-gp-overexpressing cells are less susceptible to branebrutinib treatment by determining the cytotoxicity of branebrutinib in multiple pairs of drug-sensitive parental cell lines and respective P-gp-overexpressing multidrug-resistant cell lines. As shown in Table 3, branebrutinib is equally cytotoxic to P-gp-overexpressing human ovarian NCI-ADR-RES cancer cells, human epidermal KB-V-1 cancer cells, human K562/i-S9 chronic myelogenous leukemia cells, and the corresponding drug-sensitive parental OVCAR-8, KB-3-1, and K562 cells. In addition, P-gp-transfected NIH3T3-G185 mouse fibroblast cells and MDR19-HEK293 cells, and the corresponding parental NIH3T3 and pcDNA3.1-HEK293 cells are also equally sensitive to branebrutinib treatment.
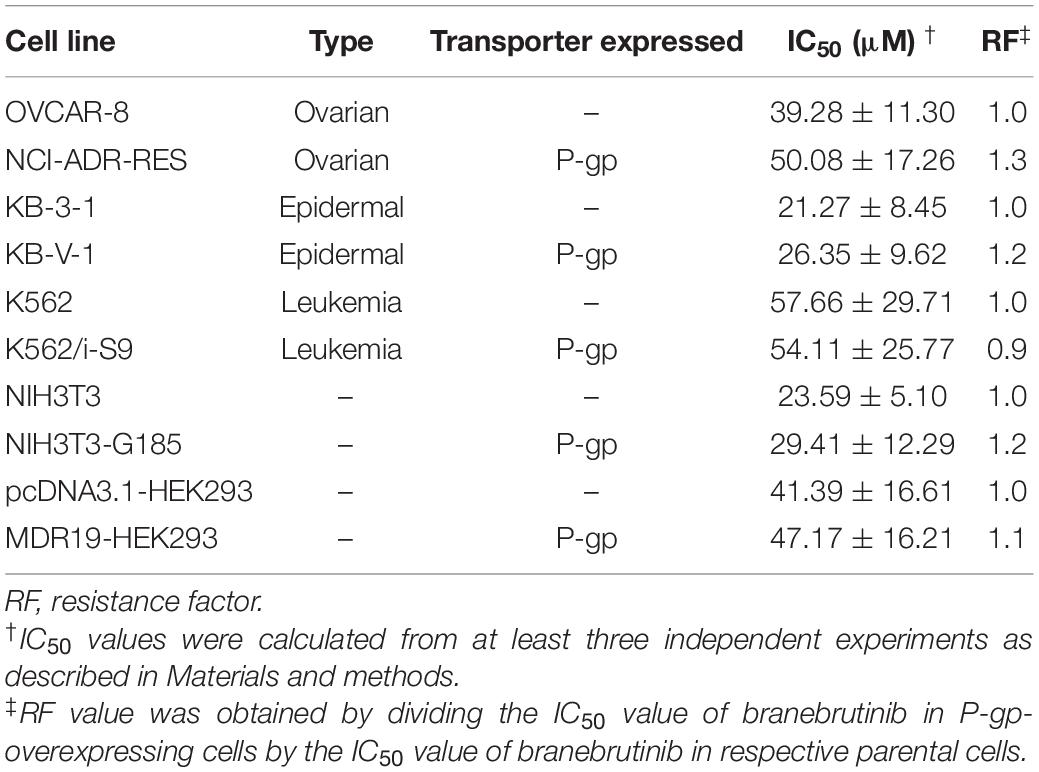
Table 3. Cytotoxicity of branebrutinib in drug-sensitive and P-glycoprotein (P-gp)-overexpressing multidrug-resistant cell lines.
Discussion
Cancer patients with poor response to conventional cytotoxic anticancer drugs, associated with the overexpression of P-gp in cancer cells, remains a major challenge in cancer chemotherapy (Gillet and Gottesman, 2010; Robey et al., 2018). While there is no effective and clinically safe inhibitors of P-gp available to date (Robey et al., 2018; Leopoldo et al., 2019; Dong et al., 2020), the results of several clinical trials have shown that cancer patients could benefit from the co-administration of conventional anticancer agents with TKIs (Geyer et al., 2006; Moore et al., 2007; Yang et al., 2013; Cetin et al., 2014; Alemany et al., 2018). Results of the combination therapy trial of gemcitabine and erlotinib were much better than monotherapy with gemcitabine in advanced pancreatic cancer patients (Moore et al., 2007; Yang et al., 2013). Similarly, results of combination therapy trial of capecitabine with lapatinib were significantly better than monotherapy with capecitabine in patients with human epidermal growth factor receptor 2 (HER2)-positive advanced breast cancer (Geyer et al., 2006; Cetin et al., 2014). More recently, encouraging results were reported in a combination therapy trial of doxorubicin with nilotinib, used to inhibit the activity of P-gp, in patients with sarcomas (Alemany et al., 2018). Together, these studies indicate that further investigation into combination therapies of multidrug-resistant cancers using conventional anticancer drugs and TKIs as modulators of P-gp is warranted. Consequently, we and others have been exploring the possibility of exploiting the polypharmacology properties of these TKIs for an additional mode of action against P-gp (Hsiao et al., 2016, 2019; Zhang et al., 2017; Wu and Fu, 2018; Wu et al., 2019, 2020a,2020b).
In the current study, we investigated the in vitro chemosensitizing effect of a BTK inhibitor branebrutinib in P-gp-overexpressing multidrug-resistant cancer cells. We determined the intrinsic toxicity of branebrutinib in several pairs of drug-sensitive and P-gp-overexpressing multidrug-resistant cell lines. Although P-gp is known to mediate resistance to numerous TKIs (Mahon et al., 2003; Hiwase et al., 2008), we found that P-gp does not confer resistance to branebrutinib in these cell lines (Table 3). Our results suggest that branebrutinib is not rapidly pumped out of cancer cells by P-gp and that the overexpression of P-gp is not likely to contribute significantly to the development of branebrutinib resistance in patients. Nevertheless, the mechanisms of resistance to branebrutinib in patients remain to be determined in clinical studies. Next, we examined the effect of branebrutinib, at sub-toxic concentrations, on P-gp-mediated resistance to anticancer drugs vincristine, paclitaxel, and colchicine. We found that branebrutinib resensitizes P-gp-overexpressing multidrug-resistant KB-V-1 and NCI-ADR-RES cancer cells, as well as HEK293 cells transfected with human P-gp, to these anticancer drugs in a concentration-dependent manner (Figure 1, Tables 1, 2). The results of branebrutinib inhibiting P-gp-mediated drug efflux (Figure 2) without altering the protein expression of P-gp (Figure 3) in KB-V-1 and NCI-ADR-RES cancer cells suggest that branebrutinib reverses P-gp-mediated MDR by blocking the drug transport function of P-gp and consequently restores the susceptibility of P-gp-overexpressing multidrug-resistant cancer cells to drug-induced apoptosis (Figure 4). These results, together with the P-gp-specific ATPase data (Figure 5) and the in silico docking analysis of branebrutinib binding to the substrate-binding site of P-gp in the inward-open conformation (Figure 6), indicate that this compound attenuates the binding of another drug substrate by interacting with numerous amino acid residues within the drug-binding pocket of P-gp (Figure 7).
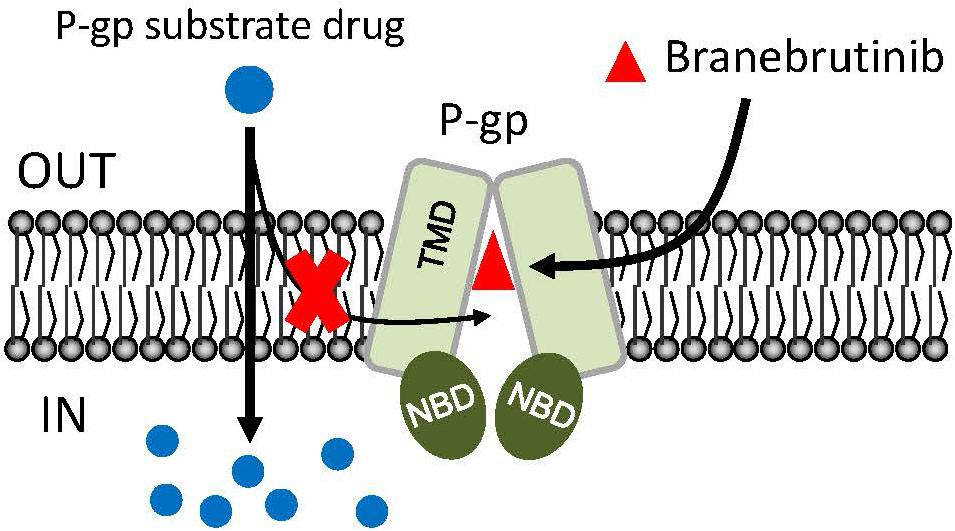
Figure 7. Simplified schematic of branebrutinib resensitizing P-glycoprotein (P-gp)-overexpressing multidrug-resistant cancer cells to anticancer drugs by blocking the drug efflux function of multidrug transporter. The intracellular concentration of a P-gp substrate drugs (blue circles) in P-gp-overexpressing cells is significantly reduced by the drug efflux function of P-gp. However, in the presence of branebrutinib (red triangles), branebrutinib outcompetes the binding of P-gp substrate drug to the same drug-binding pocket of P-gp and consequently restores the intracellular accumulation and efficacy of substrate drugs in P-gp-overexpressing multidrug-resistant cells.
In summary, we demonstrated that branebrutinib could effectively reverse P-gp-mediated MDR in cancer cells by modulating the activity of P-gp. Although the possibility of other mechanisms contributing to the resensitization of multidrug-resistant cancer cells remains, and that unexpected adverse drug interactions may occur in combination therapies (Stewart et al., 2004; Shukla et al., 2008; Libby and Hromas, 2010; Robey et al., 2018), we report here an additional action of branebrutinib that could be utilized in combination therapy with conventional anticancer drugs to treat multidrug-resistant cancers associated with the overexpression of P-gp, which should be investigated further.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
C-PW, T-HH, Y-SW, and SA designed the experiments. C-PW, Y-SW, and SA wrote the original draft manuscript and analyzed the data. C-PW and SA reviewed the manuscript. C-PW, MM, Y-SW, Y-CC, S-HH, and Y-HH performed the experiments. C-PW and SA edited the manuscript. All authors discussed the data and approved the final manuscript.
Funding
This work was supported by the Ministry of Science and Technology of Taiwan (MOST-108-2320-B-182-035 and MOST-108-2320-B-182-038-MY3) and Chang Gung Medical Research Program (BMRPC17, CMRPD1J0281, and CMRPD1K0391). SA and MM were supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.699571/full#supplementary-material
References
Agrawal, M., Abraham, J., Balis, F. M., Edgerly, M., Stein, W. D., Bates, S., et al. (2003). Increased 99mTc-sestamibi accumulation in normal liver and drug-resistant tumors after the administration of the glycoprotein inhibitor, XR9576. Clin. Cancer Res. 9, 650–656.
Alam, A., Kowal, J., Broude, E., Roninson, I., and Locher, K. P. (2019). Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. Science 363, 753–756. doi: 10.1126/science.aav7102
Alemany, R., Moura, D. S., Redondo, A., Martinez-Trufero, J., Calabuig, S., Saus, C., et al. (2018). Nilotinib as co-adjuvant treatment with doxorubicin in patients with sarcomas: a phase I trial of the Spanish Group for Research on Sarcoma. Clin. Cancer Res. 24, 5239–5249. doi: 10.1158/1078-0432.ccr-18-0851
Ambudkar, S. V. (1998). Drug-stimulatable ATPase activity in crude membranes of human MDR1-transfected mammalian cells. Methods Enzymol. 292, 504–514. doi: 10.1016/s0076-6879(98)92039-0
Ambudkar, S. V., Dey, S., Hrycyna, C. A., Ramachandra, M., Pastan, I., and Gottesman, M. M. (1999). Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu. Rev. Pharmacol. Toxicol. 39, 361–398. doi: 10.1146/annurev.pharmtox.39.1.361
Ambudkar, S. V., Kimchi-Sarfaty, C., Sauna, Z. E., and Gottesman, M. M. (2003). P-glycoprotein: from genomics to mechanism. Oncogene 22, 7468–7485. doi: 10.1038/sj.onc.1206948
Anderson, H. A., Maylock, C. A., Williams, J. A., Paweletz, C. P., Shu, H., and Shacter, E. (2003). Serum-derived protein S binds to phosphatidylserine and stimulates the phagocytosis of apoptotic cells. Nat. Immunol. 4, 87–91. doi: 10.1038/ni871
Beretta, G. L., Cassinelli, G., Pennati, M., Zuco, V., and Gatti, L. (2017). Overcoming ABC transporter-mediated multidrug resistance: the dual role of tyrosine kinase inhibitors as multitargeting agents. Eur. J. Med. Chem. 142, 271–289. doi: 10.1016/j.ejmech.2017.07.062
Catlett, I. M., Nowak, M., Kundu, S., Zheng, N., Liu, A., He, B., et al. (2020). Safety, pharmacokinetics and pharmacodynamics of branebrutinib (BMS-986195), a covalent, irreversible inhibitor of Bruton’s tyrosine kinase: randomised phase I, placebo-controlled trial in healthy participants. Br. J. Clin. Pharmacol. 86, 1849–1859. doi: 10.1111/bcp.14290
Cetin, B., Benekli, M., Turker, I., Koral, L., Ulas, A., Dane, F., et al. (2014). Lapatinib plus capecitabine for HER2-positive advanced breast cancer: a multicentre study of anatolian society of medical oncology (ASMO). J. Chemother. 26, 300–305. doi: 10.1179/1973947813y.0000000147
Cuestas, M. L., Castillo, A. I., Sosnik, A., and Mathet, V. L. (2012). Downregulation of mdr1 and abcg2 genes is a mechanism of inhibition of efflux pumps mediated by polymeric amphiphiles. Bioorg.Med.Chem.Lett. 22, 6577–6579. doi: 10.1016/j.bmcl.2012.09.012
Currier, S. J., Kane, S. E., Willingham, M. C., Cardarelli, C. O., Pastan, I., and Gottesman, M. M. (1992). Identification of residues in the first cytoplasmic loop of P-glycoprotein involved in the function of chimeric human MDR1-MDR2 transporters. J.Biol.Chem. 267, 25153–25159. doi: 10.1016/s0021-9258(19)74018-6
Dai, C. L., Tiwari, A. K., Wu, C. P., Su, X. D., Wang, S. R., Liu, D. G., et al. (2008). Lapatinib (Tykerb, GW572016) reverses multidrug resistance in cancer cells by inhibiting the activity of ATP-binding cassette subfamily B member 1 and G member 2. Cancer Res. 68, 7905–7914. doi: 10.1158/0008-5472.can-08-0499
Dong, J., Qin, Z., Zhang, W. D., Cheng, G., Yehuda, A. G., and Ashby, C. R. Jr., et al. (2020). Medicinal chemistry strategies to discover P-glycoprotein inhibitors: an update. Drug Resist. Updat. 49:100681. doi: 10.1016/j.drup.2020.100681
Eechoute, K., Sparreboom, A., Burger, H., Franke, R. M., Schiavon, G., Verweij, J., et al. (2011). Drug transporters and imatinib treatment: implications for clinical practice. Clin. Cancer Res. 17, 406–415. doi: 10.1158/1078-0432.ccr-10-2250
Geyer, C. E., Forster, J., Lindquist, D., Chan, S., Romieu, C. G., Pienkowski, T., et al. (2006). Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N. Engl. J. Med. 355, 2733–2743.
Gillet, J. P., and Gottesman, M. M. (2010). Mechanisms of multidrug resistance in cancer. Methods Mol.Biol. 596, 47–76.
Gottesman, M., and Ambudkar, S. V. (2001). Overview: ABC transporters and human disease. J. Bioenerg. Biomembr. 33, 453–458.
Gottesman, M. M., Fojo, T., and Bates, S. E. (2002). Multidrug resistance in cancer: role of ATP-dependent transporters. Nat. Rev. Cancer 2, 48–58. doi: 10.1038/nrc706
Gribar, J. J., Ramachandra, M., Hrycyna, C. A., Dey, S., and Ambudkar, S. V. (2000). Functional characterization of glycosylation-deficient human P-glycoprotein using a vaccinia virus expression system. J.Membr.Biol. 173, 203–214. doi: 10.1007/s002320001020
Hiwase, D. K., Saunders, V., Hewett, D., Frede, A., Zrim, S., Dang, P., et al. (2008). Dasatinib cellular uptake and efflux in chronic myeloid leukemia cells: therapeutic implications. Clin. Cancer Res. 14, 3881–3888. doi: 10.1158/1078-0432.ccr-07-5095
Hollo, Z., Homolya, L., Davis, C. W., and Sarkadi, B. (1994). Calcein accumulation as a fluorometric functional assay of the multidrug transporter. Biochim. Biophys. Acta 1191, 384–388. doi: 10.1016/0005-2736(94)90190-2
Hsiao, S. H., Lu, Y. J., Li, Y. Q., Huang, Y. H., Hsieh, C. H., and Wu, C. P. (2016). Osimertinib (AZD9291) attenuates the function of multidrug resistance-linked ATP-binding cassette transporter ABCB1 in vitro. Mol. Pharm. 13, 2117–2125. doi: 10.1021/acs.molpharmaceut.6b00249
Hsiao, S. H., Lusvarghi, S., Huang, Y. H., Ambudkar, S. V., Hsu, S. C., and Wu, C. P. (2019). The FLT3 inhibitor midostaurin selectively resensitizes ABCB1-overexpressing multidrug-resistant cancer cells to conventional chemotherapeutic agents. Cancer Lett. 445, 34–44. doi: 10.1016/j.canlet.2019.01.001
Hsiao, S. H., Murakami, M., Yeh, N., Li, Y. Q., Hung, T. H., Wu, Y. S., et al. (2018). The positive inotropic agent DPI-201106 selectively reverses ABCB1-mediated multidrug resistance in cancer cell lines. Cancer Lett. 434, 81–90. doi: 10.1016/j.canlet.2018.07.022
Kartner, N., Riordan, J. R., and Ling, V. (1983). Cell surface P-glycoprotein associated with multidrug resistance in mammalian cell lines. Science 221, 1285–1288.
Kovalev, A. A., Tsvetaeva, D. A., and Grudinskaja, T. V. (2013). Role of ABC-cassette transporters (MDR1, MRP1, BCRP) in the development of primary and acquired multiple drug resistance in patients with early and metastatic breast cancer. Exp. Oncol. 35, 287–290.
Leopoldo, M., Nardulli, P., Contino, M., Leonetti, F., Luurtsema, G., and Colabufo, N. A. (2019). An updated patent review on P-glycoprotein inhibitors (2011-2018). Expert. Opin. Ther. Pat. 29, 455–461. doi: 10.1080/13543776.2019.1618273
Libby, E., and Hromas, R. (2010). Dismounting the MDR horse. Blood 116, 4037–4038. doi: 10.1182/blood-2010-09-304311
Mahon, F. X., Belloc, F., Lagarde, V., Chollet, C., Moreau-Gaudry, F., Reiffers, J., et al. (2003). MDR1 gene overexpression confers resistance to imatinib mesylate in leukemia cell line models. Blood 101, 2368–2373. doi: 10.1182/blood.v101.6.2368
Mahon, F. X., Hayette, S., Lagarde, V., Belloc, F., Turcq, B., Nicolini, F., et al. (2008). Evidence that resistance to nilotinib may be due to BCR-ABL, Pgp, or Src kinase overexpression. Cancer Res. 68, 9809–9816. doi: 10.1158/0008-5472.can-08-1008
Maia, R. C., Vasconcelos, F. C., Souza, P. S., and Rumjanek, V. M. (2018). Towards Comprehension of the ABCB1/P-Glycoprotein Role in Chronic Myeloid Leukemia. Molecules 23:119. doi: 10.3390/molecules23010119
Matthews, C., Catherwood, M. A., Larkin, A. M., Clynes, M., Morris, T. C., and Alexander, H. D. (2006). MDR-1, but not MDR-3 gene expression, is associated with unmutated IgVH genes and poor prognosis chromosomal aberrations in chronic lymphocytic leukemia. Leuk. Lymphoma 47, 2308–2313. doi: 10.1080/10428190600881421
Mechetner, E. B., Schott, B., Morse, B. S., Stein, W. D., Druley, T., Davis, K. A., et al. (1997). P-glycoprotein function involves conformational transitions detectable by differential immunoreactivity. Proc. Natl. Acad. Sci. U.S.A. 94, 12908–12913. doi: 10.1073/pnas.94.24.12908
Mistry, P., Stewart, A. J., Dangerfield, W., Okiji, S., Liddle, C., Bootle, D., et al. (2001). In vitro and in vivo reversal of P-glycoprotein-mediated multidrug resistance by a novel potent modulator, XR9576. Cancer Res. 61, 749–758.
Moore, M. J., Goldstein, D., Hamm, J., Figer, A., Hecht, J. R., Gallinger, S., et al. (2007). Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 25, 1960–1966. doi: 10.1200/jco.2006.07.9525
Natarajan, K., Bhullar, J., Shukla, S., Burcu, M., Chen, Z. S., Ambudkar, S. V., et al. (2013). The Pim kinase inhibitor SGI-1776 decreases cell surface expression of P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) and drug transport by Pim-1-dependent and -independent mechanisms. Biochem.Pharmacol. 85, 514–524. doi: 10.1016/j.bcp.2012.12.006
Riordan, J. R., and Ling, V. (1979). Purification of P-glycoprotein from plasma membrane vesicles of Chinese hamster ovary cell mutants with reduced colchicine permeability. J. Biol. Chem. 254, 12701–12705. doi: 10.1016/s0021-9258(19)86370-6
Robey, R. W., Pluchino, K. M., Hall, M. D., Fojo, A. T., Bates, S. E., and Gottesman, M. M. (2018). Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 18, 452–464. doi: 10.1038/s41568-018-0005-8
Robey, R. W., Steadman, K., Polgar, O., Morisaki, K., Blayney, M., Mistry, P., et al. (2004). Pheophorbide a is a specific probe for ABCG2 function and inhibition. Cancer Res. 64, 1242–1246. doi: 10.1158/0008-5472.can-03-3298
Ross, D. D. (2000). Novel mechanisms of drug resistance in leukemia. Leukemia 14, 467–473. doi: 10.1038/sj.leu.2401694
Schwarzenbach, H. (2002). Expression of MDR1/P-glycoprotein, the multidrug resistance protein MRP, and the lung-resistance protein LRP in multiple myeloma. Med. Oncol. 19, 87–104. doi: 10.1385/mo:19:2:87
Shen, D. W., Fojo, A., Chin, J. E., Roninson, I. B., Richert, N., Pastan, I., et al. (1986). Human multidrug-resistant cell lines: increased mdr1 expression can precede gene amplification. Science 232, 643–645. doi: 10.1126/science.3457471
Shukla, S., Wu, C. P., and Ambudkar, S. V. (2008). Development of inhibitors of ATP-binding cassette drug transporters: present status and challenges. Expert Opin. Drug Metab. Toxicol. 4, 205–223. doi: 10.1517/17425255.4.2.205
Stewart, C. F., Leggas, M., Schuetz, J. D., Panetta, J. C., Cheshire, P. J., Peterson, J., et al. (2004). Gefitinib enhances the antitumor activity and oral bioavailability of irinotecan in mice. Cancer Res. 64, 7491–7499. doi: 10.1158/0008-5472.can-04-0096
Szakacs, G., Paterson, J. K., Ludwig, J. A., Booth-Genthe, C., and Gottesman, M. M. (2006). Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 5, 219–234.
Tang, C., Schafranek, L., Watkins, D. B., Parker, W. T., Moore, S., Prime, J. A., et al. (2011). Tyrosine kinase inhibitor resistance in chronic myeloid leukemia cell lines: investigating resistance pathways. Leuk.Lymphoma 52, 2139–2147. doi: 10.3109/10428194.2011.591013
Tang, S. C., Lagas, J. S., Lankheet, N. A., Poller, B., Hillebrand, M. J., Rosing, H., et al. (2012). Brain accumulation of sunitinib is restricted by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) and can be enhanced by oral elacridar and sunitinib coadministration. Int.J.Cancer. 130, 223–233. doi: 10.1002/ijc.26000
Tsubaki, M., Satou, T., Itoh, T., Imano, M., Komai, M., Nishinobo, M., et al. (2012). Overexpression of MDR1 and survivin, and decreased Bim expression mediate multidrug-resistance in multiple myeloma cells. Leuk. Res. 36, 1315–1322. doi: 10.1016/j.leukres.2012.07.003
van Hoppe, S., Jamalpoor, A., Rood, J. J. M., Wagenaar, E., Sparidans, R. W., Beijnen, J. H., et al. (2019). Brain accumulation of osimertinib and its active metabolite AZ5104 is restricted by ABCB1 (P-glycoprotein) and ABCG2 (breast cancer resistance protein). Pharmacol. Res. 146:104297. doi: 10.1016/j.phrs.2019.104297
van Hoppe, S., Rood, J. J. M., Buil, L., Wagenaar, E., Sparidans, R. W., Beijnen, J. H., et al. (2018). P-Glycoprotein (MDR1/ABCB1) restricts brain penetration of the Bruton’s tyrosine kinase inhibitor ibrutinib, while cytochrome P450-3A (CYP3A) limits its oral bioavailability. Mol. Pharm. 15, 5124–5134. doi: 10.1021/acs.molpharmaceut.8b00702
Watterson, S. H., Liu, Q., Beaudoin Bertrand, M., Batt, D. G., Li, L., Pattoli, M. A., et al. (2019). Discovery of Branebrutinib (BMS-986195): a strategy for identifying a highly potent and selective covalent inhibitor providing rapid in vivo inactivation of Bruton’s tyrosine kinase (BTK). J. Med. Chem. 62, 3228–3250.
Wu, C. P., Hsiao, S. H., Huang, Y. H., Hung, L. C., Yu, Y. J., Chang, Y. T., et al. (2020a). Sitravatinib sensitizes ABCB1- and ABCG2-overexpressing multidrug-resistant cancer cells to chemotherapeutic drugs. Cancers (Basel) 12:195. doi: 10.3390/cancers12010195
Wu, C. P., Hsiao, S. H., Sim, H. M., Luo, S. Y., Tuo, W. C., Cheng, H. W., et al. (2013). Human ABCB1 (P-glycoprotein) and ABCG2 mediate resistance to BI 2536, a potent and selective inhibitor of Polo-like kinase 1. Biochem.Pharmacol. 86, 904–913. doi: 10.1016/j.bcp.2013.08.004
Wu, C.-P., Hsieh, C.-H., and Wu, Y.-S. (2011). The emergence of drug transporter-mediated multidrug resistance to cancer chemotherapy. Mol.Pharm. 8, 1996–2011. doi: 10.1021/mp200261n
Wu, C. P., Hung, T. H., Hsiao, S. H., Huang, Y. H., Hung, L. C., Yu, Y. J., et al. (2020b). Erdafitinib resensitizes ABCB1-overexpressing multidrug-resistant cancer cells to cytotoxic anticancer drugs. Cancers (Basel) 12:1366. doi: 10.3390/cancers12061366
Wu, C. P., Lusvarghi, S., Wang, J. C., Hsiao, S. H., Huang, Y. H., Hung, T. H., et al. (2019). Avapritinib: A selective inhibitor of KIT and PDGFRalpha that reverses ABCB1 and ABCG2-mediated multidrug resistance in cancer cell lines. Mol. Pharm. 16, 3040–3052. doi: 10.1021/acs.molpharmaceut.9b00274
Wu, C. P., Shukla, S., Calcagno, A. M., Hall, M. D., Gottesman, M. M., and Ambudkar, S. V. (2007). Evidence for dual mode of action of a thiosemicarbazone, NSC73306: a potent substrate of the multidrug resistance linked ABCG2 transporter. Mol. Cancer Ther. 6, 3287–3296. doi: 10.1158/1535-7163.mct-07-2005
Wu, S., and Fu, L. (2018). Tyrosine kinase inhibitors enhanced the efficacy of conventional chemotherapeutic agent in multidrug resistant cancer cells. Mol. Cancer 17:25.
Yang, Z. Y., Yuan, J. Q., Di, M. Y., Zheng, D. Y., Chen, J. Z., Ding, H., et al. (2013). Gemcitabine plus erlotinib for advanced pancreatic cancer: a systematic review with meta-analysis. PLoS One 8:e57528. doi: 10.1371/journal.pone.0057528
Zhang, H., Patel, A., Wang, Y. J., Zhang, Y. K., Kathawala, R. J., Qiu, L. H., et al. (2017). The BTK inhibitor ibrutinib (PCI-32765) overcomes paclitaxel resistance in ABCB1- and ABCC10-overexpressing cells and tumors. Mol. Cancer Ther. 16, 1021–1030. doi: 10.1158/1535-7163.mct-16-0511
Zheng, N., Catlett, I. M., Taylor, K., Gu, H., Pattoli, M. A., Neely, R. J., et al. (2019). Determination of real time in vivo drug receptor occupancy for a covalent binding drug as a clinical pharmacodynamic biomarker by immunocapture-LC-MS/MS. Anal. Chem. 91, 8443–8452. doi: 10.1021/acs.analchem.9b01462
Keywords: multidrug resistance, chemotherapy, P-glycoprotein, ABCB1, branebrutinib, BMS-986195
Citation: Wu C-P, Murakami M, Wu Y-S, Chi Y-C, Hsiao S-H, Huang Y-H, Hung T-H and Ambudkar SV (2021) Branebrutinib (BMS-986195), a Bruton’s Tyrosine Kinase Inhibitor, Resensitizes P-Glycoprotein-Overexpressing Multidrug-Resistant Cancer Cells to Chemotherapeutic Agents. Front. Cell Dev. Biol. 9:699571. doi: 10.3389/fcell.2021.699571
Received: 23 April 2021; Accepted: 28 June 2021;
Published: 19 July 2021.
Edited by:
Zexian Liu, Sun Yat-sen University Cancer Center (SYSUCC), ChinaReviewed by:
Ilaria Genovese, University of Ferrara, ItalyAyse Banu Demir, İzmir University of Economics, Turkey
Copyright © 2021 Wu, Murakami, Wu, Chi, Hsiao, Huang, Hung and Ambudkar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chung-Pu Wu, wuchung@mail.cgu.edu.tw