Single Nucleotide Polymorphisms Associated With Gut Homeostasis Influence Risk and Age-at-Onset of Parkinson's Disease
 Anastazja M. Gorecki1,2
Anastazja M. Gorecki1,2  Megan C. Bakeberg1,3
Megan C. Bakeberg1,3  Frances Theunissen1,4
Frances Theunissen1,4  Jade E. Kenna1,3
Jade E. Kenna1,3  Madison E. Hoes1 Abigail L. Pfaff1,4
Madison E. Hoes1 Abigail L. Pfaff1,4  P. Anthony Akkari1,3,4
P. Anthony Akkari1,3,4  Sarah A. Dunlop2,5
Sarah A. Dunlop2,5  Sulev Kõks1,4
Sulev Kõks1,4  Frank L. Mastaglia1,3
Frank L. Mastaglia1,3  Ryan S. Anderton1,3,6,7*
Ryan S. Anderton1,3,6,7*- 1Perron Institute for Neurological and Translational Science, Nedlands, WA, Australia
- 2School of Biological Sciences, University of Western Australia, Crawley, WA, Australia
- 3Centre for Neuromuscular and Neurological Disorders, University of Western Australia, Nedlands, WA, Australia
- 4The Centre for Molecular Medicine and Innovative Therapeutics, Murdoch University, Murdoch, WA, Australia
- 5Minderoo Foundation, Perth, WA, Australia
- 6Institute for Health Research, University of Notre Dame Australia, Fremantle, WA, Australia
- 7School of Health Sciences, University of Notre Dame Australia, Fremantle, WA, Australia
Research is increasingly focusing on gut inflammation as a contributor to Parkinson's disease (PD). Such gut inflammation is proposed to arise from a complex interaction between various genetic, environmental, and lifestyle factors, however these factors are under-characterized. This study investigated the association between PD and single-nucleotide polymorphisms (SNPs) in genes responsible for binding of bacterial metabolites and intestinal homeostasis, which have been implicated in intestinal infections or inflammatory bowel disease. A case-control analysis was performed utilizing the following cohorts: (i) patients from the Australian Parkinson's Disease Registry (APDR) (n = 212); (ii) a Caucasian subset of the Parkinson's Progression Markers Initiative (PPMI) cohort (n = 376); (iii) a combined control group (n = 404). The following SNPs were analyzed: PGLYRP2 rs892145, PGLYRP4 rs10888557, TLR1 rs4833095, TLR2 rs3804099, TLR4 rs7873784, CD14 rs2569190, MUC1 rs4072037, MUC2 rs11825977, CLDN2 rs12008279 and rs12014762, and CLDN4 rs8629. PD risk was significantly associated with PGLYRP4 rs10888557 genotype in both cohorts. PGLYRP2 rs892145 and TLR1 rs4833095 were also associated with disease risk in the APDR cohort, and TLR2 rs3804099 and MUC2 rs11825977 genotypes in the PPMI cohort. Interactive risk effects between PGLYRP2/PGLYRP4 and PGLYRP4/TLR2 were evident in the APDR and PPMI cohorts, respectively. In the APDR cohort, the PGLYRP4 GC genotype was significantly associated with age of symptom onset, independently of gender, toxin exposure or smoking status. This study demonstrates that genetic variation in the bacterial receptor PGLYRP4 may modulate risk and age-of-onset in idiopathic PD, while variants in PGLYRP2, TLR1/2, and MUC2 may also influence PD risk. Overall, this study provides evidence to support the role of dysregulated host-microbiome signaling and gut inflammation in PD, and further investigation of these SNPs and proteins may help identify people at risk of developing PD or increase understanding of early disease mechanisms.
Introduction
Parkinson's disease (PD) is a debilitating neurodegenerative disorder with no cure. Although diagnosis and treatment typically center around motor impairments, PD is viewed as a complex, heterogeneous condition with a plethora of non-motor symptoms (Sauerbier et al., 2016; Ferreira and Massano, 2017). Notably, gastrointestinal dysfunction and the gut microbiome are increasingly studied in PD, both as disease symptoms and contributors to pathogenesis. For instance, many people with PD exhibit microbial dysbiosis (Bedarf et al., 2017; Hill-Burns et al., 2017; Gorecki et al., 2019) coupled with intestinal leakiness and inflammation (Forsyth et al., 2011; Devos et al., 2013; Clairembault et al., 2015; Schwiertz et al., 2018; Perez-Pardo et al., 2019) and future PD risk has been associated with antibiotic use (Mertsalmi et al., 2019), constipation (Adams-Carr et al., 2016), gastrointestinal infections (Nielsen et al., 2012; Nerius et al., 2019), and inflammatory bowel disease (IBD) (Villumsen et al., 2019). Furthermore, alpha-synuclein aggregation and phosphorylation throughout the enteric nervous system have been reported in people with PD (Braak et al., 2006) sometimes up to 20 years before PD diagnosis (Stokholm et al., 2016), but are not specific to PD (Visanji et al., 2015; Corbillé et al., 2016). Alpha-synuclein levels are increased throughout the enteric neurons of children with intestinal inflammation (Stolzenberg et al., 2017), adults with Crohn's disease (Prigent et al., 2019) and a high proportion of healthy individuals without a known neurodegenerative diagnosis during life (Gold et al., 2013), supporting a proposed role of alpha-synuclein in normal gut function and immunity (Barbut et al., 2019). However, truncal vagotomy reduces PD risk (Svensson et al., 2015), and rodent models support gut-to-brain transmission of alpha-synuclein aggregation via the vagus nerve in PD pathology (Kim et al., 2019; Challis et al., 2020). Consequently, the gut is implicated in early inflammatory processes that may contribute to alpha-synuclein aggregation and PD pathology in susceptible individuals (Hawkes et al., 2007; Houser and Tansey, 2017; Johnson et al., 2018), however the exact mechanisms are still unclear.
Environmental toxins (e.g., pesticides, herbicides, and heavy metals) and lifestyle factors (e.g., diet, smoking, and exercise) have been found to alter the gut microbiome and cause gut inflammation. These factors are associated with PD risk and progression, but are not always causative (Alcalay et al., 2012; Kamel et al., 2014; Fang et al., 2018; Angelopoulou et al., 2019; Marras et al., 2019). Furthermore, although several genetic mutations have been associated with autosomal dominant or recessive PD, disease presentation varies even among strongly penetrant forms of familial PD (Nishioka et al., 2006). Genome-wide association studies have identified 90 risk variants for sporadic PD (Nalls et al., 2019; Bandres-Ciga et al., 2020), though as yet they have not resulted in significant diagnostic or therapeutic translation. Notably, polymorphisms in inflammatory cytokines have previously been associated with PD risk (Hamza et al., 2011; Gao et al., 2012), and many genetic loci linked with PD risk are also associated with IBD (Lee et al., 2016; Rösler et al., 2018), a condition which also exhibits microbial dysbiosis and gut inflammation.
Rather than a single causative trigger, PD is proposed to arise from a complex interaction between various genetic, environmental and lifestyle factors which may contribute to chronic inflammation, oxidative stress, cytokine toxicity, alpha-synuclein aggregation, and eventual neuronal degeneration (Hawkes et al., 2007; Johnson et al., 2018). This is illustrated by comparison of human and rodent studies of Pink1 mutations. In humans, Pink1 mutations cause early-onset PD with almost 100% penetrance. Conversely, loss-of-function rodent models are healthy and do not develop characteristic PD-like symptoms, though intestinal infection with gram-negative bacteria in Pink1 knockout mice causes PD-like symptoms, including an autoimmune response, dopaminergic neuronal alterations and motor impairments (Matheoud et al., 2019). Moreover, candidate gene or genome-wide association studies to investigate gene-environment interactions in PD have yielded mixed results (Dick et al., 2007; Hamza et al., 2011; Gao et al., 2012; Singh et al., 2014; Biernacka et al., 2016; Lee et al., 2016; Simon et al., 2017; Rösler et al., 2018), highlighting the complex nature of PD.
As the primary exchange site with the outside world, the gut is an important mediator of gene-environment interactions, especially through the gut microbiome and intestinal epithelium. Coupled with evidence linking gut inflammation to PD pathology, genetic variants that modulate intestinal integrity or microbiome-host signaling are an intriguing target for biomarkers or new therapeutic targets for PD and have so far received little attention. Consequently, the present novel study explored gut- and IBD-related single-nucleotide polymorphisms (SNPs) in subsets of two independent PD cohorts: the Australian Parkinson's Disease Registry (APDR) and the Parkinson's Progression Markers Initiative (PPMI). Specifically, the study investigated genetic polymorphisms in peptidoglycan recognition proteins PGLYRP2 and 4; toll-like receptors TLR1, 2 and 4 and co-receptor CD14; tight-junction claudins CLDN2 and 4; and mucin glycoproteins MUC1 and MUC2, to determine if they are associated with PD risk or age of symptom onset.
Methodology
Participants
APDR Cohort
This study utilized clinical data and samples from the Perron Institute for Neurological and Translational Science Biobank, obtained from 212 Caucasians with idiopathic PD from the Australian Parkinson's Disease Registry (APDR) who were sequentially recruited from Movement Disorders Clinics at the Perron Institute for Neurological and Translational Science (Perth, Australia) between 2007 and 2019, as previously described (Bakeberg et al., 2019). All participants reported no family history of PD, and were examined by a movement disorder neurologist prior to inclusion in the study for verification of the diagnosis in accordance with the UK Brain Bank criteria for idiopathic PD (Hughes et al., 1992). The use of these samples and data for the current study was approved by a Human Research and Ethics Committee (Approval number RA/2006/073 and RA/4/20/5308), and written informed consent was obtained from all participants in accordance with the National Health and Medical Research Council guidelines.
PPMI Cohort
This study drew upon 413 people with PD from the Parkinson's Progression Markers Initiative (PPMI) Cohort. Clinical information and sequencing data pertaining to target SNPs was obtained from the PPMI database (available at www.ppmi-info.org/data). SNPs genotypes were extracted from the available variant call format (VCF) files using rs identifiers with VCFtools (Danecek et al., 2011). All individuals categorized as Black, Asian, Other or Hispanic/Latino in the PPMI database were excluded from this study to create a Caucasian cohort which reflects the APDR group and avoids variation in gene frequencies arising from ethnicity.
Control Cohort
In order to consolidate control numbers, DNA and clinical data of healthy people from three separate databases was pooled. This consisted of 59 participants from the Perron Institute for Neurological and Translational Science Biobank; 179 from the PPMI cohort; and 166 age- and gender-matched healthy controls obtained from the NINDS Repository, Coriell Institute for Medical Research (New Jersey, USA). To minimize variability and to reflect the PD cohorts, only individuals categorized as Caucasian were included in this study. Minor genotype frequency, assessed through Levene's test, did not significantly differ between controls obtained from Australian, PPMI and Coriell cohorts (p > 0.05, data not shown) and so were combined into one healthy control cohort (n = 404) for subsequent analyses.
Patient Information and Past Environmental Exposure
In addition to demographic and clinical information, APDR participants had previously reported information regarding pre-diagnosis smoking habits and toxin exposure, making this cohort enriched for information regarding environmental PD risk factors. Surveyed toxins included a variety of herbicides, pesticides, insecticides, fertilizers, heavy metals and industrial chemicals (summarized in Supplementary Table 1). Similar to previous research, participants were grouped as “Yes” for lifetime toxin exposure if they reported at least 7 days exposure prior to PD diagnosis (n = 99) or those reporting a main occupation in farming or agriculture (n = 14). Participants were grouped as “smokers” if they reported at least 6 months of regular cigarette smoking prior to their diagnosis (n = 91).
DNA Extraction
For Australian participants, DNA was extracted from blood samples or buccal swabs. Blood was collected from medial cubital vein and DNA extraction occurred at respective state pathology services, being stored at −20°C until genotyping. Buccal samples were collected using Isohelix™ DNA/RNA Buccal Swabs (Cell Projects Ltd, Kent, U.K.) and stored in −20°C until DNA extraction using QIAamp DNA mini kits (Qiagen Pty LTD., Victoria, Australia).
SNP Genotyping
SNPs previously associated with IBD and gastrointestinal integrity were investigated in this study, including immune signaling, microbial response or intestinal permeability (Table 1). APDR and Coriell samples were sent to the Australian Genome Research Facility (AGRF, Australia) for Agena Bioscience MassARRAY® genotyping on a Compact Spectrometer, conducted under blinded conditions. Assay design was conducted based on SNP sequence available from dbSNP (Supplementary Table 2). Genotyping data for PPMI cohort was obtained from the VCF files from whole genome sequencing data (available at www.ppmi-info.org/data).
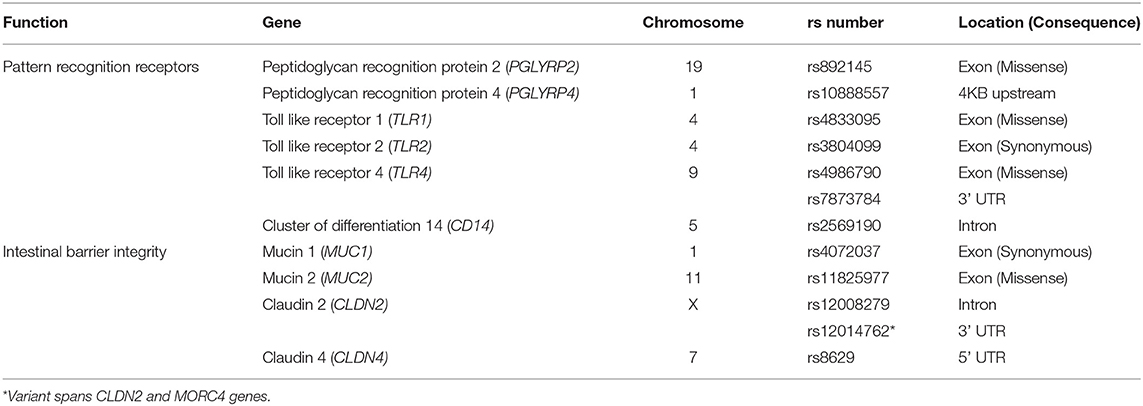
Table 1. Single-nucleotide polymorphisms investigated in this study.
Statistical Analysis
APDR and PPMI cohorts were separately compared to pooled controls using IBM-SPSS (v. 26, IBM Corporation). Significant nominal p-values of ≤ 0.05 (*) were employed. Normality was assessed utilizing Shapiro-Wilk test, and demographic information assessed through Mann-Whitney U or Chi-square tests where appropriate. When relevant, results were compared to the major genotype (underlined in all tables). To evaluate the association between SNPs and PD risk, a case-control logistic regression was performed in both a naïve (genetic variant only) and corrected models (genetic variant, gender, and age at assessment) comparing controls to APDR and controls to PPMI. Individuals were subsequently categorized by the presence of identified risk-increasing genotypes in order to calculate interactive risk effects through case-control logistic regression. Generalized linear models (GLMs) were used to investigate the association between SNPs and mean age of PD symptom onset in both cohorts (naïve model). All corrected GLM models include gender, but within PD analysis of the APDR cohort also incorporated information regarding environmental toxin exposures and lifetime smoking habits. Estimated means were calculated for significant SNPs from the GLMs, with pair-wise comparisons adjusted by Bonferroni correction for multiple comparisons.
Results
Cohort Information
Cohort demographics are summarized in Table 2. There was no significant difference in gender distribution between control, APDR and PPMI cohorts, however gender was included in all corrected models due to the presence of some X-linked SNPs. Mean age at assessment in the PPMI cohort (61.83 years ± 9.54, mean ± SD) was significantly lower than in the control (63.63 years ± 11.14) and APDR groups (65.80 years ± 8.99), and thus age at assessment was also included in subsequent corrected logistic regression models for risk. Finally, mean age of symptom onset for the PPMI cohort (59.91 years ± 9.71) was significantly later than the APDR group (57.63 years ± 10.10).
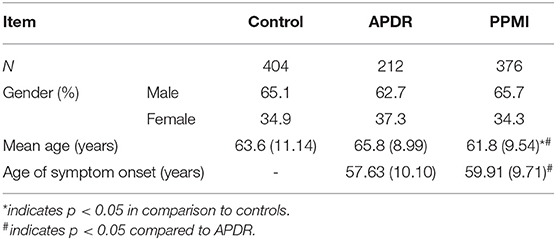
Table 2. Summary of cohort demographic information.
Influence of SNP Genotypes on PD Risk in APDR and PPMI Cohorts
Genotype frequencies of target SNPs and corrected regression models for PD risk in both APDR and PPMI are summarized in Table 3, with naïve regression models presented in Supplementary Table 3. Notably, heterozygotes of PGLYRP4 rs10888557 demonstrated significantly increased risk of PD compared to the major GG genotype in both naïve [OR (95% CI) = 1.644 (1.070–2.526), p = 0.023] and corrected models [OR = 1.632 (1.057–2.520), p = 0.027] when comparing the APDR cohort to controls. Importantly, this finding was replicated when comparing the PPMI cohort to controls, as heterozygotes of rs10888557 also demonstrate significantly increased PD risk in both naïve [OR = 1.575 (1.085–2.288), p = 0.017] and corrected models [OR = 1.596 (1.095-2.327), p = 0.015].
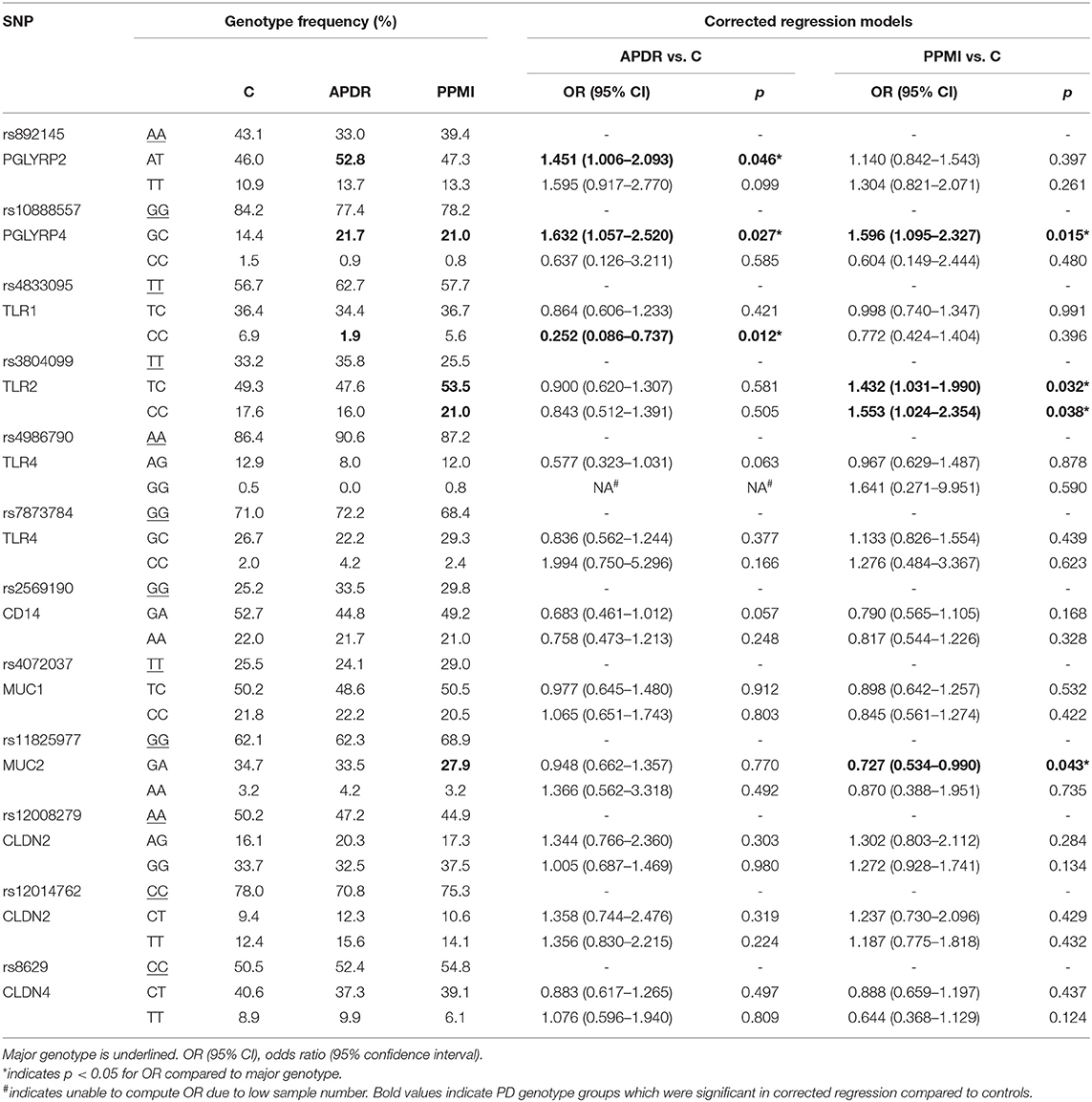
Table 3. Corrected regression models evaluating the association between SNP genotype and disease risk in two PD cohorts.
Additionally, when analyzing the APDR cohort, PGLYRP2 rs892145 AT heterozygotes demonstrated significantly higher PD risk than major AA homozygotes in both naïve [OR = 1.497 (1.041–2.152), p = 0.030] and corrected models [OR =1.451 (1.006–2.093), p = 0.046], with a greater but non-significant odds ratio with the minor TT genotype in naïve [OR = 1.638 (0.950–2.825), p = 0.076], and corrected models [OR = 1.595 (0.917–2.770), p = 0.071]. However, these results were not replicated in the PPMI cohort. Similarly, minor CC homozygotes of TLR1 rs4833095 had significantly lower PD risk than major homozygotes in the APDR cohort in both naïve [OR = 0.246 (0.084–0.717), p = 0.010] and corrected models [OR = 0.252 (0.086–0.737), p = 0.012], but this was not replicated in the PPMI analysis.
Analysis between controls and PPMI demonstrated an increasing step-wise pattern for PD risk with TLR2 rs3804099; TC heterozygotes exhibited significantly higher PD risk in naïve [OR = 1.410 (1.016–1.956), p = 0.040], and corrected regression [OR = 1.432 (1.031–1.990), p = 0.032]; a greater odds ratio was evident for minor homozygotes in naïve [1.553 (1.027–2.350), p = 0.037] and corrected models [OR = 1.553 (1.024–2.354), p = 0.038]. Finally, GA heterozygotes of MUC2 rs11825977 had significantly reduced PD risk when comparing PPMI to controls in both naïve [OR = 0.727 (0.535–0.988), p = 0.042] and corrected regression models [OR = 0.727 (0.534–0.990), p = 0.043].
Interactive Risk Effects of PGLYRP2, PGLYRP4, and TLR2 Genotypes
Additional regression models were computed to analyze the interactive effect of putative risk SNPs genotypes. When comparing APDR and controls, co-carriage of both PGLYRP2 rs892145 AT and PGLYRP4 rs10888557 GC significantly increased PD by two-fold compared to those without either risk genotype in naïve [OR = 2.030 (1.076–3.831), p = 0.029] and corrected models [OR = 2.016 (1.067–3.808), p = 0.031]. Similarly, when comparing PPMI and controls, co-carriage of both PGLYRP4 rs10888557 GC and TLR2 rs3804099 CC increased PD risk by ~1.8-fold but this was not significant in either naïve [OR = 1.835 (0.809–4.160), p = 0.146] or corrected models [OR = 1.846 (0.813–4.188), p = 0.143], likely due to the small number of individuals with both risk alleles in control (n = 10) or PD (n = 15). Subsequent analyses of TLR2 rs3804099 heterozygotes with PGLYRP4 rs10888557 GC genotype (control = 30, PD = 45) demonstrated significantly increased PD risk in naïve [OR = 1.883 (1.128–3.143), p = 0.015] and corrected models [OR = 1.890 (1.132–3.156), p = 0.015], suggesting an interactive allelic effect.
PGLYRP4 rs10888557 SNP is Associated With Age of Symptom Onset in APDR Cohort
Among the APDR cohort (n = 212), 42.0% (n = 89), and 43.4% (n = 91) of participants reported lifetime toxin exposure and smoking, respectively. GLMs were computed to determine an association between SNPs and mean age of PD symptom onset, with results from three models displayed in Table 4: naïve, corrected for gender and reported lifetime toxin exposure (toxins) and corrected for gender and lifetime smoking status (smoking). PGLYRP4 rs10888557 significantly predicted mean age of symptom onset in naïve (p = 0.029), toxin corrected (p = 0.029), and smoking corrected (p = 0.030) models. A step-wise pattern in age of symptom onset is evident upon pair-wise comparison of estimated means for rs10888557 genotypes in the naïve model, but did not withstand Bonferroni adjustment for multiple comparisons (padj): GG homozygotes (56.70 ± 0.77 years) having significantly earlier age of symptom onset than heterozygotes (60.57 ± 1.46 years, p = 0.020, padj = 0.059), and minor CC homozygotes showing a non-significant step-wise trend (66.50 ± 7.01 years, p = 0.165, padj = 0.494). The significant difference between GG and GC genotypes was present in both corrected toxin (GC = 60.36 years ± 1.47, GG = 56.51 ± 0.82 years, p = 0.021, padj = 0.062) and smoking models (GC = 60.47 ± 1.46 years, GG = 56.67 ± 0.80 years, p = 0.021, padj = 0.058), but did not withstand Bonferroni correction. Overall, mean symptom onset was ~4 years later for the GC than the GG genotype. GLMs were repeated in the PPMI cohort, and there were no significant results in naïve models or after correction for gender (Supplementary Table 4).
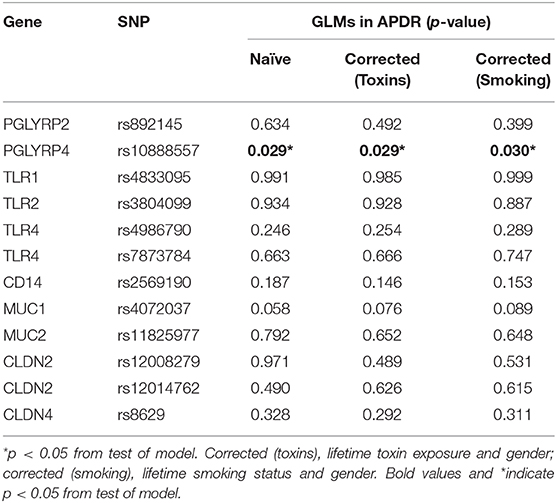
Table 4. Generalized linear models investigating target SNPs and estimated mean age of PD symptom onset in enriched APDR cohort.
Discussion
PD is increasingly considered the product of complex gene-environment interactions occurring in the gut, which are proposed to cause gastrointestinal, enteric and systemic inflammation and consequent PD pathology. Thus, it was hypothesized that a genetic predisposition for gut leakiness or inflammation, combined with environmental toxin exposure, may influence PD risk and symptom onset. Notably, this study reports a novel and robust association between PD risk and the PGLYRP4 rs10888557 SNP in two independent cohorts of PD. In addition, rs10888557 genotype was determinant of the age of symptom onset in the APDR cohort, with carriage of the GC being associated with earlier age of onset. Furthermore, polymorphisms in other microbial pattern recognition receptors and one intestinal mucin were also associated with PD risk; where PGLRYP2 rs892145 and TLR1 rs4833095 were significant in the APDR cohort, while TLR2 rs3804099 and MUC2 rs11825977 were significant in the PPMI cohort. Moreover, previously unreported interactive effects of these SNPs on disease were demonstrated in both patient cohorts.
Peptidoglycan Recognition Proteins
Notably, this study found an association between the PGLYRP4 locus rs10888557 and PD risk as well as age of symptom onset. Expressed throughout the gut and secreted into the gut lumen, peptidoglycan recognition proteins (PGLYRPs) respond to peptidoglycans and other microbial products from gram-positive and gram-negative bacteria, exerting a bactericidal function to regulate gut homeostasis and cleaving peptidoglycan through amidase function (Dziarski and Gupta, 2010). In both the APDR and PPMI cohorts, PGLYRP4 rs10888557 heterozygotes exhibited significantly increased PD risk compared to the major GG genotype. These results add to mixed literature concerning rs10888557 in PD, as CG and CC genotypes exerted a protective influence on PD risk among North Americans (Goldman et al., 2014), and no association was reported in a subsequent study in a northern Han Chinese cohort (Yu et al., 2018). The previously reported protective influence of the minor CC genotype on PD risk may contribute to the very low number of CC genotypes among people with PD and the consequent lack of statistical significance.
Coupled with the proposed role of the gut microbiome and complex-gene environment interactions in PD, these findings prompted further investigation regarding age of symptom onset incorporating self-reported exposure to environmental toxins and smoking history, factors known to influence PD risk (Kamel et al., 2014; Marras et al., 2019). In the APDR cohort, PGLYRP4 rs10888557 GG homozygotes were found to have an ~4-years earlier age of symptom onset after controlling for both toxin exposure and smoking history, possibly indicating different underlying genetic mechanisms affecting disease risk and symptom time-course. However, these results were not replicated in the PPMI cohort in naïve or corrected models. Despite similar PGLYRP4 genotype frequencies and risk associations in both cohorts, the PPMI has significantly later mean age of symptom onset than the Australian cohort and comes from a diverse international initiative. Thus, it is conceivable that associations between PGLYRP4 genotype and disease onset are masked by the more culturally and environmentally heterogeneous nature of the PPMI cohort, and larger studies with more detailed analysis of gene-environment interactions are required.
Found upstream of PGLYRP4 in a transcription factor binding site, publicly available data from the GTEx Portal (https://www.gtexportal.org/home/) indicate that rs10888557 is a significant expression quantitative trait locus (eQTL) for PGLYRP4 in various tissues, including the small intestine (terminal ileum). Additionally, variants in PGLYRP4 have been associated with onset age of Crohn's disease (Zulfiqar et al., 2013), while rs10888557 has also been associated with ulcerative colitis risk in a Greek population (Gazouli et al., 2019). However, the effect of rs10888557 on PGLYRP4 protein expression or function does not appear to have been studied. In light of the findings concerning PD risk and age of symptom onset in the current study, coupled with the notion that PGLYRP4 variants may influence immune activity in response to microbial changes in the gut, further studies are required to investigate interactions between PGLYRP4, environmental toxins, the gut microbiome and PD pathology.
Additionally, it is important to acknowledge that the SNP rs10888557 is located between PGLYRP4 and its neighboring gene S100A9. Data from NCBI (assembly GRCh38.p12) demonstrates that SNP rs10888557 is ~4,000 base pairs away from the 5' end of PGLYRP4 and ~5,000 base pairs from the 5' end of S100A9. One of the S100 calcium-binding proteins, S100A9 is a pro-inflammatory mediator with amyloidogenic properties, and has been widely implicated in inflammation, cancer and neurodegeneration. Interestingly recent studies also demonstrate a specific role of S100A9 in PD, as S100A9 is elevated and co-localizes with alpha-synuclein in PD brains ex vivo, and induces alpha-synuclein aggregation in vitro (Horvath et al., 2018). Interestingly, S100A8 and S100A9 dimers create calprotectin, where elevated fecal calprotectin levels indicate increased neutrophil migration in response to gut barrier dysfunction. Such elevated fecal calprotectin levels are evident in people with PD (Schwiertz et al., 2018; Mulak et al., 2019), and it is hypothesized that the amyloidogenic properties of calprotectin and S100A9 may contribute to fibril formation and enteric alpha-synuclein aggregation in response to gut inflammation (Mulak et al., 2019). Given the link between S100A9 and PD, further studies should investigate if rs10888557 affects S100A9 transcription factor binding or is a marker for regional variation not limited to the PGLYRP4 gene.
Found in the same peptidoglycan recognition receptor family as PGLYRP4, rs892145 in PGLYRP2 was also associated with PD risk in the APDR cohort, where heterozygotes had significantly increased risk of PD, with a similar non-significant trend evident for minor TT homozygotes in comparison to major homozygotes. This echoes a previous study demonstrating reduced PD risk with AT and AA genotypes in a North American population (Goldman et al., 2014), but was not replicated in the predominately North American PPMI PD cohort in the current study, highlighting the need for further investigation. The missense variant rs892145 is an eQTL in whole blood and liver, suggesting a possible role in modulating PGLYRP2 response to circulating pathogens arising from a leaky gut. Additionally, the expression of PGLYRP2 is induced in oral epithelial cells after bacterial exposure (Scholz et al., 2018), and thus a missense variant may influence downstream immune responses in other tissues.
TLRs
TLR1-TLR2 heterodimers recognize triacylated microbial lipoproteins from gram-negative bacteria and mycoplasma to cause an innate immune response (Burgueño and Abreu, 2020), and variants in TLR1 and TLR2 were associated with PD risk in the APDR and PPMI cohorts, respectively. Regarding TLR1 rs4833095, the CC genotype and C allele are known to be associated with increased risk of Crohn's disease and ulcerative colitis (Bank et al., 2014), and increased risk of Helicobacter pylori infection in Chinese (Yang et al., 2013) and Thai populations, with worse mucosal inflammation and altered morphology after infection in the latter (Tongtawee et al., 2016). These conditions and infections are associated with increased future PD risk (Nielsen et al., 2012; Villumsen et al., 2019). Additionally, GTEx Portal reports that rs4833095 is a significant eQTL for TLR1 in various tissues, including the colon. Contrastingly, the current study reports significantly reduced PD risk with TLR1 rs4833095 CC genotype in the APDR cohort. The minor C allele of rs4833095 impairs TLR1 signaling and subsequent inflammatory cascades (Omueti et al., 2007), while the major T allele is linked with higher levels of interleukins IL-12p40 and IL-17 (Santana et al., 2017). Thus, further studies are required to assess if an impaired TLR1 response facilitates reduced resistance to microbial pathogens, which may lead to future infection, chronic inflammation or PD pathology in the long term.
The current study demonstrates that heterozygotes and minor CC homozygotes of TLR2 rs3804099 had significantly increased PD risk in the PPMI cohort compared to healthy controls, supporting a link between infections, altered TLR2 signaling, chronic inflammation and PD. However, in contrast to the largely North American and European Caucasian PPMI cohort, the major TT allele was previously associated with increased PD risk among North-eastern Han Chinese especially among those with late-onset PD (Li et al., 2017), indicating possible geographical, cultural or ethnic differences mediating the link between TLR2 and PD pathology. The synonymous TLR2 variant rs3804099 is a significant eQTL for cultured fibroblasts, brain and esophagus tissue on GTEx, and was previously associated with susceptibility to H. pylori infection and gastric cancer pathogenesis (Mirkamandar et al., 2018; de Matos Lourenço et al., 2020). Specifically, CT and TT genotypes of rs3804099 are linked with resistance to bacterial infection due to higher TNF-a, IL-1B and IL-6 production in peripheral blood monocytes than CC homozygotes (Zhang et al., 2013). Given that TLR1 expression and TLR1/TLR2 function exhibit an age-related decline (Van Duin et al., 2007), coupled with a lack of validation for TLR1 rs4833095 and TLR2 rs3804099 in both studied cohorts, further investigations of larger cohorts are required to clarify the relationship between TLR1, TLR2, and PD risk.
Finally, heterozygotes of TLR4 rs4986790 and CD14 rs2569190 demonstrated markedly reduced PD risk [odds ratio = 0.577 (p = 0.063) and 0.683 (0.057), respectively], where a lack of significance may be due to the sample size of the cohort. Membrane-associated CD14 facilitates binding of LPS by TLR4, and co-localization of CD14 and TLR4 are necessary for LPS-mediated alterations to epithelial tight-junction proteins (Guo et al., 2013). Given increasing evidence concerning a role of LPS and TLR4 in PD (Gorecki et al., 2019, 2020; Perez-Pardo et al., 2019), further targeted studies investigating TLR4 and CD14 polymorphisms in larger cohorts are warranted.
Mucin
The current study reported reduced PD risk among MUC2 rs11825977 heterozygotes in the PPMI cohort. MUC2 is a highly glycosylated mucin secreted by goblet cells which contributes to the two mucosal layers overlying the intestinal epithelium, and is highly expressed throughout the small intestine and colon (Moehle et al., 2006). Both these mucosal layers are the first line of innate host defense to intestinal pathogens, and the outer layer is home to commensal microbes (Johansson et al., 2011). Importantly, MUC2 expression is reduced in IBD in humans, and MUC2 deficiency causes intestinal inflammation, spontaneous colitis (Van der Sluis et al., 2006) and gut dysbiosis in mice (Wu et al., 2018). Although not a significant eQTL on GTEx, the missense variant 11825977 (also known as V116M) has been linked to reduced mRNA expression of MUC2 and increased risk of Crohn's disease (Moehle et al., 2006). It is conceivable that rs11825977 influences intestinal inflammation and gut dysbiosis which could subsequently contribute to PD onset.
Limitations
The present study has a number of limitations. Firstly, as ethnicity can influence the immune system and inflammatory conditions such as IBD (Nahid et al., 2018; Gazouli et al., 2019), the study only included individuals categorized as Caucasian. However, significant genetic and cultural variation exists between Caucasians from Australia, North America and Europe. This may influence not only allele frequencies, but also environmental and lifestyle factors (e.g., occupation and diet) that in turn could alter gene expression and function (Liu et al., 2020), likely contributing to the discrepancies evident between cohorts in the current study. Further studies in larger multi-ethnic populations will therefore be important. Another limitation of the present study is the retrospective and self-report nature of toxin exposure data, which was restricted to people with PD in the APDR cohort. While the novel and robust finding of an association between PGLRYP4 polymorphism and age of symptom onset among Australians with PD was not altered when controlling for toxin exposure or lifetime smoking habits, further studies with more comprehensive information for both the PD and healthy control group may yield further significant findings. Moreover, recent studies demonstrate the importance of considering other forms of genetic variations in such association studies, particularly in the context of complex diseases such as PD (Theunissen et al., 2020). Previous studies also report that genetic variants may have regional effects beyond a single gene, so further studies of rs10888557 loci in the context of PD should include an analysis of other SNPs and structural variations (i.e., in non-coding regions) within the region of PGLYRP4 and S100A9 genes in order to investigate and potentially establish the extent of association (Roses et al., 2016; Theunissen et al., 2020).
Future Directions
Overall, the current study reports novel associations between PD and SNPs involved with gut homeostasis and immune signaling, supporting previous literature concerning gut inflammation and altered host-microbiome signaling in PD. For instance, studies combining microbial and metabolomic profiling demonstrate reduced levels of short-chain fatty acids and associated short-chain fatty acid-producing bacteria, which typically exert an anti-inflammatory influence (Unger et al., 2016; Vascellari et al., 2020). Moving forward, investigating the interaction between genes, environment and the gut microbiome through combination of genetic sequencing, microbial and metabolomics profiling may provide novel insights into the mechanisms of gut inflammation in PD, identify specific biomarkers for earlier diagnosis prior to significant neurodegeneration, and facilitate precision medicine for better disease management. Notably, genetic stratification by the gut-related SNPs identified in the current study may increase the efficacy of current gut-targeted therapeutic options for PD. For example, probiotic administration has shown promise in alleviating PD symptoms in both rodent models and people with PD (Barichella et al., 2016; Dutta et al., 2019; Tamtaji et al., 2019), and probiotic administration was particularly beneficial among children carrying high-risk SNPs for eczema (Morgan et al., 2014). Moreover, the anti-inflammatory and gut barrier-promoting effects of probiotic administration are attributed to TLR2-dependent mechanisms (Kuugbee et al., 2016; Paveljšek et al., 2020), and thus further studies are required to investigate if probiotic administration to people with pro-inflammatory TLR2 SNPs has enhanced benefit for PD symptoms. Lastly, investigation of these and other gut-related SNPs may be beneficial for other complex neurodegenerative disorders which are also associated with gut dysfunction and dysbiosis, including Alzheimer's disease, Huntington's disease, and multiple sclerosis.
Conclusion
This study aimed to elucidate risk and disease-modifying effects of anti-microbial and intestinal-related polymorphisms in PD. Importantly, we report robust significant associations between PGLYRP4 and disease risk in two independent PD cohorts, and a novel association between PGLYRP4 and age of symptom onset in an Australian PD cohort, as well as interactive risk-modifying effects of polymorphisms in the PGLYRP2, PGLRYP4, and TLR2 genes. The various SNPs which were associated with risk in the current study may confer protection from or susceptibility to complex inflammatory processes, and the mixed results evident between cohorts highlight the need for larger, prospective studies investigating genetic predispositions, environmental exposures, and lifestyles factors to examine the multifaceted nature of PD. In light of increasing evidence concerning the involvement of the gut microbiome and immune system in PD, further studies are required to investigate the functional implications of the significant variants identified in this study, which may contribute to, or exacerbate, PD pathogenesis and pathology upon interaction with other factors.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by University of Western Australia Human Research and Ethics Committee. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
AG, SD, FM, and RA: study design. MB, JK, and FM: clinical data and genetic sample collection. AG, MB, FT, MH, and PA: APDR and control DNA extraction and SNP analysis. AG, AP, and SK: PPMI data analysis. AG, MB, FT, FM, and RA: statistical analysis. AG: manuscript draft. AG, MB, SD, FM, and RA: critical revision. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the Perron Institute for Neurological and Translational Science, Parkinson's WA, the Pierce Armstrong Foundation and the Federal Cooperative Research Centre for Mental Health (CRCMH). This research was carried out while AG, MB, FT, and JK were in receipt of Australian Government Domestic Research Training Program Stipends at The University of Western Australia, and MB was also in receipt of a Richard Walter Gibbon Medical Research Scholarship.
PPMI—a public-private partnership—was funded by the Michael J. Fox Foundation for Parkinson's Research and funding partners, including Abbvie, Allergan, Amathus therapeutics, Avid Radiopharmaceuticals, Biogen Idec, Biolegend, Briston-Myers Squibb, Celgene, Denali, GE Healthcare, Genentech, GlaxoSmithKline, janssen neuroscience, Lilly, Lundbeck, Merck, Meso Scale Discovery, Pfizer, Piramal, Prevail Therapeutics, Roche, Sanofi Genzyme, Servier, Takeda, Teva, UCB, Verily, and Voyager Therapeutics.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to acknowledge the participants and all those involved with the Australian Parkinson's Disease Registry, as well as clinicians and nurses from the Movement Disorders Clinics at the Perron Institute, particularly Sue Walters and Alexa Jefferson for sample collection for DNA extraction. Additionally, data pertaining to the validation cohort used in this article were obtained from the Parkinson's Progression Markers Initiative (PPMI) database (www.ppmi-info.org/data). For up-to-date information on the study, visit www.ppmi-info.org. The extraction of PPMI data was supported by resources provided by the Pawsey Supercomputing Centre with funding from the Australian Government and the Government of Western Australia.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnagi.2020.603849/full#supplementary-material
References
Adams-Carr, K. L., Bestwick, J. P., Shribman, S., Lees, A., Schrag, A., and Noyce, A. J. (2016). Constipation preceding Parkinson's disease: a systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 87, 710–716. doi: 10.1136/jnnp-2015-311680
Alcalay, R. N., Gu, Y., Mejia-Santana, H., Cote, L., Marder, K. S., and Scarmeas, N. (2012). The association between Mediterranean diet adherence and Parkinson's disease. Mov. Disord. 27, 771–774. doi: 10.1002/mds.24918
Angelopoulou, E., Bozi, M., Simitsi, A. M., Koros, C., Antonelou, R., Papagiannakis, N., et al. (2019). The relationship between environmental factors and different Parkinson's disease subtypes in Greece: data analysis of the Hellenic Biobank of Parkinson's disease. Parkinsonism Relat. Disord. 67, 105–112. doi: 10.1016/j.parkreldis.2019.08.013
Bakeberg, M. C., Jefferson, A., Riley, M., Byrnes, M., Ghosh, S., Mastaglia, F. L., et al. (2019). Elevated serum homocysteine levels have differential gender-specific associations with motor and cognitive states in Parkinson's disease. Parkinson Dis. 2019:3124295. doi: 10.1155/2019/3124295
Bandres-Ciga, S., Diez-Fairen, M., Kim, J. J., and Singleton, A. B. (2020). Genetics of Parkinson's disease: an introspection of its journey towards precision medicine. Neurobiol. Dis. 137:104782. doi: 10.1016/j.nbd.2020.104782
Bank, S., Andersen, P. S., Burisch, J., Pedersen, N., Roug, S., Galsgaard, J., et al. (2014). Polymorphisms in the inflammatory pathway genes TLR2, TLR4, TLR9, LY96, NFKBIA, NFKB1, TNFA, TNFRSF1A, IL6R, IL10, IL23R, PTPN22, and PPARG are associated with susceptibility of inflammatory bowel disease in a Danish cohort. PLoS ONE 9:98815. doi: 10.1371/journal.pone.0098815
Barbut, D., Stolzenberg, E., and Zasloff, M. (2019). Gastrointestinal Immunity and Alpha-Synuclein. J. Parkinson's Dis. 9:S313. doi: 10.3233/JPD-191702
Barichella, M., Pacchetti, C., Bolliri, C., Cassani, E., Iorio, L., Pusani, C., et al. (2016). Probiotics and prebiotic fiber for constipation associated with Parkinson disease An RCT. Neurology 87, 1274–1280. doi: 10.1212/WNL.0000000000003127
Bedarf, J., Hildebrand, F., Coelho, L., Sunagawa, S., Bahram, M., Goeser, F., et al. (2017). Functional implications of microbial and viral gut metagenome changes in early stage L-DOPA-naïve Parkinson's disease patients. Genome Med. 9:39. doi: 10.1186/s13073-017-0428-y
Biernacka, J. M., Chung, S. J., Armasu, S. M., Anderson, K. S., Lill, C. M., Bertram, L., et al. (2016). Genome-wide gene-environment interaction analysis of pesticide exposure and risk of Parkinson's disease. Parkinsonism Relat. Disord. 32, 25–30. doi: 10.1016/j.parkreldis.2016.08.002
Braak, H., de Vos, R. A., Bohl, J., and Del Tredici, K. (2006). Gastric α-synuclein immunoreactive inclusions in Meissner's and Auerbach's plexuses in cases staged for Parkinson's disease-related brain pathology. Neurosci. Lett. 396, 67–72. doi: 10.1016/j.neulet.2005.11.012
Burgueño, J. F., and Abreu, M. T. (2020). Epithelial toll-like receptors and their role in gut homeostasis and disease. Nat. Rev. Gastroenterol. Hepatol. 17, 263–278. doi: 10.1038/s41575-019-0261-4
Challis, C., Hori, A., Sampson, T. R., Yoo, B. B., Challis, R. C., Hamilton, A. M., et al. (2020). Gut-seeded α-synuclein fibrils promote gut dysfunction and brain pathology specifically in aged mice. Nat. Neurosci. 23, 327–336. doi: 10.1038/s41593-020-0589-7
Clairembault, T. L, Leclair-Visonneau Coron, E., Bourreille, A. S, Le Dily Vavasseur, F., et al. (2015). Structural alterations of the intestinal epithelial barrier in Parkinson's disease. Acta Neuropathol. Commun. 3:12. doi: 10.1186/s40478-015-0196-0
Corbillé, A. G., Neunlist, M., and Derkinderen, P. (2016). Cross-linking for the analysis of α-synuclein in the enteric nervous system. J. Neurochemistry 139, 839–847. doi: 10.1111/jnc.13845
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
de Matos Lourenço, C., Susi, M. D., do Nascimento, M. C. A., Junior, V. S., Vila, A. P. S., Rodrigues-Flemming, G. H., et al. (2020). Characterization and strong risk association of TLR2 del-196 to-174 polymorphism and Helicobacter pylori and their influence on mRNA expression in gastric cancer. World J. Gastrointestinal Oncol. 12:535. doi: 10.4251/wjgo.v12.i5.535
Devos, D., Lebouvier, T., Lardeux, B., Biraud, M., Rouaud, T., Pouclet, H., et al. (2013). Colonic inflammation in Parkinson's disease. Neurobiol. Dis. 50, 42–48. doi: 10.1016/j.nbd.2012.09.007
Dick, F. D., De Palma, G., Ahmadi, A., Osborne, A., Scott, N. W., Prescott, G. J., et al. (2007). Gene-environment interactions in parkinsonism and Parkinson's disease: the Geoparkinson study. Occup. Environ. Med. 64, 673–680. doi: 10.1136/oem.2006.032078
Dutta, S. K., Verma, S., Jain, V., Surapaneni, B. K., Vinayek, R., Phillips, L., et al. (2019). Parkinson's disease: the emerging role of gut dysbiosis, antibiotics, probiotics, and fecal microbiota transplantation. J. Neurogastroenterol. Motility 25:363. doi: 10.5056/jnm19044
Dziarski, R., and Gupta, D. (2010). Mammalian peptidoglycan recognition proteins (PGRPs) in innate immunity. Innate Immunity 16, 168–174. doi: 10.1177/1753425910366059
Fang, X., Han, D., Cheng, Q., Zhang, P., Zhao, C., Min, J., et al. (2018). Association of levels of physical activity with risk of Parkinson disease: a systematic review and meta-analysis. JAMA Netw. Open 1:e182421. doi: 10.1001/jamanetworkopen.2018.2421
Ferreira, M., and Massano, J. (2017). An updated review of Parkinson's disease genetics and clinicopathological correlations. Acta Neurol. Scand. 135, 273–284. doi: 10.1111/ane.12616
Forsyth, C. B., Shannon, K. M., Kordower, J. H., Voigt, R. M., Shaikh, M., Jaglin, J. A., et al. (2011). Increased intestinal permeability correlates with sigmoid mucosa α-synuclein staining and endotoxin exposure markers in early Parkinson's disease. PLoS ONE 6:e28032. doi: 10.1371/journal.pone.0028032
Gao, J., Nalls, M. A., Shi, M., Joubert, B. R., Hernandez, D. G., Huang, X., et al. (2012). An exploratory analysis on gene-environment interactions for Parkinson disease. Neurobiol. Aging 33, 2528. e1–2528. e6. doi: 10.1016/j.neurobiolaging.2012.06.007
Gazouli, M., Dovrolis, N., Franke, A., Spyrou, G. M., Sechi, L. A., and Kolios, G. (2019). Differential genetic and functional background in inflammatory bowel disease phenotypes of a Greek population: a systems bioinformatics approach. Gut Pathogens 11:31. doi: 10.1186/s13099-019-0312-y
Gold, A., Turkalp, Z. T., and Munoz, D. G. (2013). Enteric α-synuclein expression is increased in Parkinson's disease but not A lzheimer's disease. Mov. Disord. 28, 237–241. doi: 10.1002/mds.25298
Goldman, S. M., Kamel, F., Ross, G. W., Jewell, S. A., Marras, C., Hoppin, J. A., et al. (2014). Peptidoglycan recognition protein genes and risk of Parkinson's disease. Mov. Disord. 29, 1171–1180. doi: 10.1002/mds.25895
Gorecki, A., Preskey, L., Bakeberg, M., Kenna, J., Gildenhuys, C., Macdougall, G., et al. (2019). Altered gut microbiome in Parkinson's disease and the influence of lipopolysaccharide in a human α-synuclein over-expressing mouse model. Front. Neurosci. 13:839. doi: 10.3389/fnins.2019.00839
Gorecki, A. M., Dunlop, S. A., Rodger, J., and Anderton, R. S. (2020). The gut-brain axis and gut inflammation in Parkinson's disease: stopping neurodegeneration at the toll gate. Exp. Opin. Therap. Targets 24, 601–604. doi: 10.1080/14728222.2020.1763956
Guo, S., Al-Sadi, R., Said, H. M., and Ma, T. Y. (2013). Lipopolysaccharide causes an increase in intestinal tight junction permeability in vitro and in vivo by inducing enterocyte membrane expression and localization of TLR-4 and CD14. Am. J. Pathol. 182, 375–387. doi: 10.1016/j.ajpath.2012.10.014
Hamza, T. H., Chen, H., Hill-Burns, E. M., Rhodes, S. L., Montimurro, J., Kay, D. M., et al. (2011). Genome-wide gene-environment study identifies glutamate receptor gene GRIN2A as a Parkinson's disease modifier gene via interaction with coffee. PLoS Genetics 7:1002237. doi: 10.1371/journal.pgen.1002237
Hawkes, C. H., Del Tredici, K., and Braak, H. (2007). Parkinson's disease: a dual-hit hypothesis. Neuropathol. Appl. Neurobiol. 33, 599–614. doi: 10.1111/j.1365-2990.2007.00874.x
Hill-Burns, E. M., Debelius, J. W., Morton, J. T., Wissemann, W. T., Lewis, M. R., Wallen, Z. D., et al. (2017). Parkinson's disease and Parkinson's disease medications have distinct signatures of the gut microbiome. Mov. Disord. 32, 739–749. doi: 10.1002/mds.26942
Horvath, I., Iashchishyn, I. A., Moskalenko, R. A., Wang, C., Wärmländer, S. K., Wallin, C., et al. (2018). Co-aggregation of pro-inflammatory S100A9 with α-synuclein in Parkinson's disease: ex vivo and in vitro studies. J. Neuroinflam. 15, 1–16. doi: 10.1186/s12974-018-1210-9
Houser, M. C., and Tansey, M. G. (2017). The gut-brain axis: is intestinal inflammation a silent driver of Parkinson's disease pathogenesis? npj Parkinson's Dis. 3:3. doi: 10.1038/s41531-016-0002-0
Hughes, A. J., Daniel, S. E., Kilford, L., and Lees, A. J. (1992). Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico-pathological study of 100 cases. J. Neurol. Neurosurg. Psychiatry 55, 181–184. doi: 10.1136/jnnp.55.3.181
Johansson, M. E., Larsson, J. M. H., and Hansson, G. C. (2011). The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host–microbial interactions. Proc. Natl. Acad. Sci. 108, 4659–4665. doi: 10.1073/pnas.1006451107
Johnson, M. E., Labrie, V., Brundin, L., and Brundin, P. (2018). Triggers, facilitators, and aggravators: redefining Parkinson's disease pathogenesis. Trends Neurosci. 42, 4–13. doi: 10.1016/j.tins.2018.09.007
Kamel, F., Goldman, S. M., Umbach, D. M., Chen, H., Richardson, G., Barber, M. R., et al. (2014). Dietary fat intake, pesticide use, and Parkinson's disease. Parkinsonism Relat. Disord. 20, 82–87. doi: 10.1016/j.parkreldis.2013.09.023
Kim, S., Kwon, S. H., Kam, T. I., Panicker, N., Karuppagounder, S. S., Lee, S., et al. (2019). Transneuronal propagation of pathologic α-synuclein from the gut to the brain models Parkinson's disease. Neuron. 103, 627–641.e7. doi: 10.1016/j.neuron.2019.05.035
Kuugbee, E. D., Shang, X., Gamallat, Y., Bamba, D., Awadasseid, A., Suliman, M. A., et al. (2016). Structural change in microbiota by a probiotic cocktail enhances the gut barrier and reduces cancer via TLR2 signaling in a rat model of colon cancer. Digest. Dis. Sci. 61, 2908–2920. doi: 10.1007/s10620-016-4238-7
Lee, P.-C., Raaschou-Nielsen, O., Lill, C. M., Bertram, L., Sinsheimer, J. S., Hansen, J., et al. (2016). Gene-environment interactions linking air pollution and inflammation in Parkinson's disease. Environ. Res. 151, 713–720. doi: 10.1016/j.envres.2016.09.006
Li, X., Xue, L., Sun, J., Sun, Y., and Xie, A. (2017). Single nucleotide polymorphisms in the toll-like receptor 2 (TLR2) gene are associated with sporadic Parkinson's disease in the North-eastern Han Chinese population. Neurosci. Lett. 656, 72–76. doi: 10.1016/j.neulet.2017.07.014
Liu, Y., Yu, X., Zhao, J., Zhang, H., Zhai, Q., and Chen, W. (2020). The role of MUC2 mucin in intestinal homeostasis and the impact of dietary components on MUC2 expression. Int. J. Biol. Macromol. 164, 884–891. doi: 10.1016/j.ijbiomac.2020.07.191
Marras, C., Canning, C. G., and Goldman, S. M. (2019). Environment, lifestyle, and Parkinson's disease: implications for prevention in the next decade. Mov. Disord. 34, 801–811. doi: 10.1002/mds.27720
Matheoud, D., Cannon, T., Voisin, A., Penttinen, A.-M., Ramet, L., Fahmy, A. M., et al. (2019). Intestinal infection triggers Parkinson's disease-like symptoms in Pink1–/– mice. Nature 571, 565–569. doi: 10.1038/s41586-019-1405-y
Mertsalmi, T. H., Pekkonen, E., and Scheperjans, F. (2019). Antibiotic exposure and risk of Parkinson's disease in finland: a nationwide case-control study. Mov. Disord. 35, 431–442. doi: 10.1002/mds.27924
Mirkamandar, E., Nemati, M., Hayatbakhsh, M. M., Bassagh, A., Khosravimashizi, A., and Jafarzadeh, A. (2018). Association of a single nucleotide polymorphism in the TLR2 gene (rs3804099), but not in the TLR4 gene (rs4986790), with Helicobacter pylori infection and peptic ulcer. Turkish J. Gastroenterol. 29:283. doi: 10.5152/tjg.2018.17484
Moehle, C., Ackermann, N., Langmann, T., Aslanidis, C., Kel, A., Kel-Margoulis, O., et al. (2006). Aberrant intestinal expression and allelic variants of mucin genes associated with inflammatory bowel disease. J. Mol. Med. 84, 1055–1066. doi: 10.1007/s00109-006-0100-2
Morgan, A. R., Han, D. Y., Wickens, K., Barthow, C., Mitchell, E. A., Stanley, T. V., et al. (2014). Differential modification of genetic susceptibility to childhood eczema by two probiotics. Clin. Exp. Allergy 44, 1255–1265. doi: 10.1111/cea.12394
Mulak, A., Koszewicz, M., Panek-Jeziorna, M., Koziorowska-Gawron, E., and Budrewicz, S. (2019). Fecal calprotectin as a marker of the gut immune system activation is elevated in Parkinson's disease. Front. Neurosci. 13:992. doi: 10.3389/fnins.2019.00992
Nahid, P., Jarlsberg, L., Kato-Maeda, M., Segal, M., Osmond, D., Gagneux, S., et al. (2018). Interplay of strain and race/ethnicity in the innate immune response to M. tuberculosis. PLoS ONE 13:195392. doi: 10.1371/journal.pone.0195392
Nalls, M. A., Blauwendraat, C., Vallerga, C. L., Heilbron, K., Bandres-Ciga, S., Chang, D., et al. (2019). Identification of novel risk loci, causal insights, and heritable risk for Parkinson's disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 18, 1091–1102. doi: 10.1016/S1474-4422(19)30320-5
Nerius, M., Doblhammer, G., and Tamgüney, G. (2019). GI infections are associated with an increased risk of Parkinson's disease. Gut 2019:318822. doi: 10.1136/gutjnl-2019-318822
Nielsen, H. H., Qiu, J., Friis, S., Wermuth, L., and Ritz, B. (2012). Treatment for Helicobacter pylori infection and risk of Parkinson's disease in Denmark. Eur. J. Neurol. 19, 864–869. doi: 10.1111/j.1468-1331.2011.03643.x
Nishioka, K., Hayashi, S., Farrer, M. J., Singleton, A. B., Yoshino, H., Imai, H., et al. (2006). Clinical heterogeneity of α-synuclein gene duplication in Parkinson's disease. Ann. Neurol. 59, 298–309. doi: 10.1002/ana.20753
Omueti, K. O., Mazur, D. J., Thompson, K. S., Lyle, E. A., and Tapping, R. I. (2007). The polymorphism P315L of human toll-like receptor 1 impairs innate immune sensing of microbial cell wall components. J. Immunol. 178, 6387–6394. doi: 10.4049/jimmunol.178.10.6387
Paveljšek, D., Ivičak-Kocjan, K., Treven, P., Benčina, M., Jerala, R., and Rogelj, I. (2020). Distinctive probiotic features share common TLR2-dependent signalling in intestinal epithelial cells. Cell. Microbiol. e13264. doi: 10.1111/cmi.13264
Perez-Pardo, P., Dodiya, H. B., Engen, P. A., Forsyth, C. B., Huschens, A. M., Shaikh, M., et al. (2019). Role of TLR4 in the gut-brain axis in Parkinson's disease: a translational study from men to mice. Gut 68, 829–843. doi: 10.1136/gutjnl-2018-316844
Prigent, A., Lionnet, A., Durieu, E., Chapelet, G., Bourreille, A., Neunlist, M., et al. (2019). Enteric alpha-synuclein expression is increased in Crohn's disease. Acta Neuropathol. 137, 359–361. doi: 10.1007/s00401-018-1943-7
Roses, A. D., Akkari, P. A., Chiba-Falek, O., Lutz, M. W., Gottschalk, W. K., Saunders, A. M., et al. (2016). Structural variants can be more informative for disease diagnostics, prognostics and translation than current SNP mapping and exon sequencing. Exp. Opin. Drug Metabol. Toxicol. 12, 135–147. doi: 10.1517/17425255.2016.1133586
Rösler, T. W., Salama, M., Shalash, A. S., Khedr, E. M., El-Tantawy, A., Fawi, G., et al. (2018). K-variant BCHE and pesticide exposure: gene-environment interactions in a case–control study of Parkinson's disease in Egypt. Sci. Rep. 8, 1–8. doi: 10.1038/s41598-018-35003-4
Santana, N. D,. L, Rêgo, J. L., Oliveira, J. M., Almeida, L.F.d., Braz, M., et al. (2017). Polymorphisms in genes TLR1, 2 and 4 are associated with differential cytokine and chemokine serum production in patients with leprosy. Mem. Inst. Oswaldo Cruz. 112, 260–268. doi: 10.1590/0074-02760160366
Sauerbier, A., Jenner, P., Todorova, A., and Chaudhuri, K. R. (2016). Non motor subtypes and Parkinson's disease. Parkinsonism Relat. Disord. 22, S41–S46. doi: 10.1016/j.parkreldis.2015.09.027
Scholz, G. M., Heath, J. E., Aw, J., and Reynolds, E. C. (2018). Regulation of the Peptidoglycan amidase PGLYRP2 in epithelial cells by interleukin-36γ. Infect. Immunity 86:18. doi: 10.1128/IAI.00384-18
Schwiertz, A., Spiegel, J., Dillmann, U., Grundmann, D. J, Bürmann, K., et al. (2018). Fecal markers of intestinal inflammation and intestinal permeability are elevated in Parkinson's disease. Parkinsonism Relat. Disord. 50, 104–107. doi: 10.1016/j.parkreldis.2018.02.022
Simon, D. K., Wu, C., Tilley, B. C., Lohmann, K., Klein, C., Payami, H., et al. (2017). Caffeine, creatine, GRIN2A and Parkinson's disease progression. J. Neurol. Sci. 375, 355–359. doi: 10.1016/j.jns.2017.02.032
Singh, N. K., Banerjee, B. D., Bala, K., Chhillar, M., and Chhillar, N. (2014). Gene-gene and gene-environment interaction on the risk of Parkinson's disease. Curr. Aging Sci. 7, 101–109. doi: 10.2174/1874609807666140805123621
Stokholm, M. G., Danielsen, E. H., Hamilton-Dutoit, S. J., and Borghammer, P. (2016). Pathological α-synuclein in gastrointestinal tissues from prodromal Parkinson disease patients. Ann. Neurol. 79, 940–949. doi: 10.1002/ana.24648
Stolzenberg, E., Berry, D., Yang, D., Lee, E. Y., Kroemer, A., Kaufman, S., et al. (2017). A role for neuronal alpha-synuclein in gastrointestinal immunity. J. Innate Immunity 9, 456–463. doi: 10.1159/000477990
Svensson, E., Horváth-Puh,ó, E., Thomsen, R. W., Djurhuus, J. C., Pedersen, L., Borghammer, P., et al. (2015). Vagotomy and subsequent risk of Parkinson's disease. Ann. Neurol. 78, 522–529. doi: 10.1002/ana.24448
Tamtaji, O. R., Taghizadeh, M., Kakhaki, R. D., Kouchaki, E., Bahmani, F., Borzabadi, S., et al. (2019). Clinical and metabolic response to probiotic administration in people with Parkinson's disease: a randomized, double-blind, placebo-controlled trial. Clin. Nutr. 38, 1031–1035. doi: 10.1016/j.clnu.2018.05.018
Theunissen, F., Flynn, L. L., Anderton, R. S., Mastaglia, F., Pytte, J., Jiang, L., et al. (2020). Structural variants may be a source of missing heritability in sALS. Frontiers in Neuroscience 14. doi: 10.3389/fnins.2020.00047
Tongtawee, T., Bartpho, T., Kaewpitoon, S., Kaewpitoon, N., Dechsukhum, C., Leeanansaksiri, W., et al. (2016). TLR1 polymorphism associations with gastric mucosa morphologic patterns on magnifying NBI endoscopy: a prospective crosssectional study. Asian Pacific J. Cancer Prevent. 17, 3391–3394. doi: 10.7314/APJCP.2016.17.3.1057
Unger, M. M., Spiegel, J., Dillmann, K.-U., Grundmann, D., Philippeit, H., Bürmann, J., et al. (2016). Short chain fatty acids and gut microbiota differ between patients with Parkinson's disease and age-matched controls. Parkinsonism Relat. Disord. 32, 66–72. doi: 10.1016/j.parkreldis.2016.08.019
Van der Sluis, M., De Koning, B. A., De Bruijn, A. C., Velcich, A., Meijerink, J. P., Van Goudoever, J. B., et al. (2006). Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 131, 117–129. doi: 10.1053/j.gastro.2006.04.020
Van Duin, D., Mohanty, S., Thomas, V., Ginter, S., Montgomery, R. R., Fikrig, E., et al. (2007). Age-associated defect in human TLR-1/2 function. J. Immunol. 178, 970–975. doi: 10.4049/jimmunol.178.2.970
Vascellari, S., Palmas, V., Melis, M., Pisanu, S., Cusano, R., Uva, P., et al. (2020). Gut microbiota and metabolome alterations associated with Parkinson's disease. Msystems 5:20. doi: 10.1128/mSystems.00561-20
Villumsen, M., Aznar, S., Pakkenberg, B., Jess, T., and Brudek, T. (2019). Inflammatory bowel disease increases the risk of Parkinson's disease: a Danish nationwide cohort study 1977–2014. Gut 68, 18–24. doi: 10.1136/gutjnl-2017-315666
Visanji, N. P., Marras, C., Kern, D. S., Al Dakheel, A., Gao, A., Liu, L. W., et al. (2015). Colonic mucosal α-synuclein lacks specificity as a biomarker for Parkinson disease. Neurology 84, 609–616. doi: 10.1212/WNL.0000000000001240
Wu, M., Wu, Y., Li, J., Bao, Y., Guo, Y., and Yang, W. (2018). The dynamic changes of gut microbiota in Muc2 deficient mice. Int. J. Mol. Sci. 19:2809. doi: 10.3390/ijms19092809
Yang, C. A., Scheibenbogen, C., Bauer, S., Kleinle, C., Wex, T., Bornschein, J., et al. (2013). A frequent Toll-like receptor 1 gene polymorphism affects NK-and T-cell IFN-γ production and is associated with Helicobacter pylori-induced gastric disease. Helicobacter 18, 13–21. doi: 10.1111/hel.12001
Yu, P.-F., Luo, X.-G., Adnan, A., Zhu, W.-Q., Zhang, S.-Y., Huo, Z.-X., et al. (2018). Association of PGLYRP and HMOX-1 polymorphism with Parkinson's disease in the northern Han Chinese population. Int. J. Clin. Exp. Med. 11, 8473–8478. Available online at: http://ijcem.com/files/ijcem0069598.pdf
Zhang, F., Gao, X.-D., Wu, W.-W., Gao, Y., Zhang, Y.-W., and Wang, S.-P. (2013). Polymorphisms in toll-like receptors 2, 4 and 5 are associated with Legionella pneumophila infection. Infection 41, 941–948. doi: 10.1007/s15010-013-0444-9
Keywords: Parkinson's disease, single nucleotide polymorphisms, gene-environment interactions, gut-brain axis, gut inflammation, toll-like receptors, peptidoglycan recognition protein
Citation: Gorecki AM, Bakeberg MC, Theunissen F, Kenna JE, Hoes ME, Pfaff AL, Akkari PA, Dunlop SA, Kõks S, Mastaglia FL and Anderton RS (2020) Single Nucleotide Polymorphisms Associated With Gut Homeostasis Influence Risk and Age-at-Onset of Parkinson's Disease. Front. Aging Neurosci. 12:603849. doi: 10.3389/fnagi.2020.603849
Received: 08 September 2020; Accepted: 20 October 2020;
Published: 17 November 2020.
Edited by:
Christian Neri, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Carlo Scialò, International School for Advanced Studies (SISSA), ItalySachchida Nand Rai, University of Allahabad, India
Copyright © 2020 Gorecki, Bakeberg, Theunissen, Kenna, Hoes, Pfaff, Akkari, Dunlop, Kõks, Mastaglia and Anderton. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ryan S. Anderton, ryan.anderton@nd.edu.au