 Guanghua Wang1,2,3
Guanghua Wang1,2,3 Yuanjin Li1,2,3Jianfeng Liu1,2,3
Yuanjin Li1,2,3Jianfeng Liu1,2,3 Biao Chen1,2,3Hongfei Su1,2,3
Biao Chen1,2,3Hongfei Su1,2,3 Jiayuan Liang1,2,3Wen Huang1,2,3
Jiayuan Liang1,2,3Wen Huang1,2,3 Kefu Yu1,2,3*
Kefu Yu1,2,3*
- 1Guangxi Key Laboratory on the Study of Coral Reefs in the South China Sea, Nanning, China
- 2Coral Reef Research Center of China, Guangxi University, Nanning, China
- 3School of Marine Sciences, Guangxi University, Nanning, China
Members of the phylum Acidobacteria are ubiquitous in various environments. Soil acidobacteria have been reported to present a variety of strategies for their success in terrestrial environments. However, owing to lack of pure culture, information on animal-associated acidobacteria are limited, except for those obtained from 16S rRNA genes. To date, only two acidobacteria have been isolated from animals, namely strain M133T obtained from coral Porites lutea and Acanthopleuribacter pedis KCTC 12899T isolated from chiton. Genomics and physiological characteristics of strain M133T and A. pedis KCTC 12899T were compared with 19 other isolates (one strain from each genus) in the phylum Acidobacteria. The results revealed that strain M133T represents a new species in a new genus in the family Acanthopleuribacteraceae. To date, these two Acanthopleuribacteraceae isolates have the largest genomes (10.85–11.79 Mb) in the phylum Acidobacteria. Horizontal gene transfer and gene duplication influenced the structure and plasticity of these large genomes. Dissimilatory nitrate reduction and abundant secondary metabolite biosynthetic gene clusters (including eicosapentaenoic acid de novo biosynthesis) are two distinct features of the Acanthopleuribacteraceae bacteria in the phylum Acidobacteria. The absence of glycoside hydrolases involved in plant polysaccharide degradation and presence of animal disease-related peptidases indicate that these bacteria have evolved to adapt to the animal hosts. In addition to low- and high-affinity respiratory oxygen reductases, enzymes for nitrate to nitrogen, and sulfhydrogenase were also detected in strain M133T, suggesting the capacity and flexibility to grow in aerobic and anaerobic environments. This study highlighted the differences in genome structure, carbohydrate and protein utilization, respiration, and secondary metabolism between animal-associated acidobacteria and other acidobacteria, especially the soil acidobacteria, displaying flexibility and versatility of the animal-associated acidobacteria in environmental adaption.
Introduction
Bacteria belonging to the phylum “Acidobacteria” are ubiquitous in various environments, including freshwater mud (Liesack et al., 1994; Zimmermann et al., 2012), hot spring microbial mats (Losey et al., 2013), metal-rich acidic waters (Falagán et al., 2017), various soils (Barns et al., 1999; Quaiser et al., 2003; Chanal et al., 2006; Vieira et al., 2017; Kalam et al., 2020), and animals (O’Connor-Sánchez et al., 2014; Liang et al., 2017; Laport et al., 2019). Soil acidobacteria, benefiting from abundant pure cultures (Kishimoto et al., 1991; Eichorst et al., 2007; Kulichevskaya et al., 2010, 2014; Pankratov and Dedysh, 2010; Okamura et al., 2011; Dedysh et al., 2012; Losey et al., 2013; Crowe et al., 2014; Tank and Bryant, 2015; Vieira et al., 2017) and genome sequencing, have been comprehensively studied in biogeographic patterns or survival strategies (Navarrete et al., 2015; Eichorst et al., 2018; Ivanova et al., 2020), metabolism of carbon (de Chaves et al., 2019) and sulfur (Hausmann et al., 2018), and benefits to plants (Kielak et al., 2016; Ivanova et al., 2020; Kalam et al., 2020). However, to date, Acanthopleuribacter pedis KCTC 12899T is the only described acidobacterium isolated from animals (chiton) (Fukunaga et al., 2008). Recently, another acidobacterial isolate, strain M133T, was isolated from stony coral Porites lutea. Bacterial species belonging to other phyla have been demonstrated to be essential for animal development, metamorphosis, nutrition, and defense against pathogens or predator (McFall-Ngai et al., 2013; Perreau and Moran, 2022). However, since pure cultures are lacking, the roles of animal-associated acidobacteria are still unclear. Therefore, this study aimed to identify the animal-associated characteristics of acidobacteria using comparative genomic and high-throughput sequencing analyses based on these two strains.
Materials and Methods
Cultivation, Genome Sequencing, and Assembly
Acanthopleuribacter pedis KCTC 12899T isolated from a chiton (Acanthopleura japonica) in Japan was obtained from the Korean Collection for Type Cultures (KCTC). This strain grows well on both Marine Agar 2216 (BD) and MA/10 [1/10 marine broth powder (BD), 3/4 natural sea water, agar powder 1.2 g/L].
Strain M133T was isolated using serial 1/10 dilution plating on MA/10 from coral Porites lutea collected from the Weizhou Island (109°08′35″ E, 21°03’42″ N), China. This strain grows well on MA/10, but not full-strength Marine Agar 2216(BD).
The whole bacterial biomass was collected from MA/10 or MB/10 [1/10 strength marine broth (BD), 3/4 natural sea water] if not indicated.
For the genome analysis, strain M133T and A. pedis KCTC 12899T were incubated in MB/10 (1/10 strength marine broth [BD], 3/4 natural sea water) at 25°C with shaking. After 5 days of incubation, the biomass was collected and used for DNA extraction. Genomic DNA was extracted using the SDS method (Lim et al., 2016). The extracted DNA was visualized using agarose gel electrophoresis and quantified using the Qubit® 2.0 Fluorometer (Thermo Scientific). The whole genome of strain M133 was sequenced using the PacBio Sequel platform and Illumina NovaSeq PE150, while the genome of A. pedis KCTC 12899T was sequenced using the Illumina NovaSeq PE150. Genomes sequencing was performed by the Beijing Novogene Bioinformatics Technology Co., Ltd. Libraries for single-molecule real-time (SMRT) sequencing were constructed with an insert size of 10 kb using the SMRT bell TM Template kit (v1.0). Meanwhile, libraries for Illumina sequencing were generated using the NEBNext® Ultra™ DNA Library Prep Kit (NEB, United States) following the manufacturer’s recommendations. For Illumina NovaSeq PE150 sequencing, raw data were filtered using readfq (v10) to obtain clean data. The specific processing steps were as follows: (1) removal of reads containing low-quality bases (mass value ≤ 20) over 40%; (2) removal of reads containing ≥ 10% N; and (3) removal of reads overlapping with adapters ≥ 15 bp. Clean data were assembled using the SOAP de novo (Luo et al., 2012), SPAdes (Bankevich et al., 2012), and Abyss (Simpson et al., 2009), and the results of these three software were integrated with CISA (Lin and Liao, 2013). For the PacBio Sequel platform, low-quality reads (less than 500 bp) were filtered, and clean reads were assembled using SMRT Link (Ardui et al., 2018) and corrected with Illumina data. Finally, the sequencing depth of A. pedis KCTC 12899T was 55 × with a Q20 of 97%. For strain M133, the PacBio sequencing depth was 236 × , the Illumina sequencing depth was 73 × with a Q20 of 98%.
Genome Annotation and Comparative Analysis
Gene search and annotation were performed using GeneMarkS (Besemer et al., 2001) and RASTtk (Aziz et al., 2008) in Kbase (Arkin et al., 2018). Metabolism pathways were annotated using the KASS server (Kanehisa et al., 2004). Annotation of COGs was performed using WebMGA (Wu et al., 2011). Peptidases were annotated using the Hotpep-protease (Bush, 2020) based on the Merops database. Carbohydrate-active enzyme annotation was conducted using the dbCAN meta server (Zhang et al., 2018). GIs and prophages were detected using the IslandViewer 4 webserver (Bertelli et al., 2017). Phaster (Arndt et al., 2016), respectively. The insert sequences were detected using IsSaga (Varani et al., 2011). The results of the detected prophages and insert sequences were manually checked with the GenBank annotations. Meanwhile, the pan-genome was calculated using OrthoMCL (2.0) with default parameters in the KBase server (Arkin et al., 2018). Duplicated genes for each genome were calculated from the pan-genome orthologous results. Intra-genome collinearity analysis was performed using MCScanX (Wang et al., 2012). Secondary metabolite biosynthetic gene clusters (BGCs) were analyzed using the online secondary metabolite search tool antiSMASH (Blin et al., 2021). Gene or gene cluster was visualized using Circos (Krzywinski et al., 2009). The average nucleotide identity was calculated using the online ANI calculator (Yoon et al., 2017). Lastly, the average amino acid identity was calculated using the EzAAI (Kim et al., 2021).
Transcriptome Analysis
H2S and N2 production pathways were checked using transcriptome analysis. For transcriptome analysis, strain M133T was firstly incubated in shaking MB/10 at 25°C. After 3 days of incubation, 50 ml of inoculum was inoculated into two bottles of 200 ml MB, respectively. Collected one bottle of M133T using centrifugation after 1 day of shaking incubation at 25°C. Then another bottle of M133T was changed to static incubation after 1 day of shaking incubation at 25°C until the emergence of black particles. Collected the black-stained biomass using centrifugation. Biomass was frozen using liquid nitrogen and stored at –70°C until RNA extraction. RNA was extracted using the RNAprep Pure Cell/Bacteria Kit (Tiangen, Beijing). Sequencing libraries were generated using NEBNext® UltraTM RNA Library Prep Kit for Illumina® (NEB, United States) following the manufacturer’s recommendations. Sequencing was performed using the Illumina Novaseq platform at the Beijing Novogene Bioinformatics Technology Co., Ltd. Clean reads were obtained after removing adapter and low quality reads. Reads containing N were removed; Reads containing low-quality bases (mass value ≤ 20) over 50% were removed. Sequencing depth was 95×, and quality score Q20 was 98%. Bowtie2-2.2.3 was used in mapping reads to reference genome (Langmead and Salzberg, 2012). Rockhopper was used to identify novel genes, operon, TSS, TTS and Cis-natural antisense transcripts (McClure et al., 2013). HTSeq was used to count the reads numbers mapped to each gene (Anders et al., 2015), and then FPKM of each gene was calculated based on the length of the gene and reads count mapped to this gene. When FPKM < 1, no expression is considered. Prior to differential gene expression analysis, for each sequenced library, the read counts were adjusted by edgeR program package (Robinson et al., 2010). Differential expression analysis of two conditions was performed using the DEGSeq R package (Wang et al., 2010).
Polyphasic Identification of Strain M133T
Taxonomic assignment of strain M133T was following the standard procedure of prokaryotic microbe identification, namely phenotypic analysis, phylogenetic analysis (Felsenstein, 1981; Saitou and Nei, 1987; Swofford, 1993; Kumar et al., 2016), phylogenomic analysis (Na et al., 2018) and chemotaxonomic analysis as done in Wang et al. (2020).
Coral Autotrophic Incubation
Coral P. lutea was collected from Weizhou Island and incubated in an aquarium at Guangxi University. For the autotrophic experiments, coral fragments were incubated in axenic natural seawater under natural light at room temperature (approximately 25°C) without feeding. During autotrophic incubation, coral fragments were washed with axenic natural seawater once a week to replace the seawater used during incubation. For the preparation of axenic natural seawater, natural seawater was sterilized at 121°C for 20 min, followed by the injection of axenic air for 20 min to restore the carbonate system.
Bacterial Composition Analysis
Coral holobiont DNA was extracted from 1 cm × 1 cm coral tissue using the TaKaRa MiniBEST Universal Genome DNA extract kit (v5.0). The bacterial community was analyzed using 16S rRNA gene libraries generated with the 338F (5′-ACTCCTACGGGAGGCAGCAG-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′) primers following the procedure described by Chen et al. (2021).
Genomes of Acidobacteria From the GenBank
Acidipila rosea DSM 103428T (GCF_004339725), “Acidisarcina polymorpha” SBC82 (GCF_003330725), Acidobacterium capsulatum ATCC 51196T (GCF_000022565), Bryocella elongata DSM 22489T (GCF_900108185), Candidatus Solibacter usitatus Ellin6076 (GCF_000014905), Edaphobacter modestus DSM 18101T (GCF_004217555), Granulicella pectinivorans DSM 21001T (GCF_900114625), Occallatibacter savannae AB23T (GCF_003131205), Silvibacterium bohemicum S15T (GCF_001006305), Terracidiphilus gabretensis S55T (GCF_0014 49115), Terriglobus roseus DSM 18391T (GCF_000265425), Terriglobus saanensis SP1PR4T (GCF_000179915), Bryobacter aggregatus MPL3T (GCF_000702445), Paludibaculum fermentans P105T (GCF_015277775), Pyrinomonas methylaliphatogenes K22T (GCF_000820845), Chloracidobacterium thermophilum BT (GCF_000226295), Holophaga foetida DSM 6591T (GCF_000242615), Geothrix fermentans DSM 14018T (GCF_000428885), Thermoanaerobaculum aquaticum MP-01T (GCF_000687145), and Luteitalea pratensis DSM 100886T (GCF_001618865).
Sequence Accession Numbers
The GenBank/EMBL/DDBJ accession numbers for the 16S rRNA gene sequence and the whole genome sequence of strain M133T are MN908335 and CP071793, respectively. The GenBank/EMBL/DDBJ accession number for the whole genome sequence of strain Acanthopleuribacter pedis KCTC 12899T is JAFREP000000000. The raw 16S rRNA gene reads of bacteria from the coral P. lutea are deposited in the NCBI Sequence Read Archive database (SRA) under BioProjects PRJNA786650 and PRJNA787388.
Results and Discussion
General Genome Features
The complete genome sequence of strain M133T is 11,786,365 bp long with a G + C content of 60.2 mol%. The genome contains three 16S-23S-5S rRNA operons and 57 tRNA genes. A total of 6,923 protein-coding genes were identified. Meanwhile, the whole genome sequence of A. pedis KCTC 12899T consists of 10,848,621 bp genome sequences obtained from 98 scaffolds, with a G + C content of 57.3 mol%. Five 5S rRNA, one 16S rRNA, one 23S rRNA, and 65 tRNA genes were identified. In total, 6,716 protein-coding genes were identified. Other acidobacterial genome sequences of type or Candidatus species with genome sizes of 2.66 to 9.97 Mb were obtained from GenBank (Table 1).
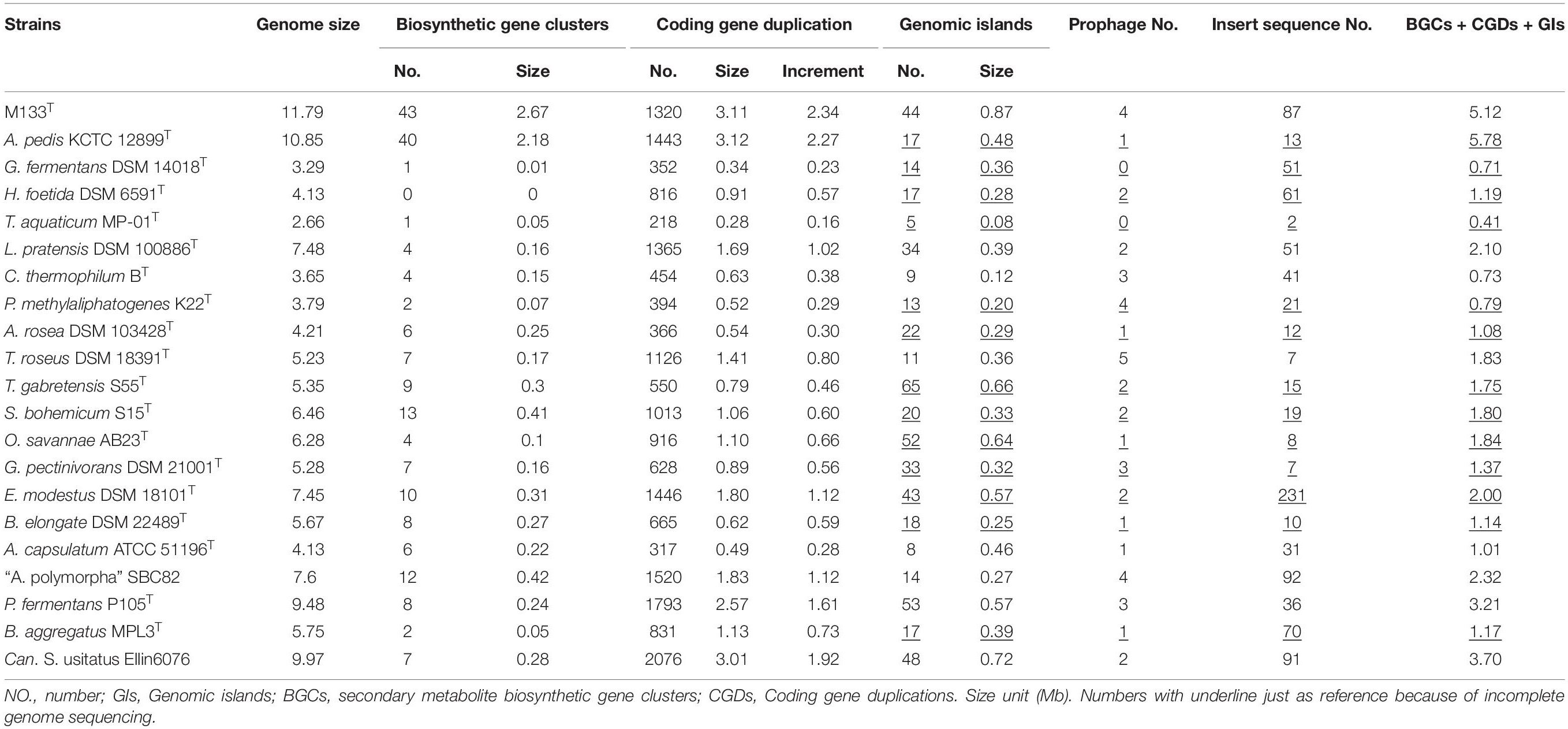
Table 1. Features of selected acidobacterial genomes.
Genome Structure and Plasticity
Based on the available sequences in GenBank, acidobacterial genome sizes range from 0.55 to 11.88 Mb, of which the completed genome sizes range from 2.32 to 11.79 Mb. The largest acidobacterial genome before this study belonged to Candidatus S. usitatus Ellin6076 (9.97 Mb) that was proposed to arise via horizontal gene transfer, followed by widespread small-scale gene duplication (Challacombe et al., 2011; Challacombe and Kuske, 2012). Thus far, strain M133 has the largest genome (11.79 Mb) among the completely sequenced acidobacteria, followed by A. pedis KCTC 12899T (10.85 Mb).
To explore the mechanism underlying large genomes, the pan-genome was calculated using 21 selected acidobacteria. Proteins from the 21 selected acidobacteria were clustered into 29,763 families, of which only 418 families belonged to the core genome, suggesting the high diversification of these acidobacteria. The genome of strain M133T contains 1,355 unique protein families (species-specific; total length of encoding genes is approximately 1.69 Mb) and 4,737 shared protein families (orthologs). The core genome of strain M133T and A. pedis KCTC 12899T is approximately 7.44 Mb, encoding 4,246 families of proteins, which is larger than most of the selected acidobacterial genomes (Table 1), suggesting that the last common ancestor of strain M133T and A. pedis KCTC 12899T had already obtained a large genome.
Horizontal gene transfer (HGT) events are important in acidobacteria for carbohydrate utilization (Naumoff and Dedysh, 2012; Naumoff, 2016), heavy metal resistance, iron uptake, secondary metabolism, and antibiotic resistance (Gonçalves and Santana, 2021). To explore HGT events, genomic islands (GIs) were explored in selected complete acidobacterial genomes (data from draft genomes were listed as references, which may underestimate the actual GIs, prophage, and insert sequence level). The quantities of GIs were highly variable among the genomes, while no habitant or taxonomic dependence was found (Table 1). The total lengths of GIs in an individual genome range from 0.08 to 0.87 Mb, accounting for 3.01–12.34% of the genome. Mobile genetic elements (MGEs), agents that affect DNA movement in HGT (Frost et al., 2005), were also analyzed. A total of 44 prophages were detected in the selected genomes, wherein 1–4 prophages were detected in an individual genome (Table 1). Interestingly, insert sequences, which were detected in all 21 genomes, were more abundant than prophages in acidobacterial genomes, ranging from 2–231 in an individual genome (Table 1). After manual verification, 44 GIs, 4 prophages, and 87 insert sequences were detected in the genome of strain M133T. The total length of 44 GIs accounted for approximately 7.38% of the genome. Structurally, all four prophages had corresponding GIs in the genome. A few insert sequences had no corresponding GIs (Figure 1), indicating that these insert sequences may be species-specific. Some prophages and insert sequences co-localized in the same genomic region (Figure 1), suggesting gene transfer and/or recombination events between mobile genetic elements (Eichorst et al., 2018).
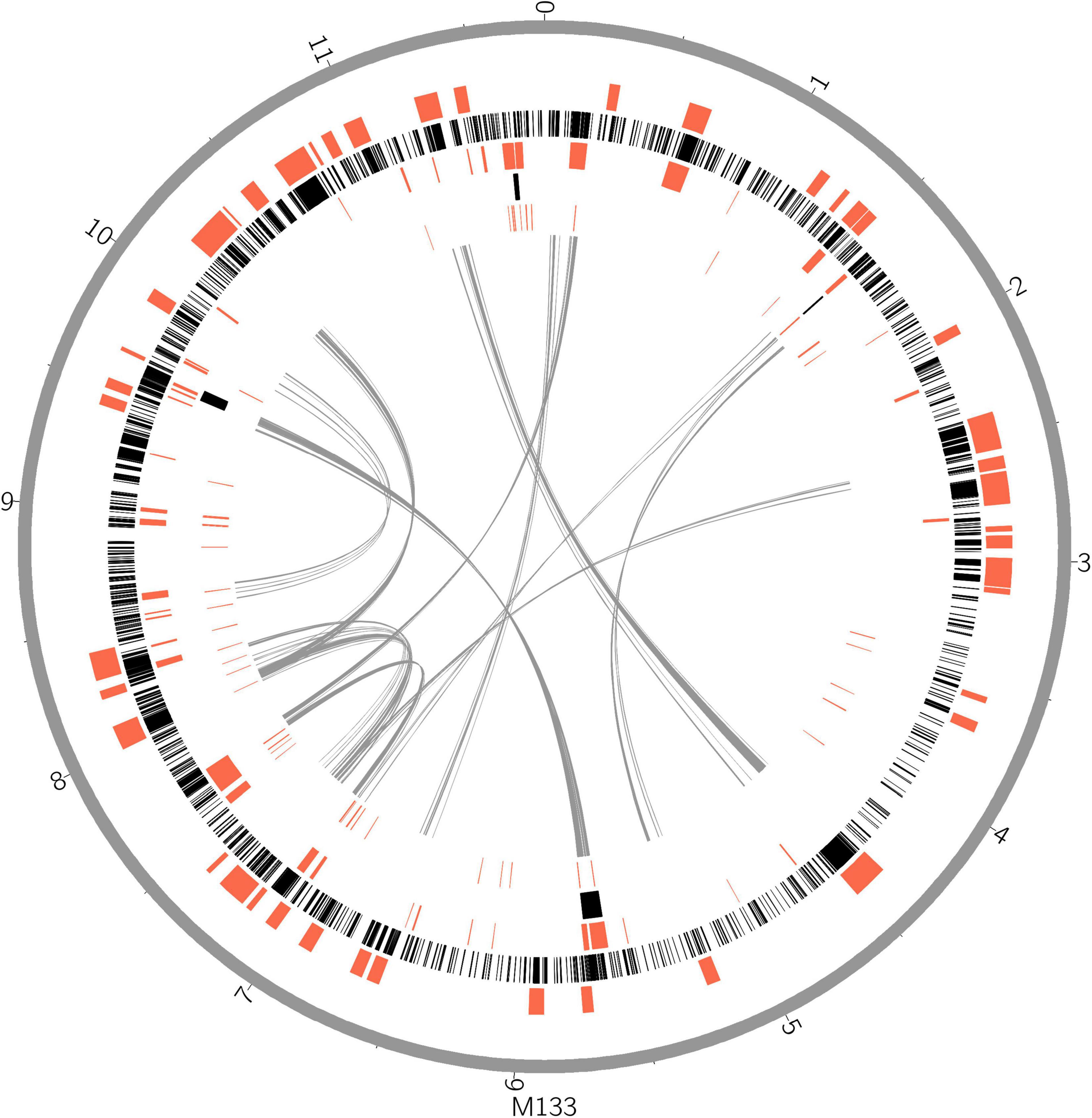
Figure 1. Horizontal gene transfer and gene duplication shaped the large genome of strain M133T. From outer circle to inner, genome scale (gray), biosynthetic gene clusters (red), coding gene duplications (black), genomic islands, insert sequences, intra genome collinearity were displayed. Genome size unit, Mb.
Gene duplications are common and highly variable among the selected acidobacterial genomes. Duplicated genes range from 218 to 2,076 for an individual genome, with a total length of 0.28–3.12 Mb, which accounts for 10.33–30.19% of the genome (Table 1). The total length of duplicated genes in the genome of strain M133T is 3.11 Mb, which accounts for 26.38% of the genome. The duplicated genes in strain M133T were detected genome-wide (Figure 1), mainly functioning in membrane transport, secondary metabolite biosynthesis, MGEs, and others (Supplementary Table 1). Furthermore, these duplicated genes suggest that there were also widespread gene duplications after HGT events in strain M133T similar to Can. S. usitatus Ellin6076 (Challacombe et al., 2011). In addition to MGEs, some species-specific genes were duplicated, such as the rRNA genes (Supplementary Figure 1). In total, three rRNA operons were detected in the genome of strain M133, with one orthologous operon with tRNA-Ile-tRNA-Ala inserted between the 16S and 23S rRNA genes, which is consistent with most of the acidobacteria except for Bryobacteraceae (tRNA-Ala-tRNA-Ile). The other two operons have lost the tRNA-Ile-tRNA-Ala insertion and are located in different strands. The 5S rRNA end of the reverse-strand operon is linked to a site-specific tyrosine recombinase, indicating that the duplicated rRNA operons were from site-specific recombination.
Biosynthetic gene clusters (BGCs) usually result from the combination of HGT, duplication, and rearrangement events during complex evolutionary processes (Lopez, 2003; Gallagher and Jensen, 2015; Boutanaev and Osbourn, 2018). The results indicate that BGCs are as large as GIs in strain M133T (Figure 1) and A. pedis KCTC 12899T, accounting for 22.6% and 20% of the genome, respectively (Table 1). For other acidobacteria, BGCs account for less than 6.5%. Some prophages and/or insert sequences were co-localized with BGCs in the same genome regions (Figure 1), suggesting that MGEs have affected the evolution of these BGCs.
The total length of GIs, duplicated genes, and BGCs (plus overlaps) is 5.12 Mb in the genome of strain M133T, which accounts for 43.43% of the genome size. Meanwhile, the total length of these in Can. S. usitatus Ellin6076 and P. fermentans P105T are 3.70 and 3.21 Mb, accounting for 37.11% and 33.86% of the genome, respectively (Table 1). Therefore, HGT and gene duplication have impacted the long-term evolution of these large genome acidobacteria.
General Metabolism Analysis
The quantity of clusters of orthologous groups (COGs) for acidobacteria ranged from 1260 to 1962, and larger genomes encoded more COGs (Supplementary Table 2). According to N class COGs of A. capsulatum ATCC 51196T (Kishimoto et al., 1991), 17 of the 21 selected acidobacteria should be motile (Supplementary Table 2), although some of them were reported to be non-motile (Liesack et al., 1994; Eichorst et al., 2007; Kulichevskaya et al., 2010, 2014; Pankratov and Dedysh, 2010; Okamura et al., 2011; Dedysh et al., 2012; Crowe et al., 2014; Vieira et al., 2017). C. thermophilum BT, G. fermentans DSM 14018T, and T. aquaticum MP-01T contain 2–7 families of N-class COGs, which are consistent with the non-motile reports (Coates et al., 1999; Losey et al., 2013; Tank and Bryant, 2015).
Meanwhile, the quantities of G-class COGs varied among habitats (Supplementary Table 2), which decreased in the sequence of soil acidobacteria, animal-associated acidobacteria (A. pedis KCTC 12899T and strain M133T), and microaerophilic or anaerobic acidobacteria (H. foetida DSM 6591T, C. thermophilum BT, G. fermentans DSM 14018T, and T. aquaticum MP-01T), indicating the adaptive carbohydrate utilization abilities of these strains (Ward et al., 2009; de Castro et al., 2013; Belova et al., 2018; Eichorst et al., 2018; Kalam et al., 2020). The Q-class of COGs (Supplementary Table 2) indicated that strain M133T and A. pedis KCTC 12899T harbors genes involved in secondary metabolite biosynthesis, transport, and catabolism compared with other acidobacteria (details are displayed in the section “Secondary Metabolism”).
Carbohydrates Metabolism
Carbohydrate-active enzymes (CAZymes) prediction results (Supplementary Table 3) revealed that facultative anaerobic P. fermentans P105T and Can. S. usitatus Ellin6076 have the most abundant families of CAZymes (111–112), followed by aerobic soil acidobacteria (75–103), animal-associated acidobacteria (56–63), and anaerobic bacteria (23–35). Glycoside hydrolases, which are involved in hydrolysis and/or rearrangement of glycosidic bonds, contributed to the differences in CAZyme abundance among the genomes (Supplementary Table 3). Soil acidobacteria have 40–71 families of glycoside hydrolases, while animal-associated and anaerobic acidobacteria have very few glycoside hydrolase families (23–24 and 7–11, respectively). These results further confirmed the outstanding carbohydrate utilization ability of soil acidobacteria (Ward et al., 2009; de Castro et al., 2013; Belova et al., 2018; Eichorst et al., 2018; Kalam et al., 2020). Interestingly, only 48 CAZymes were detected in the aerobe Bryobacter aggregatus MPL3T isolated from acidic Sphagnum peat bogs, of which 18 families belonged to glycoside hydrolases.
In this study, Acidobacteria are observed to be versatile in carbohydrate degradation. Genes encoding for chitinases (GH18, 23) involved in chitin degradation; α-amylases (GH57), glucoamylases (GH15), and glycogen/starch phosphorylases (GT35) in starch degradation; endoglucanases (GH51, 9, and 10) and β-glucosidases (GH3) in cellulose degradation, and xylanases (GH10, 51) in xylan degradation were detected in most of the acidobacterial genomes (Supplementary Table 3). Previously, some acidobacteria have been shown to degrade a range of carbohydrates. For example, soil bacteria Silvibacterium bohemicum S15T (Lladó et al., 2016) and “Acidisarcina polymorpha” SBC82 (Belova et al., 2018) have been reported to degrade starch, cellulose, chitin, xylan, and pectin. Genes encoding carbohydrate hydrolases indicated that strain M133T and A. pedis KCTC 12899T can degrade chitin (mainly from the cell wall of fungi and the exoskeleton of arthropods) using N-acetylglucosaminidase (GH20, Meekrathok et al., 2021), but not pectin (mainly from plants) using polygalacturonases (GH28) and α-rhamnosidases (CBM67, GH106).
Furthermore, acidobacteria are versatile in polysaccharide biosynthesis. Starch is synthesized by the starch synthase and 1,4-alpha-glucan branching enzyme. Genes encoding starch synthases (GT5) were detected in 20 of the selected acidobacterial genomes, excluding Geothrix fermentans DSM 14018T, while those encoding the 1,4-alpha-glucan branching enzyme (CBM48 + GH13_9) were detected in only seven genomes (Supplementary Table 3). These observations indicate that most of the acidobacteria can synthesize amylose, while only a few (including strains M133 and A. pedis KCTC 12899T) can produce glycogen. In particular, Acidobacteriaceae cannot produce glycogen.
Cellulose is important in biofilm formation and cellular water holding and is synthesized by various cellulose synthases. To date, only the genomes of A. capsulatum and Terriglobus saanensis have been reported to biosynthesize cellulose using bcs operons (Kalam et al., 2020). Genomic detection using the Kbase platform and conserved domains search in GenBank indicated that seven acidobacteria in the family Acidobacteriaceae, namely A. capsulatum ATCC 51196T, T. saanensis SP1PR4T, A. rosea DSM 103428T, B. elongate DSM 22489T, G. pectinivorans DSM 21001T, T. gabretensis S55T, and “A. polymorpha” SBC82, contain bcs operons. The distribution of the bcs operon was variable in the genus Edaphobacter, and no bcs operon was detected in the genome of E. modestus DSM 18101T. Cellulose synthase was not detected in strain M133T and A. pedis KCTC 12899T.
Peptide Degradation
According to the catalog of MEROPS peptidase, the genomes of selected acidobacteria contain 45–95 families of peptidases (Supplementary Table 4). Species from the same family have similar numbers of peptidase families. The anaerobic Holophagaceae foetida DSM 6591T has the lowest number of peptidase family (45). Acidbacteriaceae had 57–77 peptidase families, followed by animal-associated Acanthopleuribacteraceae (79–84), and Bryobacteriaceae has the highest number of peptidase families (79–95). Other acidobacteria have 62–87 families of peptidases.
Bacterial collagenase H (M09B), imelysin (M75), and MtfA peptidase (M90) were only detected in strain M133 and A. pedis KCTC 12899T (Supplementary Table 4). Moreover, collagenase (U32), subtilisin (S08A), pappalysin-1(M438), astacin (M12A), adamalysin (M12B), serralysin (M10B), fungalysin (M36), lysostaphin (M23B), immune inhibitor A peptidase (M06), thermolysin (M04), and bacteriocin-processing peptidase (C39) were detected in strain M133T and A. pedis KCTC 12899T. In particular, a high abundance of subtilisin (11–12) and thermolysin (8–11) coding sequences were detected in strain M133 and A. pedis KCTC 12899T (Supplementary Table 4). Membrane dipeptidase (M19), carnosine dipeptidase II (M20F), glutamate carboxypeptidase (M28B), aminopeptidase M29 (M29), streptogrisin A (S01E), carboxypeptidase Y (S10), SpoIVB peptidase (S55), Xaa-Pro dipeptidyl peptidase (S15), and sedolisin (S53) were absent in strain M133T and A. pedis KCTC 12899T, whereas they are widely detected in other acidobacteria.
Secondary Metabolism
Genes encoding enzymes that involved in peptide assembly, regulation, resistance, and synthesis of secondary metabolites are usually physically clustered into biosynthetic gene clusters (BGCs, Medema et al., 2015). More than 40 BGCs were detected in genomes of strain M133T and A. pedis KCTC 12899T, respectively (Table 1). According to the antiSMASH prediction, these BGCs may function in non-ribosomal peptides, polyketides, terpenes, thioamitides, arylpolyenes, and ranthipeptides biosynthesis. While other acidobacterial genomes contain only a few BGCs (0–13) (Table 1). Genes of non-ribosomal peptide synthetases and type I polyketide synthetases accounted for more than 40% of the BGCs in strain M133T and A. pedis KCTC 12899T. The exact secondary metabolites of strain M133T and A. pedis KCTC 12899T need further study.
Bacterial long-chain polyunsaturated fatty acids (PUFAs) are synthesized by multi-domain protein complexes akin to type I iterative fatty acids and polyketide synthases (Shulse and Allen, 2011). The typical PUFA-synthesizing gene cluster was discovered in marine gammaproteobacteria, in which five genes, pfaABCDE, participated in the de novo biosynthesis of PUFAs (Shulse and Allen, 2011). EPA was detected in both strain M133T and A. pedis KCTC 12899T using MIDI gas chromatography, with EPA contents of 6.2% and 12.2%, respectively (Supplementary Table 5). Among the five genes in the EPA-synthesizing gene cluster, four genes (pfaB and pfaC were annotated as one gene: pfaB/C) were detected in strain M133T and A. pedis KCTC 12899T, wherein three genes formed the cluster pfaDAB/C, while pfaE encoding for phosphopantetheinyl transferase was located far from this cluster (Supplementary Figure 2). This EPA biosynthesis cluster is much similar to those in deltaproteobacteria “Desulfococcus oleovorans” Hxd3 and Sorangium cellulosum So ce 56 (S. bulgaricum Soce 321T was reported to produce EPA, Mohr et al., 2018), but not the EPA-producing gammaproteobacteria Shewanella pealeana ATCC 700345T (Shulse and Allen, 2011). None of the pfa genes were detected in the other acidobacterial genomes.
Respiration of Acidobacteria
There are several types of respiration electron acceptors in acidobacteria, such as oxygen, nitrate, Fe (III), Mn (IV), and sulfur. Most acidobacteria, mainly Acidobacteriaceae, are aerobic or microaerobic. The A-type cytochrome c oxidase was ubiquitous in Acidobacteriaceae, while the high-oxygen affinity cbb3-type cytochrome c oxidase and cytochrome bd ubiquinol oxidase were detected in only half of the selected genomes (Supplementary Table 6). This illustrates that some of Acidobacteriaceae can respond to microoxic concentrations (Morris and Schmidt, 2013; Eichorst et al., 2018). Meanwhile, only the cbb3-type terminal oxidase has been detected in the genome of the microaerophile C. thermophilum (Tank and Bryant, 2015). Both strain M133 and A. pedis KCTC 12899T contain the A-type cytochrome c oxidase, cbb3-type cytochrome c oxidase, and cytochrome bd ubiquinol oxidase, suggesting that these two strains can survive under variable oxygen conditions.
Nitrate is an energy-efficient oxygen substitute under anaerobic conditions. However, it seems that acidobacteria are not adept at nitrate reduction. Genes for assimilatory nitrate reduction were detected in five of the 21 genomes, and those for nitrite reduction were detected in six genomes. Dissimilatory nitrate reduction genes were detected in four genomes, namely G. ferrnentans DSM 14018T, T. aquaticum MP-01T, A. pedis KCTC 12899T, and strain M133, while those for nitrite reduction were found in five genomes. These results may be due to a few assimilatory nitrate reductases involved in dissimilatory nitrogen metabolism, as suggested by Morozkina and Zvyagilskaya (2007); Eichorst et al. (2018). Denitrification genes nirS and norBC were detected in G. ferrnentans DSM 14018T, T. aquaticum MP-01T, P. fermentans P105T, Luteitalea pratensis DSM 100886T, and A. pedis KCTC 12899T, whereas the complete denitrification pathways, which included napA, nirS, norBC, and nosZ were detected only in strain M133T. Transcriptome analysis confirmed the expression of genes nirS (FPKM 2311: 1.2), norB (1364: 0.6), norC (856: 0), and nosZ (723: 1.3) under anaerobic conditions in strain M133T. API 20NE results also suggest that strain M133T can reduce nitrate to nitrogen.
Fe (III) or Mn (IV) reduction is an important anaerobic respiration pathway in acidobacteria. The outer membrane c-type cytochromes OmcA and MtrC (also known as OmcB) have been previously demonstrated to play an important role in the reduction of Fe (III) and Mn (III/IV) (Beliaev et al., 2001; Myers and Myers, 2001). OmcA and MtrC were detected in T. aquaticum MP-01T, P. fermentans P105T, and Can. S. usitatus Ellin6076. Both T. aquaticum MP-01T (Losey et al., 2013) and P. fermentans P105T (Kulichevskaya et al., 2014) have been reported to anaerobically reduce Fe (III) or Mn (IV). Fe (III) respiration in Can S. usitatus Ellin6076 needs to be confirmed using growth-based tests. The gene for MtrF, a homolog of MtrC, was detected in the genome of G. ferrnentans DSM 14018T, whereas no gene for OmcA was detected, although G. ferrnentans DSM 14018T was confirmed to use Fe (III) as an electron acceptor (Coates et al., 1999).
Oxidized sulfur compounds are relatively low-efficient electron acceptors during respiration. Genes encoding assimilatory sulfate reductases were detected in 13 of the 21 acidobacterial genomes, and those encoding dissimilatory sulfate reductases were detected in two genomes (Supplementary Table 6). Genes encoding for sulfite reductase were detected in 16 genomes, of which only one (H. foetida DSM 6591T) was dissimilatory sulfite reductase (Supplementary Table 6). Only assimilatory sulfate and sulfite reduction genes were detected in strain M133 and A. pedis KCTC 12899T.
The bifunctional enzyme sulfhydrogenase was first reported in the anaerobic archaeon Pyrococcus furiosus, and this enzyme produces sulfide hydrogen from elemental sulfur or polysulfide under anaerobic conditions (Ma et al., 1993). Complete sulfhydrogenase genes hydBGDA (Pedroni et al., 1995) and accessory genes hypCDEF were also detected in the genome of strain M133. Therefore, sulfhydrogenase may function as a secondary anaerobic respiration pathway in this strain. High nutrition, especially tryptone or other protein derivatives, promotes hydrogen sulfide production. However, the actual substrate of sulfhydrogenase in strain M133 remains unclear. In addition, the amino acid sequence of the M133 sulfhydrogenase β subunit (homolog to sulfite reductase) was searched using NCBI BLAST, and both sulfite reductase and 4Fe-4S dicluster-containing proteins were retrieved. After manually checking the RASTtk re-annotated genomes in Kbase, approximately 31 genomes were confirmed to harbor complete sulfhydrogenase genes (αβγδ) (Supplementary Table 7). Phylogenetically, these sulfhydrogenase-containing strains belonged to approximately 10 bacterial phyla, including Acidobacteria, Cyanobacteria, “Chloroflexi,” “Deinococcus-Thermus,” “Nitrospinae,” Proteobacteria (αβγδ), Bacteroidetes, Actinobacteria, Planctomycetes, and Verrucomicrobia, and two archaeon phyla. Although hydrogenase and sulfite reductase were detected in Firmicutes (Clostridium), no sulfhydrogenase was found. Three archaeon strains from Euryarchaeota and Thaumarchaeota, including Pyrococcus furiosus DSM 3638, were also found to harbor complete sulfhydrogenase genes. This universally and randomly taxonomic distribution pattern suggests that HGT may have contributed to the dissemination of sulfhydrogenase. To examine the possibility of HGT, the G + C content of hydBGDA and the corresponding genome (where the hydBGDA were extracted) were analyzed. Results indicated that the G + C content of hydBGDA is highly variable among genomes (32.84–74.08 mol%), while the G + C content difference between hydBGDA and the whole genome is usually 2–4 mol% (Supplementary Table 7). This difference between a common ancestor and the individual genomes is a long-term evolutionary result. Therefore, the existence of sulfhydrogenase in strain M133 is likely the evolutionary remnant and not a result of recent HGT.
Several strains have been reported to undergo fermentative growth (T. aquaticum MP-01, G. fermentans DSM14018, H. foetida TMBS4, and A. ailaaui PMMR2) (Liesack et al., 1994; Coates et al., 1999; Losey et al., 2013; Myers and King, 2016). However, the function of the possible fermentation enzymes remains to be confirmed (Eichorst et al., 2018). Genes involved in fermentation, such as lactate dehydrogenase, fumarate reductase/succinate dehydrogenase, and phosphate acetyltransferase, were also detected in strain M133 and A. pedis KCTC 12899T; however, no fermentation growth was detected in the API 50 CH sheet.
Acidobacteria–Animal Interaction
To date, species closely related to strain M133T (>92% 16S rRNA gene identity) have seldom been detected in the environment. To explore these allied species, GenBank databases were blasted using the 16S rRNA gene sequences of strain M133T and A. pedis KCTC 12899T. Only two operational taxonomic units (OTUs) (GU319135 and MT858037) were retrieved from the standard nucleotide database. SRA datasets for seawater and algae (Mei et al., 2019), sediment (Zhang et al., 2017; Ghate et al., 2021), coral and coral reefs (Liang et al., 2017; Pearman et al., 2019; Chen et al., 2021), and sponge (Baquiran and Conaco, 2018; Wu et al., 2018) were blasted, and the results indicated that strain M133T allied reads were occasionally detected in the datasets of the Red Sea coral reef (PRJNA479721) and the Weizhou Island coral P. lutea (PRJNA786650). This occasional appearance of strain M133T allied reads in high throughput sequencing datasets indicated that these bacteria do exist in related environments, but are usually not detected. Technically, DNA template preparation and PCR amplification can influence the outcome of certain reads. It is possible that the allied species exist in very low abundance in related habitats and/or cells are resistant to universal DNA extraction methods. Another reason may be the bias from the universal primer (Takahashi et al., 2014) and the barcode of indexed-primer (O’Donnell et al., 2016) used in PCR amplification. The exact cause of the low detection of strain M133T allied species requires further study.
To date, the hosts or habitats of the allied species of strain M133T have been limited to animals. Strain M133T was isolated from the coral P. lutea, and five reads (100% identities to strain M133T) were detected in another coral P. lutea 1 year later (PRJNA786650). Interestingly, the bacterium clone A3M_UNP0_21 (GU319135, with 98% 16S rRNA sequence identity to strain M133T) was also detected in a coral (Acropora eurystoma in Israel) (Meron et al., 2011). Meanwhile, A. pedis KCTC 12899T was isolated from the chiton Acanthopleura japonica in Japan (Fukunaga et al., 2008), and its closest bacterial clone OTU20 (MT858037, 100% identity) was detected in the gut of fish Seriola rivoliana in Mexico. Although the exact niches of the allied reads of strain M133T (92–99% to strain M133T or A. pedis KCTC 12899T) are unclear in the PVC plates deployed in the Red Sea coral reefs (Pearman et al., 2019), it is evident that benthic animal species could settle on similar autonomous reef monitoring structures (Leray and Knowlton, 2015), raising the possibility that allied reads were also from animals. In addition, some distant Acanthopleuribacteraceae reads also displayed an intimate association with coral. After three months of autotrophic incubation and weekly axenic washing, the abundance of Acanthopleuribacteraceae reads (85.9–91.7% identities to strain M133T, PRJNA787388) increased evidently in coral P. lutea, coinciding with the significant bacterial community composition shift (Supplementary Figures 3, 4). As the survivors of long-term autotrophy and repeated axenic wash, these distant reads should also be closely correlated.
In this study, it can be observed that strain M133T and A. pedis KCTC 12899T have evolved to associate with animals. Both strain M133T and A. pedis KCTC 12899T possess the ability to initiate and maintain symbiotic interactions with animals. Microbial motility and chemotaxis are pivotal for the onset and maintenance of symbiotic interactions (Raina et al., 2019). Moreover, both acidobacteria are mobile by using the flagella (Supplementary Figure 5; Fukunaga et al., 2008). Ankyrins, which was also detected in strain M133T and A. pedis KCTC 12899T, can aid bacteria in evading the eukaryotic immune system (Jahn et al., 2019) in bacteria-animal symbiosis. For intracellular symbiotic bacteria, there is usually a dramatic genome reduction (McCutcheon and Moran, 2011; Jäckle et al., 2019), so strain M133T and A. pedis KCTC 12899T are not likely to form an intracellular symbiosis with animals. The discovery of OTU20 (MT858037) in the fish gut suggests that strain M133T and A. pedis KCTC 12899T may also exist in the animal gut environment. Sulfatases can digest the highly sulfated glycans in the gut, whereas the activation of sulfatases requires post-translational modification catalyzed by radical S-adenosyl-L-methionine proteins (Benjdia et al., 2007, 2011). Both genes encoding for sulfatase and radical S-adenosyl-L-methionine proteins were detected in the genomes of strain M133T and A. pedis KCTC 12899T. The low oxygen and nitrate respiration capabilities of these bacteria further facilitate their survival and function in animal gut-like environments. The general metabolic pattern indicates that there is a positive selection resulting from long-term animal associations. Glycoside hydrolases related to plant polysaccharide degradation, such as alpha-L-rhamnosidase (GH106), endo-1,4-beta-xylanase (GH10), and alpha-galactosidase (GH27), are usually absent from strains M133T and A. pedis KCTC 12899T. In contrast, animal disease-related virulence factors, such as collagenase (Harrington, 1996; Penttinen et al., 2016), astacin (Ricard-Blum and Vallet, 2016), adamalysin (Ricard-Blum and Vallet, 2016), subtilisin (Imamura et al., 2017; Łagowski et al., 2021), and thermolysin (Kong et al., 2015; Tsaplina et al., 2020) are present in strain M133T and A. pedis KCTC 12899T.
Meanwhile, the allied species of strain M133T may benefit animal hosts in dealing with environmental fluctuations. Several bacteria have been shown to protect their animal hosts from pathogens and heat stress, such as Pseudoalteromonas sp., which inhibits coral pathogens (Vibrio sp.) using antibiotics and peptidase (Offret et al., 2016; Richards et al., 2017; Rosado et al., 2019). Ruegeria sp. inhibits the coral pathogen Vibrio coralliilyticus (Miura et al., 2019), whereas Muricauda sp. protects coral endosymbionts from thermal stress by producing zeaxanthin (Motone et al., 2020). Although the exact products of BGCs are still unclear, the large secondary metabolite biosynthesis capacities of strain M133 and A. pedis KCTC 12899T are still encouraging. According to antiSMASH known-cluster-blast, BGCs of strain M133 are very similar to the biosynthesis gene clusters of jerangolid A, oocydin A, nematophin, and microsclerodermin M. These compounds have been reported to have cytotoxic activities against fungi and gram-positive bacteria (Gerth et al., 1996; Strobel et al., 1999; Melikhova et al., 2016; Cai et al., 2017). Meanwhile, EPA accounts for 1% of the total fatty acids of the whole P. lutea holobionts; therefore, strain M133, an EPA-producer, may also benefit P. lutea holobionts in dealing with temperature and pressure variation (Valentine and Valentine, 2004).
Taxonomy of the New Isolate
The closest taxonomic neighbor of strain M133T, based on the 16S rRNA gene sequence similarity, is A. pedis KCTC 12899T, they share 92.4% identity (recommended genus demarcation is 95%, Yarza et al., 2014). The average nucleotide identity between strains M133T and KCTC 12899T is 70% (recommended genus demarcation is 74%, Barco et al., 2020). The average amino-acid identity between strains M133T and KCTC 12899T is 63.97%, which is lower than the recommended genus demarcation of 68% (Konstantinidis and Tiedje, 2005). These indices indicated that strain M133T and A. pedis KCTC 12899T belong to different genera. Phylogenetic analysis based on the 16S rRNA gene sequences using maximum-likelihood, neighbor-joining, and maximum-parsimony algorithms (Supplementary Figures 4–6) and phylogenomic analysis using 92 concatenated sequence (Figure 2) indicated that strain M133T forms a distinct branch beside A. pedis KCTC 12899T in the family Acanthopleuribacteraceae.

Figure 2. Phylogenomic tree inferred using UBCGs (concatenated alignment of 92 core genes) indicated strain M133T belongs to the family Acanthopleuribacteraceae. Gene support indices (GSIs) is given at branching points, only above 50% is displayed. Bar, 0.5 substitution per position.
Cells of strain M133T are Gram-negative, non-spore-forming, motile, aerobic rods (Supplementary Figure 7). This strain can be distinguished from A. pedis KCTC 12899T by the ability to reduce nitrate to nitrogen and the production of hydrogen sulfide. Chemotaxonomic features can also differentiate these two strains. The major cellular fatty acids (> 5%) of strain M133T are isoC15:0 (35.6%), C16:0 (29.8%), C15:0 (8.1%), and C20:5ω3c (6.2%), while those of A. pedis KCTC 12899T are C16:0 (29.6%), isoC15:0 (17.2%), isoC17:0 (12.4%), C20:5ω3c (12.2%), and C16:0N alcohol (5.8%) (Supplementary Table 5). The major polar lipids of strain M133T are phosphatidylethanolamine, phosphatidylglycerol, and diphosphatidylglycerol, which are similar to those of A. pedis KCTC 12899T (Supplementary Figure 8). Other features of strain M133T are listed in Table 2 and the new taxon description.
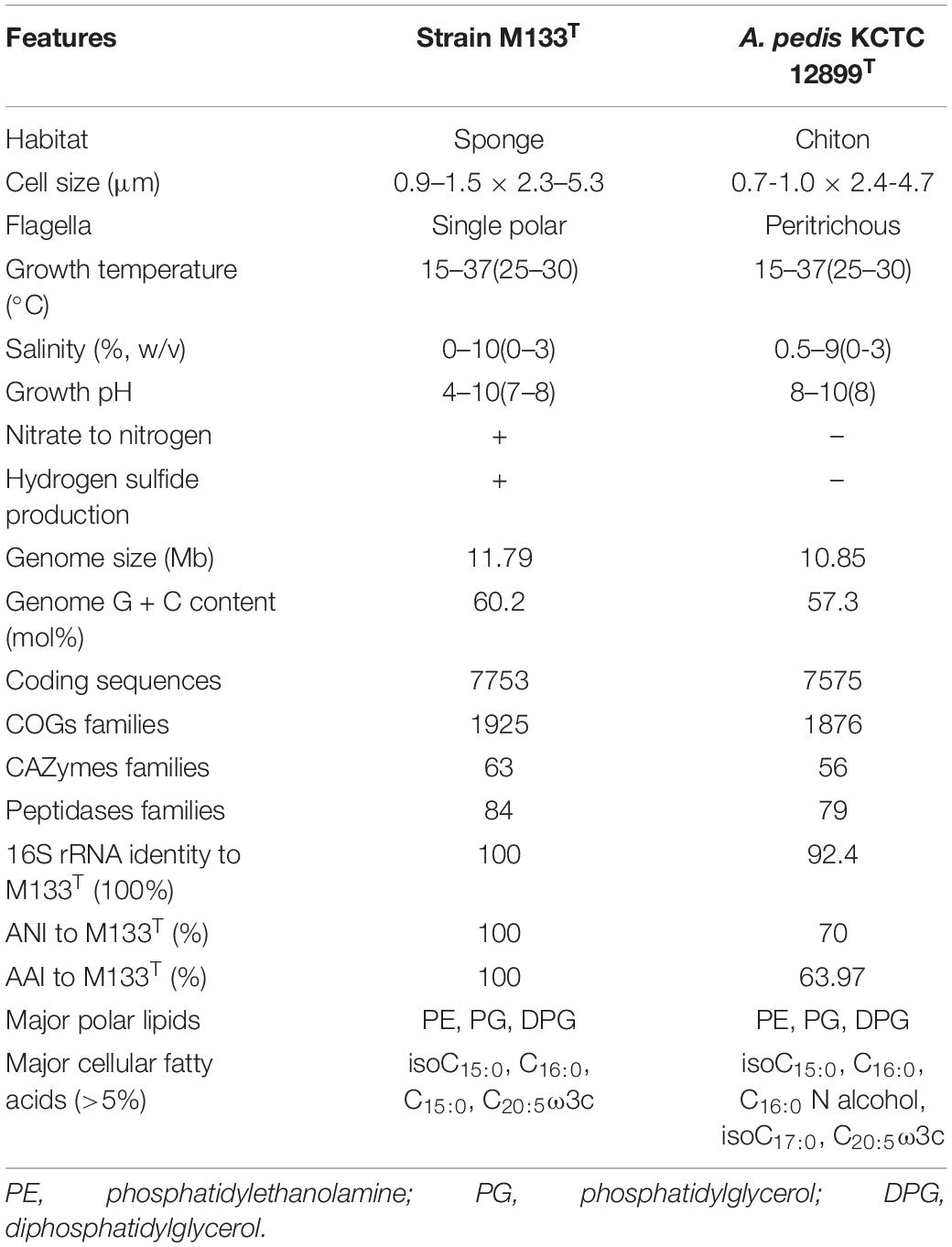
Table 2. Characteristics distinguish strain M133T from Acanthopleuribacter pedis KCTC 12899T.
In summary, phylogenomic, phylogenetic, chemotaxonomic and phenotypic differences to the closest type strain A. pedis KCTC 12899T indicated that strain M133T represents a new species in a new genus in the family Acanthopleuribacteraceae, for which Sulfidibacter corallicola gen. nov., sp. nov. is proposed.
Description of Sulfidibacter gen. nov
Sulfidibacter (sul.fi.di.bac’ter. N.L. neut. n. sulfidum, sulfide; N.L. masc. n. bacter, rod; N. L. masc. n. Sulfidibacter, sulfide producing rod).
Cells are Gram-stain negative, non-spore-forming, motile, aerobic rods. Catalase and oxidase are positive. Nitrate reduction is positive. Major respiratory quinone are menaquinone 6 and 7 (MK6 & 7). Major cellular fatty acids are isoC15:0, C16:0, C15:0 and C20:5ω3c. Major polar lipids are phosphatidylethanolamine, phosphatidylglycerol, and diphosphatidylglycerol.
Type species is Sulfidibacter corallicola.
Description of Sulfidibacter corallicola sp. nov
Sulfidibacter corallicola (co.ral.li′co.la. L. neut. n. corallum, coral; L. masc. suff. -cola, inhabitant dweller; N.L. n. corallicola, coral-dweller).
Cells have following features in addition to the genus description. Cells are usually 2.3–5.3 μm in length and 0.9–1.5 μm in width. Cells are motile by single polar flagellum. Colonies on marine agar are circular, smooth and yellow in color. H2S can be produced when incubated in high strength of marine agar or marine broth. Cells can grow at 15–37°C, pH4-10 under 0–10% NaCl (w/v). Starch, tween20, 40 and 60 are degraded. Nitrate is reduced to nitrogen. Arginine dihydrolase, urease, β-glucosidase, β-galactosidase and protease are positive in API 20NE test. Alkaline phosphatase, esterase C4, esterase C8, lipase C14, leucine arylamidase, valine arylamidase, cystine arylamidase, trypsin, chymotrypsin, acid phosphatase, naphthol-AS-BI-phosphohydrolase and N-acetyl-β-glucosaminidase are positive in API ZYM test. D-maltose, D-trehalose, D-cellobiose, gentiobiose, sucrose, turanose, D-salicin, N-acetyl-β-glucosamine, N-acetyl-β-galactosamine, N-acetyl neuraminic acid, D-glucose, D-fucose, L-fucose, inosine, D-arabitol, myo-inositol, glycerol, glucose-6-PO4, D-fructose-6-PO4, gelatin, L-aspartic acid, L-glutamic acid, L-histidine, L-pyroglutamic acid, L-serine, D-galacturonic acid, D-gluconic acid, D-glucuronic acid, glucuronamide, quinic acid, D-saccharic acid, D-lactic acid methyl ester, L-lactic acid, citric acid, α-keto-glutaric acid, D-malic acid, Tween40, γ-amino-butyric acid, β-hydroxy-butyric acid, α-keto-butyric acid, acetic acid and formic acid are oxidized in Biolog GenIII microplate. The cellular fatty acids also contain C14:0, isoC17:0, isoC11:0, C14:1ω7c, isoC13:0. The polar lipids also contain one phospholipid, five unidentified ninhydrin-positive lipids, and three unidentified polar lipids.
Type strain M133T (=MCCC 1K03775T = KCTC 72445T), was isolated from coral Porites lutea, China. Genome accession in GenBank is CP071793, and the DNA G + C content is 60.2 mol%.
Conclusion
Species belonging to phylum Acidobacteria are ubiquitous among animals, yet their ecological role remains unclear. In this study, comparative genomic and high-throughput sequencing analyses indicated that Acanthopleuribacteraceae is a distinct branch of the phylum Acidobacteria, features a large genome that harbors genes associated with secondary metabolic production, tolerance to oxygen fluctuation, and intimate animal association. Phylogenetic, phenotypic, and taxonomic analysis indicated that strain M133T represents a new species in a new genus in the family Acanthopleuribacteraceae. To date, strain M133T and A. pedis KCTC 12899T are the only two acidobacterial isolates obtained from animals, which are not sufficient to disclose the roles of acidobacteria in animals. Therefore, pure cultures of acidobacteria from animals are urgently needed. For strain M133T and A. pedis KCTC 12899T, their exact ecological niches, community volume, and the form and material they interact with animals need further study.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, CP071793; https://www.ncbi.nlm.nih.gov/genbank/, JAFREP000000000.
Author Contributions
GW and KY derived the idea for the whole project. YL and JFL performed the experiments. GW performed the bioinformatics analysis and wrote the manuscript. BC and WH provided the samples. BC, HS, and JYL joined discussion of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the National Key Research and Development Program of China (2018YFD0900803); the National Natural Sciences Foundation of China (42090041, 42030502, and 41866004); and the Science and Technology Project of Guangxi (AD17129063, AA17204074, and AA18242026).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful to professor Aharon Oren for his suggestions in new species nomenclature. We also acknowledge to editor and reviewers for your constructive comments in increasing the quality of this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.778535/full#supplementary-material
References
Anders, S., Pyl, P. T., and Huber, W. (2015). HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. doi: 10.1093/bioinformatics/btu638
Ardui, S., Ameur, A., Vermeesch, J. R., and Hestand, M. S. (2018). Single molecule real-time (SMRT) sequencing comes of age: applications and utilities for medical diagnostics. Nucleic Acids Res. 46, 2159–2168. doi: 10.1093/nar/gky066
Arkin, A. P., Cottingham, R. W., Henry, C. S., Harris, N. L., Stevens, R. L., Maslov, S., et al. (2018). KBase: the United States Department of Energy Systems Biology Knowledgebase. Nat. Biotechnol. 36, 566–569. doi: 10.1038/nbt.4163
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Baquiran, J., and Conaco, C. (2018). Sponge-microbe partnerships are stable under eutrophication pressure from mariculture. Mar. Pollut Bull. 136, 125–134. doi: 10.1016/j.marpolbul.2018.09.011
Barco, R. A., Garrity, G. M., Scott, J. J., Amend, J. P., Nealson, K. H., and Emerson, D. (2020). A Genus Definition for Bacteria and Archaea Based on a Standard Genome Relatedness Index. mBio 11, e02475–19. doi: 10.1128/mBio.02475-19
Barns, S. M., Takala, S. L., and Kuske, C. R. (1999). Wide distribution and diversity of members of the bacterial kingdom Acidobacterium in the environment. Appl. Environ. Microbiol. 65, 1731–1737. doi: 10.1128/AEM.65.4.1731-1737.1999
Beliaev, A. S., Saffarini, D. A., McLaughlin, J. L., and Hunnicutt, D. (2001). MtrC, an outer membrane decahaem c cytochrome required for metal reduction in Shewanella putrefaciens MR-1. Mol. Microbiol. 39, 722–730. doi: 10.1046/j.1365-2958.2001.02257.x
Belova, S. E., Ravin, N. V., Pankratov, T. A., Rakitin, A. L., Ivanova, A. A., Beletsky, A. V., et al. (2018). Hydrolytic Capabilities as a Key to Environmental Success: chitinolytic and Cellulolytic Acidobacteria From Acidic Sub-arctic Soils and Boreal Peatlands. Front. Microbiol. 9:2775. doi: 10.3389/fmicb.2018.02775
Benjdia, A., Leprince, J., Guillot, A., Vaudry, H., Rabot, S., and Berteau, O. (2007). Anaerobic sulfatase-maturating enzymes: radical SAM enzymes able to catalyze in vitro sulfatase post-translational modification. J. Am. Chem. Soc. 129, 3462–3463. doi: 10.1021/ja067175e
Benjdia, A., Martens, E. C., Gordon, J. I., and Berteau, O. (2011). Sulfatases and a radical S-adenosyl-L-methionine (AdoMet) enzyme are key for mucosal foraging and fitness of the prominent human gut symbiont, Bacteroides thetaiotaomicron. J. Biol. Chem. 286, 25973–25982. doi: 10.1074/jbc.M111.228841
Bertelli, C., Laird, M. R., Williams, K. P., Simon Fraser University Research Computing Group, Lau, B. Y., Hoad, G., et al. (2017). IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45, W30–W35. doi: 10.1093/nar/gkx343
Besemer, J., Lomsadze, A., and Borodovsky, M. (2001). GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 29, 2607–2618. doi: 10.1093/nar/29.12.2607
Blin, K., Shaw, S., Kloosterman, A. M., Charlop-Powers, Z., van Wezel, G. P., Medema, M. H., et al. (2021). antiSMASH 6.0: improving cluster detection and comparison capabilities. Nucleic Acids Res. 49, W29–W35. doi: 10.1093/nar/gkab335
Boutanaev, A. M., and Osbourn, A. E. (2018). Multigenome analysis implicates miniature inverted-repeat transposable elements (MITEs) in metabolic diversification in eudicots. Proc. Natl. Acad. Sci. U. S. A. 115, E6650–E6658. doi: 10.1073/pnas.1721318115
Bush, P. K. (2020). Accurate, automatic annotation of peptidases with hotpep-protease. Green Chem. Eng. 1, 124–130. doi: 10.1016/j.gce.2020.11.008
Cai, X., Challinor, V. L., Zhao, L., Reimer, D., Adihou, H., Grün, P., et al. (2017). Biosynthesis of the Antibiotic Nematophin and Its Elongated Derivatives in Entomopathogenic Bacteria. Org. Lett. 19, 806–809. doi: 10.1021/acs.orglett.6b03796
Challacombe, J., and Kuske, C. (2012). Mobile genetic elements in the bacterial phylum Acidobacteria. Mob. Genet. Elements 2, 179–183. doi: 10.4161/mge.21943
Challacombe, J. F., Eichorst, S. A., Hauser, L., Land, M., Xie, G., and Kuske, C. R. (2011). Biological consequences of ancient gene acquisition and duplication in the large genome of Candidatus Solibacter usitatus Ellin6076. PLoS One 6:e24882. doi: 10.1371/journal.pone.0024882
Chanal, A., Chapon, V., Benzerara, K., Barakat, M., Christen, R., Achouak, W., et al. (2006). The desert of Tataouine: an extreme environment that hosts a wide diversity of microorganisms and radiotolerant bacteria. Environ. Microbiol. 8, 514–525. doi: 10.1111/j.1462-2920.2005.00921.x
Chen, B., Yu, K., Liao, Z., Yu, X., Qin, Z., Liang, J., et al. (2021). Microbiome community and complexity indicate environmental gradient acclimatisation and potential microbial interaction of endemic coral holobionts in the South China Sea. Sci. Total Environ. 765:142690. doi: 10.1016/j.scitotenv.2020.142690
Coates, J. D., Ellis, D. J., Gaw, C. V., and Lovley, D. R. (1999). Geothrix fermentans gen. nov., sp. nov., a novel Fe(III)-reducing bacterium from a hydrocarbon-contaminated aquifer. Int. J. Syst. Bacteriol. 49, 1615–1622. doi: 10.1099/00207713-49-4-1615
Crowe, M. A., Power, J. F., Morgan, X. C., Dunfield, P. F., Lagutin, K., Rijpstra, W., et al. (2014). Pyrinomonas methylaliphatogenes gen. nov., sp. nov., a novel group 4 thermophilic member of the phylum Acidobacteria from geothermal soils. Int. J. Syst. Evol. Microbiol. 64, 220–227. doi: 10.1099/ijs.0.055079-0
de Castro, V. H., Schroeder, L. F., Quirino, B. F., Kruger, R. H., and Barreto, C. C. (2013). Acidobacteria from oligotrophic soil from the Cerrado can grow in a wide range of carbon source concentrations. Can. J. Microbiol. 59, 746–753. doi: 10.1139/cjm-2013-0331
de Chaves, M. G., Silva, G., Rossetto, R., Edwards, R. A., Tsai, S. M., and Navarrete, A. A. (2019). Acidobacteria Subgroups and Their Metabolic Potential for Carbon Degradation in Sugarcane Soil Amended With Vinasse and Nitrogen Fertilizers. Front. Microbiol. 10:1680. doi: 10.3389/fmicb.2019.01680
Dedysh, S. N., Kulichevskaya, I. S., Serkebaeva, Y. M., Mityaeva, M. A., Sorokin, V. V., Suzina, N. E., et al. (2012). Bryocella elongata gen. nov., sp. nov., a member of subdivision 1 of the Acidobacteria isolated from a methanotrophic enrichment culture, and emended description of Edaphobacter aggregans Koch 2008. Int. J. Syst. Evol. Microbiol. 62, 654–664. doi: 10.1099/ijs.0.031898-0
Eichorst, S. A., Breznak, J. A., and Schmidt, T. M. (2007). Isolation and characterization of soil bacteria that define Terriglobus gen. nov., in the phylum Acidobacteria. Appl. Environ. Microbiol. 73, 2708–2717. doi: 10.1128/AEM.02140-06
Eichorst, S. A., Trojan, D., Roux, S., Herbold, C., Rattei, T., and Woebken, D. (2018). Genomic insights into the Acidobacteria reveal strategies for their success in terrestrial environments. Environ. Microbiol. 20, 1041–1063. doi: 10.1111/1462-2920.14043
Falagán, C., Foesel, B., and Johnson, B. (2017). Acidicapsa ferrireducens sp. nov., Acidicapsa acidiphila sp. nov., and Granulicella acidiphila sp. nov.: novel acidobacteria isolated from metal-rich acidic waters. Extremophiles 21, 459–469. doi: 10.1007/s00792-017-0916-4
Felsenstein, J. (1981). Evolutionary trees from DNA sequences: a maximum likelihood approach. J. Mol. Evol. 17, 368–376. doi: 10.1007/BF01734359
Frost, L. S., Leplae, R., Summers, A. O., and Toussaint, A. (2005). Mobile genetic elements: the agents of open source evolution. Nat. Rev. Microbiol. 3, 722–732. doi: 10.1038/nrmicro1235
Fukunaga, Y., Kurahashi, M., Yanagi, K., Yokota, A., and Harayama, S. (2008). Acanthopleuribacter pedis gen. nov., sp. nov., a marine bacterium isolated from a chiton, and description of Acanthopleuribacteraceae fam. nov., Acanthopleuribacterales ord. nov., Holophagaceae fam. nov., Holophagales ord. nov. and Holophagae classis nov. in the phylum ‘Acidobacteria’. Int. J. Syst. Evol. Microbiol. 58, 2597–2601. doi: 10.1099/ijs.0.65589-0
Gallagher, K. A., and Jensen, P. R. (2015). Genomic insights into the evolution of hybrid isoprenoid biosynthetic gene clusters in the MAR4 marine streptomycete clade. BMC Genomics 16:960. doi: 10.1186/s12864-015-2110-3
Gerth, K., Washausen, P., Hofle, G., Irschik, H., and Reichenbach, H. (1996). The jerangolids: a family of new antifungal compounds from Sorangium cellulosum (Myxobacteria). Production, physico-chemical and biological properties of jerangolid A. J. Antibiot. 49, 71–75. doi: 10.7164/antibiotics.49.71
Ghate, S. D., Shastry, R. P., Arun, A. B., and Rekha, P. D. (2021). Unraveling the bacterial community composition across aquatic sediments in the Southwestern coast of India by employing high-throughput 16S rRNA gene sequencing. Reg. Stud. Mar. Sci. 46:101890. doi: 10.1016/j.rsma.2021.101890
Gonçalves, O. S., and Santana, M. F. (2021). The coexistence of monopartite integrative and conjugative elements in the genomes of Acidobacteria. Gene 777:145476. doi: 10.1016/j.gene.2021.145476
Harrington, D. J. (1996). Bacterial collagenases and collagen-degrading enzymes and their potential role in human disease. Infect. Immun. 64, 1885–1891. doi: 10.1128/iai.64.6.1885-1891.1996
Hausmann, B., Pelikan, C., Herbold, C. W., Köstlbacher, S., Albertsen, M., Eichorst, S. A., et al. (2018). Peatland Acidobacteria with a dissimilatory sulfur metabolism. ISME J. 12, 1729–1742. doi: 10.1038/s41396-018-0077-1
Imamura, T., Murakami, Y., and Nitta, H. (2017). Aeromonas sobria serine protease (ASP): a subtilisin family endopeptidase with multiple virulence activities. Biol. Chem. 398, 1055–1068. doi: 10.1515/hsz-2016-0344
Ivanova, A. A., Zhelezova, A. D., Chernov, T. I., and Dedysh, S. N. (2020). Linking ecology and systematics of acidobacteria: distinct habitat preferences of the Acidobacteriia and Blastocatellia in tundra soils. PLoS One 15:e0230157. doi: 10.1371/journal.pone.0230157
Jäckle, O., Seah, B., Tietjen, M., Leisch, N., Liebeke, M., Kleiner, M., et al. (2019). Chemosynthetic symbiont with a drastically reduced genome serves as primary energy storage in the marine flatworm Paracatenula. Proc. Natl. Acad. Sci. U. S. A. 116, 8505–8514. doi: 10.1073/pnas.1818995116
Jahn, M. T., Arkhipova, K., Markert, S. M., Stigloher, C., Lachnit, T., Pita, L., et al. (2019). A Phage Protein Aids Bacterial Symbionts in Eukaryote Immune Evasion. Cell Host Microbe 26, 542–550.e5. doi: 10.1016/j.chom.2019.08.019
Kalam, S., Basu, A., Ahmad, I., Sayyed, R. Z., El-Enshasy, H. A., Dailin, D. J., et al. (2020). Recent Understanding of Soil Acidobacteria and Their Ecological Significance: a Critical Review. Front. Microbiol. 11:580024. doi: 10.3389/fmicb.2020.580024
Kanehisa, M., Goto, S., Kawashima, S., Okuno, Y., and Hattori, M. (2004). The KEGG resource for deciphering the genome. Nucleic Acids Res. 32, D277–D280. doi: 10.1093/nar/gkh063
Kielak, A. M., Cipriano, M. A., and Kuramae, E. E. (2016). Acidobacteria strains from subdivision 1 act as plant growth-promoting bacteria. Arch. Microbiol. 198, 987–993. doi: 10.1007/s00203-016-1260-2
Kim, D., Park, S., and Chun, J. (2021). Introducing EzAAI: a pipeline for high throughput calculations of prokaryotic average amino acid identity. J. Microbiol. 59, 476–480. doi: 10.1007/s12275-021-1154-0
Kishimoto, N., Kosako, Y., and Tano, T. (1991). Acidobacterium capsulatum gen. nov., sp. nov.: an acidophilic chemoorganotrophic bacterium containing menaquinone from acidic mineral environment. Curr. Microbiol. 22, 1–7. doi: 10.1007/BF02106205
Kong, L., Lu, A., Guan, J., Yang, B., Li, M., Hillyer, J. F., et al. (2015). Thermolysin damages animal life through degradation of plasma proteins enhanced by rapid cleavage of serpins and activation of proteases. Arch. Insect Biochem. Physiol. 88, 64–84. doi: 10.1002/arch.21178
Konstantinidis, K. T., and Tiedje, J. M. (2005). Towards a genome-based taxonomy for prokaryotes. J. Bacteriol. 187, 6258–6264. doi: 10.1128/JB.187.18.6258-6264.2005
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Kulichevskaya, I. S., Suzina, N. E., Liesack, W., and Dedysh, S. N. (2010). Bryobacter aggregatus gen. nov., sp. nov., a peat-inhabiting, aerobic chemo-organotroph from subdivision 3 of the Acidobacteria. Int. J. Syst. Evol. Microbiol. 60, 301–306. doi: 10.1099/ijs.0.013250-0
Kulichevskaya, I. S., Suzina, N. E., Rijpstra, W., Damsté, J., and Dedysh, S. N. (2014). Paludibaculum fermentans gen. nov., sp. nov., a facultative anaerobe capable of dissimilatory iron reduction from subdivision 3 of the Acidobacteria. Int. J. Syst. Evol. Microbiol. 64, 2857–2864. doi: 10.1099/ijs.0.066175-0
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Łagowski, D., Gnat, S., Nowakiewicz, A., and Osińska, M. (2021). Assessment of the subtilisin gene profile in Trichophyton verrucosum isolated from human and animal dermatophytoses in two-stage multiplex PCR. J. Appl. Microbiol. 131, 300–306. doi: 10.1111/jam.14942
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Laport, M. S., Pinheiro, U., and Rachid, C. (2019). Freshwater Sponge Tubella variabilis Presents Richer Microbiota Than Marine Sponge Species. Front. Microbiol. 10:2799. doi: 10.3389/fmicb.2019.02799
Leray, M., and Knowlton, N. (2015). DNA barcoding and metabarcoding of standardized samples reveal patterns of marine benthic diversity. Proc. Natl. Acad. Sci. U. S. A. 112, 2076–2081. doi: 10.1073/pnas.1424997112
Liang, J., Yu, K., Wang, Y., Huang, X., Huang, W., Qin, Z., et al. (2017). Distinct bacterial communities associated with massive and branching scleractinian corals and potential linkages to coral susceptibility to thermal or cold stress. Front. Microbiol. 8:979. doi: 10.3389/fmicb.2017.00979
Liesack, W., Bak, F., Kreft, J. U., and Stackebrandt, E. (1994). Holophaga foetida gen. nov., sp. nov., a new, homoacetogenic bacterium degrading methoxylated aromatic compounds. Arch. Microbiol. 162, 85–90. doi: 10.1007/BF00264378
Lim, H. J., Lee, E. H., Yoon, Y., Chua, B., and Son, A. (2016). Portable lysis apparatus for rapid single-step DNA extraction of Bacillus subtilis. J. Appl. Microbiol. 120, 379–387. doi: 10.1111/jam.13011
Lin, S. H., and Liao, Y. C. (2013). CISA: contig integrator for sequence assembly of bacterial genomes. PLoS One 8:e60843. doi: 10.1371/journal.pone.0060843
Lladó, S., Benada, O., Cajthaml, T., Baldrian, P., and García-Fraile, P. (2016). Silvibacterium bohemicum gen. nov. sp. nov., an acidobacterium isolated from coniferous soil in the Bohemian Forest National Park. Syst. Appl. Microbiol. 39, 14–19. doi: 10.1016/j.syapm.2015.12.005
Lopez, J. V. (2003). Naturally mosaic operons for secondary metabolite biosynthesis: variability and putative horizontal transfer of discrete catalytic domains of the epothilone polyketide synthase locus. Mol. Genet. Genomics 270, 420–431. doi: 10.1007/s00438-003-0937-9
Losey, N. A., Stevenson, B. S., Busse, H. J., Damsté, J., Rijpstra, W., Rudd, S., et al. (2013). Thermoanaerobaculum aquaticum gen. nov., sp. nov., the first cultivated member of Acidobacteria subdivision 23, isolated from a hot spring. Int. J. Syst. Evol. Microbiol. 63, 4149–4157. doi: 10.1099/ijs.0.051425-0
Luo, R., Liu, B., Xie, Y., Li, Z., Huang, W., Yuan, J., et al. (2012). SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1:18. doi: 10.1186/2047-217X-1-18
Ma, K., Schicho, R. N., Kelly, R. M., and Adams, M. W. (1993). Hydrogenase of the hyperthermophile Pyrococcus furiosus is an elemental sulfur reductase or sulfhydrogenase: evidence for a sulfur-reducing hydrogenase ancestor. Proc. Natl. Acad. Sci. U. S. A. 90, 5341–5344. doi: 10.1073/pnas.90.11.5341
McClure, R., Balasubramanian, D., Sun, Y., Bobrovskyy, M., Sumby, P., Genco, C. A., et al. (2013). Computational analysis of bacterial RNA-Seq data. Nucleic Acids Res. 41:e140. doi: 10.1093/nar/gkt444
McCutcheon, J. P., and Moran, N. A. (2011). Extreme genome reduction in symbiotic bacteria. Nat. Rev. Microbiol. 10, 13–26. doi: 10.1038/nrmicro2670
McFall-Ngai, M., Hadfield, M. G., Bosch, T. C., Carey, H. V., Domazet-Lošo, T., Douglas, A. E., et al. (2013). Animals in a bacterial world, a new imperative for the life sciences. Proc. Natl. Acad. Sci. U. S. A. 110, 3229–3236. doi: 10.1073/pnas.1218525110
Medema, M. H., Kottmann, R., Yilmaz, P., Cummings, M., Biggins, J. B., Blin, K., et al. (2015). Minimum Information about a Biosynthetic Gene cluster. Nat. Chem. Biol. 11, 625–631. doi: 10.1038/nchembio.1890
Meekrathok, P., Bürger, M., Porfetye, A. T., Kumsaoad, S., Aunkham, A., Vetter, I. R., et al. (2021). Structural basis of chitin utilization by a GH20 β-N-acetylglucosaminidase from Vibrio campbellii strain ATCC BAA-1116. Acta Crystallogr. D Struct. Biol. 77, 674–689. doi: 10.1107/S2059798321002771
Mei, X., Wu, C., Zhao, J., Yan, T., and Jiang, P. (2019). Community Structure of Bacteria Associated With Drifting Sargassum horneri, the Causative Species of Golden Tide in the Yellow Sea. Front. Microbiol. 10:1192. doi: 10.3389/fmicb.2019.01192
Melikhova, E. Y., Pullin, R. D., Winter, C., and Donohoe, T. J. (2016). Dehydromicrosclerodermin B and Microsclerodermin J: total Synthesis and Structural Revision. Angew. Chem. Int. Ed. Engl. 55, 9753–9757. doi: 10.1002/anie.201604764
Meron, D., Atias, E., Iasur Kruh, L., Elifantz, H., Minz, D., Fine, M., et al. (2011). The impact of reduced pH on the microbial community of the coral Acropora eurystoma. ISME J. 5, 51–60. doi: 10.1038/ismej.2010.102
Miura, N., Motone, K., Takagi, T., Aburaya, S., Watanabe, S., Aoki, W., et al. (2019). Ruegeria sp. Strains Isolated from the Reef-Building Coral Galaxea fascicularis Inhibit Growth of the Temperature-Dependent Pathogen Vibrio coralliilyticus. Mar. Biotechnol. 21, 1–8. doi: 10.1007/s10126-018-9853-1
Mohr, K. I., Wolf, C., Nübel, U., Szafrańska, A. K., Steglich, M., Hennessen, F., et al. (2018). A polyphasic approach leads to seven new species of the cellulose-decomposing genus Sorangium, Sorangium ambruticinum sp. nov., Sorangium arenae sp. nov., Sorangium bulgaricum sp. nov., Sorangium dawidii sp. nov., Sorangium kenyense sp. nov., Sorangium orientale sp. nov. and Sorangium reichenbachii sp. nov. Int. J. Syst. Evol. Microbiol. 68, 3576–3586. doi: 10.1099/ijsem.0.003034
Morozkina, E. V., and Zvyagilskaya, R. A. (2007). Nitrate reductases: structure, functions, and effect of stress factors. Biochemistry 72, 1151–1160. doi: 10.1134/s0006297907100124
Morris, R. L., and Schmidt, T. M. (2013). Shallow breathing: bacterial life at low O2. Nat. Rev. Microbiol. 11, 205–212. doi: 10.1038/nrmicro2970
Motone, K., Takagi, T., Aburaya, S., Miura, N., Aoki, W., and Ueda, M. (2020). A Zeaxanthin-Producing Bacterium Isolated from the Algal Phycosphere Protects Coral Endosymbionts from Environmental Stress. mBio 11, e01019–19. doi: 10.1128/mBio.01019-19
Myers, J. M., and Myers, C. R. (2001). Role for outer membrane cytochromes OmcA and OmcB of Shewanella putrefaciens MR-1 in reduction of manganese dioxide. Appl. Environ. Microbiol. 67, 260–269. doi: 10.1128/AEM.67.1.260-269.2001
Myers, M. R., and King, G. M. (2016). Isolation and characterization of Acidobacterium ailaaui sp. nov., a novel member of Acidobacteria subdivision 1, from a geothermally heated Hawaiian microbial mat. Int. J. Syst. Evol. Microbiol. 66, 5328–5335. doi: 10.1099/ijsem.0.001516
Na, S. I., Kim, Y. O., Yoon, S. H., Ha, S. M., Baek, I., and Chun, J. (2018). UBCG: up-to-date bacterial core gene set and pipeline for phylogenomic tree reconstruction. J. Microbiol. 56, 280–285. doi: 10.1007/s12275-018-8014-6
Naumoff, D. G. (2016). GH10 Family of Glycoside Hydrolases: structure and Evolutionary Connections. Mol. Biol. 50, 151–160. doi: 10.7868/S0026898415060208
Naumoff, D. G., and Dedysh, S. N. (2012). Lateral gene transfer between the Bacteroidetes and Acidobacteria: the case of α-L-rhamnosidases. FEBS Lett. 586, 3843–3851. doi: 10.1016/j.febslet.2012.09.005
Navarrete, A. A., Venturini, A. M., Meyer, K. M., Klein, A. M., Tiedje, J. M., Bohannan, B. J., et al. (2015). Differential Response of Acidobacteria Subgroups to Forest-to-Pasture Conversion and Their Biogeographic Patterns in the Western Brazilian Amazon. Front. Microbiol. 6:1443. doi: 10.3389/fmicb.2015.01443
O’Connor-Sánchez, A., Rivera-Domínguez, A. J., Santos-Briones, C., López-Aguiar, L. K., Peña-Ramírez, Y. J., and Prieto-Davo, A. (2014). Acidobacteria appear to dominate the microbiome of two sympatric Caribbean Sponges and one Zoanthid. Biol. Res. 47:67. doi: 10.1186/0717-6287-47-67
O’Donnell, J. L., Kelly, R. P., Lowell, N. C., and Port, J. A. (2016). Indexed PCR Primers Induce Template-Specific Bias in Large-Scale DNA Sequencing Studies. PLoS One 11:e0148698. doi: 10.1371/journal.pone.0148698
Offret, C., Desriac, F., Le Chevalier, P., Mounier, J., Jégou, C., and Fleury, Y. (2016). Spotlight on Antimicrobial Metabolites from the Marine Bacteria Pseudoalteromonas: chemodiversity and Ecological Significance. Mar. Drugs 14:129. doi: 10.3390/md14070129
Okamura, K., Kawai, A., Yamada, T., and Hiraishi, A. (2011). Acidipila rosea gen. nov., sp. nov., an acidophilic chemoorganotrophic bacterium belonging to the phylum Acidobacteria. FEMS Microbiol. Lett. 317, 138–142. doi: 10.1111/j.1574-6968.2011.02224.x
Pankratov, T. A., and Dedysh, S. N. (2010). Granulicella paludicola gen. nov., sp. nov., Granulicella pectinivorans sp. nov., Granulicella aggregans sp. nov. and Granulicella rosea sp. nov., acidophilic, polymer-degrading acidobacteria from Sphagnum peat bogs. Int. J. Syst. Evol. Microbiol. 60, 2951–2959. doi: 10.1099/ijs.0.021824-0
Pearman, J. K., Aylagas, E., Voolstra, C. R., Anlauf, H., Villalobos, R., and Carvalho, S. (2019). Disentangling the complex microbial community of coral reefs using standardized Autonomous Reef Monitoring Structures (ARMS). Mol. Ecol. 28, 3496–3507. doi: 10.1111/mec.15167
Pedroni, P., Della Volpe, A., Galli, G., Mura, G. M., Pratesi, C., and Grandi, G. (1995). Characterization of the locus encoding the [Ni-Fe] sulfhydrogenase from the archaeon Pyrococcus furiosus: evidence for a relationship to bacterial sulfite reductases. Microbiology 141, 449–458. doi: 10.1099/13500872-141-2-449
Penttinen, R., Kinnula, H., Lipponen, A., Bamford, J. K. H., and Sundberg, L. (2016). High Nutrient Concentration Can Induce Virulence Factor Expression and Cause Higher Virulence in an Environmentally Transmitted Pathogen. Microb. Ecol. 72, 955–964. doi: 10.1007/s00248-016-0781-1
Perreau, J., and Moran, N. A. (2022). Genetic innovations in animal-microbe symbioses. Nat. Rev. Genet. 23, 23–39. doi: 10.1038/s41576-021-00395-z
Quaiser, A., Ochsenreiter, T., Lanz, C., Schuster, S. C., Treusch, A. H., Eck, J., et al. (2003). Acidobacteria form a coherent but highly diverse group within the bacterial domain: evidence from environmental genomics. Mol. Microbiol. 50, 563–575. doi: 10.1046/j.1365-2958.2003.03707.x
Raina, J. B., Fernandez, V., Lambert, B., Stocker, R., and Seymour, J. R. (2019). The role of microbial motility and chemotaxis in symbiosis. Nat. Rev. Microbiol. 17, 284–294. doi: 10.1038/s41579-019-0182-9
Ricard-Blum, S., and Vallet, S. D. (2016). Proteases decode the extracellular matrix cryptome. Biochimie 122, 300–313. doi: 10.1016/j.biochi.2015.09.016
Richards, G. P., Watson, M. A., Needleman, D. S., Uknalis, J., Boyd, E. F., and Fay, J. P. (2017). Mechanisms for Pseudoalteromonas piscicida-Induced Killing of Vibrios and Other Bacterial Pathogens. Appl. Environ. Microbiol. 83, e00175–17. doi: 10.1128/AEM.00175-17
Robinson, M. D., McCarthy, D. J., and Smyth, G. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi: 10.1093/bioinformatics/btp616
Rosado, P. M., Leite, D., Duarte, G., Chaloub, R. M., Jospin, G., Nunes da Rocha, U., et al. (2019). Marine probiotics: increasing coral resistance to bleaching through microbiome manipulation. ISME J. 13, 921–936. doi: 10.1038/s41396-018-0323-6
Saitou, N., and Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425. doi: 10.1093/oxfordjournals.molbev.a040454
Shulse, C. N., and Allen, E. E. (2011). Widespread occurrence of secondary lipid biosynthesis potential in microbial lineages. PLoS One 6:e20146. doi: 10.1371/journal.pone.0020146
Simpson, J. T., Wong, K., Jackman, S. D., Schein, J. E., Jones, S. J., and Birol, I. (2009). ABySS: a parallel assembler for short read sequence data. Genome Res. 19, 1117–1123. doi: 10.1101/gr.089532.108
Strobel, G., Li, J. Y., Sugawara, F., Koshino, H., Harper, J., and Hess, W. M. (1999). Oocydin A, a chlorinated macrocyclic lactone with potent anti-oomycete activity from Serratia marcescens. Microbiology 145, 3557–3564. doi: 10.1099/00221287-145-12-3557
Swofford, D. L. (1993). PAUP: Phylogenetic analysis using parsimony, version 3.1.1. Champaign: Illinois Natural History Survey.
Takahashi, S., Tomita, J., Nishioka, K., Hisada, T., and Nishijima, M. (2014). Development of a prokaryotic universal primer for simultaneous analysis of Bacteria and Archaea using next-generation sequencing. PLoS One 9:e105592. doi: 10.1371/journal.pone.0105592
Tank, M., and Bryant, D. A. (2015). Chloracidobacterium thermophilum gen. nov., sp. nov.: an anoxygenic microaerophilic chlorophotoheterotrophic acidobacterium. Int. J. Syst. Evol. Microbiol. 65, 1426–1430. doi: 10.1099/ijs.0.000113
Tsaplina, O., Demidyuk, I., Artamonova, T., Khodorkovsky, M., and Khaitlina, S. (2020). Cleavage of the outer membrane protein OmpX by protealysin regulates Serratia proteamaculans invasion. FEBS Lett. 594, 3095–3107. doi: 10.1002/1873-3468.13897
Valentine, R. C., and Valentine, D. L. (2004). Omega-3 fatty acids in cellular membranes: a unified concept. Prog. Lipid Res. 43, 383–402. doi: 10.1016/j.plipres.2004.05.004
Varani, A. M., Siguier, P., Gourbeyre, E., Charneau, V., and Chandler, M. (2011). ISsaga is an ensemble of web-based methods for high throughput identification and semi-automatic annotation of insertion sequences in prokaryotic genomes. Genome Biol. 12:R30. doi: 10.1186/gb-2011-12-3-r30
Vieira, S., Luckner, M., Wanner, G., and Overmann, J. (2017). Luteitalea pratensis gen. nov., sp. nov. a new member of subdivision 6 Acidobacteria isolated from temperate grassland soil. Int. J. Syst. Evol. Microbiol. 67, 1408–1414. doi: 10.1099/ijsem.0.001827
Wang, G., Dang, G., Xu, S., Liu, J., Su, H., Liang, J., et al. (2020). Aliikangiella coralliicola sp. nov., a bacterium isolated from coral Porites lutea, and proposal of Pleioneaceae fam. nov. to accommodate Pleionea and Aliikangiella. Int. J. Syst. Evol. Microbiol. 70, 5880–5887. doi: 10.1099/ijsem.0.004489
Wang, L., Feng, Z., Wang, X., Wang, X., and Zhang, X. (2010). DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26, 136–138. doi: 10.1093/bioinformatics/btp612
Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40:e49. doi: 10.1093/nar/gkr1293
Ward, N. L., Challacombe, J. F., Janssen, P. H., Henrissat, B., Coutinho, P. M., Wu, M., et al. (2009). Three genomes from the phylum Acidobacteria provide insight into the lifestyles of these microorganisms in soils. Appl. Environ. Microbiol. 75, 2046–2056. doi: 10.1128/AEM.02294-08
Wu, S., Ou, H., Liu, T., Wang, D., and Zhao, J. (2018). Structure and dynamics of microbiomes associated with the marine sponge Tedania sp. during its life cycle. FEMS Microbiol. Ecol. 94:5. doi: 10.1093/femsec/fiy055
Wu, S., Zhu, Z., Fu, L., Niu, B., and Li, W. (2011). WebMGA: a customizable web server for fast metagenomic sequence analysis. BMC Genomics 12:444. doi: 10.1186/1471-2164-12-444
Yarza, P., Yilmaz, P., Pruesse, E., Glöckner, F. O., Ludwig, W., Schleifer, K. H., et al. (2014). Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat. Rev. Microbiol. 12, 635–645. doi: 10.1038/nrmicro3330
Yoon, S. H., Ha, S. M., Lim, J. M., Kwon, S. J., and Chun, J. (2017). A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie van Leeuwenhoek 110, 1281–1286. doi: 10.1007/s10482-017-0844-4
Zhang, H., Yohe, T., Huang, L., Entwistle, S., Wu, P., Yang, Z., et al. (2018). dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 46, W95–W101. doi: 10.1093/nar/gky418
Zhang, Y., Wang, X., Zhen, Y., Mi, T., He, H., and Yu, Z. (2017). Microbial Diversity and Community Structure of Sulfate-Reducing and Sulfur-Oxidizing Bacteria in Sediment Cores from the East China Sea. Front. Microbiol. 8:2133. doi: 10.3389/fmicb.2017.02133
Keywords: animal acidobacteria, Acanthopleuribacteraceae, sulfhydrogenase, eicosapentaenoic acid (EPA), Sulfidibacter corallicola
Citation: Wang G, Li Y, Liu J, Chen B, Su H, Liang J, Huang W and Yu K (2022) Comparative Genomics Reveal the Animal-Associated Features of the Acanthopleuribacteraceae Bacteria, and Description of Sulfidibacter corallicola gen. nov., sp., nov. Front. Microbiol. 13:778535. doi: 10.3389/fmicb.2022.778535
Received: 17 September 2021; Accepted: 07 January 2022;
Published: 31 January 2022.
Edited by:
Acacio Aparecido Navarrete, Federal University of Mato Grosso do Sul, BrazilReviewed by:
Cristine Chaves Barreto, Catholic University of Brasília (UCB), BrazilVinicius Abreu, University of São Paulo, Brazil
Copyright © 2022 Wang, Li, Liu, Chen, Su, Liang, Huang and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kefu Yu, kefuyu@scsio.ac.cn