 Fenghua Tian
Fenghua Tian Changtian Li
Changtian Li Yu Li
Yu Li- 1Department of Plant Pathology, College of Agriculture, Guizhou University, Guiyang, China
- 2Engineering Research Center of Chinese Ministry of Education for Edible and Medicinal Fungi, Jilin Agricultural University, Changchun, China
Yuanmo [Sarcomyxa edulis (Y.C. Dai, Niemelä & G.F. Qin) T. Saito, Tonouchi & T. Harada] is an important edible and medicinal mushroom endemic to Northeastern China. Here we report the de novo sequencing and assembly of the S. edulis genome using single-molecule real-time sequencing technology. The whole genome was approximately 35.65 Mb, with a G + C content of 48.31%. Genome assembly generated 41 contigs with an N50 length of 1,772,559 bp. The genome comprised 9,364 annotated protein-coding genes, many of which encoded enzymes involved in the modification, biosynthesis, and degradation of glycoconjugates and carbohydrates or enzymes predicted to be involved in the biosynthesis of secondary metabolites such as terpene, type I polyketide, siderophore, and fatty acids, which are responsible for the pharmacodynamic activities of S. edulis. We also identified genes encoding 1,3-β-glucan synthase and endo-1,3(4)-β-glucanase, which are involved in polysaccharide and uridine diphosphate glucose biosynthesis. Phylogenetic and comparative analyses of Basidiomycota fungi based on a single-copy orthologous protein indicated that the Sarcomyxa genus is an independent group that evolved from the Pleurotaceae family. The annotated whole-genome sequence of S. edulis can serve as a reference for investigations of bioactive compounds with medicinal value and the development and commercial production of superior S. edulis varieties.
Introduction
Sarcomyxa edulis (Y.C. Dai, Niemelä & G.F. Qin) T. Saito, Tonouchi & T. Harada is a fungus that is native to the temperate regions of Northeastern China, Northern Japan, United States, and Russian Far East (Jin et al., 2001; Dai et al., 2003; Saito et al., 2014). It is commonly known as the late oyster and is also called “Yuanmo,” “Huangmo,” or “Dongmo” in Chinese and “Mukitake” in Japanese. The fruiting body of S. edulis is fan-shaped, which is similar to Ganoderma lucidum (“Lingzhi”), but because of its yellow color, it is also known as “Huanglinggu” in China.
Sarcomyxa edulis is an edible and medicinal fungus (Imazeki et al., 1988; Li, 1991; Pan, 1995) that is prized for its nutritional value, unique aroma, delicate flavor, and meaty texture as well as its medicinal properties (Li, 1991; Ma et al., 1991; Pan, 1995; Inafuku et al., 2012; Kim et al., 2012; Inoue et al., 2013; Li et al., 2013, 2016; Zhang et al., 2014). In traditional Chinese medicine, S. edulis is prepared as an alcohol decoction to treat stomachache and other ailments.
Sarcomyxa edulis was first described and assigned to the genus Panellus (Dai et al., 2003) through a comparison with the terribly bitter mushrooms from Finland to the specimens of China. S. edulis is under the same genus as Sarcomyxa serotina. As the type species, the taxonomic status of S. serotina has changed multiple times from the Agaricus genus to Pleurotus, Sarcomyxa, Hohenbuehelia, and finally Panellus (Dai et al., 2003). Phylogenetic studies have shown that S. serotina and S. edulis are excluded from the Panellus clade. A maximum likelihood (ML) tree constructed based on the D1/D2 region of the large subunit of the 28S ribosomal RNA gene showed that the Sarcomyxa genus formed a clade that was independent of Mycena and Panellus (Matheny et al., 2006; Saito et al., 2014). Thus, the classification of Sarcomyxa is controversial.
Sarcomyxa edulis was domesticated in the last decade in China; like Hericium erinaceus, it is an important specialty mushroom cultivated in Northern China, the largest production region. The commercial cultivation of S. edulis is profitable because of high demand. However, production has markedly declined in recent years as a result of diseases and the lack of resistant varieties.
Knowledge of the biology and evolution of S. edulis is limited because molecular-level information from the genome is lacking. The availability of a whole-genome sequence can clarify the taxonomy of S. edulis and aid future breeding efforts to improve this species for commercial cultivation. To this end, in the present study, we sequenced and annotated the genome of a monokaryotic strain of S. edulis and carried out a phylogenetic analysis that included 32 sequenced fungi, with the aim of establishing the taxonomic status of S. edulis and identifying secondary metabolites of medicinal importance.
Materials and Methods
Sarcomyxa edulis Specimens
Sarcomyxa edulis basidiomes were collected from maple wood in Antu County, Jilin Province, China. Specimen 2016092521 was identified by morphologic and molecular analyses (Dai et al., 2003; Saito et al., 2014). The specimen was deposited in the herbarium of the Culture Center of Mycophyta in Jilin Agricultural University under accession number CCMJ18024 (Figure 1). The monokaryotic strain SE1 was germinated from one of the spores of specimen 2016092521 (Choi et al., 1999; Senanayake et al., 2020) and used for whole-genome sequencing. The mycelia of SE1 were cultured on an improved potato dextrose agar medium containing 5% wheat bran for 10 days at 24°C in the dark and collected for genome sequencing.

Figure 1. Fruiting body of Sarcomyxa edulis. (a) Developmental stages of S. edulis. (b) Mature S. edulis.
Whole-Genome Sequencing
Total DNA of S. edulis strain SE1 was extracted using the NuClear Plant Genomic DNA kit (Tiangen Biotech, Beijing, China). The DNA was detected by agarose gel electrophoresis and quantified with a Qubit fluorometer (Thermo Fisher Scientific, Waltham, MA, United States). The whole genome of SE1 was sequenced using PacBio RSII Single Molecule Real-Time (SMRT) technology, which generated 20-kb SMRTbell libraries. High-throughput sequencing on an HiSeq PE150 system (Illumina, San Diego, CA, United States) was carried out to polish the DNA sequence; additionally, paired-end read libraries were obtained by sequencing 350-bp inserts.
Custom Novogene pipelines (Beijing, China) were used to filter the PacBio RSII reads; those of low complexity and quality were filtered out with SMRT portal v3.2.0, with default parameters (Berlin et al., 2015; Koren and Phillippy, 2015), and the resultant clean reads were de novo assembled into a continuous contig with no gaps. Pilon v1.22 was used to polish the assembled long reads with the clean Illumina short reads (Walker et al., 2014). Genome completeness was assessed using Benchmarking Universal Single-Copy Orthologs (v4.1.4; Simão et al., 2015). RepeatMasker version open-4.0.5 was used to detect and annotate dispersed repeat sequences (Saha et al., 2008). Tandem repeat (TR) sequences were analyzed with Tandem Repeat Finder v4.07b (Benson, 1999). rRNA sequences were predicted with rRNAmmer v1.2 (Lagesen et al., 2007), and tRNA genes and tRNA secondary structures were predicted with tRNAscan-SE v1.3 (Lowe and Eddy, 1997). Non-coding RNAs including small (s)RNAs, micro (mi)RNAs, and small nuclear (sn)RNAs were annotated with Rfam (Gardner et al., 2009; Nawrocki et al., 2009) using default parameters (Cui et al., 2016). Ab initio and homology-based gene prediction methods were used to annotate the repeat masked SE1 genome assembly. For homolog-based gene prediction, the protein sequences of Agaricus bisporus (Morin et al., 2012), Coprinopsis cinerea (Stajich et al., 2010), Pleurotus ostreatus (Qu et al., 2016), and Schizophyllum commune (Ohm et al., 2010) were downloaded from the National Center for Biotechnology Information (NCBI) database and aligned using tBLASTn. Subsequently, mapping results were merged, and gene structures were predicted using GeneWise (v. 2.2.0; Birney et al., 2004). Ab initio genes were predicted using Augustus (Stanke et al., 2006), Genescan (Burge and Karlin, 1997), GlimmerHMM (Majoros et al., 2004), and SNAP (Korf, 2004). The protein-coding genes predicted from the ab initio and homology-based gene prediction methods were integrated using GLEAN (Elsik et al., 2007).
Gene Annotation and Functional Analysis
In order to determine the functions of the predicted genes, we compared homologous genes to protein and nucleotide sequences in the general functional BLAST databases. The basic steps of functional annotation were as follows: (1) the predicted gene protein sequences were compared with the functional databases by DIAMOND (with an e-value ≤ 10–5; Buchfink et al., 2014) and (2) filtering of alignment results: for the alignment results of each sequence, select the alignment result with the highest score (default identity ≥ 40%, coverage ≥ 40%) for annotation (Altschul et al., 1990).
Six gene and protein databases were used to predict gene functions including the NCBI Non-redundant Protein Database (nr; Li et al., 2002), Kyoto Encyclopedia of Genes and Genomes (KEGG; Kanehisa et al., 2004, 2006), Gene Ontology (GO; Ashburner et al., 2000), Eukaryotic Orthologous Groups (KOG; Tatusov et al., 2003), and Transporter Classification Database (TCDB; Saier et al., 2014).
Secondary metabolites were annotated using the antiSMASH (fungiSMASH option) database1 and NaPDoS2 (Ziemert et al., 2012; Weber et al., 2015; Blin et al., 2017). To validate the predicted results, the obtained gene clusters were manually verified. Each gene model within the database was searched with BLASTP and TBLASTN algorithms (Grigoriev et al., 2012). For polyketide synthase (PKS)/non-ribosomal peptide synthetase (NRPS) analysis, we selected a query type and entered the sequence data to identify ketosynthase KS and/or condensation C domains using the NaPDos pipeline3. Carbohydrate-active enzymes (CAZymes) were determined using the dbCAN 2 meta server (Zhang et al., 2018). Cytochrome P450 monooxygenase (P450) analysis was performed by searching a reference dataset4 using the BLASTP program (Nelson, 2009).
Phylogenetic Tree Construction
In addition to S. edulis, 31 fungal species in the Basidiomycota and Ascomycota phyla were included in the phylogenetic analysis. Protein sequence data for four taxa were downloaded from the Joint Genome Institute genome database (Bao et al., 2013; Aronsen and Læssøe, 2016; Castanera et al., 2017), and sequences for the other 27 taxa were obtained from the NCBI database (Supplementary Table 1). BLASTP was used to compare the 32 species (Chen et al., 2016). OrthoMCL software (Li et al., 2003) and all-versus-all BLASTP were used with the parameters (e-value ≤ 1e-15, coverage ≥ 50%) to identify orthologous groups. Single-copy orthologs were extracted by Perl script (command line parameters of Gblocks: Gblocks proteins.fasta –b4 = 5 –b5 = h.), and sequence alignments were analyzed with MAFFT v7.158b (Katoh and Standley, 2013). ProTest was used to generate an optimal base substitution model with ML and neighbor-joining (NJ; Castresana, 2000; Miller et al., 2010). Several fossil calibration points fixed were used as references: the most recent common ancestor (MRCA) of P. ostreatus, S. commune, Lentinula edodes, Panellus stipticus, Mycena albidolilacea, Termitomyces sp., Hypsizygus marmoreus, Volvariella volvacea, A. bisporus var. bisporus, A. bisporus var. burnettii, Leucoagaricus sp., C. cinerea, Laccaria bicolor, Hypholoma sublateritium, Hebeloma cylindrosporum, Gymnopilus dilepis, and Galerina marginata diverged 123 million years ago (MYA), while the MRCAs of other taxonomic groups diverged at the following time points: Serpula lacrymans and Coniophora olivacea, 104 MYA; Ganoderma sinens, Postia placenta, Wolfiporia cocos, Trametes versicolor, and Dichomitus squalens, 122 MYA; Tremella mesenterica and Cryptococcus neoformans, 153 MYA; and Ustilago maydis, 273 MYA (Floudas et al., 2012). The final phylogenetic tree was constructed using RAxML (Stamatakis, 2014).
Results
Identification of Specimen 2016092521
Specimen 2016092521 was identified as S. edulis. It is mild-tasting with ventricose cystidia, while the related species S. serotina is very bitter-tasting with hymenial cystidia. The basidiospores of the specimen are slightly longer than in S. serotina, 4.5–6.0 μm × 1.0–1.3 μm (Supplementary Figure 1). Maximum parsimony phylogenetic tree based on ITS gene sequences showed that specimen 2016092521 clusters with S. edulis and is distinct to S. serotina (Supplementary Figure 2).
Features of the S. edulis Genome
In total, 5,318 Mb of clean data were obtained, from which a 35.65-Mb assembly was obtained. The genome consisted of 41 contigs with N50 of 1,772,559 bp, N90 of 554,178 bp, and 48.31% G + C content (Table 1). A BLAST search of repeat sequences yielded 1,371,373 bp, covering 3.85% of the SE1 genome; meanwhile, interspersed nuclear elements and TRs accounted for 1.79 and 2.05% of the genome, respectively. Approximately 1.53% of the genome was long terminal repeats, 0.09% was DNA transposons, and 0.16% was long interspersed nuclear elements. The proportion of TRs in the assembled genome was 1.36%, while minisatellite and microsatellite DNA accounted for 0.60 and 0.01% of the genome, respectively.

Table 1. General features of the Sarcomyxa edulis genome.
Functional Annotation
There were 9,364 gene models predicted in the different databases with a total sequence length of 15,135,900 bp, accounting for 42.45% of the whole genome with an average sequence length of 1,616 bp. We predicted 143 tRNAs (13,216 bp), 14 rRNAs (29,757 bp), 37 snRNAs (3,044 bp), 143 miRNAs (8,449 bp), and six sRNAs (359 bp; Supplementary Table 2). Among the 143 tRNAs, 21 were putative pseudogenes and 122 were anticodon tRNAs corresponding to the 20 common amino acid codons.
Annotation was performed with the NCBI nr, KEGG, KOG, TCDB, GO, P450, Secretory Protein, and CAZy databases (Table 2 and Supplementary Table 3). In the nr database, 4,855 non-redundant proteins found in S. edulis had most matching with six fungi, accounting for 67.6% of total nr predicted proteins (Figure 2). These were similarly annotated in six species—namely, G. marginata (PRJNA207683), L. bicolor (PRJNA19043), P. ostreatus (PRJNA476433), S. lacrymans (PRJNA412961), Moniliophthora roreri (PRJNA279170), and Jaapia argillacea (PRJNA207685).
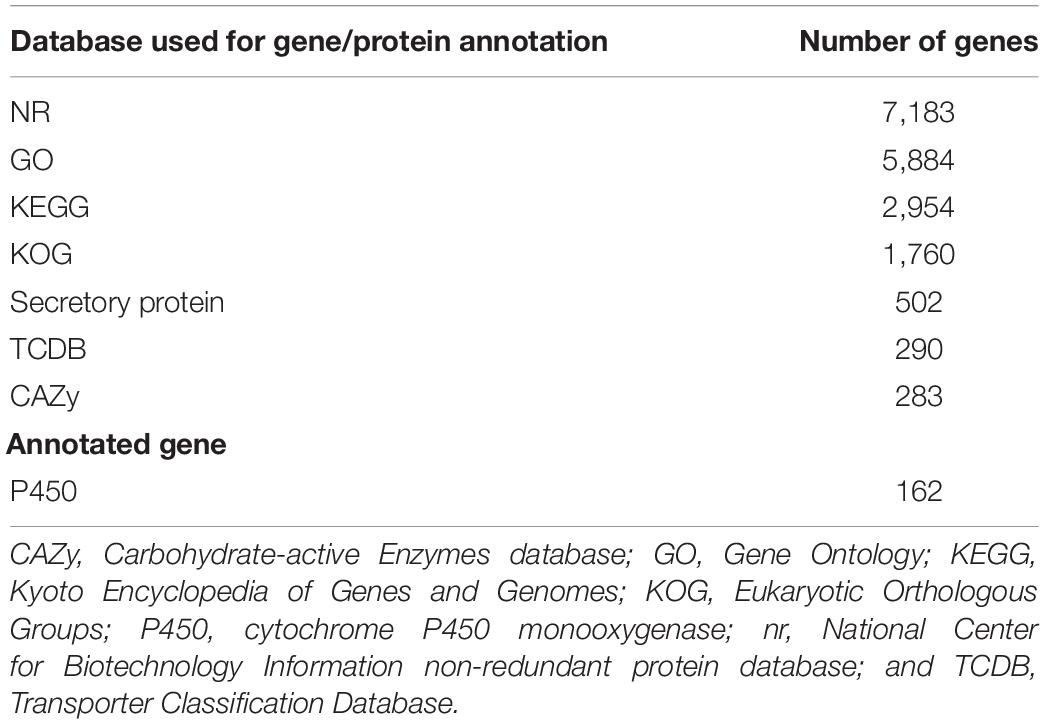
Table 2. Summary of Sarcomyxa edulis gene annotations.
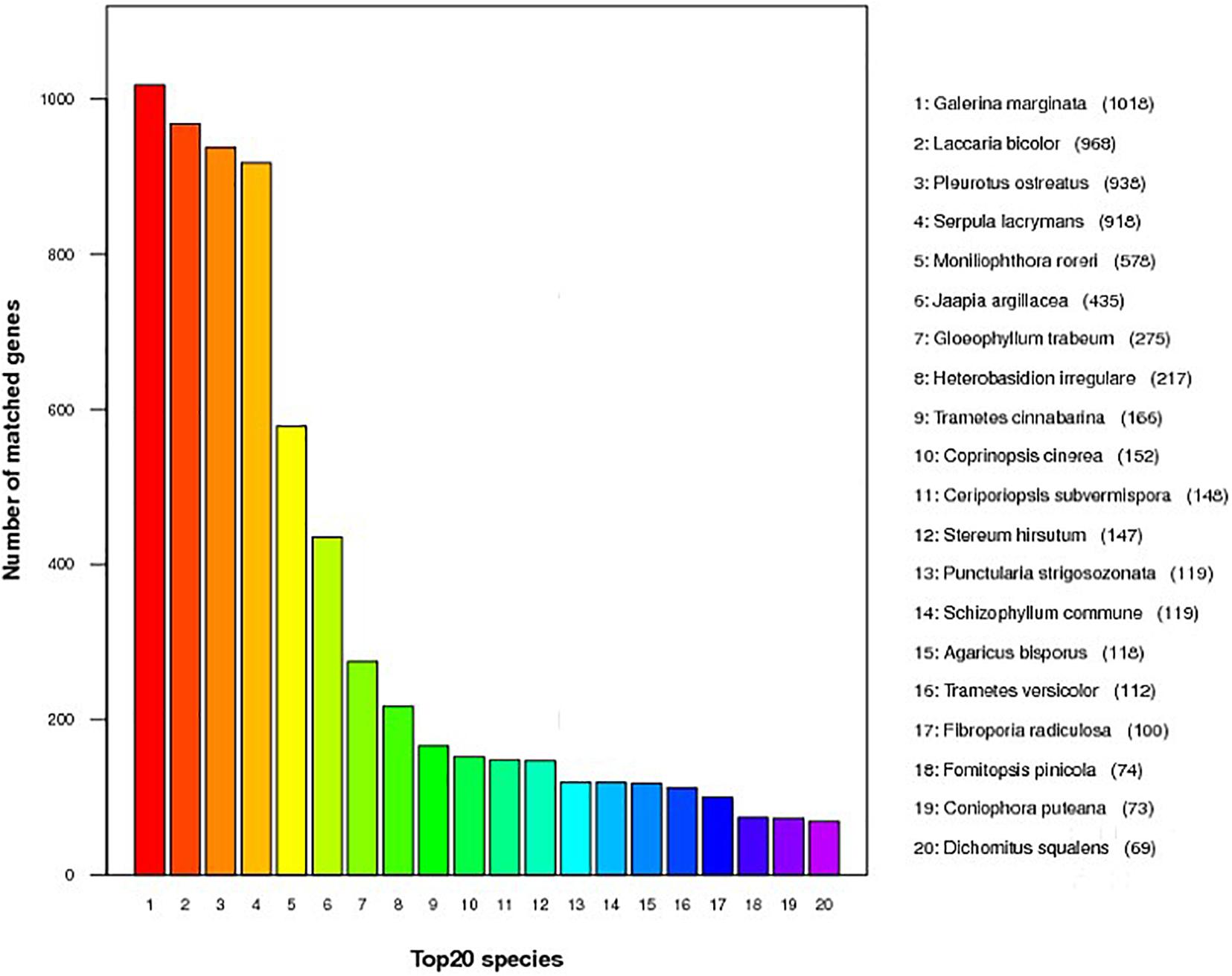
Figure 2. Predicted proteins from Sarcomyxa edulis genome to the National Center for Biotechnology Information non-redundant protein database among different fungal species.
We assigned 1,760 proteins (18.80% of 9,364, the total annotated predicted proteins) to NCBI KOG categories (Figure 3). The “posttranslational modification, protein turnover, chaperones” category had the most enriched genes (230), followed by “general function prediction only” (190), “translation, ribosomal structure and biogenesis” (188), “energy production and conversion” (163), “amino acid transport and metabolism” (125), “RNA processing and modification” (124), “signal transduction mechanisms” (113), and “intracellular trafficking, secretion, and vesicular transport” (106). The representation of genes related to protein and energy metabolism could reflect the capacity of S. edulis to absorb and transform nutrients from a variety of substrates.
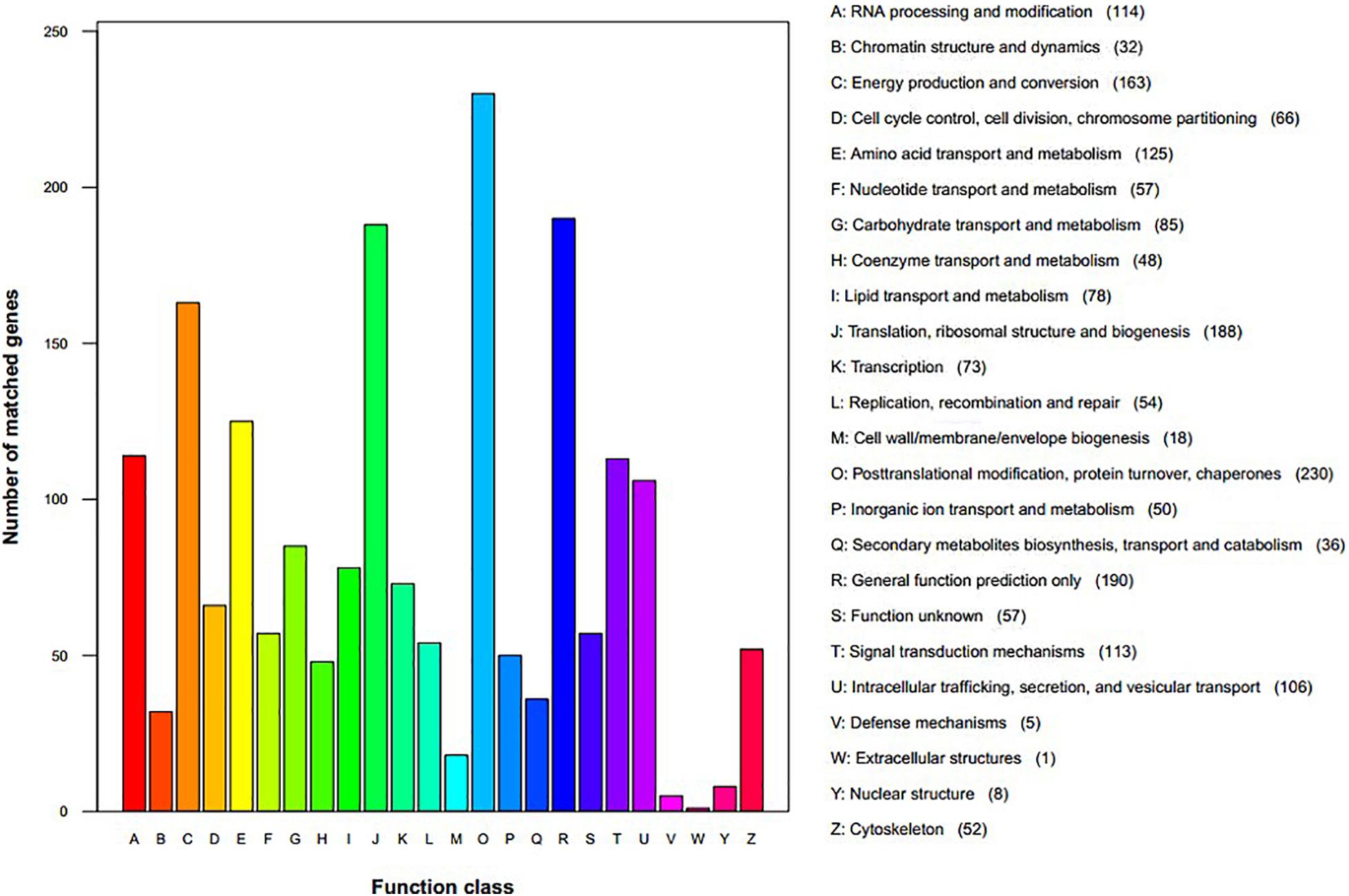
Figure 3. Eukaryotic Orthologous Groups functional classification of Sarcomyxa edulis proteins.
We mapped the predicted genes to the KEGG database and assigned functional classifications to 2,954 (31.55%, 9,364 gene models; Figure 4). Some categories related to metabolism and biosynthesis were highly enriched, including “purine metabolism,” “oxidative phosphorylation,” “butanoate metabolism,” “fructose and mannose metabolism,” “starch and sucrose metabolism,” “amino sugar and nucleotide sugar metabolism,” “arginine and proline metabolism,” “pyrimidine metabolism,” and “chloroalkane and chloroalkene degradation.”
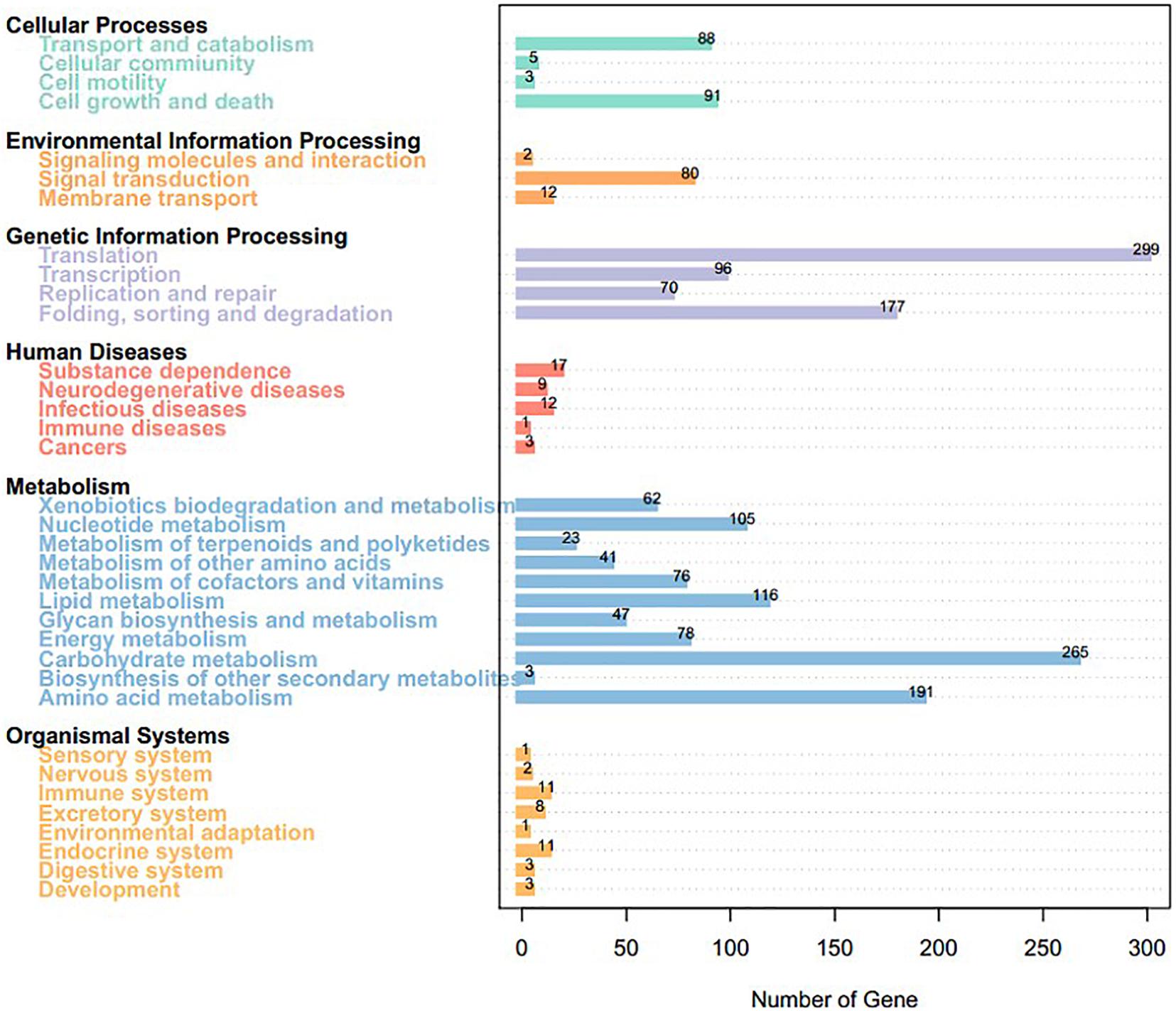
Figure 4. Kyoto Encyclopedia of Genes and Genomes pathway annotation of Sarcomyxa edulis genes.
We used the TCDB to perform protein domain analysis and assigned 290 putative transport proteins to seven functional classes including “accessory factors involved in transport,” “channels/pores,” “electrochemical potential-driven transporters,” “group translocators,” “incompletely characterized transport systems,” “primary active transporters,” and “transmembrane electron carriers” (Figure 5). The top two enriched categories were “porters (uniporters, symporters, antiporters)” and “P–P bond hydrolysis-driven transporters.”
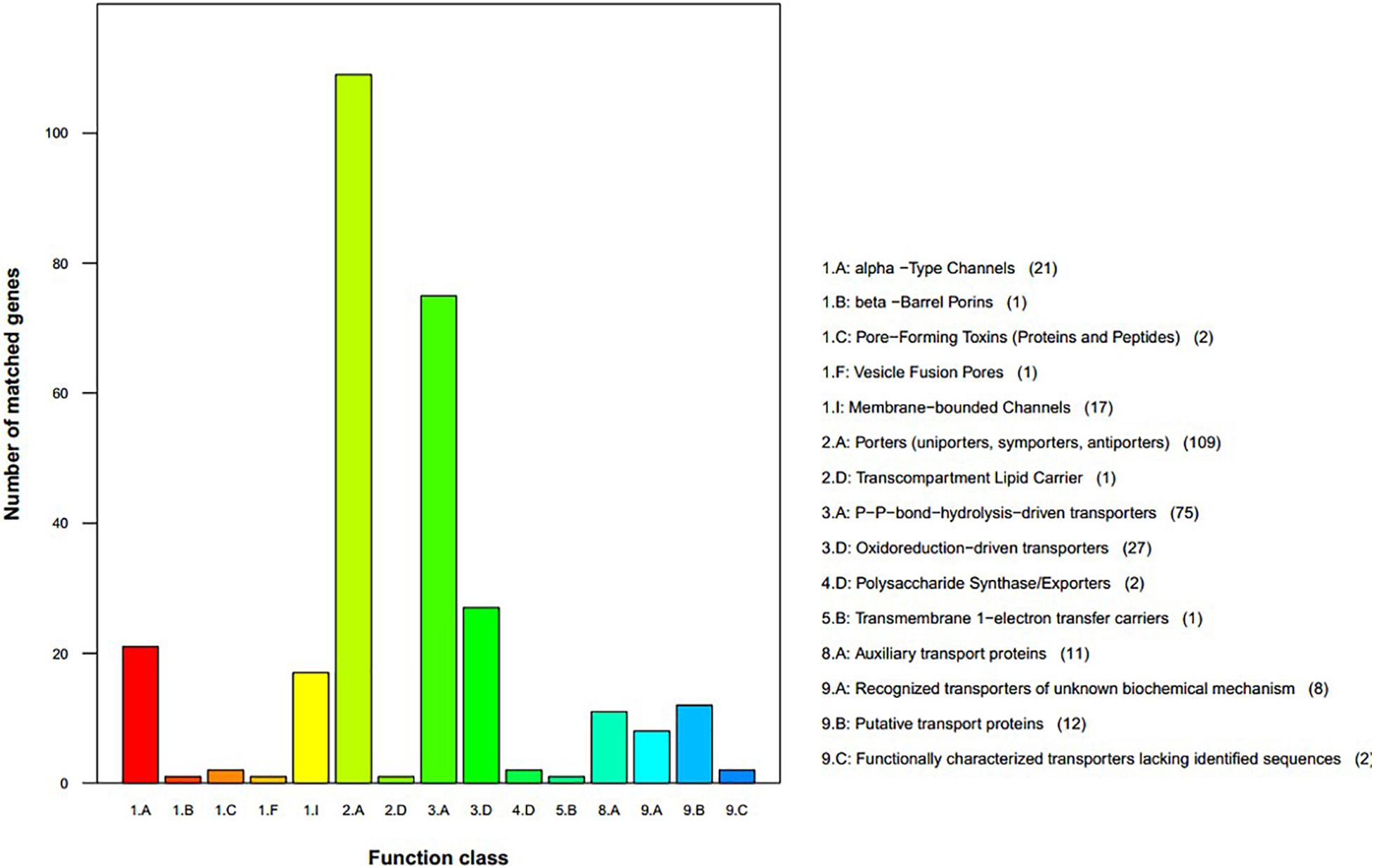
Figure 5. Transporter Classification Database functional annotation of Sarcomyxa edulis genes.
In terms of GO functional classes, we predicted 5,884 proteins that accounted for 62.84% of the total predicted proteins. The most highly enriched GO terms in S. edulis were “cell,” “cell part,” “cellular process,” “catalytic activity,” “metabolic process,” and “binding” (Figure 6).
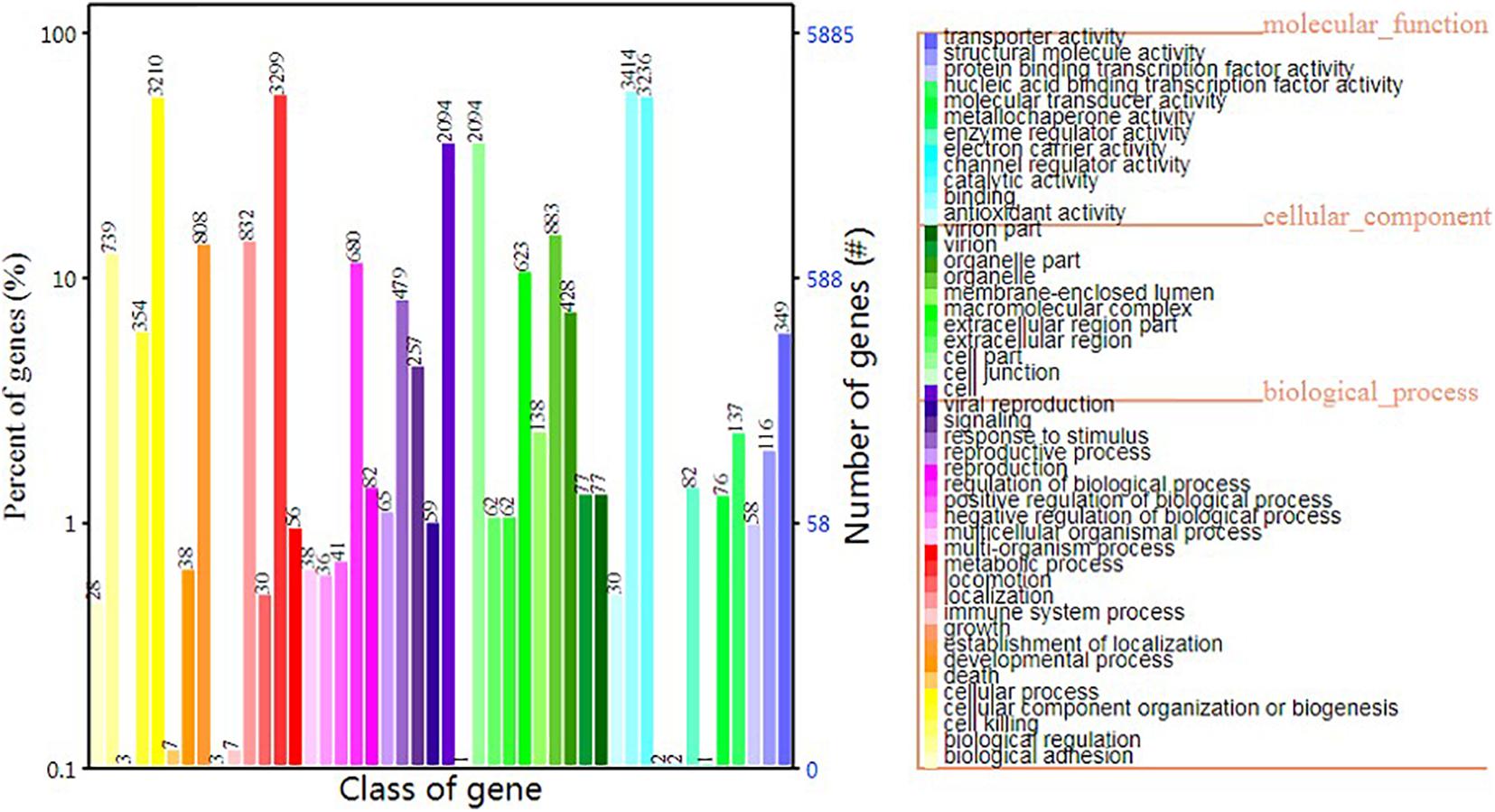
Figure 6. Gene Ontology functional annotation of Sarcomyxa edulis genes.
Cytochrome P450 (CYP) is a superfamily of hemoproteins that use heme as a cofactor. CYPs have various substrates in different enzymatic reactions and are present in all kingdoms. We identified 83 putative CYP genes in S. edulis through a BLAST search and classified them into 34 families (Supplementary Table 4). The CYP5144 family had the highest number of enriched genes (14), followed by CYP5037 (13).
The CAZymes play important roles in the degradation of renewable lignocelluloses to provide carbohydrates for fungal growth, development, and reproduction (Xie et al., 2018). We determined CAZymes in the SE1 genome using the dbCAN 2 meta server. A total of 313 CAZyme-encoding gene models were assigned, including 93 superfamilies, viz., six carbohydrate esterases, 41 glycoside hydrolases, 23 glycosyltransferases, 11 carbohydrate-binding modules, eight auxiliary activities, and four polysaccharide lyases (Table 3). There were 63 AA (auxiliary activities) genes in the SE1 genome, including 13 AA1 (multicopper oxidase), 10 AA2 (lignin-modifying peroxidase), 20 AA3 (glucose-methanol-choline oxidoreductase including cellobiose dehydrogenase, arylalcohol oxidase/glucose oxidase, alcohol oxidase, and pyranose oxidase), five AA5 (copper radical oxidase), two AA6 (1,4-benzoquinone reductase), one AA8 (cellobiose dehydrogenase), and 11 AA9 (lytic polysaccharide monooxygenase) genes.
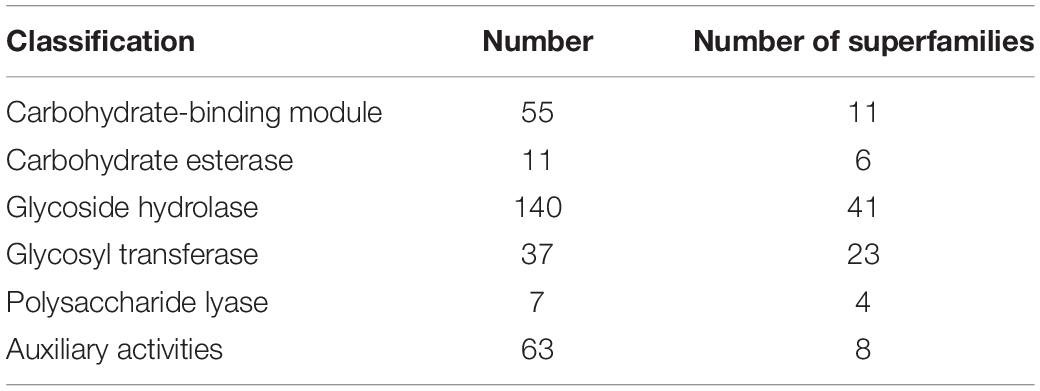
Table 3. Carbohydrate-active enzyme annotation results.
We further investigated genes involved in the biosynthesis of secondary metabolites in S. edulis based on those identified in previous studies and found genes encoding fatty acid synthetases, NRPS, PKS, siderophore synthetases, and terpene synthases (Schwarzer et al., 2003; Finking and Marahiel, 2004; Sims and Schmidt, 2008; Chen et al., 2012; Lackner et al., 2012; Quin et al., 2014) that were often in contiguous gene clusters (Keller et al., 2006). Using the fungiSMASH database, we identified 39 secondary metabolite gene clusters in the SE1 genome, including four terpenes, one NRPS, one T1PKS, one siderophore, and one fatty acid gene cluster (Supplementary Table 5). Unlike primary metabolism, secondary metabolism does not participate in the growth and development of an organism but instead promotes its survival in a given environment, for example, by enhancing the defense response to pathogens. A Natural Product Domain Seeker analysis identified two C domains (C2 bacitracin_DCL and C1 cyclosporin_dual) and four KS domains (naphthopyrone_iterative, avermectin_modular, myxothiazol_modular, and epothilone_modular) in the SE1 genome.
Phylogenetic Analysis of S. edulis
A phylogenetic tree was constructed based on single-copy orthologous protein genes from 32 species of fungus, including 30 from the phylum Basidiomycota and two from the phylum Ascomycota serving as outgroups (Figure 7). The result showed that the Sarcomyxa genus formed a distinct clade to Panellus and Mycena in the Mycenaceae family. The topology suggested that Sarcomyxa was also independent of Pleurotaceae. Fungi in the Basidiomycota and Polyporales clades were clearly separated from those in the Agaricales clade, with Polyporales diverging before Agaricales. Notably, the Agaricales order followed a certain evolutionary pattern from “on log” to “on ground” growth.
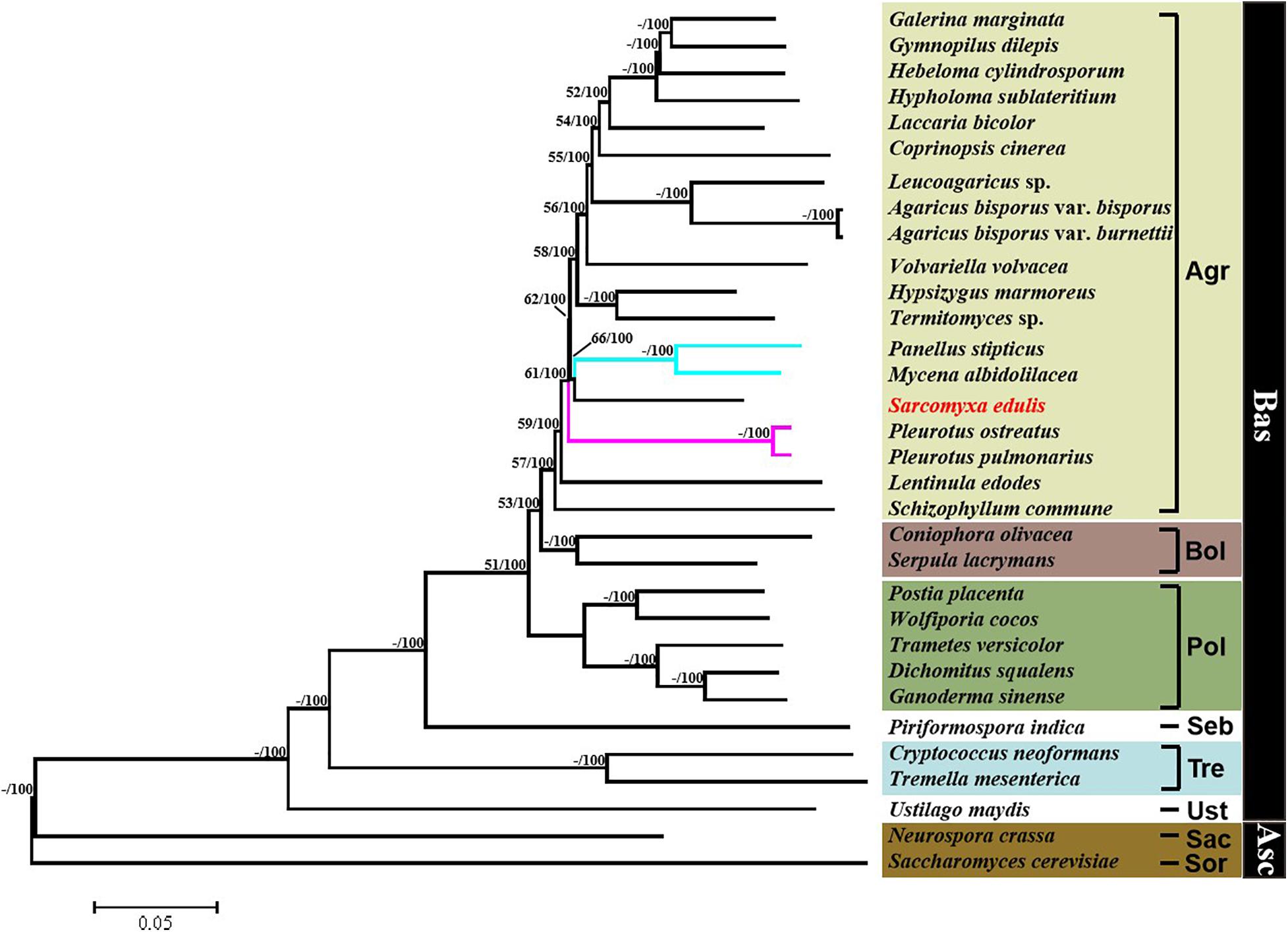
Figure 7. Phylogenetic tree of Sarcomyxa edulis and 31 other fungal species. Maximum likelihood and neighbor-joining above 50% were placed close to topological nodes and separated by “/”. The bootstrap values below 50% were labeled with “-”. Asc, Ascomycota; Bas, Basidiomycota; Sor, Sordariales; Sac, Saccharomycetales; Ust, Ustilaginales; Tre, Tremellales; Seb, Sebacinales; Pol, Polyporales; Bol, Boletales; and Aga, Agaricales.
Discussion
CAZyme Analysis
GHs, GTs, PLs, CEs, and AAs—which catalyze the modification, biosynthesis, or degradation of glycoconjugates and carbohydrates—were the main CAZymes in the SE1 genome. CMBs were non-catalytic modules that appended to the enzymes stated above. There were twice as many GH genes as there were GT genes; this may be related to the lignocellulose degradation capacity of S. edulis that is necessary for its survival. Polysaccharide decomposition is more important for S. edulis than anabolism. S. edulis had fewer genes related to initial lignin degradation than the average number present in Basidiomycota fungi (Supplementary Table 6; Yuan et al., 2019). The protein products of CE, GH, and PL superfamilies are involved in the breakdown of the polysaccharide portion of plant cell walls, which mainly consists of pectin, cellulose, and hemicellulose and are known as cell wall-degrading enzymes (Ospina-Giraldo et al., 2010; Yap et al., 2014). S. edulis had fewer candidate CAZymes than other edible fungi, but it could make better use of hardwood. Therefore, the mechanism of substance degradation of S. edulis needs to be further explored.
Secondary Metabolite Analysis
Medicinal fungi have been the focus of many pharmacologic studies because of their secondary metabolites, which have antioxidant, antitumor, and antimicrobial properties. S. edulis has shown promising pharmacodynamic activities (Li, 1991; Inafuku et al., 2012; Kim et al., 2012; Inoue et al., 2013; Li et al., 2013, 2016; Zhang et al., 2014). Type I PKSs constitute a family of multifunctional proteins with high molecular weight and multiple catalytic domains that play an important role in the biosynthesis of reducing macrolide polyketides (Fujii et al., 2001). The chemical properties of predicted type I PKS genes in the SE1 genome have been characterized. Two C domains on contig 7—bacitracin and cyclosporine—were identified. Bacitracin is a branched cyclic peptide antibiotic active against Gram-positive bacteria and acts by binding to undecaprenyl pyrophosphate—a lipid carrier of cell wall precursors—and interfering with cell wall and peptidoglycan biosynthesis. Furthermore, bacitracin has been used for the prevention and treatment of skin and ophthalmic diseases (Ishihara et al., 2002; Hiron et al., 2011). As an immunosuppressant that is administered orally or via injection, cyclosporine is used to treat nephrotic syndrome (Cattran et al., 2007), psoriasis (Kumar et al., 2016), and keratoconjunctivitis sicca (dry eyes; Kasper et al., 2017). Four KS domains were also identified: naphthopyrone (KS4 on contig 12), avermectin (KS1 on contig 17), myxothiazol (KS2 on contig 17), and epothilone (KS3 on contig 17). Naphthopyrone prevents advanced glycation end product formation (Lee et al., 2006), while avermectin belongs to a family of macrocyclic lactones and functions as a biological pesticide that is highly active against a variety of nematodes. Avermectin has been widely used as an antiparasitic agent in aquaculture (Sheng et al., 2015). Myxothiazol is an antibiotic that inhibits ubiquinol. Epothilones are a class of water-soluble compounds similar to taxanes and with anticancer potential that has shown greater efficacy than paclitaxel in P-glycoprotein-expressing multidrug-resistant cell lines (Kowalski et al., 1997). At present, research on the active components of S. edulis are mainly focused on polysaccharides and lipids. However, study on PKS has not been reported. Therefore, the active constituents of the mushroom are still waiting to be tapped.
Biosynthesis of Polysaccharides in S. edulis
The major types of bioactive compound with medicinal properties in S. edulis are polysaccharides such as β-D-glucan, (1→6)-β-D-glucose, and (1→3)-β-D-glucan, which have relatively high bioactivity (Ma et al., 1991). The biosynthesis of uridine diphosphate glucose—the precursor of these polysaccharides—is mediated by 1,3beta-glucan synthase and endo-1,3(4)-beta-glucanase. Two GT48 and 20 GH16 were annotated, which were present in S. edulis (Supplementary Table 7). The 20 GH16 genes were mainly predicted to be endo-1,3beta-glucanase (EC 3.2.1.39), endo-1,3(4)-beta-glucanase (EC 3.2.1.6), and endo-beta-1,3-galactanase (EC 3.2.1.181). Compared with edible and medicinal fungi, the number of GH16 in S. edulis is more than that in Auricularia heimuer and W. cocos but less than that in Ganoderma lingzhi. The two GT48 genes were predicted to be 1,3-beta-glucan synthase (EC 2.4.1.34). The same number of genes in GT48 was also reported in those mushrooms that contain high polysaccharide active components and have good medicinal value such as A. heimuer, A. bisporus, G. lingzhi, P. ostreatus, V. volvacea, and W. cocos.
Phylogenetic Analysis of S. edulis
Sarcomyxa edulis is an important edible and medicinal fungus that belongs to the order Agaricus, but the taxonomy of this genus is still controversial. In order to comprehensively analyze the relationship between S. edulis and related species and genera, 32 fungal species from Basidiomycota and Ascomycota were used in the phylogenetic analysis. The whole genome sequences of one to two representative species from different groups of edible and medicinal fungi and several adjacent genera of Sarcomyxa were analyzed to show the correctness of the analysis results.
Phylogenetic analyses of the concatenated dataset using ML methods resulted in an identical and well-supported topology in all alignment strategies compared to the study of Lin et al. (2013). It can reasonably model the large stochastic processes with lower variance. Even with very short sequences, it may outperform alternative methods such as parsimony or distance methods. It also has an explicit evolutionary model for data and better accounting for branch length (Mishler, 2006). For example, Li et al. (2021) and Xu et al. (2020) used the ML algorithm in RAxML to construct a genome-based phylogenetic tree. In this study, based on the ML/NJ phylogenetic analysis of S. edulis and 31 other species of fungus, it was shown that the Sarcomyxa genus is a distinct group. This is consistent with the previous classification of Sarcomyxa as a clade that is independent of and emerged before the Mycena and Panellus genera based on a ML tree constructed using the D1/D2 region of S. edulis comb. nov. and other species (Saito et al., 2014). A study of the major clades of the Agaricales order arrived at a similar conclusion based on a maximum probability tree that combined rpb1, rpb1-intron2, rpb2, 18S, 25S, and 5.8S nucleotide sequences (Matheny et al., 2006). Additionally, we found that Basidiomycota, Polyporales, and Agaricales constituted distinct clades. The evolution of the Agaricales order followed a trend from “on log” to “on ground” growth.
We also observed a transition from white rot to brown rot in the Polyporales clade over the course of evolution. This is in accordance with a previous analysis of lignin-degrading peroxidases in 31 fungal genomes, which showed that the lignin degradation mechanism of white rot fungi was retained in the early evolution of Agaricomycetes, while the loss of lignin peroxidases led to the emergence of brown rot fungi exhibiting a non-ligninolytic mode of wood decay (Floudas et al., 2012; Supplementary Table 1).
The comparative analyses of secondary metabolites revealed that S. edulis harbors terpene and PKS biosynthesis-associated genes. The annotated whole-genome sequence of S. edulis can serve as a reference for identifying genes related to the synthesis of bioactive compounds with medicinal or nutritional value and the development and commercial production of superior S. edulis varieties.
Data Availability Statement
The original contributions presented in the study are publicly available. This data can be found here: The genome sequence data and assembly reported in this paper are associated with NCBI BioProject: PRJNA483858 and BioSample: SAMN09748201 in GenBank. The Whole Genome Shotgun (WGS) number is INSDC: QUOL00000000.1).
Author Contributions
FT contributed to conceptualization, methodology, software, data analysis, investigation, resources, data curation, and manuscript writing—original draft preparation. CL contributed to data validation, supervision, and project administration. CL and YL contributed to writing—review and editing. YL contributed to visualization and funding acquisition. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by the National Natural Science Foundation of China (NSFC: 32000013), National Key Research and Development Program of China (2020YFD1000304), Program of Introducing Talents of Discipline to Universities (D17014), and National Key Research and Development Program of China Grant (2017YFD0601002).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Francis Martin and Christoffer Bugge Harder and the 1000 Fungal Genome consortium for access to unpublished genome data. The genome sequence data were produced by the United States Department of Energy Joint Genome Institute in collaboration with the user community, and we would like to thank the National Center for Biotechnology Information for the genomic data provided. We thank the Mushroom Base of Jilin Agricultural University for assistance with cultivation.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.652324/full#supplementary-material
Supplementary Table 1 | Summary of fungi analyzed in this study and origin of their genomes.
Supplementary Table 2 | Genes predicted to be involved in non-coding RNA in Sarcomyxa edulis.
Supplementary Table 3 | Sarcomyxa edulis gene annotations.
Supplementary Table 4 | Putative CYP gene families of Sarcomyxa edulis.
Supplementary Table 5 | Genes predicted to be involved in the secondary metabolism of Sarcomyxa edulis.
Supplementary Table 6 | Distribution of the CAZyme family genes in different fungi.
Supplementary Table 7 | Genes predicted to be involved in polysaccharide biosynthesis in Sarcomyxa edulis.
Footnotes
- ^ http://antismash.secondarymetabolites.org
- ^ http://napdos.ucsd.edu
- ^ http://napdos.ucsd.edu/napdos_home.html
- ^ http://drnelson.uthsc.edu/CytochromeP450.html
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Aronsen, A., and Læssøe, T. (2016). “The genus Mycena s.l,” in The Fungi of Northern Europe, eds T. G. Frøslev, T. Læssøe, J. H. Petersen, and S. A. Elborne (Copenhagen, DK: Danish Mycological Society), 373.
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene ontology: tool for the unification of biology. Nat. Genet. 25, 25–29. doi: 10.1038/75556
Bao, D. P., Gong, M., Zheng, H. J., Chen, M. J., Zhang, L., Wang, H., et al. (2013). Sequencing and comparative analysis of the straw mushroom (Volvariella volvacea) genome. PLoS One 8:e58294. doi: 10.1371/journal.pone.0058294
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Berlin, K., Koren, S., Chin, C. S., Drake, J., Landolin, J. M., and Phillippy, A. M. (2015). Assembling large genomes with single-molecule sequencing and locality sensitive hashing. Nat. Biotechnol. 33, 623–630. doi: 10.1038/nbt.3238
Birney, E., Clamp, M., and Durbin, R. (2004). Genewise and genomewise. Genome Res. 14, 988–995. doi: 10.1101/gr.1865504
Blin, K., Wolf, T., Chevrette, M. G., Lu, X., Schwalen, C. J., Kautsar, S. A., et al. (2017). AntiSMASH 4.0-improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res. 45, W36–W41. doi: 10.1093/nar/gkx319
Buchfink, B., Xie, C., and Huson, D. H. (2014). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Burge, C., and Karlin, S. (1997). Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268, 78–94. doi: 10.1006/jmbi.1997.0951
Castanera, R., Pérez, G., López-Varas, L., Amselem, J., LaButti, K., Singan, V., et al. (2017). Comparative genomics of Coniophora olivacea reveals different patterns of genome expansion in Boletales. BMC Genomics 18:883. doi: 10.1186/s12864-017-4243-z
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Cattran, D. C., Alexopoulos, E., Heering, P., Hoyer, P. F., Johnston, A., Meyrier, A., et al. (2007). Cyclosporin in idiopathic glomerular disease associated with the nephrotic syndrome: workshop recommendations. Kidney Int. 72, 1429–1447. doi: 10.1038/sj.ki.5002553
Chen, L. F., Gong, Y. H., Cai, Y. L., Liu, W., Zhou, Y., Xiao, Y., et al. (2016). Genome sequence of the edible cultivated mushroom Lentinula edodes (Shiitake) reveals insights into lignocellulose degradation. PLoS One 11:e0160336. doi: 10.1371/journal.pone.0160336
Chen, W., Zhu, P., He, S., Jin, H., and Yan, X. (2012). Nonribosomal peptides synthetases gene clusters and core domain in Pseudoalteromonas sp. NJ631. Acta Microbiol. Sin. 52, 1531–1539.
Choi, Y. W., Hyde, K. D., and Ho, W. H. (1999). Single spore isolation of fungi. Fungal Divers. 3, 29–38.
Cui, X. F., Lu, Z. W., Wang, S., Wang, J. J. Y., and Gao, X. (2016). CMsearch: simultaneous exploration of protein sequence space and structure space improves not only protein homology detection but also protein structure prediction. Bioinformatics 32, 332–340. doi: 10.1093/bioinformatics/btw271
Dai, Y. C., Niemela, T., and Qin, G. F. (2003). Changbai wood-rotting fungi. a new pleurotoid species Panellus edulis. Ann. Bot. Fenn. 40, 107–112.
Elsik, C. G., Mackey, A. J., Reese, J. T., Milshina, N. V., Roos, D. S., and Weinstock, G. M. (2007). Creating a honey bee consensus gene set. Genome Biol. 8:R13. doi: 10.1186/gb-2007-8-1-r13
Finking, R., and Marahiel, M. A. (2004). Biosynthesis of nonribosomal peptides. Annu. Rev. Microbiol. 58, 453–488. doi: 10.1146/annurev.micro.58.030603.123615
Floudas, D., Binder, M., Riley, R., Barry, K., Blanchette, R. A., Henrissat, B., et al. (2012). The paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science 336, 1715–1719. doi: 10.1126/science.1221748
Fujii, I., Watanabe, A., Sankawa, U., and Ebizuka, Y. (2001). Identification of claisen cyclase domain in fungal polyketide synthase WA, a naphthopyrone synthase of Aspergillus nidulans. Chem. Biol. 8, 189–197. doi: 10.1016/S1074-5521(00)90068-1
Gardner, P. P., Daub, J., Tate, J. G., Nawrocki, E. P., Kolbe, D. L., Lindgreen, S., et al. (2009). Rfam: updates to the RNA families database. Nucleic Acids Res. 37(suppl. 1), D136–D140. doi: 10.1093/nar/gkn766
Grigoriev, I. V., Nordberg, H. P., Shabalov, I., Aerts, A., Cantor, M., Goodstein, D., et al. (2012). The genome portal of the department of energy joint genome institute. Nucleic Acids Res. 40, D26–D32. doi: 10.1093/nar/gkr947
Hiron, A., Falord, M., Valle, J., Débarbouillé, M., and Msadek, T. (2011). Bacitracin and nisin resistance in Staphylococcus aureus: a novel pathway involving the BraS/BraR two-component system (SA2417/SA2418) and both the BraD/BraE and VraD/VraE ABC transporters. Mol. Microbiol. 81, 602–622. doi: 10.1111/j.1365-2958.2011.07735.x
Inafuku, M., Nagao, K., Nomura, S., Shirouchi, B., Inoue, N., Nagamori, N., et al. (2012). Protective effects of fractional extracts from Panellus serotinus on non-alcoholic fatty liver disease in obese, diabetic db/db mice. Br. J. Nutr. 107, 639–646. doi: 10.1017/S0007114511003485
Inoue, N., Inafuku, M., Shirouchi, B., Nagao, K., and Yanagita, T. (2013). Effect of mukitake mushroom (Panellus serotinus) on the pathogenesis of lipid abnormalities in obese, diabetic ob/ob mice. Lipids Health Dis. 12:18. doi: 10.1186/1476-511X-12-18
Ishihara, H., Takoh, M., Nishibayashi, R., and Sato, A. (2002). Distribution and variation of bacitracin synthetase gene sequences in laboratory stock strains of Bacillus licheniformis. Curr. Microbiol. 45, 18–23. doi: 10.1007/s00284-001-0041-5
Jin, J., Hughes, K. W., and Petersen, R. H. (2001). Phylogenetic relationships of Panellus (Agaricales) and related species based on morphology and ribosomal large subunit DNA sequences. Mycotaxon 79, 7–21.
Kanehisa, M., Goto, S., Hattori, M., Aoki-Kinoshita, K. F., Itoh, M., Kawashima, S., et al. (2006). From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 34(suppl. 1), D354–D357. doi: 10.1093/nar/gkj102
Kanehisa, M., Goto, S., Kawashima, S., Okuno, Y., and Hattori, M. (2004). The KEGG resource for deciphering the genome. Nucleic Acids Res. 32(suppl. 1), D277–D280. doi: 10.1093/nar/gkh063
Kasper, M. A. A. S. A., Farahat, H. G., Al-Morsy, O. A., and Alhagaa, A. A. (2017). Comparative study using cyclosporine a 0.05% eye drops, autologous serum 20% eye drops, and combined use of cyclosporine A 0.05% eye drops and autologous serum 20% eye drops in the treatment of moderate to severe dry eye. Menoufia Med. J. 2, 525–531. doi: 10.4103/1110-2098.215461
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Keller, N. P., Turner, G., and Bennett, J. W. (2006). Fungal secondary metabolism-from biochemistry to genomics. Nat. Rev. Microbiol. 3, 937–947. doi: 10.1038/nrmicro1286
Kim, J. H., Lee, J. S., Lee, K. R., Shim, M. J., Lee, M. W., Shin, P. G., et al. (2012). Immunomodulating and antitumor activities of Panellus serotinus polysaccharides. Mycobiology 40, 181–188. doi: 10.5941/MYCO.2012.40.3.181
Koren, S., and Phillippy, A. M. (2015). One chromosome, one contig: complete microbial genomes from long-read sequencing and assembly. Curr. Opin. Microbiol. 23, 110–120. doi: 10.1016/j.mib.2014.11.014
Korf, I. (2004). Gene finding in novel genomes. BMC Bioinformatics 5:59. doi: 10.1186/1471-2105-5-59
Kowalski, R. J., Giannakakou, P., and Hamel, E. (1997). Activities of the microtubule-stabilizing agents epothilones A and B with purified tubulin and in cells resistant to paclitaxel (Taxol®). J. Biol. Chem. 272, 2534–2541. doi: 10.1074/jbc.272.4.2534
Kumar, R., Dogra, S., Amarji, B., Singh, B., Kumar, S., Vinay, K., et al. (2016). Efficacy of novel topical liposomal formulation of cyclosporine in mild to moderate stable plaque psoriasis: a randomized clinical trial. JAMA Dermatol. 152, 807–815. doi: 10.1001/jamadermatol.2016.0859
Lackner, G., Misiek, M., Braesel, J., and Hoffmeister, D. (2012). Genome mining reveals the evolutionary origin and biosynthetic potential of basidiomycete polyketide synthases. Fungal Genet. Biol. 49, 996–1003. doi: 10.1016/j.fgb.2012.09.009
Lagesen, K., Hallin, P. F., Rodland, E. A., Staerfeldt, H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Lee, G. Y., Jang, D. S., Lee, Y. M., Kim, J. M., and Kim, J. S. (2006). Naphthopyrone glucosides from the seeds of Cassia tora with inhibitory activity on advanced glycation end products (AGEs) formation. Arch. Pharm. Res. 29, 587–590. doi: 10.1007/BF02969270
Li, C., Guo, Z. J., Zhou, S. Y., Han, Y. Q., Zhang, M. M., Peng, Y., et al. (2021). Evolutionary and genomic comparisons of hybrid uninucleate and nonhybrid Rhizoctonia fungi. Commun. Biol. 4:201. doi: 10.1038/s42003-021-01724-y
Li, L., Stoeckert, C. J. Jr., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Li, R. G. (1991). The Fungal Flora of Jilin Province. Changchun: Northeastern Normal University Press.
Li, W. Z., Jaroszewski, L., and Godzik, A. (2002). Tolerating some redundancy significantly speeds up clustering of large protein databases. Bioinformatics 18, 77–82. doi: 10.1093/bioinformatics/18.1.77
Li, X. Y., Wang, L., Wang, Y., and Xiong, Z. H. (2016). Effect of drying method on physicochemical properties and antioxidant activities of Hohenbuehelia serotina polysaccharides. Process Biochem. 51, 1100–1108. doi: 10.1016/j.procbio.2016.05.006
Li, X. Y., Wang, Z. Y., and Wang, L. (2013). Polysaccharide of Hohenbuehelia serotina as a defense against damage by whole-body gamma irradiation of mice. Carbohyd. Polym. 94, 829–835. doi: 10.1016/j.carbpol.2013.02.015
Lin, Y., Hu, F., Tang, J. J., and Moret, B. M. E. (2013). Maximum likelihood phylogenetic reconstruction from high-resolution whole-genome data and a tree of 68 eukaryotes. Pacific Symposium on Biocomputing, Switzerland, 285–296. doi: 10.1142/97898144479730028
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.955
Ma, Y., Mizino, T., and Ito, H. (1991). Antitumor activity of some polysaccharides isolated from a Chinese mushroom, “Huangmo” the fruiting body of Hohenbuehelia serotina. Agric. Biol. Chem. 55, 2701–2710. doi: 10.1080/00021369.1991.10871044
Majoros, W. H., Pertea, M., and Salzberg, S. L. (2004). TigrScan and glimmerhmm: two open source ab initio eukaryotic gene-fifinders. Bioinformatics 20, 2878–2879. doi: 10.1093/bioinformatics/bth315
Matheny, P. B., Curtis, J. M., Hofstetter, V., Aime, M. C., Moncalvo, J. M., Ge, Z. W., et al. (2006). Major clades of Agaricales: a multilocus phylogenetic overview. Mycologia 98, 982–995. doi: 10.1080/15572536.2006.11832627
Miller, M. A., Pfeiffer, W., and Schwartz, T. (2010). “Creating the CIPRES science gateway for inference of large phylogenetic trees,” in Proceedings of the Gateway Computing Environments Workshop (GCE), (New Orleans, LA: IEEE Computer Society).
Mishler, B. D. (2006). Integrative Biology 200A “Principles of Phylogenetics”. Berkeley, CA: University of California.
Morin, E., Kohler, A., Baker, A. R., Foulongne-Oriol, M., Lombard, V., Nagy, L. G., et al. (2012). Genome sequence of the button mushroom Agaricus bisporus reveals mechanisms governing adaptation to a humic-rich ecological niche. Proc. Natl. Acad. Sci. U.S.A. 110, 4146–4148. doi: 10.1073/pnas.1206847109
Nawrocki, E. P., Kolbe, D. L., and Eddy, S. R. (2009). Infernal 1.0: inference of RNA alignments. Bioinformatics 25, 1335–1337. doi: 10.1093/bioinformatics/btp157
Nelson, D. R. (2009). The cytochrome P450 homepage. Hum. Genomics 4, 59–65. doi: 10.1186/1479-7364-4-1-59
Ohm, R. A., de Jong, J. F., Lugones, L. G., Aerts, A., Kothe, E., Stajich, J. E., et al. (2010). Genome sequence of the model mushroom Schizophyllum commune. Nat. Biotechnol. 28, 957–963. doi: 10.1038/nbt.1643
Ospina-Giraldo, M. D., Griffith, J. G., Laird, E. W., and Mingora, C. (2010). The CAZyome of Phytophthora spp.: a comprehensive analysis of the gene complement coding for carbohydrate-active enzymes in species of the genus Phytophthora. BMC Genomics 11:525. doi: 10.1186/1471-2164-11-525
Pan, X. R. (1995). Macrofungi of the Xiaoxingan Mts. Harbin: Northeastern Forestry University. Press.
Qu, J., Zhao, M., Hsiang, T., Feng, X., Zhang, J., and Huang, C. (2016). Identification and characterization of small noncoding RNAs in genome sequences of the edible fungus Pleurotus ostreatus. BioMed. Res. Int. 2016:2503023. doi: 10.1155/2016/2503023
Quin, M. B., Flynn, C. M., and Schmidt-Dannert, C. (2014). Traversing the fungal terpenome. Nat. Prod. Rep. 31, 1449–1473. doi: 10.1039/C4NP00075G
Saha, S., Bridges, S., Magbanua, Z. V., and Peterson, D. G. (2008). Empirical comparison of ab initio repeat finding programs. Nucleic Acids Res. 36, 2284–2294. doi: 10.1093/nar/gkn064
Saier, M. H. Jr., Reddy, V. S., Tamang, D. G., and Västermark, Å (2014). The transporter classification database. Nucleic Acids Res. 42, D251–D258. doi: 10.1093/nar/gkt1097
Saito, T., Tonouchi, A., and Harada, Y. (2014). Biological characteristics and molecular phylogeny of Sarcomyxa edulis comb. nov. and S. serotina. Jap. J. Mycol. 55, 19–28.
Schwarzer, D., Finking, R., and Marahiel, M. A. (2003). Nonribosomal peptides: from genes to products. Nat. Prod. Rep. 20, 275–287. doi: 10.1039/B111145K
Senanayake, I. C., Rathnayaka, A. R., Marasinghe, D. S., Calabon, M. S., Gentekaki, E., Lee, H. B., et al. (2020). Morphological approaches in studying fungi: collection, examination, isolation, sporulation and preservation. Mycosphere 11, 2678–2754. doi: 10.5943/mycosphere/11/1/20
Sheng, W. B., Li, W., Zhang, G. X., Tong, Y. B., Liu, Z. Y., and Jia, X. (2015). Study on the UV-shielding and controlled-release properties of a polydopamine coating for avermectin. New J. Chem. 39, 2752–2757. doi: 10.1039/C4NJ01744G
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with singlecopy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Sims, J. W., and Schmidt, E. W. (2008). Thioesterase-like role for fungal PKS-NRPS hybrid reductive domains. J. Am. Chem. Soc. 130, 11149–11155. doi: 10.1021/ja803078z
Stajich, J. E., Wilke, S. K., Ahrén, D., Au, C. H., Birren, B. W., Borodovsky, M., et al. (2010). Insights into evolution of multicellular fungi from the assembled chromosomes of the mushroom Coprinopsis cinerea (Coprinus cinereus). Proc. Natl. Acad. Sci. U.S.A. 107, 11889–11894. doi: 10.1073/pnas.1003391107
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stanke, M., Keller, O., Gunduz, I., Hayes, A., Waack, S., and Morgenstern, B. (2006). AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439. doi: 10.1093/nar/gkl200
Tatusov, R. L., Fedorova, N. D., Jackson, J. D., and Natale, D. A. (2003). The COG database: an updated version includes eukaryotes. BMC bioinformatics 4:41. doi: 10.1186/1471-2105-4-41
Walker, B. J., Abeel, T., Shea, T., Priest, M., and Earl, A. M. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963
Weber, T., Blin, K., Duddela, S., Krug, D., Kim, H. U., Bruccoleri, R. E., et al. (2015). Antismash 3.0-a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 43, W237–W243. doi: 10.1093/nar/gkv437
Xie, C. L., Gong, W. B., Zhu, Z. H., Li, Y., Hu, Z. X., and Peng, Y. D. (2018). Comparative transcriptomics of Pleurotus eryngii reveals blue-light regulation of carbohydrate-active enzymes (CAZymes) expression at primordium differentiated into fruiting body stage. Genomics 110, 201–209. doi: 10.1016/j.ygeno.2017.09.012
Xu, R., Liu, X. C., Peng, B., Liu, P. B., Li, Z., Dai, Y. T., et al. (2020). Genomic features of Cladobotryum dendroides, which causes cobweb disease in edible mushrooms, and identification of genes related to pathogenicity and mycoparasitism. Pathogens 9:232. doi: 10.3390/pathogens9030232
Yap, H. Y. Y., Chooi, Y. H., Firdaus-Raih, M., Fung, S. Y., Ng, S. T., Tan, C. S., et al. (2014). The genome of the tiger milk mushroom, Lignosus rhinocerotis, provides insights into the genetic basis of its medicinal properties. BMC Genomics 15:635. doi: 10.1186/1471-2164-15-635
Yuan, Y., Wu, F., Si, J., Zhao, Y. F., and Dai, Y. C. (2019). Whole genome sequence of Auricularia heimuer (Basidiomycota, Fungi), the third most important cultivated mushroom worldwide. Genomics 111, 50–58. doi: 10.1016/j.ygeno.2017.12.013
Zhang, H., Yohe, T., Huang, L., Entwistle, S., Wu, P., Yang, Z., et al. (2018). dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 46, W95–W101. doi: 10.1093/nar/gky418
Zhang, R., Zhao, L. Y., Wang, H. X., and Ng, T. B. (2014). A novel ribonuclease with antiproliferative activity toward leukemia and lymphoma cells and HIV-1 reverse transcriptase inhibitory activity from the mushroom, Hohenbuehelia serotina. Int. J. Mol. Med. 33, 209–214. doi: 10.3892/ijmm.2013.1553
Keywords: edible-medicinal mushroom, lignin-degrading peroxidases, phylogenetic analyses, Sarcomyxa edulis, secondary metabolism
Citation: Tian F, Li C and Li Y (2021) Genomic Analysis of Sarcomyxa edulis Reveals the Basis of Its Medicinal Properties and Evolutionary Relationships. Front. Microbiol. 12:652324. doi: 10.3389/fmicb.2021.652324
Received: 12 January 2021; Accepted: 26 May 2021;
Published: 01 July 2021.
Edited by:
Peter Edward Mortimer, Kunming Institute of Botany, Chinese Academy of Sciences, ChinaReviewed by:
Shanshan Liu, The First Affiliated Hospital of Bengbu Medical College, ChinaRajesh Jeewon, University of Mauritius, Mauritius
Copyright © 2021 Tian, Li and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yu Li, fungi966@126.com; Changtian Li, lct@jlau.edu.cn