
Profiling the structural determinants of aminoketone derivatives as hNET and hDAT reuptake inhibitors by field-based QSAR based on molecular docking
Abstract
BACKGROUND:
Bupropion, one of the dual norepinephrine and dopamine reuptake inhibitors (NDRIs), is an aminoketone derivative performed effect in improving cognitive function for depression. However, its therapeutic effect is unsatisfactory due to poor clinical response, and there are only few derivatives in pre-clinical settings.
OBJECTIVE:
This work attempted to elucidate the essential structural features for the activity and designed a series of novel derivatives with good inhibitive ability, pharmacokinetic and medicinal chemistry properties.
METHODS:
The field-based QSAR of aminoketone derivatives of two targets were established based on docking poses, and the essential structural properties for designing novel compounds were supplied by comparing contour maps.
RESULTS:
The selected two models performed good predictability and reliability with R
CONCLUSIONS:
Bulky groups in R

1.Introduction
Depression is a common psychiatric disorder, affecting more than 350 million people around the world [1, 2, 3, 4]. Cognitive dysfunctions as a core complication of depression, leads to serious problems in executive functioning, attention, learning processing speed and memory [5, 6, 7]. These functional impairments often are remitted as the mood symptoms improved, but still residual after remission of mood symptoms, which reduce the work abilities of patients [5, 8, 9]. These resulted in the crippling economic burden including not only direct costs for treatment, but also sizeable indirect costs for the absenteeism and poor productivity in the work [5, 10]. As the World Health Organization predicts, depression will be the leading cause of disease burden worldwide by 2030 [1, 5]. Thus, recovering cognitive dysfunctions has been identified as the novel therapeutic strategy for depression [5, 11, 12].
As reported, prefrontal cortex (PFC), hippocampus, nucleus accumbens (NAc), amygdala, and ventral striatum were the common neuropathological platform for depression and cognitive dysfunction due to depression [4, 13, 14, 15]. Neuropsychological and imaging evidence indicated the PFC plays a critical role in the control of cognitive function [14, 16, 17, 18]. In the course of treatment, some improving cognitive function drugs preferentially targeted to PFC catecholamines, and the striatum and NAc might contribute to their therapeutic efficacy [14]. In these brain areas, the dysfunctional catecholaminergic signaling is the posited etiology of depression and related cognitive dysfunction especially for the catecholamine neurotransmitters norepinephrine (NE) and dopamine (DA), which are required for proper prefrontal functions acting as coordination with each other [15, 19, 20]. Moreover, the transporters of norepinephrine (hNET) and dopamine (hDAT) are distributed in PFC, striatum and nucleus accumbens, and hDAT is sparse and hNET is high density in the PFC while are opposite in striatum and nucleus accumbens [14, 16]. Meanwhile, the hNET displays a high affinity for DA and plays a prominent role in DA clearance [16]. Thus selective NE reuptake inhibitors (sNRIs) merely could elevate both NE and DA in the PFC but with minimal effects on striatal and NAc DA [14], and selective DA reuptake inhibitors (sDRIs) mainly effected on striatal and NAc DA. In contrast, dual NE and DA reuptake inhibitors (NDRIs) performed more efficacy because they could impact on NE and DA level not only in PFC but also in striatal and NAc [20], and which of them with hNET
Recently, there are 4 NDRIs (Dexmethylphenidate, Dextroamphetamine, Dextromethamphetamine, Bupropion) used to improve cognitive dysfunction. The first three of them are reported to remit the cognitive dysfunction reduced by Attention-Deficit Hyperactivity Disorder (ADHD), but the risk of abuse potential and addictive for them limit their widespread use [15, 21, 22]. The last one bupropion, an aminoketone derivative is phase 4 clinical study in improving cognitive function for depression patients [5, 23]. However, its therapeutic effect is unsatisfactory attributed to the poorer clinical response [24]. Besides bupropion, none aminoketone derivatives of NDRIs antidepressants are currently in clinical trial, and only few are in pre-clinical.
In order to satisfy the strong need for antidepressants with improved cognitive dysfunction and enhanced efficacy, this work explored the Quantitative Structure-Activity Relationship (QSAR) characteristics of aminoketone derivatives based on the potent poses by molecular docking, which could be utilized as structural guidance for assessing and rationally designing more efficacious NDRIs candidates with hNET
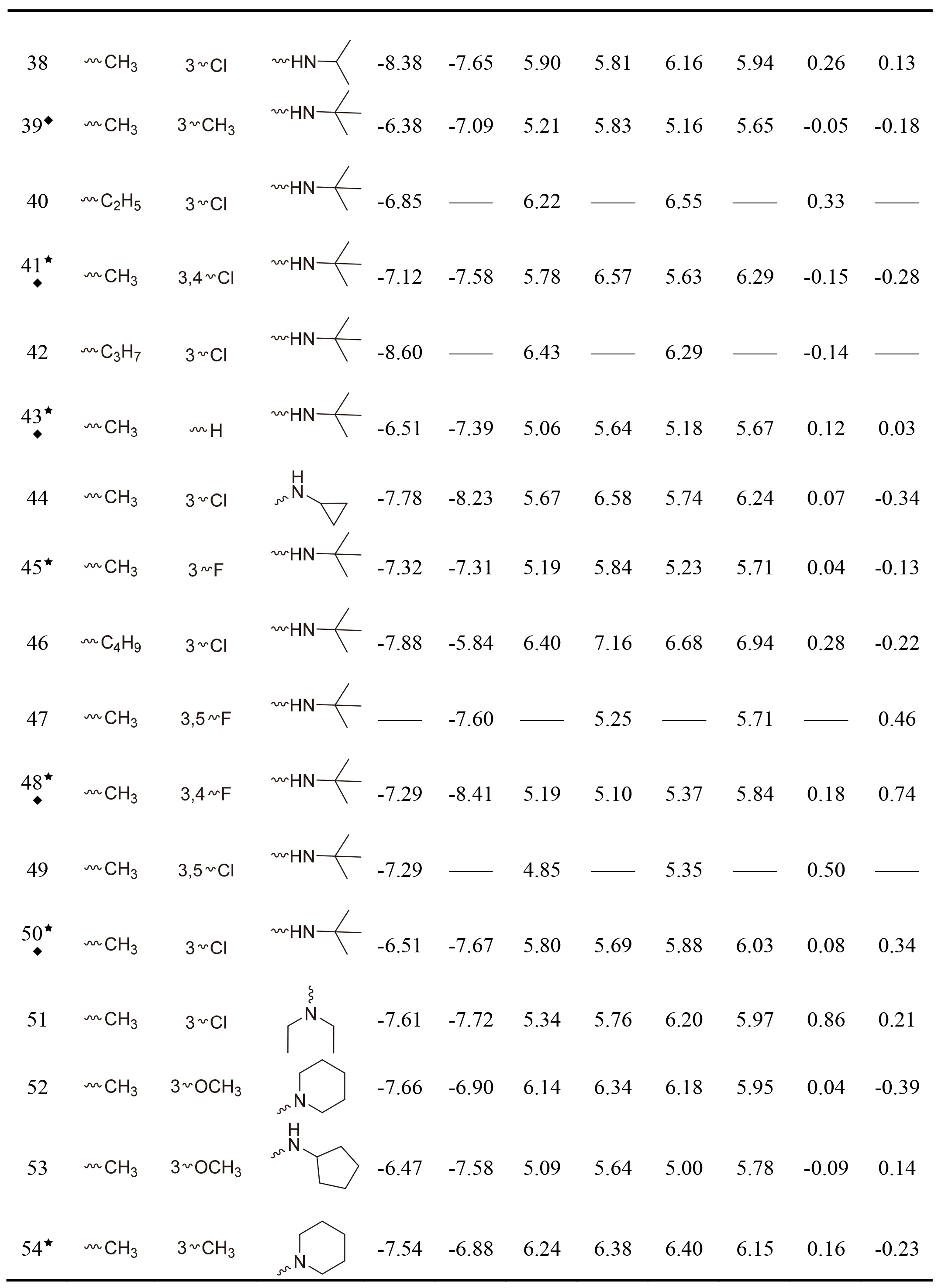
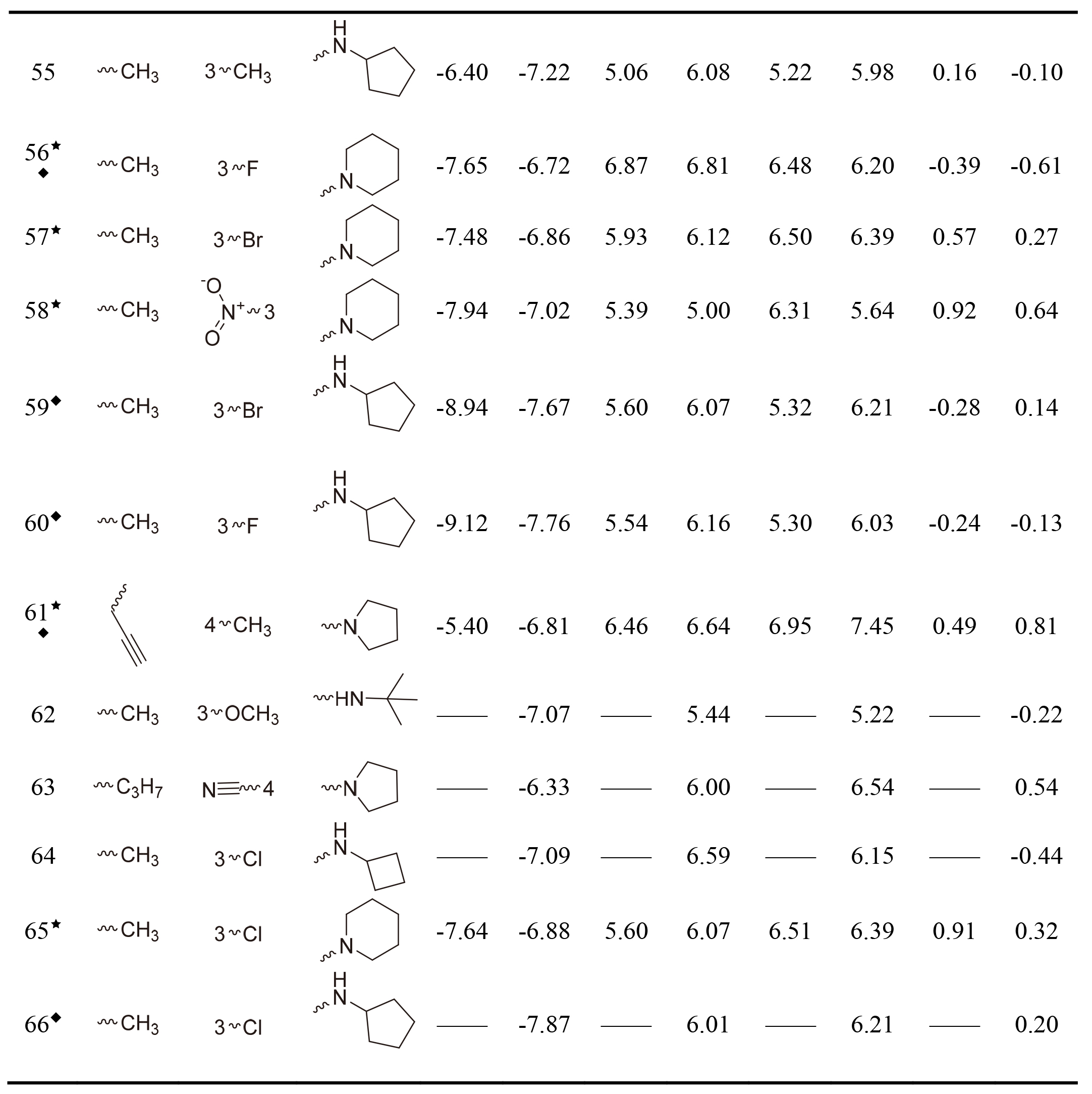
Table 1
The Glide gscore, experimental and predicted activities in data sets for selected QSAR models of hNET and hDAT
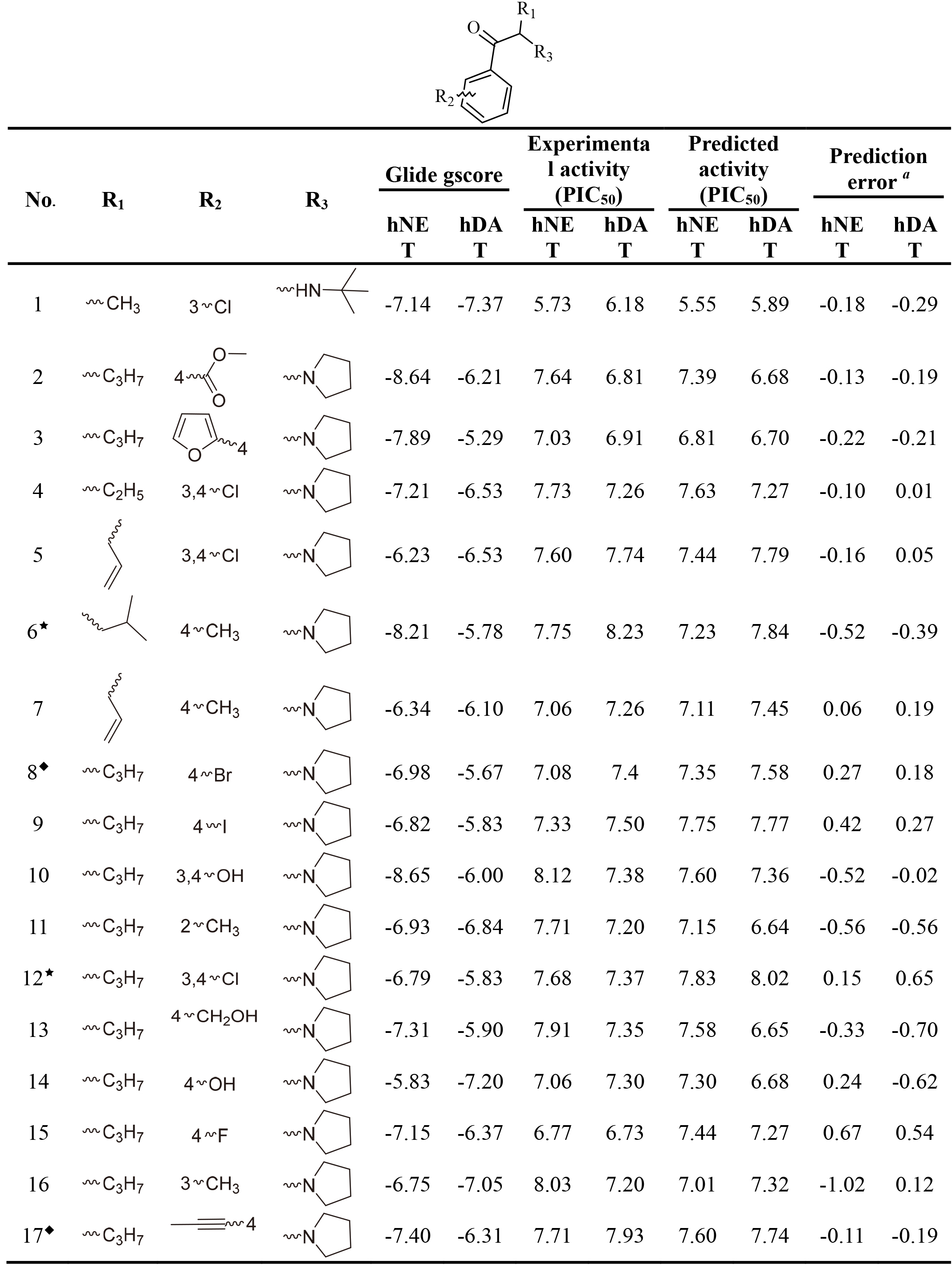
|
|
Table 1, continued |

|
|
Table 1, continued |
|
|
Table 1, continued |
|
2.Materials and methods
2.1Datasets preparation
This work collected 60 and 63 aminoketone derivatives with active data of hNET and hDAT from CHEMBL database [25] for field-based QSAR study as shown in Table 1. The datasets were randomly assigned 70% into training set of 42 molecules for hNET and 44 molecules for hDAT to generate QSAR models, and the rest of them as test sets were used to validate. The process of dividing datasets for both models should be considered the uniform distribution of compounds with as enough structural diversity and wide range activities as possible. The training and test sets of both models are demonstrated in Table 1.
2.2Homology modeling and molecular docking
The potent poses of compounds for each target were identified based on the complexes of target-ligand achieved by molecular docking and prime minimization. Since there is no crystal structural data reported in Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB) [26], the 3D structures of hNET and hDAT need to be constructed by homology modeling performed in SWISS-MODEL workspace [27]. The template used to construct models was confirmed according to sequence identity with targets by sequence alignment using ClustalW2 [23, 28, 29], and the stereochemical quality of built structures were checked by Ramachandran plot referenced our previous work [30, 31, 32].
The process of molecular docking was carried out in Maestro [33]. The first detailed work, active compounds of hNET and hDAT were collected from CHEMBL, and then their structures were preprocessed by the LigPrep [34] using OPLS-2005 force field [35]. The ionized state was assigned by Epik [36] at pH value of 7.0
2.3Field-based QSAR models construction and validation
Field-based QSAR studies were performed on Field-based QSAR module [39] in Maestro [33]. The potent poses were superimposed to bupropion by SMARTS method based on common scaffold in Flexible ligand alignment module. The field-based QSAR models were constructed by training sets at the extended Gaussian field, and the maximum PLS factors was set 5 for hNET and hDAT. The best models for each target were identified by statistical robustness, and their stabilities and predictive abilities were validated by the testing sets with leave-one-out (LOO) cross-validation methods. The most important statistics are the test set statistics including RMSE, Q
2.4ADME/T and chemical synthetize properties predicted for candidates
In order to obtain effective drugs, this work evaluated the ADME/T properties of the designed NDRIs by the pkCSM (http://biosig.unimelb.edu.au/pkcsm/prediction) and ADMETlab online server (http://admet.scbdd.com/calcpre/index/). On the SwissADME online server (http://www.swissadme.ch/index.php), the problematic fragments in novel candidates were identified by the complementary pattern recognition method of PAINS (pan assay interference compounds) and Brenk filter. Moreover, the synthetic accessibilities (SA) were evaluated to demonstrate the synthetic complexities for molecules. A molecule with SA score closer to 10 was considered difficult to synthetic, while was considered as easy with closer to 1.
3.Results and discussion
3.1Homology modeling and validation for hNET and hDAT
The X-ray crystal structures of Drosophila melanogaster dopamine transporter (dDAT, PDB ID: 4XPH) showed high sequence identity with hNET (60.98%) and hDAT (55.83%), and showed higher sequence identity in binding site as the same as our previous work (69.05% and 78.57% for hNET and hDAT, respectively) [31]. Homology models of hNET and hDAT were constructed by dDAT as template, and the stereochemical quality and accuracy of models were illustrated by Ramachandran plot. Residues of models in allowed regions were 99.8% and 100% for hNET and hDAT respectively, indicating the reasonability of obtained models.
3.2The selected the potent poses by molecular docking and MM-GBSA prime
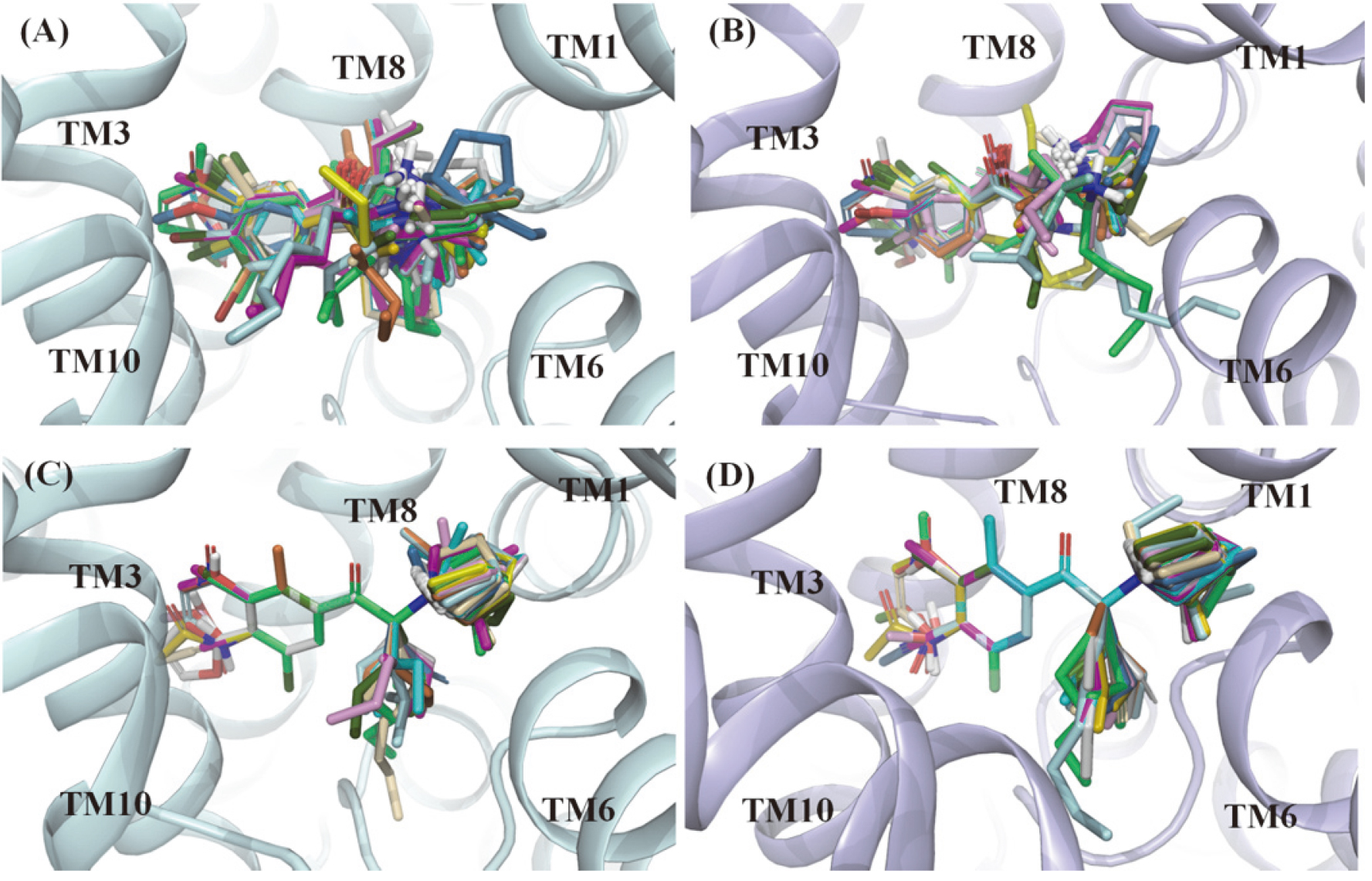
In this work, re-docking of 4XPH was first performed to validate the credibility of docking protocol. The binding pose of DCP in dDAT achieved by molecular docking was close to the original conformation with 0.474 of root mean square deviation (RMSD), indicating the validity of the docking protocol. Then, all active molecules were docked in each target using the same parameter settings as re-docking, and the obtained all complexes were minimized by MM-GBSA prime. Finally, the potent pose of bupropion was identified as the same as our previous work [31], including the electrostatic interaction between ammonium moiety and Asp75/Asp79 in hNET/hDAT recognition, and hydrophobic interactions formed in corresponding sub-sites for the other two groups. For other molecules, the docking poses were selected based on the docking score and the orientation of bupropion. Figure 1A and B show all selected poses gathered in the binding site surrounded by TM1, 3, 6, 8 and 10.
Figure 1.
Docking poses (A and B) and superimposed poses (C and D) template as bupropion of NDRIs compounds in the binding pocket of hNET and hDAT. The hNET (palecyan) and hDAT (lightblue) were shown in ribbon representation.
3.3Constructing and assessing of filed-based QSAR models for hNET and hDAT
All selected poses for hNET and hDAT were superimposed template as bupropion as demonstrated in Fig. 1C and D. In this work, 10 filed-based QSAR models were constructed using superimposed training sets compounds for hNET and hDAT, and the reliabilities of models were assessed by testing sets. All QSAR statistics are collected in Table 2.
Table 2
PLS statistics of hNET and hDAT field-based QSAR models
| Factors | SD | R | R | R | Stability |
|
| RMSE | Q | Pearson-r |
|---|---|---|---|---|---|---|---|---|---|---|
| hNET QSAR models | ||||||||||
| 1 | 0.6121 | 0.6143 | 0.5168 | 0.1873 | 0.987 | 63.7 | 8.38e-010 | 0.51 | 0.6882 | 0.8458 |
| 2 | 0.4662 | 0.7818 | 0.5428 | 0.3378 | 0.906 | 69.9 | 1.28e-013 | 0.50 | 0.6985 | 0.8836 |
| 3 | 0.3944 | 0.8479 | 0.5873 | 0.4553 | 0.880 | 70.6 | 1.35e-015 | 0.47 | 0.7352 | 0.8776 |
| 4 | 0.3251 | 0.8993 | 0.6186 | 0.5658 | 0.842 | 82.7 | 6.29e-018 | 0.46 | 0.7469 | 0.8830 |
| 5 | 0.2841 | 0.9252 | 0.6150 | 0.6456 | 0.808 | 89.1 | 3.04e-019 | 0.39 | 0.8199 | 0.9145 |
| hDAT QSAR models | ||||||||||
| 1 | 0.5559 | 0.4800 | 0.3669 | 0.2074 | 0.984 | 38.8 | 1.88e-007 | 0.43 | 0.6582 | 0.8134 |
| 2 | 0.4580 | 0.6554 | 0.4602 | 0.3296 | 0.956 | 39.0 | 3.27e-010 | 0.46 | 0.6015 | 0.7784 |
| 3 | 0.3497 | 0.8040 | 0.5107 | 0.4613 | 0.880 | 54.7 | 3.24e-014 | 0.45 | 0.6266 | 0.8072 |
| 4 | 0.2751 | 0.8817 | 0.5727 | 0.5402 | 0.826 | 72.7 | 1.52e-017 | 0.49 | 0.5471 | 0.7644 |
| 5 | 0.2407 | 0.9118 | 0.5981 | 0.6056 | 0.798 | 78.6 | 5.55e-019 | 0.53 | 0.4804 | 0.7239 |
For QSAR models of hNET, the values of R
Figure 2.
Correlation plots of the predicted and experimental PIC
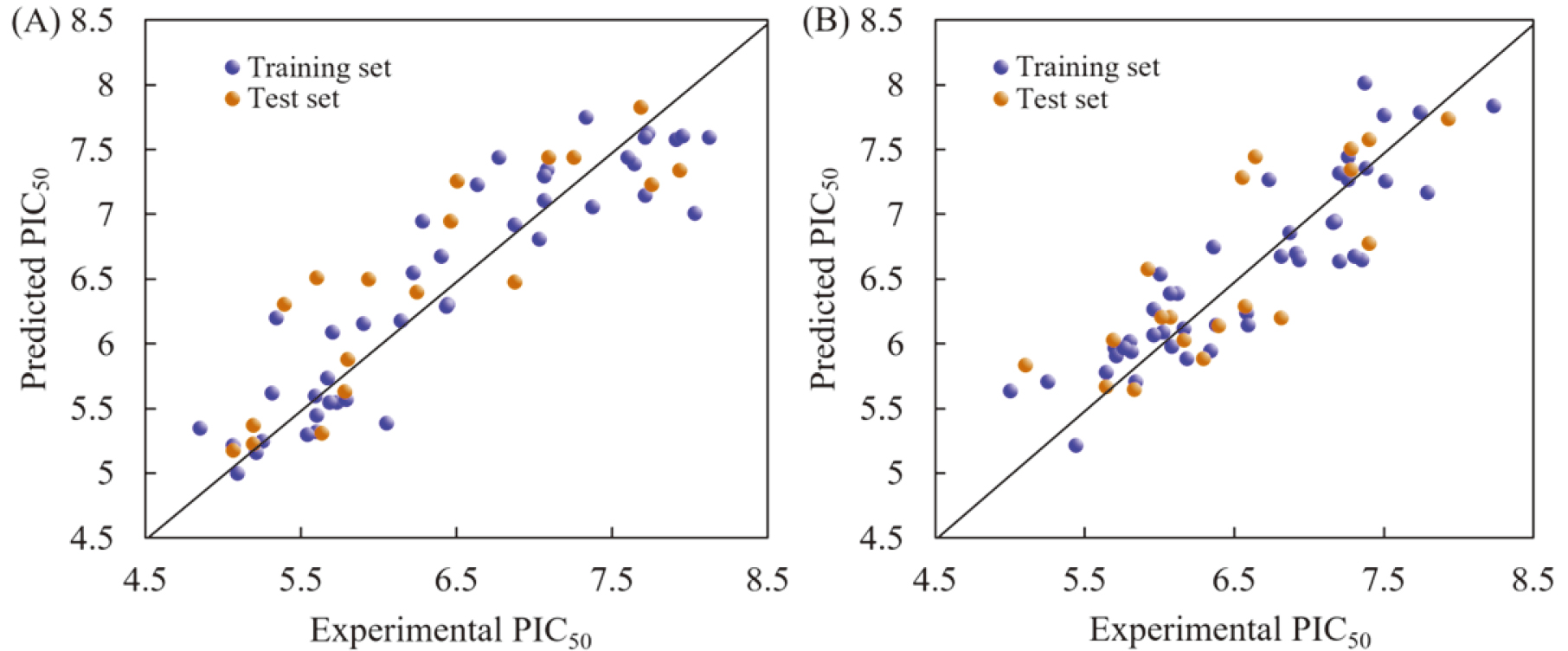
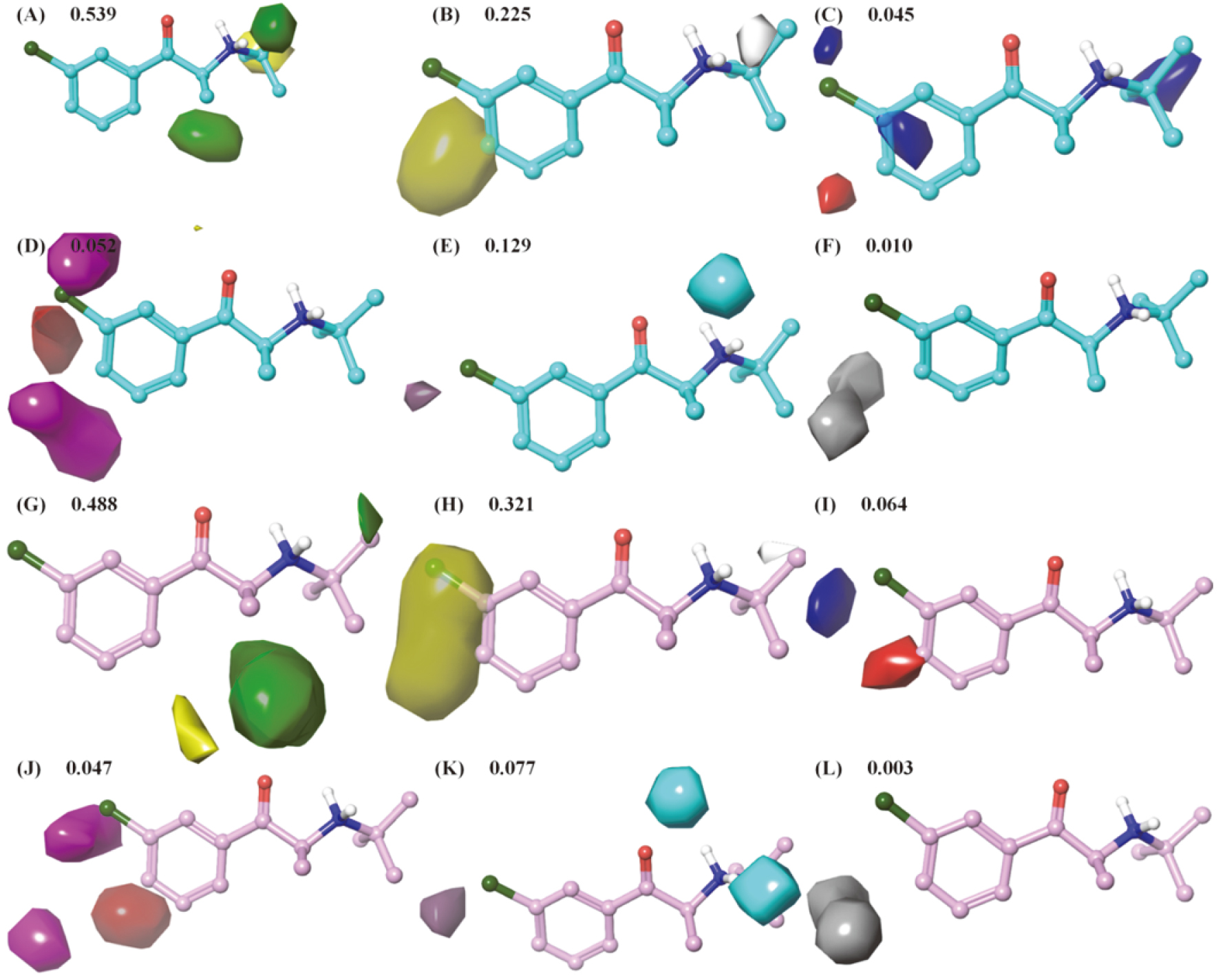
Figure 3.
3D contour maps of 6 extended Gaussian fields referenced bupropion for hNET and hDAT field-based QSAR models. A-F and G-L were steric, hydrophobic, electrostatic, hydrogen bond acceptor, hydrogen bond donor and aromatic ring field contour maps based on bupropion in hNET (cyan) and hDAT (pink) models, respectively. The colors of favorable and unfavorable regions were green and yellow for steric fields, yellow and white for hydrophobic field, blue and red for electrostatic fields, red and magenta for hydrogen bond acceptor, purple and cyan for hydrogen bond donor and orange and gray for aromatic ring field, respectively.
3.4Contour maps analyses of QSAR models for hNET and hDAT
3D contour maps of 6 extended Gaussian fields referenced bupropion (compound 1) are illustrated in Fig. 3, which provide information about the contribution of substituents on phenyl-aminoketone scaffold to the activity for each target. As shown in Fig. 3, steric and hydrophobic fields performed more potent contribution to activity than other 4 fields with 0.539 and 0.225 respectively for hNET, and with 0.484 and 0.323 respectively for hDAT. Thus, the volume and hydrophobicity of substituents on phenyl-aminoketone played major roles in regulating activity.
From the steric contour maps (Fig. 3A), there were 2 green and 1 yellow contour regions around aminoketone moiety, with green meaning bulky groups are favored and yellow meaning bulky groups are disfavored. The green contour maps at R
The yellow contour map of hydrophobicity was mainly sited at R
The electrostatic field contour maps are illustrated in Fig. 3C and I. The regions in red surrounded at para-position of R
The H-bond acceptor contour maps (Fig. 3D and J) closed to R
Considering all discussed above, this work supplied the graphical description of the structural features for designing novel NDRIs. In order to improve the activity, researchers could replace the methyl by the bulky groups with suitable carbon chain at R
Table 3
The predicted activities of designed novel derivatives by selected hNET and hDAT QSAR models
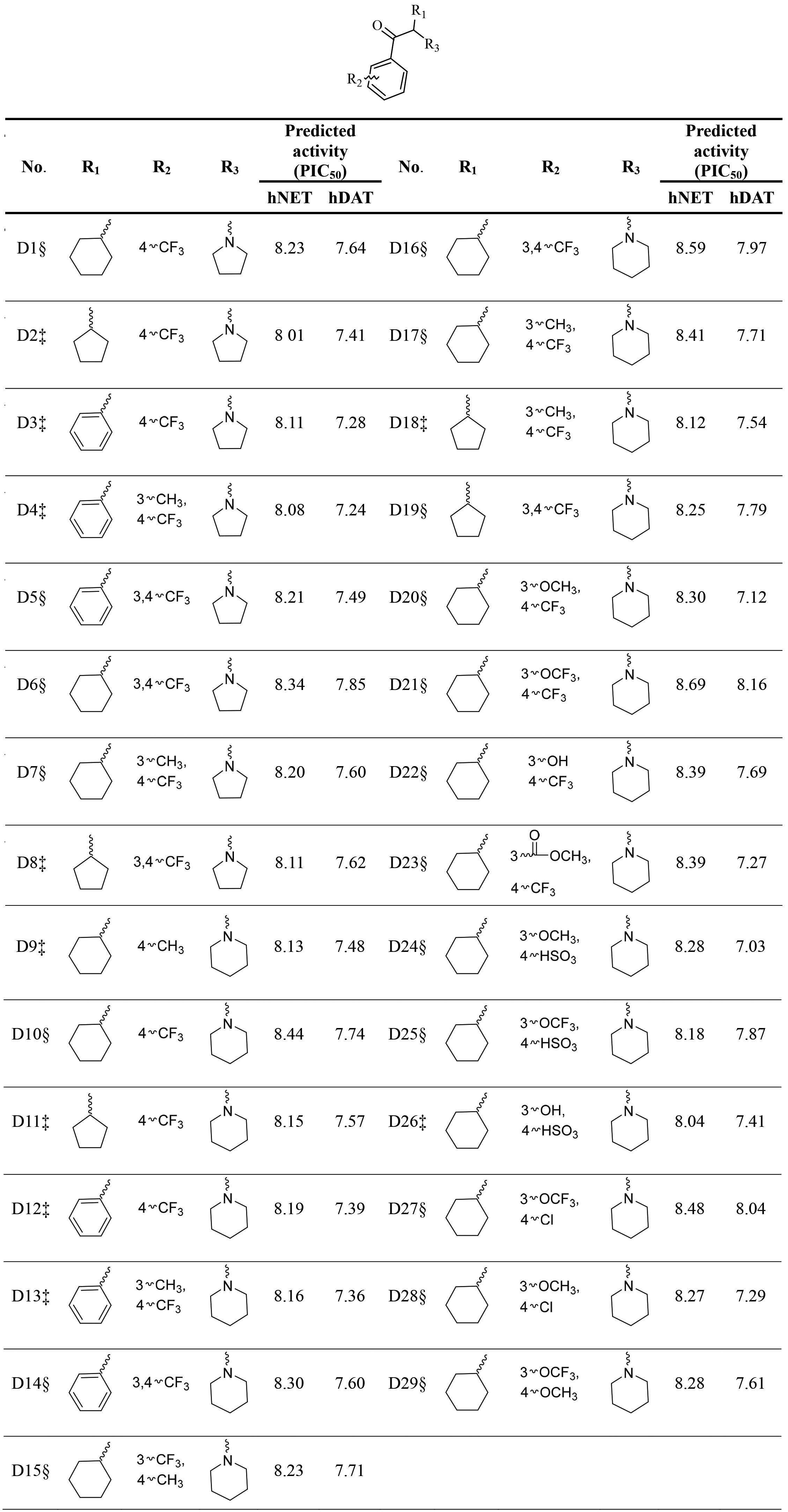
|
Table 4
ADME/T and medicinal chemistry friendliness properties of 29 designed molecules with high activities
| No | Absorption | Distribution | Metabolism | Excretion | Toxicity | Medicinal chemistry | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HIA | VDss | BBB | CNS | 2D6 | 3A4 | 1A2 | 1A2 | 2C19 | 2C9 | 2D6 | 3A4 | CLtot | AMES | Hepato toxicity | Skin sensitization | PAIN | Brenk | SA | |
| Substrate | Inhibitor | ||||||||||||||||||
| D1 | 92.35 | 1.10 | 0.55 | Yes | Yes | Yes | No | No | No | Yes | No | 1.03 | No | Yes | No | 0 | 0 | 2.85 | |
| D2 | 95.22 | 1.00 | 0.60 | No | Yes | Yes | No | No | No | Yes | No | 1.09 | No | No | No | 0 | 0 | 2.74 | |
| D3 | 93.02 | 1.08 | 0.71 | Yes | Yes | Yes | Yes | No | No | Yes | No | 1.00 | No | Yes | No | 0 | 0 | 2.61 | |
| D4 | 93.46 | 1.13 | 0.72 | Yes | Yes | Yes | Yes | No | No | Yes | No | 0.97 | No | Yes | No | 0 | 0 | 2.76 | |
| D5 | 90.67 | 0.95 | 0.66 | Yes | Yes | No | No | No | No | Yes | No | 0.71 | No | Yes | No | 0 | 0 | 2.91 | |
| D6 | 90.08 | 0.86 | 0.51 | No | Yes | No | No | No | No | No | No | 0.73 | No | Yes | No | 0 | 0 | 3.15 | |
| D7 | 92.87 | 1.09 | 0.56 | Yes | Yes | Yes | No | No | No | Yes | No | 0.99 | No | Yes | No | 0 | 0 | 3.00 | |
| D8 | 92.71 | 0.82 | 0.59 | No | Yes | No | No | No | No | Yes | Yes | 0.78 | No | Yes | No | 0 | 0 | 3.04 | |
| D9 | 94.34 | 1.34 | 0.57 | Yes | Yes | No | No | No | No | Yes | No | 1.14 | No | Yes | Yes | 0 | 0 | 2.78 | |
| D10 | 91.96 | 1.12 | 0.54 | Yes | Yes | Yes | No | No | No | Yes | No | 0.97 | No | Yes | No | 0 | 0 | 2.96 | |
| D11 | 94.83 | 1.02 | 0.59 | No | Yes | Yes | No | No | No | Yes | No | 1.05 | No | No | No | 0 | 0 | 2.85 | |
| D12 | 92.63 | 1.12 | 0.69 | Yes | Yes | Yes | Yes | No | No | Yes | No | 0.95 | No | Yes | No | 0 | 0 | 2.71 | |
| D13 | 93.07 | 1.16 | 0.70 | Yes | Yes | Yes | Yes | No | No | Yes | No | 0.92 | No | Yes | No | 0 | 0 | 2.86 | |
| D14 | 90.28 | 0.98 | 0.65 | Yes | Yes | Yes | No | No | No | Yes | No | 0.66 | No | Yes | No | 0 | 0 | 3.01 | |
| D15 | 92.88 | 0.81 | 0.69 | Yes | No | Yes | No | No | No | Yes | No | 0.92 | No | Yes | No | 0 | 0 | 3.17 | |
| D16 | 89.69 | 0.88 | 0.49 | No | Yes | No | No | No | No | No | No | 0.68 | No | Yes | No | 0 | 0 | 3.26 | |
| D17 | 92.49 | 1.11 | 0.55 | Yes | Yes | Yes | No | No | No | Yes | No | 0.94 | No | Yes | No | 0 | 0 | 3.11 | |
| D18 | 95.12 | 1.04 | 0.60 | No | Yes | Yes | No | No | No | Yes | No | 1.01 | No | No | No | 0 | 0 | 3.00 | |
| D19 | 92.32 | 0.85 | 0.57 | No | Yes | No | No | No | No | Yes | Yes | 0.73 | No | No | No | 0 | 0 | 3.15 | |
| D20 | 95.57 | 0.81 | 0.61 | No | Yes | Yes | No | No | No | Yes | Yes | 0.95 | No | No | No | 0 | 0 | 3.28 | |
| D21 | 89.06 | 0.59 | 0.48 | No | Yes | No | No | No | No | No | Yes | 0.44 | No | No | No | 0 | 0 | 3.58 | |
| D22 | 91.38 | 0.96 | 0.16 | Yes | Yes | No | No | No | No | Yes | No | 0.90 | No | No | No | 0 | 0 | 3.18 | |
| D23 | 92.32 | 0.74 | No | Yes | Yes | No | No | No | No | Yes | 0.91 | No | No | No | 0 | 0 | 3.58 | ||
| D24 | 96.19 | 0.83 | 0.72 | No | No | Yes | Yes | No | No | Yes | No | 1.13 | No | No | No | 0 | 0 | 3.16 | |
| D25 | 92.33 | 0.58 | 0.76 | No | No | Yes | No | No | No | No | No | 0.69 | No | No | No | 0 | 0 | 3.38 | |
| D26 | 94.57 | 1.04 | 0.21 | No | No | No | No | No | No | No | No | 1.16 | No | No | No | 0 | 0 | 2.97 | |
| D27 | 90.87 | 0.52 | 0.74 | No | No | Yes | No | No | No | No | No | 0.69 | No | No | No | 0 | 0 | 3.33 | |
| D28 | 94.73 | 0.77 | 0.68 | No | No | Yes | No | No | No | Yes | No | 1.12 | No | No | No | 0 | 0 | 3.17 | |
| D29 | 91.61 | 0.68 | 0.44 | No | Yes | Yes | No | No | No | No | No | 0.76 | No | No | No | 0 | 0 | 3.54 | |
3.5Design of novel derivatives
According to the information based on compound 1 provided by Field-based QSAR models, firstly this work replaced methyl of R
3.6ADME/T profiles and medicinal chemistry friendliness properties of candidates
The ADME/T properties and medicinal chemistry of 29 high active candidates were predicted using pre-ADMET web servers, and the results are shown in Table 4. As results, the rang of HIA was from 89.056% to 98.477%, indicating that all candidates performed high absorption properties from intestine. The VDss of most molecules was between 0.71 and 2.81 L/kg, which meant that they were evenly distributed in the plasma and in tissues, while compounds D21, D25, D27 and D29 with low 0.71 L/kg were distributed more in the plasma rather than in tissues. There was no compound to be considered as poorly distributed to the brain and unable to penetrate the CNS according BBB and CNS permeability. For metabolism, all designed molecules were predicted as substrates for the CYP450 2D6/3A4/1A2 subtype, which indicated they could be metabolized by P450 isoforms. In addition, compound D6, D16, D25, D26, D27 and D29 could not inhibit the CYP450 family, whereas the other compounds might inhibit the CYP450 1A2/2C19/2C9/2D6/3A4 subtype. The predicted total clearance of all compounds demonstrated they could be cleared by combing hepatic and renal tissue. Based on the predicted the toxicity, some candidates would be harmful for the liver, and only a few compounds were likely to be associated with skin sensitization, and none of them performed mutagenic potency. Finally, as shown the medicinal chemistry friendliness properties in Table 4, there was no problematic fragment in novel compounds assessing by PAINS and Brenk filter. The SA of all designed candidates ranged from 2.85 to 3.58, indicating the low synthetic complexity of structure and high synthetic feasibility. Taking all predicted ADMET properties and medicinal chemistry profiles into consideration, the designed 29 novel derivatives could be served as lead compounds for further development of NDRIs.
4.Conclusions
This work established firstly field-based QSAR models of aminoketone derivatives as hNET and hDAT reuptake inhibitors based on the potent poses by molecular docking, and the best models performed good predictability in in/external validations and reliability. According to the contour maps of QSAR models for hNET and hDAT, three strategies were proposed to improve the activity: (I) the bulky groups with suitable carbon chain at R
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant number 81903544); the Incubation Fund of National scientific research in Huanghuai University (grant number XKPY-2018008); and the Key Scientific Research Projects of the Universities in Henan Province (grant number 19A150033).
Conflict of interest
The authors declare that they have no conflict of interest.
References
[1] | Madhav KC, Sherchand SP, Sherchan S. Association between screen time and depression among US adults. Prev Med Rep. (2017) ; 8: : 67. doi: 10.1016/j.pmedr.2017.08.005. |
[2] | Zhang Y, Zheng GX, Fu TT, Hong JJ, Li FC, Yao XJ, et al. The binding mode of vilazodone in the human serotonin transporter elucidated by ligand docking and molecular dynamics simulations. Phys Chem Chem Phys. (2020) ; 22: (9): 5132. doi: 10.1039/c9cp05764a. |
[3] | Xue WW, Fu TT, Zheng GX, Tu G, Zhang Y, Yang FY, et al. Recent advances and challenges of the drugs acting on monoamine transporters. Curr Med Chem. (2020) ; 27: (23): 3830. doi: 10.2174/0929867325666181009123218. |
[4] | Xue WW, Wang PP, Tu G, Yang FY, Zheng GX, Li XF, et al. Computational identification of the binding mechanism of a triple reuptake inhibitor amitifadine for the treatment of major depressive disorder. Phys Chem Chem Phys. (2018) ; 20: (9): 6606. doi: 10.1039/c7cp07869b. |
[5] | Baune BT, Brignone M, Larsen KG. A Network Meta-Analysis comparing effects of various antidepressant classes on the Digit Symbol Substitution Test (DSST) as a measure of cognitive dysfunction in patients with major depressive disorder. Int J Neuropsychopharmacol. (2018) ; 21: (2): 97. doi: 10.1093/ijnp/pyx070. |
[6] | Oliveira MR, Chenet AL, Duarte AR, Scaini G, Quevedo J. Molecular mechanisms underlying the anti-depressant effects of resveratrol: a review. Mol Neurobiol. (2018) ; 55: (6): 4543. doi: 10.1007/s12035-017-0680-6. |
[7] | Fu TT, Zheng GX, Tu G, Yang FY, Chen YZ, Yao XJ, et al. Exploring the binding mechanism of metabotropic glutamate receptor 5 negative allosteric modulators in clinical trials by molecular dynamics simulations. Acs Chem Neurosci. (2018) ; 9: (6): 4192. doi: 10.1021/acschemneuro.8b00059. |
[8] | Schaefer JD, Scult MA, Caspi A, Arseneault L, Belsky DW, Hariri AR, et al. Is low cognitive functioning a predictor or consequence of major depressive disorder? A test in two longitudinal birth cohorts. Dev Psychopathol. (2017) ; 1. doi: 10.1017/S095457941700164X. |
[9] | Targum SD, Wedel PC, Fava M. Changes in cognitive symptoms after a buspirone-melatonin combination treatment for Major Depressive Disorder. J Psychiatr Res. (2015) ; 68: : 392. doi: 10.1016/j.jpsychires.2015.04.024. |
[10] | Ekman M, Granstrom O, Omerov S, Jacob J, Landen M. The societal cost of depression: evidence from 10,000 Swedish patients in psychiatric care. J Affect Disord. (2013) ; 150: (3): 790. doi: 10.1016/j.jad.2013.03.003. |
[11] | Bortolato B, Miskowiak KW, Kohler CA, Maes M, Fernandes BS, Berk M, et al. Cognitive remission: a novel objective for the treatment of major depression? BMC Med. (2016) ; 14: : 9. doi: 10.1186/s12916-016-0560-3. |
[12] | Salagre E, Sole B, Tomioka Y, Fernandes BS, Hidalgo-Mazzei D, Garriga M, et al. Treatment of neurocognitive symptoms in unipolar depression: a systematic review and future perspectives. J Affect Disord. (2017) ; 221: : 205. doi: 10.1016/j.jad.2017.06.034. |
[13] | Levada OA, Troyan AS. Insulin-like growth factor-1: a possible marker for emotional and cognitive disturbances, and treatment effectiveness in major depressive disorder. Ann Gen Psychiatry. (2017) ; 16: : 38. doi: 10.1186/s12991-017-0161-3. |
[14] | Schmeichel BE, Berridge CW. Neurocircuitry underlying the preferential sensitivity of prefrontal catecholamines to low-dose psychostimulants. Neuropsychopharmacology. (2013) ; 38: (6): 1078. doi: 10.1038/npp.2013.6. |
[15] | Bymaster FP, Golembiowska K, Kowalska M, Choi YK, Tarazi FI. Pharmacological characterization of the norepinephrine and dopamine reuptake inhibitor EB-1020: implications for treatment of attention-deficit hyperactivity disorder. Synapse. (2012) ; 66: (6): 522. doi: 10.1002/syn.21538. |
[16] | Spencer RC, Devilbiss DM, Berridge CW. The cognition-enhancing effects of psychostimulants involve direct action in the prefrontal cortex. Biol Psychiatry. (2015) ; 77: (11): 940. doi: 10.1016/j.biopsych.2014.09.013. |
[17] | Miceli M, Gronier B. Psychostimulants and atomoxetine alter the electrophysiological activity of prefrontal cortex neurons, interaction with catecholamine and glutamate NMDA receptors. Psychopharmacology. (2015) ; 232: (12): 2191. doi: 10.1007/s00213-014-3849-y. |
[18] | Xue WW, Wang PP, Li B, Li YH, Xu XF, Yang FY, et al. Identification of the inhibitory mechanism of FDA approved selective serotonin reuptake inhibitors: an insight from molecular dynamics simulation study. Phys Chem Chem Phys. (2016) ; 18: (4): 3260. doi: 10.1039/c5cp05771j. |
[19] | Park JE, Song C, Choi K, Sim T, Moon B, Roh EJ. Synthesis and biological evaluation of novel 3,4-diaryl lactam derivatives as triple reuptake inhibitors. Bioorg Med Chem Lett. (2013) ; 23: (20): 5515. doi: 10.1016/j.bmcl.2013.08.062. |
[20] | Xing B, Li YC, Gao WJ. Norepinephrine versus dopamine and their interaction in modulating synaptic function in the prefrontal cortex. Brain Res. (2016) ; 1641: (Pt B): 217. doi: 10.1016/j.brainres.2016.01.005. |
[21] | Pliszka S, Issues AWGoQ. Practice parameter for the assessment and treatment of children and adolescents with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. (2007) ; 46: (7): 894. doi: 10.1097/chi.0b013e318054e724. |
[22] | Schmeichel BE, Zemlan FP, Berridge CW. A selective dopamine reuptake inhibitor improves prefrontal cortex-dependent cognitive function: potential relevance to attention deficit hyperactivity disorder. Neuropharmacology. (2013) ; 64: : 321. doi: 10.1016/j.neuropharm.2012.07.005. |
[23] | Wang PP, Zhang XY, Fu TT, Li S, Li B, Xue WW, et al. Differentiating physicochemical properties between addictive and nonaddictive ADHD drugs revealed by molecular dynamics simulation studies. ACS Chem Neurosci. (2017) ; 8: (6): 1416. doi: 10.1021/acschemneuro.7b00173. |
[24] | Bruder GE, Alvarenga JE, Alschuler D, Abraham K, Keilp JG, Hellerstein DJ, et al. Neurocognitive predictors of antidepressant clinical response. J Affect Disord. (2014) ; 166: : 108. doi: 10.1016/j.jad.2014.04.057. |
[25] | Gaulton A, Hersey A, Nowotka M, Bento AP, Chambers J, Mendez D, et al. The ChEMBL database in 2017. Nucleic Acids Res. (2017) ; 45: (D1): D945. doi: 10.1093/nar/gkw1074. |
[26] | Goodsell DS, Zardecki C, Di Costanzo L, Duarte JM, Hudson BP, Persikova I, et al. RCSB protein data bank: enabling biomedical research and drug discovery. Protein Sci. (2020) ; 29: (1): 52. doi: 10.1002/pro.3730. |
[27] | Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. (2018) ; 46: (W1): W296. doi: 10.1093/nar/gky427. |
[28] | Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics. (2007) ; 23: (21): 2947. doi: 10.1093/bioinformatics/btm404. |
[29] | Zheng GX, Xue WW, Yang FY, Zhang Y, Chen YZ, Yao XJ, et al. Revealing vilazodone’s binding mechanism underlying its partial agonism to the 5-HT1A receptor in the treatment of major depressive disorder. Phys Chem Chem Phys. (2017) ; 19: (42): 28885. doi: 10.1039/c7cp05688e. |
[30] | Wang PP, Yang FY, Yang H, Xu XF, Liu D, Xue WW, et al. Identification of dual active agents targeting 5-HT1A and SERT by combinatorial virtual screening methods. Biomed Mater Eng. (2015) ; 26: (Suppl 1): S2233. doi: 10.3233/BME-151529. |
[31] | Wang PP, Fu TT, Zhang XY, Yang FY, Zheng GX, Xue WW, et al. Differentiating physicochemical properties between NDRIs and sNRIs clinically important for the treatment of ADHD. Biochim Biophys Acta Gen Subj. (2017) ; 1861: (11 Pt A): 2766. doi: 10.1016/j.bbagen.2017.07.022. |
[32] | Zheng GX, Yang FY, Fu TT, Tu G, Chen YZ, Yao XJ, et al. Computational characterization of the selective inhibition of human norepinephrine and serotonin transporters by an escitalopram scaffold. Phys Chem Chem Phys. (2018) ; 20: (46): 29513. doi: 10.1039/c8cp06232c. |
[33] | Schrödinger Release 2018-2: Maestro, Schrödinger, LLC, New York, (2018) . |
[34] | Schrödinger Release 2018-2: LigPrep, Schrödinger, LLC, New York, (2018) . |
[35] | Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J Phys Chem B. (2001) ; 105: : 6474. doi: 10.1021/jp003919d. |
[36] | Schrödinger Release 2018-2: Epik, Schrödinger, LLC, New York, (2018) . |
[37] | Schrödinger Release 2018-2: Glide, Schrödinger, LLC, New York, (2018) . |
[38] | Schrödinger Release 2018-2: Prime, Schrödinger, LLC, New York, (2018) . |
[39] | Schrödinger Release 2018-2: Field-based QSAR, Schrödinger, LLC, New York, (2018) . |
[40] | Zheng GX, Xue WW, Wang PP, Yang FY, Li B, Li XF, et al. Exploring the Inhibitory Mechanism of Approved Selective Norepinephrine reuptake inhibitors and reboxetine enantiomers by molecular dynamics study. Sci Rep-Uk. (2016) ; 6: : 26883. doi: 10.1038/Srep26883. |

