Review Article
Lack of applicability of the Enterocyte Chloride ion secretion paradigm to the Pathology of Cystic Fibrosis

Michael L Lucas*
Institute of Life Sciences, School of Medicine, Veterinary & Life Sciences, Glasgow University, Glasgow, G12 8QQ Scotland, UK
*Address for Correspondence: Michael L Lucas, Institute of Life Sciences, School of Medicine, Veterinary & Life Sciences, Glasgow University, Glasgow, G12 8QQ Scotland, UK, Tel: 441413304494; Email: Michael.Lucas@glasgow.ac.uk
Dates: Submitted: 20 March 2017; Approved: 20 October 2017; Published: 23 October 2017
How to cite this article: Lucas ML. Lack of applicability of the Enterocyte Chloride ion secretion paradigm to the Pathology of Cystic Fibrosis. Arch Asthma Allergy Immunol. 2017; 1: 061-085. DOI: 10.29328/journal.aaai.1001007
Copyright License: © 2017 Lucas ML. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Summary
This review examines of the concept of a defective chloride channel in epithelial cells being a major cause of cystic fibrotic pathophysiology. The central concept of the defective chloride ion channel paradigm is that faulty CFTR protein or failed delivery of CFTR protein to the mucosal membrane of epithelial cells is the basis of cystic fibrosis. Defective placement or function of CFTR prevents hydration of bronchial mucus that is normally caused by epithelial cells; these are capable through chloride ion secretion of transporting fluid to the mucosal surface. This concept relies heavily on a paradigm taken from intestinal physiology-namely that the intestinal epithelial cell secretes chloride ion and fluid and that this has conferred heterozygote selective advantage in carriers of the cystic fibrosis gene. This present review examines the evidence for that hypothesis and assembles evidence from past studies that it is the smooth muscle cell that is of greater relevance. This review does not aim to provide an overview of current research into cystic fibrosis. The intention is to provide an overview of past research that led to the concept of a failure of epithelial cells to hydrate bronchial mucus because of compromised CFTR function. It is important to present all past evidence for aspects of the chloride secretion hypothesis and its associated heterozygote advantage concept so that the important evidential milestones can be re-assessed.
Introduction
This historical approach to past CF research shows that the evidence for the mucus hydration view is slender and can be called into question as providing adequate proof. This approach also indicates that associated smooth muscle cell pathophysiology in CF has been overlooked but may prove to be the more productive hypothesis. Present views on the pathophysiology of cystic fibrotic lung function have drawn heavily on developments within the field of intestinal secretion physiology, having assumed that the current paradigm of enterocyte secretion is, in almost all respects, the correct one for explaining how secretory diarrhoeal disease occurs. This model for secretion assumes that microbes and associated pathogens act on normally occurring fluid secretion from the enterocytes by pathologically enhancing chloride ion secretion, which in turn due to osmotic imbalance at the brush ZDFSVVVXV compels the movement of fluid into the lumen. The enterocytes are purported to have a normally occurring chloride secretion mechanism, in addition to their undoubted ability to absorb fluid. Within the brush ZDFSVVVXV of the enterocyte is a chloride channel (the cystic fibrosis transmembrane regulator or CFTR protein) that participates in a putative chloride secretion process. As the CFTR channel is defective in CFTR patients, for various reasons to be discussed, aspects of cystic fibrosis pathology became inextricably intertwined with small intestinal secretory diseases because of the common chloride ion channel in the affected lung and intestinal cell types. Particular weight was given to the possibility that heterozygote advantage in cystic fibrosis could be explained by a defective CFTR channel. Since CFTR mediated chloride ion secretion is now assumed to underpin most forms of secretory diarrhoeal disease, a damaged CFTR channel would minimise or event prevent fluid secretion in intestinal diarrhoeal disease. The inability of CF gene heterozygotes to respond to specific lethal pathogens would be consistent with this conferring an indisputable heterozygote advantage. This lack of fluid secretion was a satisfying explanation for the pulmonary pathophysiology of cystic fibrosis and also purported to explain how life threatening diarrhoeal disease could arise, particularly in a disease such as cholera.
The inference from small intestinal studies was that CFTR heterozygote intestinal tissue responded less to secretory stimuli and for that reason, a similar phenomenon might occur in cystic fibrotic patients. Translated into cystic fibrotic pathology, a failure to secrete fluid by analogous mucosal cells in the bronchial epithelium would mean a failure to lubricate bronchial mucus and more importantly make the viscous mucus more difficult to clear. This plus alteration in thickness of local fluid films would either reduce airflow or allow identical rates of air flow to be achieved only through greater work done to overcome the resistance to flow. The intellectual linkage between intestinal fluid secretion with production of sticky mucus in cystic fibrosis through the common theme of epithelial cell fluid secretion is an interesting example of a sub-hypothesis providing apparent support for a larger more inclusive hypothesis: a set of observations on cystic fibrotic tissues apparently adds substantial weight to the intestinal secretion paradigm. Falsification of the cystic fibrotic sub-hypothesis i.e. cessation of fluid secretion in the CF epithelium is not fatal to the larger intestinal secretion paradigm. However, the converse phenomenon of falsification of the intestinal secretion model of chloride secretion through CFTR channels completely discredits current views on why the CF gene persists in the general population. This is not simply an intellectual question of academic interest only: it implies that the search for effective therapeutic agents for CF is not soundly based on correct conclusions about intestinal secretion. The difficulty for CF research is that the enterocyte secretion hypothesis for diarrhoeal disease is eminently challengeable [1]. If this is true, then several current directions within CF research are based on the faulty view that a very prominent aspect of CF pathology originates from the bronchial epithelial cells’ inability to secrete fluid. If the intestinal chloride ion secretion paradigm is false, it is evident that the causes of life threatening diarrhoeal disease lie outside of the epithelial cell. It is also probable that the focus on restoration of the defective chloride ion channel within epithelial tissues is unlikely to be a successful means of improving either secretion in the intestine or poor lung function in cystic fibrosis. A recent focus on gene delivery to respiratory epithelium to improve the defective CF channel had led to minor improvement in forced expiratory volume [2] and will continue to disappoint, if the central tenet of fluid secretion through chloride ion secretion is fundamentally wrong.
To explain why an alternative view is possible both for intestinal pathology in diarrhoeal disease and lung pathology in CF, it is necessary to describe what the intestinal chloride ion secretion model is and how it has overshadowed an older paradigm, based on vascular smooth muscle and not epithelial changes in disease states. It can then be seen that the smooth muscle model for intestinal secretion and rectification by CFTR acting agents is applicable both to the respiratory and the intestinal tract. Not only will it be argued that smooth muscle constriction is an equally valid explanation for respiratory and intestinal pathology, it will be seen that the smooth muscle model is the better paradigm since it encompasses observations which have been left uncommented on in the secretion literature. Finally, recent work on CFTR enhancers is briefly reviewed and the suggestion noted that these latest compounds may also be acting on smooth muscle rather than epithelial cells.
The enterocyte chloride ion secretion hypothesis as a basis for secretory diarrhoeal disease
The current concept of fluid movement from the serosal side of the enterocyte into the lumen is essentially a biochemical intellectual construct since it relies on the internal energy from the epithelial cell to power ion flows across the cell. The ion flows themselves result from specific carriers and channels that move ions in accordance with the formalism of enzyme kinetics as expected from the canonical foundations of biochemistry. Since prior to the enterocyte secretion view, the absorption of nutrients, sodium ion and fluid movement was so well explained by a cellular based view, it is perhaps not so surprising that an explanation for secretion of fluid should also have been based on a biochemical model. However, in this case, it is evident that biochemistry based models cannot transcend the short-comings of all explanations being cellular based and biochemical in origin. In this case, an explanation must be sought in the physical forces that cause mass transport, as is often the case in physiology. A more likely explanation is that of mass transport of fluid because of physical forces acting within tissues, although it must be acknowledged that the transported fluid should have the same composition as it had at its origin. A likely explanation for secretion of fluids of differing composition is that extruded fluid is operated on by the absorptive ion uptake mechanisms that are undoubtedly present in the enterocytes.
The purely biochemical model requires that chloride ion enters the enterocyte cell interior and leaves it through a mucosally sited chloride channel (Figure 1), assuming that the electrochemical gradient is correctly aligned for chloride ion extrusion into the lumen of the small intestine. The Na+/K+/2Cl- co-transporter (NKCC1 or NKCC2 depending on the isoform) in the serosal ZDFSVVVXV allows entry of chloride ion into the cell. This entry is energetically dependent on the ionic gradients for the three ions and it is likely that the sodium ion: potassium ion ATPase, Na+/K+ ATPase, provides a sodium ion gradient aligned for entry into the cell to pull chloride and potassium ions into the cell interior. Stoicheiometry dictates that two chloride ions enter the cell for every sodium ion returning via the triporter and hence the intracellular chloride ion concentration should build up.
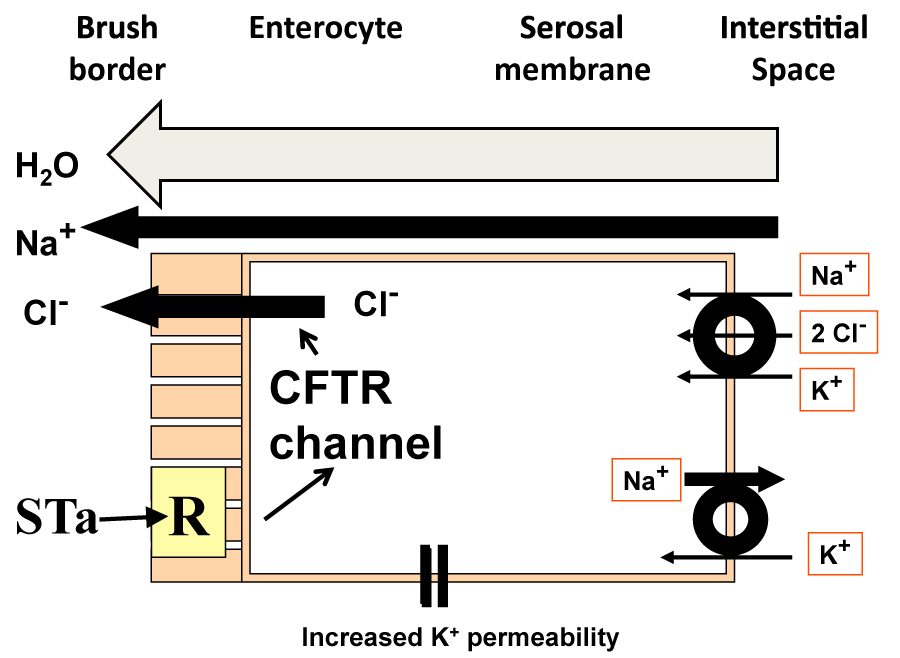
Figure 1:
Given that the chloride ion should distribute across the mucosal membrane in accordance with the electrochemical gradient, the assumption is that at equilibrium, the tendency for chloride ion to enter the cell from the lumen because of the adverse chemical gradient is exactly balanced by the tendency for the chloride ion to exit through the CFTR channel. When chloride ions enter the enterocyte via the triporter, the assumption is that the concentration is slightly above equilibrium and chloride ion will be extruded as electrochemical equilibrium is restored. From this, it can be seen that it is important for chloride ion extrusion that the cell membrane potential difference is maintained, a function attributed to serosal membrane potassium ion permeability. Given the presence of the biochemical machinery within the cell, routes for chloride ion translocation exist from the serosal to the mucosal surface of the enterocyte, together with appropriately balanced electrochemical forces that should allow chloride ion transmission to the mucosal surface. The extrusion of chloride ion together with sodium ion presumably through the paracellular pathways causes an accumulation of solute near the enterocyte brush ZDFSVVVXV. This provides an osmotic pressure pulling fluid through the lateral spaces, ultimately from the sub-epithelial interstitial space. As a result, chloride ion and water move into the lumen. If this process can be enhanced by enterotoxins, then this would form the basis for enhanced intestinal secretion in secretory diarrhoeal disease. In the diagram, a receptor, R, for heat stable E. coli enterotoxin (STa) is present in the brush ZDFSVVVXV. The enterocyte secretion view envisages that STa fits into the luminal receptor and elevates intracellular second messenger concentration, in this case cyclic GMP. This in turn, just as cAMP is thought to do for cholera toxin, acts via protein kinases on the CFTR channel causing it to become more permeable. Given no change in electrochemical gradients across the luminal membrane, it is possible that more chloride ions are extruded per unit electrochemical gradient and therefore more fluid is secreted.
While intellectually self-contained and satisfying within the constraints of in vitro work, the model does not survive challenge when confronted with observations from in vivo experiments. The canon of in vitro techniques used to measure 'secretion' such as isotope fluxes and electrical measurements are methodologically flawed [3]. In vivo experiments not using permeable isotopes to measure fluid secretion indicate that i) E. coli STa cannot cause secretion [4] despite a wide spread expectation that it should and that ii) potassium channel blockers have no effect on secretion [5]. These and many other observations in the historical literature on the causes of secretory diarrhoea point to an alternative view that has been dormant in the secretion literature but can accommodate the data from the enterocyte secretion model whilst being itself a concept based on physical rather than chemical forces across the mucosa.
The intestinal arteriolar vasodilatation hypothesis as a basis for secretory diarrhoeal disease
An alternative model for fluid secretion is one that has repeatedly been put forward but has always been overshadowed by the epithelial cell fluid secretion model. This repeated intrusion of the physical forces model is often simply ignored. Its most convincing manifestation was as late as 1971 after the finding that when small intestine was perfused in vivo with cholera toxin, the intestine secreted profusely. When that tissue was removed and made into everted sacs, the tissue did not secrete or rather could not secrete in vitro, presumably because of the absence of the necessary physical forces across the mucosa [6]. The physical model is presented (Figure 2) together with the biochemical model to facilitate comparison.
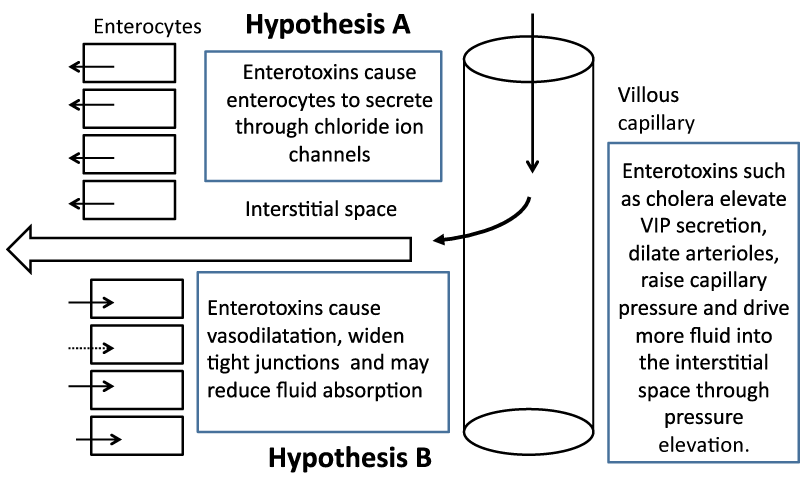
Figure 2:
The first hypothesis (A) requires that enterotoxins cause enterocytes to secrete through mucosal chloride ion channels after exposure of the tissue to bacterial enterotoxins. The second hypothesis (B) relies on the fact that the villous capillary is not completely impermeable to fluid and that fluid moves into the interstitial space. It does so because of the Starling forces that exist across all capillaries in that an excess of intraluminal capillary pressure over interstitial fluid pressure pushes fluid into the sub-epithelial space and greater colloid osmotic pressure of the blood pulls fluid back into the capillary. A steady volume state is achieved when the loss of fluid into the lumen is balanced by the tendency for interstitial fluid to move into the capillary. If there is a small net driving force for fluid movement into the lumen, this would be enhanced by increases in the tight junction widths. It should be remembered that an increase in tight junction width can also serve to promote fluid absorption if the luminal pressure, even if transiently, exceeds interstitial fluid pressure as it may do in the contracting intestine. If enterotoxins reduced fluid absorption, the model predicts increased seepage of fluid into the lumen since some of the exudate would presumably be reabsorbed by absorption.
Compromise of absorption would undoubtedly lead to diarrhoea facilitated by seepage of interstitial fluid. Copious and life threatening diarrhoea at the rate that occurs during a cholera episode is likely to be caused by profound vasodilatation. It is a physiological and somewhat counterintuitive fact that capillary pressure increases if there is vaso-dilatation of the immediately preceding arteriole. The increase in vasodilatation causes the capillary pressure to rise from average values of 15 mmHg to whatever the arterial pressure is on the capillary side of the arteriole and may rise to 40 mmHg or more. This increase in hydrostatic pressure forces fluid into the interstitial space and increases fluid secretion into the lumen by physical means. If accessory toxins increase epithelial and capillary endothelium paracellular pathways, as well as inhibit intestinal fluid absorption mechanisms, then this would lead to catastrophic fluid loss, essentially powered by the mechanical force of the heartbeat. It is unlikely that chemical reactions could match this degree of force or effect changes in interstitial volume in such a short time frame.
Evidence for this mechanism has come from experiments in vivo that examined fluid movement with changes in mean arterial pressure in small intestine that was rendered unable to absorb fluid [7]. This was achieved by using perfusates that were devoid of sodium ion and also contained ethyl-isopropyl amiloride. This was to prevent sodium ion absorption and hence fluid absorption from any exudate seeping to the luminal surface and bringing with it sodium ion for subsequent absorption. In these experiments fluid secretion occurred as arterial blood pressure fell, reached a maximum and then reverted to fluid absorption, exactly as predicted by changes in the Starling forces. Model A cannot accommodate any effect of arterial pressure since it does not incorporate physical forces into the model. In seems very likely that intestinal secretion is caused by smooth muscle events in the vasculature. If further such supportive evidence becomes available in the form of intracapillary pressure measurements during enterotoxin exposure and the enterocyte fluid secretion hypothesis is conclusively shown to be false, it is highly likely that pathophysiological changes in the lung in cystic fibrosis are also being caused by changes in bronchial or vascular smooth muscle tone.
The context and structure of the present review of the relevance of two models for intestinal secretion to cystic fibrosis heterozygote advantage
The context: Present views on compromised lung function in cystic fibrosis assume that bronchial epithelial cells in common with intestinal epithelium have the ability to secrete fluid. Further that this secretion optimises the fluidity of lung mucus. The failure to secrete fluid to hydrate lung mucus is a large factor in the poor lung function in cystic fibrosis. This disadvantage is counterbalanced by the fact that the failure to secrete has a positive outcome when the cystic fibrotic gut is challenged by cholera or other secretion causing toxins. This heterozygote advantage explanation has increasingly to confront the view that intestinal secretion mediated by chloride transport through the CFTR channel is not how secretory diarrhoeal disease arises. Fluid secretion does occur in some forms of secretory diarrhoeal disease but it is just as likely to be mediated by the Starling forces across intestinal capillaries and not by the enterocytes. This has consequences for symptom relief and restoration of function in cystic fibrosis suffers. If the underlying cause of the malady has been incorrectly identified, then attempts to remedy it by reference to the present intestinal secretory paradigm must fail or at least succeed only by chance. It is instructive to realise that although the enterocyte chloride secretion model has been dominant for over forty years, no specific intestinal channel blocker has been invented and is in use today to interrupt secretory diarrhoeal disease. There has been the development of enkephalinase inhibitors but arguably owing their emergence to a historical knowledge of opiate pharmacology. CFTR restoring agents show promise but it is not clear that their site of action is the enterocyte.
The relevance: The focus on an epithelial secretion model incorporated from the field of intestinal secretion may deflect attention from other areas of merit for correction of CF pathology such as smooth muscle physiology. An approach based on correction of faulty bronchial epithelial cell CFTR channels is to repair them by pharmacological intervention or by genetic engineering. However, interruption of channel activity is desirable in diarrhoeal disease but not necessarily so in CF if a poorly functioning channel confers heterozygote advantage. Correction of this mutation is the basis for much of current research into CF since repair of the CFTR channel by gene transfer therapy should again allow secretion and make mucus less viscous and difficult to shift, if the intestinal parallel is valid. Identification of how fluid secretion is achieved in the intestine and whether the enterocyte is the correct target tissue for anti-diarrhoeal compounds is all the more pressing since clinical trials and key experiments to rectify CF defects by genetic engineering appear to be disappointing. In recent clinical trials, [2] involving inhalation of a gene transfer vehicle, it is arguable that lung function improved not at all or only trivially as assessed by forced expiratory volume measurements in cystic fibrotics. While the claim was made that gene therapy had ‘stabilised’ the CF patients, an alternative and pessimistic but valid interpretation is that the ‘placebo’ treatment of usual therapeutic measures itself caused some deterioration in lung function and the gene therapy did nothing.
For this reason, the evidence for a selective advantage to CF heterozygotes has been examined together with a review of the work done to date on intestinal manifestations of CF disease. In particular, the likelihood that the selective advantage is one of muted secretion in cystic fibrosis through the enterocyte CFTR channels is reviewed and found to be unlikely. An alternative explanation for the findings in CF is proposed-namely that hitherto unremarked but increasingly emphasised changes in vascular and small intestine smooth muscle may make CF tissue incapable of vasodilatation and hence also unable to secrete fluid. This review interprets data from CF studies in the light of poor vasodilatory and perhaps generalised poor smooth muscle function. Vasodilatation is a credible mechanism for diarrhoeal fluid secretion in normal individuals challenged by enterotoxins and lack of vasodilatation is a good explanation for lack of secretion in CF patients. The fact that there may be a relative lack of vasodilatation in CF and a generalised smooth muscle pathology based on CFTR mutations indicates that there are very many other possibilities for the heterozygote advantage, none of which need involve the intestinal epithelium. Dysfunction in cystic fibrosis sufferers is reviewed necessarily in the context of evidence for the fluid secretion model in cystic fibrotic intestine and refractoriness to some forms of diarrhoeal disease. This makes many demands on the reader who is familiar with respiratory physiology but not necessarily with intestinal physiology and pathophysiology. In addition, evidence for the existing and also for the intended replacement secretion model is drawn from several sources as diverse as population genetics and the mathematics of fluid flow in tubes. For that reason, the essential points of each section are given here in prećis form, so that the basic theme can firstly be appreciated and then the specific detail and review of the evidence considered in greater detail in the indicated sections.
Prećis of salient points in the following sections of this review
• Cystic fibrosis pathology is thought to be related mainly to the defective cystic fibrotic transmembrane regulator (CFTR) channel.
• The assumed function of this channel is chloride ion and water secretion from lung tissue, resulting in failure to hydrate mucus when channel function is impaired by a deleterious mutation.
• Repair of this channel in lung epithelium is regarded as a desirable therapeutic aim.
• A difficulty with this approach is the reliance on the correctness of the enterocyte chloride secretion model to explain diarrhoeal secretory disease and its incorporation into the CF model of poor lung function.
• Gene transfer therapies have to date produced only very minor, possibly even negligible improvements in lung function.
• If enterocyte chloride ion secretion does not occur, it is also likely that it does not occur in epithelial tissue in the respiratory system and cannot be modified to effect fluid secretion.
• The persistence of the cystic fibrosis gene in the population is proposed to be the conferral of heterozygote selective advantage. This view is largely based on family size considerations and a conjecture that secretory diarrhoeal diseases, particularly cholera, may not occur as frequently in CF heterozygotes.
• The human evidence for heterozygote advantage is based on family size which is an act of human choice and not selection pressure. There is also no evidence for a selective advantage stemming from refractoriness to cholera.
• Early work on CF tissue points to a chloride ion transport abnormality but work on human heterozygotes points to a chloride absorption defect and not a secretion defect.
• Mucus is tenacious in the intestine, evidenced by difficult to reconcile unstirred layer measurements with slightly better glucose absorption measurements.
• Overlooked older histology indicates swollen villi in CF sufferers indicative of unrestrained absorption in the intestine not a failure to secrete fluid. In animal studies, thicker smooth muscle is found in cftr knockouts.
• Mathematical modelling of lung function shows increased damping and stiffness in cf mouse knockouts, i.e. physical factors associated with airways alteration.
• Past observations indicate a failure in cystic fibrotic subjects to vasodilate, also a smooth muscle property. The most recent generation of CFTR channel inhibitors may also act on smooth muscle.
• A view that harmonises lung and intestinal pathology in cystic fibrosis is that both intestinal and vascular smooth muscle is thicker in CF, making the vasculature resistant to vaso-dilatation.
• The selective advantage in this case could be partial or multifactorial and relate to a failure to vasodilate. This would give some advantage in those diarrhoeal diseases that involve bacteria producing vasoactive agents that cause the tissues to vasodilate e.g. cholera toxin and VIP production.
• Other forms of selective advantage are conceivable based on refractoriness to vasodilatation e.g. cerebral vascular events.
• CF lung pathology may be more likely to respond to restoration of lung vasculature or bronchial smooth muscle ability to alter muscle tone.
• A more recent approach to CF therapy involves chloride channel activators but these also seem to involve alterations to bronchial smooth muscle and vascular smooth muscle
Review of the evidence for the epithelial focus of cystic fibrosis and the associated selective advantage hypothesis
Preface: The purpose of this main body of the review is to critique the evidence for the incorporation of the enterocyte secretion model into views on cystic fibrotic pathology. The persistence of the cf gene in the population is thought to arise through heterozygote advantage. If the adopted epithelial cell chloride secretion model is found to be false, then it is also evident that the reasons for heterozygote advantage cannot be referred back to the mutated cftr channel in the epithelium of lung tissue. The case is made for smooth muscle dysfunction being the essence of cystic fibrotic disease.
Introduction: Cystic fibrosis is an autosomal recessive disease that is carried by between 1 to 2 % of the Caucasian population and is therefore a common genetic disorder. The features of the disease include secretion of viscous mucus and, with that, a propensity to lung disease because respiratory mucus is difficult to shift from the lungs. Pancreatic enzyme and electrolyte secretion is affected which leads to upper gastrointestinal tract acidity as gastric acid is no longer neutralized, together with a need for pancreatic enzyme supplementation. There are also abnormal aspects to sweat gland secretions since cystic fibrotic sweat contains more chloride ion than usual. This ability to secrete sweat with a high chloride ion concentration forms the basis of a test for the occurrence of cystic fibrosis.
Defective chloride channel (CFTR) in cystic fibrosis
Heterozygote advantage concept: Selective advantage requires that there must be some form of selection pressure that favours the survival of certain genes. Otherwise, unfavourable genes put the carriers of such genes at such disadvantage that either they or their offspring have lesser survival rates than the normal population. This is particularly evident when a lethal gene is autosomal and dominant. A familiar example is the sickling gene which allows red cells to collapse onto malarial parasites in the blood of the heterozygote form giving heterozygote advantage in a malarial area.
The genetic defect: In the late 1980s, a genetic defect associated with cystic fibrosis was identified as coming from chromosome 7, the gene product of which was a defective chloride channel protein [8,9]. The channel is named CFTR (cystic fibrosis transmembrane regulator) because it regulates other ion transport channels. CFTR is a ubiquitous chloride channel protein that is found in many tissues but research at first focussed on the intestinal effects of CFTR since its discovery. In part, this is probably because of the dominating presence of the enterocyte secretion hypothesis in the field of intestinal secretion and the perceived likelihood of a similar role in lung epithelium to explain CF pathology. Two things are evident in the above example. With a lethal gene, the percentage of carriers in the population must inevitably be small, so small that any decline in allele frequency is difficult to detect unless there is massive screening of entire populations on a repeated basis, with allowance made for new entrants into the gene pool from outside the catchment area. It is therefore difficult to verify a decline in frequency of a rare lethal gene. The second point is that there has presumably to be an identifiable selection pressure for there to be selective advantage for the heterozygotes. When a population moves away from the selection factor e.g. when Africans are transported to America out of the malaria infested regions, the heterozygote advantage should disappear.
Likelihood of a selective advantage to heterozygote carriers of the CF gene
Evidence for selective advantage: As a recessively inherited trait, the cystic fibrotic gene should exit from the gene pool if the carriers of the trait fail to reproduce or die before reaching the age of reproduction. Given the present day incidence of the cystic fibrotic gene, it is possible to estimate from Hardy-Weinberg calculations that a mutation giving the defective gene may have arisen relatively recently in human history. This could arise as a spontaneous mutation, as recently as twenty five or so generations back, as late as 1100 AD, perhaps later given the life expectancy of the Middle Ages. On the other hand, it is also possible that the defective gene remains in the gene pool at a given frequency because the asymptomatic carriers of one defective allele have a survival advantage. The apparently higher than expected frequency of the CF gene in people of European or Caucasian origin has led to the search for the root cause of that selection pressure. In particular, there have been attempts to demonstrate the fact of selective advantage by showing the existence of increased frequency of descendants in people with cystic fibrotic lineage. The argument runs that there will be more people in such families having a cystic fibrosis sufferer in a previous generation when compared to the normal population. The grandchildren of grandparents who had a cystic fibrotic child should be more numerous than those of grandparents who did not, since the offspring should or at least might show some propensity to survive adult or childhood diseases or hazards.
The proposed evidence for selective advantage of the heterozygotes therefore mainly resides in two very similar studies of families with a cystic fibrotic child and those without, in an attempt to assess the outcomes for the descendants. In one study [10], families with a CF child were asked to participate and also to name for participation purposes, two comparable families from their friendship group, without a CF child. This device was intended to standardise for age, social grouping and living conditions on the basis that these families would not be too dissimilar or not be subjected to very different environments. When this was done, it became evident that one striking difference was that cystic fibrosis families had larger family sizes. This was statistically significant and indicated a mean difference of about 0.6 people per family. This conclusion from this Australian study was confirmed by a contemporary one in the US which ranked the families by family size. Unlike the Australian study [10], which provided an overall mean difference, the US study [11] compared by family size so that the size distribution was also known. The family size was looked for in the grandparental generation since they were parents who had had a CF child and were both heterozygotes for the cystic fibrotic gene. It was clear that, on average, combining all families over the size range, there were more offspring in the grandparental generation of the CF families group, the difference being about 0.91 children per family. On average, the size of the families was also larger. The normal modal value was two children per family but was three children in the antecedents of the CF families.
In general, the greater number of offspring arose through larger family size. This can be tested by applying the standard χ2 test to the various family size contingencies. The range of families was large, extending to family sizes in the low teens but these were size contingencies with relatively low frequencies of occurrence of one or two families. In accordance with recommended practise, contingency size should be selected to have four or more occurrences in each contingency. When this is done, difference in distribution by the χ2 test was shown to be highly significant, (p<0.01) meaning that the family size was larger in the CF antecedent generation. The greater preponderance of siblings in the preceding generation has been assumed to be proof of selective advantage. The appropriate calculations can then be done to estimate the extent of advantage in the face of selection pressure, as was indeed done [11] in accordance with the principles of quantitative genetics. However, this may indicate the extent of advantage only if it is accepted that the fact of excess births is an aspect of evolutionary advantage. Are there no other explanations?
With most forms of selective advantage, one might anticipate the unknowing and unself-conscious hand of evolution directing survival in the form of an advantage at the cellular level, such as a biochemical mechanism or the collapse of red blood cells onto a parasite, where there can also be no question of the involvement of conscious thought. In contrast, human family size is an act of human volition, involving the choice to have children. It is therefore not at all self-evident that the larger families’ sizes are a genuine manifestation of selective advantage.
Ancestral residence in a diarrhoeal disease endemic area: In the case of cystic fibrosis, after the discovery of the defective CFTR channel and its incorporation into the theoretical mechanism for fluid secretion, it was proposed that there was a selective advantage to not secreting fluid. If the population under consideration resided in an area where life threatening diarrhoeal diseases were present, the carriers of the dysfunctional CFTR channel would enjoy a selective advantage. The people in question would be those of European or at least Caucasian ancestry since they have the highest allele frequency and their ancestors should have lived in a diarrhoeal disease endemic area. The defective CFTR channel was initially proposed to be a response to cholera [12] and would require that the Caucasian ancestors lived near the source of cholera. Epidemics of cholera such as those that spread throughout Europe in previous centuries are generally agreed to have started close to the Bay of Bengal [13] where the disease is endemic, though not all would agree with the premise that cholera is solely of Asiatic origin [14]. In order to acquire a selective advantage, the Caucasian population should have resided for many generations near the Bay of Bengal. This question of prolonged residency is a problematical question for population geneticists and also for linguists interested in the spread of language. Accompanying the recognition that the Indo-Aryan languages have spread throughout the world is the associated but vexed problem of the geographical origin of the Indo-Aryan language speakers. Some authorities [15] place their origin in Anatolia in eastern Turkey, other authorities [16] provide evidence for their origin being in the Pontic-Caspian region near southern Russia. There is also the associated problem of what Caucasian means because it is essentially a euphemistic term for white people, assumed to be a sub-set of the Indo-Aryan population. There is also the caveat that the spread of language does not necessarily correspond to the spread of a migrating people.
On balance, it is likely that most Indo-Aryans did not reside near the Bay of Bengal for a long period of time. A subset of Indo-Aryans migrated south but was unlikely to have entered the region until 3000 BC or later. It seems that the fractional split in the Indo-Aryans and their late entry of some of them into the cholera endemic area makes it unlikely that cholera was the relevant selection pressure. At a later date, the view arose [17] that cholera was unlikely to have been the selection pressure. Exposure to E. coli enterotoxins was substituted as the likely factor as humans have been interacting with E. coli for longer. The problem with this modified hypothesis is that there is no obvious group or population that is particularly affected or even unaffected by E. coli enterotoxins. The selection pressure would be present everywhere with no obvious epicentre or reason for any particular population slowly to derive a selective advantage.
Studies on Indo-Aryan migration makes cholera an unlikely selection pressure
Possible selection pressures: There is the additional objection that the heat stable form of E. coli enterotoxin is not secretory and does not require a chloride channel to effect lack of fluid absorption, [18], although at present, few researchers in the field would agree with that view. If this is accepted, then the argument for the intestinal aspect of selection pressure becomes even weaker since only E. coli enterotoxins such as LT would be secretory. Enterotoxins that remove cells or alter tight junction permeability would also not constitute a pressure against which a defective chloride channel could provide an advantage. However, selective advantage can still be proposed if one discards the idea of CFTR being involved in secretion into the intestine (i.e. abandonment of the enterocyte secretion hypothesis) and one proposes instead a survival advantage to cystic fibrotics heterozygotes because of improved lung or smooth muscle function.
Improved lung function or altered smooth muscle physiology may be the selection pressure
Diagnosis of cystic fibrosis: Earlier work on cystic fibrosis investigated the feasibility of diagnosis without intrusive clinical function tests. This concentrated on measurement of skin chloride ion concentrations in the sweat and also testing blood samples. It had long been the received wisdom of midwives that cystic fibrosis could be detected by licking a new born baby’s forehead and checking for excessive saltiness. From this observation developed the diagnostic procedure of screening sweat in paediatric patients for excessive chloride ion concentrations. Normal sweat contains 16 mM chloride ion and 23 mM sodium ion while cystic fibrotic sweat contains 99 mM chloride ion and 101 mM sodium ion [19]. This itself seems to be an anomalous occurrence given that dysfunctional CFTR should be associated with failed chloride secretion in cystic fibrotic children. This was perhaps the first indication that the problem with possessing dysfunctional CFTR is not the failure to secrete but the failure to absorb through the broken channel. This alternative view would be consistent with physical secretion of isotonic fluid, with subsequent retrieval of cations and anions by absorption mechanisms, as occurs in the renal tubules. It is likely that sweat is initially an exudate of plasma secreted because of hydrostatic pressure gradients across the gland membranes with re-absorption of electrolyte content within the sweat ducts or secretory coil in order to provide a hypotonic secretion for evaporation at the skin surface. This model of isotonic exudation followed by selective retrieval of sodium and chloride ions would resemble that occurring during salivary secretion where again at low salivary flow rates, the secreted fluid is hypotonic, perhaps reflecting again the physiological imperative for a land dwelling mammal not to lose salt. High chloride concentration in the sweat might then reflect a failure to re-absorb chloride ion resulting in a high concentration in the secreted sweat.
An intestinal factor: Attempts to diagnose cystic fibrosis have included taking blood from a patient, spinning the red cells down and measuring the fall in short circuit current in intestinal tissue in vitro exposed to the serum. Cystic fibrotic serum reduced short-circuit current in rat jejunum [20] an effect that could be overcome by heating the serum. The action on short-circuit current was confirmed [21] for heterozygote and homozygote serum, although in a larger study, both heterozygote and normal blood serum reduced the short circuit current by 40% but the homozygote serum gave higher values with the median value clustered at closer to 65% inhibition [22]. This heat labile factor did not seem to be important when considering the possibility of inhibiting glucose uptake as assessed in vesicles [23], perhaps indicating that CF serum will act only on ion channels.
Experiments on in vitro intestinal function in cystic fibrotic tissue
In vitro experiments: Intestinal tissue taken during surgical operations to relieve intestinal obstructions can be investigated for intestinal function using the venerated in vitro techniques of short circuit current and isotope flux measurements, neither of which technique is fit for the purpose of detecting secretion [3,4]. In an Ussing chamber, small intestine from non-CF tissue demonstrated increased short circuit current after theophylline, prostaglandin E2, A23187 and bethanechol exposure while CF tissue did not. Similarly, in colonic tissue, theophylline and A23187 were able to elevate short-circuit current in non-CF but not in CF tissue. Reprising the situation with serum factor experiments, both CF and non-CF tissues demonstrated an increase in short-circuit current on perfusion with glucose, indicating that sodium ion-dependent glucose uptake was not inhibited [24]. Preliminary studies in a small group of biopsies showed that CF jejunum did not respond electrically to luminal acetylcholine or to PGE2 whilst retaining a muted response to glucose perfusion [25]. Similar short-circuit current studies showed an absence of a voltage response across the jejunum in CF patients when pilocarpine and PGE2 were given. Normal tissue experienced a rise in short-circuit current after dibutyryl AMP and A23187 exposure but the response in CF tissue was absent or very small [26]. Curiously, despite recording large intraluminal pressure waves in cystic fibrotic and normal jejuna, only the normal jejunum responded with changes in transmural voltage, implying the inability to mount a secretory response because of a lack of functioning chloride channel. The likelihood of CF tissue not receiving afferent information about pressure events or something unusual concerning CF smooth muscle [27] was not considered at the time.
Isotope fluxes in CF tissue failed to demonstrate the changes that were characteristic of non-CF tissue, particularly the increase in luminally directed isotopic chloride flux after theophylline exposure. Similar refractoriness of short-circuit current in CF tissue after 3-iso-butyl-methylxanthine (IBMX) exposure was confirmed, as well as the lack of enhancement in the luminally directed chloride (Jsm) flux in CF tissue [28]. In the former study, it is noteworthy that the chosen ‘secretagogues’ are all capable of causing vascular smooth muscle relaxation, which is relevant to the alternative explanation of the origin of the short circuit current expressed here.
Experiments on secretory function using CFTR knock-out mice
Knock out experiments: With the discovery of the defective CFTR gene product stemming from chromosome 7, this area has been targeted to provide heterozygous and homozygous knock-out mice for CFTR for comparison of secretory function with that of wild type mice. In humans, the defect concerning CFTR is its lack of functioning, mainly by a failure to be inserted into the luminal membrane but also by partial lack of function because of permeability defects, despite correct insertion into membranes. Mice that have had CFTR production capacity deleted from their DNA are models therefore of cystic fibrosis sufferers but are not exactly analogous, given the fact the human cystic fibrosis is characterised by mutations of the gene rather than absence. Given the ability to knock out genes in mice, investigations were initiated on the effect of an absent CFTR channel on the ability to secrete fluid, since, in accordance with the enterocyte secretion model, cftr (-/-) mice should not be able to secrete fluid in response to putatively known stimuli of intestinal secretion.
Short-circuit current studies: Using the ΔF508 mutant colon from heterozygous mouse, the short-circuit current responses (a proxy for secretion) were no different in CF and in normal mouse tissue to stimulation by VIP and by forskolin [29] neither were the short-circuit current responses to cholera toxin. Short circuit increases were identical after isoprenaline, E. coli STa enterotoxin, guanylin, lysyl-bradykinin and carbachol. Frusemide action was also not different in the two types of tissue. As previously, increase in short-circuit current was assumed to be chloride secretion and this is clearly not the case with STa exposure since no net secretion is ever found with this enterotoxin [18]. Again, these observations may be of greater relevance to what causes the rise in short circuit current. All these compounds either cause smooth muscle vasodilatation (VIP, forskolin, isoprenaline, bradykinin and carbachol) or inhibit sodium ion: hydrogen ion exchange and subsequent luminal acidification (STa, carbachol, guanylin), hence the increase in short-circuit current is again consistent with the vasodilatation reflex that has been proposed [30]. If one accepts, without reservation, the chloride ion secretion hypothesis, no advantage seemed to be conferred by the ΔF508 CF mutation.
An interesting aspect of short-circuit current findings in CF mice [28] is the likelihood that some if not all of the current is an inwardly directed electrogenic sodium ion current and not outwardly directed chloride ion current. This only becomes apparent when the electroneutral sodium ion absorption mechanism is compromised. Amiloride does little to short-circuit current in mouse colon prior to guanylin stimulation but it moderately inhibits the rise in short-circuit current after guanylin is administered. In the absence of chloride ion, the effect of guanylin is attenuated. In CF mouse colon also, amiloride also reduced the increment in current detectable after guanylin addition. In CF tissue as well as normal tissue, part of the short-circuit current increase occurs because guanylin has acted on the NHE: 3 system and ‘can cause electrogenic sodium absorption’ [31].
In other words, when the NHE: 3 exchanger is functioning, guanylin inhibits it and associated fluid absorption, with amiloride having no effect. When this electrically silent mechanism for fluid absorption is inhibited by guanylin, short circuit current increases because an electrogenic mechanism comes into existence; this is now inhibitable by amiloride whereas it wasn’t before.
Short-circuit current studies in knock-out mice confirm the ‘refractoriness’ in CF intestine to secretion, when this is equated to change in short-circuit current. Undoubtedly, short-circuit current does change but this is likely to be stimulation of an electrogenic sodium ion uptake current and not chloride ion secretion.
In vivo ligated loop studies: When fluid movement was measured in another mouse model by a fluid accumulation ratio method, cholera toxin caused a large luminal fluid to gut mass ratio in normal mice. There was a low ratio in homozygous mice and an intermediate value for heterozygote mice. It should be noted that short-circuit current was increased in the CFTR(+/-) mice but not as much as the CFTR (+/+) did [32]. Similarly, the actual values of fluid retained and gut weight are not given. While these observations are often taken as evidence for enterocyte chloride ion secretion, intercellular extrusion of fluid from the vasculature into the lumen also explains these findings. In this case, there would be a heterozygote advantage in not being able to extrude fluid into the lumen, with the cause of this residing within vascular smooth muscle.
In vivo studies show reduced secretion after exposure to cholera toxin in cf mice. This is consistent with reduced secretion through the chloride ion channel but an argument with greater validity is the refractoriness of CF smooth muscle to relaxation.
Aspects of histology: A curious aspect of the histology of cftrtm1Unc mouse [33]; (Figure 1) is that while there are mucus deposits in dilated crypts, an unremarked feature is the width of the villi in the cftr deleted mouse which is considerably wider (156 um v 100 um in normal mice) at the mid-villous point and the lesser intra-villous space (10um v 30 um in normal mice), with the width at the villous at the tips being almost doubled. Similar widening of the villus is just discernible in the cftr mutants in gene delivery studies [34]. This is consistent with intestinal tissue having a large interstitial fluid volume due to increased rates of intestinal fluid absorption, decreased exit of fluid into the vasculature or a combination of both. It may be that tenacious mucus that blocks the crypt lumina prevents the translocation of guanylin to the villous tips to lower fluid absorption by inhibiting the NHE: 3 exchanger. Hence, the intestinal obstruction problems in cftr defective mice may be caused by uninhibited fluid and therefore excessive absorption rather than the usual conjectured failure of fluid secretion. A non-functioning cftr channel may be a way of reducing excessive absorption to make mucus less viscous.
Later in vivo perfusion experiments on cystic fibrotic patients
Human studies: More recent studies have involved intestinal perfusion of the jejunum of cystic fibrotic patients with low chloride ion containing perfusate in order to maximize the potential for getting information about chloride ion movement. The lumen was found to be slightly positive in CF and non CF patients but became negative on perfusion with theophylline in the non-CF patients but was unaffected in CF patients. According to the authors [35], increased lumen negativity (increased serosal surface positivity) can only be explained by increased anion secretion or decreased (sic) cation absorption. In non-CF control patients, theophylline increased water secretion but did not do so in the CF patient group. Luminal chloride ion concentrations were also higher in the control group and rose on theophylline stimulation but were low in the CF patients and remained so despite theophylline administration. This is again consistent with the enterocyte section hypothesis but does not add substantial proof because the flows of fluid and the lack in CF might also arise from changes in vascular tone.
Testing the heterozygote advantage hypothesis: It was evident from perfusion studies that abnormal passive chloride absorption in cystic fibrosis jejunum did not agree with the classic view of a chloride secretory defect [36]. The question of heterozygote selective advantage was therefore addressed directly by intestinal perfusion in cystic fibrotic ΔF508 heterozygotes perfused principally with an isotonic sodium chloride and sodium sulphate solution. The heterozygote CF group secreted as much fluid as the normal group, also when secretion was stimulated by prostaglandin E1. Homozygous CF patients did not secrete fluid significantly after PGE1 perfusion. In this case [37], human heterozygotes do not show the heterozygote advantage that they are supposed to have and which seems to be evident in the mouse knock-out model. When normal balanced electrolyte solutions were perfused, containing glucose, glucose was absorbed at the same rate in normal and heterozygote CF subjects but there was no sodium or chloride ion absorption in cystic fibrotic patients and increased fluid absorption was only one third of control values. This, together with differences in mannitol induced potential differences, indicated that the known lack of chloride ion permeability acts to prevent normal fluid absorption and is not associated with increased fluid secretion. The malfunctioning chloride channel in CF seems therefore to be a poorly functioning chloride channel that does not allow complete chloride absorption. In conclusion, there is not only a lack of evidence for heterozygote advantage in CF , there is also now evidence against it and an alternative view that CF mutations lead to a damaged chloride uptake rather than a secretory process.
Earlier work on rectal potential difference in cystic fibrotic patients [38] confirms this view. Rectal transmural voltage difference was about -41 mV lumen negative in normal tissues and -21 mV in cystic fibrotic patients. This value fell by about 30 mV in the normal subjects and by a similar amount in CF, on perfusion with low chloride ion containing perfusate, enough to change the polarity of the CF transmural voltage. With STa, the enterotoxin that has no discernible secretory action [4] if fluid transport is appropriately measured by volume recovery, transmural potential difference increased in normal subjects but not at all in CF patients. This is consistent with STa inhibiting electroneutral NHE:3 exchange and inducing an underlying electrogenic method of absorption of sodium ion - with this not possible in CF because of the broken CFTR channel that prevents chloride ion uptake in association with sodium ion uptake. This event takes place after the interruption of normal sodium ion uptake by STa since both CF and normal tissue display increases in tissue cGMP content after STa exposure. The fact that enhanced chloride ion uptake takes place after STa challenge is clearly seen in non-cystic fibrotic cultured colonocytes. On exposure to STa, chloride uptake into these cells increases threefold and can be inhibited by furosemide and DPC, the chloride channel blocker. If these compounds blocked NKCC1, only reduced chloride ion uptake might be expected. If STa caused chloride ion secretion, uptake of chloride ion might reasonably be expected to be reduced but was not. These observations are entirely consistent with those of Fordtran’s group who convincingly demonstrated a failing chloride uptake mechanism, not an enhanced chloride ion secretory mechanism in cystic fibrotic heterozygote patients. While increased chloride ion uptake was detected in the colonocytes, the authors themselves [38] concluded that human colonocytes secrete chloride ion (summary paragraph) in response to STa, emphasising the increase but neglecting the direction of that movement i.e. into not out of the colonocytes as required by secretion.
Findings in CF patients in support of the tenacious mucus concept
Evidence of viscous mucus: Most CF research has focused on the CFTR channel and its likely role in the prevention of chloride ion secretion as proposed by the doctrine of chloride ion secretion emanating from enterocytes and also the likely selective advantage that disruption of the secretory channel might confer. Sometimes, research work is reported that emphasizes the fact that secreted mucus alters histological appearance or the physiological outcomes of absorption experiments. CF mice require a 6% polyethylene glycol supplemented electrolyte solution i.e. hypertonic saline, in order to ensure greater survival rates and prevent death from intestinal obstruction from which particular genotypes suffer [33,39]. Where intestinal perfusion experiments are done in CF patients, a minor but noteworthy aspect of the process is that perfusion catheters become clogged in all CF patients, though only in 1 of 9 control subjects, highlighting the very viscous mucus in the CF patients [35]. In histological slides made of CF biopsies, the lumina of the crypts are dilated with PAS positive mucus like material [40]. In this paper, oedema within the villi is stated (text to Figure 1) but without further comment in the discussion. Oedema might be unusual in a tissue that was incapable of secreting but would not be discordant in a tissue where absorption from the lumen was unregulated. This might arise if fluid absorption was no longer a controllable event through local movement of guanylin from the crypts to the villous tip cell. Sticky mucus might prevent the diffusion of this local paracrine agent to the target cells. Similar concentrations of mucus were also found in the ileal and colonic crypt lumina of cftr knockout mice [34].
Unstirred layer thickness and glucose uptake: Viscous mucus within the crypts might explain the occasional observation of slightly enhanced substrate absorption. Enhanced glucose and alanine transport was claimed for CF intestine [41] on the basis of electrical measurement but confirmed by direct measurements in perfused intestine [42]. Here, the Km for glucose absorption was found to be lower in CF than in normal intestine and the unstirred layer thickness was found to be considerably smaller in CF. Curiously, the authors argue that abnormal mucus over the tips of the villi causes thinning of unstirred layer thickness.
Unstirred layer thickness is deemed to be the thickness of an aqueous layer above the villi bounded by the villi on one side and a notional stirred: unstirred boundary at the other end. Substances have first to diffuse across this layer in order to reach the absorbing epithelial cells. Typical values for the length may represent this aqueous layer thickness and to a lesser extent the gap between the villi extending down towards the crypts. Unstirred layer thickness is determined by an electrical method of solution change where altered bulk phase sodium ion concentration causes diffusion into or out of the unstirred layer and then beyond and probably down the length of the villi. There is no reason to assume that solute diffusion stops when the advancing solute front has reached the tips of the villi and that there is no onward diffusion down into the intravillus space. Hence, unstirred layer length measurements are probably composites of villus length and unstirred layer above the tips.
If the cystic fibrotic mucus, known to be present in the crypts, is a tenacious barrier to diffusion, then diffusion down the intervillus space is probably prevented to a certain extent, thereby shortening the apparent unstirred layer length. It is notable that the difference in unstirred layer length was estimated to be 780 for normal subjects minus - 546 or 234 um, close to the length of a villus which would be unusual. Cystic fibrotic villi would have to be visibly shorter than normal villi if this were true. In addition, higher substrate concentrations obtain at the absorbing villus tip sites and would not be lower than normal in CF because of diffusion past and away from the tips down the intervillus space. Substrate would therefore cross the aqueous region to reach the villi but in the case of CF tissue with inspissated mucus, the solute front would pass only very slowly down the intervillous space, making it seem like the diffusion path was shorter. It is highly likely that these observations result from the tenacious mucus that is associated with CF and result from an increase, not from any decrease in diffusion barriers.
Consequences of tenacious mucus
The viscous mucus associated with cystic fibrosis may be the cause of the various problems associated with CF since difficult to dislodge mucus impacts on any flow down ducts and indeed movement in the trachea. It may be that the mutational defect in cystic fibrosis has survival value precisely because the ion channel is defective in CF or not inserted into the membrane. This would imply that CFTR is an important chloride channel that is required for uptake of fluid and not secretion. Where fluid uptake continued uninterrupted, it would lead to the production of viscous mucus that would make lung and pancreatic function abnormal because of blockage of ducts and difficulty in shifting mucus in the lung. A mutation in the CFTR channel could presumably reduce the rate of fluid absorption and allow the mucus to be less viscous than would otherwise be the case.
Relief through interrupted absorption: Some studies in cftr mutant mice support this concept. Where fluid uptake mechanisms are compromised, there is increased survival of CF mice; in cftr (-/-) knockout mice, with intact NHE3, survival time is poor with most not surviving beyond six weeks. Double knock-out cftr (-/-) and Nhe3 (-/-) mice have 75% survival rates at eight weeks, demonstrating clearly that inability to absorb fluid protects against cystic fibrotic pathology [43].
It is possible that the essential pathology of CF is pathological mucus formation in the lung and that mutation in CFTR prevents fluid absorption. The selective advantage to defective or otherwise inoperative CFTR could be that fluid absorption in the lung bronchioles is inhibited and mucus is kept more hydrated than it otherwise might be. This might explain why there are very many mutations in CFTR because the mutations confer survival. This implies that lung function is improved slightly but the alteration in CFTR causes coincidental changes in sweat and saliva secretion that are not as life threatening as the impaired lung function, were chloride absorption to be left unchecked.
Observations on the identification of defective CFTR protein as the key aspect of cystic fibrosis
Review of the genetic research: At the present state in cystic fibrosis research, it is arguable that the concept of selective advantage of the heterozygote condition in the intestine through reduced chloride secretion has been tested and shown to be very unlikely. Associated phenomena related to defective CFTR and the intestine seem to be related more to short-circuit current measurements that could have their origin remote from the enterocyte cells. If that is so, then a reason must be found for why a consistent finding in CF studies is the refractoriness to substances that stimulate short circuit current in normal tissues. Before providing a possible reason, there may be some merit in reviewing the background to the identification of CFTR protein as the essence of the problem of cystic fibrosis, and with that, the identification of chromosome 7 as the source of the disorder. Identification of the relevant chromosome was done by the process of detection of restriction fragment length polymorphisms which do not necessarily occur in genes. Some forms of RFLPs are inherited with particular diseases. This need not arise because the restriction site was created by the disease state but apparently only because the sequence is very close to the affected gene [44]. In addition, samples of cDNA were created from mRNA from affected cells such as sweat glands. The cDNA prepared from mRNA from an affected cell is the nucleotide sequence that encodes the relevant protein but not necessarily sequences that have been suppressed in that particular cell. It may be that the focus has been on a mutation in CFTR that allows survival beyond the uterus instead of the essential gene mutation that is the cause of CF and which might turn out to be a gene involved in mucus production.
Conclusion
There are aspects of CF phenotype that seem to be widespread in transporting cells wherever CFTR is present and there is no doubt that it is an important chloride channel, involved in sodium chloride transport. However, heterozygote advantage is unlikely to take the form of absence of secretion. Just as likely is a hypothesized protection against bronchial asthma [45]. There are undoubtedly aspects of the intestinal phenotype that merit further investigation such as the apparent lack of diarrhoeal symptoms coupled with the presence of higher anti-body titres in cystic fibrotics in whom C. difficile bacteria are present in the intestine [46,47]. These isolated observations are pertinent to the hypothesis developed below of an inability of CF vascular smooth muscle to vasodilate to the same extent as normal vascular smooth muscle. It is well known that combinations of C. difficile toxins A & B cause genuine net fluid secretion into the intestinal lumen as well as exudate containing red blood cells, implying vasodilatation and increased capillary pressure forcing both plasma and formed elements of the blood into the lumen. A restricted ability for arteriolar vasodilatation would be an advantage when exposed to C. difficile and would explain the higher anti-body titres without obvious diarrhoeal symptoms. The current linkage of defective CFTR to poor secretion is unlikely given the lack of secretion in human heterozygotes. The link that does exist, (the notable exception of the very muted response to cholera toxin in CF mice), is only tenuously linked to enterocyte secretion through a rigid interpretation that changes in chloride ion movement alone are reflected in increased short-circuit current. What is proposed in the next section is an alternative explanation for the lack of secretion in CF patients based on pathological alterations in vascular smooth muscle that are consistent with the intestinal capillary filtration model of fluid secretion.
Respiration abnormalities in cftr (-/-) mice
A major difficulty with cftr knock-out mouse models of cystic fibrosis has been the lack of evidence for a lung phenotype in the cftr (-/-) mouse. This may be because the anticipated phenotypic alterations were not what was eventually found. Static compliance of lungs assessed through lung pressure-volume relationships in cftr knock-out mice was found to be less than normal in the cftr (-/-) mouse but enhanced in the cftr (+/-) mouse [48]. Static lung compliance is a lumped parameter of all elements in the tissue that may reduce an increase in volume for a given increment in pressure. Dynamic properties were also measured by imposing a sinusoidal forcing function that altered lung volume. Impedance, elastance, inertance and viscance are the preferred terms used in lung physiology to indicate time varying resistance, elasticity, inertia and viscosity. The total impedance of the lung was measured and resolved into the separate components of airways resistance and airways inertance, combined with tissue damping (viscance) and stiffness (elastance) factors that also contribute to the total impedance.
Measurement of these latter properties relies on a mathematical treatment [49] of the pressure transients that occur on lung volume change. The unit impulse response treatment was extended to include forced sinusoidal variations in volume that precede by a given phase angle, the induced changes in pressure, so that pressure change will lag the imposed volume change. A later treatment, using Hildebrandt’s [49] derivation but using the integrated form of the Laplace transformation of time dependent equations [50] provides a compact solution that includes a term for viscance (G) and elastance (H). The Hildebrandt and the Hantos treatments of the factors involved in airways resistance are attempts to resolve individual contributions by making use of solutions to the fundamental problem of a volume based sinusoidal forcing function acting on a system that exhibits approximately simple harmonic motion that is damped. Hence, this allows for hysteresis effects and the loss of energy that should lead to decay of simple harmonic motion but for the repeated imposition of a volume change.
Neglect of certain terms in Hildebrandt’s treatments of the inflation of the lung and the balloon gives, for the lung, what has come to be called the constant-phase model. Hildebrandt found empirically that the relationship of the pressure change per unit volume change was logarithmic in the lung but linear for the balloon. Using the constant-phase model for the lung, Hantos and colleagues detected an almost twofold increase in damping in the dog lung and a third more increase in elastance after intravenous histamine infusion. There was also a doubling of airways resistance, as might be expected after contraction of bronchial smooth muscle. The increase in damping and stiffness is also presumably because the lung smooth muscle is in a more contracted state.
Using this model, Cohen and colleagues [48] showed that not only was the static compliance of the cftr (-/-) mouse lung reduced but also that the dynamically determined damping and stiffness coefficients were double the normal value and possibly also the airways resistance was increased. Heterozygote mice in contrast had slightly less dramatic reductions in dynamic airways resistance (impedance), damping and stiffness. It would be interesting to know whether the bronchial smooth muscle was thicker in cftr (-/-) mice and thinner than normal in the cftr (-/+) mice. The authors conjecture that this could be the heterozygote advantage that allows cystic fibrosis to persist in the population. However, given the lack of evidence for a demonstrable selective advantage based on population data, this question cannot be unequivocally answered. Nevertheless, even if this confers no selective advantage, the increased damping and stiffness reinforces the idea that a central aspect of cystic fibrosis pathology might be alteration in smooth muscle structure, somehow related to the defective chloride channel, perhaps related more to altered smooth muscle physiology than to inhibited enterocyte fluid secretion.
Evidence for changes in intestinal and vascular smooth muscle vasomotion in cystic fibrosis
A noteworthy but unremarked aspect of cftr (-/-) animals models of cystic fibrosis is the change in smooth muscle structure. In cftr mouse mutants, a threefold increase in circular muscle size was noted in the mouse jejunum [51]. Although no histology was presented, wall tension of mouse aorta rings was found to be three times higher in cftr (-/-) mice when compared with wild type control mice, together with very impaired muscular relaxation after VIP exposure [52]. Inhibition by CFTR (inh)-172 inhibitor of sphingosine-1-phosphate enhanced myogenic vasoconstriction occurs in hamster resistance arteries, again implying impaired vasodilatation [53]. Corroborative evidences comes also from human studies in which higher doses of the vasodilator isoprenaline were required in the hand veins of CF patients to achieve relaxation compared with normal subjects. In addition, the maximal venodilatation achieved was less in CF patients [54]. The apparent increase in smooth muscle thickness is also seen in VIP knock-out mice both in the lung and the intestine [55], together with interference with CFTR localisation.
The concept that failure of dilatation of smooth muscle occurs in cystic fibrosis sufferers or if not physiological difficulties in relaxation of smooth muscle, then thickening of the smooth muscle to prevent dilatation has been occasionally recorded in the literature on CF. A high percentage of portal hypertension occurs [56] in CF sufferers as well as carotid body hypertrophy [57], lung compliance is less in terms of change in volume per unit change in inflation pressure [48] in the lung of heterozygote and homozygote cftr (-/-) mice and intestinal circular muscle mass is greater [51], all pointing to physical or physiological reasons why smooth muscle does not relax normally in the CF state.
Reduced relaxation of pulmonary arteries was found in CF patients [58] from tissue obtained at heart lung transplant. CFTR was determined to be involved in normal bronchodilatation with contraction inhibited by CFTRinh-172, the CFTR channel blocker, indicating the involvement of the CFTR channel in normal constriction of the smooth muscle of the trachea. Innervation of the trachea was less in knock-out mice as was the muscle tone of the trachea [59]. Intestinal circular muscle is less co-ordinated in cftr knockout mice and is also refractory to carbachol and curiously also to depolarisation caused by addition of KCl [60]. In addition, increased tissue levels of prostaglandins were found. In aortic rings from mice, the vascular tone is higher in cftr knockouts, again implying an inability to undergo smooth muscle relaxation and in this case, cftr (-/-) tissue responses to 80 mM KCl were also much higher than wild type responses [52]. Disrupting the CFTR gene reduces the response of vascular smooth muscle to vasoactive agents but also prevents some forms of vasodilatation.
The Cl– equilibrium potential is more positive than the resting membrane potential enabling Cl to be a depolarizing ion upon activation of a Cl – conductance channel but does not seem to fit in with the physiological role of CFTR in vascular smooth muscle [61]. The change in membrane potential towards depolarization is associated with contraction but does not accord with the findings that CFTR is needed for contraction and CFTR inhibitors prevent vascular smooth muscle relaxation [52]. What is evident is that CFTR plays an important role in smooth muscle contraction and its compromised action in CF patients is likely to be the reason for lack of vascular and other smooth muscle relaxation.
The action of CTFTR in the enterocyte may be a by-product and not a matter of central concern in the pathology of cystic fibrosis
One interpretation of these findings is that vascular smooth muscle is less able to undergo relaxation in CF sufferers. This could be due to refractoriness to dilatatory stimuli but also there is the possibility that larger circular smooth muscle mass does not allow vasodilatation. This interpretation fits in with the findings in CF knockout mice of their refractoriness to cholera and their lesser ability to respond to intestinal challenge by bacterial enterotoxins. If the smooth muscle of CF arteries cannot easily achieve a state of vasodilatation, then this would severely reduce fluid secretion into the lumen, as has been shown by the work of Gabriel and colleagues [32]. In addition, a case can also made for the origin of the increased short-circuit after enterotoxin derangement being in the intestinal vasculature [30]. If the vasculature in CF cannot adequately vasodilate, then the stimulus for provoking an increase in short-circuit current would be severely muted [31], not necessarily absent but attenuated since active neurogenic vasodilatation might not occur but some expansion of the internal diameter through physical forces might still be possible even if the arterioles were thicker than normal and had less compliance.
The concept of fluid secretion and also the signal for changing short-circuit current originating in the vascular smooth muscle harmonises with work on the effect of PGE1 and misoprostol on intestinal function. In isolated T84 cells grown to confluence, prostaglandin E1 alters the pattern of ion movement and enhances bidirectional chloride ion permeation but more particularly towards the luminal bathing fluid [62]. Evidently there must be prostaglandin receptors in or on the cells and there is an undoubted in vitro action on ion transport by the cells with significant increases in short-circuit current.
In humans, in vivo, the PGE1 analogue, misoprostol, changes fluid absorption to secretion and changes the measured potential difference [63]. In cystic fibrotic subjects, misoprostol reduces net absorption to zero but there is neither net secretion nor a change in potential difference. The conventional explanation is that PGE1 is secretory and the change in electrical events should reflect this but for the dysfunctional chloride channel in CF heterozygotes. However, the data also fit well with the idea that an anti-absorptive action of PGE1 stops fluid absorption i.e. reduces it to zero and a separate vasodilator effect on arterioles causes’ secretion. In CF, the anti-absorptive action is present and reduces net absorption to zero. Note that if CF protects against secretion and this action is via the chloride channel, there is no particular reason why fluid absorption should be zero; it should prevent PGE1 from having any effect at all and net absorption should arguably be close to control values and yet this does not occur. The alternative explanation that CF arterioles cannot vasodilate offers the explanation that the anti-absorptive effect of PGE1 continues but CF arterioles cannot dilate and therefore there is no net secretory action of PGE1 on CF intestine. Similarly, there are no changes in electrical potential difference because there are normally initiated by a change in interstitial volume causing vasodilatation but the CF vasculature dilates poorly, if at all, and therefore there is no change in electrical signal.
An alternative explanation for the reduced ability in CF to secrete after cholera challenge and to mount a short-circuit current response after STa could be that the smooth muscle is overdeveloped in CF and vasodilatation is very muted.
Recent evidence for smooth muscle involvement in CF from clinical trials with lumacaftor and ivacaftor
The concept of smooth muscle physiology being a cause of cystic fibrotic dysfunction as indicated here, has received support from the recent introduction of new drugs, ivacaftor and lumacaftor, that facilitate introduction of CFTR into membranes or repair the existing CFTR. A recent review of the effects of these drugs [64] has drawn attention to the improvement in the forced expiratory volume after one second, FEV1, in patients with CF but with caveats about the type of mutation that the patient has. In patients with the G551-D mutation, ivacaftor gave on average a 10% increase in respiratory function based on forced expiration measurements [65,66], not confirmed in patients having class 2 mutations [67]. Where there is no insertion of CFTR into the cell membrane, it seems that these new drugs are less impressive. In the appropriate mutation, improvements in FEV1 of the order of 15% have been detected [68]. Importantly for the smooth muscle concept detailed here, non-invasive pulse wave velocity measurements were lower after ivacaftor treatment, indicative of vascular smooth muscle relaxation. In addition, there was increased airways distensibility, both events showing a general dysfunction in this mutation of smooth muscle physiology. The responding G551-D mutation is a small fraction of the CF pool. However, there may be an underlying smooth muscle abnormality in F508 deletions that cannot be treated by these newer drugs but might be by drugs that facilitate insertion of CFTR channels into smooth muscle cell membrane.
Recent clinical trials have indicated that improvement in forced expiratory volume is seen in particular CF deletions and points to smooth muscle involvement in CF
Synopsis
1. Secretion in the small intestine may not be mediated by chloride ion secretion by the enterocyte but by vasodilatation of intestinal arterioles raising intracapillary and therefore interstitial fluid pressure and extruding fluid by physical force into the intestine in accordance with Starlings equation for fluid movement across the capillary. If a valid model, then this makes deleterious mutation of the enterocyte chloride ion channel irrelevant to the concept of selective advantage but it may show instead that net fluid absorption is hindered by a defective chloride ion channel.
2. The common occurrence of CFTR in smooth muscle cells as well as in the enterocytes makes it possible that agents that affect fluid movement are also acting on the vascular smooth muscle. There is evidence that CF smooth muscle does not undergo relaxation. It is this aspect of CF pathology that may explain the selective advantage but it is evident that failure to secrete fluid in response to bacterial challenge may be just an associated phenomenon. Important aspects of CF pathology may not be well illuminated by inferences drawn from the enterocyte secretion model.
References
- Lucas ML. Diarrhoeal disease through enterocyte secretion: a doctrine untroubled by proof. Exp Physiol. 2010; 95: 479-484. Ref.: https://goo.gl/3fu4M7
- Alton EWFW, Armstrong DK, Ashby D, Bayfield KJ, Bilton D, et al. A randomised, double blind, placebo-controlled trial of repeated nebulisation of non-viral cystic fibrosis transmembrane conductance regulator (CFTR) gene therapy in patients with cystic fibrosis. Efficiency and Mechanism Evaluation. 2016. Ref.: https://goo.gl/o5M6Ud
- Lucas ML. Amendments to the theory underlying Ussing chamber data of chloride ion secretion after bacterial enterotoxin exposure. J Theor Biol. 2005; 234: 21-37. Ref.: https://goo.gl/ZJd863
- Lucas ML. A reconsideration of the evidence for Escherichia coli STa (heat stable) enterotoxin driven fluid secretion: a new view of STa action and a new paradigm for fluid absorption. J Appl Microbiol. 2001; 90: 7-26. Ref.: https://goo.gl/vhVnKT
- Lucas ML, Gilligan LC, Whitelaw CC, Wynne PJ, Morrison JD. Lack of restoration in vivo by K+-channel modulators of jejunal fluid absorption after heat stable Escherichia coli enterotoxin (STa) challenge J Trop Med. 2011; Article ID 853686: 7. Ref: https://goo.gl/92bfe1
- Strombeck DR. The production of intestinal fluid by cholera toxin in the rat. Proc Soc exp Biol Med. 1972; 140: 297-303. Ref: https://goo.gl/vSkb5k
- Lucas ML, Morrison JD. An investigation into the relationship between small intestinal fluid secretion and systemic arterial blood pressure in the anaesthetized rat. Physiological Reports. 2015;3, e12407, 1-14. Ref: https://goo.gl/VgESw6
- Tsuji LC, Buchwald M, Barker D, Braman JC, Knowlton R, et al. Cystic fibrosis locus defined by a genetically linked polymorphic DNA marker. Science. 1985; 230:1054-1057. Ref.: https://goo.gl/pkvGxi
- Rommens JM, Iannuzzi MC, Keren BS, Drumm ML, Melmer G, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989; 245: 1059-1064. Ref.: https://goo.gl/bS1fNg
- Danks DM, Allan J, Anderson CM. A genetic study of fibrocystic disease of the pancreas. Ann Human Genet (Lond). 1965; 28: 323-356. Ref.: https://goo.gl/f76C63
- Knudson AG Jr, Wayne L, Hallett WY. On the selective advantage of cystic fibrosis heterozygote. Amer J Hum Genet. 1967; 19: 388-392. Ref.: https://goo.gl/TH4ij7
- Quinton PM. Abnormalities in electrolyte secretion in cystic fibrosis Eds: Quinton PM, Martinez RM, Hopfer U. San Francisco Press, San Francisco, USA. 1982; 53-76.
- Barua D. A history of cholera. Cholera. 1992; 1-36. Ref.: https://goo.gl/kKp2dC
- De SN. Cholera. Oliver & Boyd. Edinburgh & London. UK. 1961.
- Renfrew C. Archaeology and language; the puzzle of Indo-European origins. University of Chicago Press, Chicago, USA. 1987.
- Mallory JP. In search of the indo-europeans: language, archaeology and myth; Thames & Hudson. 1989.
- Quinton PM. Physiological basis of cystic fibrosis: a historical perspective. Physiol Rev. 1999; 79: 3-22. Ref.: https://goo.gl/pTRbza
- Lucas ML, Thom MM, Bradley JM, O’Reilly NF, McIlvenny TJ, et al. Escherichia coli heat stable (STa) enterotoxin and the upper small intestine: lack of evidence in vivo for net fluid secretion. J mem Biol. 2005; 206: 29-42. Ref.: https://goo.gl/EQjUHY
- Di Sant’Agnese PA, Powell GF. The eccrine sweat defect in cystic fibrosis of the pancreas (mucoviscidosis). Ann NY Acad Sci. 1962; 93: 555-599. Ref.: https://goo.gl/V6W6UR
- Araki H, Field M, Shwachman H. A new assay for cystic fibrosis factor: effects of sera from patients with cystic fibrosis on the in vivo electrical properties of rat jejunum. Pediatr Res. 1975; 9: 932-934. Ref.: https://goo.gl/bmFZHi
- Gilmore JP, Davis M, Gibbs GE. Influence of cystic fibrotic and heterozygous serum on rat jejunum. Proc Soc Exp Biol Med. 1978; 157: 70-74. Ref.: https://goo.gl/jNnHeG
- Tucker RD, Gibbs GE, Christensen MB. Cystic fibrosis serum effect on short circuit current of rat jejunum. Pediatr Res. 1979; 13: 1371-1374. Ref.: https://goo.gl/Un1rzd
- Will PC, Boat TF, Hopfer U. Evidence against a specific effect of serum from patients with a cystic fibrosis on sodium-dependent glucose transport in the rat jejunum. Pediatr Res. 1979; 13: 1129-1133. Ref.: https://goo.gl/ZLfyRM
- Berschneider HM, Knowles MR, Azizkhan RG, Boucher RC, Tobey NA, et al. Altered intestinal chloride transport in cystic fibrosis. FASEB J. 1988; 2: 2625-2629. Ref.: https://goo.gl/aB6qKJ
- Taylor CJ, Baxter P, Hardcastle J, Hardcastle PT. Absence of secretory response in jejunal biopsy samples from children with cystic fibrosis. Lancet. 1987; 107-108. Ref.: https://goo.gl/ZDnDDC
- Taylor CJ, Baxter P, Hardcastle J, Hardcastle PT. Failure to induce secretion in jejunal biopsies from children with cystic fibrosis. Gut. 1988; 29: 107-108. Ref.: https://goo.gl/AD3pLW
- Baxter P, Wilson AJ, Read NW, Hardcastle J, Hardcastle PT, et al. Abnormal jejunal potential difference in cystic fibrosis. Lancet. 1989; 464-466. Ref.: https://goo.gl/L5xG34
- O’Loughlin EV, Hunt DM, Gaskin KJ, Stiel D, Bruzuszcak IM, et al. Abnormal epithelial transport in cystic fibrosis jejunum. Am J Physiol. 1991; 260: 758-763. Ref.: https://goo.gl/LZcFW1
- Cuthbert AW, Halstead J, Ratcliff R, Colledge WH, Evans MJ. The genetic advantage hypothesis in cystic fibrosis heterozygotes: a murine study. J Physiol. 1995; 482: 449-454. Ref.: https://goo.gl/7DsAnS
- Lucas ML. An alternative explanation for the occurrence of short circuit current increases in the small intestine following challenge by bacterial enterotoxins. Med Hypotheses. 2013; 81: 601-606. Ref.: https://goo.gl/1yyi6p
- Cuthbert AW, Hickman ME, MacVinish LJ, Evans MJ, Colledge WH, et al. Chloride secretion in response to guanylin in colonic epithelia from normal and transgenic cystic fibrosis mice. Br J Pharmacol. 1994; 112: 31-36. Ref.: https://goo.gl/US1nns
- Gabriel SE, Brigman KN, Koller BH, Boucher RC, Stutts MJ. Cystic Fibrosis Heterozygote Resistance to Cholera Toxin in the Cystic Fibrosis Mouse Model. Science. 1994; 266: 107-109. Ref.: https://goo.gl/5KaAp4
- Grubb BR, Gabriel SE. Intestinal physiology and pathology in gene-targeted mouse models of cystic fibrosis. Am J Physiol. 1997; 273: 258-266. Ref.: https://goo.gl/VLg6jk
- Zhou L, Dey CR, Wert SE, Duvall MD, Frizzell RA, et al. Correction of lethal intestinal defect in a mouse model of cystic fibrosis by human CFTR. Science. 1994; 266: 1705-1709. Ref.: https://goo.gl/sp9v1F
- Teune TM, Timmers-Reker AJM, Bouquet J, Bijman J, De Jonge HR, et al. In vivo measurement of chloride and water secretion in the jejunum of cystic fibrosis patients. Pediatr Res. 1996; 40: 522-527. Ref.: https://goo.gl/o7PhYQ
- Russo MA, Högenauer C, Coates SW Jr, Santa Ana CA, Porter JL, et al. Abnormal passive chloride absorption in cystic fibrosis jejunum functionally opposes the classic chloride secretory defect. J Clin Invest. 2003; 112: 118-124. Ref.: https://goo.gl/rZyhDt
- Högenauer C, Santa Ana CA, Porter JL, Millard M, Gelfand A, et al. Active intestinal chloride secretion in human carriers of cystic fibrosis mutations: an evaluation of the hypothesis that heterozygotes have subnormal active intestinal chloride secretion. Am J Hum Genet. 2000; 6: 1422-1427. Ref.: https://goo.gl/Z7GCf3
- Goldstein JL, Sahi J, Bhuva M, Layden TJ, Rao MC. Escherichia coli heat-stable enterotoxin-mediated colonic Cl- secretion is absent in cystic fibrosis. Gastroenterology. 1994; 107: 950-956. Ref.: https://goo.gl/Zb9wah
- Grubb BR. Ion transport across the jejunum in normal and cystic fibrotic mice. Am J Physiol. 1995; 268: 505-513. Ref.: https://goo.gl/Bg5fZx
- Antonowicz I, Lebenthal E, Schwachman H. Dissacharidase activities in small intestinal mucosa in patients with cystic fibrosis. J Pediatr. 1978; 92: 214-219. Ref.: https://goo.gl/pUKXxp
- Baxter P, Goldhill J, Hardcastle J, Hardcastle PT, Taylor CJ. Enhanced intestinal glucose and alanine transport in cystic fibrosis. Gut. 1990; 31: 817-820. https://goo.gl/LWQC87
- Frase LL, Strickland AD, Kachel GW, Krejs GJ. Enhanced glucose absorption in the jejunum of patients with cystic fibrosis. Gastroenterology. 1985; 88: 478-484. Ref.: https://goo.gl/7T33Kx
- Bradford EM, Sartor MA, Gawenis LR, Clarke LL, Shull GE. Reduced NHE3-mediated Na+ absorption increases survival and decreases the incidence of intestinal obstructions in cystic fibrosis mice. Am J Physiol. 2009; 29: 886-898. Ref.: https://goo.gl/FwSK2P
- Drlica K. Understanding DNA and gene cloning: A guide for the curious. Wiley & Sons, New York, USA. 1997.
- Schroeder SA, Gaughan DM, Swift M. Protection against bronchial asthma by CFTR delta F508 mutation: A heterozygote advantage in cystic fibrosis. Nat Med. 1995; 1: 703-705. Ref.: https://goo.gl/d7bvbr
- Peach SL, Boriello SP, Gaya H, Barclay FE, Welch AR. Asymptomatic carriage of Clostridium difficile in patients with cystic fibrosis. J Clin Pathol. 1986; 39: 1013-1018. Ref.: https://goo.gl/KodCR8
- Monaghan TM, Robins A, Knox A, Sewell HF, Mahida YR. Circulating antibody and memory B-cell responses to C. difficile toxins A and B in patients with C. difficile-associated diarrhoea, inflammatory bowel disease and cystic fibrosis. Plos One. 2013; 8: 74452. Ref.: https://goo.gl/LjpNww
- Cohen JC, Lundblad LKA, Bates JHT, Levitsky M, Larson JE. The “Goldilocks Effect” in cystic fibrosis: identification of a lung phenotype in the cftr knockout and heterozygous mouse. BMC Genet. 2004; 5: 21. Ref.: https://goo.gl/f6KR6o
- Hildebrandt J. Comparison of mathematical models for cat lung and viscoelastic balloon derived by Laplace transform methods from pressure-volume data. Bull Math Biophys. 1969; 31: 651-667. Ref.: https://goo.gl/YvnnoM
- Hantos Z, Daroczy B, Suki B, Nagy S, Fredberg JJ. Input impedance and peripheral inhomogeneity of dog lungs. J Appl Physiol. 1992; 72: 168-178. Ref.: https://goo.gl/7wCMCe
- Risse PA, Kachmar L, Matusovsky OS, Novali M, Gil FR, et al. Ileal smooth muscle dysfunction and remodeling in cystic fibrosis. Am J Physiol Gastrointest Liver Physiol. 2012; 303: 1-8. Ref.: https://goo.gl/AUkByv
- Robert R, Norez C, Becq F. Disruption of CFTR chloride channel alters mechanical properties and cAMP-dependent Cl- transport of mouse aortic smooth muscle cells. J Physiol. 2005; 568: 483-495. Ref.: https://goo.gl/NdSMYm
- Peter BF, Lidington D, Harada A, Bolz HJ, Vogel L, et al. Role of sphingosine-1-phosphate phosphohydrolase 1 in the regulation of resistance artery tone. Circ Res. 2008; 103: 315-324. Ref.: https://goo.gl/N9XALj
- Eichler HG, Eichler I, Lewiston N, Blaschke TF, Hoffman BB. Responsiveness of superficial hand veins to adrenergic stimuli in patients with cystic fibrosis. Clin Sci (Lond). 1989; 76: 283-287. Ref.: https://goo.gl/Pm9BBU
- Alcolado NG, Conrad DJ, Poroca D, Alshafie W, Chappe FG, et al. Cystic fibrosis transmembrane conductance regulator dysfunction in VIP knockout mice. Am J Physiol Cell Physiol. 2014; 307: 195-207. Ref.: https://goo.gl/dk4XZB
- Efrati O, Barak A, Modan-Moses D, Augarten A, Vilozni D, et al. Liver cirrhosis and portal hypertension in cystic fibrosis. Eur J Gastroenterol Hepatol. 2003; 15: 1073-1078. Ref.: https://goo.gl/T16n1X
- Lack EE. Carotid body hypertrophy in patients with cystic fibrosis and congenital cyanotic heart disease. Hum Pathol. 1977; 8: 39-50. Ref.: https://goo.gl/pZpuaz
- Dinh Xuan AT, Higenbottam TW, Pepke-Zaba J, Clelland C, Wallwork J. Reduced endothelium-dependent relaxation of cystic fibrosis pulmonary arteries. Eur J Pharmacol. 1989; 163: 401-403. Ref.: https://goo.gl/Bc5E2Z
- Pan J, Luk C, Kent G, Lutz E, Yeger H. Pulmonary neuroendocrine cells airway innervation and smooth muscle are altered in cftr null mice. Am J Respir Cell Mol Biol. 2006; 35: 320-326. Ref.: https://goo.gl/c7Na3w
- De Lisle RC, Sewell R, Meldi L. Enteric circular muscle dysfunction in the cystic fibrosis mouse small intestine. Neurogastroenterol Motil. 2010; 22: 341-387. Ref.: https://goo.gl/C68g2E
- Matchkov VV, Dam VS, Bødtkjer DMB, Aalkjær C. Transport and function of chloride in vascular smooth muscles. J Vasc Res. 2013; 50: 69-87. Ref.: https://goo.gl/qi4vKN
- Weyer A, Huott P, Liu W, McRoberts JA, Dharmsathaphorn K. Chloride secretory mechanism induced by prostaglandin-E1 in a colonic epithelial cell line. J Clin Invest. 1985; 76: 1828-1836. Ref.: https://goo.gl/Gq4VK5
- Coates SW, Hoegenauer C, Santa Ana CA, Rosenblatt RL, Emmett M, et al. Inhibition of neutral sodium ion absorption in patients with cystic fibrosis. Gastroenterol. 2004; 127: 65-72.
- Hubert D, Bui S, Marguet C, Colomb-Jung V, Murris-Espin M, et al. Nouvelles therapeutiique de la mucoviscidose ciblant le gene ou la protein CFTR. Revue des Maladies Respiratoires. 2016. Ref.: https://goo.gl/DKpJGn
- Ramsey BW, Davies J, McElvaney NG. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011; 365: 1663-1672. Ref.: https://goo.gl/NvCXRJ
- Davies JC, Wainwright CE, Canny GJ, Chilvers MA, Howenstine MS, et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am J Respir Crit Care Med. 2013; 187: 1219-1225. Ref.: https://goo.gl/AVkpYk
- Flume PA, Liou TG, Borowitz DS, et al. Ivacaftor in subjects with cystic fibrosis who are homogenous for the F508del-CFTR mutation. Chest. 2012; 142: 718-724.
- Adam RJ, Hisert KB, Dodd JD, Grogan B, Launspach JL, et al. Acute administration of ivacaftor to people with cystic fibrosis and a G551D-CFTR mutation reveals smooth muscle abnormalities. JCI Insight. 2016; 1: 86183. Ref.: https://goo.gl/5AzTiq