


Mutations in the glucokinase (GK) gene cause defects in blood glucose homeostasis. In some cases (V62M and G72R), the phenotype cannot be explained by altered enzyme kinetics or protein instability. We used transient and stable expression of green fluorescent protein (GFP) GK chimaeras in MIN6 β-cells to study the phenotype defect of V62M and G72R. GK activity in lysates of MIN6 cell lines stably expressing wild-type or mutant GFP GK showed the expected affinity for glucose and response to pharmacological activators, indicating the expression of catalytically active enzymes. MIN6 cells stably expressing GFP V62M or GFP G72R had a lower GK activity–to–GK immunoreactivity ratio and GK activity–to–GK mRNA ratio but not GK immunoreactivity–to–GK mRNA ratio than wild-type GFP GK. Heterologous expression of liver 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFK2/FDP2) in cell lines increased GK activity for wild-type GK and V62M but not for G72R, whereas expression of liver GK regulatory protein (GKRP) increased GK activity for wild type but not V62M or G72R. Lack of interaction of these mutants with GKRP was also evident in hepatocyte transfections from the lack of nuclear accumulation. These results suggest that cellular loss of GK catalytic activity rather than impaired translation or enhanced protein degradation may account for the hyperglycemia in subjects with V62M and G72R mutations.
Glucokinase (GK) (hexokinase IV) is the predominant hexokinase expressed in hepatocytes and in pancreatic β-cells, where it functions as the glucose sensor for insulin secretion (1,2). It is also expressed in cells of the gut, hypothalamus, and anterior pituitary (3–5). The role of GK in the control of blood glucose homeostasis is supported by the impact of naturally occurring mutations, which cause either diabetes (GK maturity-onset diabetes of the young [MODY] and GK permanent neonatal diabetes mellitus) or persistent hyperinsulinemic hypoglycemia of infancy (1,6).
GK is regulated by both transcriptional and posttranscriptional mechanisms (7). Posttranscriptional regulation differs in hepatocytes and pancreatic β-cells, as demonstrated by the different subcellular location of GK. In hepatocytes, regulation involves an inhibitory regulatory protein (GKRP) that functions as a nuclear receptor, sequestering GK in an inactive state in the nucleus at low glucose (8–10). An increase in extracellular glucose concentration causes translocation of GK to the cytoplasm, enabling rapid stimulation of glucose phosphorylation. Translocation of GK between the nucleus and the cytoplasm is also dependent on other proteins that function as cytoplasmic receptors or shuttling proteins, such as the bifunctional protein phosphofructo-2-kinase (PFK2)/fructose-2,6-bisphosphate (FDP2), which catalyzes the formation and degradation of FDP2 (11,12).
Islet β-cells do not express hepatic GKRP, and, accordingly, GK does not accumulate in the nucleus (5,10,13). Whether they express a truncated form of GKRP lacking the nuclear localizing signal (14) is not known. PFK2/FDP2 serves as a GK binding protein in β-cells and has a role in the regulation of GK activity (11,15,16). Additional binding proteins of GK have also been identified in β-cells, including neuronal nitric oxide and others (17–19).
To date, ∼200 naturally occurring GK mutations have been identified that cause either diabetes or hypoglycemia (1,20). Detailed kinetic analysis is available for about one-fifth of these (20,21), and, while in most cases the defective glycemia can be explained by the enzyme kinetics, some mutations that cosegregate with hyperglycemia in GK MODY have near-normal enzyme kinetics or even a mild increase in affinity for glucose. Some mutants (H137R, V203A, M298K, S263P, E300K, V367M, and C213R) are enzymatically unstable in heat-stability assays (21,22), while the mutant E300K also showed protein instability when expressed in pancreatic β-HC9 cells using an adenoviral expression system (23).
Two mutants that are of particular interest are V62M and G72R (24,25). These mutants segregate with hyperglycemia in three families but are moderately activating and show only mild thermolability. Both mutants show lack of inhibition by liver GKRP and lack of activation by pharmacological GK activators that bind to the novel allosteric site (26). Because liver GKRP is not expressed in pancreatic β-cells and no physiological ligands have been identified that mimic the action of pharmacological activators, no explanation is available as yet for the mechanism by which V62M and G72R cause hyperglycemia.
The aim of this study was to develop a cell-based approach to investigate the activity and expression of GK MODY mutants, such as V62M and G72R, where the clinical phenotype cannot be explained by the kinetic properties of the enzyme.
RESEARCH DESIGN AND METHODS
Generation of constructs.
Constructs for human wild-type pancreatic β-cell GK were generated with the green fluorescent protein (GFP) tag at either the NH2- or COOH-terminal. For generation of NH2-terminal GFP-tagged GK, GK was excised from the pT73.36His vector (kindly provided by K. Brocklehurst, AstraZeneca, Cheshire, U.K.) using the BamHI/XhoI restriction sites for ligation into BglII/SalI of the pEGFP-C1 vector (Gibco Invitrogen, Paisley, U.K.). For generation of COOH-terminal–tagged GK, GK was excised from the pT73.36His vector using the BamHI/NotI restriction sites for ligation into the PCR3 vector (Promega, Southampton, U.K.), with the addition of a COOH-terminal GFP tag generated by PCR. The mutant constructs glutathione S-transferase (GST) V62M and G72R were excised from the pGEX-3X plasmid and inserted into the pEGFP-C3 vector for the addition of the NH2-terminal GFP tag as above. Correct insertion into the plasmids was confirmed by DNA sequencing.
Cell culture and treatment with adenoviral vectors.
MIN6 cells (p20–27) were cultured in Dulbecco's modified Eagle's medium containing 15% (vol/vol) fetal bovine serum. Adenoviral vectors for expression of untagged GK (Ad-LGK), GKRP (Ad-GKRP), and PFK2/FDP2 (Ad-PFK2/FDP2) were previously described (27–29). MIN6 monolayers were incubated with the adenoviral vectors for 4–6 h and then cultured for 24–48 h to allow protein expression.
Transfection and generation of stable cell lines.
For transient transfection, MIN6 were transfected with 1–3 μg plasmid DNA and 1–3 μl Lipofectamine 2000 for 8 h and then cultured for 24–48 h. Stable cell lines were generated after transient transfection by selection with 300 μg/ml G418 for 3–4 weeks. For nonclonal cell lines (GFP GK and GK GFP), cells were cultured in G418-containing medium. For clonal cell lines (GFP GK, GFP V62M, and GFP G72R), clones were isolated and expanded in the presence of 300 μg/ml G418. Clones with appropriate expression of GFP GK were selected for further study, based on GK activity, immunoreactivity, and human GK mRNA expression. The clones used in this study were WT3B and WT10B for GFP GK, V62M 3C for GFP V62M, and G72R 25A for GFP G72R. For transient transfection of hepatocytes, freshly isolated rat hepatocytes (30) were transfected 3 h after attachment with 4 μg plasmid DNA and 12 μl jet-PEI-Gal (Qbiogene) and cultured for 24 h.
GK activity, immunoreactivity, and kinetics.
GK activity of MIN6 and GFP-tagged GK cell lines were determined on 13,000g supernatants as previously described (31). Glucose phosphorylating activity was determined at 0.5 and 50 mmol/l glucose, representing low-Km and total hexokinase activity, respectively, and GK activity was determined from the difference between total hexokinases and low Km hexokinases. GK immunoreactivity was determined by SDS-PAGE and immunoblotting with a GK antibody (H-88; Santa Cruz Biotechnology, Santa Cruz, CA), as previously described (31). For kinetic analysis, GK activity was determined at the indicated concentrations of glucose and a GK activator (GKA) (GKA1 [32], a gift from M. Coghlan; AstraZeneca). The S0.5 for glucose was determined from Hill plots.
Real-time RT-PCR.
Cellular RNA was extracted using TRIzol (Invitrogen) and treated with DNaseI (Roche, East Sussex, U.K.). Single-strand cDNA was synthesized from 1 μg total RNA with random hexamers and Superscript II (Invitrogen). Real-time RT-PCR was performed in a total volume of 10 μl containing 20 ng of reverse-transcribed total RNA for determination of murine GK, human GK, and cyclophilin; 5 μmol/l primers (murine GK: forward 5′-GCA GAA GGG AAC AAC ATC GT, reverse 5′-CAC ATT CTG CAT CTC CTC CA; Human GK: forward 5′-ATC TCC GAC TTC CTG GAC AA, reverse 5′-CAC TCG GTA TTG ACG CAC AT; and cyclophilin: forward 5′-ATG GCA CTG GTG GCA AGT CC-3′, reverse 5′-TTG CCA TTC CTG GAC CCA AA-3′ [33]), 2 mmol/l MgCl2, and 1 μl Sybr Green (Roche). The reactions were carried out in capillaries in a Light Cycler (Roche), with initial denaturation at 95°C for 10 min followed by 40 cycles consisting of 95°C for 15 s, 58°C for 7 s, and 72°C for 15 s. Murine and human GK mRNA were expressed relative to cyclophilin mRNA and as a percentage of control.
Isolation of mouse pancreatic islets.
Pancreatic islets were isolated from male C57/BL6 mice. The pancreas was shaken with 1.5 mg/ml liberase (Roche) for 6 min with further digestion of 0.75 mg/ml liberase for 2–6 min. Overdigestion was prevented by washing in Krebs-Ringer phosphate buffer (10 mmol/l HEPES [pH 7.4], 90 mmol/l NaCl, 5 mmol/l NaHCO3, 4.8 mmol/l KCl, 0.7 mmol/l KH2PO4, 0.6 mmol/l MgSO4, 2.5 mmol/l 1M CaCl2, 0.1% BSA, and 5.5 mmol/l glucose). Islets were picked and cultured overnight in RPMI.
Insulin secretion.
MIN6 monolayers were cultured in 24-well plates and washed with Krebs-Ringer buffer (34). They were preincubated for 30 min in glucose-free Krebs-Ringer buffer followed by 1 h incubation at the indicated concentrations of glucose and GKA (35) (a gift from J. McCormack, OSI Prosidion, Oxford, U.K.). After an overnight culture, islets were preincubated with Krebs-Ringer phosphate buffer (three islets/200 μl) at 3 mmol/l glucose for 30 min, followed by 2 h at the indicated concentrations of glucose and GKA. Medium insulin was determined using a rat insulin assay kit (Mercodia, Uppsala, Sweden).
Microscopy.
Cells cultured on coverslips were washed with PBS and fixed using 4% paraformaldehyde in PBS. Immunostaining for endogenous GK and insulin was as previously described (31). Cells were visualized using a Leica TCS-SP2-UV microscope with an ×63, NA 1.3 oil immersion objective or an LSM Zeiss microscope with an ×63 1.4 oil-immersion objective.
Statistical analysis.
Statistical analysis was conducted using ANOVA followed by the Bonferroni test using the Prism analysis program.
RESULTS
GFP-tagged GK is catalytically active when expressed in MIN6.
Transient transfection of MIN6 with GK tagged with GFP at either the NH2- or COOH-terminal was associated with immunoreactivity to GK at 62–64 kDa, as expected, and with a 7- to 10-fold increase in GK activity, confirming that GFP-tagged GK is catalytically active (Fig. 1A). Because fluorescence microscopy showed large intercellular heterogeneity in GFP expression after transient transfection, we generated stable nonclonal cell lines by G418 selection. GK activity conferred by GFP-tagged GK was stable during early passages (<p9) but declined in later passages (Fig. 1B). Likewise, human GK-mRNA decreased with progressive passage (five- to eightfold between p9 and p19). Expression of endogenous (murine) GK-mRNA was similar in the GK GFP and GFP GK cell lines (125 and 93%, respectively) as in nontransfected MIN6. The affinity of GK for glucose (mmol/l) was similar in extracts of GK GFP and GFP GK cells (8.9 ± 0.3 and 8.9 ± 0.8, respectively) as in MIN6 overexpressing untagged GK with an adenoviral vector (8.5 ± 0.6). It was similarly increased by a GKA (32) in the cell lines (2.3 ± 0.3 and 2.1 ± 0.1) as in the untransfected MIN6 (2.8 ± 0.3), indicating that the GFP tag does not compromise affinity for glucose or activation by a GKA.
Effects of GK activation and overexpression on insulin secretion.
Glucose-induced insulin secretion was similar in GK GFP and GFP GK cell lines as in untransfected early passage MIN6 (Fig. 2A). Insulin secretion in β-cells is dependent on GK activity (36) and on the differentiated state, which in MIN6 declines with passage (37). Clonal cell lines transfected with GFP alone were considered an inappropriate control because they showed a greater increase in low Km hexokinase with increasing passage than the GFP GK cell lines, indicating greater dedifferentiation (results not shown). Conversely, untransfected early passage MIN6 may represent a more differentiated state than the GFP GK cell lines, which could explain the similar glucose-induced insulin secretion (Fig. 2A). To investigate the effect of GK activity and concentration on insulin secretion independently of passage number or the differentiated state of MIN6, we compared the effect of titrated adenoviral overexpression of untagged GK with pharmacological GK activation. Whereas GK overexpression had little effect on insulin secretion irrespective of the glucose concentration (Fig. 2A), the GKA caused a large stimulation at 3 mmol/l glucose (Fig. 2B). Similar results were obtained when insulin secretion was determined in mouse islets. The GKA caused a 2.6-fold stimulation (P < 0.01), whereas GK overexpression (confirmed by immunoblotting) had no significant effect (Fig. 2C).
Subcellular location of GFP-tagged GK chimeras in MIN6 and hepatocytes.
Colocalization of GK with insulin granules in pancreatic β-cells has been shown by immunofluorescence microscopy (38–41) (Fig. 3A), electron microscopy (38,41), and subcellular fractionation (31). To determine whether GFP-tagged GK colocalizes with insulin granules, GFP GK cell lines and MIN6 cells transiently transfected with GFP GK were stained for insulin (red) and visualized by confocal microscopy. Endogenous GK showed clear colocalization with endogenous insulin (Fig. 3A), whereas GFP GK transiently expressed showed negligible colocalization (Fig. 3B) and stably expressed GFP GK showed partial colocalization with insulin (Fig. 3C).
When the GK GFP and GFP GK constructs were transiently transfected into hepatocytes, both proteins accumulated in the nucleus during incubation with 5 mmol/l glucose (results not shown), indicating that the GFP tag does not interfere with binding to nuclear GKRP, in agreement with previous findings with an NH2-terminal GFP tag (42).
Effect of MG132 on expression of GFP-tagged GK chimeras.
We tested whether the decrease in transgene expression in late passages could be prevented by inhibition of proteosomal degradation using MG132. MG132 had no effect in either untransfected MIN6 or MIN6 overexpressing untagged GK; however, it increased GK activity and immunoreactivity (64 kDa) in the GK GFP cell lines (Fig. 4A). To test whether this increase in GK activity and immunoreactivity was due to inhibition of protein degradation, we determined the effect of MG132 in the presence of cycloheximide to inhibit protein synthesis. Although GK activity decreased in the presence of cycloheximide, this decrease was not prevented by MG132, excluding an effect on protein degradation (Fig. 4B). MG132 caused a large increase in human GK mRNA in the GFP GK cell lines (100- to 200-fold) (Fig. 4C). This was abolished by the transcriptional inhibitor, actinomycin D (Fig. 4C), indicating that MG132 increased transgene expression. MG132 was used in the rest of the study to increase transgene expression.
Pharmacological activation of GK.
Previous enzyme kinetic studies on purified GST-GK fusion proteins showed that V62M and G72R mutants have a lower S0.5 for glucose than wild-type GK and were not activated by a GKA (24,25). We determined the affinity for glucose of GFP V62M and GFP G72R in cell extracts of the clonal cell lines precultured with MG132 (see above). Both mutants had a lower S0.5 for glucose than wild type (wild type, 9.0 ± 1.0; V62M, 5.2 ± 0.4; G72R, 4.6 ± 1.0 mmol/l), and neither was significantly activated by the GKA (32) (Fig. 5A–C). There was no effect of MG132 on the affinity for glucose (Fig. 5D).
GK activity and immunoreactivity of GFP V62M and GFP G72R in clonal cell lines.
The catalytic activity and GK immunoreactivity of the mutants were determined in the clonal cell lines GFP V62M and GFP G72R and compared with two clonal cell lines expressing wild-type GFP GK (WT3B and WT10B). GK activity, immunoreactivity, and mRNA were determined after preculture with or without MG132, which caused a large increase in transgene mRNA, GK protein, and activity (Fig. 6A–C). GFP V62M and GFP G72R had a lower GK activity than the wild-type clones (Fig. 6D) when expressed relative to either immunoreactivity or mRNA (Fig. 6E and F). There was no significant difference in GK immunoreactivity relative to mRNA levels for either mutant compared with wild-type clones, suggesting instability of catalytic activity but not immunoreactive protein.
Transient transfection studies with GFP V62M and GFP G72R.
The activity-to-immunoreactivity ratio was also determined after transient transfection of MIN6 with varying plasmid amounts of GFP GK (wild type), GFP V62M, and GFP G72R (Fig. 7A and B). Plots of GK activity against immunoreactivity showed a lower activity-to-immunoreactivity ratio for both GFP V62M and GFP G72R compared with wild-type GFP GK (Fig. 7C), which is consistent with the findings from the clonal cell lines.
The effect of the GKA (35) on insulin secretion was determined in MIN6 that were either untransfected or transiently transfected with the GFP constructs. The GKA increased (P < 0.05) insulin secretion at 5 mmol/l glucose in all four tested conditions (untransfected, 1.7 ± 0.2 to 2.7 ± 0.1; GFP GK, 1.8 to ± 0.1 to 3.6 ± 0.1; GFP V62M, 1.7 ± 0.1 to 3.0 ± 0.1; and GFP G72R, 1.5 ± 0.1 to 3.7 ± 0.2 μg · h−1 · mg protein−1).
Stabilization of GK by liver PFK2/FDP2 and GKRP.
The GK binding proteins PFK2/FDP2 and GKRP modulate GK activity in pancreatic β-cells and liver, respectively. We therefore tested whether the mutants show altered activation or stabilization when these proteins are heterologously expressed in MIN6 cells. Expression of PFK2/FDP2 using an adenoviral vector in the clonal cell lines increased GK activity in the wild-type clones and in GFP V62M but not in GFP G72R or in the nontransfected cells (Fig. 8A). The lack of effect in the nontransfected cells may be due to a saturating effect of the endogenous PFK2/FDP2 on endogenous GK.
Overexpression of the liver GKRP protein resulted in a small but significant increase in GK activity in the wild-type GFP GK but not in the mutant cell lines or in untransfected MIN6 (Fig. 8B). Interaction of the mutants with GKRP was also tested by transient transfection of the GFP constructs in hepatocytes. Unlike GFP GK, which accumulated in the nucleus at 5 mmol/l glucose and translocated to the cytoplasm at 25 mmol/l glucose, neither GFP V62M nor GFP G72R accumulated in the nucleus at 5 mmol/l glucose (Fig. 8C).
DISCUSSION
Mutations in the GK gene identified in MODY type 2 subjects are associated with a similar phenotype of elevated fasting blood glucose (>5.5 mmol/l) and a 2-h increment in blood glucose after an oral glucose tolerance test of ∼2 mmol/l, irrespective of whether the mutation severely compromises GK kinetics or has negligible effect or is mildly activating (21,24,25). The apparent paradox of elevated blood glucose in MODY type 2 subjects with mildly activating GK mutations is as yet unexplained. In this study, we used MIN6 cells either transiently transfected or stably expressing GFP GK chimaeras to study the expression of the MODY mutants V62M and G72R, which are mildly activating. The limitations of transient transfection are the intercellular heterogeneity of expression, which necessitates high levels of transgene transfection and also the variability in mRNA and protein expression with time. Conversely, the limitation of cell lines is the progressive loss in a differentiated state with increasing passage, which may differ between wild-type and mutant clones. A complementary approach of both transient and stable transfection methods identifies possible artifacts that may be inherent in either technique.
In this study, we show that GFP-tagged GK expressed either transiently in MIN6 cells or after clonal selection with G418 is catalytically active and shows similar kinetic properties as the untagged enzyme or recombinant GST-GK fusion enzymes (43). Accordingly, GFP-tagged GK is predicted to be functional when expressed in MIN6. However, although clonal cell lines expressing GFP-tagged GK had increased GK activity, they showed similar glucose-induced insulin secretion as untransfected MIN6. This cannot be attributed to mistargeting of the GFP-tagged GK because similar results were obtained when untagged GK was overexpressed with the adenoviral vector in MIN6. The latter finding is not a unique property of the MIN6 β-cell model because lack of stimulation of insulin secretion after GK overexpression was also reported in rat islets (27). In this study, we show that in both mouse islets and MIN6 cells that there is lack of stimulation of insulin secretion by titrated GK overexpression, despite stimulation by a pharmacological GKA. A tentative explanation is that coupling between GK activity and stimulation of insulin secretion is dependent on subcellular location or targeting through association with binding proteins. Saturation of these receptors by endogenous GK may explain the lack of stimulation of insulin secretion by GK overexpression, despite stimulation by the GKA, which mimics the effect of elevated glucose.
We could not detect a difference in subcellular localization between mutants and wild type. It is noteworthy, however, that colocalization between GK and insulin in MIN6 cells is most clearly observed at endogenous levels of GK (31,38,39). Expression of wild-type GFP GK resulted in the accumulation of GK in the cytoplasm with partial colocalization with the insulin granules. This may be explained by saturation of GK binding proteins similar to saturation of GKRP in hepatocytes when GK is overexpressed twofold, which results in a marked decrease in nuclear/cytoplasmic distribution (44). Although we could not detect a difference in colocalization between mutants and wild type, we also cannot unequivocally rule out a difference in targeting.
In this study, we show, using clonal cell lines expressing mutant (V62M and G72R) or wild-type GFP GK, that the GK activity-to-immunoreactivity ratio is lower for mutants than for wild-type GK, despite a similar protein expression based on immunoreactivity-to-mRNA ratio. The transient transfection experiments in MIN6 also confirmed a lower activity-to-immunoreactivity ratio for V62M and G72R (60 and 85% decrease in the slope of the linear plots, respectively). Two possible mechanisms can be considered for the lower specific activity of the mutants. First, GK binding proteins have a positive effect on the wild-type enzyme by either an “activation” mechanism, as proposed for PFK/FDP2, or by protecting GK from inactivation by oxidation of thiol groups (45,46). Second, the mutants are more unstable than the wild type in the cellular environment because their conformation makes them more susceptible to inactivation by oxidation of thiol groups or other mechanisms. The lack of catalytic instability of the mutants when tested in thermal stability assays using the purified recombinant enzyme (24,25) (C.A., L.A., unpublished observations) argues against the latter possibility.
A role for binding proteins in stabilizing GK activity has been clearly demonstrated in liver. Although GKRP was initially identified as an inhibitor and nuclear receptor for GK (8–10), studies on GKRP−/− mice established a major role for this protein in stabilizing hepatic GK at the posttranscritpional level (47,48). However, less is known about stabilizing proteins in pancreatic β-cells. Furthermore, whether MIN6 cells express the full complement of GK binding proteins as pancreatic islets is unknown. The present finding that heterologous expression of the liver protein GKRP in β-cells increased GK activity for GFP GK wild type but not V62M or G72R is consistent with the lack of inhibition of catalytic activity of the recombinant mutants by GKRP (24,25) but is also indicative of a high catalytic or protein instability of wild-type GK in β-cells. The lack of effect on GK activity of GKRP overexpression in untransfected MIN6 indicates a role for endogenous proteins in β-cells that may have a homologous or analogous function to GKRP. A role for PFK2/FDP2 in activating GK in β-cells has been demonstrated (11,15,16). In this study, expression of PFK2/FDP2 increased GK activity in cells expressing wild-type GFP GK and GFP V62M but not in nontransfected cells. This supports a role for the endogenous PFK2/FDP2 in MIN6 cells, which was also confirmed by downregulation of PFK2/FDP2 by siRNA in MIN6 (C.A., L.A., unpublished information). The lack of effect of PFK2/FDP2 on GK activity in cells expressing GFP G72R suggests that this mutation affects the interaction with PFK2/FDP2 and indicates that the decrease in catalytic activity/immunoreactivity for G72R is at least in part explained by lack of activation by PFK2/FDP2.
Although the V62M and G72R mutants are relatively unresponsive to activation by pharmacological GKAs compared with wild-type GK (24,25), insulin secretion in MIN6 cells overexpressing GFP V62M or GFP G72R was stimulated by the GKA to a similar extent as in untransfected MIN6 or in cells expressing wild-type GFP GK. This shows that the mutants do not have a dominant-negative effect or attenuate insulin secretion. Accordingly, the explanation for the phenotype of these mutations is the low catalytic activity.
In summary, we show in this study using a cell-based approach that cellular instability of GK catalytic activity in pancreatic β-cells rather than increased mRNA degradation, impaired translation, or enhanced protein degradation may be the key factor that accounts for the hyperglycemia associated with the V62M and G72R mutations.
![FIG. 1. Transient and stable expression of GFP GKand GK GFP in MIN6 cells. A: MIN6 were transiently transfected with GK GFP and GFP GK. After 48 h, GK activity (expressed as percentage of nontransfected cells [NT]) and immunoreactivity (50 and 62–64 kDa) were determined. Results are representative of two experiments. B–D: Nonclonal GK GFP or GFP GK cell lines. B: GK activity at increasing passage number. C and D: GK activity at varying glucose cocentrations without (C dashed lines; D □) or with (C solid lines; D ▪) 5 μmol/l GKA (32) in GK GFP and GFP GK cell lines and MIN6 cells that were either untreated or pretreated (18 h) with Ad-LGK for expression of untagged liver GK (+GK). C: GK activity as percentage of 30 mmol/l glucose. The effects of GKA were significant for all four cell types: *P < 0.05 and **P < 0.01. D: GK activity at 30 mmol/l glucose *P < 0.05, **P < 0.01 relative to untreated MIN6, n = 6.](https://ada.silverchair-cdn.com/ada/content_public/journal/diabetes/56/7/10.2337_db06-1151/2/m_zdb0070749080001.jpeg?Expires=1716299642&Signature=trpAhj15z2HCDpcFIUY31ScNaOhIQGCAEOzzEG2n2GI12ppkSk8Vr~V5503IsRZ~wF19Z3wDu-FIvYreZs~VKelvIeqaRykLmDgsTis3LLffPVKZ8Y0gqushyahs-IzjRRPP3TKHJy-1nRr02XAjk5fVtYMkeP5K5C7PwvbC0AEEf4F95quyyIprTSaasHLoWQVA8bNaKaUK1U8eTuxK5Pgu~fHaAMzv87UXpFZk~-Xvu7Krez9IWmTBPNKl~rHRc6sRRpjiars0BEQASb8L7lLdQzMnTzbRZ12bcy-ZftI5t4AaKcpDY5lQOs0gc8TUxOTBihsVqehurIsmQUK-8Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Transient and stable expression of GFP GKand GK GFP in MIN6 cells. A: MIN6 were transiently transfected with GK GFP and GFP GK. After 48 h, GK activity (expressed as percentage of nontransfected cells [NT]) and immunoreactivity (50 and 62–64 kDa) were determined. Results are representative of two experiments. B–D: Nonclonal GK GFP or GFP GK cell lines. B: GK activity at increasing passage number. C and D: GK activity at varying glucose cocentrations without (C dashed lines; D □) or with (C solid lines; D ▪) 5 μmol/l GKA (32) in GK GFP and GFP GK cell lines and MIN6 cells that were either untreated or pretreated (18 h) with Ad-LGK for expression of untagged liver GK (+GK). C: GK activity as percentage of 30 mmol/l glucose. The effects of GKA were significant for all four cell types: *P < 0.05 and **P < 0.01. D: GK activity at 30 mmol/l glucose *P < 0.05, **P < 0.01 relative to untreated MIN6, n = 6.
Transient and stable expression of GFP GKand GK GFP in MIN6 cells. A: MIN6 were transiently transfected with GK GFP and GFP GK. After 48 h, GK activity (expressed as percentage of nontransfected cells [NT]) and immunoreactivity (50 and 62–64 kDa) were determined. Results are representative of two experiments. B–D: Nonclonal GK GFP or GFP GK cell lines. B: GK activity at increasing passage number. C and D: GK activity at varying glucose cocentrations without (C dashed lines; D □) or with (C solid lines; D ▪) 5 μmol/l GKA (32) in GK GFP and GFP GK cell lines and MIN6 cells that were either untreated or pretreated (18 h) with Ad-LGK for expression of untagged liver GK (+GK). C: GK activity as percentage of 30 mmol/l glucose. The effects of GKA were significant for all four cell types: *P < 0.05 and **P < 0.01. D: GK activity at 30 mmol/l glucose *P < 0.05, **P < 0.01 relative to untreated MIN6, n = 6.

Effect of GK overexpression or activation on insulin secretion. A: Insulin secretion was determined in untreated MIN6 cells (×), MIN6 cells overexpressing GK by pretreatment with Ad-LGK (○), GK GFP transfected cells (□), and GFP GK transfected cells (▵) and expressed as μg/h per mg protein. n = 3–5. B: Insulin secretion was determined at 3 mmol/l glucose in MIN6 treated with 10 μmol/l of GKA (35) or MIN6 cells pretreated with increasing titers of Ad-LGK for overexpression of GK by 2.6-, 3.8-, and 5.8-fold relative to untreated cells (1). Insulin secretion is expressed as fold increase relative to control (1). Results are representative of two experiments. C: Insulin secretion was determined at 3, 5, and 25 mmol/l glucose in mouse islets either pretreated (18 h) with Ad-LGK (+GK) or incubated with 10 μmol/l of GKA (+GKA). Insulin secretion is expressed as fold increase relative to control. ***P < 0.005 relative to untreated at 5 mmol/l, n = 2.
Effect of GK overexpression or activation on insulin secretion. A: Insulin secretion was determined in untreated MIN6 cells (×), MIN6 cells overexpressing GK by pretreatment with Ad-LGK (○), GK GFP transfected cells (□), and GFP GK transfected cells (▵) and expressed as μg/h per mg protein. n = 3–5. B: Insulin secretion was determined at 3 mmol/l glucose in MIN6 treated with 10 μmol/l of GKA (35) or MIN6 cells pretreated with increasing titers of Ad-LGK for overexpression of GK by 2.6-, 3.8-, and 5.8-fold relative to untreated cells (1). Insulin secretion is expressed as fold increase relative to control (1). Results are representative of two experiments. C: Insulin secretion was determined at 3, 5, and 25 mmol/l glucose in mouse islets either pretreated (18 h) with Ad-LGK (+GK) or incubated with 10 μmol/l of GKA (+GKA). Insulin secretion is expressed as fold increase relative to control. ***P < 0.005 relative to untreated at 5 mmol/l, n = 2.

Subcellular location of GFP-tagged GK chimeras in MIN6. A: Untreated MIN6 cells were fixed and immunostained for GK (fluorescein isothiocyanate green) and insulin (Texas Red). B: MIN6 cells transiently transfected with GFP GK (green) immunostained for insulin (red). C: Stable GFP GK (green) cells immunostained for insulin (red). Yellow staining indicates colocalization.
Subcellular location of GFP-tagged GK chimeras in MIN6. A: Untreated MIN6 cells were fixed and immunostained for GK (fluorescein isothiocyanate green) and insulin (Texas Red). B: MIN6 cells transiently transfected with GFP GK (green) immunostained for insulin (red). C: Stable GFP GK (green) cells immunostained for insulin (red). Yellow staining indicates colocalization.
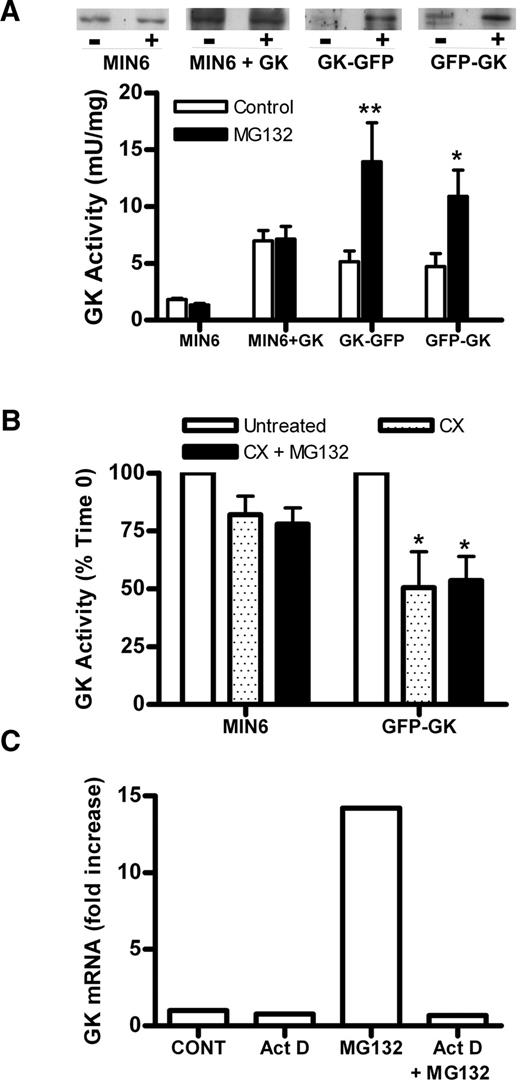
Effect of MG132 on expression of GFP-tagged chimeras. A: GK activity was determined after culture for 18 h without (□) or with (▪) 10 μmol/l MG132 in GK GFP and GFP GK cells and MIN6 cells either untreated or treated with Ad-LGK (+GK), n = 7. GK immunoreactivity is representative of seven experiments. *P < 0.05 and **P < 0.01 effect of MG132. B: Untreated MIN6 and MIN6 cells transiently transfected with GFP GK were incubated without (□) or with 5 μmol/l cycloheximide and without (□) or with 10 μmol/l MG132 (▪) for 4–6 h. *P < 0.05 relative to untreated cells. n = 2. C: Clonal cell lines expressing GFP GK (WT10B) were incubated with 0.4 μg/ml actinomycin D (Act D), 10 μmol/l MG132, or both for 3 h, n = 2.
Effect of MG132 on expression of GFP-tagged chimeras. A: GK activity was determined after culture for 18 h without (□) or with (▪) 10 μmol/l MG132 in GK GFP and GFP GK cells and MIN6 cells either untreated or treated with Ad-LGK (+GK), n = 7. GK immunoreactivity is representative of seven experiments. *P < 0.05 and **P < 0.01 effect of MG132. B: Untreated MIN6 and MIN6 cells transiently transfected with GFP GK were incubated without (□) or with 5 μmol/l cycloheximide and without (□) or with 10 μmol/l MG132 (▪) for 4–6 h. *P < 0.05 relative to untreated cells. n = 2. C: Clonal cell lines expressing GFP GK (WT10B) were incubated with 0.4 μg/ml actinomycin D (Act D), 10 μmol/l MG132, or both for 3 h, n = 2.

GK kinetics and activation by a GKA in GFP GK, GFP V62M, and GFP G72R clonal cell lines. Clonal cell lines GFP GK clone WT3B (A), GFP V62M (B), and GFP G72R (C) were incubated with 10 μmol/l MG132 for 18 h. Untreated MIN6 cells (D) were incubated without (squares) or with (triangles) 10 μmol/l MG132 for 18 h. GK activity was determined at varying glucose concentration (0.5–30 mmol/l) on 13,000g supernatants in the absence (open) or presence (solid) of 5 μmol/l GKA and is expressed as percentage 30 mmol/l glucose. Means ± SE, n = 6 (A–C) or n = 5 (D). *P < 0.05 and **P < 0.01, effect of GKA.
GK kinetics and activation by a GKA in GFP GK, GFP V62M, and GFP G72R clonal cell lines. Clonal cell lines GFP GK clone WT3B (A), GFP V62M (B), and GFP G72R (C) were incubated with 10 μmol/l MG132 for 18 h. Untreated MIN6 cells (D) were incubated without (squares) or with (triangles) 10 μmol/l MG132 for 18 h. GK activity was determined at varying glucose concentration (0.5–30 mmol/l) on 13,000g supernatants in the absence (open) or presence (solid) of 5 μmol/l GKA and is expressed as percentage 30 mmol/l glucose. Means ± SE, n = 6 (A–C) or n = 5 (D). *P < 0.05 and **P < 0.01, effect of GKA.

GK-mRNA, immunoreactivity, and activity in clonal cell lines GFP GK, GFP V62M, and GFP G72R. Clonal cell lines GFP GK (clones WT3B and WT10B), GFP V62M, and GFP G72R were generated by G418 selection. The cells were preincubated for 18 h without (□) or with (▪) 10 μmol/l MG132, and GK mRNA, immunoreactivity, and activity were determined. A: Human GK mRNA/cyclophilin mRNA expressed as fold increase in the presence of MG132. B: GK immunoreactivity (RDU) immunoblot is representative of four experiments. C: GK activity. D: GK immunoreactivity–to–mRNA ratio. E: GK activity–to–mRNA ratio. F: GK activity–to–immunoreactivity ratio. Ratios in D–F are normalized to WT3B. Means ± SE, n = 4, *P < 0.05 and **P < 0.01 relative to both WT3B and WT10B in the absence or presence of MG132.
GK-mRNA, immunoreactivity, and activity in clonal cell lines GFP GK, GFP V62M, and GFP G72R. Clonal cell lines GFP GK (clones WT3B and WT10B), GFP V62M, and GFP G72R were generated by G418 selection. The cells were preincubated for 18 h without (□) or with (▪) 10 μmol/l MG132, and GK mRNA, immunoreactivity, and activity were determined. A: Human GK mRNA/cyclophilin mRNA expressed as fold increase in the presence of MG132. B: GK immunoreactivity (RDU) immunoblot is representative of four experiments. C: GK activity. D: GK immunoreactivity–to–mRNA ratio. E: GK activity–to–mRNA ratio. F: GK activity–to–immunoreactivity ratio. Ratios in D–F are normalized to WT3B. Means ± SE, n = 4, *P < 0.05 and **P < 0.01 relative to both WT3B and WT10B in the absence or presence of MG132.

GK activity and immunoreactivity in MIN6 cells transiently transfected with GFP GK, GFP V62M, and GFP G72R. MIN6 cells were transfected with varying amounts of plasmid: 1 μg (□), 2 μg (▪), and 3 μg ( ▒) of GFP GK (WT), GFP V62M, and GFP G72R and cultured for 48 h. A: GK activity. B: Immunoreactivity (RDU). Immunoblot is representative of three experiments. C: GK activity vs. immunoreactivity, n = 3.
GK activity and immunoreactivity in MIN6 cells transiently transfected with GFP GK, GFP V62M, and GFP G72R. MIN6 cells were transfected with varying amounts of plasmid: 1 μg (□), 2 μg (▪), and 3 μg ( ▒) of GFP GK (WT), GFP V62M, and GFP G72R and cultured for 48 h. A: GK activity. B: Immunoreactivity (RDU). Immunoblot is representative of three experiments. C: GK activity vs. immunoreactivity, n = 3.
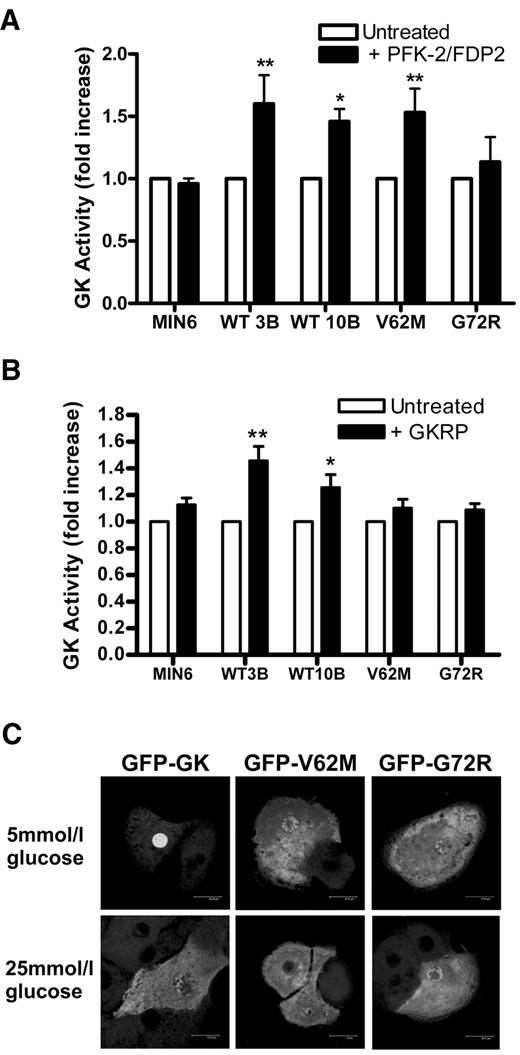
Interaction of GFP GK, GFP V62M, and GFP G72R with PFK2/FDP2 and GKRP. A: MIN6 cells and clonal cell lines GFP GK (clones WT3B and WT10B), GFP V62M, and GFP G72R were either untreated (□) or pretreated with Ad-PFK2/FDP2 (▪). GK activity is expressed as percentage untreated control. *P < 0.05 and **P < 0.01, effect of PFK2/FDP2. Means ± SE, n = 5. B: MIN6 and clonal cell lines GFP GK (clones WT3B and WT10B), GFP V62M, and GFP G72R were either untreated (□) or pretreated with Ad-GKRP (▪). GK activity is expressed as percentage untreated control. *P < 0.05 and **P < 0.01, effect of GKRP. Means ± SE, n = 5. C: Primary hepatocytes were transfected with plasmids GFP GK, GFP V62M, or GFP G72R and cultured for 18 h at 5 mmol/l glucose. Cells were then incubated for 3 h at either 5 or 25 mmol/l glucose before acetone fixation. Fluorescence was visualized by confocal microscopy. Images are representative of five experiments.
Interaction of GFP GK, GFP V62M, and GFP G72R with PFK2/FDP2 and GKRP. A: MIN6 cells and clonal cell lines GFP GK (clones WT3B and WT10B), GFP V62M, and GFP G72R were either untreated (□) or pretreated with Ad-PFK2/FDP2 (▪). GK activity is expressed as percentage untreated control. *P < 0.05 and **P < 0.01, effect of PFK2/FDP2. Means ± SE, n = 5. B: MIN6 and clonal cell lines GFP GK (clones WT3B and WT10B), GFP V62M, and GFP G72R were either untreated (□) or pretreated with Ad-GKRP (▪). GK activity is expressed as percentage untreated control. *P < 0.05 and **P < 0.01, effect of GKRP. Means ± SE, n = 5. C: Primary hepatocytes were transfected with plasmids GFP GK, GFP V62M, or GFP G72R and cultured for 18 h at 5 mmol/l glucose. Cells were then incubated for 3 h at either 5 or 25 mmol/l glucose before acetone fixation. Fluorescence was visualized by confocal microscopy. Images are representative of five experiments.
Published ahead of print at http://diabetes.diabetesjournals.org on 27 March 2007. DOI: 10.2337/db06-1151.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Article Information
This work was funded by grants from Diabetes UK (BDA RD01/0002364 and RD04/0002738) and the Medical Research Council (G0100348 and G0501543).
We thank Dr. Jun-ichi Miyazaki for MIN6 cells.