ABSTRACT
Gerstmann-Sträussler-Scheinker is a genetic prion disease and the most common mutation is p.Pro102Leu. We report clinical, molecular and neuropathological data of seven individuals, belonging to two unrelated Brazilian kindreds, carrying the p.Pro102Leu. Marked differences among patients were observed regarding age at onset, disease duration and clinical presentation. In the first kindred, two patients had rapidly progressive dementia and three exhibited predominantly ataxic phenotypes with variable ages of onset and disease duration. In this family, age at disease onset in the mother and daughter differed by 39 years. In the second kindred, different phenotypes were also reported and earlier ages of onset were associated with 129 heterozygosis. No differences were associated with apoE genotype. In these kindreds, the codon 129 polymorphism could not explain the clinical variability and 129 heterozygosis was associated with earlier disease onset. Neuropathological examination in two patients confirmed the presence of typical plaques and PrPsc immunopositivity.
Gerstmann-Sträussler-Scheinker disease; prion diseases; prions
RESUMO
A doença de Gerstmann-Sträussler-Scheinker é uma doença priônica genética, cuja mutação mais frequente é p.Pro102Leu. Descrevem-se dados clínicos, moleculares e neuropatológicos de sete indivíduos em duas famílias não relacionadas com p.Pro102Leu. Diferenças notáveis entre os pacientes em relação à idade de início, duração da doença e apresentação clínica foram encontradas. Na primeira família, dois pacientes apresentaram demência rapidamente progressiva e três apresentaram fenótipo de ataxia com idade variáveis de início e duração da doença. Nesta família, a idade de início entre mãe e filha diferiu em 39 anos. Na segunda família, fenótipos diferentes foram observados e idades precoces de início dos sintomas foram associadas à heterozigose no códon 129. Não houve diferença em relação ao genótipo do gene da apoE. O genótipo do códon 129 não foi responsável pela variabilidade clínica; heterozigose no códon 129 esteve associada ao início precoce da doença. O exame neuropatológico em dois pacientes confirmou presença de placas típicas e imunohistoquímica para PrPsc.
doença de Gerstmann-Sträussler-Scheinker; doenças de prion; príons
Human spongiform encephalopathies, or prion diseases, are rare fatal neurodegenerative conditions associated with conversion of prion protein (PrPC) to its misfolded pathogenic isoform (PrPsc)11. Prusiner SB. Prions. Proc Natl Acad Sci USA. 1998;95(23):13363-83. https://doi.org/10.1073/pnas.95.23.13363
https://doi.org/10.1073/pnas.95.23.13363...
. According to their etiology, prion diseases can be classified as sporadic, genetic or acquired by infection and their estimated annual incidence is approximately one per million individuals22. Holman RC, Belay ED, Christensen KY, Maddox RA, Minino AM, Folkema AM et al. Human prion diseases in the United States. PLoS One;2010;5(1):e8521. https://doi.org/10.1371/journal.pone.0008521
https://doi.org/10.1371/journal.pone.000...
,33. Puoti G, Bizzi A, Forloni G, Safar JG, Tagliavini F, Gambetti P. Sporadic human prion diseases: molecular insights and diagnosis. Lancet Neurol. 2012;11(7):618-28. https://doi.org/10.1016/S1474-4422(12)70063-7
https://doi.org/10.1016/S1474-4422(12)70...
,44. Wadsworth JD, Hill AF, Beck JA, Collinge J. Molecular and clinical classification of human prion disease. Br Med Bull. 2003;66(1):241-54. https://doi.org/10.1093/bmb/66.1.241
https://doi.org/10.1093/bmb/66.1.241...
.
Genetic forms of prion diseases are inherited as an autosomal dominant trait and associated with mutations in the prion protein gene (PRNP), accounting for around 15% of cases55. Ladogana A, Puopolo M, Croes EA, Budka H, Jarius C, Collins S et al. Mortality from Creutzfeldt-Jakob disease and related disorders in Europe, Australia, and Canada. Neurology. 2005;64(9):1586-91. https://doi.org/10.1212/01.WNL.0000160117.56690.B2
https://doi.org/10.1212/01.WNL.000016011...
. The PRNP mutations lead to three major phenotypes: Creutzfeldt-Jakob disease (CJD), Gerstmann-Sträussler-Scheinker (GSS) disease and familial fatal insomnia44. Wadsworth JD, Hill AF, Beck JA, Collinge J. Molecular and clinical classification of human prion disease. Br Med Bull. 2003;66(1):241-54. https://doi.org/10.1093/bmb/66.1.241
https://doi.org/10.1093/bmb/66.1.241...
.
Gerstmann-Sträussler-Scheinker disease was first reported in an Austrian family in 1936 and its main clinical features are slowly progressive cerebellar ataxia associated with cognitive decline66. Gerstmann J, Straussler E, Scheinker J. Über eine eigenartige hereditär-familiäre Erkrankung des Zentralnervensystems. Z Neurol. 1936;154:736-62.. The molecular basis of GSS was identified in 1989, when the substitution of a proline at position 102 for leucine (p.Pro102Leu) in the PRNP gene was recognized in two families77. Hsiao K, Baker HF, Crow TJ, Poulter M, Owen F, Terwilliger JD et al. Linkage of a prion protein missense variant to Gerstmann-Sträussler syndrome. Nature. 1989;338(6213):342-5. https://doi.org/10.1038/338342a0
https://doi.org/10.1038/338342a0...
; this same variant was subsequently identified in the original Austrian family88. Kretzschmar HA, Honold G, Seitelberger F, Feucht M, Wessely P, Mehraein P et al. Prion protein mutation in family first reported by Gerstmann, Sträussler and Scheinker. Lancet. 1991;337(8750):1160. https://doi.org/10.1016/0140-6736(91)92826-N
https://doi.org/10.1016/0140-6736(91)928...
. A number of other mutations associated with GSS were also reported, predominantly missense and nonsense pathogenic variants located at codons 105, 117, 131, 187, 198, 202, 212, 217, and 232, or copy number variation in octapeptide repeats99. Collins S, McLean CA, Masters CL. Gerstmann-Sträussler-Scheinker syndrome, fatal familial insomnia, and kuru: a review of these less common human transmissible spongiform encephalopathies. J Clin Neurosci. 2001;8(5):387-97. https://doi.org/10.1054/jocn.2001.0919
https://doi.org/10.1054/jocn.2001.0919...
. The common variant in the PRNP gene, p.Met129Val, might act as a disease modifier in prion disease, but alone is not solely responsible for this condition. Clinical and pathological phenotypic variability among individuals with the same mutation had previously been reported worldwide1010. Arata H, Takashima H, Hirano R, Tomimitsu H, Machigashira K, Izumi K et al. Early clinical signs and imaging findings in Gerstmann-Sträussler-Scheinker syndrome (Pro102Leu). Neurology. 2006;66(11):1672-8. https://doi.org/10.1212/01.wnl.0000218211.85675.18
https://doi.org/10.1212/01.wnl.000021821...
,1111. Hainfellner JA, Brantner-Inthaler S, Cervenáková L, Brown P, Kitamoto T, Tateishi J et al. The original Gerstmann-Sträussler-Scheinker family of Austria: divergent clinicopathological phenotypes but constant PrP genotype. Brain Pathol. 1995;5(3):201-11. https://doi.org/10.1111/j.1750-3639.1995.tb00596.x
https://doi.org/10.1111/j.1750-3639.1995...
,1212. Piccardo P, Dlouhy SR, Lievens PM, Young K, Bird TD, Nochlin D et al. Phenotypic variability of Gerstmann-Sträussler-Scheinker disease is associated with prion protein heterogeneity. J Neuropathol Exp Neurol. 1998;57(10):979-88. https://doi.org/10.1097/00005072-199810000-00010
https://doi.org/10.1097/00005072-1998100...
,1313. Riudavets MA, Sraka MA, Schultz M, Rojas E, Martinetto H, Begué C et al. Gerstmann-Sträussler-Scheinker syndrome with variable phenotype in a new kindred with PRNP-P102L mutation. Brain Pathol. 2014;24(2):142-7. https://doi.org/10.1111/bpa.12083
https://doi.org/10.1111/bpa.12083...
,1414. Webb TE, Poulter M, Beck J, Uphill J, Adamson G, Campbell T et al. Phenotypic heterogeneity and genetic modification of P102L inherited prion disease in an international series. Brain. 2008;131(10):2632-46. https://doi.org/10.1093/brain/awn202
https://doi.org/10.1093/brain/awn202...
. Several factors may contribute to this variability, such as the genotype for the variant p.Met129Val and differences in molecular prion strains1414. Webb TE, Poulter M, Beck J, Uphill J, Adamson G, Campbell T et al. Phenotypic heterogeneity and genetic modification of P102L inherited prion disease in an international series. Brain. 2008;131(10):2632-46. https://doi.org/10.1093/brain/awn202
https://doi.org/10.1093/brain/awn202...
,1515. Mead S. Prion disease genetics. Eur J Hum Genet. 2006;14(3):273-81. https://doi.org/10.1038/sj.ejhg.5201544
https://doi.org/10.1038/sj.ejhg.5201544...
,1616. Wadsworth JD, Joiner S, Linehan JM, Cooper S, Powell C, Mallinson G et al. Phenotypic heterogeneity in inherited prion disease (P102L) is associated with differential propagation of protease-resistant wild-type and mutant prion protein. Brain. 2006;129(6):1557-69. https://doi.org/10.1093/brain/awl076
https://doi.org/10.1093/brain/awl076...
. A study conducted in the largest reported GSS disease kindred confirmed marked phenotypic variability and suggested that the p.Met129Val variant genotype was associated with it1414. Webb TE, Poulter M, Beck J, Uphill J, Adamson G, Campbell T et al. Phenotypic heterogeneity and genetic modification of P102L inherited prion disease in an international series. Brain. 2008;131(10):2632-46. https://doi.org/10.1093/brain/awn202
https://doi.org/10.1093/brain/awn202...
.
In the present study, we report two unrelated Brazilian families with GSS disease due to a p.Pro102Leu mutation. Clinical, molecular and/or pathological data of five cases in one family and two cases in another family are reported. Age at onset ranged from 25 to 66 years old and disease duration ranged from eight months to 15 years.
METHODS
Patients
Two individuals from Brazilian families with clinical symptoms suggestive of prion disease were first evaluated, and GSS disease confirmed based on the finding of the variant p.Pro102Leu in one allele of the PRNP gene. Based on this diagnosis, data from other family members were collected and prospective follow-up performed. Where possible, family members were interviewed, patients examined and blood samples taken for molecular analysis after informed consent. Additional investigations, such as brain MRI, protein 14.3.3 in cerebrospinal fluid (CSF) and an electroencephalogram were performed when possible. This study was approved by the Institutional Ethics Committee and participating families signed the informed consent form.
Genetic analysis
The DNA was extracted according to standard protocols using the Wizard Genomic DNA Purification Kit®. The PRNP coding region amplification and sequencing were performed adopting previously published procedures1717. Castro RM, Landemberger MC, Walz R, Carlotti CG Jr, Huang N, Cunha DR et al. High capacity and low cost detection of prion protein gene variant alleles by denaturing HPLC. J Neurosci Methods. 2004;139(2):263-9. https://doi.org/10.1016/j.jneumeth.2004.05.001
https://doi.org/10.1016/j.jneumeth.2004....
. In order to investigate the segregation of the variants p.Pro102Leu and p.Met129Val, PCR products of the complete coding region of PRNP were cloned using pGEM®-T Easy Vector Systems (Promega) and then sequenced. For apolipoprotein E (apoE) gene analysis, allele-specific real-time PCR was performed1818. Calero O, Hortigüela R, Bullido MJ, Calero M. Apolipoprotein E genotyping method by real time PCR, a fast and cost-effective alternative to the TaqMan and FRET assays. J Neurosci Methods. 2009;183(2):238-40. https://doi.org/10.1016/j.jneumeth.2009.06.033
https://doi.org/10.1016/j.jneumeth.2009....
.
Brain tissue analyses
Formalin-fixed fragments of the cerebral cortex, basal ganglia, hippocampus and cerebellum were immersed in 95% formic acid, and then processed for paraffin embedding. Histological sections were stained with H&E and Congo Red. Antigen retrieval was performed after tissue exposure to 10mM citrate buffer, pH6.0 for 40 minutes at 96oC. Immunohistochemical studies were performed using anti-PrPC monoclonal antibody (clone PrP3F4;DAKO) followed by Polymer Secondary System (Novolink, Leica). Unfortunately, there was no availability of frozen unfixed brain tissue for western blot analysis of protease-resistant prion protein.
RESULTS
Clinical descriptions
Family A
In this family, there were five affected individuals (pedigree shown in Figure 1). Clinical and/or pathological data of five patients over two generations are given in Table 1. In the first generation, the father of three affected individuals died after the seventh decade of life while the mother died in her forties, without symptoms of GSS disease. Three siblings from the second generation, aged from 61 to 69 years old, have no symptoms to-date. Fifteen relatives of the third generation, aged from 27 to 51 years old, are also asymptomatic.
Patient IIIa - 1 (index)
Disease onset occurred at the age of 27 years with rapid cognitive decline progressing to severe dementia in two years. Gait instability and dysarthria emerged after five years. Neurologic examination seven years after onset showed severe dementia, grasping reflex, parkinsonian gait with bilateral support, hyperactive tendon reflexes with Babinski sign, and plastic rigidity in the upper limbs. Myoclonus occurred eight months later. She died 15 years after the first symptoms.
Family B
This Brazilian family is from Portuguese descent and had six probably-affected individuals (pedigree shown in Figure 1). The first known case was the paternal grandfather of the proband (Ib - 1). He died before the age of 40 with a disease that rendered him dependent for several years. In the next generation, there were reports of at least three individuals who died at around the fourth decade of life, with similar features. The father of the proband (IIb - 1) died at age 33 after eight years of disease, with gait and speech impairment and cognitive decline. In the third generation, two patients were evaluated. Clinical data for these two patients are given in Table 1.
Patient IIIb – 1
Disease onset occurred at age 35 with cognitive decline and balance deficit. Severe dementia and abasia occurred three years later. Neurologic examination three years after onset revealed severe dementia with frontal syndrome and global cerebellar ataxia. Spasticity, brisk tendon reflexes in all four limbs and Babinski sign were also present.
The age of onset in the two reported families ranged from 27 to 66 years old. The duration of the disease ranged from eight months to 15 years. The clinical diagnosis of possible CJD was initially proposed in two patients. Typical GSS features were observed in four patients (IIa-2, IIa-3, IIIa-2 and IIIb-2).
Molecular data
The p.Pro102Leu mutation was found in all six patients (IIa-2, IIa-3, IIIa-1, IIIa-2, IIIb-1 and IIIb-2) who underwent genetic analyses. This investigation was not possible in one patient (IIa-1). Four patients were heterozygote at codon 129 (methionine/valine, Met129Val) and two were homozygote for methionine. All patients from family A were e3/e3 and all patients from family B were e2/e3 for apoE gene (Table 1).
Neuropathological data
Neuropathological study was performed in three of the five patients from family A.
Spongiform encephalopathy was found on frontal lobe biopsy of patient IIa-1. At the time immunohistochemistry for PrPSc was not available, and the slides and blocks could not be retrieved for further studies.
The study of patient IIIa-1 showed that focal neuronal loss, spongiosis and gliosis were minimal, both in the cerebral and the cerebellar cortex. However, there were multicentric plaques in the molecular layer of the cerebellum (Figure 2) and deep regions of the temporal cortex, including regions of the hippocampus, adjacent to pyramidal cells and granular cells of the dentate fascia (Figure 3). The basal ganglia, thalami, midbrain, pons, and medulla were normal.
Patient IIIa-1. Multicentric plaque adjacent to granular cells of the dentate fascia of the hippocampus.
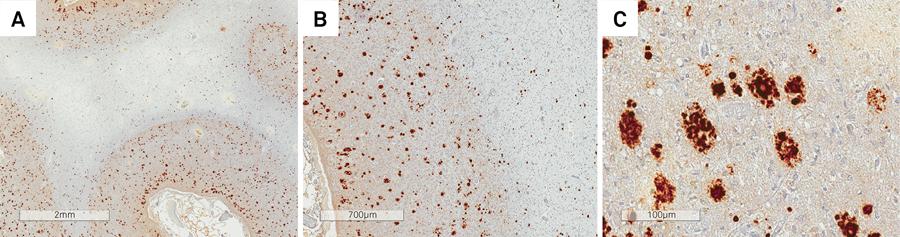
In patient IIIa-2, there was severe diffuse cerebral and cerebellar cortical atrophy, in addition to extensive involvement of the subjacent white matter, which was rarefied and collapsed, in some areas containing small cavities (Figure 4). The basal ganglia and thalami were also extremely reduced in size. There was extensive neuronal loss, reactive gliosis and spongiosis, both in the cerebral and cerebellar cortex, with extensive Purkinje and granular cell loss. The subjacent white matter was rarefied, with hardly any myelin (Figures 5 and 6). Multicentric plaques, immunopositive for PrPSc (Figure 7), some of them containing amyloid material, confirmed with Congo Red staining, were observed in the cerebral and cerebellar cortex.
Patient IIIa-2. Severe diffuse cerebral (a) and cerebellar (b) cortical atrophy, in addition to extensive involvement of the subjacent white matter.
(A, B and C) Patient IIIa-2. Multicentric plaques, immunopositive for PrPSc observed in the cerebral cortex.
Patient IIIa-2. Cerebellar white matter was rarefied (A) and the cortex contained multicentric plaques (B).
DISCUSSION
The p.Pro102Leu variant is the most frequent mutation associated with GSS and has been reported in a number of kindreds from Austria, Germany, China, Japan, UK, Italy, Argentina and USA families1414. Webb TE, Poulter M, Beck J, Uphill J, Adamson G, Campbell T et al. Phenotypic heterogeneity and genetic modification of P102L inherited prion disease in an international series. Brain. 2008;131(10):2632-46. https://doi.org/10.1093/brain/awn202
https://doi.org/10.1093/brain/awn202...
,1919. Doh-ura K, Tateishi J, Sasaki H, Kitamoto T, Sakaki Y. Pro→Leu change at position 102 of prion protein is the most common but not the sole mutation related to Gerstmann-Sträussler-Scheinker syndrome. Biochem Biophys Res Commun. 1989;163(2):974-9. https://doi.org/10.1016/0006-291X(89)92317-6
https://doi.org/10.1016/0006-291X(89)923...
,2020. Kovács GG, Puopolo M, Ladogana A, Pocchiari M, Budka H, Duijn C et al. Genetic prion disease: the EUROCJD experience. Hum Genet. 2005;118(2):166-74. https://doi.org/10.1007/s00439-005-0020-1
https://doi.org/10.1007/s00439-005-0020-...
.
Classically, GSS with the p.Pro102Leu mutation is characterized by slowly progressive cerebellar ataxia with symptom onset in the fifth or sixth decade of life. During the course of the disease, pyramidal signs and cognitive decline are common. Movement disorders, typically myoclonus and athetosis, may occur. Seizures, deafness and cranial nerve palsies number among the less common features99. Collins S, McLean CA, Masters CL. Gerstmann-Sträussler-Scheinker syndrome, fatal familial insomnia, and kuru: a review of these less common human transmissible spongiform encephalopathies. J Clin Neurosci. 2001;8(5):387-97. https://doi.org/10.1054/jocn.2001.0919
https://doi.org/10.1054/jocn.2001.0919...
. Mean survival of GSS is from five to seven years, longer than seen in other prion diseases. Of the seven individuals from two families reported herein, four died, within eight months, four, seven and 15 years of disease onset, respectively. Survival of longer than 10 years is uncommon and the longest reported disease duration, so far, was 13 years99. Collins S, McLean CA, Masters CL. Gerstmann-Sträussler-Scheinker syndrome, fatal familial insomnia, and kuru: a review of these less common human transmissible spongiform encephalopathies. J Clin Neurosci. 2001;8(5):387-97. https://doi.org/10.1054/jocn.2001.0919
https://doi.org/10.1054/jocn.2001.0919...
.
The clinical phenotype of GSS disease ranges from ataxia followed by dementia to primary cognitive decline, while the disease progression rate also varies considerably. Phenotypic variability has been reported in other series and appears to be unrelated to the p.Met129Val variant genotype2121. Barbanti P, Fabbrini G, Salvatore M, Petraroli R, Cardone F, Maras B et al. Polymorphism at codon 129 or codon 219 of PRNP and clinical heterogeneity in a previously unreported family with Gerstmann-Sträussler-Scheinker disease (PrP-P102L mutation). Neurology. 1996;47(3):734-41. https://doi.org/10.1212/WNL.47.3.734
https://doi.org/10.1212/WNL.47.3.734...
. Patients IIIa - 1 and IIIb - 1 had cognitive decline as the first symptom and exhibited very early onset, which is unusual but consistent with some other previous reports1313. Riudavets MA, Sraka MA, Schultz M, Rojas E, Martinetto H, Begué C et al. Gerstmann-Sträussler-Scheinker syndrome with variable phenotype in a new kindred with PRNP-P102L mutation. Brain Pathol. 2014;24(2):142-7. https://doi.org/10.1111/bpa.12083
https://doi.org/10.1111/bpa.12083...
. One patient presented with clinical features of CJD (patient IIa - 1), while another had rapidly-progressively dementia with a long survival (patient IIIa - 1). Many studies support the existence of three dementia phenotypes associated with the p.Pro102Leu variant: rapid global dementia resembling CJD2121. Barbanti P, Fabbrini G, Salvatore M, Petraroli R, Cardone F, Maras B et al. Polymorphism at codon 129 or codon 219 of PRNP and clinical heterogeneity in a previously unreported family with Gerstmann-Sträussler-Scheinker disease (PrP-P102L mutation). Neurology. 1996;47(3):734-41. https://doi.org/10.1212/WNL.47.3.734
https://doi.org/10.1212/WNL.47.3.734...
,2222. Majtényi C, Brown P, Cervenáková L, Goldfarb LG, Tateishi J. A three-sister sibship of Gerstmann-Sträussler-Scheinker disease with a CJD phenotype. Neurology. 2000;54(11):2133-7. https://doi.org/10.1212/WNL.54.11.2133
https://doi.org/10.1212/WNL.54.11.2133...
, slowly progressive cognitive decline2323. Ishizawa K, Komori T, Shimazu T, Yamamoto T, Kitamoto T, Shimazu K et al. Hyperphosphorylated tau deposition parallels prion protein burden in a case of Gerstmann-Sträussler-Scheinker syndrome P102L mutation complicated with dementia. Acta Neuropathol. 2002;104(4):342-30. https://doi.org/10.1007/s00401-002-0547-3
https://doi.org/10.1007/s00401-002-0547-...
and frontotemporal type dementia2424. Giovagnoli AR, Di Fede G, Aresi A, Reati F, Rossi G, Tagliavini F. Atypical frontotemporal dementia as a new clinical phenotype of Gerstmann-Straussler-Scheinker disease with the PrP-P102L mutation: description of a previously unreported Italian family. Neurol Sci. 2008;29(6):405-10. https://doi.org/10.1007/s10072-008-1025-z
https://doi.org/10.1007/s10072-008-1025-...
.
In the largest published investigation involving 84 GSS individuals belonging to the same extended family, methionine homozygosity at codon 129 was associated with early-onset disease while other combinations were not associated with disease duration1414. Webb TE, Poulter M, Beck J, Uphill J, Adamson G, Campbell T et al. Phenotypic heterogeneity and genetic modification of P102L inherited prion disease in an international series. Brain. 2008;131(10):2632-46. https://doi.org/10.1093/brain/awn202
https://doi.org/10.1093/brain/awn202...
. In contrast to this recent data, the earliest clinical onsets in the present study occurred in four patients (IIIa - 1, IIIa - 2, IIIb - 1 and IIIb – 2) heterozygous at codon 129, and the p.Pro102Leu mutation was located in the 129M allele for family A and the 129V allele in family B. Besides the age at onset and duration of the disease, other phenotypic differences were observed among the patients described.
Similar clinical contrasts, with different phenotypes within the same family, have been reported worldwide. The causes of phenotypic variability within a family with GSS mutation are unknown. The polymorphism at codon 129 alone is probably not the only explanation for the pathologic and clinical heterogeneity in GSS (codon 102 mutation)2121. Barbanti P, Fabbrini G, Salvatore M, Petraroli R, Cardone F, Maras B et al. Polymorphism at codon 129 or codon 219 of PRNP and clinical heterogeneity in a previously unreported family with Gerstmann-Sträussler-Scheinker disease (PrP-P102L mutation). Neurology. 1996;47(3):734-41. https://doi.org/10.1212/WNL.47.3.734
https://doi.org/10.1212/WNL.47.3.734...
. However, they are of particular interest in the study of prion diseases since they point to other, as yet unknown, factors that may contribute to the diverse phenotype associated with the same mutation at PRNP. In family A, disease symptoms first manifested at 27 years old in the index case, whereas, in her mother the symptoms were only reported at the age of 66.
Another genetic factor that could explain some clinical heterogeneity in prion diseases is the apoE polymorphism. Conflicting results for the association of sporadic CJD and the presence of apoE e4 have been published. Although apoE and PRNP may represent effect size of risk association in patients with sporadic CJD and e2 carriers have late onset disease2525. Calero O, Bullido MJ, Clarimón J, Frank-García A, Martínez-Martín P, Lleó A et al. Genetic cross-interaction between APOE and PRNP in sporadic Alzheimer’s and Creutzfeldt-Jakob diseases. PLoS One. 2011;6(7):e22090. https://doi.org/10.1371/journal.pone.0022090
https://doi.org/10.1371/journal.pone.002...
, few data supports that apoE is not a risk factor for p.Pro102Leu GSS2626. Chapman J, Cervenáková L, Petersen RB, Lee HS, Estupinan J, Richardson S et al. APOE in non-Alzheimer amyloidoses: transmissible spongiform encephalopathies. Neurology. 1998;51(2):548-53. https://doi.org/10.1212/WNL.51.2.548
https://doi.org/10.1212/WNL.51.2.548...
. However, e4 carriers had delayed age of onset by 10 years in the largest GSS kindred studied so far1414. Webb TE, Poulter M, Beck J, Uphill J, Adamson G, Campbell T et al. Phenotypic heterogeneity and genetic modification of P102L inherited prion disease in an international series. Brain. 2008;131(10):2632-46. https://doi.org/10.1093/brain/awn202
https://doi.org/10.1093/brain/awn202...
. In our study, there were no e4 carriers and no difference in apoE polymorphism among patients in the same family. Indeed, these polymorphisms could not explain the clinical variability in the kindreds we are describing.
The presence of CSF 14-3-3 and the occurrence of periodic synchronous discharge is unusual in GSS patients and may be confounding factors in some cases, leading to CJD misdiagnosis1414. Webb TE, Poulter M, Beck J, Uphill J, Adamson G, Campbell T et al. Phenotypic heterogeneity and genetic modification of P102L inherited prion disease in an international series. Brain. 2008;131(10):2632-46. https://doi.org/10.1093/brain/awn202
https://doi.org/10.1093/brain/awn202...
. In our sample, all the patients had an electroencephalogram, but none of them had periodic synchronous discharge. Two patients had CSF analysis, but they were negative for 14-3-3.
The pathognomonic neuropathological characteristic of GSS is the presence of multicentric amyloid plaques with positive PrPc immunoreactivity. Other pathologic features that can be found in GSS include gliosis, neuronal loss, neurofibrillary tangles and spongiform changes2727. Ellison D, Love S, Chimelli L et al. Neuropathology: a reference text of CNS pathology. 3rd ed. Lonson: Elsevier-Mosby; 2013.. The degree of spongiform change is variable, and an absence of spongiosis in brain tissue, which is uncommon in prion diseases, has been reported1111. Hainfellner JA, Brantner-Inthaler S, Cervenáková L, Brown P, Kitamoto T, Tateishi J et al. The original Gerstmann-Sträussler-Scheinker family of Austria: divergent clinicopathological phenotypes but constant PrP genotype. Brain Pathol. 1995;5(3):201-11. https://doi.org/10.1111/j.1750-3639.1995.tb00596.x
https://doi.org/10.1111/j.1750-3639.1995...
. White matter degeneration, as observed in one of the present cases, may reflect the degree of neuronal loss, rapidly progressive course and/or longer survival in some cases and is described as the panencephalopathic form of prion disease2828. Jansen C, Head MW, Rozemuller AJ, Ironside JW. Panencephalopathic Creutzfeldt-Jakob disease in the Netherlands and the UK: clinical and pathological characteristics of nine patients. Neuropathol Appl Neurobiol. 2009;35(3):272-82. https://doi.org/10.1111/j.1365-2990.2008.01004a.x
https://doi.org/10.1111/j.1365-2990.2008...
, and has been previously reported in one patient with GSS2929. Iwasaki Y, Mori K, Ito M, Nokura K, Tatsumi S, Mimuro M et al. Gerstmann-Straeussler-Scheinker disease with P102L prion protein gene mutation presenting with rapidly progressive clinical course. Clin Neuropathol. 2014;33(5):344-53. https://doi.org/10.5414/NP300733
https://doi.org/10.5414/NP300733...
.
There is evidence that, besides the mutant isoform, the protease-resistant wild-type PrP may also be found in cerebral tissue of patients with GSS1616. Wadsworth JD, Joiner S, Linehan JM, Cooper S, Powell C, Mallinson G et al. Phenotypic heterogeneity in inherited prion disease (P102L) is associated with differential propagation of protease-resistant wild-type and mutant prion protein. Brain. 2006;129(6):1557-69. https://doi.org/10.1093/brain/awl076
https://doi.org/10.1093/brain/awl076...
. The frequency and spatial distribution of both PrPs could offer another explanation for the variability in clinical and pathological findings between individuals in the same family1616. Wadsworth JD, Joiner S, Linehan JM, Cooper S, Powell C, Mallinson G et al. Phenotypic heterogeneity in inherited prion disease (P102L) is associated with differential propagation of protease-resistant wild-type and mutant prion protein. Brain. 2006;129(6):1557-69. https://doi.org/10.1093/brain/awl076
https://doi.org/10.1093/brain/awl076...
. Atypical p.Pro102Leu phenotypes are associated with high levels of protease-resistant wild-type PrP in few cases. In the present study, we could neither verify the distribution of different forms of PrP, nor of the two abnormal fragments of ~21-30 kDa or 8 kDa.
In family A, third-generation patients presented with symptoms earlier than second-generation, suggesting genetic anticipation. However, a recent report ruled out this genetic phenomenon in prion diseases3030. Minikel EV, Zerr I, Collins SJ, Ponto C, Boyd A, Klug G et al. Ascertainment bias causes false signal of anticipation in genetic prion disease. Am J Hum Genet. 2014;95(4):371-82. https://doi.org/10.1016/j.ajhg.2014.09.003
https://doi.org/10.1016/j.ajhg.2014.09.0...
.
It is noteworthy that MRI findings also differ between GSS patients. Neuroimaging studies are typically described as normal or with non-specific atrophy of cerebral hemispheres and/or cerebellum99. Collins S, McLean CA, Masters CL. Gerstmann-Sträussler-Scheinker syndrome, fatal familial insomnia, and kuru: a review of these less common human transmissible spongiform encephalopathies. J Clin Neurosci. 2001;8(5):387-97. https://doi.org/10.1054/jocn.2001.0919
https://doi.org/10.1054/jocn.2001.0919...
. There are, however, few reports of cortical and basal ganglia hyperintensities on diffusion-weighted-MRI during the disease course3131. Chi NF, Lee YC, Lu YC, Wu HM, Soong BW. Transmissible spongiform encephalopathies with P102L mutation of PRNP manifesting different phenotypes: clinical, neuroimaging, and electrophysiological studies in Chinese kindred in Taiwan. J Neurol. 2010;257(2):191-7. https://doi.org/10.1007/s00415-009-5290-4
https://doi.org/10.1007/s00415-009-5290-...
,3232. Irisawa M, Amanuma M, Kozawa E, Kimura F, Araki N. A case of Gerstmann-Sträussler-Scheinker syndrome. Magn Reson Med Sci. 2007;6(1):53-7. https://doi.org/10.2463/mrms.6.53
https://doi.org/10.2463/mrms.6.53...
, as disclosed in patient IIa-2.
In conclusion, the two Brazilian families with GSS associated with p.Pro102Leu mutation showed similar clinical heterogeneity to other cases described worldwide. However, these results failed to corroborate the hypothesis that phenotype variation is attributable to polymorphism at codon 1291414. Webb TE, Poulter M, Beck J, Uphill J, Adamson G, Campbell T et al. Phenotypic heterogeneity and genetic modification of P102L inherited prion disease in an international series. Brain. 2008;131(10):2632-46. https://doi.org/10.1093/brain/awn202
https://doi.org/10.1093/brain/awn202...
,1515. Mead S. Prion disease genetics. Eur J Hum Genet. 2006;14(3):273-81. https://doi.org/10.1038/sj.ejhg.5201544
https://doi.org/10.1038/sj.ejhg.5201544...
. Indeed, genomic and epigenetic variations within this family warrant further investigation.
ACKNOWLEDGMENT
The authors thank to Professor Pedro Piccardo for the valuable contribution and revision of the manuscript.
References
-
1Prusiner SB. Prions. Proc Natl Acad Sci USA. 1998;95(23):13363-83. https://doi.org/10.1073/pnas.95.23.13363
» https://doi.org/10.1073/pnas.95.23.13363 -
2Holman RC, Belay ED, Christensen KY, Maddox RA, Minino AM, Folkema AM et al. Human prion diseases in the United States. PLoS One;2010;5(1):e8521. https://doi.org/10.1371/journal.pone.0008521
» https://doi.org/10.1371/journal.pone.0008521 -
3Puoti G, Bizzi A, Forloni G, Safar JG, Tagliavini F, Gambetti P. Sporadic human prion diseases: molecular insights and diagnosis. Lancet Neurol. 2012;11(7):618-28. https://doi.org/10.1016/S1474-4422(12)70063-7
» https://doi.org/10.1016/S1474-4422(12)70063-7 -
4Wadsworth JD, Hill AF, Beck JA, Collinge J. Molecular and clinical classification of human prion disease. Br Med Bull. 2003;66(1):241-54. https://doi.org/10.1093/bmb/66.1.241
» https://doi.org/10.1093/bmb/66.1.241 -
5Ladogana A, Puopolo M, Croes EA, Budka H, Jarius C, Collins S et al. Mortality from Creutzfeldt-Jakob disease and related disorders in Europe, Australia, and Canada. Neurology. 2005;64(9):1586-91. https://doi.org/10.1212/01.WNL.0000160117.56690.B2
» https://doi.org/10.1212/01.WNL.0000160117.56690.B2 -
6Gerstmann J, Straussler E, Scheinker J. Über eine eigenartige hereditär-familiäre Erkrankung des Zentralnervensystems. Z Neurol. 1936;154:736-62.
-
7Hsiao K, Baker HF, Crow TJ, Poulter M, Owen F, Terwilliger JD et al. Linkage of a prion protein missense variant to Gerstmann-Sträussler syndrome. Nature. 1989;338(6213):342-5. https://doi.org/10.1038/338342a0
» https://doi.org/10.1038/338342a0 -
8Kretzschmar HA, Honold G, Seitelberger F, Feucht M, Wessely P, Mehraein P et al. Prion protein mutation in family first reported by Gerstmann, Sträussler and Scheinker. Lancet. 1991;337(8750):1160. https://doi.org/10.1016/0140-6736(91)92826-N
» https://doi.org/10.1016/0140-6736(91)92826-N -
9Collins S, McLean CA, Masters CL. Gerstmann-Sträussler-Scheinker syndrome, fatal familial insomnia, and kuru: a review of these less common human transmissible spongiform encephalopathies. J Clin Neurosci. 2001;8(5):387-97. https://doi.org/10.1054/jocn.2001.0919
» https://doi.org/10.1054/jocn.2001.0919 -
10Arata H, Takashima H, Hirano R, Tomimitsu H, Machigashira K, Izumi K et al. Early clinical signs and imaging findings in Gerstmann-Sträussler-Scheinker syndrome (Pro102Leu). Neurology. 2006;66(11):1672-8. https://doi.org/10.1212/01.wnl.0000218211.85675.18
» https://doi.org/10.1212/01.wnl.0000218211.85675.18 -
11Hainfellner JA, Brantner-Inthaler S, Cervenáková L, Brown P, Kitamoto T, Tateishi J et al. The original Gerstmann-Sträussler-Scheinker family of Austria: divergent clinicopathological phenotypes but constant PrP genotype. Brain Pathol. 1995;5(3):201-11. https://doi.org/10.1111/j.1750-3639.1995.tb00596.x
» https://doi.org/10.1111/j.1750-3639.1995.tb00596.x -
12Piccardo P, Dlouhy SR, Lievens PM, Young K, Bird TD, Nochlin D et al. Phenotypic variability of Gerstmann-Sträussler-Scheinker disease is associated with prion protein heterogeneity. J Neuropathol Exp Neurol. 1998;57(10):979-88. https://doi.org/10.1097/00005072-199810000-00010
» https://doi.org/10.1097/00005072-199810000-00010 -
13Riudavets MA, Sraka MA, Schultz M, Rojas E, Martinetto H, Begué C et al. Gerstmann-Sträussler-Scheinker syndrome with variable phenotype in a new kindred with PRNP-P102L mutation. Brain Pathol. 2014;24(2):142-7. https://doi.org/10.1111/bpa.12083
» https://doi.org/10.1111/bpa.12083 -
14Webb TE, Poulter M, Beck J, Uphill J, Adamson G, Campbell T et al. Phenotypic heterogeneity and genetic modification of P102L inherited prion disease in an international series. Brain. 2008;131(10):2632-46. https://doi.org/10.1093/brain/awn202
» https://doi.org/10.1093/brain/awn202 -
15Mead S. Prion disease genetics. Eur J Hum Genet. 2006;14(3):273-81. https://doi.org/10.1038/sj.ejhg.5201544
» https://doi.org/10.1038/sj.ejhg.5201544 -
16Wadsworth JD, Joiner S, Linehan JM, Cooper S, Powell C, Mallinson G et al. Phenotypic heterogeneity in inherited prion disease (P102L) is associated with differential propagation of protease-resistant wild-type and mutant prion protein. Brain. 2006;129(6):1557-69. https://doi.org/10.1093/brain/awl076
» https://doi.org/10.1093/brain/awl076 -
17Castro RM, Landemberger MC, Walz R, Carlotti CG Jr, Huang N, Cunha DR et al. High capacity and low cost detection of prion protein gene variant alleles by denaturing HPLC. J Neurosci Methods. 2004;139(2):263-9. https://doi.org/10.1016/j.jneumeth.2004.05.001
» https://doi.org/10.1016/j.jneumeth.2004.05.001 -
18Calero O, Hortigüela R, Bullido MJ, Calero M. Apolipoprotein E genotyping method by real time PCR, a fast and cost-effective alternative to the TaqMan and FRET assays. J Neurosci Methods. 2009;183(2):238-40. https://doi.org/10.1016/j.jneumeth.2009.06.033
» https://doi.org/10.1016/j.jneumeth.2009.06.033 -
19Doh-ura K, Tateishi J, Sasaki H, Kitamoto T, Sakaki Y. Pro→Leu change at position 102 of prion protein is the most common but not the sole mutation related to Gerstmann-Sträussler-Scheinker syndrome. Biochem Biophys Res Commun. 1989;163(2):974-9. https://doi.org/10.1016/0006-291X(89)92317-6
» https://doi.org/10.1016/0006-291X(89)92317-6 -
20Kovács GG, Puopolo M, Ladogana A, Pocchiari M, Budka H, Duijn C et al. Genetic prion disease: the EUROCJD experience. Hum Genet. 2005;118(2):166-74. https://doi.org/10.1007/s00439-005-0020-1
» https://doi.org/10.1007/s00439-005-0020-1 -
21Barbanti P, Fabbrini G, Salvatore M, Petraroli R, Cardone F, Maras B et al. Polymorphism at codon 129 or codon 219 of PRNP and clinical heterogeneity in a previously unreported family with Gerstmann-Sträussler-Scheinker disease (PrP-P102L mutation). Neurology. 1996;47(3):734-41. https://doi.org/10.1212/WNL.47.3.734
» https://doi.org/10.1212/WNL.47.3.734 -
22Majtényi C, Brown P, Cervenáková L, Goldfarb LG, Tateishi J. A three-sister sibship of Gerstmann-Sträussler-Scheinker disease with a CJD phenotype. Neurology. 2000;54(11):2133-7. https://doi.org/10.1212/WNL.54.11.2133
» https://doi.org/10.1212/WNL.54.11.2133 -
23Ishizawa K, Komori T, Shimazu T, Yamamoto T, Kitamoto T, Shimazu K et al. Hyperphosphorylated tau deposition parallels prion protein burden in a case of Gerstmann-Sträussler-Scheinker syndrome P102L mutation complicated with dementia. Acta Neuropathol. 2002;104(4):342-30. https://doi.org/10.1007/s00401-002-0547-3
» https://doi.org/10.1007/s00401-002-0547-3 -
24Giovagnoli AR, Di Fede G, Aresi A, Reati F, Rossi G, Tagliavini F. Atypical frontotemporal dementia as a new clinical phenotype of Gerstmann-Straussler-Scheinker disease with the PrP-P102L mutation: description of a previously unreported Italian family. Neurol Sci. 2008;29(6):405-10. https://doi.org/10.1007/s10072-008-1025-z
» https://doi.org/10.1007/s10072-008-1025-z -
25Calero O, Bullido MJ, Clarimón J, Frank-García A, Martínez-Martín P, Lleó A et al. Genetic cross-interaction between APOE and PRNP in sporadic Alzheimer’s and Creutzfeldt-Jakob diseases. PLoS One. 2011;6(7):e22090. https://doi.org/10.1371/journal.pone.0022090
» https://doi.org/10.1371/journal.pone.0022090 -
26Chapman J, Cervenáková L, Petersen RB, Lee HS, Estupinan J, Richardson S et al. APOE in non-Alzheimer amyloidoses: transmissible spongiform encephalopathies. Neurology. 1998;51(2):548-53. https://doi.org/10.1212/WNL.51.2.548
» https://doi.org/10.1212/WNL.51.2.548 -
27Ellison D, Love S, Chimelli L et al. Neuropathology: a reference text of CNS pathology. 3rd ed. Lonson: Elsevier-Mosby; 2013.
-
28Jansen C, Head MW, Rozemuller AJ, Ironside JW. Panencephalopathic Creutzfeldt-Jakob disease in the Netherlands and the UK: clinical and pathological characteristics of nine patients. Neuropathol Appl Neurobiol. 2009;35(3):272-82. https://doi.org/10.1111/j.1365-2990.2008.01004a.x
» https://doi.org/10.1111/j.1365-2990.2008.01004a.x -
29Iwasaki Y, Mori K, Ito M, Nokura K, Tatsumi S, Mimuro M et al. Gerstmann-Straeussler-Scheinker disease with P102L prion protein gene mutation presenting with rapidly progressive clinical course. Clin Neuropathol. 2014;33(5):344-53. https://doi.org/10.5414/NP300733
» https://doi.org/10.5414/NP300733 -
30Minikel EV, Zerr I, Collins SJ, Ponto C, Boyd A, Klug G et al. Ascertainment bias causes false signal of anticipation in genetic prion disease. Am J Hum Genet. 2014;95(4):371-82. https://doi.org/10.1016/j.ajhg.2014.09.003
» https://doi.org/10.1016/j.ajhg.2014.09.003 -
31Chi NF, Lee YC, Lu YC, Wu HM, Soong BW. Transmissible spongiform encephalopathies with P102L mutation of PRNP manifesting different phenotypes: clinical, neuroimaging, and electrophysiological studies in Chinese kindred in Taiwan. J Neurol. 2010;257(2):191-7. https://doi.org/10.1007/s00415-009-5290-4
» https://doi.org/10.1007/s00415-009-5290-4 -
32Irisawa M, Amanuma M, Kozawa E, Kimura F, Araki N. A case of Gerstmann-Sträussler-Scheinker syndrome. Magn Reson Med Sci. 2007;6(1):53-7. https://doi.org/10.2463/mrms.6.53
» https://doi.org/10.2463/mrms.6.53
Publication Dates
-
Publication in this collection
June 2017
History
-
Received
03 Feb 2017 -
Accepted
15 Feb 2017