










Heart failure is a global health challenge, affecting 1–2% of the population and up to an estimated 64 million people worldwide.1,2 In the UK, just under 1 million people have heart failure, with approximately 350 new diagnoses each year per 100,000 population.3 The lifetime risk of developing heart failure at 55 years of age is 33% for men and 28% for women.4 The heart failure population is heterogenous with 17 aetiologies defined in the Lancet Global Burden of Disease studies.5 Ischaemic heart disease predominates, accounting for 40–50% of cases, with hypertensive and valvular disease accounting for a further 15%.6,7 About one-third of heart failure patients are labelled as having idiopathic dilated cardiomyopathy (DCM). Global studies highlight a significantly higher burden of non-ischaemic heart failure outside of western Europe and the US.8–10
Patients attending a heart failure clinic undergo a clinical history and examination as per clinical practice guidelines. They will have a 12-lead ECG, echocardiography and serological tests to determine full blood count, renal function, thyroid function and selected cardiac biomarkers. Patients are classified using left ventricular ejection fraction (LVEF) into having heart failure with reduced ejection fraction (HFrEF; LVEF <40%), mid-range ejection fraction (HFmrEF; LVEF 40–49%) or preserved ejection fraction (HFpEF; LVEF ≥50%).11 For patients with a reduced or mid-range EF, further investigation aims to rule out ischaemic, valvular and hypertensive heart disease. Where these tests are negative, patients are frequently given a diagnosis of idiopathic DCM, with emphasis on prompt initiation and uptitration of evidence-based pharmacological treatment and consideration of device therapy.
In recent years, this approach has been augmented by improved community heart failure services and the introduction of newer pharmacological therapies.12–14 However, trends in survival rates have shown only a modest improvement, particularly when compared to fields such as oncology.15
Current diagnostic strategies fail to identify the presence of rare disease in the heart failure population. The EU defines rare disease as one that is present in fewer than 1 in 2,000 of the population. However, with as many as 8,000 recognised rare diseases, the cumulative burden of rare disease affects more than 30 million people in Europe alone. Those affected frequently suffer from delays in diagnosis with a significant impact on quality of life and, potentially, prognosis.16,17
Rarer causes of common presentations are seldom considered in everyday clinical practice. The diagnostic process often places too much emphasis on clinical traits such as LVEF, to the exclusion of a more nuanced approach. Traditional rhetoric argues that delineation of the idiopathic DCM population is superfluous to their treatment, as elucidation of aetiology will not lead to a deviation from standard management protocols. Increasingly, it is recognised that identification of an underlying genetic, inflammatory or infiltrative cause of heart failure can have profound implications for the patient and – where hereditary – their relatives.18–20
Genetic Cardiomyopathies
A significant proportion of people with idiopathic DCM have familial disease, with estimates typically in the region of 30–50%.21,22 A large scale meta-analysis, including 23 studies published from 1980 to 2010, resulted in a combined prevalence estimate of 23% with a large range (2–65%), highlighting significant heterogeneity across studies.21 The estimated prevalence of familial DCM increased with publication year, suggesting that greater awareness and access to screening has increased the number of recognised cases. A recent study identified familial disease in 26% of idiopathic DCM patients listed for cardiac transplantation.23
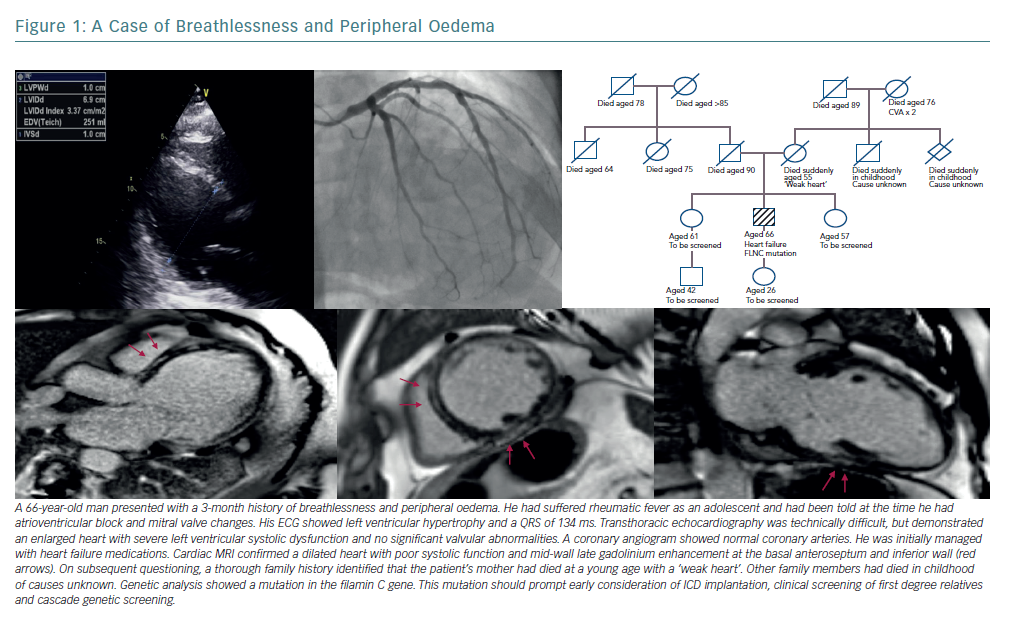
Using these data, we can conservatively estimate that 10% of an undifferentiated heart failure population may have familial disease, emphasising the importance of a detailed family history.24 This has implications for patients and their families and should prompt consideration of family screening and genetic testing.25,26 Panel-based, next-generation sequencing has shown a pathogenic or likely pathogenic variant in almost 50% of patients with familial DCM and 25% in those with sporadic disease.27,28 The yield from genetic testing will change with a better understanding of variant pathogenicity and the discovery of new disease genes.
Identification of a definite genetic cause can have profound implications for the patient and their family (Figure 1). Mutations in the gene coding for lamins A and C (LMNA) are well recognised as predictors of a malignant clinical course with progressive conduction disease and arrhythmia, even where left ventricular systolic impairment is mild.29 Patients with an LMNA mutation must be considered for prophylactic ICD implantation at an early stage of disease.30 Filamin C (FLNC) mutations are similarly predictive of a high risk of arrhythmogenic complications. A recent study has found that 85% of DCM patients with truncating FLNC variants had either ventricular arrhythmias or sudden cardiac death.31,32 Pathogenic variants in SCN5A (encoding for the cardiac sodium channel), RMB20 (encoding for the RNA binding motif protein 20) and BAG3 (encoding for an antiapoptotic protein on the sarcomere Z-disc) have also been identified as carrying an increased risk of ventricular arrhythmia or progressive heart failure.33–37
Myocarditis
Regardless of aetiology, myocardial injury often prompts an inflammatory response and cardiac autoantibodies are found in high levels in patients with both ischaemic and non-ischaemic end-stage heart failure.38–41 However, care should be taken to differentiate between cardiac injury leading to inflammation and cardiac dysfunction driven by inflammation. The term inflammatory cardiomyopathy has been used to describe myocarditis that leads to impaired function. Causes of myocarditis are numerous, with viral infection being the most common and parvovirus B19, human herpes virus 6 and enteroviruses the most frequently implicated.42,43 Bacteria, parasites, protozoa and many toxic agents and autoimmune disorders also contribute.44
Histological evidence for myocarditis in patients with DCM and heart failure is frequently reported. In a study of 1,278 patients with congestive cardiac failure and DCM, 24% had an underlying diagnosis confirmed on endomyocardial biopsy that could be attributed to an inflammatory cause.45 Inflammatory disease may be particularly common in patients with idiopathic DCM and ventricular arrhythmia. A study of 103 non-ischaemic cardiomyopathy patients presenting with monomorphic or polymorphic ventricular tachycardia, or with premature ventricular complexes, found that just under 50% had ongoing focal myocardial inflammation on fluorodeoxyglucose PET (FDG-PET). Immunosuppressive therapy was associated with an improvement in ejection fraction, particularly in those identified at an early stage of the disease process. A third of those with a positive scan were subsequently diagnosed with sarcoidosis by tissue biopsy.46
The prevalence of myocarditis remains elusive due to the heterogeneity of aetiologies and modes of presentation. In approximately 50% of cases, acute myocarditis resolves in 2–4 weeks. However, up to 25% develop persistent cardiac dysfunction and 12–25% may acutely deteriorate or progress to end-stage DCM.47 Histological studies suggest that between 10–52% of patients with acute viral myocarditis progress to a DCM and biopsy-based analysis shows that viral persistence is associated with progressive LV dysfunction.48–51 Recent data suggest that more than half of patients presenting with acute lymphocytic myocarditis and an LVEF <50% suffer persistent left ventricular systolic dysfunction.52 Immunohistological analysis of biopsy material from patients with chronic DCM shows that 40% have evidence of myocardial inflammation.53 The subclinical nature of myocardial inflammation is well recognised in children and adults.54–56
That inflammatory cardiomyopathy exists within the heart failure population seems irrefutable. Identifying these patients offers the opportunity to significantly alter the course of the disease. For example, immunosuppression in patients with virus-negative, chronic inflammatory cardiomyopathy can result in a mean improvement in LVEF from 26% to 46%.57 It also improves rates of transplant-free survival.58 Independent of virus-status, DCM patients with increased human leukocyte antigen (HLA) system expression have also demonstrated significant improvements in LVEF with immunosuppression.53
Systemic Autoimmune Disorders
Systemic autoimmune disorders frequently affect the heart and autoimmunity plays a pivotal role in myocarditis and in DCM.59 Systemic lupus erythematosus (SLE) is associated with numerous cardiac disorders and carries a high risk of developing myocarditis, with clinical detection rates in the region of 3–15%.60 African-American people are particularly at risk.61 The inflammatory process is driven by immune complex and complement deposition in the myocardium leading to cardiac dysfunction, DCM and heart failure. SLE may present with myocarditis or with heart failure, which can rapidly progress.62,63 A low threshold of suspicion must be maintained, with awareness that cardiac biomarkers and ECG results may be normal.64 An echocardiogram may demonstrate regional or global ventricular dysfunction, with strain techniques offering earlier diagnosis.65 Cardiac MRI can detect subclinical inflammation in SLE.66,67 Recommended treatment is corticosteroids and immunosuppressive agents such as cyclophosphamide.68 Myocarditis in SLE is associated with lower survival rates.61
Rheumatoid arthritis and systemic sclerosis-associated myocarditis are diagnosed less frequently. However, cardiac MRI studies of these patient populations suggest a high prevalence of myocardial involvement, with half of patients demonstrating evidence of myocardial fibrosis with late gadolinium enhancement.69,70 Cardiac involvement is found in 27–47% of patients with eosinophilic granulomatosis with polyangiitis, previously called Churg-Strauss syndrome, with myocarditis one of the most common cardiac manifestations.71–73 Myocarditis is also seen in Behcet’s disease, Takayasu arteritis, dermatomyositis and polymyositis. It tends to be associated with poorer outcomes and treatment is with steroids and immunosuppression.44,68
Giant Cell Myocarditis
Giant cell myocarditis is a disease that predominantly affects young people. It typically presents with acute heart failure and ventricular arrhythmias. Approximately 20% of patients have a pre-existing autoimmune disorder and the severe inflammation appears to be mediated predominantly by a T-cell response. Early diagnosis is key as median survival without transplant or immunosuppression is less than 3 months.44
Sarcoidosis
Sarcoidosis is an inflammatory, non-caseating granulomatous disease affecting multiple organ systems including the skin, eyes, lungs, heart and the nervous system. The incidence rate in the US has been reported at 10.9 per 100,000 in white people and 35.5 per 100,000 in black people.74 Higher incidence rates are reported in people from Scandinavia and Japan.75 Cardiac manifestations are seen in only 2–5% of patients with systemic sarcoidosis but there is a significantly higher proportion of subclinical cardiac disease.76,77 At autopsy, cardiac involvement was observed in 14% of white people, 21% of black people and 68% of Japanese people with sarcoidosis.78 The aetiology is poorly understood. Environmental exposures and infectious organisms have been implicated and the involvement of genetic factors is supported by twin studies and familial clustering.79
Cardiac sarcoidosis is frequently asymptomatic but may present with conduction defects, arrhythmias or heart failure. Clinical clues include bilateral hilar lymphadenopathy on chest X-ray and elevated levels of serum angiotensin converting enzyme and serum calcium. The ECG may highlight conduction defects or repolarisation abnormalities, but is abnormal in less than 10% of people with clinically silent disease.74 An echocardiogram with longitudinal strain parameters is useful in identifying cardiac involvement and in predicting major cardiovascular events.80,81 Imaging techniques such as FDG-PET and cardiac MRI with gadolinium contrast offer a high degree of diagnostic accuracy, but diagnostic criteria either fail to recognise the benefits of advanced imaging or rely heavily on biopsy results.82–85
Corticosteroids remain the mainstay of treatment for cardiac sarcoidosis. Data is sparse and conflicting, but indicate a beneficial effect on left ventricular function, reduced heart failure admissions and improved long-term clinical outcomes.86,87 Early diagnosis and initiation of therapy is important and titration of immunosuppression can be guided by FDG-PET.87–89
Cardiac Infiltration
Heart failure with preserved ejection fraction remains poorly understood. However, recent evidence suggests that there is a subset of cardiomyopathies caused by cardiac amyloidosis in this patient group. Moreover, better understanding of these conditions is yielding disease-modifying treatments, placing a greater emphasis on early identification of these patients.
The term amyloidosis describes a condition in which structurally unstable proteins misfold and aggregate to form fibrils that are deposited in organs and tissues. The misfolding and deposition of two proteins results in the vast majority of cardiac amyloidosis: immunoglobulin light chain (AL) and transthyretin (ATTR). Transthyretin can be further classified into wild type (ATTRwt) and that due to a mutation in the gene encoding transthyretin (ATTRm).90
Systemic Light-chain Amyloidosis
Systemic AL amyloidosis is a rare disease with a yearly incidence estimated at 3 per million in the UK and 8.9 per million, or 3,000 new cases per year, in the US.91,92 Approximately 30–50% of sufferers have cardiac involvement with 5% having isolated cardiac involvement. The average time from initial symptoms to diagnosis is 2 years, with nearly a third of patients requiring review by five physicians before receiving a diagnosis of amyloidosis.90 Cardiologists were consulted more than haematologists, nephrologists and oncologists, but rarely made the diagnosis.93
Proteinuria or hepatomegaly in combination with heart failure should raise the suspicion of AL amyloidosis.94 Macroglossia and periorbital purpura are highly specific but present only in a minority of cases.90 Patients with significant cardiac involvement are unable to tolerate the preferred treatment of high-dose chemotherapy with autologous stem cell transplantation, emphasising the importance of early diagnosis.95 Mortality at 6 months remains high at 24%, but is improving as patients are diagnosed earlier with less severe cardiac involvement.96
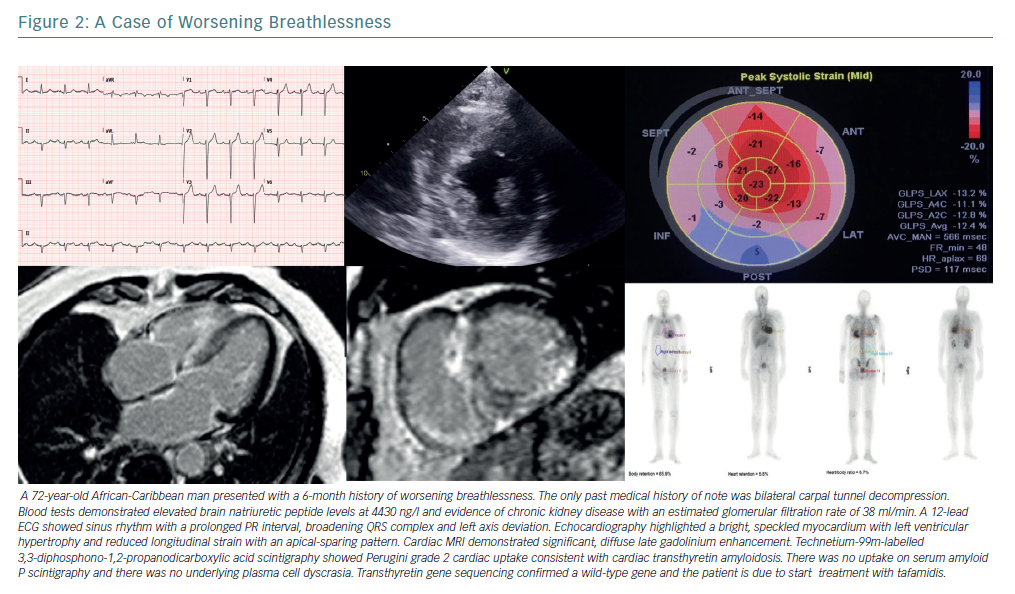
Transthyretin Amyloidosis
ATTR amyloidosis is a multisystem disorder caused by aggregation of TTR amyloid fibrils.97–100 TTR cardiomyopathy (TTR-CM) is usually a late-onset disease found in older men, with patients typically presenting aged 60 years or older.101 Symptoms include dyspnoea, fatigue and orthostatic hypotension. Signs of heart failure predominate in TTR-CM, often with a preserved ejection fraction. Clinical clues to its presence involve extra-cardiac amyloid deposition including carpal tunnel syndrome and peripheral neuropathy (Figure 2).102
TTR-CM can be hereditary or non-hereditary. The non-hereditary form of the disease is caused by aggregation of ATTRwt. The hereditary form of the disease can be caused by more than 100 mutations in the TTR gene, of which at least 22 are associated predominantly with TTR-CM.103–5 The point mutation valine 122 to isoleucine (Val122Ile) is the most common TTR-CM variant, with a prevalence of about 3–4% in the African-American population. The proportion that develop clinical disease is much smaller.106–108
In patients with ATTRwt-CM, median survival ranges from 26–67 months from diagnosis and 72 months from symptom onset. Patients with Val122Ile ATTRm-CM have a median survival time from diagnosis ranging from 36 months to 43 months. Death in most patients with TTR-CM is from sudden death or progressive heart failure.109,110
Recent data suggest that the prevalence of TTR-CM in older people has been under-appreciated. Autopsy data demonstrate that 25% of adults older than 80 years have TTR amyloid deposition in the myocardium, and 32% of patients aged 75 years or older with HFpEF demonstrate myocardial TTR amyloid deposits.111,112 Technetium-99m-labelled 3,3-diphosphono-1,2-propanodicarboxylic acid (99mTc-DPD) scintigraphy reliably detects TTR-CM. Its use highlights a prevalence of 13% of TTR-CM in patients admitted to hospital with HFpEF.113–116
Effective treatment may soon be available for TTR-CM. TTR is a 55-kD protein with a tetrameric structure that transports thyroxine and vitamin A complex. Amyloidogenesis occurs when the tetrameric structure dissociates resulting in the formation of intermediates that then deposit and polymerise as amyloid fibrils in the myocardium. The discovery of a stabilising polymorphism in the TTR gene has led to the discovery of tafamidis, a nonsteroidal anti-inflammatory drug benzoxazole derivative that adheres to the thyroxine binding sites and inhibits tetramer dissociation. Tafamadis has been shown to be superior to placebo in reducing the risk of death and cardiovascular-related hospitalisations and it reduces the decline in functional capacity and quality of life.117
Disorders of Metabolism
Anderson-Fabry Disease
Anderson-Fabry disease (AFD) is an X-linked recessive disorder caused by mutations in the GLA gene leading to reduced or absent activity of the enzyme alpha-galactosidase A. This deficiency leads to accumulation of glycosphingolipids in lysosomes in various tissues, including the heart. It results in myocardial remodelling and left ventricular hypertrophy and manifests as systolic and diastolic dysfunction, valvular abnormalities and conduction disease.
Presentation of AFD is typically in the first decade, with gastrointestinal symptoms, angiokeratomas or pain caused by a small-fibre neuropathy. However, up to 10% of patients can present with cardiac involvement, highlighting the importance of adopting a low-threshold of suspicion, particularly for patients with left ventricular hypertrophy.118,119 As many as 10% of patients with AFD develop severe heart failure (defined by New York Heart Association classification ≥3) and cardiac disease is the major cause of death, accounting for 38% of all-cause mortality.120,121 Enzyme replacement therapy is the standard treatment for AFD, aiming to compensate for reduced endogenous alpha-galactosidase activity with treatment leading to improved cardiac outcomes. It is readily apparent that enzyme replacement therapy started at a younger age leads to better outcomes illustrating the importance of identifying these patients at an early stage in the disease process.122–125
Iron-overload Cardiomyopathy
Iron-overload cardiomyopathy (IOC) should be considered in any patient with unexplained heart failure. It can be caused by a primary haemochromatosis where iron accumulation occurs through increased gastrointestinal iron absorption, or by secondary iron-overload, most commonly due to a high frequency of blood transfusions.94 The pathophysiology is complex and not simply related to myocardial iron accumulation with immunoinflammatory, molecular and genetic factors implicated.126
The early stages of IOC are characterised by left ventricular diastolic dysfunction with restrictive physiology.127,128 Progression can lead to a dilated phenotype with biventricular dilatation and systolic dysfunction or to a restrictive phenotype with preservation of LVEF, pulmonary hypertension and right ventricular dilatation.129–131 It can also present with acute heart failure.132
Serum ferritin and transferrin saturation should be included in the initial investigation of any patient with newly diagnosed heart failure.11 Echocardiography may help with ventricular assessment and an estimation of pulmonary pressures, however cardiac MRI with T2* is able to quantitatively assess myocardial iron content and predict progression to heart failure within one year.133,134 Identification of patients with an IOC is imperative as both iron removal using phlebotomy and chelation therapy have been shown to improve heart function.135–138 With the introduction of chelation therapies and MRI with T2* there has been a significant improvement in mortality rates, predominantly driven by reductions in death due to cardiac iron overload.139 Early initiation of therapy is therefore crucial.
Improving the Detection of Actionable Diagnoses
The definition of heart failure rests on a constellation of typical symptoms and an underlying structural or functional cardiac abnormality leading to deranged cardiac physiology. This definition captures a patient cohort that, historically, have been managed with a simple treatment algorithm; a ‘one-size-fits-all’ approach. A third of these patients are categorised as suffering an idiopathic DCM. We contend that rare but readily identifiable aetiologies reside within this group and, more importantly, that they are actionable diagnoses leading to targeted therapies.
The diagnosis of rare disease among people with heart failure may be challenging, particularly where resources are constrained and access to specialist cardiac investigations limited. Diagnoses are often made following a conspicuous event such as unexplained syncope or sudden death in a relative. On other occasions, diagnoses are delayed and are reached only with retrospective recognition of atypical features.
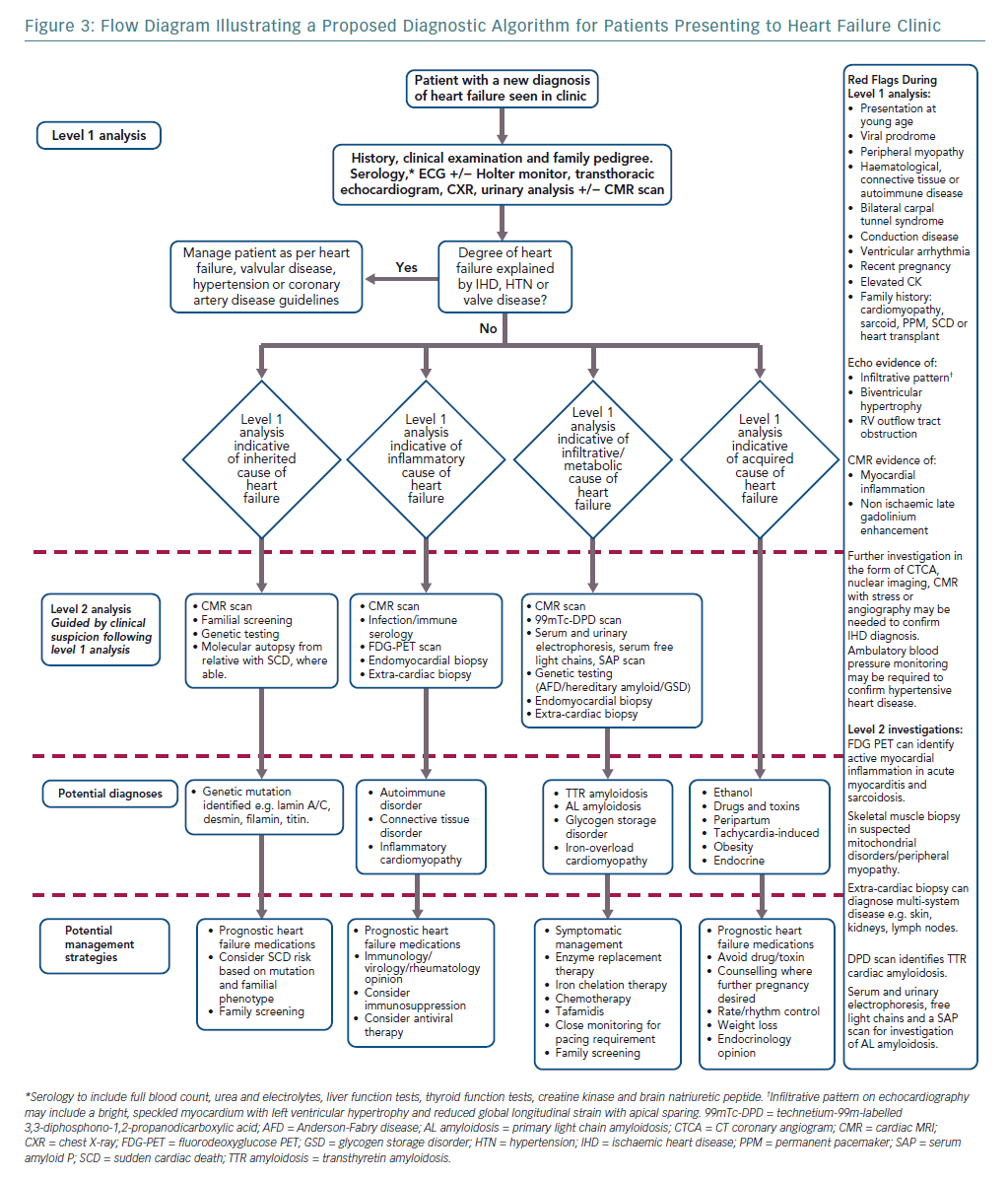
Elucidating the rarer causes of heart failure may need specialised knowledge or access to investigations such as genetic testing. We argue, however, that a diagnostic ‘red flag’ approach to clinical assessment in the heart failure clinic, combined with routinely available tests, can act as an initial filter to more readily identify uncommon disease. This approach begins with a thorough, hypothesis-driven clinical and family history and incorporates basic serology, an ECG and baseline imaging (Figure 3).18 Ischaemic, valvular and hypertensive heart disease should be managed as per guidelines.140–142 Acquired and reversible causes of heart failure such as alcohol excess and prolonged tachycardia are readily identifiable with thorough clinical assessment and routine investigations.11 Cardio-oncology is a growing sub-specialty, with increasing recognition of the cardiotoxic effects of both established chemotherapy agents and newer immunotherapies and targeted therapies. Left ventricular dysfunction and heart failure are relatively common and serious side-effects of cancer treatment.143
After preliminary assessment, patients without an obvious cause for heart failure can be directed to one of three investigative streams: genetic, inflammatory or infiltrative/metabolic.24,144,145 Awareness of dual pathologies is important as, for example, hypertension may coexist with infiltrative disease in a patient with left ventricular hypertrophy.146
Precision Medicine in Heart Failure
NHS England embraces the concept of personalised, precision medicine recognising that “complex diseases should no longer be considered a single entity” and that “different subtypes of patients within a given condition can be identified and treatment can be tailored to the underlying cause”. It urges more precise diagnoses and emphasises the usefulness of genomic medicine.147
The field of oncology offers a paradigm of the precision medicine approach. Where previously cytotoxic treatments with severe side-effects were the norm, we now see therapies designed to precisely target cancer cells. This is chiefly through two methods: pathway-targeted therapy and immunotherapy.148 These approaches seem particularly well-suited to future treatment of inflammatory and infiltrative heart disease.
The one-size-fits-all approach continues to prevail in the modern management of heart failure. This article highlights the diverse pathogeneses of the heart failure syndrome and how it could benefit from a precision medicine approach. The underlying biology of the conditions outlined is increasingly well described, resulting in the development of new therapies that offer significant clinical benefit, particularly when instituted at an early stage in the disease process. Furthermore, these conditions, traditionally thought of as rare, are more common than first thought and, in combination, account for a significant burden of disease.
We offer a diagnostic algorithm that aims to co-opt principles of precision medicine into heart failure clinics. If adopted, we hypothesise that the population prevalence of idiopathic DCM would decline as patients are given aetiology-specific diagnoses with accompanying disease-modifying treatments.