

Abstract
Ventral tegmental area dopamine neurons control reward-driven learning, and their dysregulation can lead to psychiatric disorders. Tonic and phasic activity of these dopaminergic neurons depends on cholinergic tone and activation of nicotinic acetylcholine receptors (nAChRs), particularly those containing the β2 subunit (β2*-nAChRs). Nuclear peroxisome proliferator-activated receptors type-α (PPARα) tonically regulate β2*-nAChRs and thereby control dopamine neuron firing activity. However, it is unknown how and when PPARα endogenous ligands are synthesized by dopamine cells. Using ex vivo and in vivo electrophysiological techniques combined with biochemical and behavioral analysis, we show that activation of α7-nAChRs increases in the rat VTA both the tyrosine phosphorylation of the β2 subunit of nAChRs and the levels of two PPARα endogenous ligands in a Ca2+-dependent manner. Accordingly, in vivo production of endogenous PPARα ligands, triggered by α7-nAChR activation, blocks in rats nicotine-induced increased firing activity of dopamine neurons and displays antidepressant-like properties. These data demonstrate that endogenous PPARα ligands are effectors of α7-nAChRs and that their neuromodulatory properties depend on phosphorylation of β2*-nAChRs on VTA dopamine cells. This reveals an autoinhibitory mechanism aimed at reducing dopamine cell overexcitation engaged during hypercholinergic drive. Our results unveil important physiological functions of nAChR/PPARα signaling in dopamine neurons and how behavioral output can change after modifications of this signaling pathway. Overall, the present study suggests PPARα as new therapeutic targets for disorders associated with unbalanced dopamine–acetylcholine systems.
Introduction
Ventral tegmental area (VTA) dopamine neurons form the mesocorticolimbic pathway (Albanese and Minciacchi, 1983). They play a role in processing emotionally salient information (Lapish et al., 2007; Mark et al., 2011; Wang and Tsien, 2011) by changing their firing pattern and activity (Wightman and Robinson, 2002). The VTA dopamine cell firing pattern is controlled by extrinsic afferents, among which cholinergic inputs from laterodorsal tegmental nucleus play a critical role (Lodge and Grace, 2006) through activation of nicotinic acetylcholine receptors (nAChRs) (Schilström et al., 2003; Mameli-Engvall et al., 2006). Two major forms of nAChRs are expressed on dopamine cell somata (Yang et al., 2009): high-affinity β2*-nAChRs and low-affinity α7-nAChRs (Clarke et al., 1985; Séguéla et al., 1993; Klink et al., 2001; Wooltorton et al., 2003). Whereas β2*-nAChRs on VTA dopamine cells are required for their transition from tonic to phasic activity (Mameli-Engvall et al., 2006) and for nicotine's reinforcing properties (Picciotto et al., 1998), the functional relevance of somatodendritic α7-nAChRs remains elusive.
We have previously shown that β2*-nAChRs on VTA dopamine cells are bidirectionally regulated by peroxisome proliferator-activated receptors type-α (PPARα) (Melis et al., 2010). In fact, β2*-nAChRs are required for PPARα to decrease both dopamine cell activity and VTA net output in vitro, as well as nicotine-induced increased locomotion in vivo. Accordingly, nicotine's effects on the dopamine system are suppressed by synthetic PPARα agonists (Melis et al., 2008; Mascia et al., 2011; Panlilio et al., 2012). An interaction between PPARα and β2*-nAChRs in VTA dopamine cells was then postulated, informed by evidence of PPARα as modulators of VTA dopamine neuronal activity through activation of tyrosine kinases and phosphatases (Melis et al., 2008, 2010). Thus, given that β2*-nAChRs switch dopamine cells from a resting to an excited state (Mameli-Engvall et al., 2006), PPARα ligands, by negatively modulating β2*-nAChRs, contribute to endogenous mechanisms controlling VTA dopamine cell excitability (Melis et al., 2010). Particularly, the role of endogenous PPARα ligands was revealed after increased cholinergic drive through nAChRs, suggestive of a reciprocal control between acetylcholine and PPARα (Melis et al., 2010).
Endocannabinoid-related N-acylethanolamines (NAEs), such as the anorectic oleoylethanolamide (OEA) (Rodríguez de Fonseca et al., 2001; Fu et al., 2003) and the anti-inflammatory palmitoylethanolamide (PEA) (Lo Verme et al., 2005), are endogenous PPARα ligands. These lipids are signaling molecules involved in the regulation of diverse physiologic functions (Pistis and Melis, 2010). The mechanisms accounting for PPARα ligand synthesis in dopamine neurons are unknown, whereas it has been shown that NAE key synthesizing enzymes are Ca2+-dependent (Astarita et al., 2008), and NAE formation in cortical neurons requires enhanced cholinergic tone (Stella and Piomelli, 2001). Therefore, we hypothesized that low-affinity Ca2+-permeable α7-nAChRs (Bertrand et al., 1993) could participate in the regulation of NAE levels in dopamine cells after enhanced cholinergic tone. To test this hypothesis, we investigated the role of α7-nAChRs in regulating endogenous PPARα ligand levels, and the resulting modulation of dopamine neuron activity, by using a multidisciplinary approach that involved biochemical and electrophysiological techniques, as well as behavioral analysis.
Materials and Methods
All procedures were performed in accordance with the Guidelines for the Care and Use of Mammals in Neuroscience and Behavioral Research (National Research Council, 2004) and EEC Council Directives (219/1990 and 220/1990). We made all efforts to minimize pain and suffering and to reduce the number of animals used. Animals were housed in groups of three to six in standard conditions of temperature (21 ± 1°C) and humidity (60%) under a 12 h light/dark cycle (lights on at 7:00 A.M.) with food and water available ad libitum.
Electrophysiological studies
Ex vivo.
Whole-cell patch-clamp recordings from Sprague Dawley rat VTA dopamine cells were similar to those described previously (Melis et al., 2008). Briefly, male rats (14–21 d; Harlan) were anesthetized with halothane and killed. A block of tissue containing the midbrain was rapidly dissected and sliced in the horizontal plane (300 μm) with a vibratome (VT1000S; Leica) in an ice-cold, low-Ca2+ solution containing (in mm) 126 NaCl, 1.6 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 0.625 CaCl2, 18 NaHCO3, and 11 glucose. Slices were transferred to a holding chamber with artificial CSF (ACSF; 37°C) saturated with 95% O2 and 5% CO2 containing (in mm) 126 NaCl, 1.6 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 18 NaHCO3, and 11 glucose. Slices (two per animal) were allowed to recover for at least 1 h before being placed (as hemislices) in the recording chamber and superfused with the ACSF (37°C) saturated with 95% O2 and 5% CO2. Cells were visualized with an upright microscope with infrared illumination (Axioskop FS 2 plus; Zeiss), and whole-cell current-clamp recordings (one per hemislice) were made by using an Axopatch 200B amplifier (Molecular Devices). Current-clamp experiments were made with electrodes filled with a solution containing the following (in mm): 144 KCl, 10 HEPES, 3.45 BAPTA, 1 CaCl2, 2.5 Mg2ATP, and 0.25 Mg2GTP (pH 7.2–7.4, 275–285 mOsm). Biocytin (0.2%) was added to the internal solution to mark the recorded neuron for later cytochemical characterization. However, as reported previously by Zhang et al. (2010), recording from neurons with this intracellular solution for longer than 3 min impairs immunocytochemical detection of tyrosine hydroxylase (TH). Therefore, we performed a set of experiments with a K-gluconate internal solution because all recorded neurons strongly express TH immunocytochemical signal regardless of recording duration (Margolis et al., 2010). The K-gluconate internal solution contained the following (in mm): 133.5 K-gluconate, 1.8 NaCl, 10 HEPES, 0.05 EGTA, 1.7 MgCl2, 2 Mg2ATP, and 0.4 Mg2GTP (pH 7.2–7.4, 275–285 mOsm). All experiments were begun only after series resistance had stabilized (typically 15–40 MΩ). Data were filtered at 2 kHz, digitized at 10 kHz, and collected on-line with acquisition software (pClamp 8.2; Molecular Devices). Dopamine neurons were identified according to the already published criteria (Melis et al., 2007, 2008): cell morphology and anatomical location (i.e., medial to the medial terminal nucleus of the accessory optic tract), large hyperpolarization activated current (Ih > 100 pA), slow pacemaker-like firing rate (<5 Hz), and long action potential duration (>2 ms). Each slice received only a single drug exposure. Drugs were applied in known concentrations to the superfusion medium. Nicotine, EGTA, neostigmine, and atropine were dissolved in distilled water, whereas all the other drugs were dissolved in DMSO. The final concentration of DMSO was <0.01%. Action potential frequency was analyzed off-line with MiniAnalysis software (Synaptosoft). The averaged action potential frequency within the first minute immediately before drug administration was taken as baseline, and the averaged frequency for the 1 min period centered on the peak response was taken for drug effect.
In vivo.
Male Sprague Dawley rats (63–90 d; Harlan) were anesthetized with urethane (1300 mg/kg, i.p.) and placed in a stereotaxic apparatus (Kopf) with their femoral vein cannulated for intravenous administration of pharmacological agents. Thereafter, the scalp was retracted and one burr hole was drilled above the VTA (AP, −6.0 mm from bregma; lateral, 0.3–0.6 mm from midline) for the placement of a recording electrode. VTA was localized according to the stereotaxic atlas of Paxinos and Watson (1997). Single-unit activity of neurons located in VTA (ventral, 7.0–8.0 mm from the cortical surface) was recorded extracellularly with glass micropipettes filled with 2% pontamine sky blue dissolved in 0.5 m sodium acetate (impedance, 2–5 MΩ). Single-unit activity was filtered (bandpass, 500–5000 Hz), and individual spikes were isolated by means of a window discriminator (Digitimer), displayed on a digital storage oscilloscope (TDS 3012; Tektronics). Experiments were sampled on-line and off-line with Spike2 software (Cambridge Electronic Design) by a computer connected to the CED 1401 interface (Cambridge Electronic Design). Since only one cell was recorded per rat, single units were isolated and identified according to the already published criteria (Melis et al., 2008). VTA dopamine neurons were selected when all criteria for identification were fulfilled: firing rate, <10 Hz; duration of action potential, >2.5 ms as measured from start to end; inhibitory responses to hindpaw pinching (Lecca et al., 2012).
Behavioral study: forced swim test
Male Sprague Dawley rats (60–90 d; Harlan) were used. All behavioral experiments were conducted between 10:00 A.M. and 3:00 P.M.
The forced swim test was performed in accordance with a previously described procedure (Castagne et al., 2011). As an index of a depressive-like status, the following behavioral parameter was measured: immobility, which is time (seconds) spent by the animal floating in the water performing the minimum amount of movements with the anterior paws, to maintain its head above the water surface.
Biochemical studies
Western blotting.
Male Sprague Dawley rats (63–90 d; Harlan) were administered PNU282987 (PNU; 3 mg/kg, i.p.), MK886 (1 mg/kg, i.p.), WY14643 (WY; 40 mg/kg, i.p.), or vehicle (5 ml/kg, i.p.) 30 min before decapitation. The VTA was dissected immediately and frozen in liquid nitrogen. Frozen samples of VTA were sonicated in cell lysis buffer (50 mm TRIS, pH 7.4, 250 mm NaCl, 5 mm EDTA, 50 mm NaF, 1 mm sodium orthovanadate, 1% Triton X-100, 0.02% NaN3) containing 1 mm phenylmethylsulfonyl fluoride and protease inhibitor mixture. Protein concentrations of the lysates were measured by the Bio-Rad DC Protein Assay. β2 subunit protein was immunoprecipitated from whole-cell lysates using a rabbit polyclonal antibody raised against a recombinant protein corresponding to amino acids 342–433 of the human β2 subunit (sc-11372, Santa Cruz Biotechnology). Antibodies were coupled to protein A Dynabeads (Invitrogen) using 5 μg of anti-β2 antibody by rotating the mixture for 10 min at room temperature. Beads were washed twice in PBS, and the antibody-conjugated beads were incubated with 250 μg (VTA) of protein lysate for 10 min at 4°C, followed by three washing steps in PBS supplemented with 0.1% Tween 20 (Sigma-Aldrich). Bound protein was eluted with 4× XT sample buffer (Bio-Rad) and a reducing agent (Bio-Rad). The immunoprecipitates were separated on an XT Criterion 10% gel (Bio-Rad) with 1× XT MOPS running buffer (Bio-Rad) for 1 h at 175 V (constant) and subsequently electro-transferred to a nitrocellulose membrane at 400 mA (constant) for 1 h. The membranes were incubated with 4G10 anti-phosphotyrosine (PY) antibodies (Millipore). Chemiluminescence was detected and quantified with the Versa Doc 1000 Imaging System (Bio-Rad). Samples from control and treated rats were immunoblotted and analyzed together. To control for equal loading, blots incubated with a phospho-tyrosine antibody were reprobed with the nonphosphorylated β2 subunit nAChR antibody. Moreover, the amount of phosphorylated receptor was normalized to the amount of total nAchR. Values obtained from treated rats were calculated as percentages of control values.
N-Acylethanolamine quantification.
Midbrain slices containing the VTA from Sprague Dawley rats (29–33 d; Harlan) were prepared as described previously (Melis et al., 2006). After recovery, slices underwent different pharmacological treatments and were then frozen. Frozen slices were homogenized and extracted with 50 mm chloroform/methanol/Tris-HCl, pH 7.5 (2:1:1, v/v), containing internal deuterated standards for anandamide (AEA), PEA, and OEA quantification by isotope dilution ([2H]8AEA, [2H]4 PEA, [2H]4 OEA; Cayman Chemical). The lipid-containing organic phase was dried down, weighed, and prepurified by open-bed chromatography on a silica gel. Fractions were obtained by eluting the column with 90:10 (v/v) chloroform/methanol. AEA, PEA, and OEA were quantified by liquid chromatography–atmospheric pressure chemical ionization–mass spectrometry [1100 HPLC system (Agilent Technologies) equipped with MS Detector 6110 single quadruple] and using selected ion monitoring at M + 1 values for the four compounds and their deuterated homologs, as described previously (Di Marzo et al., 2001; Piscitelli et al., 2011).
Immunohistochemistry
After recordings, slices containing biocytin fills were fixed in 4% paraformaldehyde in 0.1 m PBS, pH 7.4, for 3 h and washed three times with PBS, pH 7.4. Preblocking of tissue sections was performed with 10% normal donkey serum (NDS), 1% bovine serum albumin (BSA), and 0.2% Triton X-100 in PBS for 1 h at room temperature. Sections were then incubated for 24 h at 4°C with a mouse monoclonal anti-TH antibody (1:400; Millipore) in PBS containing 0.2% Triton X-100, 0.1% BSA, and 1% NDS. Then, after being washed in PBS/0.2% Triton X-100, sections were incubated for 2 h at room temperature with Alexa Fluor 488-labeled donkey anti-mouse IgG (1:500; Invitrogen) and strepdavidin Alexa FluorR 594 (1:1000; Invitrogen) for 2 h in the dark at room temperature. Sections were then rinsed and mounted on slides using VectaShield antifade mounting media (Vector Laboratories) and visualized using an Olympus IX 61 microscope. Images were taken with a 12-bit cooled F View II camera (Olympus). Color compositions were made using images of single antibodies as RGB channels. After being captured on the computer, images were analyzed using the Cell P AnalySIS software module.
Statistical analysis
All the numerical data are given as mean ± SEM. Data were compared and analyzed by using two-way ANOVA for repeated measures (treatment × time), or one-way ANOVA or Student's t test for repeated measures when appropriate. Post hoc multiple comparisons were made using either the Dunnett's test or the Bonferroni's test. The significance level was established at p < 0.05.
Drugs
Nicotine [(−)-nicotine hydrogen tartrate], EGTA (tetrasodium salt), biocytin, neostigmine bromide, and atropine sulfate were purchased from Sigma-Aldrich. MK886, WY14643, PNU282987, GW6471, methyllycaconitine, dihydro-β-erythroidine hydrobromide, and amitriptyline were purchased from Tocris. URB597 (URB) was purchased from Cayman Chemical. For the in vivo experiments, MK886 was dissolved in 10% Tween 80, 20% DMSO, and 70% distilled water and administered (5 ml/kg, i.p.) 1 h before testing. PNU282987 was dissolved in saline solution and administered (5 ml/kg, i.p.) 30 min before testing. Amitriptyline was dissolved in saline solution and administered (5 ml/kg, i.p.) 1 h before testing.
Results
N-Acylethanolamine formation in the midbrain requires α7-nAChR-mediated Ca2+ entry
In cortical neurons, NAE formation depends on enhanced cholinergic tone and increased cytosolic Ca2+ (Stella and Piomelli, 2001). The levels of endogenous PPARα ligands (i.e., PEA and OEA) were measured in midbrain slices in which endogenous acetylcholine actions at nAChRs were enhanced and isolated by incubating the slices with the acetylcholinesterase inhibitor neostigmine (2 μm, 5 min) and the muscarinic receptor antagonist atropine (5 μm, 5 min). We found that the levels of OEA and PEA were selectively increased [neo+atro: PEA, 593.1 ± 34.2 pmol/g tissue weight, p = 0.0001, t test; OEA, 739.2 ± 97.4 pmol/g tissue weight, p = 0.0001, t test; AEA, 78.9 ± 8.7 pmol/g tissue weight, p = 0.2, t test; n = 5 for all groups; Table 1, Fig. 1A]. Since key synthesizing enzymes for OEA and PEA are Ca2+ dependent (Astarita et al., 2008) and low-affinity α7-nAChRs are Ca2+ permeable (Bertrand et al., 1993), they could participate in NAE formation in the midbrain. To test this hypothesis, NAE levels were measured after activation of α7-nAChRs in midbrain slices. The α7-nAChR agonist PNU282987 (PNU, 50 nm, 5 min) increased levels of OEA (265.2 ± 34.8 pmol/g tissue weight; p < 0.001, t test; n = 5; Table 1, Fig. 1B) and PEA (295.5 ± 37.1 pmol/g tissue weight; p < 0.001, t test; n = 5; Table 1, Fig. 1B) but not of AEA (69.5 ± 8.1 pmol/g tissue weight; p = 0.3, t test; n = 5; Table 1, Fig. 1B). The effect of PNU282987 was Ca2+-dependent, as the levels of PEA and OEA were not modified in the presence of the Ca2+ chelator EGTA (5 mm; PNU+EGTA: PEA, 166.3 ± 15.8 pmol/g tissue weight, p < 0.01; OEA, 158.4 ± 17.1 pmol/g tissue weight, p < 0.05, t test; AEA: 44.6 ± 5.9 pmol/g tissue weight, p > 0.05; F(1,16) = 9.27; n = 5 for all groups; two-way ANOVA followed by the Bonferroni's test; Table 1, Fig. 1B).
Data are means ± SEM and were compared by two-way ANOVA, followed by the Bonferroni's test

Enhanced cholinergic tone increases levels of endogenous PPARα agonists. A, The levels of endogenous PPARα ligands (PEA and OEA), but not of the endocannabinoid anandamide (AEA), are increased by enhancing acetylcholine activation of nAChRs (neo+atro: p = 0.0001 for both PEA and OEA, t test; n = 5 for all groups). B, α7-nAChR activation by PNU enhances PEA (p = 0.007, t test; n = 5) and OEA (p = 0.04, t test; n = 5) levels in Ca2+-dependent manner (PNU+EGTA: p = 0.01 and p = 0.02 for PEA and OEA, respectively; t test). C, Representative immunoblots and summarizing bar graph showing that PNU282987 (3 mg/kg, i.p.) causes an increase in β2 subunit phosphorylation in rat VTA homogenates that is blocked by the PPARα antagonist MK886 (1 mg/kg, i.p.). D, Representative immunoblots and summarizing bar graph showing that the PPARα agonist WY (40 mg/kg, i.p.) increases β2 subunit phosphorylation in rat VTA homogenates. For each experiment in C and D, tissue lysates containing the same amount of total proteins were subjected to immunoprecipitation (IP) with anti-β2 AChR antibody and were immunoblotted with anti-PY antibody. The IPs were run on the same immunoblots and analyzed together. The blots were stripped and reprobed with anti-β2 AChR antibody to normalize for protein loading. Band size, 50 kD. Numbers in bars indicate n values. Data are expressed as mean ± SEM. *p < 0.05; **p < 0.001; ***p < 0.0001; #p < 0.05; PNU+MK versus PNU. veh, Vehicle.
We previously suggested that endogenous PPARα ligands act as modulators of β2*-nAChR function in VTA dopamine cells through a nongenomic mechanism involving activation of tyrosine kinases and phosphatases (Melis et al., 2008, 2010). To study the molecular basis of such an interaction, we analyzed phosphorylation of β2 subunits with Western blot analysis after in vivo exposure to the α7-nAChR agonist PNU (3 mg/kg, i.p.): an increased immunoreactivity of β2 subunits phosphorylated on a tyrosine residue in VTA total homogenates was observed in PNU-treated animals (139.1 ± 9.8%; n = 9; one-way ANOVA followed by Bonferroni's test; F(2,24)=8.9; p = 0.001; Fig. 1C). This effect was blocked by pretreatment with the PPARα antagonist MK886 (1 mg/kg, i.p., 30 min before PNU treatment; 112.4 ± 6.1%; n = 9; one-way ANOVA followed by Bonferroni's test; F(2,24)=8.9; p = 0.001; Fig. 1C) and mimicked by the synthetic PPARα agonist WY (40 mg/kg, i.p., 15 min before tissue collection; 143.6 ± 3.7%; n = 8; t = 2.78; p = 0.01; Fig. 1D), further substantiating the constitutive interaction between PPARα and β2*-nAChRs in the VTA.
Enhanced cholinergic drive to α7-nAChRs modulates dopamine neuronal activity through activation of PPARα
We next assessed whether pharmacological manipulations resulting in increased levels of endogenous ligands of PPARα in midbrain slices would affect VTA dopamine neuronal activity. We performed whole-cell current-clamp recordings from rat dopamine cells in the lateral portion of the VTA under the same experimental conditions (see above). To isolate cholinergic responses, experiments were performed in the presence of CNQX (10 μm), d-AP-5 (100 μm), and picrotoxin (100 μm) to block AMPA, NMDA, and GABAA receptor-mediated responses, respectively. When endogenous acetylcholine actions at nAChRs were enhanced, by applying both the acetylcholinesterase inhibitor neostigmine (2 μm, 3 min) and the muscarinic antagonist atropine (5 μm, 3 min), we observed an increased dopamine cell firing rate (neo+atro, n = 5; Table 2) consistent with an endogenous cholinergic tone in the midbrain slice preparation (Melis et al., 2010; Mao et al., 2011). We found that subsequent blockade of α7-nAChRs with methyllycaconitine (MLA), at a concentration that per se had no effect on the dopamine cell firing rate (3 nm, 5 min) (Melis et al., 2010), enhanced dopamine neuronal activity (+MLA: 284.6 ± 36.9%, t = 5.2, n = 5, p = 0.02, paired t test; Fig. 2A, Table 2). These effects are related to elevated ACh levels in the slice, since a lower concentration of neostigmine (200 nm; +atropine, 5 μm, 3 min) was unable to enhance the dopamine neuronal firing rate (93.3 ± 7%, t = 1.2, n = 8, p = 0.34, paired t test; Table 2). Moreover, increased firing rate after enhanced ACh in the slice and blockade of α7-nAChRs occurred through activation of α4β2-nAChRs, since the effect was fully blocked in the presence of the α4β2-nAChR antagonist dihydro-β-erythroidine (1 μm, 5 min; +DHβE: 82.1 ± 18.6%, t = 5.1, n = 5, p = 0.035, paired t test; Fig. 2B, Table 2). These results suggest that ACh acting at α7-nAChRs exert a negative control on the firing rate of dopamine neurons through β2*-nAChRs. Additionally, when the actions of endogenous ligands at PPARα were blocked by the PPARα antagonist MK886, at a concentration that had no effect on firing frequency per se (Melis et al., 2008, 2010), VTA dopamine cells underwent an aberrant excitation (+MK886: 548.1 ± 21.2%, F(19,80) = 1.78, n = 5, p = 0.03, one way ANOVA; MK886+MLA vs MLA: F(1,20) = 4.86, n = 5, p = 0.05, two-way ANOVA; Fig. 2C, Table 2). Notably, the effects of hypercholinergic tone occurring through activation of PPARα do not require activation of muscarinic AChRs (+neo+MLA vs neo+MLA+MK886: t = 3.4, n = 5, p = 0.07; Fig. 2D, Table 2).
Summary of the effects of different drugs acting at nAChRs and PPARα on VTA dopamine neuron's firing rate

Enhanced cholinergic tone to α7-nAChRs modulates VTA dopamine cell activity via PPARα. A, Ex vivo, the dopamine cell firing rate enhances during increased cholinergic tone at β2*-nAChRs (neo+atro+MLA). Left, Current-clamp traces of a dopamine neuron before (basal) and during enhanced cholinergic tone (neo+atro) at β2*-nAChRs (neo+atro+MLA). Right, the effects of enhanced cholinergic tone at β2*-nAChRs on dopamine neurons are represented in a bar graph form. B, Bar graph depicting that the effects of increased cholinergic input onto the dopamine cell firing rate depend upon activation of β2*-nAChRs because DHβE fully blocks such effects. C, Left, Current-clamp traces of a dopamine neuron before (basal), during enhanced cholinergic tone at β2*-nAChRs (neo+atro+MLA), and once PPARα are blocked (+MK886). Right, Time course of the effect of neo+atro+MLA alone (shaded area) or together with MK886 (black bar) on dopamine neuron activity. D, The synthesis of endogenous PPARα ligands after raises in ACh and their consequent effects on the dopamine firing rate are not mediated by muscarinic receptor activation (neo+MLA). E, Removal of extracellular Ca2+ (EGTA+neo+atro) enhances the dopamine cell firing rate, which is further increased by blockade of α7-nAChRs with MLA. Left, Current-clamp traces of a dopamine neuron before (basal) and during enhanced cholinergic tone in the presence of Ca2+ chelator EGTA (+EGTA) and antagonist of α7-nAChRs (+MLA). In the central panel, the peak effects are represented in a bar graph form. Right, Time course of EGTA (shaded area) alone or in combination with neo+atro (thick black line) and MLA and MK886 is represented. The black bars represent time of MLA and MK886 application. Numbers in bars indicate n values. Data are expressed as mean ± SEM. *p < 0.05; **p < 0.001.
To test the hypothesis that Ca2+ mediates the effects of α7-nAChRs on firing rate, EGTA (5 mm, 3 min) was perfused. EGTA had no effect on the dopamine cell firing rate per se (Fig. 2E) but augmented dopamine neuron activity once cholinergic tone was elevated (neo+atro+EGTA: 221.1 ± 16.6%, t = 8.09, n = 6, p = 0.007, paired t test; EGTA: 85 ± 25.5%, t = 0.78, n = 6, p = 0.23, paired t test; Fig. 2E, Table 3). These effects on firing activity further increased in the presence of MLA (3 min; 293.2 ± 33.3%, t = 6.13, n = 6, p = 0.02, paired t test; Fig. 2E, Table 3).
Summary of the effects of different drugs acting at nAChRs and PPARα on VTA dopamine neuron's firing rate
Given that the α7-nAChR agonist PNU282987 increases both the levels of endogenous PPARα ligands and phosphorylation of β2 subunits in the VTA, we next tested whether bath application of PNU282987 (50 nm, 5 min) was able to affect the dopamine neuronal firing rate through PPARα. PNU282987 reduced dopamine cell spontaneous activity (50.4 ± 5.6%; t = 4.88; n = 5; p = 0.02, paired t test; Fig. 3A,B, Table 3), and this effect was blocked by two structurally different PPARα antagonists MK886 (113.6 ± 4.3%; F(1,133) = 12.84; n = 5; p = 0.009, two-way ANOVA followed by Bonferroni's test; Fig. 3A, Table 3) and GW6471 (100 nm; 118.4 ± 4.3%; F(1,133)=11.97; n = 5; p = 0.01, two-way ANOVA followed by Bonferroni's test; Fig. 3A).

PPARα activation downstream α7-nAChRs alters excitability of VTA dopamine neurons. A, Left, α7-nAChR agonist PNU reduces dopamine cell activity through synthesis of PPARα ligands. Right, Time course of the effects of PNU282987 (shaded area) in the absence (thick red line) or presence of the PPARα antagonists MK886 (thick dark gray line) and GW6471 (thick light gray line). B, Left, Effects of α7-nAChR agonist PNU282987 are not enhanced by pharmacological inhibition of FAAH enzyme through application of URB. Right, Time course of the effects of PNU282987 (shaded area) in the absence (thick red line) or presence (thick dark gray line) of FAAH inhibitor URB597 applied through the recording pipette ([URB]i). Numbers in bars indicate n values. Data are expressed as mean ± SEM. Thick and dashed lines represent means and SEM, respectively. *p < 0.05; **p < 0.001. C, Determination of the dopamine phenotype using immunohistochemistry. Ca, Cb, Examples of rat VTA neurons in which relatively long (>20 min) whole-cell recordings were made with a KCl (a) and K-gluconate (b) internal solution. Post hoc immunocytochemical detection revealed that example neuron a (white arrow indicating presumably a false negative) was TH(−) and example neuron b was TH(+). Neurons that were whole-cell patch clamped were backfilled by including biocytin in the recording pipette (red). TH immunohistochemistry routinely labeled the TH-positive neurons (green) when the whole-cell recordings were made with the K-gluconate internal solution. Scale bar, 20 μm.
NAEs are mainly degraded by the fatty acid amide hydrolase (FAAH) (Ueda et al., 2000) localized in the postsynaptic compartment (Gulyas et al., 2004). We next tested whether pharmacological blockade of FAAH would potentiate α7-nAChR-mediated response: dopamine cells were filled with URB597 (100 nm) to rapidly inactivate FAAH, and PNU282987 was then applied (50 nm, 5 min). Figure 3B shows that, in the presence of URB597, the effect PNU282987 on dopamine cell firing activity was not enhanced (URB+PNU, ∼40% of basal firing rate; F(1,133)=0.95; n = 5; p = 0.5, two-way ANOVA followed by Bonferroni's test vs PNU alone; Fig. 3B, Table 3). Since we used a concentration of URB597 that is able to prevent nicotine-induced excitation of VTA dopamine neurons (data not shown), in agreement with our previous observation (Melis et al., 2008), we can rule out the possibility that URB597 was not effective in blocking FAAH. Alternatively, URB597 ineffectiveness on enhancing PNU-induced effects on dopamine neuronal activity might result from the actions of the diverse FAAH substrates. In particular, AEA exerts stimulating effects on the firing rate of dopamine cells through activation of TRPV1 receptors (Melis et al., 2008), which might counterbalance, thereby masking, the effects produced by increased levels of OEA and PEA via PPARα.
To determine whether the neurons were indeed dopaminergic, we filled neurons with biocytin while recording the spontaneous activity and subsequently processed slices for TH. In agreement with Zhang et al. (2010), we found that the probability of observing a positive immunohistochemical TH signal varied with the length (>3 min) of whole-cell patch-clamp recordings with a KCl internal solution, thereby leading to a false negative (Fig. 3Ca). Therefore, the experiments in Figure 3B were performed with a K-gluconate internal solution while targeting the same neuronal population. Under these conditions, we did not observe any loss of TH signal (Fig. 3Cb), in agreement with Margolis et al. (2010). Thus, changes in excitability downstream of α7-nAChRs mediated by activation of PPARα are indeed observed in VTA dopamine-containing neurons.
α7-nAChR activation blocks the stimulating properties of nicotine in vivo through PPARα activation
As shown previously, PPARα activation blocks nicotine-induced excitation of dopamine cells in vivo (Melis et al., 2008, 2010; Mascia et al., 2011; Panlilio et al., 2012). Given our present findings that α7-nAChR activation increases the levels of endogenous PPARα ligands, one would predict that α7-nAChR activation would produce a similar effect on nicotine-induced excitation of dopamine neurons in vivo. Indeed, we found that PNU (0.5 mg/kg, i.v., 4 min before nicotine) fully prevented nicotine-induced excitation of dopamine neurons (F(1,9) = 32.25; p = 0.003, two-way ANOVA followed by Bonferroni's test; n = 5; Fig. 4A,B). When rats were pretreated with the PPARα antagonist MK886 (0.1 mg/kg, i.p., 1 h before nicotine), which itself had no effect on the dopamine cell firing rate (3.8 ± 0.4 Hz vs vehicle, 3.6 ± 0.5 Hz; n = 5 for both groups), PNU282987 was no longer able to prevent nicotine from increasing dopamine neuron frequency (F(1,9 = 15; p = 0.004, two-way ANOVA followed by Bonferroni's test; n = 5; Fig. 4A,B).

Effects of α7-nAChR activation on excitation of VTA dopamine neurons by nicotine. A, Representative firing rate histograms showing effects of intravenous nicotine (NIC; injected at arrowhead) on discharge activity of individual VTA dopamine neurons recorded from anesthetized rats. Top, Typical response to 0.2 mg/kg nicotine in control conditions after intravenous injection of vehicle (veh; 4 min before nicotine). Middle, Lack of effect of α7-nAChR agonist PNU (0.5 mg/kg, i.v.; 4 min before nicotine) on spontaneous firing rate of dopamine neurons. Notably, PNU282987 prevents stimulating effects of nicotine. Bottom, The effect of nicotine is restored in a MK886-pretreated animal (0.1 mg/kg, i.p., 1 h before beginning of recording session). B, Graph illustrating the time course of nicotine effects on firing rate of VTA dopamine neurons. Nicotine-induced excitation of dopamine neurons is abolished by PNU282987 but restored by MK886. Data are expressed as mean ± SEM. n = 5 for all groups. Shaded regions beyond dashed lines indicate time after nicotine administration.
Activation of α7-nAChRs has antidepressant-like properties through PPARα activation
Blockade or desensitization of β2*-nAChRs may exert antidepressant activity in rodent models of depression (Caldarone et al., 2004; Mineur et al., 2011), thus supporting the hypothesis that cholinergic transmission may contribute to the pathophysiology of depression (Janowsky et al., 1972; Mineur and Picciotto, 2010; Philip et al., 2010, 2012). On the other hand, activation of α7-nAChRs displays antidepressant-like properties, thus stirring controversy in terms of whether either activation or blockade of nAChRs are therapeutically useful in the treatment of depressive disorders (Mineur and Picciotto, 2010; Philip et al., 2010). Remarkably, PEA has also been shown to possess antidepressant-like effects (Yu et al., 2011). Given our present findings linking α7-nAChRs to endogenous PPARα ligands and β2*-nAChRs, we tested whether α7-nAChR activation has antidepressant-like actions through PPARα in the rat forced swim test, which is predictive of antidepressant-like activity (Porsolt et al., 1977). PNU-treated (3 mg/kg, i.p.) rats showed significant reductions in immobility (F(4,39) = 46.61; p < 0.0001, one-way ANOVA; n = 8; Fig. 5) that were similar to those seen with the currently used antidepressant amitriptyline (15 mg/kg, i.p.; n = 8). Notably, PNU282987-induced reduction of immobility was PPARα mediated since it was fully blocked by the PPARα antagonist MK886 (1 mg/kg, i.p.; F(4,39) = 46.61; p < 0.0001, one-way ANOVA; n = 8; Fig. 5).

Effects of α7-nAChR activation on rat forced swim test require PPARα activation. The effect of α7-nAChR agonist PNU282987 (3 mg/kg, i.p.) in the forced swim test are PPARα 32 mediated (***p < 0.001, PNU vs corresponding vehicle, veh+MK886, and PNU+MK886 groups; ***p < 0.001, amitriptyline group vs all groups). Data are expressed as mean ± SEM. Numbers in bars indicate n values.
Discussion
Our findings reveal a novel form of self-regulation of VTA dopamine cells triggered by low-affinity α7-nAChR activation and Ca2+-dependent synthesis of endogenous PPARα ligands targeted to the same dopamine neurons. PPARα ligands, as effectors of α7-nAChRs, act as intrinsic modulators of cholinergic transmission and alter dopamine cell excitability, contributing to acetylcholine effects on mood control. This novel form of self-regulation mediated by PPARα is selectively activated during increased cholinergic transmission at nAChRs and can be mimicked by α7-nAChR activation. Ergo, the attenuated response to acetylcholine during excessive cholinergic drive provides a fine modulation of the mesocorticolimbic dopamine pathway at a single-cell level, where phosphorylation of β2*-nAChRs is the effector mechanism (Fig. 6).

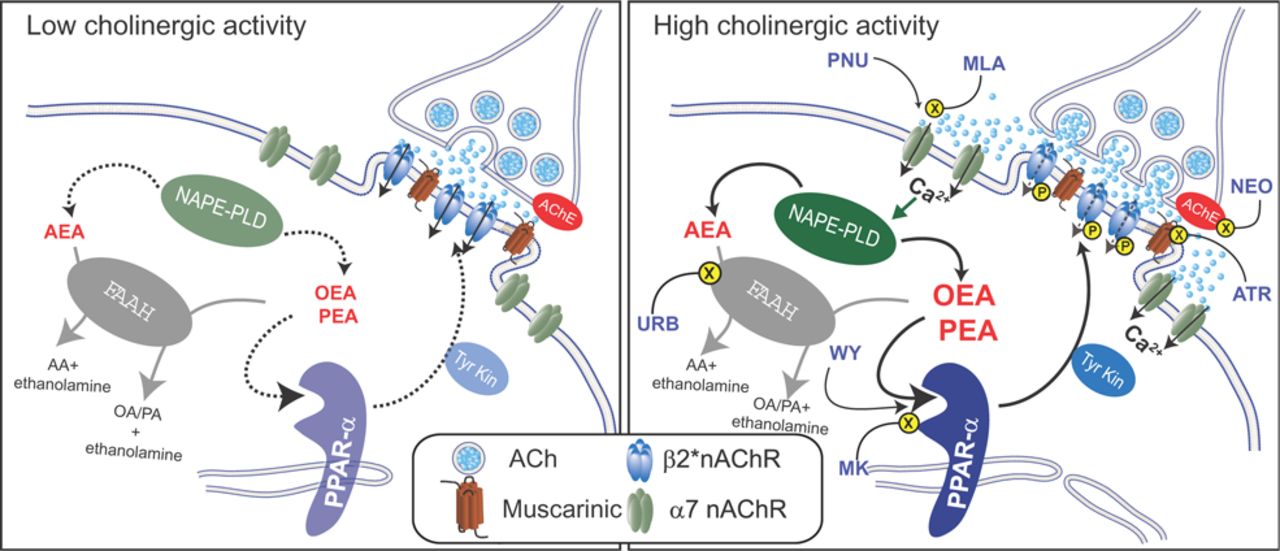
Endogenous PPARα ligands act as brake during high cholinergic tone on dopamine cells. Schematic diagram illustrating the proposed mechanism of PPARα-induced regulation of cholinergic transmission. Left, During low cholinergic activity, acetylcholine (ACh) preferentially binds to high-affinity β2*nAChRs. Production of endogenous PPARα ligands, the N-acylethanolamines AEA, OEA, and PEA, by the Ca2+-sensitive NAPE-PLD remains low and does not trigger PPARα-mediated modulation of β2*-nAChRs. Right, When acetylcholine transmission is potentiated, i.e., by blocking acetylcholinesterase (AChE) with neostigmine (NEO), Ca2+-permeable low-affinity α7-nAChRs are activated. α7-nAChRs are located extrasynaptically on the somatodendritic region of dopamine neurons (Jones and Wonnacott, 2004). α7-nAChRs are also activated by the selective agonist PNU and are blocked by MLA. α7-nAChR-mediated increase in intracellular Ca2+ stimulates N-acyl phosphatidylethanolamine phospholipase D (NAPE-PLD), leading to the production of OEA and PEA (and AEA). These molecules, in turn, activate PPARα that exerts negative modulation of β2*-nAChRs operated by a tyrosine kinase-mediated phosphorylation (P). Phosphorylation of β2*nAChRs might reduce responses to acetylcholine or promote rapid internalization of these receptors. The effects of endogenous PPARα ligands are mimicked by the synthetic agonist WY and blocked by the PPARα antagonist MK886 (MK). Increases in levels of N-acylethanolmines can also be triggered by blockade of FAAH by URB597. FAAH is the major inactivating enzyme for OEA, PEA and AEA and converts these molecules in ethanolamine and oleic acid (OA), palmitic acid (PA), and arachidonic acid (AA), respectively. However, in this case, AEA might counteract the effects of OEA and PEA by activating TRPV1 (Melis et al., 2008).
Our observations substantially support a constitutive interaction between PPARα and β2*-nAChRs in VTA dopamine neurons (Melis et al., 2010), consistent with PPARα expression within TH-positive neurons (Plaza-Zabala et al., 2010) in the midbrain (Kainu et al., 1994; Cullingford et al., 1998; Galan-Rodriguez et al., 2009). However, the abundance of constitutive levels of OEA and PEA with respect to AEA is not remarkable, since it results from different incorporation of NAE precursors into phospholipids. In fact, palmitic and oleic acids are mainly esterified in the sn-1 position, whereas arachidonic acid is esterified in position sn-2 of phospholipids. As a result, since NAEs derive from fatty acids in the sn-1 position, OEA and PEA are more abundant than AEA, and the midbrain is no exception to this rule (Melis et al., 2013). What is remarkable is a cell's ability to regulate NAE levels under different conditions, such as excessive input drive or metabolic state. Our results show that downstream of α7-nAChR activation and subsequent Ca2+ entry, dopamine cells can selectively increase OEA and PEA levels and thereby prevent their own aberrant excitation, a mechanism that was unmasked pharmacologically through blockade of either α7-nAChRs or PPARα.
Somato-dendritic β2*-nAChRs play a crucial role in controlling the state of activity of VTA dopamine cells (Mameli-Engvall et al., 2006). Dopamine neurons in vivo may exhibit three different types of phasic firing. However, in α7-nAChR knock-out animals, only the most excited state remains. We therefore propose that α7-nAChRs, via Ca2+ influx, OEA and PEA synthesis, and activation of PPARα, act as possible switches in the tonic/phasic transition regulated by these receptors.
Our study also provides alternative cellular mechanisms elucidating previously reported phenomena, such as the evidence that nicotine patches reduce symptoms of depression in nonsmoking depressed patients (Salín-Pascual et al., 1996). Strangely, both activation of α7-nAChRs and inhibition of β2*-nAChRs contribute to nicotine antidepressant properties (Coe et al., 2005; Mihalak et al., 2006; Mineur and Picciotto, 2010). In fact, desensitization of β2*-nAChRs after chronic nicotine administration is critical for its antidepressant-like properties (Gentry and Lukas, 2002; Shytle et al., 2002; Paradiso and Steinbach, 2003; Mineur and Picciotto, 2009). Accordingly, partial agonists at β2*-nAChRs show robust antidepressant-like properties (Mineur et al., 2011; Philip et al., 2012). In this context, the antidepressant-like properties of PNU282987 (Andreasen and Redrobe, 2009) might be explained by its ability to increase the levels of PEA and OEA, activate PPARα, and phosphorylate β2*-nAChRs. Accordingly, PEA possesses antidepressant-like activity in mice (Yu et al., 2011). Consequently, our findings broaden the already wide variety of PPARα functions, mainly known to regulate nutrient metabolism and energy homeostasis. Nevertheless, it is worth mentioning that PEA (Normast) is already effective in models of stroke and other CNS traumata (Koch et al., 2011; Schomacher et al., 2012). Thus, as α7-nAChR effectors, PEA and OEA may have a homeostatic role and be the link through which α7-nAChR stimulation proves beneficial in pathological states such as (neuro)inflammation, depressive states, and neurotoxicity. As such, the present study also provides a foundation to explain anorectic, anti-inflammatory, and antidepressant properties of nicotine, as well as of α7-nAChR agonists (Avena and Rada, 2012; Bencherif et al., 2011; Lakhan and Kirchgessner, 2011). Activation of PPARα signaling may prove valuable in disorders or conditions associated with dysfunction of dopamine–ACh interplay like stress, drug addiction, schizophrenia, and depressive states. Particularly, given that relapse to nicotine is often related to stress and acute withdrawal, where functional upregulated β2*-nAChRs play a key role (Schwartz and Kellar, 1983; Marks et al., 1985), targeting PPARα might represent a promising therapeutic approach in nicotine relapse prevention and, ultimately, to help overcome nicotine addiction. Accordingly, synthetic PPARα agonists not only decrease nicotine taking in experienced animals but also prevent the relapse-inducing effects of reexposure to nicotine, as well as nicotine-associated cues after a period of abstinence (Panlilio et al., 2012). Additionally, given the antidepressant-like activity of FAAH inhibitors (Gobbi et al., 2005; Bortolato et al., 2007; Adamczyk et al., 2008; Umathe et al., 2011), PPARα signaling may provide a useful alternative target for novel antidepressant drugs. Finally, activation of either PPARα or α7-nAChRs has proven to be effective in preclinical models of schizophrenia (Pichat et al., 2007; Acker et al., 2008; Kucinski et al., 2012; Rolland et al., 2012; Thomsen et al., 2010; Zanaletti et al., 2012), thus supporting the involvement of PPARα and α7-nAChRs in the pathophysiology of this disease (Freedman et al., 2000; Newhouse et al., 2004; Costa et al., 2013).
Whereas targeting PPARα is promising as a novel therapeutic approach to treat psychiatric disorders such as nicotine addiction and depressive states, α7-nAChR activation might be exploited to overcome current limitations of PEA in clinical use, mainly its poor bioavailability.
Footnotes
This work was supported by the “Regione Autonoma della Sardegna, Assessorato alla Programmazione”: grants for basic research (Legge Regionale 7/2007 to W.F.), through the program “Bursaries for Young Researchers” (Legge Regionale 7/2007 to S.L. and A.L.), and through the program “Visiting Professor” (to B.S.). This research was also supported by the Italian Ministry of University (Grant PRIN 2009: 200928EEX4 to M.P.) and “Fondazione Banco di Sardegna.” We thank William T. Dunn III for proofreading this manuscript.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Miriam Melis, Department of Biomedical Sciences, University of Cagliari, Cittadella Universitaria, 09042 Monserrato (CA), Italy. myriam{at}unica.it





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}