

Abstract
Recent studies have demonstrated protective roles for autophagy in various neurodegenerative disorders, including the polyglutamine diseases; however, the role of autophagy in retinal degeneration has remained unclear. Accumulation of activated rhodopsin in some Drosophila mutants leads to retinal degeneration, and although it is known that activated rhodopsin is degraded in endosomal pathways in normal photoreceptor cells, the contribution of autophagy to rhodopsin regulation has remained elusive. This study reveals that activated rhodopsin is degraded by autophagy in collaboration with endosomal pathways to prevent retinal degeneration. Light-dependent retinal degeneration in the Drosophila visual system is caused by the knockdown or mutation of autophagy-essential components, such as autophagy-related protein 7 and 8 (atg-7/atg-8), or genes essential for PE (phosphatidylethanolamine) biogenesis and autophagosome formation, including Phosphatidylserine decarboxylase (Psd) and CDP-ethanolamine:diacylglycerol ethanolaminephosphotransferase (Ept). The knockdown of atg-7/8 or Psd/Ept produced an increase in the amount of rhodopsin localized to Rab7-positive late endosomes. This rhodopsin accumulation, followed by retinal degeneration, was suppressed by overexpression of Rab7, which accelerated the endosomal degradation pathway. These results indicate a degree of cross talk between the autophagic and endosomal/lysosomal pathways. Importantly, a reduction in rhodopsin levels rescued Psd knockdown-induced retinal degeneration. Additionally, the Psd knockdown-induced retinal degeneration phenotype was enhanced by Ppt1 inactivation, which causes infantile neuronal ceroid lipofuscinosis, implying that autophagy plays a significant role in its pathogenesis. Collectively, the current data reveal that autophagy suppresses light-dependent retinal degeneration in collaboration with the endosomal degradation pathway and that rhodopsin is a key substrate for autophagic degradation in this context.
Introduction
Inherited retinal degenerative disorders are pathologically and genetically heterogeneous (Hartong et al., 2006; Michaelides et al., 2006) and are characterized by the premature and progressive death of rod and cone photoreceptors. Several genes are involved in retinal diseases in humans and in animal models (Michaelides et al., 2006; Knust, 2007; Karan et al., 2008), and, of these, the rhodopsin gene is most frequently mutated. Approximately 25% of the autosomal dominant retinitis pigmentosa cases in humans result from various mutations of the rhodopsin gene; thus, understanding rhodopsin regulation will further our knowledge of retinal disease pathogenesis.
In normal photoreceptors, rhodopsin is transported through early and late endosomes and multivesicular bodies (MVBs) before delivery to lysosomes for degradation (Alloway et al., 2000; Xu et al., 2004; Acharya et al., 2008; Chinchore et al., 2009). Defects in genes involved in this lysosomal trafficking pathway, such as Sunglasses (Sun) (Xu et al., 2004), or the vacuolar protein sorting (vps) mutants, including carnation/vps33 and light/vps41 (Chinchore et al., 2009), result in retinal degeneration in Drosophila, attributable to the aberrant accumulation of rhodopsin. Thus, efficient rhodopsin degradation appears to be crucial for the viability of photoreceptor cells. However, it is still unclear whether rhodopsin degradation is controlled solely by the lysosomal pathway or whether other mechanisms, such as autophagy, are also involved.
The autophagy pathway mediates the degradation of cytoplasmic proteins, protein aggregates, and organelles on nutrient starvation (Todde et al., 2009). These cell materials are enclosed by a double-membrane structure, the autophagosome, which fuses with endosomes, MVBs, or lysosomes to enable their degradation. Autophagosome formation requires the Atg8-PE conjugate, a product of ubiquitin-like conjugation reactions, in which the ubiquitin-like protein, Atg8, is added to phosphatidylethanolamine (PE), a plasma membrane lipid component, by Atg7 and Atg3 (which function like E1 and E2 enzymes, respectively). PE is synthesized by phosphatidylserine decarboxylase (Psd) and CDP-ethanolamine:diacylglycerol ethanolaminephosphotransferase (Ept) in mitochondria and endoplasmic reticulum (ER), respectively. Although the yeast Psd mutation reduces Atg8p recruitment to the pre-autophagosomal structure (Nebauer et al., 2007), there is no evidence that Psd is required for autophagy in higher organisms.
Previous studies have shown that autophagy plays a neuroprotective role in the CNS. Knock-out mice lacking Atg5 or Atg7, and the Drosophila atg7 mutant display neuronal degeneration in aged animal brains (Hara et al., 2006; Komatsu et al., 2006; Juhász et al., 2007), probably because of the accumulation of toxic oligomers or large aggregates of soluble, misfolded proteins. Additionally, autophagy inactivation enhances neurodegeneration caused by polyglutamine-containing proteins such as Huntingtin. Although the ubiquitin–proteasome pathway normally degrades these cytoplasmic, misfolded proteins, there is evidence that autophagy acts as a compensatory degradation pathway when the ubiquitin–proteasome pathway is impaired (Pandey et al., 2007). However, it is unclear whether autophagy also compensates for the endosomal/lysosomal pathway during membrane protein degradation.
This study provides the first evidence that degradation of the major rhodopsin, Rhodopsin 1 (Rh1), occurs via cross talk between the autophagy and endosomal degradation pathways, and that autophagy acts as a compensatory pathway when the endosomal/lysosomal pathway is impaired.
Materials and Methods
Drosophila strains.
The Drosophila strains used in this study included the following: GMR-GAL4, UAS-gfp-RNAi, UAS-Psd-RNA#1, UAS-Psd-RNAi#2, UAS-fab1-RNAi, UAS-Ept-RNAi#1, UAS-Ept-RNAi#2, UAS-cpt-RNAi, UAS-ptdss1-RNAi, UAS-atg8a-RNAi, UAS-atg7-RNAi, UAS-Ppt1-RNAi, y1w67c23 P{EPgy2}CG5991EY03559 (UAS-Psd), w-, CS, y1 P{SUPor-P}Atg8aKG07569/FM7c, w y hs-gfp-atg8b, ninaE-atg1-myc;ninaE-atg1-myc, UAS-atg6-myc, tublin-yfp-rab7, w Df(1)446-20, and w Ppt1S77F. The UAS-Psd-RNAi#2, UAS-Ept-RNAi#2, UAS-atg8a-RNAi, and UAS-Ppt1-RNAi strains were purchased from the Vienna Drosophila RNAi Center (Vienna, Austria). UAS-Psd and y1 P{SUPor-P}Atg8aKG07569/FM7c flies were purchased from the Bloomington Drosophila Stock Center (Indiana University, Bloomington, IN), and w y hs-gfp-atg8b and UAS-atg6-myc were kindly provided by Dr. Thomas P. Neufeld (University of Minnesota, Minneapolis, MN). The ninaE-atg1-myc and tublin-yfp-rab7 flies were a kind gift from Dr. Bruce A. Edgar (Basic Sciences Division, Fred Hutchinson Cancer Research, Seattle, WA) and Dr. Suzanne Eaton (Max Planck Institute of Molecular Cell Biology and Genetics, Dresden, Germany), respectively. The w Df(1)446-20 and w Ppt1S77F flies were a kind gift from Dr. Robert L. Glaser (New York State Department of Health, Albany, NY). The targeted sites of each UAS-RNAi line were as follows (number indicates the distance from the start codon): UAS-Psd-RNAi#1, 2-503; UAS-Psd-RNAi#2, 751-1145; UAS-fab1-RNAi, 532-1031; UAS-Ept-RNAi#1, 13-512; UAS-Ept-RNAi#2, 963-1222; UAS-cpt-RNAi, 555-1045; UAS-ptdss1-RNAi, 366-865; UAS-atg8a-RNAi, 55-354; UAS-atg7-RNAi, 201-700; and UAS-Ppt1-RNAi, 90-399. All of the 19-mers in the targeting sequences were compared with the other targeting sequences using dsCheck (Naito et al., 2005).
Antibodies.
Rhodopsin 1 and Chaoptin mouse monoclonal antibodies and a DE-cadherin rat monoclonal antibody were obtained from the Developmental Studies Hybridoma Bank (University of Iowa, Iowa City, IA). Rat anti-α-tubulin polyclonal antibodies were purchased from Oxford Biotechnology. Rabbit anti-Myc polyclonal antibodies (A-14) and mouse anti-green fluorescent protein (GFP) monoclonal antibodies (B-2) were purchased from Santa Cruz Biotechnology. Rabbit anti-Rab7 polyclonal antibodies and rabbit anti-Arl8 polyclonal antibodies were a kind gift from Dr. Akira Nakamura (Riken CDB, Kobe, Japan) and Dr. Sean Munro (Medical Research Council–Laboratory of Molecular Biology, Cambridge, UK), respectively. Rabbit anti-GM130 antibodies were produced as previously described (Yano et al., 2005).
Reverse transcription–PCR.
Before the extraction of total RNA using the three methods described below, flies were reared in the dark at 27°C. Method A was as follows: The extraction of total RNA and preparation of cDNA from retinal slices was performed using a Power SYBR Green Cells-to-CTTM kit (Ambion). Briefly, 10 retinal slices were collected from adult flies by carefully peeling the compound eyes from the adult heads using tweezers and suspending them in 50 μl of lysis solution with DNaseI. To produce cDNA, 10 μl of the resulting lysate was added to 40 μl of RT MasterMix and incubated at 37°C for 60 min, and then at 95°C for 5 min. An aliquot of 0.5–5 μl of the cDNA solution and specific oligonucleotide primer pairs was then added to the reaction mixture with TaKaRa Ex Taq (TaKaRa) and PCR was performed according to the following program for 33–38 cycles: 94°C for 30 s, 58°C for 30 s, and 72°C for 1 min. The volume of cDNA solution was normalized using control (gapdh) bands between samples of knockdown flies and control (gfp-RNAi) flies. Method B was as follows: Retinal slices were collected from 100 adult flies by excising the compound eyes followed by the removal of adherent brains. Total RNA was then extracted using an RNeasy Mini Kit (QIAGEN), and the purity of the samples and RNA content was measured using a UV photometer. An aliquot of 140 ng of total RNA was reverse transcribed using SuperScript III Reverse Transcriptase (Invitrogen) and oligo-dT primers in a final volume of 25 μl. This reaction was used as a template for subsequent PCR. Amplifications were performed using the program described above for 31–37 cycles. Method C was as follows: Total RNA from 50 adult fly heads was prepared as described in method B, cDNA was synthesized from 500 ng of total RNA, and PCR was performed for 31 cycles also as described in method B. The sequences of the primers were as follows: Psd forward primer-1 (F-1), 5′-ATGGATGCCGATCTGAAGAC-3′; Psd reverse primer-1 (R-1), 5′-GGTGCTGCCCATATTGAACT-3′; Psd forward primer-2 (F-2), 5′-GGCAATCTCAGACAGGTGGT-3′; Psd reverse primer-2 (R-2), 5′-GCAGACTGGGAGGCAAGTAG-3′; Ept forward primer, 5′-ATGGATGCCGATCTGAAGAC-3′; Ept reverse primer, 5′-GGTGCTGCCCATATTGAACT-3′; atg8a forward primer, 5′-ATTGAGGAAAACCGCAGGA-3′; atg8a reverse primer, 5′-GCATTCGCACGGATCAATTA-3′; ptdss1 forward primer, 5′-ATCCACCACAACGGGTAAAA-3′; ptdss1 reverse primer, 5′-AACCGCGAGGTAGACAAATG-3′; fab1 forward primer, 5′-CAGCAATCAGCAAGGTGAAA-3′; fab1 reverse primer, 5′-CCAAGTACCAAGACGCCATT-3′; cpt forward primer, 5′-ACGAGCTTCCTCAGCGTCTA-3′; cpt reverse primer, 5′-GCTTCGAAACCCCAATCATA-3′; atg7 forward primer, 5′-TGACCCGGTTCTTTGTTCTC-3′; atg7 reverse primer, 5′-CTTTGGCTAAGATCGCAAGC-3′; gapdh forward primer, 5′-ATCGTCGAGGGTCTGATGAC-3′; and gapdh reverse primer, 5′-ACCGAACTCGTTGTCGTACC-3′. The Psd F-1/Psd R-1 and Psd F-2/Psd R-2 primer pairs were used for reverse transcription–PCR analysis of Psd-RNAi#1 flies and Psd-RNAi#2 flies, respectively.
Immunohistochemistry.
Immunolabeling of dissected Drosophila retinas was performed as follows, and eight to nine flies were quantified in each genotype: adult heads were dissected and sliced in half in fixation buffer [2% paraformaldehyde (PFA), 0.1% Triton X-100 in PBS, or 4% PFA, 0.06% Triton X-100 in PBS]. These half-heads were then placed on ice for 30 min in fixation buffer, and retinal slices were prepared by cutting compound eyes horizontally using dissecting scissors (Vannas Dissecting; World Precision Instruments). Dissected retinas were then fixed in fixation buffer on ice for 30 min and incubated with primary antibodies in blocking buffer (0.1% BSA, 0.1% Triton X-100 in PBS) at room temperature for 2 h. This was followed by incubation with fluorescent-labeled secondary antibodies (Alx-488/Alx-546-conjugated anti-mouse/rat/rabbit IgG; Invitrogen; or anti-mouse IgG Cy5-conjugate; Millipore Bioscience Research Reagents) and Alx-633-conjugated phalloidin (Molecular Probes) in blocking buffer at room temperature for 2 h. The fluorescently labeled slices were transferred onto slides, mounted in 20 μl of 70% glycerol in PBS using an 18 × 18 mm2 coverslip and a spacer between the coverslip and slide, and analyzed using a confocal microscope (FluoView FV500; Olympus) equipped with an UPlanApo 100×/1.35 oil-immersion objective. Image processing and analysis were performed with a FluoView FV500 confocal laser-scanning microscope (Olympus) and Adobe Photoshop CS.
Transmission electron microscopy.
Drosophila heads were cut longitudinally, fixed in a mixture of 2% PFA and 2% glutaraldehyde in 0.1 m cacodylate buffer, pH 7.4, at room temperature for several hours, washed with 8% sucrose in 0.1 m cacodylate buffer, and then postfixed with 1% osmium tetroxide in 0.1 m cacodylate buffer for 2 h at 4°C. After several washes in distilled water, the specimens were stained with 0.5% aqueous uranyl acetate for 1 h, dehydrated with ethanol, and embedded in Epon 812. Ultrathin sections were cut with an ultramicrotome (Ultracut UCT; Leica), collected on copper slot grids, stained with uranyl acetate/lead citrate, and observed using a transmission electron microscope (JEM-1230; JEOL).
Immuno-electron microscopy.
Drosophila heads were cut longitudinally, fixed in PLP fixation buffer (4% PFA, 10 mm NaIO4, 75 mm lysine, 37.5 mm phosphate buffer, pH 7.4) at room temperature for 1 h, and incubated at 4°C with 10% sucrose–PBS for 30 min, 20% sucrose–PBS for 1 h, and 30% sucrose–PBS overnight. Drosophila heads were then embedded in OCT compound, and 10 μm sections were cut using a cryostat (HM500-OM; Carl Zeiss), blocked with blocking buffer (0.1% saponin, 5% normal goat serum, PBS, pH 7.4) at room temperature for 30 min, immunolabeled with mouse anti-GFP monoclonal antibodies in blocking buffer at 4°C overnight, washed with PBS, and then incubated with secondary antibodies (biotin-conjugated goat anti-mouse antibodies) in blocking buffer at 4°C overnight. The avidin–biotin peroxidase method (Vectastain Elite ABC kit; Vector Laboratories) was used to detect GFP-Atg8b immunoreactivity. The tissue was then washed with PBS at 4°C and incubated with 0.05% diaminobenzidine (DAB) in PBS at room temperature for 30 min, and the peroxidase reaction was developed in 0.05% DAB and 0.03% H2O2 for 5 min. At this point, the immunolabeled sections were also analyzed using differential interference contrast microscopy (see Fig. 5B). For electron microscopy, the developed slices were washed with PBS, postfixed with 2% OsO4, 0.1 m phosphate buffer, pH 7.4, at room temperature for 1 h, washed with H2O, dehydrated in a graded ethanol series, and embedded in Epon 812. Ultrathin sections were cut and examined under an electron microscope.
MitoTracker labeling.
Ommatidia were isolated from adult Drosophila heads essentially as described previously (Hardie, 1991). For MitoTracker labeling, isolated ommatidia were incubated with 100 nm MitoTracker Red CMXRos (Invitrogen) in Schneider's medium at room temperature for 30 min, washed with PBS, incubated again with Schneider's medium at room temperature for 30 min, and fixed with 4% PFA in PBS at 37°C for 20 min. The ommatidia were then washed with PBS, mounted in PBS containing TOPRO3 (Molecular Probes), and analyzed using a confocal microscope as described above.
Immunoblotting.
Adult fly heads were homogenized in lysis buffer [20 mm Tris-HCl, pH 7.4, 0.15 m NaCl, 1% Triton X-100, 0.5% DOC (sodium deoxycholate), and 0.1% SDS containing a protease inhibitor mixture; Roche Diagnostics], sonicated, and centrifuged at 15,000 rpm at 4°C for 20 min. The supernatant was collected as the adult head lysate. These lysates were immunoblotted with mouse anti-Rh1 or rat anti-α-tubulin primary antibodies followed by HRP-conjugated anti-mouse/rat antibodies (Pierce). Blots were developed using SuperSignal West Pico Chemiluminescent Substrate (Pierce).
Urea–SDS-PAGE.
Retinal cell extract was collected from 100 adult flies by excising the compound eyes followed by the removal of adherent brains and homogenization with lysis buffer. Then, 20 μg of retinal cell lysate was subjected to SDS-PAGE containing 6 m urea.
Retinoid-free medium.
Retinoid-free medium (Sang's Medium) was prepared according to a protocol developed by Dr. William S. Stark (Saint Louis University, St. Louis, MO; http://starklab.slu.edu/Erg/tutorial.htm). To generate retinoid-deprived flies, the flies were reared from egg to adult on retinoid-free medium.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling assay.
Adult fly heads were obtained and cut from the occipital foramen to the dorsal edge of the head, and fixed in 4% PFA, 0.1% Triton X-100 in PBS. The heads were then immersed in 12% sucrose, 0.1% Triton X-100 in PBS at 4°C for 8 h, and embedded in OCT compound. Frozen sections (20 μm thick) were prepared using a cryostat as described above and the terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) assay was performed using an Apo-BrdU In Situ DNA Fragmentation Assay kit (BioVision) according to the manufacturer's instructions.
Light exposure experiments.
Flies were reared from the egg to third larval stage at 25°C under normal light exposure conditions and then reared until adulthood at 25°C in the dark. Hatched flies were then exposed to room light (4000 lux at 26–26.5°C or 8000 lux at 27–27.5°C) for 1–15 d as noted in each experiment. Flies reared in the dark at 26–26.5°C or at 27–27.5°C were prepared synchronously as controls (dark-reared flies) for the analysis of light-exposed flies.
Statistics.
All statistical analyses were performed using the Microsoft Excel 2004 analysis toolpack. Significant differences among sample means were determined using Welch's t test.
Results
Knockdown of Psd in the Drosophila eye results in light-dependent retinal degeneration
To search for novel genes involved in retinal degeneration, we screened a library of RNA interference (RNAi)-inducible Drosophila strains by examining the formation of photoreceptor cells. The RNAi-inducible inverted repeat (IR) strains in this library harbor inverted repeats of Drosophila genes downstream of yeast upstream activating sequences (UASs) that are activated by the yeast Gal4 transcription factor (Nishihara et al., 2004). Crossing these IR strains with the eye-specific Gal4 driver strain (GMR-Gal4) leads to the eye-specific knockdown of the corresponding gene. In our present screen, sectioned eyes of the knockdown flies were stained with anti-Rh1, anti-Chp, or with phalloidin, which detects the presence of actin fibers in the rhabdomere. From this analysis, we found that knockdown of the Drosophila Psd gene (CG5991) resulted in the intracellular accumulation of Rh1 and the progressive destruction of photoreceptor cells under normal light conditions (Fig. 1A).

A Psd knockdown in Drosophila causes light-dependent retinal degeneration after intracellular Rh1 accumulation. A, Immunolabeling of the dissected retinas of control (gfp-RNAi) and Psd-RNAi#1 flies reared under normal light exposure (NL) (30∼300 lux) for 3 or 17 d. The dissected retinas were immunolabeled with antibodies as indicated in each panel. In the flies reared for 3 d, abnormal Rh1 accumulation was found in the photoreceptors of the Psd-RNAi#1 variants (arrows). In the flies reared for 17 d, most of the rhabdomeres (labeled with phalloidin) were lost in the retinas of the Psd-RNAi#1 flies. Scale bar, 10 μm. B, Knockdown of Psd causes light-dependent ommatidial degeneration. Bi, Dissected retinas from dark-reared or light-exposed flies were triply stained (Rh1, Cad, and phalloidin). A large number of Rh1-positive puncta (arrows) were found in the photoreceptors of Psd-RNAi#1 flies after light exposure. Disrupted ommatidia (showing partial or complete loss of a phalloidin-positive rhabdomere; arrowheads) were found after light exposure for 5 d. Scale bar, 10 μm. Bii, Statistical analysis of Rh1-positive puncta after light exposure. The number of Rh1-positive puncta per ommatidium was counted for 150 ommatidia in two independent experiments. The values shown are the mean ± SD. ***p < 0.001, t test. C, A Psd knockdown causes light-dependent retinal cell death. Ci, gfp-RNAi and Psd-RNAi#1 flies were reared under dark or light conditions as indicated in each panel, and a TUNEL assay was performed. In the retinas of light-exposed Psd-RNAi#1 flies, TUNEL-positive nuclei were found near the apical surface (arrows) and near to the retinal floor (arrowheads). Scale bars, 20 μm. Cii, Statistical analysis of the TUNEL-positive nuclei shown in Ci. The percentage of TUNEL-positive nuclei located from the apical surface to the retinal floor (indicated by the asterisks in the merged images) is shown. Each value was calculated from 16 frozen sections taken from eight flies. The values shown are the mean ± SD. ***p < 0.001, t test. BrdU, 5-Bromodeoxyuridine; PI, propidium iodide.
Retinal degeneration processes can be classified according to their light dependency. Defects in photoreceptor development or in rhodopsin folding/transport cause light-independent degeneration, whereas excessive rhodopsin internalization, the intracellular accumulation of rhodopsin, or the dysregulation of Ca2+ levels in photoreceptor cells cause light-dependent degradation (Alloway et al., 2000; Kiselev et al., 2000; Raghu et al., 2000; Webel et al., 2000; Orem and Dolph, 2002a; Orem et al., 2006). In this study, the knockdown of the Psd gene resulted in the loss of photoreceptor cells under light-exposed conditions, but showed only very marginal defects in dark-reared conditions (Fig. 1B). This suggested that the Psd protein is required for the normal processing of rhodopsin after its activation.
This light-dependent degeneration of the Drosophila retina allowed us to ascertain the primary defects in the Psd knockdown cells. To this end, sectioned eyes were stained with an anti-Rh1 antibody, or with phalloidin, to analyze the time-dependent degradation processes that occur after light exposure. Larvae and pupae were grown in the dark and the hatched flies were exposed to light for a specific period. The Psd knockdown flies, reared in the dark for 5 d, showed normal rhabdomeres and no intracellular accumulation of Rh1 (Fig. 1B). The same normal phenotype was also observed in control photoreceptor cells in which GFP dsRNA had been induced (Fig. 1B). In contrast, Psd knockdown flies exposed to light for 2.5 d, showed an accumulation of Rh1 in comparatively large dotlike structures within the photoreceptor cells, but showed only marginal defects in the rhabdomeres (Fig. 1B). However, light exposure for 5 d caused severe defects in the rhabdomeres of these flies (Fig. 1B), and these defects were associated with apoptotic cell death as revealed by positive TUNEL staining (Fig. 1C). Hereafter, this rhabdomere defect was used as a histological marker of retinal degeneration in Drosophila. Collectively, these data clearly indicated that the intracellular accumulation of Rh1 was the primary defect in Psd knockdown flies.
To identify the compartment in which Rh1 accumulates after the knockdown of Psd, sectioned eyes were costained with several markers for these bodies. In the knockdown photoreceptor cells, accumulated Rh1 colocalized with Rab7, a late endosomal marker (Fig. 2A,C), but not with Avl8, a lysosomal marker (Fig. 2B), indicating that Rh1 accumulates in late endosomes in Psd knockdown retinas in a light-dependent manner.

A Psd knockdown in Drosophila causes Rh1 accumulation in Rab7-positive endosomes in a light-dependent manner. A, B, Flies were reared under light exposure (8000 lux) for 8 d, and dissected retinas were stained as indicated in each panel. Rh1-positive large vesicles were found in the retinas of Psd-RNAi#2 flies (arrows) after light exposure. These vesicles colocalized with Rab7 (A, arrows) but not with Arl8 (B, arrows). Scale bars, 10 μm. C, Statistical analysis of both Rh1- and Rab7-positive vesicles in the retinas of the Psd knockdown flies after light exposure for 8–10 d. Left, Number of both Rh1- and Rab7-positive vesicles per area was calculated from 10 to 14 images (4 to 7 × 103 μm2/image) obtained from two to three independent experiments. The number of vesicles observed was significantly greater in Psd-RNAi flies compared with the control (gfp-RNAi). Values are the mean ± SD. **p < 0.01, t test. Right, Cross sections of the Rh1-negative or Rh1-positive areas in Rab7-positive vesicles after light exposure for 8–10 d were measured. Both Rh1- and Rab7-positive vesicles observed in Psd-RNAi flies were significantly larger than those in the control (gfp-RNAi flies). Approximately 100 vesicles from two or three independent experiments were analyzed. Values are the mean ± SEM. ***p < 0.001, t test.
The possibility that the observed retinal defects were attributable to an off-target effect of dsRNA was excluded by two experiments. In the first of these, a knockdown using a second set of dsRNAs corresponding to a different region of Psd was also found to cause the intracellular accumulation of Rh1 and late-onset retinal degradation (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). There were neither overlapping, nor homologous sequences between the first and second sets of dsRNAs (see Materials and Methods). In the second experiment, coexpression of wild-type Psd with the targeting dsRNAs resulted in the partial rescue of retinal degeneration (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). In addition, the levels of mRNAs for the targeted genes in the Psd knockdown eyes decreased compared with the wild-type levels (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). The knockdown efficiencies of the dsRNAs used in this study were also examined and found to be sufficiently high in all cases.
The Psd knockdown did not cause severe mitochondrial dysfunction in retinal cells
The mouse Psd mutant exhibits deformed mitochondria (Steenbergen et al., 2005). Given that mitochondrial dysfunction can cause neural degeneration in mammals (Schapira, 2008), it is possible that the knockdown of Psd in Drosophila eyes may also induce retinal degeneration through mitochondrial dysfunction. To examine this possibility, we studied the morphology and function of the mitochondria in Psd knockdown cells. Morphological studies using electron microscopy revealed that the shapes and numbers of mitochondria were indistinguishable between the control and knockdown cells (Fig. 3A). In addition, mitochondrial function was also examined by comparing the uptake of the dye, MitiTrackerRed CMXRos, in control and knockdown cells. The uptake of this dye, whose accumulation depends on mitochondrial membrane potential (Brown et al., 2006), was indistinguishable between control and Psd knockdown photoreceptor cells that were exposed to light for 2.5 d (Fig. 3B) and showed light-induced accumulation of Rh1 (Fig. 1B). These data suggested that mitochondrial dysfunction is not related to Rh1 accumulation and retinal degeneration in Psd knockdown cells. However, light exposure for 5 d resulted in severe retinal degeneration (Fig. 1B) and a slight reduction in the uptake of the dye (Fig. 3C). This late-onset defect of mitochondria may be a secondary effect of retinal degeneration or other defects caused by Psd knockdown.

The Psd knockdown did not cause major mitochondrial dysfunction in retinal cells. A, Transmission electron micrographs of gfp-RNAi and Psd-RNAi#1 retinas after dark rearing for 3 d. The morphology of the mitochondria (arrows) from both retina types did not show obvious differences. Scale bars, 2 μm. Mitochondria are highlighted in the insets. Scale bars, 0.5 μm. B, C, MitoTracker Red CMXRos labeling of cultured ommatidia collected from gfp-RNAi and Psd-RNAi#1 flies. Nuclei were labeled with TOPRO3. Bi, Ommatidia collected from flies reared in the dark or exposed to light for 2.5 d. Bii, Statistical analysis. Signal intensity of MitoTracker Red CMXRos-positive vesicles were measured from 10 to 15 ommatidia. Ten to 15 MitoTracker Red CMXRos-positive vesicles were measured meticulously from the end to the central region of each ommatidium (n = 150). The values shown are the mean ± SEM. Ci, Ommatidia collected from flies reared in the dark or exposed to light for 5 d. Cii, Statistical analysis. Signal intensities of MitoTracker Red CMXRos-positive vesicles were measured as shown in Bii. MitoTracker signals in Psd-RNAi#1 flies did not decrease after dark rearing for 5 d, but showed a slight decrease after light exposure for 5 d compared with gfp-RNAi flies. The values shown are the mean ± SEM. ***p < 0.001, t test. Scale bars: Bi, Ci, 5 μm.
Ept knockdown also causes defective Rh1 transport and light-dependent retinal degradation
As shown above, Psd is required for the viability of photoreceptor cells in the Drosophila retina. Since Psd generates PE through the decarboxylation of phosphatidylserine (PS), we questioned whether a reduction in PE levels could be responsible for retinal degeneration. PE is synthesized in two major pathways, the PS decarboxylation pathway and the CDP-ethanolamine pathway (Fig. 4A). Ept catalyzes the final reaction step in the CDP-ethanolamine pathway in the ER. In this study, the knockdown of Ept caused Rh1 accumulation in Rab7-positive late endosomes and light-dependent retinal degeneration (Fig. 4B,C), as was also observed in the Psd knockdown eyes. The same phenotype was also caused by the knockdown of Ept using a dsRNA corresponding to a different region of the Ept gene (supplemental Fig. S4, available at www.jneurosci.org as supplemental material), excluding the possibility that off-target RNAi effects were responsible.

The suppression of PE synthesis causes light-dependent retinal degeneration. A, Schematic representation of the Drosophila PE synthetic pathway according to the KEGG database (Kanehisa et al., 2008). Bi, Bii, An Ept knockdown also causes light-dependent retinal degeneration. Flies were reared, and dissected retinas were triply stained as indicated in each panel. Severe disruption of the ommatidia (Bi, arrows) was evident after light exposure for 10 d. Rh1-positive large vesicles were detected and observed to colocalize with Rab7 (Bii, arrows) after light exposure for 8 d. Scale bars, 10 μm. C, Statistical analysis of both Rh1- and Rab7-positive vesicles in the retinas of Ept knockdown flies after light exposure for 8–10 d. Each value was calculated as shown in Figure 2C. Left, Number of both Rh1- and Rab7-positive vesicles per area. Values are the mean ± SD. **p < 0.01, t test. Right, Cross section of an area of Rab7-positive vesicles. Values are the mean ± SEM. ***p < 0.001, t test. D, The double knockdown of Psd and Ept enhances light-dependent retinal degeneration. Flies were reared and dissected retinas were triply stained as indicated at the top of the panels. Disrupted ommatidia (arrows) were evident in Psd-RNAi#1/Ept-RNAi#1 retinas. Scale bars, 10 μm. Right panel, Statistical analysis of retinal degeneration. Dissected retinal slices from eight flies were labeled as shown in the left panels, and the percentage of disrupted ommatidia per slice was calculated. More than nine slices obtained from two independent experiments were analyzed. The values shown are the mean ± SD. ***p < 0.001, t test.
In addition, we found that a genetic interaction exists between Psd and Ept. A weak induction of dsRNAs for Psd or Ept produced almost normal photoreceptors (Fig. 4D); however, under the same conditions, the induction of dsRNAs for both Psd and Ept caused severe degeneration of photoreceptors after light exposure for 4 d. Furthermore, neither Rh1 accumulation, nor retinal degeneration was caused by the knockdown of diacylglycerol cholinephosphotransferase (cpt, CG7149) or CDP-diacylglycerol-serine O-phosphatidyltransferase (ptdss1, CG4825) (supplemental Fig. S5, available at www.jneurosci.org as supplemental material), which are essential for the production of PC (phosphatidylcholine) and PS, respectively (Fig. 4A). Together, these results demonstrated that retinal degeneration caused by Psd or Ept knockdown was attributable to a reduction in PE.
Autophagosomes containing Rh1 are deformed by Psd knockdown
Since PE is an essential component of autophagosomes, autophagosome formation was examined in Psd knockdown photoreceptor cells. To detect autophagosomes in situ, we expressed the following autophagosomal/autolysosomal markers in photoreceptor cells: GFP-conjugated Atg8b (GFP-Atg8b), Myc-tagged Atg1 (Atg1-Myc), and Myc-tagged Atg6 (Atg6-Myc) (Scott et al., 2004; Tekinay et al., 2006; Sun et al., 2008; Venkatachalam et al., 2008). Whereas only small punctate areas, positive for both GFP-Atg8b and Atg1-Myc, were detected under dark-reared conditions, these double-positive structures were much larger after exposure to light (Fig. 5A). Immuno-electron-microscopic analysis, using an anti-GFP antibody, revealed that these larger structures, which were also positive for Atg6-Myc (Fig. 6), were vesicles containing residual membranous structures in their lumina (Fig. 5B, arrows), some of which were double-membrane vesicles. This EM data strongly suggested that these structures were autophagosomes/autolysosomes. In addition, we found that, under light-exposed conditions, these autophagosomes/autolysosomes were positive for Rh1 (Figs. 5C, 6), but not for the Golgi marker, GM130 (Fig. 5C).

Autophagosome formation is dependent on Psd. A, Left, Retinas of hs-gfp-atg8b/GMR-Gal4; UAS-atg1-myc/+ flies stained with anti-GFP, anti-Myc, and phalloidin. Flies were incubated at 37°C for 50 min and reared in the dark or exposed to light for 6 d. The incubation at 37°C was repeated twice every 3 d. Whereas structures positive for GFP-Atg8b and Atg1-Myc were small (arrowheads in top panels) under dark conditions, they became larger after light exposure. Two types were detected: large punctate structures (arrows in bottom panels) and ring-shaped structures (arrowheads in bottom panels). Right, Cross-sectional area of GFP-atg8b and Atg1-Myc-positive vesicles (n = 100, 2 independent experiments). Values represent the mean ± SEM. ***p < 0.001, t test. Scale bars, 10 μm. B, Immunodetection of GFP-Atg8b-positive structures by conventional (left) or electron (center, right) microscopy. Large structures (arrows in left panel) are revealed as vesicles containing residual membrane structures in their lumina (arrows in center and right panels), some of which are double-membrane vesicles. Rhabdomeres are indicated by yellow circles (left) or the letter “R” (center, right). Scale bars: left, 10 μm; center, right, 1 μm. C, Double staining of retinas of control (hs-gfp-atg8b) flies after heat shock followed by light exposure (8000 lux) for 6 d. GFP-atg8b-positive autophagosomes/autolysosomes (arrows and arrowheads) were costained with anti-Rh1 and phalloidin (top panels, arrows), but not with anti-GM130 (bottom panels). D, Double staining of retinas of Psd knockdown (hs-gfp-atg8b+Psd-RNAi#1) flies treated as shown in C. The GFP-atg8b-positive vesicles (arrows) were smaller than those observed in control flies (shown in C) and did not include Rh1 (arrowheads) or phalloidin. Scale bars: C, D, 10 μm. E, Statistical analysis of GFP-atg8b-positive vesicles shown in C and D. Left, Cross-sectional area containing GFP-atg8b-positive vesicles (n = 80–100, 2 independent experiments). Right, Rh1 signal within the range of GFP-Atg8b-positive vesicles (n = 80–100, 2 independent experiments). Values are the mean ± SEM. ***p < 0.001, t test. F, Immunodetection of GFP-Atg8b in Psd knockdown retinas. Anti-GFP signals were dispersed in some cytoplasmic domains (arrows). G, PE conjugation of GFP-Atg8b in control (hs-gfp-atg8b) and Psd knockdown (hs-gfp-atg8b+Psd-RNAi#1) retinas. Flies were heat shocked and reared as indicated in A and then retinal cell extracts were subjected to urea–SDS-PAGE followed by immunoblotting using anti-GFP antibodies. The amount of PE-conjugated GFP-Atg8b was increased in control flies by light exposure, and this increase was suppressed by Psd knockdown. *GFP-Atg8b; **GFP-Atg8b-PE. Scale bars, 1 μm.
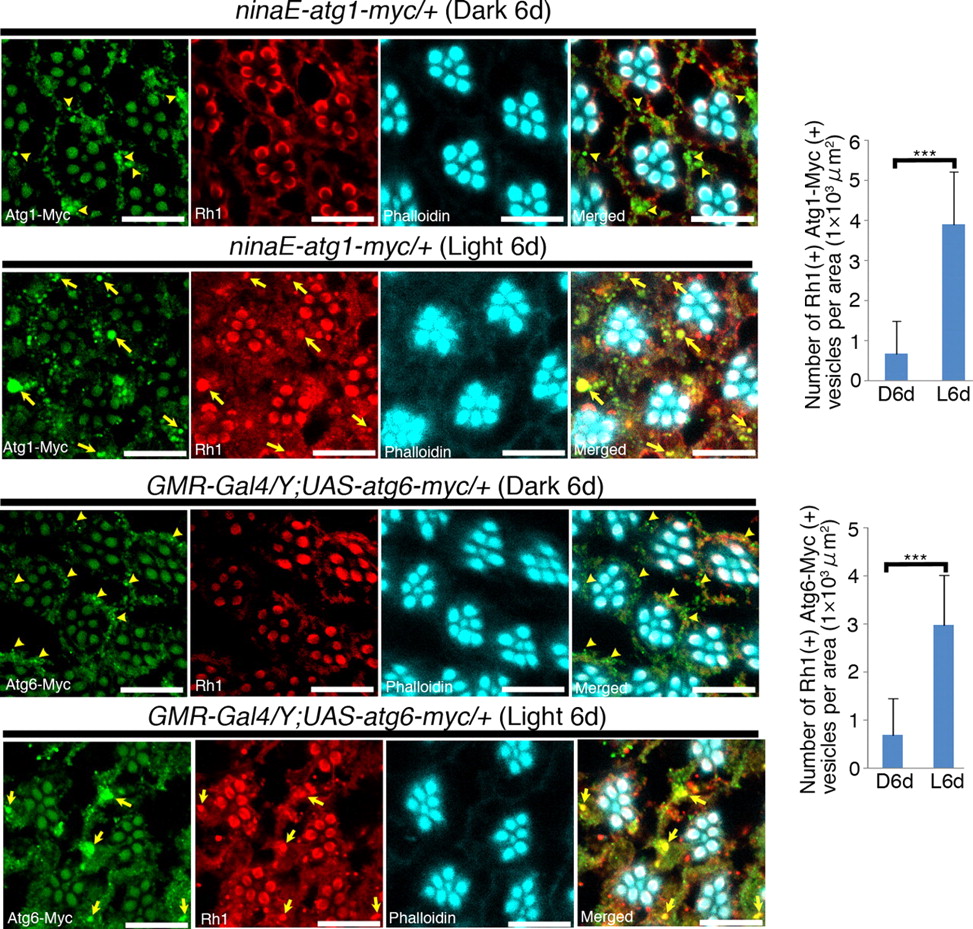
Light-dependent accumulation of Rh1 in Atg1- and Atg6-positive autophagosomes/autolysosomes. Left panels, Atg1-Myc- or Atg6-Myc-expressing flies were reared in the dark or under light exposure (8000 lux) for 6 d, and dissected retinas were triply stained as indicated in each panel. In retinas of dark-reared flies, both Atg1-Myc and Atg6-Myc were localized to the small punctate structures (arrowheads). In retinas of light-exposed flies, both Atg1-Myc and Atg6-Myc colocalized with Rh1 in large vesicles (arrows). Right, Statistical analysis of Atg1 and Rh1-positive (top) or Atg6 and Rh1-positive (bottom) vesicles shown in left panels. Each value was calculated as shown in Figure 2C. The number of both Atg1/Atg6 and Rh1-positive vesicles was significantly increased in light-exposed flies. Values are the mean ± SD. ***p < 0.001, t test. Scale bars, 10 μm.
The large autophagosomes/autolysosomes were not formed in Psd knockdown cells and only small structures remained that contained little Rh1 (Fig. 5D,E). Consistently, immuno-electron-microscopic studies confirmed that the double-membrane vesicles were rarely detected in the Psd knockdown cells and revealed cytoplasmic domains stained by anti-GFP antibody (Fig. 5F, arrows). These anti-GFP-positive domains may correspond to the small punctate signals in Figure 5D (arrows). This reduction in autophagosome size and number was consistent with previous reports, which showed that the number and size of autophagosomes were also reduced in Drosophila atg7 and atg3 mutants (Juhász et al., 2003, 2007).
Next, we investigated the molecular basis for the deformation of autophagosomes in Psd knockdown cells. Given that the PE-conjugated form of Atg8 is essential for autophagosome formation in yeast and mammals, we analyzed the amount of Atg8-PE present in cells biochemically. Lysates from control and Psd knockdown retinas were subjected to urea–SDS-PAGE, since free Atg8 and PE-conjugated Atg8 migrate at different rates (Kirisako et al., 2000; Amar et al., 2006). Consistent with the light-induced formation of autophagosomes, levels of Atg8-PE were elevated by light exposure in the control retinas (Fig. 5G). This elevation was not observed in the knockdown cells (Fig. 5G), suggesting that Psd is required for autophagosome formation since it is involved in the synthesis of PE, the substrate of Atg8-PE.
An atg8a mutation or knockdown of atg8a or atg7 causes Rh1 accumulation and retinal degeneration
Given that both autophagosome deformation and Rh1 accumulation result from Psd knockdown, we examined whether autophagy is required for the efficient degradation of Rh1. Since Atg8 and Atg7 are essential for autophagosome formation, we analyzed the following mutant and knockdown flies: atg8a mutant (atg8aKG07596) and RNAi-inducible IR strains, atg8a and atg7. In atg8aKG07596 mutant eyes, Rh1 accumulated in compartments that were Rab7-positive and phalloidin-negative (Fig. 7B,E); this was also observed in the Psd and Ept knockdown cells. The atg8aKG07596 animals had very poor health and the escaped adults died before retinal degeneration occurred. Hence, we focused our analysis on the knockdown flies. We observed Rh1 accumulation and severe retinal degeneration in the Rab7-positive endosomes of the atg8a or atg7 knockdown photoreceptor cells (Fig. 7D,E; supplemental Fig. S6, available at www.jneurosci.org as supplemental material). In addition, these phenotypes were not induced under dark-reared conditions (Fig. 7B,D; supplemental Fig. S6A, available at www.jneurosci.org as supplemental material). Together, we concluded from our data that autophagy is required for efficient Rh1 degradation under light exposure and that impaired autophagy results in retinal degeneration.

The suppression of atg8a causes Rh1 accumulation in Rab7-positive endosomes. A–D, Immunostaining of dissected retinas. The flies were treated and dissected retinas were triply stained as indicated in each panel. A, CS (Canton-S, wild-type) flies. B, atg8aKG07569 flies. Rh1 accumulation was found in Rab7-positive endosomes (arrows) after light exposure. C, gfp-RNAi flies. D, atg8a-RNAi flies. After light exposure for 8 d, Rh1-positive large vesicles were found in all of the ommatidia and colocalized with Rab7 (arrows). After light exposure for 15 d, the reduction or loss of rhabdomeres (arrowheads) was found throughout the range of ommatidia. Scale bars: A–D, 10 μm. E, Statistical analysis of Rh1- and Rab7-positive vesicles after light exposure (5 d for CS and atg8aKG07569 flies, and 8–10 d for gfp-RNAi and atg8a-RNAi flies). Left, Number of Rh1- and Rab7-positive vesicles per area was calculated as shown in Figure 2C. Values are the mean ± SD. ***p < 0.001, t test. Right, Cross section of the area containing Rab7-positive vesicles observed after light exposure was calculated as shown in Figure 2C. Values are the mean ± SEM. ***p < 0.001, t test.
A reduction in rhodopsin expression rescued retinal degeneration caused by Psd knockdown
Given that impairment of the autophagic pathway resulted in the intracellular accumulation of Rh1 followed by retinal degeneration, we wanted to examine whether the main cause of this phenotype was the accumulation of excess Rh1. The expression of rhodopsins is reduced when Drosophila is grown in retinoid-free medium (Fig. 8A,B) (Picking et al., 1996; Orem and Dolph, 2002b), and the Psd knockdown flies grown in retinoid-free medium showed neither intracellular accumulation of Rh1, nor retinal degeneration under light-exposed conditions, whereas flies cultured in normal medium showed severe retinal degeneration (Fig. 8A). In addition, TUNEL staining revealed that no cell death occurred in the Psd knockdown flies grown in retinoid-free medium (Fig. 8C). The retinal degeneration caused by the Ept knockdown was also suppressed under retinoid-free conditions (Fig. 8D).

The suppression of Rh1 synthesis rescues light-dependent retinal degeneration in Psd-RNAi flies. A, Triple staining of dissected retinas from gfp-RNAi and Psd-RNAi#1 flies. Flies were reared on either normal medium (left panels) or retinoid-free medium (right panels) for 5 d under light exposure (4000 lux). Psd-RNAi flies, deprived of retinoids, showed resistance to light-dependent retinal degeneration. Scale bars, 10 μm. B, Immunoblotting of adult head lysates collected from gfp-RNAi and Psd-RNAi#1 flies reared on normal medium (N) or retinoid-free medium (R) in the dark. Lysate from one-half of the harvested adult heads was immunoblotted with anti-Rh1 (Rh1) (top panel) or anti-α-tubulin (T) (bottom panel) antibodies. C, Retinoid deprivation suppresses the light-dependent retinal cell death of Psd-RNAi#1 flies. Ci, gfp-RNAi and Psd-RNAi#1 flies were reared on normal medium (as shown in Fig. 1Ci) or retinoid-free medium (this figure) under light exposure at 4000 lux for 5 d. A TUNEL assay was then performed as described in Figure 1C. The retinal cell death observed in light-exposed Psd-RNAi#1 flies was completely suppressed by retinoid deprivation. Scale bars, 20 μm. Cii, Statistical analysis of TUNEL-positive nuclei. The percentage of TUNEL-positive nuclei located from the apical surface to the retinal floor (shown by asterisks in the merged images in Ci) was calculated as in Figure 1Cii. The values are the mean ± SD. ***p < 0.001, t test. D, Triple staining of the dissected retinas of Ept-RNAi#1 flies reared on either normal medium (left panels) or retinoid-free medium (right panels) for 10 d under light exposure (8000 lux). The Ept-RNAi flies, deprived of retinoid, showed resistance to light-dependent retinal degeneration. Scale bars, 10 μm. E, The light-dependent retinal degeneration observed in Psd knockdown flies was rescued by an rh1 mutation, ninaE. Psd-RNAi#1 or Psd-RNAi#1+ninaE5 flies were reared and triply stained as indicated in each panel. The severe impairment of ommatidia caused by the Psd knockdown after light exposure was completely suppressed by the rh1 mutation. Scale bars, 10 μm.
The expression of Rh1 was also reduced by introduction of the rh1 mutant allele, ninaE5, which reduced the levels of Rh1 to between 3 and 13% of that of the wild type (flybase; http://flybase.org/reports/FBgn0002940.html). Light-dependent retinal degeneration in the Psd knockdown photoreceptor cells was also clearly suppressed by the introduction of the ninaE5 mutation (Fig. 8E). These results suggested that PE reduction induces retinal degeneration through the excess accumulation of rhodopsin.
The results of another series of experiments supported this conclusion. Rh1 is inactivated by red light (∼580 nm) (Kiselev et al., 2000), whereas treatment with blue light (∼480 nm) followed by red light exposure causes transient activation but no internalization of Rh1. In our current analyses, we found that blue light exposure for 10 min followed by 1 h of darkness caused internalization of Rh1 in the Psd knockdown flies (Fig. 9). When such flies were reared in the dark for >5 d, the photoreceptors became severely deformed (Fig. 9). In contrast, blue light exposure followed by 10 min of red light exposure blocked Rh1 internalization and no retinal degradation occurred even after rearing in the dark for an additional 5 d (Fig. 9). These results further suggested that the internalization of Rh1 is responsible for the underlying retinal degeneration caused by the impairment of the autophagic pathway.

Blue/red light exposure reversibly induces retinal degeneration in Psd knockdown flies. The gfp-RNAi and Psd-RNAi#1 flies were treated as indicated in each panel and dissected retinas were triply stained (Rh1, Cad, and phalloidin). Rh1-positive puncta, possibly originating from internalized Rh1, were found in both types of flies (arrows) after blue light exposure (22,000 lux) for 10 min followed by 1 h of darkness (BL→D1h). The ommatidia of Psd-RNAi#1 flies were severely impaired (arrowheads) after blue light exposure for 10 min followed by 5 d in the dark (BL→D5d). The retinal degeneration induced by blue light exposure was suppressed by subsequent red light exposure (4000 lux) for 10 min (BL→RL→D5d). Scale bars, 10 μm.
Overexpression of Rab7 rescues retinal degeneration caused by impairment of the autophagic pathway
The internalized Rh1 accumulated in the Rab7-positive endosomes because of the impairment of the autophagic pathway, suggesting cross talk between the autophagic and endosomal degradation pathways. To test this theory, we examined whether retinal degeneration caused by impaired autophagy was rescued by use of the endosomal degradation pathway. We overexpressed Rab7, which accelerates late endosome to lysosome trafficking of proteins requiring degradation (Entchev et al., 2000; Seto et al., 2002), in the Psd and atg8a knockdown photoreceptor cells. The overexpression of Rab7 caused a significant reduction in the accumulation of Rh1 (Fig. 10A,B) and rescued retinal degeneration in light-exposed Psd knockdown flies (Fig. 10C). We found consistently that the accumulation of excess Rh1 in Rab7-positive endosomes in the atg8a mutant and knockdown cells, and the light-dependent retinal degeneration seen in atg8 knockdown flies, were also suppressed by Rab7 overexpression (Fig. 10D–F). These data suggested that the autophagic and endosomal pathways cooperatively degrade excess Rh1 that is internalized by light exposure.

Overexpression of Rab7 suppresses light-dependent retinal degeneration in autophagy deficient flies. A, Rab7 expression suppresses Rh1 accumulation in late endosomes caused by Psd knockdown. The Psd-RNAi#1 (top panels) flies and Psd-RNAi#1 flies carrying the rab7 gene (bottom panels) were reared in the dark or exposed to light (8000 lux) for 3 d. The retinas were triply stained as shown in each panel. The number and size of large Rh1-positive late endosomes (arrows) shown in light-exposed Psd-RNAi#1 retinas were clearly reduced by Rab7 expression. B, Statistical analysis of Rh1- and Rab7-positive vesicles after light exposure for 3 d. Left, Number of Rh1- and Rab7-positive vesicles per area was calculated as shown in Figure 2C. Values are the mean ± SD. ***p < 0.001, t test. Right, Cross-sectional area of Rh1- and Rab7-positive vesicles. Values are the mean ± SEM. ***p < 0.001, t test. C, Rab7 expression suppresses light-dependent retinal degeneration in Psd-RNAi#1 retinas. Flies were reared in the dark or exposed to light (8000 lux) for 8 d, and the retinas were doubly stained as shown in each panel. Ommatidial deformation caused by Psd knockdown under light-exposed conditions was completely suppressed by Rab7 expression. D, Rab7 expression suppresses Rh1 accumulation in late endosomes caused by the atg8a mutation (atg8aKG07569)/knockdown (atg8-RNAi). The atg8a mutant/knockdown flies (top panels) and atg8a mutant/knockdown flies bearing the rab7 gene (bottom panels) were reared in the dark, or exposed to light (8000 lux) for 5 or 12 d, and the retinas were triply stained as shown in each panel. The number and size of large Rh1-positive late endosomes (arrows) shown in light-exposed atg8a mutant/knockdown retinas were reduced by Rab7 expression (arrowheads). E, Statistical analysis of Rh1- and Rab7-positive vesicles after light exposure for 5 or 12 d. Each value was calculated as shown in B. Values are the mean ± SD and the mean ± SEM in left and right graphs, respectively. *p < 0.05, ***p < 0.001, t test. F, Rab7 expression suppresses light-dependent retinal degeneration caused by atg8a knockdown. Flies were reared in the dark or exposed to light (8000 lux) for 12 d, and the retinas were doubly stained as shown in each panel. The retinal degeneration caused by atg8a knockdown after light exposure was suppressed by Rab7 expression. The arrows indicate Rh1-positive large vesicles. Scale bars: A, C, D, F, 10 μm.
The suppression of Ppt1 causes retinal degeneration in Drosophila and this defect is enhanced synergistically by Psd knockdown
Since it is known that Psd interacts genetically with the Drosophila ortholog of human PPT1, a gene that is mutated in the pediatric neurodegenerative disease known as infantile neuronal ceroid lipofuscinosis (INCL) (Glaser et al., 2003; Hickey et al., 2006), we expected that palmitoyl-protein thioesterase 1 (Ppt1) in Drosophila would be essential for the viability of photoreceptor cells. Therefore, we analyzed the phenotype of the Ppt1 knockdown. In Ppt1 knockdown cells, light exposure induced Rh1 accumulation in the Rab7-positive late endosome, and this was followed by retinal degeneration (Fig. 11A; supplemental Fig. S7, available at www.jneurosci.org as supplemental material) associated with cell death (Fig. 11B). However, these events were suppressed when the knockdown flies were grown in retinoid-free medium (Fig. 11A–C), suggesting that Rh1 accumulation is the cause of the degeneration. In addition, light-dependent degeneration also occurred in the Ppt1 mutants, Df(1)446-20 and Ppt1S77F (supplemental Fig. S8, available at www.jneurosci.org as supplemental material), excluding the possibility that off-target effects of RNAi were responsible for the observed anomalies. Collectively, therefore, our data indicated that Ppt1 is essential for photoreceptor viability in Drosophila.
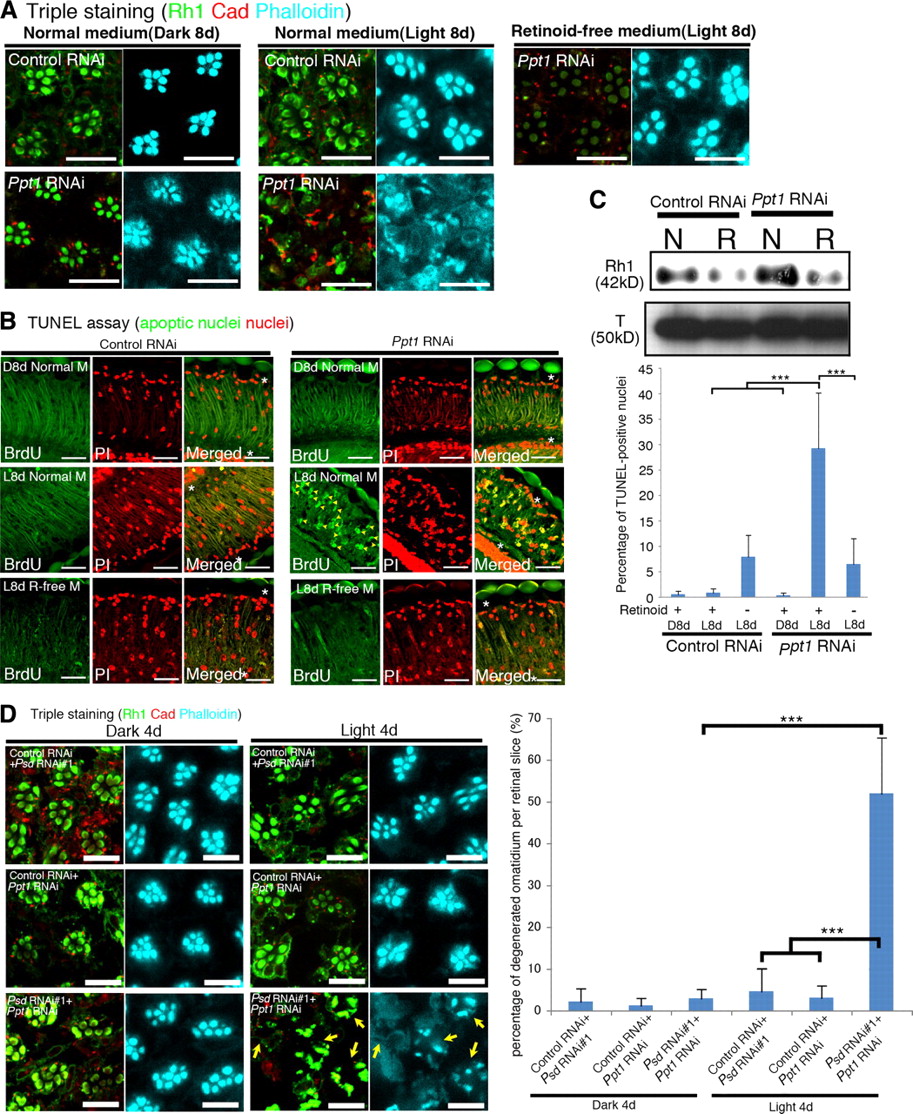
The Ppt1 knockdown causes light-dependent retinal degeneration and genetically interacts with the Psd knockdown. A, Triple staining of the dissected retinas of flies bearing two copies of gfp-RNAi (gfp-RNAi/gfp-RNAi) or Ppt1-RNAi (Ppt1-RNAi/Ppt1-RNAi). Flies were reared on either normal medium (left and middle panels) or retinoid-free medium (right panels) for 8 d under dark-reared (left panels) or light-exposed (8000 lux, middle and right panels) conditions. The Ppt1-RNAi/Ppt1-RNAi flies, deprived of retinoids, showed resistance to light-dependent retinal degeneration. Scale bars, 10 μm. B, A TUNEL assay was then performed as described in Figure 1Ci. Light-dependent photoreceptor cell death in Ppt1-RNAi/Ppt1-RNAi flies (arrowheads in panels L8d Normal M) was suppressed by retinoid deprivation (R-free M). Scale bars, 20 μm. C, Top panels, Immunoblotting of adult head lysates collected from gfp-RNAi/gfp-RNAi and Ppt1-RNAi/Ppt1-RNAi flies. Flies were treated and immunoblotting was performed as shown in Figure 8B. Bottom panel, Statistical analysis of TUNEL-positive nuclei. The percentage of TUNEL-positive nuclei from the apical surface to the retinal floor (indicated by asterisks in the merged images) was calculated as in Figure 1Ci. The values are the mean ± SD. ***p < 0.001, t test. D, Left, Flies were dark-reared or light-exposed (4000 lux) for 4 d, and the dissected retinas were triply stained (Rh1, Cad, and phalloidin). Impaired ommatidia (arrows) were detected in Psd-RNAi/Ppt1-RNAi retinas. Scale bars, 20 μm. Right, Statistical analysis of the disruption in the retinas of double-knockdown Psd and Ppt1 flies. The percentage of disrupted ommatidia per slice was calculated as shown in Figure 4D. Values are the mean ± SD. ***p < 0.001, t test.
The genetic interaction between the Psd and Ppt1 loss-of-function alleles was examined because a previous study described an interaction between overexpressed Ppt1 and Psd (Buff et al., 2007). To test this, dsRNAs for Psd and Ppt1 were induced simultaneously using weak conditions under which expression of each one of the dsRNAs did not cause severe defects (Fig. 11D). However, the simultaneous induction of both dsRNAs produced severe defects and a count of the degenerated ommatidia per retinal slice clearly revealed the synergistic enhancement of retinal degeneration (Fig. 11D). These results suggest that Psd and Ppt1 undergo a genetic interaction under conditions of reduced expression.
Discussion
Degradation of rhodopsin by autophagy
In this Drosophila study, we have demonstrated that Rh1, a major rhodopsin, is degraded by autophagy, in addition to the endosomal degradation pathway, and that defects in the autophagic pathway cause light-dependent retinal degeneration. PE, a lipid found in the membrane of autophagosomes, is synthesized by Psd in mitochondria and by Ept in the ER and nuclear envelope (Choi et al., 2005; Vance, 2008). Using an eye-specific knockdown screen with an inducible RNAi library, we identified Psd as an essential factor involved in the prevention of light-dependent retinal degeneration in Drosophila. Psd-deficient mice die at around embryonic day 9, before neural cells differentiate (Steenbergen et al., 2005), and it is possible that this lethality might be attributable to mitochondrial defects, since fibroblasts established from Psd-deficient mice exhibit fragmented, aberrantly shaped mitochondria. Given that mitochondrial defects have been implicated in neurodegeneration, such defects might also contribute to the retinal degeneration observed in the Psd knockdown flies in this study. However, an Ept knockdown, which reduced PE production, but did not affect mitochondrial function, also showed light-dependent retinal degeneration. Moreover, the morphology of mitochondria in the photoreceptor cells of dark-reared Psd knockdown flies appeared normal in our electron-microscopic analyses and the membrane potential was normal in these ommatidia even after light exposure for 2.5 d. Therefore, these results strongly suggested that the Psd knockdown causes retinal degeneration through defective PE production and autophagy, rather than through mitochondrial defects.
The retinal degeneration phenotype in Psd or Ept knockdown flies was rescued when Rh1 expression was reduced in a ninaE mutant background, or under retinoid-free conditions. Moreover, exposure to blue light followed by red light exposure blocked the internalization of Rh1 and suppressed the retinal degeneration phenotype in the Psd knockdown eyes. These results strongly supported the notion that the intracellular accumulation of Rh1 is the cause of the retinal degeneration in these flies.
A very recent study by Wang et al. (2009) has shown, independently, that light-dependent retinal degeneration is induced by the hyperactivation of target of rapamycin (TOR) protein kinase, or by an atg7 mutation. The authors also found that inhibition of TOR or inducing Atg1 expression suppressed both the retinal degeneration caused by ectopic expression of the mutant Huntingtin protein (which carries a long polyQ stretch) and phospholipase C (norpA)-mediated accumulation of the cytoplasmic rhodopsin– arrestin complex. However, it remains unclear whether autophagy is required for the suppression of polyQ- or norpA-induced retinal degeneration. It is also unclear whether rhodopsin is degraded directly by the autophagosome/autolysosome. In this study, we found that Rh1 is required for light-induced retinal degeneration in Psd knockdown flies and that Rh1 is incorporated into the autophagosome in normal photoreceptor cells. These results clearly indicate that rhodopsin is degraded through autophagy and is a key target of the autophagy-dependent degradation responsible for the suppression of retinal degeneration.
The processes involved in autophagosome formation, revealed by studies using yeast and mammalian cells, appear to be conserved in Drosophila. For example, Drosophila atg7 and atg3 mutations produce a significant reduction in the number and size of autophagosomes/autolysosomes in larval fat bodies and midgets (Juhász et al., 2003, 2007), a phenomenon also seen in yeast mutants and mammalian cells (Nakatogawa et al., 2007; Sou et al., 2008). However, the significance of the conjugation of PE to Atg8 had not been directly represented in Drosophila. Our study shows that the knockdown of Psd impaired both PE-Atg8 conjugation and autophagosome formation, providing direct evidence that PE conjugation to Atg8 is essential for autophagosome formation in Drosophila. Moreover, we found that the amount of PE-Atg8 was elevated by exposure to light in Drosophila photoreceptor cells. This light-induced formation of autophagosomes has also been reported in rat photoreceptor cells (Remé et al., 1999), suggesting that light-dependent regulation of autophagy in photoreceptor cells may be conserved in metazoa.
The endosomal/lysosomal pathway is necessary for autophagy-dependent degradation, since autophagosomes must fuse to late endosomes (and to MVBs and lysosomes). Defects in the endosomal/lysosomal pathway do indeed hamper autophagy-dependent degradation (Shirahama et al., 1997; Nara et al., 2002; Besteiro et al., 2006; Filimonenko et al., 2007; Lee et al., 2007; Rusten et al., 2007); however, it was unknown whether defects in the autophagic pathway also affected the endosomal degradation pathway. In this study, the mutation and/or knockdown of atg, Psd, and Ept caused the abnormal accumulation of Rh1 in the Rab7-positive late endosome (Fig. 12). On exposure to light, increased amounts of Rh1 are transported to the late endosome and MVBs. In addition to conventional lysosomal activities, this excess Rh1 may also be degraded by autolysosomes that fuse to the late endosome or MVBs, since Rh1 was detected in Atg8b-GFP-positive autolysosomes after light exposure. These results suggested that autophagy plays an essential role in rhodopsin degradation in cooperation with the endosomal degradation pathway. It will be interesting, in future studies, to determine how this compensatory degradation pathway is selected under stress conditions or in pathological contexts.

A schematic model of the cross talk between the endosomal and autophagy pathways during light-induced Rh1 degradation. A, In control flies, Rh1 is internalized and degraded by the endosomal pathway (left). The autophagy pathway (right) plays a compensatory role during Rh1 degradation, possibly through the fusion of autophagosomes with Rh1-positive MVB/late endosomes. B, In atg7-, atg8a-, Psd-, or Ept-deficient flies, autophagosome formation is impaired. This causes the accumulation of Rh1 in MVB/late endosomes after light exposure as evidenced by the impairment of the divergent pathway shown in A. The Rh1 accumulation in MVB/late endosomes eventually leads to photoreceptor cell death/apoptosis, and this can be suppressed by the overexpression of Rab7, which promotes late endosome to lysosome trafficking.
Insight into INCL, a disease caused by Ppt1 mutation
INCL is caused by mutations in the Ppt1 gene (Vesa et al., 1995), which encodes the soluble lysosomal enzyme, Ppt1, and is known to interact genetically with the Psd gene (Buff et al., 2007). The pathological features of INCL manifest as normal early development in the first year followed by various neural defects, including blindness (Haltia et al., 1973). However, it is not clear how the loss of Ppt1 activity causes retinal degeneration. It has been reported that Ppt1 and Psd interact genetically when they are overexpressed (Buff et al., 2007). Moreover, in this study, we have demonstrated that Ppt1 mutants show light-dependent retinal degeneration and that there is a genetic interaction between these genes when they are downregulated. Interestingly, the Ppt1 knock-out mouse exhibits lysosomal dysfunction (Virmani et al., 2005). Since retinal degeneration in the Psd knockdown fly is caused by the intracellular accumulation of rhodopsins, this may also underlie the retinal degeneration that occurs in Drosophila and humans as a result of a Ppt1 disruption. A severe form of human autosomal dominant retinitis pigmentosa is caused by Arg135 mutant rhodopsins that accumulate in aberrant endosomes (Chuang et al., 2004). It will thus be necessary in future studies to determine whether rhodopsins also accumulate in the photoreceptor cells of INCL patients.
In summary, this study underlines the importance of the interplay between the autophagic and endosomal/lysosomal pathways in protecting light-induced retinal degeneration through the efficient degradation of activated rhodopsin. In addition, we have provided genetic evidence that the autophagic pathway influences the degenerative defects caused by Ppt1, although the mechanism is unclear. This report will help to shed light on the mechanisms behind human retinal degenerative diseases, since it is possible that cross talk between the autophagic and endosomal/lysosomal pathways might also be conserved in humans.
Footnotes
-
This work was supported by Ministry of Education, Culture, Sports, Science, and Technology of Japan Grant-in-Aid 60280575 (S.G.) and National Institute of Genetics Cooperative Research Program Grants-in-Aid 2008-A69 and 2009-A54 (S.G., E.S.). We thank Drs. R. L. Glaser, A. Nakamura, S. Munro, and T. P. Neufeld for providing reagents.
- Correspondence should be addressed to Satoshi Goto, Department of Physiology, School of Medicine, Keio University, 35 Shinanomachi, Shinjuku-ku, Tokyo 160-8582, Japan. sgoto{at}a7.keio.jp





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}