

Abstract
Cortical efferents growing in the same environment diverge early in development. The expression of particular transcription factors dictates the trajectories taken, presumably by regulating responsiveness to guidance cues via cellular mechanisms that are not yet known. Here, we show that cortical neurons that are dissociated and grown in culture maintain their cell type-specific identities defined by the expression of transcription factors. Using this model system, we sought to identify and characterize mechanisms that are recruited to produce cell type-specific responses to Semaphorin 3A (Sema3A), a guidance cue that would be presented similarly to cortical axons in vivo. Axons from presumptive corticofugal neurons lacking the transcription factor Satb2 and expressing Ctip2 or Tbr1 respond far more robustly to Sema3A than those from presumptive callosal neurons expressing Satb2. Both populations of axons express similar levels of Sema3A receptors (neuropilin-1, cell adhesion molecule L1, and plexinA4), but significantly, axons from neurons lacking Satb2 internalize more Sema3A, and they do so via a raft-mediated endocytic pathway. We used an in silico approach to identify the endocytosis effector flotillin-1 as a Sema3A signaling candidate. We tested the contributions of flotillin-1 to Sema3A endocytosis and signaling, and show that raft-mediated Sema3A endocytosis is defined by and depends on the recruitment of flotillin-1, which mediates LIM domain kinase activation and regulates axon responsiveness to Sema3A in presumptive corticofugal axons.
Introduction
In the cerebral cortex, pyramidal neurons initiate their axonal trajectories beneath the cortical plate contemporaneous with neural migration. The path that axons take and their final destination characterize distinct neuron types, such that populations of pyramidal neurons that send axons across the corpus callosum to innervate contralateral cortical targets are distinct from those that send axons to subcortical targets. Thus, cortical axons within a shared environment must display differential sensitivities to cues to generate distinct trajectories.
Semaphorin3A (Sema3A) is a potent guidance cue that can repel cortical axons (Bagnard et al., 1998; Castellani et al., 2000). Its localization within the developing neuroepithelium suggests that all cortical projections are exposed to the cue (Skaliora et al., 1998), but in vitro assays show that responses are not uniform (Mintz et al., 2008), and in vivo analyses suggest that responses may change over the course of development (Behar et al., 1996; Taniguchi et al., 1997; Campbell et al., 2001; Sibbe et al., 2007; Chen et al., 2008b). In cortical neurons, to transmit repelling signals to the cytoskeleton, the Sema3A receptor neuropilin-1 (Npn1) uses a plexin-A4 (PlxA4) coreceptor (Tamagnone et al., 1999; Suto et al., 2005; Yaron et al., 2005). PlxA activation can initiate a variety of signals that converge on actin severing or depolymerization (Aizawa et al., 2001; Terman et al., 2002; Toyofuku et al., 2005). The actions of additional coreceptors are required to generate full responsiveness (Maness and Schachner, 2007; Law et al., 2008), and in cortical neurons Sema3A-mediated repulsion requires Npn1 binding to the cell adhesion molecule L1 (L1CAM) (Castellani et al., 2000). The signaling pathways initiated by L1CAM in response to Sema3A include a focal adhesion kinase–mitogen-activated protein kinase cascade (Bechara et al., 2008) and regulated interactions between ERM proteins and the actin cytoskeleton (Mintz et al., 2008). Differential receptor/coreceptor expression would be an obvious mechanism for generating cell type-specific responses, but mRNAs encoding Npn1, L1CAM, and PlxA4 are expressed in nearly all developing cortical neurons, making this unlikely (Perälä et al., 2005; Morita et al., 2006) (GenePaint database, Allen Brain Atlas). Alternate mechanisms have not been identified.
Several cell types can selectively internalize receptors to regulate responsiveness to ligands, including guidance cues (Hong et al., 1999; Piper et al., 2005). We hypothesized that receptor internalization could be used selectively to gate responsiveness to Sema3A in different populations of cortical axons. We took advantage of previous work that has defined cortical neuron populations based on the selective expression of transcription factors (Arlotta et al., 2005; Britanova et al., 2005) to generate an in vitro assay in which we could identify cell type-specific responses to Sema3A and test the underlying mechanisms. We show that axons from cortical neurons lacking the callosal projection neuron marker Satb2 are more responsive to Sema3A and that this selective response requires Sema3A internalization via a lipid raft and flotillin-dependent, dynamin-independent pathway that activates LIM domain kinase (LIMK). These findings extend earlier studies documenting the significance of membrane microdomains for guidance receptor signaling (Guirland et al., 2004) and further define an adaptable molecular pathway that can be used to generate cell type-specific responses.
Materials and Methods
Dissociated cortical cultures.
All use of animals conformed to guidelines established by Mount Sinai's Institutional Animal Care and Use Committee and those of the National Institutes of Health. Cortical neurons were cultured as previously described (Mintz et al., 2008). Briefly, pregnant Sprague Dawley rats were killed by CO2 exposure, and embryos were removed from the uterus at embryonic day 18 (E18). The presumptive embryonic somatosensory cortex was dissected out in cold BSS (1× HBSS, 1% HEPES, 1% PenStrep) and digested for 15 min at 37°C in 0.25% trypsin (Invitrogen). The tissue was then triturated to a single-cell suspension and plated on poly-l-lysine-coated glass coverslips at a density between 20,000 and 40,000 cells per coverslip. Neurons were allowed to adhere for ∼3 h in media containing 10% fetal bovine serum, and then they were transferred to Neurobasal Media containing B27 (Invitrogen) or NS21 (Chen et al., 2008c).
Ex utero electroporation.
Rat embryos were removed from the uterus at either E15 or E17, and a mix of yellow fluorescent protein (YFP) plasmid and Fast Green (1:20) was injected in the right lateral ventricle using a pulled glass electrode. Approximately 5 μg of DNA was injected in each brain. Four 50 ms pulses of 40 V were applied at 950 ms intervals to the head of the embryo with the positive electrode placed on the right side. The somatosensory cortex was then dissected in cold BSS and cultured as described above (adapted from Hand et al., 2005).
Plasmids and RNA interference.
Except for ex utero electroporation (EUE) experiments, plasmids and RNA interference were introduced into cortical neurons immediately after dissociation using the Amaxa Nucleoporator. The following plasmids were used: YFP: pEYFP-N1 (BD Biosciences); dynamin K44A in the plasma encoding enhanced green fluorescent protein (GFP) vector (gift from Dr. P. DeCamilli, Yale University School of Medicine, New Haven, CT) (Takei et al., 1999); and Flot1 short hairpin RNAs (shRNAs) (AGAGAAGGCCCAGCTGATCAT, CCTGACCTCAGCCAATAAGAT, and AGCTACACTTTGAAGGATATT) were purchased from SA Biosciences. The control sequence was GGAATCTCATTCGATGCATAC. The plasmid also expresses GFP to detect transfected cells.
Reagents and antibodies.
Recombinant mouse Sema3A fused to a human Fc fragment was purchased from R&D Systems (see below, for concentrations used). Filipin, monodansyl cadaverine (MDC), and dynasore (Sigma) were used at 1 μg/ml, 10 μm, and 80 μm, respectively. The Src family kinase inhibitor SU6656 (Sigma) was used at 10 μm. Phalloidin, conjugated with Oregon Green, Texas Red, or Alexa 647 fluorophores, was purchased from Invitrogen and used at 2 U/ml; 655 nm Q-dot conjugated F(ab) anti-human-Fc was purchased from Invitrogen and used as described below.
The following primary antibodies were used: mouse anti-Satb2 (Abcam,1:400); rabbit anti-Ctip2 (Novus Biologicals, 1:100); chick anti-GFP (recognizes YFP as well; Aves, 1:4000); rabbit anti-Npn-1 (Zymed, 1:100); rabbit anti-PlxA4 (Abcam, 1:50); mouse anti-L1CAM (Abcam, 1:500); rabbit anti-flotillin-1 (Abcam, 1:100); mouse anti-flotillin-1 (BD Biosciences, 1:50); mouse anti-flotillin-2 (BD Biosciences, 1:50); rabbit anti-LIMK (Abcam, 1:100); and phospho-LIMK (Abcam, 1:50). All fluorophore-conjugated secondary antibodies were from Jackson Immunoresearch and were used at dilutions between 1:200 and 1:400.
Collapse assay.
For the live collapse assay, neurons were plated in glass-bottom dishes and imaged on an Olympus inverted microscope equipped with a live-cell imaging chamber that maintained the environment at 37°C and 5% CO2. After the chamber equilibrated, control media was added and growth cones imaged for 30 min followed by 100 μg of Sema3A-Fc for another 30 min. The neurons were then fixed and processed for Satb2 immunolabeling, as described below. Movies were analyzed and quantified blind to the results of post hoc immunocytochemistry. Growth cones that retracted or collapsed only during the Sema3A exposure but not during the initial control incubation were considered collapsed. For fixed preparations, an equal number of coverslips were incubated in control media or in 100 μg of Sema3A-Fc media for 30 min, then fixed and immunolabeled. The coverslips were imaged on a Zeiss Axiophot microscope and the collapsed growth cones counted blind to the conditions. Specific inhibitors were added 15 min before and remained during the exposure to Sema3A.
Patterned substrates.
Alternating stripes of Sema3A and control Fc fragment were prepared on glass coverslips that had been coated first with 1 mg/ml poly-l-lysine and then with anti-human Fc antibody (Sigma, B3773, 1:1000). Silicon matrices with alternating 100 μm channels were stamped on top of the coated coverslips, and a solution of 4 μg/ml recombinant Sema3A-fused human Fc was injected in the channels of this matrix (purchased from S. Lang, Max Planck Institute, Tübingen, Germany) (Knöll et al., 2007). The fusion protein was allowed to bind the substrate overnight at 4°C, and then the matrix was removed. After washing, the coverslips were incubated with 10 μg/ml solution of Cy3 conjugated human Fc fragment for 2 h at room temperature (RT). The fluorescent Fc fragment binds to the unoccupied anti-Fc antibodies, highlighting the interstripes. For clarity, the colors were inversed in the final images such that the Sema3A stripes appear red and the control stripes appear black.
Sema3A-quantum dot uptake.
The 100 μg/ml recombinant mouse Sema3A–human Fc chimera was mixed 10:1 with a 1 μm solution of quantum dot (Qdot)-conjugated anti-human Fab fragment, and incubated for 1–3 h at RT in the dark. The mix was then resuspended in home media for a final concentration of 4 μg/ml Sema3A. Cortical neurons plated in glass-bottom dishes were first washed in warm L15 media and then incubated with ∼75 μl of Sema3A–Qdot mix for 30 min at 37°. When endocytosis was blocked, specific inhibitors were added 15 min before and during the entire incubation with Qdot-conjugated Sema3A. Neurons were then washed once with warm L15 (for 1 min) and then acid stripped with pH 2 DMEM (Invitrogen) for 1–2 min (Yap et al., 2008a). The neurons were washed three more times with warm L15 media (Invitrogen) and then fixed with 4% paraformaldehyde (PFA) and 4% sucrose for 5 min. They were placed in cold PBS and imaged soon thereafter.
Immunolabeling.
Cultured neurons were fixed in 4% PFA, 4% sucrose for 10 min at RT or 2% PFA, 2% sucrose for 20 min (nonpermeabilizing conditions) (Kamiguchi and Lemmon, 2000; Wisco et al., 2003). For intracellular antigens, neurons were permeabilized with 0.1% Triton X-100 (Sigma) for 1 min at RT. Nonspecific binding was blocked by incubation in 10% bovine serum albumin (BSA) (Sigma) for 1 h at RT. Primary antibodies were diluted in 1% BSA to the appropriate concentration and incubated for 12 h at 4°C or 2–3 h at RT. After washing the coverslips in PBS, fluorophore-conjugated secondary antibodies were diluted in PBS and added for 1 h at RT in the dark. The coverslips were then washed and mounted on slides using Mowiol.
Embryonic brains were collected at E14.5, fixed in fresh 4% PFA for 12 h at 4°C, and cryoprotected in 30% sucrose. The 50-μm-thick sections were cut on a cryostat and mounted on charged slides. The sections were blocked in 3% BSA or 3% normal goat serum (NGS) for 1 h at RT. Primary antibodies were then added in 0.5% BSA or NGS for 12–48 h at 4°C. The sections were washed three times in PBS, and fluorophore-conjugated secondary antibodies were added at the appropriate concentration for 1 h at RT. After washing in PBS, the sections were coverslipped in Vectashield (Vector Laboratories).
Image acquisition.
Images were acquired on a Zeiss 510 laser scanning confocal microscope, unless specified otherwise. The objectives used were 20×, 40×, and 100×, depending on the assay; larger fields were acquired using a tiling function. For measurements of axonal length, images were imported into Neurolucida, axons were traced in their entirety, and the measurements were analyzed using Neuroexplorer (Microbrightfield). For Ctip2-overexpressing neurons, axons were measured in ImageJ (Rasband, 1997–2009). All statistical analyses were made using Prism.
Results
Cortical projection neurons maintain their identities in dissociated cultures
Projection neurons sharing particular pathways and targets can be identified in vivo by their expression of particular transcription factors. Satb2 is expressed in the majority of callosal projection neurons, while Ctip2 or Tbr1 are expressed in neurons sending axons to subcortical targets (Arlotta et al., 2005; Chen et al., 2005; Alcamo et al., 2008; Britanova et al., 2008; Chen et al., 2008a). To assess whether cortical neurons maintain their identities in culture, we immunolabeled for Satb2 and Ctip2 at the time of plating and after 2, 4, or 6 d in culture. The percentage of neurons immunolabeled for Satb2 (Satb2pos) steadily increases from 26% immediately after plating to a maximum of 62% at 4 d in vitro (DIV) after which it rapidly diminishes (Fig. 1 A,B). Satb2pos neurons show little to no labeling for either Ctip2 or Tbr1, which are expressed in a much lower proportion of cortical neurons (20 and 12% at 4 DIV, respectively) (Fig. 1 A,B). The gradual increase in Satb2 expression in postmitotic neurons, its exclusion from neurons labeled for Ctip2 or Tbr1, and its subsequent loss closely resemble in vivo observations (Alcamo et al., 2008; Britanova et al., 2008). The interval defining stable Satb2 expression, between 2 and 4 DIV, was used for all subsequent experiments.

Cortical neurons maintain their identity in dissociated cultures. A , Confocal images of cortical neurons grown in culture, fixed at 0, 2, 4, or 6 DIV and then immunostained for Satb2 (blue) and Ctip2 (red). Phalloidin staining shows actin filaments in green. Very few neurons expressing Satb2 have detectable levels of Ctip2 labeling. B , Bar graph plots the percentage of neurons positive for Satb2 (blue) or Ctip2 (red). Between 2 and 4 DIV, ∼25% are not labeled by either Satb2 or Ctip2. Error bars represent SEM (n = 3 experiments). C , D , Confocal images of cultured cortical neurons in which populations of deep ( C ) or superficial layer neurons ( D ) were labeled by EUE of a YFP expression vector at either E15 or E17, respectively. Neurons were immunostained for Satb2 (blue), Ctip2 (red) and YFP (green). E , Equal proportions of E15 electroporated neurons label for Satb2 or Ctip2, with little overlap. About 80% of E17 electroporated neurons are labeled for Satb2. Scale bars: A , 100 μm; (in D ) C , D , 50 μm.
All axons projecting corticofugally originate in deep cortical layers 5 and 6, whereas neurons having callosal projections reside mainly in superficial layers 2/3 and in 5a. We asked whether transcription factor expression in cultured neurons also reflects appropriate laminar destiny. To visualize lamina-specific cortical neurons in culture, ex utero electroporation (EUE) (see Materials and Methods) was used to deliver YFP into progenitors of the ventricular zone at E15.5, when layer 5 neurons are born in the rat somatosensory cortex, or E17.5, when layer 2/3 neurons are born (Bayer and Altman, 1990; Koester and O'Leary, 1994); cortices were dissociated immediately thereafter, cultured for 3 d, and then immunolabeled for transcription factors. Quantitative analysis indicates that half of the E15-born neurons express Satb2 (Fig. 1 C,E), and many neurons lacking Satb2 (Satb2neg) express Ctip2. In contrast, nearly 80% of the E17-born neurons express Satb2 (Fig. 1 D,E), and none express Ctip2 alone. These results are consistent with observations in vivo (Britanova et al., 2008). Thus, cortical neurons express and continue to develop key cell type-specific characteristics when grown in culture, providing a tractable model system in which cell biological mechanisms contributing to cell type-specific responses can be identified and evaluated.
Satb2neg neurons respond more robustly to Sema3A
We asked whether Sema3A differentially affects axonal growth from cortical neuron populations defined by Satb2. Layer 5 neurons were labeled with YFP using EUE at E15, dissociated and exposed to Sema3A for 72 h, and then immunolabeled for Satb2. Axonal growth was compared quantitatively by tracing axons in their entirety. The data show that E15-born Satb2neg neurons have significantly shorter axons when grown in the presence of Sema3A compared with control conditions (Fig. 2 A,B) and, consistent with previous work (Dent et al., 2004), axon branching is diminished (30% reduction in branch number relative to control). In contrast, Sema3A has no significant effect on the length of axons originating from E17-born neurons, the vast majority of which express Satb2 (ANOVA, p > 0.05; n = 50).
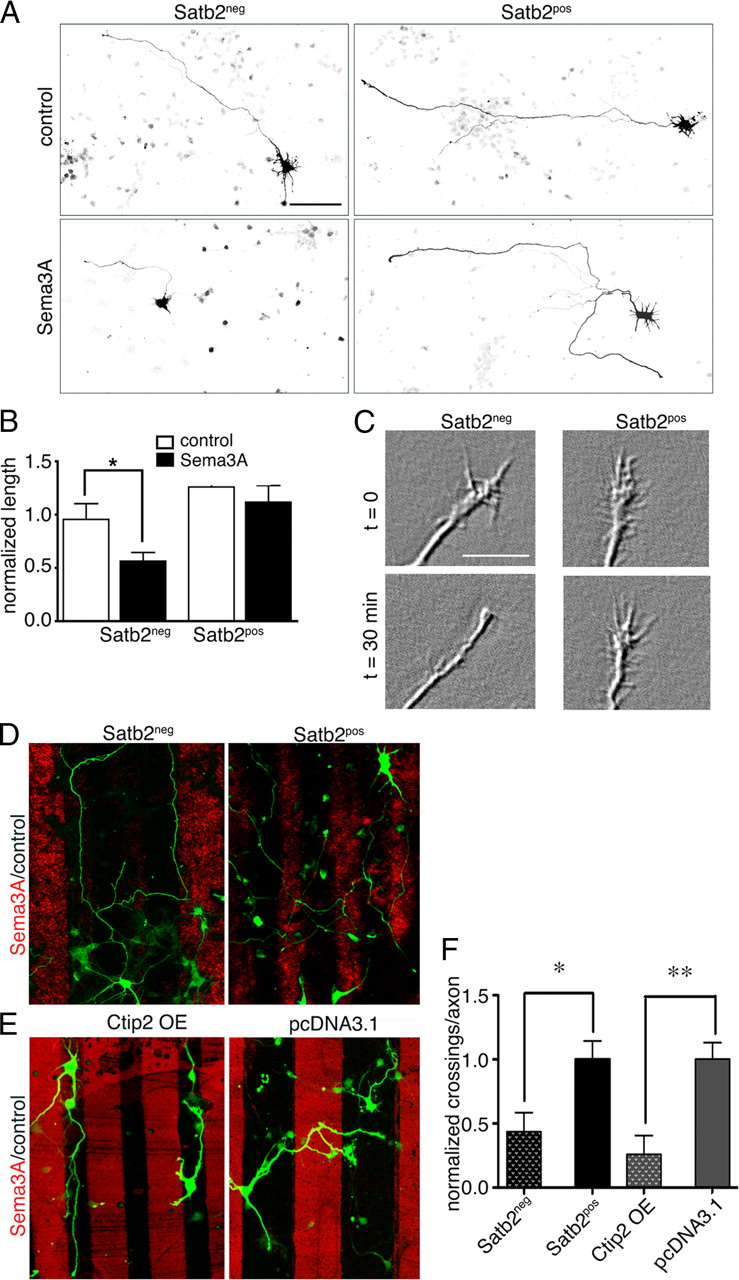
Sema3A triggers distinct responses in two populations of cortical neurons. A , Inverted fluorescence microscope images of E15 neurons expressing YFP and grown in either control or Sema3A media, and then fixed and labeled for Satb2 (data not shown). Axons from Sema3A-exposed Satb2neg neurons are notably shorter. Scale bar, 100 μm. B , Axonal length measurements from neurons treated as in A were normalized to the mean values of Satb2neg controls. Satb2neg neurons display significant axon growth inhibition (n = 26 axons; t test, p = 0.035), while axons of Satb2pos neurons have similar lengths in control and Sema3A conditions (n = 29 axons; t test, p = 0.6; *significant difference). C , Differential interference contrast images are representative snapshots of the same axonal growth cones taken after 30 min in control media (t = 0) and after 30 min in Sema3A. After imaging, neurons were fixed and immunolabeled for Satb2 (data not shown). Quantitative analysis showed that 78.75 ± 12.46% of Satb2neg axonal growth cones collapse in response to Sema3A, whereas only 37.03 ± 8.06% of growth cones from Satb2pos neurons collapse (n > 35 growth cones per group; t test, *p = 0.015). Scale bar, 10 μm. D , Confocal images show GFP-labeled neurons plated on stripes of Sema3A (red) alternating with stripes of control Fc (black), and then fixed and immunostained for Satb2 (data not shown). Axons of Satb2neg neurons (left) avoid Sema3A stripes, whereas axons of Satb2pos neurons appear to ignore the borders between Sema3A and control Fc stripes. The examples shown display stripes of different widths, but results were similar with wide or thin stripes. Magnification as in A . E , As in D , with neurons transfected with Ctip2 or control (pcDNA3.1). F , Results from multiple stripe experiments were quantified as the number of border crossings per axon, normalized to the values for Satb2pos axons ( D : n = 50 axons; t test, *p = 0.01) or for control, pcDNA3.1 expressing axons ( E : n = 47 axons; t test, **p = 0.003). Error bars represent SEM.
Acute exposure to Sema3A can produce growth cone collapse (Luo et al., 1993). To compare growth cone collapse in Satb2pos and Satb2neg neurons, axonal growth cones were imaged for 30 min before and following addition of Sema3A-Fc, after which cell bodies were immunolabeled post hoc for Satb2. A significantly greater proportion of Satb2neg neurons show growth cone collapse (Fig. 2 C).
Sema3A binds chondroitin sulfate proteoglycans and thus may be presented to neurons as part of the extracellular matrix rather than as a soluble cue (de Wit and Verhaagen, 2003). To test whether cortical axon populations respond differentially to a substrate of Sema3A, we plated neurons on stripes of Sema3A-Fc alternating with control Fc stripes. A comparison of the number of border crossings shows that axons of Satb2neg neurons avoid Sema3A stripes, while axons of Satb2pos neurons grow equally well on Sema3A and control stripes (Fig. 2 D,F).
The data indicate that axons of Satb2neg neurons respond more robustly to the repulsive effects of Sema3A than axons of Satb2pos neurons. We next tested whether this difference is dictated by the restricted expression of transcription factors. Overexpression of Satb2 would be predicted to reduce responsiveness to Sema3A, but it also increases cell death and could not be used for this. In mice lacking Satb2, Ctip2 expression is permitted in layer 2/3 neurons, many of which send axons inappropriately to subcortical targets (Alcamo et al., 2008; Britanova et al., 2008). With this in mind, we asked whether Ctip2 overexpression in a mixed population of cortical neurons (Satb2pos and Satb2neg) would increase responsiveness to Sema3A. The data show that Ctip2 overexpression increases axon outgrowth over 72 h and that Sema3A abrogates this effect (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Moreover, in a stripe assay we see that axons of Ctip2-overexpressing neurons avoid Sema3A stripes (Fig. 2 E,F). Thus, Ctip2 expression enhances axon responsiveness to Sema3A.
All cortical neurons express similar levels of Sema3A receptors
Guidance cue responsiveness can be regulated by selective expression of receptors and coreceptors (Hong et al., 1999; Liu et al., 2005; Chauvet et al., 2007). While all cortical neurons express Npn1, PlexA4, and L1CAM mRNAs, we asked whether subcellular distribution of the receptors could alter responsiveness. Immunocytochemistry (Fig. 3 A–C) and mean fluorescence intensity measurements (data not shown) show that growth cones of Satb2pos and Satb2neg neurons express similar levels of Npn1, PlxA4, and L1CAM receptors.

Subunits of the Sema3A receptor complex are expressed at similar levels in cortical axonal growth cones. A–E , Confocal images showing immunolabeling for total levels of Npn1 ( A ), PlxA4 ( B ), L1CAM ( C ), and for surface levels of Npn1 ( D ) and L1CAM ( E ) in axonal growth cones from Satb2neg and Satb2pos neurons (nuclear Satb2 staining not shown). Top panels of each group show immunostaining in green as an overlay with F-actin in purple to demarcate growth cone. Bottom panels show an inverted black-and-white view of the immunolabeling only. Scale bar, 5 μm.
Although the total pool of receptors is similar, their availability on the growth cone surface may differ. However, surface labeling for Npn1 and L1CAM was also similar in Satb2pos and Satb2neg axons (Fig. 3 D,E). It remains possible that plexinA4 is differentially maintained at the surface, but we were unable to detect reliable plexinA4 labeling under nonpermeabilizing conditions.
Axonal internalization of Sema3A correlates with responsiveness
Sema3A induces endocytosis (Fournier et al., 2000; Jurney et al., 2002; Castellani et al., 2004; Piper et al., 2005), so we asked whether different cortical neuron populations could be differentiated based on their ability to internalize Sema3A. To examine Sema3A internalization directly, we tagged recombinant Sema3A-hFc with a Qdot-conjugated Fab fragment that recognizes the hFc tail. Neurons were incubated with Sema3A-Qdots, and the internalized pool was visualized selectively by stripping surface ligands with an acidic wash. Growth cones, dendrites, and cell bodies all show internalized fluorescent puncta, which are heterogeneous in shape and size. To compare populations, we used EUE to label layer 5 or layer 2/3 neurons and then quantified the level of internalized Sema3A in growth cones. E15-born neurons internalize twice the level of Sema3A-Qdots as E17-born neurons (Fig. 4 A,B,G). To confirm that the enhanced internalization capacity resides in layer 5 neurons lacking Satb2, neurons were immunolabeled post hoc for Satb2, Ctip2, or Tbr1. As expected, Satb2neg neurons internalize significantly more Sema3A than Satb2pos neurons (supplemental Fig. 2A,B, available at www.jneurosci.org as supplemental material), and these Satb2neg neurons are either corticospinal or corticothalamic projecting neurons as they express Ctip2 or Tbr1, respectively (supplemental Fig. 2C,D, available at www.jneurosci.org as supplemental material).

Sema3A internalization is distinct between deep and superficial layer neurons. A–F , Images show internalized Sema3A-Qdots (black) in masks of growth cones defined by YFP expression in neurons labeled by EUE (green). The top row ( A , C , E ) are growth cones from E15-born neurons and the bottom row, from E17-born neurons. Treatment conditions are indicated at the top of each column. Sema3A internalized/growth cone area by E15 EUE neurons ( A ) is greater than in growth cones of E17 EUE neurons ( B ) and prevented by filipin treatment. Scale bar: A (for A–F ), 10 μm. G , Bar graph compares internalization following MDC (dotted bars) and filipin (hatched bars) in E15- and E17-born neurons. E15-born neurons internalize significantly more Sema3A. For E15-born neurons, filipin significantly reduces internalization (one-way ANOVA, p = 0.02; n = 27 growth cones from 3 experiments). For E17-born neurons, both MDC and filipin significantly reduce internalization (one-way ANOVA, p = 0.002; n = 25 growth cones from 3 experiments). Bonferroni's multiple-comparison test was used to identify the sources of significant differences (*). H , Images of growth cone masks based on phalloidin-staining (blue) and surface bound Sema3A-Qdots (black) in Satb2neg and Satb2pos neurons. I , Bar graph shows that filipin treatment prevents growth cone collapse in response to Sema3A (one-way ANOVA, p = 0.001; n = 4 experiments; *significant difference in Bonferroni's post-test). J , Bar graph shows that MDC does not increase the collapse of growth cones in Satb2pos neurons exposed to Sema3A (n = 3 experiments; t test, p > 0.05; one-way ANOVA showed no differences between control and MDC groups). Error bars represent SEM.
Despite the similar distribution of Sema3A receptors in all cortical growth cones, it is possible that Satb2neg neurons can bind more Sema3A. To test this, Sema3A–Qdot binding was assessed in neurons that were cooled to 12°C, which prevents internalization. Satb2neg growth cones bind a modestly greater amount of Sema3A than Satb2pos growth cones, but this difference is not significant (1.0 ± 0.12 vs 0.71 ± 0.2, respectively; t test, p = 0.17; n = 9 per group). More notable is that surface-bound Sema3A appears more diffuse in Satb2neg neurons compared with Satb2pos neurons (Fig. 4 H). This observation suggests that Sema3A receptors might be differentially clustered and/or distributed within membrane domains of growth cones.
Raft-mediated endocytosis is the principal mode of Sema3A internalization in responsive growth cones
Mammalian cells employ distinct endocytic paths to compartmentalize cargo within contexts that are significant for downstream signaling, recycling, or degradation (Nichols and Lippincott-Schwartz, 2001; Pelkmans and Helenius, 2002; Conner and Schmid, 2003). To identify the route taken by Sema3A, we compared the two most prominent pathways: clathrin-mediated endocytosis (CME) and raft-mediated endocytosis (RME). We first confirmed that the two paths are present in cortical neurons by comparing internalization of rhodamine-tagged Transferrin, a marker for CME with FITC-tagged cholera toxin B, which binds GM1 gangliosides and is used to tag sphingolipid-enriched membrane rafts. As expected, there are endosomes tagged with Transferrin alone or Cholera toxin alone, in addition to a population tagged with both (Pelkmans et al., 2004) (supplemental Fig. 3, available at www.jneurosci.org as supplemental material).
We next tested the impact of RME or CME blockade on Sema3A internalization. In axonal growth cones of E15-born neurons, filipin, an inhibitor of RME, dramatically reduces levels of internalized Sema3A (Fig. 4 A,C,G), while MDC, an inhibitor of CME, has virtually no effect (Fig. 4 A,E,G). In contrast, in growth cones of E17-born neurons, which internalize much less Sema3A overall, both filipin and MDC decrease levels of internalized Sema3A, suggesting that both endocytic routes contribute equally (Fig. 4 B,D,F,G).
These findings suggest that RME may be responsible for the repelling effects of Sema3A. To test this, we assayed Sema3A-mediated growth cone collapse in neurons exposed to filipin. Consistent with a critical role for RME, filipin prevents collapse (Fig. 4 I) (Guirland et al., 2004).
Since CME contributes to Sema3A internalization in layer 2/3 neurons, we asked whether it counters the impact of RME. However, the data show that blocking CME in the less responsive Satb2pos neurons does not enhance Sema3A-mediated growth cone collapse (Fig. 4 J). Taken together, these data indicate that Sema3A internalization via RME is required for its repulsive effects in Satb2neg neurons.
Molecular mechanisms controlling the raft-mediated internalization of Sema3A
To identify proteins that could contribute to Sema3A internalization, we first assembled a list of proteins known to function downstream of Sema3A, consisting of Npn1, L1CAM, and PlxAs (Fig. 5 A, in red). Using Genes2networks (Berger et al., 2007), we used this seed list to find previously reported protein–protein interactions identified experimentally in mammalian cells that would “connect” the seed list proteins through additional intermediate proteins. This approach generated a list of potential one-step intermediate interactions between members of the seed list. The candidate proteins (Fig. 5 A, in yellow and blue) were ordered according to their calculated z-scores, which were computed based on the candidate protein links in the seed subnetwork compared with the candidate's known previously reported direct physical protein interactions (supplemental Table 1, available at www.jneurosci.org as supplemental material). The higher the z-score of a protein, the more likely the protein is to function specifically downstream of Sema3A. Of the 33 proteins identified having z-scores >2.5, two proteins stood out because of their known role in endocytosis: flotillin-1 and -2, also known as Reggie-2 and -1 (Bickel et al., 1997; Lang et al., 1998) (z-scores, 3.9 and 6.2, respectively). Flotillins are members of the SPFH domain family of proteins, which bind lipid rafts. Significantly, Glebov et al. (2006) have shown that in HeLa cells flotillins define and are required for a caveolin-independent RME pathway (Nichols and Lippincott-Schwartz, 2001; Frick et al., 2007).

Flotillin-1 and -2 are novel candidates downstream of Sema3A and are immunodetected in cortical axons in vivo and in vitro. A , In silico built network of Sema3A seed list (red) and novel candidate genes with high z-score (yellow) or lower z-score (blue). Flotillin-1 and -2 are magnified on the left. B–E , Confocal images of immunohistochemiical staining for flotillin-1 ( B , C ) and -2 ( D , E ) in E14.5 mouse cerebral cortex. Flotillins (green) are detected in phalloidin-labeled tracts and processes (in purple, red arrows) in the intermediate zone (IZ) and the internal capsule (IC). Boxed regions in A and D are shown in inverted black-and-white images in C and E . Scale bars: B , D , 200 μm; C , E , 100 μm. F , G , Confocal images of dissociated cortical neurons. Flotillin-1 ( F ), and -2 ( G ) (both green and black in adjacent, inverse image) are detected in small puncta in the filopodia and lamellipodia of axonal growth cones (delineated by F-actin labeling shown in purple) as well as axonal shafts. Scale bars, 5 μm.
To determine whether developing cortical axons express flotillins, we immunolabeled E14.5 neocortex for flotillin-1 or -2. Immunolabeling for both flotillins can be seen throughout the neocortex. The overlying pia and blood vessels show the highest levels of immunolabeling, while lower levels are observed throughout the cortical plate, intermediate zone, and ventricular zone. In the intermediate zone and internal capsule, where corticocortical and corticofugal axons travel, flotillin labeling colocalizes with F-actin-labeled fibers (Fig. 5 B–E). In axonal growth cones of dissociated neurons, where flotillin localization can be examined at a higher resolution, labeling is primarily punctate (Fig. 5 F,G).
Flotillin-1 responds to Sema3A stimulation
In non-neuronal cells, flotillins relocalize from smaller patches on the plasmalemma to larger and brighter clusters in endosomal compartments in response to epidermal growth factor (EGF) (Neumann-Giesen et al., 2007; Riento et al., 2009). Thus, to determine whether flotillins respond similarly to Sema3A, we used an imaging-based approach. In Satb2neg neurons, flotillin-1 appears less clustered than flotillin-2 in unexposed growth cones. In response to Sema3A, only flotillin-1 responds, becoming more clustered (Fig. 6 A). In axons from Satb2pos neurons, the distribution of both flotillins appears similar to Satb2neg growth cones, but neither changes in response to Sema3A (Fig. 6 A). These observations were analyzed quantitatively by plotting the mean intensity vs the area labeled in growth cones, and the data show that Sema3A selectively increases the appearance of low-to-medium-intensity clusters of flotillin-1 in Satb2neg neurons (Fig. 6 B).

Sema3A increases flotillin-1 clustering in growth cones of Satb2neg but not Satb2pos neurons. A , Confocal images of axonal growth cones from Satb2neg and Satb2pos neurons treated with control media or Sema3A and immunolabeled for flotillin-1 (green in overlay and black in adjacent, inverse image) and flotillin-2 (red in overlay and black in adjacent, inverse image) and stained with phalloidin (F-actin; blue). B , Histograms plot flotillin labeling/growth cone area relative to intensity for control (white circles) and Sema3A exposure (black circles). To reduce artifact, data acquisition was limited to intensities >30 arbitrary units (a.u.) and to better illustrate the relevant changes in the histogram, the upper limit was reduced to 170 a.u. (from 250 a.u.). In growth cones of Satb2neg neurons (left two graphs), the mean intensity of flotillin-1 clusters increases after stimulation with Sema3A (red arrow) (n = 25 growth cones from 3 experiments; p < 0.01, two-way ANOVA). Flotillin-2 clustering does not significantly change under the same conditions (p > 0.05). In growth cones of Satb2pos neurons (right two graphs), neither flotillin-1 nor -2 labeling changes after Sema3A exposure (n = 18 growth cones from 3 experiments; p > 0.05, two-way ANOVA). C , In growth cones of Satb2neg neurons treated with filipin, confocal images and analysis (as in A , B ) show that the mean intensity of flotillin-1 clusters is similar between control and Sema3A-treated neurons (n = 20 growth cones from 2 experiments; p > 0.05, two-way ANOVA). D , In growth cones of Satb2neg neurons treated with MDC, Sema3A is still capable of increasing the intensity/number of flotillin-1 puncta compared with control media (n = 17 growth cones from 2 experiments; p < 0.05, two-way ANOVA). Scale bar: A (for A , C , D ), 10 μm.
The Sema3A-stimulated increase in fluorescence is prevented by preincubation with filipin (Fig. 6 C) and is unchanged by MDC (Fig. 6 D), consistent with flotillin-1 recruitment to cholesterol-enriched membrane microdomains. Based on previous work, the increased flotillin-1 labeling most likely arises from the clustering of a pre-existing pool (Neumann-Giesen et al., 2007; Riento et al., 2009), but it is also possible that flotillin is newly synthesized. To test this, we assayed Sema3A-dependent clustering when protein translation was blocked by anisomycin. Anisomycin had no effect (supplemental Fig. 4A,B,E,F, available at www.jneurosci.org as supplemental material).
In HeLa cells, EGF-mediated flotillin recruitment requires the activation of Fyn kinase (Neumann-Giesen et al., 2007; Riento et al., 2009). Since Fyn activation is also essential for Sema3A function (Morita et al., 2006), we asked whether exposure to the Src family kinase inhibitor SU6656 would alter flotillin-1 recruitment in response to Sema3A. Flotillin-1, but not flotillin-2, acquires a diffuse expression in cortical axons in response to SU6656, and exposure to Sema3A only modestly increases its clustered appearance from this low baseline level (supplemental Fig. 4A–D, available at www.jneurosci.org as supplemental material). These data suggest that the higher capacity of Satb2neg neurons to internalize Sema3A via a raft-mediated pathway likely results from their ability to cluster flotillin-1 in a Src-dependent manner in response to Sema3A.
Flotillin-1 is required for Sema3A internalization
Since flotillin-1 responds robustly to Sema3A in Satb2neg neurons, we asked whether it is required for Sema3A internalization. To test this, we knocked down flotillin-1 using an equal mix of three shRNAs, each of which target different regions of the mRNA (see Materials and Methods) and together significantly decrease flotillin-1 levels to 48% of that in neurons expressing control shRNAs (supplemental Fig. 5A–C, available at www.jneurosci.org as supplemental material). Flotillin-1 knockdown significantly decreases Sema3A internalization in growth cones to 35% of control values (Fig. 7 A,B).

Flotillin-1 is required for the dynamin-independent, ERM-dependent endocytosis of Sema3A. A , Images show that flotillin knockdown reduces the amount of internalized Sema3A (in black) compared with growth cones expressing control shRNAs. Growth cone boundaries are delineated by masks (green). Addition of dynasore 5 min before and during incubation with Sema3A appears to modestly decrease uptake, but flotillin shRNA more dramatically decreases Sema3A endocytosis. B , Bar graph compares mean area internalized Sema3A per growth cone area, normalized to controls (n = 34 growth cones from 2 experiments, p = 0.0058, one-way ANOVA; *p < 0.05 in a Bonferroni post-test). C , NEz inhibits Sema3A internalization and flotillin knockdown does not add significantly to this effect. D , Bar graph as in B (n = 34 growth cones from 2 experiments; p = 0.0031, one-way ANOVA; *p < 0.05 in a Bonferroni post-test). Scale bar: A (for A , C ), 10 μm. Error bars represent SEM.
Previous work suggests that flotillin-defined RME does not require dynamin-mediated fission (Glebov et al., 2006). To test whether the RME pathway used by Sema3A is also dynamin independent, we treated neurons with a specific and rapidly acting small molecule inhibitor of dynamin, dynasore (Macia et al., 2006), and then quantified Sema3A-Qdot internalization. Dynasore treatment produced a modest, but insignificant, decrease in Sema3A internalization (Fig. 7 A,B, and data not shown), supporting that this pathway is largely independent of dynamin. When dynasore was combined with flotillin-1 shRNAs, there was a further, but not statistically significant, reduction in Sema3A internalization (Fig. 7 A,B). Together, these data support that Sema3A uses a flotillin-dependent, but dynamin-independent, endocytic pathway in growth cones from Satb2neg neurons.
We have previously shown that ERMs can also regulate internalization of Npn1 and L1CAM in response to Sema3A, most likely via their association with the intracellular tail of L1CAM (Mintz et al., 2008). To test whether flotillin-1-mediated endocytosis functions in the same pathway as ERMs, we compared Sema3A internalization in growth cones expressing an ERM dominant negative, NEz, which acts as a pan-ERM dominant negative (Algrain et al., 1993; Dickson et al., 2002), alone or together with flotillin-1 shRNAs. As expected, NEz greatly reduces Sema3A internalization (Fig. 7 C,D). Combined with flotillin knockdown, there is a similar reduction in Sema3A internalization. The absence of an additive effect suggests that flotillin and ERMs function in the same pathway controlling the magnitude of Sema3A internalization (Fig. 7 C,D).
Flotillin-1 mediates signaling to the cytoskeleton and axonal responsiveness to Sema3A
The data suggest that flotillin-1-mediated Sema3A internalization could be important for mediating the repulsive effects of Sema3A in axons from Satb2neg neurons. To test this, we assayed axonal growth inhibition in response to Sema3A in Satb2neg neurons expressing flotillin-1 shRNAs. The data show that axonal growth inhibition is prevented (Fig. 8 A,B). Flotillin-1 knockdown also significantly inhibited Sema3A-mediated growth cone collapse (57.8 ± 3% vs 38.5 ± 3%, respectively; n = 2 experiments; t test, p < 0.01), and in neurons growing on alternating stripes of Sema3A and control substrates, flotillin-1 knockdown produced a modest increase in the frequency of border crossings per axon (1.7 ± 0.4 vs 2.1 ± 0.3, respectively; n = 2 experiments; 15 per group).

Flotillin-1 knockdown decreases Sema3A-mediated axon responses and LIMK activation. A , Inverted fluorescence microscope images of Satb2neg neurons (Satb2 labeling not shown) transfected with either control or Flot1 shRNAs and grown in control or Sema3A media for 72 h. Scale bar, 80 μm. B , Quantitative comparison of total axon length normalized to control data (tracings of 25 neurons transfected with control shRNA (*p = 0.0475, t test) and 32 neurons transfected with Flot1 shRNA mix (p = 0.8673, t test). C , D , Sema3A stimulation results in a nearly threefold increase in the levels of detectable T508-phosphorylated LIMK1 (purple) in Satb2neg growth cones expressing a control shRNA. Scale bar, 10 μm. In growth cones expressing flotillin-1 shRNAs, the increase is attenuated and not significant (n = 36 growth cones from 2 experiments; **p = 0.0022, one-way ANOVA and Bonferroni post-test). E , The levels of total LIMK do not change significantly (p = 0.4). Error bars represent SEM.
To mediate repulsion, flotillin-based RME would be anticipated to initiate signaling pathways to the growth cone cytoskeleton. It has been shown previously that Sema3A-mediated growth cone collapse in dorsal root ganglia requires LIMK-1-mediated inactivation of cofilin (Aizawa et al., 2001). LIM kinases are positively regulated by phosphorylation, so we first asked whether Sema3A stimulates LIMK phosphorylation in growth cones from Satb2neg cortical neurons. As expected, 5 min exposure to Sema3A increases phospho-LIMK labeling by nearly threefold, while showing no change in labeling for total LIMK (Fig. 8 C–E). Flotillin-1 knockdown reduces this response by a third. Together, the data show that in presumptive corticofugal axons, flotillin-1 is required for the internalization of Sema3A-generating endosomes that signal to the cytoskeleton via LIMK activation and thus can initiate axonal responsiveness.
Discussion
While it has long been appreciated that axons can take different trajectories while growing in a similar environment (Dodd and Jessell, 1988; Fishell and Hanashima, 2008), the cellular mechanisms that drive such differential responses are poorly understood. Here, we show that cortical neurons grown in culture retain their cell type-specific identities and thus, can be used as a tractable model system to investigate the mechanisms mediating cell type-selective responses. Using this system, we show that axons extending from cortical neurons lacking the transcription factor Satb2 respond more robustly to Sema3A in a variety of assays than those that express Satb2. This differential responsiveness cannot be accounted for by differences in Sema3A receptor expression. Rather, the data show that axons from Satb2neg neurons use RME to internalize more Sema3A. We identify flotillin-1 as a Sema3A signaling intermediate that defines this raft-mediated pathway, and show that it can regulate LIMK activity and is essential for generating Sema3A responses in cortical neurons.
Response selectivity
Axonal growth cones employ a variety of mechanisms to mediate spatial, temporal, and cell type-specific responses. Most previously identified mechanisms regulate the number or type of receptors available on the surface. For example, PKC activation in hippocampal neurons leads to the selective internalization of the UNC5H1 receptor for Netrin, converting Netrin-based repulsion to attraction (Williams et al., 2003); in the developing spinal cord, commissural axons increase their expression of Robo1 receptors and become sensitive to the repelling effects of midline Slits after crossing the midline (Kidd et al., 1998; Zou et al., 2000; Reeber et al., 2008). In contrast, cortical axons express similar levels of Sema3A receptors but use different modes of internalization.
In many cases, cell type specificity or graded responses can be directly attributed to the selective or graded expression of the transcription factors that control the expression of receptors or ligands (Barbieri et al., 2002; Mui et al., 2002; Herrera et al., 2003; Pak et al., 2004; Polleux et al., 2007; Lee et al., 2008). While we show that Satb2 expression correlates with decreased sensitivity, and Ctip2 overexpression increases axon sensitivity to Sema3A, we also provide data showing similar levels of Npn1, L1CAM, and PlxA4 receptors in all cortical axons. Since we observe that membrane-bound Sema3A is more diffuse in axons of Satb2neg neurons, it is possible that in responsive axons, receptors form combinations at the plasma membrane that are more likely to reside in lipid rafts and/or be internalized via RME. These data suggest that Satb2 and Ctip2 regulate the expression of proteins affecting plasma membrane organization, of binding proteins that alter receptor distribution within membrane microdomains, or of signaling proteins that act downstream of receptor binding. It will be important in future experiments to identify the relevant transcriptional targets.
Sema3A has been shown previously to repel cortical axons (Bagnard et al., 1998; Polleux et al., 1998), but before the current study it was not clear how different cortical populations respond to this cue. Knockout studies for Sema3A and its receptors offer valuable but sometimes confounding information on cortical axon guidance (Behar et al., 1996; Dahme et al., 1997; Taniguchi et al., 1997; Demyanenko et al., 1999; Gu et al., 2003; Suto et al., 2005; Yaron et al., 2005; Sibbe et al., 2007). In these models, the manipulation is broad and chronic, allowing for non-cell-autonomous effects and for homeostatic compensation. These shortcomings have made it difficult to parse roles for particular guidance cues in generating structures that rely on multiple simultaneous cues (like the neocortex). Thus, while it has been demonstrated that cortical axons are attracted by Netrin-1 (expressed in the basal telencephalon) and repelled by Semaphorin 5B (ventricular zone), Draxin (cortical plate), and Slit1 and Slit2 (basal telencephalon) (Metin et al., 1997; Richards et al., 1997; Bagri et al., 2002; Lett et al., 2009), the selective influence of these cues on molecularly defined neuronal populations at different stages of development is poorly understood.
Endocytosis
In general, cells use several distinct modes of endocytosis to efficiently control the spatial and temporal propagation of signals from the plasma membrane to intracellular effectors. There is substantial evidence for a large number of endocytic paths that differ in their targets, the effectors and adaptors used, and their regulation (Pelkmans et al., 2001; Pelkmans and Helenius, 2002; Yap et al., 2008b). In retinotectal axons, CME is required for growth cone adaptation to Netrin-1 and to Sema3A, and for responsiveness to Ephrins (Cowan et al., 2005; Piper et al., 2005). The function of RME in axon guidance has received comparatively little attention, although, significantly, localization within membrane microdomains can segregate guidance cue response pathways (Marquardt et al., 2005), and guidance cue receptors, including Npn1, can translocate to lipid rafts following ligand binding, an event that is essential for the appropriate response (Guirland et al., 2004; Ibanez, 2004).
Axonal growth cones of Satb2neg neurons internalize Sema3A via a pathway that is dependent on lipid raft integrity and on expression of flotillin-1, a resident protein of rafts that actively participates in membrane bending and internalization, independent of clathrin, dynamin, and caveolin-1 (Glebov et al., 2006). This mechanism is inherently adaptable as it permits neurons to intrinsically control signaling downstream of a guidance cue by an internalization switch. This strategy would allow neurons to generate relevant signals in distinct cellular compartments and at different developmental stages, and therefore to employ the same cues and receptors for different outcomes. Satb2pos neurons offer an excellent example. While their axons do not respond to Sema3A, they probably employ Npn1 receptors to respond to Sema3C (Piper et al., 2005; Niquille et al., 2009). At the same time, their cell bodies and apical processes use Npn1 for radial migration in the cortical plate, most likely in response to Sema3A secreted by the marginal zone (Chen et al., 2008b). It will be interesting to determine whether internalization of Npn1 is required for these additional functions.
Relationship between RME and CME
In previous work, we showed that ERM proteins could regulate L1CAM and Npn1 internalization in cortical axons (Mintz et al., 2008), and in Cos cells it has been shown that Npn1 internalization requires L1CAM (Castellani et al., 2004). Since the binding domains for ERMs and the clathrin adapter AP2 on L1CAM partially overlap (Kamiguchi et al., 1998; Cheng et al., 2005; Sakurai et al., 2008), our interpretation was that ERMs most likely prevent CME. However, despite many attempts, we did not detect competition between the binding of ERM and AP2 to L1CAM, and the current data show that ERMs function in the same pathway as flotillin-1. In non-neuronal cells ERM proteins can associate with lipid rafts and control the translocation of their transmembrane partners to these membrane domains (Gupta et al., 2006; Chakrabandhu et al., 2007), suggesting that ERMs function upstream of flotillin-1 to promote Npn1 translocation to lipid rafts (Guirland et al., 2004).
Sema3A can also induce CME (Piper et al., 2005), and blocking CME with MDC reduces Sema3A-mediated growth cone collapse in cortical neurons (Mintz et al., 2008). However, since we do not detect significant uptake of Sema3A via the CME pathway in responsive growth cones, we hypothesize that Sema3A engages CME indirectly. Cross talk between RME and CME has been described in other cell types (Pelkmans et al., 2004), and while speculative, our data fit a model (Fig. 9) in which RME of Sema3A leads to LIMK-1 activation, Cofilin inactivation (Aizawa et al., 2001), and the stabilization of a pool of subplasmalemmal F-actin necessary for CME (Merrifield et al., 2002; Römer et al., 2010). CME could remove adhesive sites, an event that precedes the plexin-initiated depolymerization of F-actin, but is equally important for growth cone detachment during retraction (Mikule et al., 2002; Terman et al., 2002; Hung et al., 2010).

Model for the role of RME in the response of corticofugal axons to Sema3A. Sema3A stimulation triggers the recruitment of its receptors to lipid rafts (Guirland et al., 2004) and their consequent internalization in flotillin-1-positive endosomes. This step leads to the activation of LIMK, which inactivates cofilin-1. Cofilin-1 regulation of the actin cytoskeleton stimulates CME of adhesion sites (Chao and Kunz, 2009), allowing the detachment and retraction of the growth cone.
Footnotes
-
This work was supported by National Institutes of Health Grants NS050634 and AA014898.
- Correspondence should be addressed to Deanna L. Benson, Fishberg Department of Neuroscience, Box 1065, The Mount Sinai School of Medicine, 1425 Madison Avenue, New York, NY 10029. deanna.benson{at}mssm.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}