





Insulin resistance in type 2 diabetes is due to impaired stimulation of the glucose transport system in muscle and fat. Different defects are operative in these two target tissues because glucose transporter 4 (GLUT 4) expression is normal in muscle but markedly reduced in fat. In muscle, GLUT 4 is redistributed to a dense membrane compartment, and insulin-mediated translocation to plasma membrane (PM) is impaired. Whether similar trafficking defects are operative in human fat is unknown. Therefore, we studied subcellular localization of GLUT4 and insulin-regulated aminopeptidase (IRAP; also referred to as vp165 or gp160), which is a constituent of GLUT4 vesicles and also translocates to PM in response to insulin. Subcutaneous fat was obtained from eight normoglycemic control subjects (body mass index, 29 ± 2 kg/m2) and eight type 2 diabetic patients (body mass index, 30 ± 1 kg/m2; fasting glucose, 14 ± 1 mm). In adipocytes isolated from diabetics, the basal 3-O-methylglucose transport rate was decreased by 50% compared with controls (7.1 ± 2.9 vs. 14.1 ± 3.7 mmol/mm2 surface area/min), and there was no increase in response to maximal insulin (7.9 ± 2.7 vs. 44.5 ± 9.2 in controls). In membrane subfractions from controls, insulin led to a marked increase of IRAP in the PM from 0.103 ± 0.04 to 1.00± 0.33 relative units/mg protein, concomitant with an 18% decrease in low-density microsomes and no change in high-density microsomes (HDM). In type 2 diabetes, IRAP overall expression in adipocytes was similar to that in controls; however, two abnormalities were observed. First, in basal cells, IRAP was redistributed away from low-density microsomes, and more IRAP was recovered in HDM (1.2-fold) and PM (4.4-fold) from diabetics compared with controls. Second, IRAP recruitment to PM by maximal insulin was markedly impaired. GLUT4 was depleted in all membrane subfractions (43–67%) in diabetes, and there was no increase in PM GLUT4 in response to insulin. Type 2 diabetes did not affect the fractionation of marker enzymes. We conclude that in human adipocytes: 1) IRAP is expressed and translocates to PM in response to insulin; 2) GLUT4 depletion involves all membrane subfractions in type 2 diabetes, although cellular levels of IRAP are normal; and 3) in type 2 diabetes, IRAP accumulates in membrane vesicles cofractionating with HDM and PM under basal conditions, and insulin-mediated recruitment to PM is impaired. Therefore, in type 2 diabetes, adipocytes express defects in trafficking of GLUT4/IRAP-containing vesicles similar to those causing insulin resistance in skeletal muscle.
PERIPHERAL INSULIN RESISTANCE is a key factor in the pathogenesis of type 2 diabetes mellitus (1) and involves defects in the glucose transport system in both skeletal muscle (2) and adipose tissue (3). Insulin stimulates glucose transport via net translocation of glucose transporter 4 (GLUT4) proteins from an intracellular depot to the cell surface. In fat cells, GLUT4 expression is markedly reduced in type 2 diabetes and obesity (4–6). Although other defects may exist in insulin action pathways, GLUT4 depletion appears to be the major functional cause of insulin resistance in fat due to the profound degree of GLUT4 loss and its central role in transport stimulation. However, in skeletal muscle, GLUT4 levels were found to be normal in multiple insulin-resistant disease states, including obesity, type 2 diabetes, impaired glucose tolerance, and gestational diabetes (7, 8). More recent data have indicated that insulin resistance in skeletal muscle is the result of impaired translocation of intracellular GLUT4 to sarcolemma (9–11). Such a translocation defect could arise either from defects in insulin signal transduction (12) or defects in GLUT4 traffic/targeting that are intrinsic to the glucose transport effector system.
We have recently obtained evidence that defects within the glucose transport apparatus contribute to impaired GLUT4 translocation in insulin-resistant patients. In skeletal muscle from insulin-resistant subjects, GLUT4 was redistributed to dense membrane vesicles under basal conditions (11). Furthermore, accumulation of GLUT4 in this dense membrane compartment was associated with a marked impairment in translocation in response to insulin. A similar pattern of abnormalities was observed in insulin-resistant omental adipocytes from women with gestational diabetes (13). A much greater proportion of cellular GLUT4 was localized in membrane vesicles[ (cofractionating with plasma membrane (PM) and high-density microsomes (HDM)] in basal cells compared with that in nondiabetic gravidas, and there was little or no GLUT4 recruitment from microsomes to PM in response to insulin. In both muscle and adipocytes, the abnormal subcellular distribution of GLUT4 that was observed under basal conditions could not be explained by abnormalities in insulin signal transduction. On the basis of these data, we proposed that human insulin resistance involves a defect in GLUT4 traffic/targeting leading to accumulation in a dense membrane compartment under basal conditions and that this abnormality is linked to the inability of insulin to recruit GLUT4 to the cell surface.
In type 2 diabetes, then, the current notion is that different mechanisms of insulin resistance are operative in muscle, where defects in GLUT4 trafficking/translocation and signaling predominate, and fat, where pretranslational mechanisms markedly reduce GLUT4 expression. However, whether fat also expresses defects in GLUT4 vesicle trafficking/translocation in type 2 diabetes has not been studied. One consideration is that the profound reduction in GLUT4 expression could make it difficult to accurately study changes in subcellular localization (4). These studies were less problematic in omental adipocytes in gestational diabetes, which contain a normal cellular complement of GLUT4 in approximately half of these patients (13). To help address this issue in type 2 diabetes, we studied both GLUT4 and a zinc-dependent membrane aminopeptidase, referred to as insulin-regulated aminopeptidase (IRAP), vp165 (14), or gp160 (15), which is another protein constituent of the intracellular vesicles that sequester GLUT4 in muscle and fat cells. In rat adipocytes, IRAP colocalizes in GLUT4 vesicles and is translocated to the PM in response to insulin (14–21). In the current study, we assessed subcellular localization, expression, and translocation of IRAP and GLUT4 in human adipocytes. Our goal was to examine whether IRAP could be used to identify abnormalities in subcellular localization of GLUT4/IRAP-containing vesicles in adipocytes from type 2 diabetic patients with low GLUT4 expression.
Subjects and Methods
Experimental subjects
The clinical characteristics of the study group are listed in Table 1. The study population consisted of 16 subjects, including a normoglycemic control subgroup with 8 subjects (5 female and 3 male; mean age, 32 yr) and a subgroup of 8 type 2 diabetic patients (4 female and 4 male; mean age, 40 yr). Two parameters demonstrated that the degree of adiposity was similar between the control and diabetic subgroups. First, body mass index was similar (P = NS) in control subjects (29 ± 2 kg/m2) and in patients with type 2 diabetes (30 ± 1 kg/m2). Second, mean adipocyte size, assessed in isolated sc adipocytes that were obtained by biopsy from the anterior abdominal wall (see below), was comparable (P = NS) in controls (625 ± 143 pl) and type 2 diabetic patients (612 ± 40 pl). Each member of the control subgroup was demonstrated to have normal glucose tolerance after a standard 75-g oral glucose tolerance test (data not shown). The type 2 diabetic patients were overtly hyperglycemic. Two of the diabetics were newly diagnosed, and six were treated with insulin but had been withdrawn from all exogenous insulin and followed as outpatients for 3 weeks at the time of study. All subjects studied were chemically and/or clinically euthyroid and without renal, hepatic, or cardiac disease. The study was approved by the Institutional Review Board, and written informed consent was obtained from all subjects.
Clinical characteristics
| Subgroups | Gender | Age (yr) | BMI (kg/m2) | Fasting glucose (mm) | Fasting insulin (pm) | Adipocyte mean volume (pl) |
|---|---|---|---|---|---|---|
| Controls | 5F /3M | 32 ± 2 | 29 ± 2 | 5.2 ± 0.2 | 84 ± 18 | 625 ± 143 |
| Type 2 diabetics | 4F /4M | 40 ± 3 | 30 ± 1 | 13.8 ± 1.4 | 90 ± 24 | 612 ± 40 |
| Subgroups | Gender | Age (yr) | BMI (kg/m2) | Fasting glucose (mm) | Fasting insulin (pm) | Adipocyte mean volume (pl) |
|---|---|---|---|---|---|---|
| Controls | 5F /3M | 32 ± 2 | 29 ± 2 | 5.2 ± 0.2 | 84 ± 18 | 625 ± 143 |
| Type 2 diabetics | 4F /4M | 40 ± 3 | 30 ± 1 | 13.8 ± 1.4 | 90 ± 24 | 612 ± 40 |
F, Females; M, males.
Clinical characteristics
| Subgroups | Gender | Age (yr) | BMI (kg/m2) | Fasting glucose (mm) | Fasting insulin (pm) | Adipocyte mean volume (pl) |
|---|---|---|---|---|---|---|
| Controls | 5F /3M | 32 ± 2 | 29 ± 2 | 5.2 ± 0.2 | 84 ± 18 | 625 ± 143 |
| Type 2 diabetics | 4F /4M | 40 ± 3 | 30 ± 1 | 13.8 ± 1.4 | 90 ± 24 | 612 ± 40 |
| Subgroups | Gender | Age (yr) | BMI (kg/m2) | Fasting glucose (mm) | Fasting insulin (pm) | Adipocyte mean volume (pl) |
|---|---|---|---|---|---|---|
| Controls | 5F /3M | 32 ± 2 | 29 ± 2 | 5.2 ± 0.2 | 84 ± 18 | 625 ± 143 |
| Type 2 diabetics | 4F /4M | 40 ± 3 | 30 ± 1 | 13.8 ± 1.4 | 90 ± 24 | 612 ± 40 |
F, Females; M, males.
Materials and methods
Preparation of adipocytes and subcellular membrane fractions. All subjects were nihil per os after midnight, and in the morning, sc adipose tissue was obtained by open biopsy of the anterior abdominal wall using local anesthesia, as previously described (4, 5). The biopsy procedure yielded approximately 20 g of sc fat, which was placed immediately in a Krebs-Ringer phosphate buffer (buffer A) at pH 7.4 containing 20 mm HEPES, 2.5 mm NaH2PO4, 4% BSA (fraction V), and 5 mmd-glucose. The specimens were brought quickly to laboratory, trimmed of connective tissue, and weighed. Subcutaneous fat from each subject was minced with fine scissors, and isolated adipocytes were obtained by gently shaking the tissue in polypropylene containers at 37 C for 1 h in buffer A containing 1.5 mg/ml collagenase (Cooper, Freehold, NJ), as described previously (4, 5, 13). Then, the adipocytes were filtered and washed in buffer A without glucose and collagenase, and a small aliquot was removed for immediate cell sizing, counting, and glucose transport assays. In each subject, the adipocyte diameter of 200 cells was measured using a cell-counting chamber and eyepiece micrometer, and the mean value was used to calculate adipocyte volume, assuming that the cells were spherical. Adipocyte counts were performed in a hemocytometer, and cell viability assessed by trypan blue exclusion was greater than 98%.
Isolated adipocytes from each subject were used to study the subcellular localization of GLUT4 and IRAP under basal and insulin-stimulated conditions. Equal portions of isolated cells were incubated in buffer A (37 C) in the absence and presence of 16.7 nm insulin (100 ng/ml) for 30 min; these conditions maximally stimulate the glucose transport system in adipocytes from humans with and without type 2 diabetes (3). Then, the adipocytes were homogenized at 17 C in pH 7.4 buffer B containing 20 mm HEPES, 255 mm sucrose, 1 mm EDTA, and protease inhibitors including 5 μg/ml leupeptin, 1 μg/ml pepstatin, 5 μg/ml aprotinin, and 400 μm phenylmethylsulfonyl fluoride in a glass homogenizer with Teflon pestle (Arthur H. Thomas Co., Philadelphia, PA). PM, low-density microsome (LDM), and HDM subfractions were prepared from both basal and insulin-stimulated adipocytes using a differential ultracentrifugation protocol as described previously (4, 5, 13). The resulting PM and microsomal pellets were resuspended in buffer B (1–3 mg protein/ml) and stored frozen at −80 C. To assess the yield and purity of subfractions, organelle marker enzymes were measured in homogenate, PM, HDM, and LDM, including 5′-nucleotidase, using the method of Avruch and Wallach (22); uridine 5′-diphosphate-galactose/N-acetylglucosamine galactosyl-transferase, using the method of Fleisher (23); and rotenone-insensitive nicotinamide adenine dinucleotide-cytochrome c reductase, according to Dallner et al. (24), as described previously (4, 5, 13, 25, 26).
Glucose transport assays. Intact isolated adipocytes were incubated in the absence and presence of 16.7 nm insulin (100 ng/ml) for 45 min at 37 C. Initial rates of 3-O-methylglucose uptake were assayed then by a modification of the method of Whitesell and Gliemann (27) as described previously by Foley et al. (28). In each experiment, glucose transport rate was assessed as the mean of quadruplicate determinations, and the distribution space for radiolabeled 3-O-methylglucose in the presence of 0.3 mm phloretin was used to correct for nonspecific carryover of radioactivity with cells and the uptake of hexose by simple diffusion.
Immunoblot analysis. Membrane proteins (100 μg) were solubilized in Laemmli sample buffer (29), resolved by SDS-PAGE on gels containing 6% polyacrylamide, and then electrophoretically transferred to nitrocellulose filters (30). Immunological detection of GLUT4 and IRAP was accomplished as described previously (4, 8, 11, 13). The GLUT4-specific antibody was raised in rabbits against the 12-amino acid carboxyl-terminal of rat GLUT4 (East Acres Biologicals, Southbridge, MA) and was used at 1:400 dilution. Nitrocellulose filters (immunoblots) were incubated with GLUT4 antibodies followed by 125I-protein A. The IRAP (or vp165) affinity-purified antibody was raised in rabbits against the N-terminal 109 amino acids (cytoplasmic domain) of rat IRAP as a GST-fusion protein (14). Immunoreactivity for IRAP was identified by an enhanced chemiluminescence system (Amersham Pharmacia Biotech, Arlington Heights, IL). Immunoreactive proteins were quantified by autoradiography and scanning densitometry (Molecular Dynamics, Inc., Sunnyvale, CA). Parallel experiments were performed to show that each signal in individual subjects was in the linear range of correlation between signal density and amount of protein loaded on gels. A quantitative comparison among the different gels was made possible by including five internal standard lanes on each gel; these lanes contained total postnuclear membrane samples (100 μg) from two human fat biopsies and 12.5, 50, and 100 μg of total rat muscle membrane proteins.
Other assays and statistics. Plasma glucose was measured by the glucose oxidase method with a glucose analyzer (YSI 2300, YSI, Inc., Yellow Springs, OH). Serum insulin levels were measured using a microparticle enzyme immunoassay kit (Abbott Diagnostics, Chicago, IL). Protein was measured by the Coomassie brilliant blue method described by Bradford (31) with crystalline BSA as the standard. Data are means ± se. When comparing results in controls and diabetics, statistical significance was determined using the t test; when comparing three or more means, significance was established using ANOVA and Fisher protected least significant difference test for pairwise differences.
Results
Glucose transport activity
In control and type 2 diabetic subjects, basal and maximally insulin-stimulated rates of 3-O-methylglucose transport were measured in sc fat adipocytes. Mean values for each subject group are shown in Fig. 1 normalized per cell surface area. When compared with normoglycemic control subjects, the basal glucose transport rate per cell surface area was decreased by approximately 50% in type 2 diabetic patients. With maximal insulin stimulation, there was a 3.2-fold increase over basal in controls (from 14.1 ± 3.7 to 44.5 ± 9.2 mmol/mm2·min; P < 0.01), but there was no change in diabetic patients (P = NS). These data indicate that the glucose transport system is resistant to insulin in type 2 diabetes.
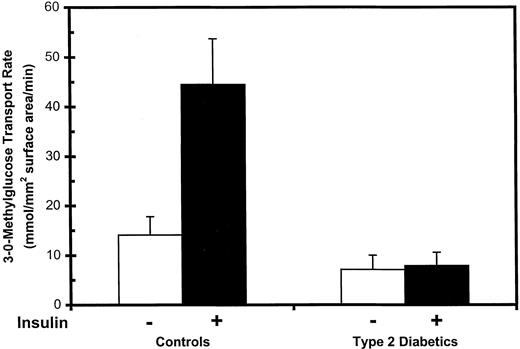
Glucose transport rates in isolated human adipocytes. Subcutaneous abdominal wall adipose tissue was obtained by open biopsy from normoglycemic controls and untreated patients with type 2 diabetes. Isolated adipocytes were prepared, and initial rates of 3-O-methyglucose uptake were measured under basal conditions and after stimulation by maximal insulin. Transport rates were normalized per cell surface area, and data are the means ± se of eight controls and eight type 2 diabetics.
Subcellular localization of GLUT4/IRAP
Adipocytes from control and type 2 diabetic subjects were incubated in the absence and presence of a maximal insulin concentration, homogenized, and processed by differential centrifugation to obtain PM, LDM, and HDM subcellular membrane fractions. To assess subcellular localization of IRAP and GLUT4, membrane fractions were subjected to SDS-PAGE and immunoblot analyses. For illustration, Fig. 2 shows an immunoblot for IRAP in membrane subfractions from a control subject, and mean data in controls and diabetics are shown in Fig. 3, A and B, for GLUT4 and IRAP, respectively. In control subjects, insulin stimulation increased PM GLUT4 from 0.49 ± 0.23 U/mg protein in basal cells to 1.00± 0.33 U/mg, concomitant with a 32% reduction in LDM (from 1.92± 0.44 to 1.31 ± 0.41 U/mg; P < 0.05). In type 2 diabetes, GLUT4 levels were markedly depleted in both PM and LDM, and there was no significant translocation from LDM to PM in response to insulin (PM and LDL levels in insulin-treated cells were similar to respective levels in basal cells; P = NS).
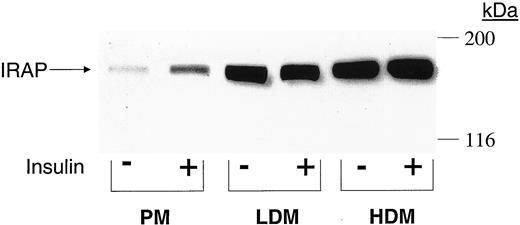
Immunological detection of IRAP in human adipocyte membrane subfractions. Basal and insulin-stimulated adipocytes from a control subject were homogenized and processed to obtain PM, HDM, and LDM subfractions. Equal amounts of membrane protein (100 μg) were resolved by SDS-PAGE, transferred to filters, and reacted with anti-IRAP antibodies, followed by an enhanced chemiluminescence detection system. Migration of molecular weight markers is shown on the right in kilodaltons. The figure is a typical autoradiogram.
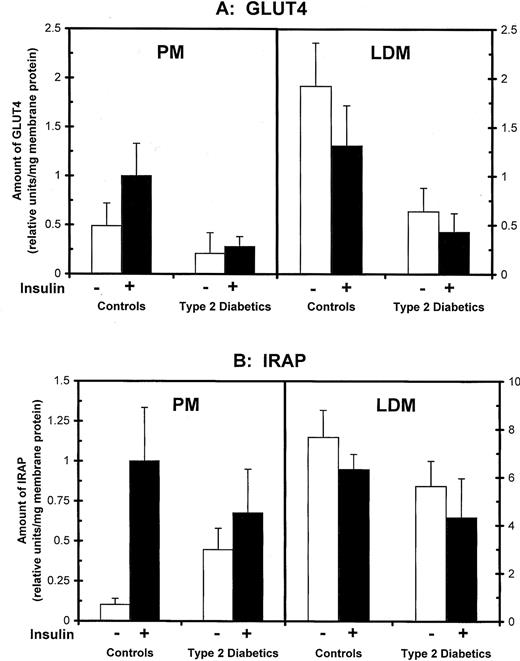
Effects of type 2 diabetes on GLUT4 glucose transporters and IRAP in adipocyte membrane fractions. Isolated adipocytes were obtained from control and type 2 diabetic subjects, incubated in the absence and presence of maximal insulin, and processed to obtain PM and LDM fractions. Membrane proteins were analyzed on immunoblots using GLUT4- (A) and IRAP- (B) specific antibodies and quantified by autoradiography and densitometry. Data are the means± se of eight controls and eight type 2 diabetic patients.
The cellular content of IRAP was unaffected by diabetes. IRAP levels in adipocyte homogenates were similar in control and diabetic subgroups (P = NS; data not shown). In controls, insulin stimulation led to a 10-fold increase in PM IRAP (from 0.103 ± 0.037 to 1.000 ± 0.334; P < 0.01) concomitant with a 17.5% decrease in the LDM fraction (P = NS), as shown in Fig. 3B. The pattern in type 2 diabetes was different in two aspects. First, in basal cells, the amount of IRAP recovered in the PM fraction was 4.3 times higher in diabetics (P < 0.01) compared with control subjects (0.447 ± 0.133 vs. 0.103 ± 0.037, respectively). Second, the ability of maximal insulin to increase IRAP in PM was abolished (P = NS). Both the fold increase and absolute increase in PM IRAP was assessed in individual subjects, and the mean values in type 2 diabetes were minimal in comparison with the control subgroup (Fig. 4, A and B).
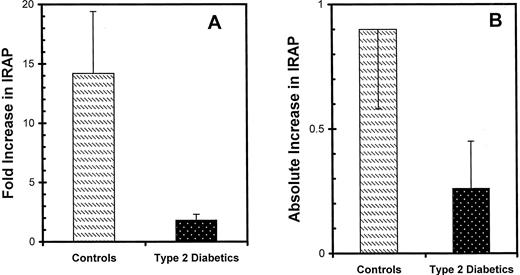
Effects of type 2 diabetes on the ability of insulin to increase IRAP in the PM in human adipocytes. PM subfractions were obtained from isolated adipocytes that were preincubated in the absence and presence of insulin. PM IRAP was quantified by immunoblot analysis as described for Fig. 3. Both the fold increase above basal (A) and the absolute increase over basal (B) were calculated in individual subjects, and the data are the means ± se in eight controls and eight type 2 diabetics.
In basal cells, greater recovery of IRAP in the PM fraction could represent either more IRAP in cell surface membranes or redistribution of IRAP to intracellular vesicles that cofractionate with PMs. Although the data do not rigorously address which of these alternatives is true, we favor the latter possibility based on findings in the HDM fraction, shown in Fig. 5, A and B. As was observed in PM, more IRAP was recovered in this microsomal fraction in basal cells from diabetic patients (8.4 ± 0.7) than in controls (7.0 ± 1.1), although the difference did not achieve statistical significance. More importantly, it was clear that cellular depletion of GLUT4 in diabetes did not involve all cellular compartments equally. GLUT4 in HDM was reduced by only 43% in type 2 diabetes compared with controls (Fig. 5B), and this was less than the 60% decrement in PM and 67% decrement in LDM (Fig. 3B). Thus, in the context of overall cellular GLUT4 depletion, there appeared to be a relative redistribution of GLUT4 (and possibly IRAP) to denser microsomal membranes in type 2 diabetes.
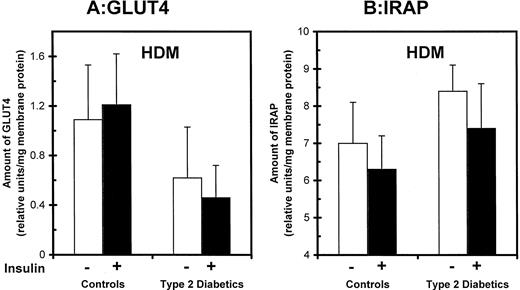
Effects of type 2 diabetes on GLUT4 and IRAP levels in high-density microsome (HDM) subfractions in human adipocytes. HDMs were obtained from isolated adipocytes that were preincubated in the absence and presence of maximal insulin. Relative levels of GLUT4 (A) and IRAP (B) were quantified by immunoblot analyses as described for Fig. 3. The data are the means ± se in eight controls and eight type 2 diabetics.
Differences in the subcellular localization of GLUT4 or IRAP could not be explained on the basis of nonspecific changes in the fractionation of cells or in the recovery of membrane protein. We measured specific activities of 5′-nucleotidase as a marker for PMs (22), galactosyl-transferase as a marker for the Golgi apparatus (23), and cytochrome c reductase as a marker for endoplasmic reticulum (24) in the cell homogenate and in PM, HDM, and LDM fractions. The degree to which marker enzymes were enriched (over homogenate) in each of the membrane subfractions was comparable between controls and diabetics and in basal and insulin-treated cells, as shown in Table 2. Therefore, as we have repeatedly demonstrated in previous studies (4, 5, 13, 25, 26), neither diabetes nor acute insulin stimulation alters the fractionation of these organelle markers in adipocytes. These data indicate that alterations in subcellular localization are specific for IRAP and GLUT4.
Distribution of organelle marker enzyme activities in membrane subfractions from human adipocytes
| Membrane fraction | Fold increase in specific activity over homogenatea | ||
|---|---|---|---|
| 5′-Nucleotidase | Galactosyl-transferase | Cytochrome c reductase | |
| Controls | |||
| PM | 7.8 ± 2.6 | 1.4 ± 0.4 | 0.5 ± 0.1 |
| LDM | 1.3 ± 0.4 | 8.8 ± 1.9 | 3.8 ± 0.9 |
| HDM | 3.3 ± 1.3 | 4.3 ± 1.0 | 4.5 ± 0.8 |
| Type 2 diabetics | |||
| PM | 8.9 ± 3.2 | 1.3 ± 0.4 | 0.7 ± 0.1 |
| LDM | 2.5 ± 0.7 | 6.7 ± 1.1 | 3.8 ± 1.0 |
| HDM | 5.0 ± 1.2 | 3.3 ± 0.1 | 4.6 ± 0.8 |
| Membrane fraction | Fold increase in specific activity over homogenatea | ||
|---|---|---|---|
| 5′-Nucleotidase | Galactosyl-transferase | Cytochrome c reductase | |
| Controls | |||
| PM | 7.8 ± 2.6 | 1.4 ± 0.4 | 0.5 ± 0.1 |
| LDM | 1.3 ± 0.4 | 8.8 ± 1.9 | 3.8 ± 0.9 |
| HDM | 3.3 ± 1.3 | 4.3 ± 1.0 | 4.5 ± 0.8 |
| Type 2 diabetics | |||
| PM | 8.9 ± 3.2 | 1.3 ± 0.4 | 0.7 ± 0.1 |
| LDM | 2.5 ± 0.7 | 6.7 ± 1.1 | 3.8 ± 1.0 |
| HDM | 5.0 ± 1.2 | 3.3 ± 0.1 | 4.6 ± 0.8 |
Bold numbers correspond to the membrane fraction that contains the highest degree of enrichment for the respective organelle marker enzyme. 5′-Nucleotidase is a marker for PM, galactosyl-transferase is a marker for the Golgi apparatus, and cytochrome c reductase is a marker for endoplasmic reticulum.
Distribution of organelle marker enzyme activities in membrane subfractions from human adipocytes
| Membrane fraction | Fold increase in specific activity over homogenatea | ||
|---|---|---|---|
| 5′-Nucleotidase | Galactosyl-transferase | Cytochrome c reductase | |
| Controls | |||
| PM | 7.8 ± 2.6 | 1.4 ± 0.4 | 0.5 ± 0.1 |
| LDM | 1.3 ± 0.4 | 8.8 ± 1.9 | 3.8 ± 0.9 |
| HDM | 3.3 ± 1.3 | 4.3 ± 1.0 | 4.5 ± 0.8 |
| Type 2 diabetics | |||
| PM | 8.9 ± 3.2 | 1.3 ± 0.4 | 0.7 ± 0.1 |
| LDM | 2.5 ± 0.7 | 6.7 ± 1.1 | 3.8 ± 1.0 |
| HDM | 5.0 ± 1.2 | 3.3 ± 0.1 | 4.6 ± 0.8 |
| Membrane fraction | Fold increase in specific activity over homogenatea | ||
|---|---|---|---|
| 5′-Nucleotidase | Galactosyl-transferase | Cytochrome c reductase | |
| Controls | |||
| PM | 7.8 ± 2.6 | 1.4 ± 0.4 | 0.5 ± 0.1 |
| LDM | 1.3 ± 0.4 | 8.8 ± 1.9 | 3.8 ± 0.9 |
| HDM | 3.3 ± 1.3 | 4.3 ± 1.0 | 4.5 ± 0.8 |
| Type 2 diabetics | |||
| PM | 8.9 ± 3.2 | 1.3 ± 0.4 | 0.7 ± 0.1 |
| LDM | 2.5 ± 0.7 | 6.7 ± 1.1 | 3.8 ± 1.0 |
| HDM | 5.0 ± 1.2 | 3.3 ± 0.1 | 4.6 ± 0.8 |
Bold numbers correspond to the membrane fraction that contains the highest degree of enrichment for the respective organelle marker enzyme. 5′-Nucleotidase is a marker for PM, galactosyl-transferase is a marker for the Golgi apparatus, and cytochrome c reductase is a marker for endoplasmic reticulum.
Discussion
We have tested the hypothesis that defects in GLUT4 trafficking and translocation are operative in insulin-resistant adipocytes in type 2 diabetes. Because pretranslational events lead to severe GLUT4 depletion in adipocytes from these diabetic patients (4), we also assessed IRAP, another protein constituent of GLUT4 vesicles, in membrane subfractions. Although IRAP has not been studied previously in human adipocytes, several groups of investigators had shown in rat adipocytes and differentiated 3T3-L1 cells that a large proportion of IRAP is localized to GLUT4 vesicles residing in LDM under basal conditions and that intracellular IRAP translocates to PM in response to insulin (14–16, 18–21). Furthermore, in rat adipocytes (14, 15), there is minimal IRAP in PM from basal cells, and the fold increase in PM IRAP in response to insulin is greater than that for GLUT4. The current data demonstrate that IRAP exhibits similar characteristics in human adipocytes isolated from normoglycemic controls and that, unlike GLUT4, diabetes does not affect cellular IRAP protein expression. Thus, type 2 diabetes appears to affect only the cellular expression of GLUT4, and not the biogenesis of GLUT4/IRAP-containing vesicles in adipocytes. These observations supported the study of IRAP as a marker to assess subcellular localization and translocation of GLUT4/IRAP vesicles in adipocytes from type 2 diabetic patients.
We observed two distinct abnormalities in subcellular IRAP localization in diabetic patients compared with controls. The first abnormality pertains exclusively to basal cells. In basal cells, 4.3-fold more IRAP was recovered in vesicles cofractionating with PM in type 2 diabetes. This component of IRAP was presumably the result of redistribution away from the LDM fraction in which IRAP was depleted by 27% relative to that in controls. Because the vast majority of cellular IRAP is located in microsomes, the relatively small decrement in LDM could be responsible for the large increment in PM. The redistribution of IRAP from LDM to higher density membrane vesicles in type 2 diabetes may involve HDM as well, because there was a tendency for an IRAP increase in the HDM fraction (20% higher in diabetics vs. controls) that did not achieve statistical significance. However, observations with GLUT4 provided stronger support for this notion. Although GLUT4 depletion involved all membrane subfractions in type 2 diabetes, GLUT4 loss was relatively less in HDM, suggestive of a relative redistribution of GLUT4 to this membrane compartment. Thus, in basal adipocytes from type 2 diabetic patients, greater proportions of IRAP and GLUT4 are recovered in dense membrane vesicles cofractionating in the HDM and PM fractions.
The second distinct abnormality in type 2 diabetes was observed in insulin-stimulated cells. In controls, maximal insulin produced a large 14-fold increase in PM IRAP as a result of translocation from LDM (Fig. 4). In contrast, both the absolute increase and fold increase in PM IRAP over basal levels were insignificant in type 2 diabetes, and translocation of IRAP was not apparent. Similarly, insulin stimulation resulted in a 2-fold increase in PM GLUT4 in controls, whereas there was no translocation of GLUT4 from LDM to PM in type 2 diabetes. Thus, the adipocyte glucose transport system in type 2 diabetes contains defects in both GLUT4 expression and insulin-mediated translocation of GLUT4/IRAP-containing vesicles. The latter defect has not been demonstrated previously in adipocytes from type 2 diabetic patients.
This combination of abnormalities in the subcellular localization of IRAP in basal cells, together with the defect in translocation in insulin-stimulated cells, is analogous to defects in GLUT4 trafficking and translocation observed in human skeletal muscle (11). In muscle biopsied under basal conditions, both GLUT4 and IRAP were redistributed to membrane vesicles with higher buoyant density in insulin-resistant subjects with and without type 2 diabetes compared with insulin-sensitive controls. This abnormality in GLUT4 targeting or trafficking evident in basal muscle was coupled with impaired translocation of GLUT4 vesicles after insulin stimulation. Furthermore, we have observed similar abnormalities in GLUT4 in omental adipocytes from women with gestational diabetes (13). Based on current and previous data, we propose that a defect in GLUT4 trafficking/targeting causes human insulin resistance and is common to both skeletal muscle and adipocytes. This defect leads to GLUT4 accumulation in or targeting to a dense membrane compartment under basal conditions from which transporters are unable to be recruited to cell surface membranes. This mechanism of insulin resistance is operative in adipocytes but is relegated to secondary importance by a marked reduction in GLUT4 expression in type 2 diabetes. However, this mechanism predominates in skeletal muscle in which normal expression of GLUT4 is maintained.
The abnormalities in type 2 diabetes are characteristics of diabetes and not obesity. One reason is that mean body mass index was similar between the type 2 diabetes and control subgroups. Furthermore, whereas insulin resistance in skeletal muscle can vary dramatically among nondiabetic individuals independent of obesity (32), insulin resistance in adipocytes is directly proportional to adipocyte size (4, 5, 33). Again, mean adipocyte size was similar between the two subgroups. Thus, the results are relevant to the more pronounced degree of insulin resistance due to diabetes per se, as reflected by lower basal and absent stimulation of glucose transport activity normalized per adipocyte surface area. The question remains as to whether the trafficking/translocation defects are induced secondarily by hyperglycemia or constitute more primary abnormalities in type 2 diabetes. In previous studies using rat adipocytes, treatment with high glucose or glucosamine induced a translocation block with retention of GLUT4 in LDM (25) and a redistribution of IRAP but not GLUT4 to HDM (34). The relevance of this to human adipocytes is not clear because, in the current study, the hyperglycemic environment in type 2 diabetic patients was accompanied by a translocation defect, together with apparent redistribution in basal cells of both GLUT4 and IRAP to HDM and PM. Also, a similar combination of defects was observed in muscle from normoglycemic insulin-resistant subjects in which effects of high glucose would not be an issue (11). Thus, the extent to which defects in trafficking and translocation are primary or induced by hyperglycemia will require additional study. An additional point is that, in both basal and insulin-stimulated adipocytes, abnormalities in subcellular localization were specific for IRAP and GLUT4. Organelle marker enzymes were not affected by diabetes and indicated that there were no major differences in fractionation and recovery of organelle membranes in the various membrane subfractions.
Published data indicate that GLUT4 trafficking involves both a constitutive exocytotic/endocytotic recycling pathway and sorting to an inducible tubulo-vesicular compartment that contains the insulin-responsive translocating pool (16, 17, 34–36). Abnormal GLUT4/IRAP redistribution to a dense membrane compartment could result from either a block at the level of the sorting step leading to accumulation in the proximal endosomal pool(s) that is a normal component of the GLUT4 trafficking pathway or defective sorting to an alternative compartment (with physicochemical properties similar to PM/HDM) from which insulin is unable to recruit GLUT4 to the cell surface. The data do not favor a block in translocation between the inducible compartment and the cell surface because this would predictably increase the amount of GLUT4 and/or IRAP recovered in LDM (which contains the inducible pool) and could not readily explain the observed increase in the PM and HDM fractions. Rather, the data seem to favor accumulation of GLUT4 in an endosomal pool that cofractionates with PM and HDM. In normal adipocytes, the majority of IRAP resides in the inducible pool of GLUT4 vesicles (16) in which it is immediately sorted with GLUT4 after internalization (37). In human insulin resistance, we are suggesting that sorting of IRAP and GLUT4 to the inducible pool could be impaired with accumulation in endosomes. It is also possible that, in diabetes, GLUT4 and IRAP may no longer be colocalized in the same vesicles. This latter scenario would still be indicative of a trafficking defect in insulin-resistant cells, although IRAP would no longer serve as a marker for GLUT4 vesicles. In any event, these predictions are speculative and require further study. What we have demonstrated in the current manuscript is that the subcellular distribution of IRAP and GLUT4 in adipocytes from type 2 diabetic patients is different from that in controls under basal conditions, and there is no translocation in response to insulin.
The biochemical basis of defects in GLUT4/IRAP subcellular localization remains to be defined. A translocation defect per se could result from a defect in insulin signal transduction or lie intrinsic to the glucose transporter effector system. Although signal transduction abnormalities may be contributory (38), it should be emphasized that the translocation defect is associated with accumulation of IRAP in dense membrane vesicles in basal cells. Thus, the defect in cellular targeting or trafficking of IRAP is independent of the acute effects of insulin and could not be explained by defects in insulin signaling. The implication is that the abnormality in basal GLUT4/IRAP targeting/trafficking is a distinct defect intrinsic to the glucose transport system, which is then mechanistically linked to impaired translocation. These data in humans are in contrast to other known cellular (25) and animal models (39, 40) of insulin resistance that involve isolated translocation defects without evidence that GLUT4 trafficking is disturbed under basal conditions. GLUT4 vesicle trafficking is highly regulated, is multicompartmental with constitutive and inducible compartments, and involves multiple proteins that also direct exocytosis/endocytosis of synaptic neurosecretory vesicles (41–43). It is possible that defects at any of several steps could alter trafficking/targeting of GLUT4 to produce the novel pattern of abnormalities observed in human patients. Elucidation of these defects in both adipose and muscle tissues would have important implications for both treatment and prevention of peripheral insulin resistance and type 2 diabetes.
Acknowledgements
We gratefully acknowledge Barbara Wojciechowski, M.S., for assistance with graphics and statistics.
This work was supported by NIH Grants DK-38764 and DK-25336, by the Medical Research Service of the Department of Veterans Affairs, and by the Medical University of South Carolina General Clinical Research Center (M01-RR-1070).
Abbreviations:
- GLUT4,
Glucose transporter 4;
- HDM,
high-density microsome;
- IRAP,
insulin-regulated aminopeptidase;
- LDM,
low-density microsome;
- PM,
plasma membrane.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}