
Abstract
Process simulation facilitates scale-up of hot-melt extrusion (HME) and enhances proper understanding of the underlying critical process parameters. However, performing numeric simulations requires profound knowledge of the employed materials’ properties. For example, an accurate description of the compounds’ melt rheology is paramount for proper simulations. Hence, sample preparation needs to be optimized to yield results as predictive as possible. To identify the optimal preparation method for small amplitude oscillatory shear (SAOS) rheological measurements, binary mixtures of hydroxypropylmethylcellulose acetate succinate or methacrylic acid ethyl acrylate copolymer (Eudragit L100-55) together with the model drugs celecoxib and ketoconazole were prepared. The physical powder mixtures were introduced into the SAOS as a compressed tablet or a disk prepared via vacuum compression molding (VCM). Simulations with the derived parameters were conducted and compared to lab-scale extrusion trials. VCM was identified as the ideal preparation method resulting in the highest similarity between simulated and experimental values, while simulation based on conventional powder-based methods insufficiently described the HME process.
Similar content being viewed by others


INTRODUCTION
A common approach to overcome poor aqueous solubility and related limited bioavailability is the formation of amorphous solid dispersions (ASDs). Among feasible manufacturing processes, the advantages of hot-melt extrusion contain high-throughput and easy as well as versatile downstream options (1,2,3). While it is crucial to establish and utilize screening processes to determine a suitable polymer for the respective API in early development, it is paramount to simultaneously begin early process development for future scale-up procedures. Selection of quasi-adiabatic conditions during early process development in lab scale would ease the process of scale-up; however, finding the right and applicable quasi-adiabatic setup is complex and tedious and can be material-consuming (4).
Establishing an extrusion process begins with determining feed rate, screw speed, geometry/D ratio, screw configuration, and the general temperature profile across the length of the barrel. Although this might seem easily accomplishable, every aspect is affected by various variables and some of them are interlinked (5,6,7). For instance, the feed rate directly determines the residence time of the product in the extruder, but the residence time is also influenced by the screw design, where restrictive elements cause a longer residence time (8,9,10). Moreover, restrictive elements can induce different shear forces, leading to different energy inputs into the melt and consequently to possible temperature peaks, so called hot spots, within the extrusion process (11, 12). These hot spots could lead to degradation of thermally sensitive API or polymers, if not addressed through an adaption of process parameters affecting the SME, such as feed rate, screw speed, screw configuration, and/or the general temperature profile of the barrel (12, 13). Additionally, these interactions are shifting in relevance among the various scales, complicating the scale-up process even more (14).
This variability in parameter impact obstructs full experimental description as the process intricacies remain unknown. To increase the understanding of the process, 1D simulations can serve as a fairly easy to use option (15,16,17). This is especially important in early formulation processes in small scale, as viscous dissipation can be easily compensated by conducted heat or cooling from the barrel, which is not the case at larger scales; i.e., adiabatic conditions by geometric scale from small scale can be easily overlooked (15, 18, 19).
Simulations allow saving experiments during a scale-up and at the same time the process understanding is further improved. However, to perform simulations, it is necessary to have a broad understanding of the components’ material properties like melt rheology, glass transition temperature (Tg), heat capacity, and the plasticizing effect of dissolved API as well as the API solubility in the polymer matrix. Especially for polymers, which recently caught interest for HME, such as methacrylic acid ethyl acrylate copolymer (Eudragit L100-55) or hydroxypropylmethylcellulose acetate succinate (HPMCAS), only limited data are available and usually not in combination with the respective API and/or at the relevant drug-load, especially considering their melt rheology (20, 21). However, it is crucial to describe the product rheologically, since the rheological properties of the polymer melt have a major impact on most of the important parameters of extrusion, such as the SME (22, 23). This may be due to the fact that the samples under investigation must closely resemble the expected HME product to ensure reliable differential scanning calorimetry (DSC) and rheological results. This resemblance typically includes formation of a completely amorphous, homogeneous, and single-phase system (24). This cannot generally be assumed for simple powder samples, especially if the inherent viscosity of the polymer is high and the drug-load is low. In such a case, dissolution and homogenous distribution of API in polymer without any shear are hindered (25).
A promising method to address this issue is the preparation of samples by vacuum compression molding (VCM) (26). This method enables creation of homogenous, amorphous, single-phase samples allowing a reliable determination of the sample’s glass transition temperature (Tg) (27). Likewise, the geometry of the product can be decided by the geometry of the VCM tool used, which is of particular interest for plate-plate small amplitude oscillatory shear (SAOS) rheometers, since trimming for the measurements became obsolete and the measurements overall led to more accurate results as demonstrated by Treffer et al. (26). In addition, the tool allows the production of samples without sample loss and air entrapment, which further enhances accuracy (26). Thus, generation of samples that could be reliably used for rheological and thermal measurements might be facilitated and subsequently could be used as accurate input for simulations.
This work aims to investigate the extent of different sample preparation methods, namely the VCM tool (meltprep samples) and normal powder samples as well as their comprimates, on the results of the thermal and rheological characterization of API-polymer blends. As API/polymer models celecoxib (CXB)/HPMCAS and ketoconazole (KTZ)/L100-55 were selected for their high melt viscosity (25), i.e., high susceptibility on sample preparation for SAOS measurements. Subsequently, further impact on the simulation of the HME processes was assessed. Finally, the accuracy of the formulation-dependent simulations was validated by experimental hot-melt extrusion.
MATERIALS AND METHODS
Material
Eudragit L100-55 (Tg: 117.71°C, molecular weight: 320,000 g*mol−1, degradation temperature 205°C (weight loss > 1%) (25)) was kindly donated by Evonik (Darmstadt, Germany) and HPMCAS LG (Tg: 117.15°C, molecular weight: 18,000 g*mol−1, degradation temperature: 265°C (weight loss > 1%) (25)) by Shin-Etsu (Tokyo, Japan), respectively. Celecoxib (melting point: 164.95°C, Tg: 58.45°C, molecular weight: 381.37 g*mol−1, degradation temperature: 352.1°C (28)) was purchased from Kekule Pharma Limited (India), and ketoconazole (153.55°C, Tg: 42.52°C, molecular weight: 531.43 g*mol−1, degradation temperature: 363.0°C (29)) from Fagron (Glinde, Germany).
Methods
Melt Rheology
An oscillatory rheometer (HAAKE MARS III, Thermo Scientific, Karlsruhe, Germany) equipped with a plate-plate geometry (d = 20 mm) was used. All experiments were conducted in controlled deformation AutoStrain (CDAutoStrain) mode after equilibration of the samples for 10 min at the starting temperature with a gap height between 1.2 and 1.8 mm, dependent on the sample specimen height. HPMCAS and HPMCAS/CXB mixtures (0.1, 0.2, 0.3, 0.4, and 0.5 w/w CXB) were measured using the following parameters: temperature range 140–180°C, 10°C step width, amplitude range 1.0–1.2% at 0.1–10 Hz. Frequency sweeps with HPMCAS were carried out starting from 140 to 180°C with 10°C steps and an amplitude of 1.0% and 1.2% was used for plain HPMCAS- and CXB-containing samples, respectively. In the case of plain L100-55, frequency sweeps were performed from 145 to 165°C with 5°C step and an amplitude of 1.0% and 0.5% was used for plain L100-55– and KTZ-containing samples (0.1, 0.2, 0.3, 0.4, and 0.5 w/w KTZ). All frequency sweeps were executed with a frequency starting from 10 Hz (62.83 rad/s) to 0.1 Hz (0.63 rad/s). The amplitude for each blend and/or polymer was determined to be in the linear viscoelastic range (LVR) via amplitude sweeps, beforehand. At last, a horizontal time–temperature superposition shifting operation was applied (see Fig. 1) resulting in master curves shifted to 145°C for neat L100-55 and ASDs thereof and 160°C for neat HPMCAS and ASDs thereof. As the resulting master curve(s) were in the power law region, the following equation was employed for fitting of the data:

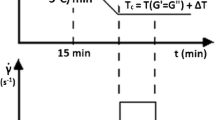
General approach of determining the required model parameters. The consistency (K, graph b) and the power law index (n, graph b) are identified with the help of a shifted master curve (graph b). The master curve emerges from shifting measurement segments at different temperatures but within the same frequency range (graph a). The various segments are shifted to the reference temperature (T0) utilizing the empirical relation of Williams-Landel-Ferry. In the end, a master curve at the reference temperature with a wide frequency range arises (graph b). Additionally, temperature dependency of flow can be described by a model originating from Arrhenius (graph d). Results of temperature sweep (graph c), covering the desired temperature range, are displayed in a manner that activation energy of flow can be readily determined by employing the equation shown in graph d
where K is the consistency index (Pa*s), γ is the shear rate (s−1), and n is the power law index. To allow an expression of temperature dependency for the simulations, the Arrhenius activation energy of flow (EA) needs to be known. Temperature sweeps with an underlying cooling rate of 2°C/min, a fixed frequency of 1 Hz (6.23 rad/s), and the reported amplitude were conducted. Neat L100-55 and mixtures were evaluated from 160 to 120°C and HPMCAS and mixtures from 180 to 120°C. Determination of EA was carried out by employing the following Eq. (30) after displaying the data as described in Fig. 1:
where η* is the measured complex viscosity (Pa*s), A is the pre-exponential factor, R is the gas constant (8.314 J*K−1*mol−1), T is the respective temperature (K), and EA is the slope of the resulting Arrhenius plot multiplied by R.
Tableting
A pneumo-hydraulic tablet press (FlexiTab®, Röltgen, Solingen, Germany) equipped with 20-mm flat faced punches was used for the preparation of flat tablets for use as starting specimen for the determination of rheological properties of powder samples. The powder was filled manually into the die. The target height of the tablets was 1.5 to 1.8 mm, and the smallest possible compression pressure was used that still resulted in mechanically stable samples.
Vacuum Compression Molding/Meltprep
Samples for rheological measurements were prepared as described by Treffer et al. (26). In brief, a setup with a diameter of 20 mm and complete vacuum was used, resulting in a compaction pressure of 2.5 bar. A total of 500–600 mg of polymer or blend was needed to achieve suitable heights of 1.3 to 1.6 mm. L100-55 and mixtures were molded at 160°C for 15 min whereas HPMCAS and mixtures were molded at 180°C for 12 min.
Porosity Determination
The particle density (ρp) of the powder blends was determined utilizing the helium pycnometer BELPYCNO L by Microtrac Retsch GmbH (Haan, Germany). Samples were analyzed after five cleaning cycles with a restrictive pressure of 0.2 bar stopping after five good cycles (standard deviation less than 0.1%) up to a maximum of 50 cycles. All measurements were conducted in triplicate.
Envelope density (ρb) of the VCM disks and tablets was calculated utilizing the following equation:
where m is the mass and V is the volume of the specimen. The volume can be calculated employing the measured height as well as a set diameter of the die.
Finally, the porosity of the specimen can be determined by employing the following equation:
where ε is the porosity, ρb describes the envelope density, and ρp the particle density.
Extrusion
A co-rotating twin-screw extruder ZE 12 from Three-Tec GmbH (Seon, Switzerland) with a functional length of 25:1 L/D, 12 mm screws, 2 mm die, and a maximum torque of 15 Nm was used. The screw configuration and barrel partitioning are shown in Fig. 2. The screw speed was kept constant at 100 rpm for all experiments. The specific settings for all blends are shown in Table I. The torque was recorded for calculation of the specific mechanical energy (SME). For the SME calculation, the following equation was employed:

Extruder and screw configuration used for simulation and experimental trials. Blue segments represent conveying elements. Red and green segments correspond to kneading elements
where ns is the screw speed (rpm), \(\dot{m}\) is the feed rate (kg/h), and τ is the measured torque (Nm) with a subtracted idling torque of 1.8 Nm.
Statistical Analysis
Testing for statistical significance was conducted with the Prism 8’s t-test, corrected for multiple comparisons using the Holm-Sidak method.
Simulation
The simulation software Ludovic® V6.0 PharmaEdition (Sciences Computers Consultants, Saint Etienne, France) was used to compute the flow conditions of hot-melt extrusion processes. It is a one-dimensional simulation for flow modeling and allows the calculation of various parameters along the screw profile. The model assumes non-isothermal flow conditions and an instantaneous melting prior to the first restrictive screw element. Due to the unknown filling ratio at the beginning of the extruder, the computation starts at the die and proceeds backwards in an iterative procedure. In brief, after calculation of the required pressure to allow the melt to pass the die, the needed temperature at the die is calculated depending on the rheological and thermal properties of the used compound. Thereupon, the respective properties and outputs for each element (from die to the first conveying element) will be calculated in an iterative manner (30). To allow the comparison of the impact onto simulation outcome of either input parameters derived from powder samples or meltprep samples, key output parameters of the simulation were reviewed. For this purpose, the parameters specific mechanical energy (SME), mean residence time (MRT), and melt temperature at the die were measured for every extrusion trial and compared to the simulated counterpart. Furthermore, simulated melt temperatures at the kneading elements were also compared in terms of influence of melt rheology, based on either meltprep or compressed powder data.
DSC
DSC experiments were carried out on a Mettler-Toledo DSC 2 equipped with a nitrogen cooling system (Mettler-Toledo, Gießen, Germany). The glass transition temperatures (Tgs) of the different preparation and measuring setups were determined using a multi-frequency temperature modulation (TOPEM® mode) with an underlying heating rate of 2 K/min, a pulse height of 1 K, and a constant nitrogen purge (30 mL/min).
Every blend of each polymer was prepared in three different ways: mixed powder sample, molded sample via VCM, and milled extrudate, if achievable. Furthermore, the powder samples were measured in a conventional method and a “Meltprep-like” method. The conventional method consisted of a heating cycle from 25 to 160°C (L100-55) or 180°C (HPMCAS) with an underlying heating rate of 10 K/min and a cooling step to 25°C. The Meltprep-like method consisted of the conventional method and an isothermal step, mimicking the settings used for VCM samples as reported earlier. Afterwards, the samples were measured in TOPEM-mode from 25 to 160°C (L100-55) or 180°C (HPMCAS) and examined for Tg(s).
RESULTS
DSC
In order to evaluate the impact of sample preparation, DSC measurements for powder, VCM, and extrudate samples were conducted as previously described and analyzed regarding heat capacity (see Supplementary Data, Fig. 8) and Tg (Table II), as these values allow a straightforward and suitable comparison of thermal properties and are further needed for simulation. The Tg results are summarized in Table II. It is noticeable that Tg values determined for extrudate and meltprep samples are indistinguishable. For example, it turned out that extruded samples containing 30% KTZ in L100-55 resulted in a measured Tg of 90.80°C while the meltprep samples exhibited a Tg of 90.98°C. In contrast, Tg of powder samples as well as samples with prolonged annealing differed from the values obtained for extrudates. This phenomenon was particularly visible at lower drug-loads (10–30%) with the determined Tg deviating notably from extrudates; e.g., a difference of 10°C was measured for 10% CXB in HPMCAS (110.42°C pressed vs. 100.47°C extrudate). Furthermore, several powder samples displayed two Tgs even when prolonged annealing was applied, namely 20% CXB and 30%, 40%, and 40% KTZ blends (see Fig. 3 for one example of each polymer). In each instance, two Tgs, one closer to the polymer and one closer to pure API, were visible for powder samples, independent of the DSC method used, whereas the Tgs of the meltprep sample and the extrudate were indistinguishable. No Tgs for 10% and 20% KTZ in L100-55 extrudate were obtained as these blends were not possible to extrude.

Differential scanning calorimetry (DSC) thermograms of two exemplary drug-containing mixtures. a 20% celecoxib in HPMCAS and b 40% ketoconazole in L100-55. The different employed methods to possibly create an amorphous solid dispersion (ASD), as well as the neat polymer and active pharmaceutical ingredient (API), are shown. Arrows indicate the location of glass transition temperature(s)
Porosity
The porosity of the specimen used for rheological measurements was determined as previously described. In this case, the determined porosity of VCM samples was in the range of 2.01 to 3.44% while pressed samples possessed a porosity ranging from 27.77 to 40.52%, revealing a difference between the two preparation methods regarding porosity by a factor of approximately ten (Supplementary Data, Table IV).
Rheology
The melt rheology is one of the key input parameters for simulation with the Ludovic simulation software. To facilitate simulation, a description of melt rheology, i.e., a rheological model, must be employed for both shear dependency and temperature dependency. The results are summarized Fig. 4. Regarding consistency, the determined consistency values of meltprep samples were higher, independent of polymer and drug-load. In the case of CXB blends, an increased drug-load led to a decreasing deviation between the two sample preparation methods, ultimately resulting in an indistinguishable consistency value starting from a drug-load of 30% CXB. For power law index, no differences between powder samples and meltprep samples could be observed for HPMCAS blends (Fig. 4b). The only exception is the power law index (n) of 50% CXB/HPMCAS mixtures, where tablet specimen (n = 0.3813 ± 0.060) differed significantly (p < 0.01) from the meltprep samples (n = 0.4849 ± 0.028). Generally, higher standard deviations were obtained when fitting the measurements of powder samples except for 10% CXB/HPMCAS, where the meltprep sample exhibited a higher standard deviation (coefficient of variation up to 33.45 and 12.16% for tablet and meltprep specimen, respectively).

Comparison of rheological parameters derived from utilizing the power law fit for the respective master curve of the two sample preparation methods used. The comparison for the HPMCAS and celecoxib (CXB) mixtures is shown in a (consistency) and b (power law index) while for L100-55 and ketoconazole mixtures, comparisons are shown in c (consistency) and d (power law index). The master curves containing CXB were employed with a reference temperature of 160°C and for KTZ mixtures the reference temperature was 145°C. p < 0.05; **p < 0.01; ***p < 0.001
Blends consisting of L100-55 did not show the same trends as the HPMCAS blends. Pronounced deviations regarding consistency between powder and meltprep samples were observable with no distinct increase or decrease in deviation with rising drug-load. Furthermore, comparing the power law index of each L100-55 blend (Fig. 4d), a difference between powder and meltprep samples was observed. In total, a trend of differing power law index with higher drug-load for meltprep samples in comparison to powder samples was visible. This was especially prominent for the sample with 40% and 50% drug-load. While the power law index of powder samples followed the trend of decreasing power law index with increasing drug-load, meltprep samples displayed a slight reversal of trend at the 40% drug-load mark.
Extrusion: Simulation vs. Experimental
The SME and MRT results of extrusion trials containing HPMCAS are visualized in Fig. 5, divided into trials with a feed rate of 2 g/min (a and b) and a feed rate of 3 g/min (c and d). The MRT of experimental trials were significantly higher than the simulated MRT, if derived from input parameters of pressed specimen. In contrast, simulations with input parameters derived from meltprep samples predominantly led to values not significantly deviating from experimentally determined MRT. For example, while experimental MRT of 10% CXB at 2 g/min was determined to be 361 s ± 23 s, simulation computed a MRT of 363 s (meltprep) and 261.5 s (pressed) in the latter case deviating significantly (p < 0.001) from experimental values. This discrepancy between experimental MRT and simulated (pressed) MRT was also evident if a feed rate of 3 g/min was used; e.g., extrusion trials of 20% CXB had a MRT of 244.33 s ± 9.87 s and simulations determined MRT values of 186.1 s for pressed and 259.5 s for meltprep input parameters. However, there is one exception where both types of simulations resulted in unsignificant differences between the simulated and experimental MRT, namely 50% CXB/HPMCAS with a freed rate of 3 g/min.

Comparison of mean residence time (MRT) and specific mechanical energy (SME) of HPMCAS and CXB mixtures with a feed rate of 2 g/min (A and B) and 3 g/min (C and D, respectively). Experimental results were compared to simulations with input parameters derived either from vacuum compression molded samples (meltprep) or from comprimates (pressed). p < 0.05; **p < 0.01; ***p < 0.001
The trend that experimental data turned out to be higher than the determined counterpart by simulation was also observed in the determination of SME. Again, the SME was better described when working with simulations that used meltprep input parameters as a basis. For example, experimental SME of every blend could be described particularly well at a feed rate of 2 g/min by simulations (meltprep) as no significant deviations from experimental values were observed. In contrast, each simulation with input parameters derived from pressed samples resulted in SME values significantly deviating from experimental SME (at least p < 0.01). The difference between experimental and simulated SME was particularly small for 40% CXB (2 g/min) 261.14 ± 6.50 kWh/t experimental versus 263 kWh/t simulated. For all blends other than 10% and 50% at 3 g/min, simulations (meltprep) led to SME values insignificantly deviating from experimental determined SME. In contrast, simulations with data of pressed samples generally led to significant (p < 0.001) deviations from experimental values with exception for 30% and 40% CXB.
A similar pattern was observed when comparing SME and MRT for the L100-55 blends. The comparison for L100-55 trials and their comparison is summarized in Fig. 6. In this case, MRT values in simulations (pressed) differed significantly (p < 0.001) in every case compared to experimental results. This was not the case for simulations (meltprep), where simulated MRT values were identical to experimental values in all cases. In contrast, simulated SME values were only equivalent for simulations with meltprep inputs for trials with 3 g/min feed rate. In all other cases, simulated SME values differed significantly (p < 0.001) from experimental SME, whereby simulation (pressed) led to considerably lower SME values.

Comparison of mean residence time (MRT) and specific mechanical energy (SME) of L100-55 and KTZ mixtures with a feed rate of 2 g/min (a and b) and 3 g/min (c and d), respectively. Experimental results were compared to simulations with input parameters derived either from vacuum compression molded samples (meltprep) or from comprimates (pressed). p < 0.05; **p < 0.01; ***p < 0.001
Another important factor of hot-melt extrusion is the temperature of the melt across the extrusion process. As measurements within the extruder are difficult to measure, the measured melt temperature at the die was compared to simulated data. The comparison is summarized in Table V (see Supplementary Data). In this context, simulated melt temperature at the die was comparable to experimental determined values, if meltprep input parameters were employed. Deviations less than 5% were achievable for all simulations (meltprep) except for 50% KTZ at 3 g/min where, nevertheless, the deviation was less than 10%. In contrast, simulations (pressed) generally led to temperature values deviating more pronounced to experimental values, as only the simulations of 10% CXB at 2 g/min and 10–30% CXB at 3 g/min resulted in values deviating less than 5%.
In addition, the simulated temperature profiles along the extruder were compared between pressed and meltprep input parameters. The comparison was projected to the peak melt temperature at the second kneading zone. The results are summarized in Table III; in addition, the residence time for the extrusion process is exemplified in Fig. 7. The segment with the highest melting temperature accounted for about half of the total residence time in the extruder. At the same time, the different input parameters led to different peak temperatures in this segment. The phenomenon repeated that simulations (meltprep) predicted higher temperatures. In this context, the simulations of 30% KTZ and 40% KTZ were particularly striking. While simulation with input parameters originating from pressed samples led to predicted temperatures of 188.6 and 172.9°C, respectively, simulations (meltprep) predicted temperatures of 210.3 and 196.5°C, respectively. Thermogravimetric data (see Supplementary Data, Fig. 9) suggested that thermal decay of the respective polymers was readily apparent starting from 205°C (weight loss > 1%) for L100-55 and from 265°C (weight loss > 1%) for HPMCAS (25).

Exemplary presentation of simulated melt temperature of either simulations based on meltprep or pressed sample input parameters along the extrusion process. The simulated total cumulative residence time along the screw was added. The example shows the simulation of 10% CXB in HPMCAS with a feed rate of 2 g/min
DISCUSSION
Rheology
In general, the DSC data (Table II; Fig. 3) indicated that neither conventional nor prolonged annealing methods could reliably achieve single-phase systems for powder samples. Since a homogeneous, single-phase system exclusively formed at high drug-load, viscosity was likely the limiting factor for homogeneous ASD formation. Exceeding a certain viscosity, the API cannot homogenously distribute by diffusion only into the polymer matrix within the applied timeframe. Exceptions were observed at drug-loads starting from at least 40% for HPMCAS and 50% for L100-55 blends, indicating that a specific amount of solid-state plasticizer was necessary to reduce the impact of viscosity onto the dissolution rate of the API into the polymer matrix. At lower drug contents, several or no Tgs were identifiable.
Consequently, it must be assumed that rheology measurements with compressed powder samples did not form a single-phase homogeneous system during the measurement either. Additionally, it is questionable whether in situ formation of an ASD during the SAOS experiment is completed at low temperature and/or low shear, as prolonged annealing of powder samples did not result in single-phase systems, either. Therefore, the whole rheological model and the determined parameters are debatable, as requirements previously mentioned were not met. These findings would be in line with a hypothesis by Bhujbal et al. that melt viscosity above 300 cP would result in impeded ASD generation (31). In contrast, DSC data from meltprep samples showed explicit Tgs corresponding to those of extrudates, which emphasizes a homogeneous single-phase system, thus indicating that the applied compaction force of the VCM tool in combination with heat resulted in sufficiently shear thinning behavior. This means that for the determination of a Tg for blends with high viscosity polymers or polymers possessing a yield point (and low drug-load), sample preparation by VCM is indispensable; otherwise, no ASD can be formed and no Tg or one not corresponding to the respective ASD can be determined. Also, samples prepared by VCM meet the requirements (amorphous, single-phase system) to apply the TTS to generate a master curve, whereas compressed powder samples do not meet the requirements (24).
Regarding the rheological parameters, determined pressed samples generally showed lower consistencies than the meltprep samples. As meltprep samples presumably contain API completely dissolved into the polymer matrix, the API is available as a solid-state plasticizer. In contrast, for pressed samples this cannot be assumed and hence, an explanation for the lower determined consistency of pressed samples remains. A possible explanation would be that as some mixtures formed measurable drug-rich phases (two Tgs measurable; see Table II), these drug-rich phases acted as voids for the polymer chains. The voids would allow the chains to escape the friction of other chains by slipping into the drug-rich phases and consequently lowering the needed shear forces for movement (3). Finally, this led to a lower viscosity measured. This phenomenon could also be applied to possible voids caused by air entrapment (32). Due to the high viscosity of the polymers, possibly trapped air (bubbles) cannot escape. If one compares the porosity of powder compacts and meltprep samples, the porosity of compacts was about 8 times higher than that of meltprep samples. Since, in contrast to the normal compression of powder, the application of the VCM tool produced samples with no visible air inclusions, it was likely that the difference in consistency was likely caused by entrapped air. Entrapped air would act, similar to drug-rich phases, as voids into which polymer chains could slip, ultimately leading lower needed shear forces for deformation.
As the drug-load increased, the measured consistencies became more and more similar, comparable to the formation of a homogeneous system during DSC measurements. The resulting lower viscosity possibly allowed air to escape from the sample. However, since the values determined for the pressed samples were still somewhat below the meltprep samples, air probably did not escape completely. This difference was higher for L100-55 blends than for HPMCAS blends further supporting this hypothesis, as a fundamentally higher viscosity was measurable for L100-55. Consequently, the viscosity of the blend was also higher and more likely to prevent air from escaping, so adequate sample preparation became more important as the viscosity of the polymer was higher.
Simulation
When analyzing the MRT data, for input parameters of the pressed samples, the MRT turned out to be too low in all cases compared to experimentally determined MRT. This was not the case when meltprep input parameters were used. As for simulations, the MRT depends on the assumed density of the melt if feed rate is kept constant (33). Since determined densities are about 25% lower for pressed samples, approximately 25% lower simulated MRT compared to simulations with meltprep input parameters were expectable. The MRT data from the meltprep simulations were similar to the experimental data. These values were at the same time as, if not closer to, the experimental data as simulations previously published based on apparent density (34). Thus, it is possible to use the VCM tool to prepare samples and determine density via a simple geometry-based calculation and utilize this for a descriptive simulation.
One of the most important parameters of extrusion is the SME, as it can be used during scale-up of processes (15). It is therefore important that the SME is described particularly accurately by the simulation, so that experiments during scale-up can be saved by simulations. SME is determined by the rheological parameters of the product in interaction with the residence time of the material (7). Since rheological parameters of pressed samples were distorted by entrapped air, no accurate simulation of SME with pressed input parameters was possible. In contrast, simulated SME with meltprep input parameters led to results similar to the experimental SME. Due to the higher viscosity, a higher necessary shear force and, in conjunction with the MRT, longer residence times resulted in higher calculated SME. In the present comparative data, it is noticeable that if there were deviations larger than 10%, the simulations computed lower SME than experimentally determined. This can most likely be explained by an overestimated gearbox efficiency of the used extruder, since the simulations usually yielded lower values even with small deviations. Additionally, special features of the respective mixture or compound could be an explanation for the deviation between experimental and simulated SME, e.g., minuscule degradation during extrusion or circumstantial high moisture of the blend. It is noticeable that at lower feed rate and declining drug-load the L100-55 simulations deviated increasingly from the experimental values. Reported degradation of L100-55 above 205°C and predicted melt temperatures of over 210°C starting from 30% drug-load downwards emphasize the visible degradation. The resulting vapors changed the process to such an extent that the modeling boundaries were exceeded, and simulation was no longer fitting. A possible explanation would be that the vapors lead to sticking of the powder, especially in the first third of the screws, which resulted in a larger screw diameter and caused the screws to rub against each other and thus leading to an increased measured torque. At the same time, the vapors produced could also be the reason why the measured temperature at the nozzle during the runs, where degradation was visible, was lower than the simulated temperature. In contrast, for runs where no degradation was visually visible, the simulated values (meltprep) were comparable to the measured values.
CONCLUSION
In contrast to Tg values from powder samples, Tgs from VCM and extrudate samples were identical. Subsequently, vacuum compression molding was the superior sample preparation method for compounds exhibiting high melt viscosity. Furthermore, the difference between tablet and VCM samples was also apparent in melt rheology, density, and porosity. Ultimately, determining blend properties and utilizing them as input parameters in 1D-simulation led to converging simulations of the performed extrusion trials when the VCM preparation method was used. In this context, a simple volumetric density determination of VCM samples served as an adequate input parameter for prediction of MRT, eliminating the need for a time-consuming determination via helium pycnometry. Additionally, employing melt rheology from VCM samples enabled predictive simulation of SME and melt temperature profiles of each HME trial. The improved melt temperature profile prediction could be confirmed using a lowly plasticized L100-55 blend as a temperature indicator due to pronounced degradation at temperatures exceeding 205°C.
References
Breitenbach J. Melt extrusion: from process to drug delivery technology. Eur J Pharm Biopharm. 2002;54(2):107–17.
Repka MA, Shah S, Lu J, Maddineni S, Morott J, Patwardhan K, et al. Melt extrusion: process to product. Expert Opin Drug Deliv. 2012;9(1):105–25.
Stanković M, Frijlink HW, Hinrichs WLJ. Polymeric formulations for drug release prepared by hot melt extrusion: application and characterization. Drug Discov Today. 2015;20(7):812–23.
Agrawal AM, Dudhedia MS, Zimny E. Hot melt extrusion: development of an amorphous solid dispersion for an insoluble drug from mini-scale to clinical scale. AAPS PharmSciTech. 2016;17(1):133–47.
Patil H, Tiwari RV, Repka MA. Hot-melt extrusion: from theory to application in pharmaceutical formulation. AAPS PharmSciTech. 2016;17(1):20–42.
Hanada M, Jermain SV, Lu X, Su Y, Williams RO. Predicting physical stability of ternary amorphous solid dispersions using specific mechanical energy in a hot melt extrusion process. Int J Pharm. 2018;548(1):571–85.
Haser A, Huang S, Listro T, White D, Zhang F. An approach for chemical stability during melt extrusion of a drug substance with a high melting point. Int J Pharm. 2017;524(1):55–64.
Emin MA, Schuchmann HP. Analysis of the dispersive mixing efficiency in a twin-screw extrusion processing of starch based matrix. J Food Eng. 2013;115(1):132–43.
Nakamichi K, Nakano T, Yasuura H, Izumi S, Kawashima Y. The role of the kneading paddle and the effects of screw revolution speed and water content on the preparation of solid dispersions using a twin-screw extruder. Int J Pharm. 2002;241(2):203–11.
Bauer H, Matić J, Evans RC, Gryczke A, Ketterhagen W, Sinha K, et al. Determining local residence time distributions in twin-screw extruder elements via smoothed particle hydrodynamics. Chem Eng Sci. 2022;247: 117029.
Alexy P, Lacı́k I, Šimková B, Bakoš D, Prónayová N, Liptaj T, et al. Effect of melt processing on thermo-mechanical degradation of poly(vinyl alcohol)s. Polym Degrad Stab. 2004;85(2):823–30.
Huang S, O’Donnell KP, Delpon de Vaux SM, O’Brien J, Stutzman J, Williams RO. Processing thermally labile drugs by hot-melt extrusion: the lesson with gliclazide. Eur J Pharm Biopharm. 2017;119:56–67.
Evans RC, Kyeremateng SO, Asmus L, Degenhardt M, Rosenberg J, Wagner KG. Development and performance of a highly sensitive model formulation based on torasemide to enhance hot-melt extrusion process understanding and process development. AAPS PharmSciTech. 2018;19(4):1592–605.
Wesholowski J, Hoppe K, Nickel K, Muehlenfeld C, Thommes M. Scale-Up of pharmaceutical hot-melt-extrusion: process optimization and transfer. Eur J Pharm Biopharm. 2019;142:396–404.
Zecevic DE, Evans RC, Paulsen K, Wagner KG. From benchtop to pilot scale–experimental study and computational assessment of a hot-melt extrusion scale-up of a solid dispersion of dipyridamole and copovidone. Int J Pharm. 2018;537(1):132–9.
Matić J, Witschnigg A, Zagler M, Eder S, Khinast J. A novel in silico scale-up approach for hot melt extrusion processes. Chem Eng Sci. 2019;204:257–69.
Schittny A, Ogawa H, Huwyler J, Puchkov M. A combined mathematical model linking the formation of amorphous solid dispersions with hot-melt-extrusion process parameters. Eur J Pharm Biopharm. 2018;132:127–45.
Evans RC, Bochmann ES, Kyeremateng SO, Gryczke A, Wagner KG. Holistic QbD approach for hot-melt extrusion process design space evaluation: linking materials science, experimentation and process modeling. Eur J Pharm Biopharm. 2019;141:149–60.
Van Renterghem J, Van de Steene S, Digkas T, Richter M, Vervaet C, De Beer T. Assessment of volumetric scale-up law for processing of a sustained release formulation on co-rotating hot-melt extruders. Int J Pharm. 2019;569: 118587.
Bochmann ES, Neumann D, Gryczke A, Wagner KG. Micro-scale solubility assessments and prediction models for active pharmaceutical ingredients in polymeric matrices. Eur J Pharm Biopharm. 2019;141:111–20.
Rask MB, Knopp MM, Olesen NE, Holm R, Rades T. Comparison of two DSC-based methods to predict drug-polymer solubility. Int J Pharm. 2018;540(1):98–105.
Lu J, Obara S, Ioannidis N, Suwardie J, Gogos C, Kikuchi S. Understanding the processing window of hypromellose acetate succinate for hot-melt extrusion, Part I: Polymer characterization and hot-melt extrusion. Adv Polym Technol. 2018;37(1):154–66.
Thompson SA, Williams RO. Specific mechanical energy – an essential parameter in the processing of amorphous solid dispersions. Adv Drug Deliv Rev. 2021;173:374–93.
Hajikarimi P, Moghadas Nejad F. Chapter 5 - Time–temperature superposition. In: Hajikarimi P, Moghadas Nejad F, editors. Applications of viscoelasticity [Internet]. Elsevier; 2021 [cited 2022 Jan 16]. p. 83–105. Available from: https://www.sciencedirect.com/science/article/pii/B9780128212103000061
Monschke M, Wagner KG. Amorphous solid dispersions of weak bases with pH-dependent soluble polymers to overcome limited bioavailability due to gastric pH variability – An in-vitro approach. Int J Pharm. 2019;564:162–70.
Treffer D, Troiss A, Khinast J. A novel tool to standardize rheology testing of molten polymers for pharmaceutical applications. Int J Pharm. 2015;495(1):474–81.
Shadambikar G, Kipping T, Di-Gallo N, Elia AG, Knüttel AN, Treffer D, et al. Vacuum compression molding as a screening tool to investigate carrier suitability for hot-melt extrusion formulations. Pharmaceutics. 2020;12(11):1019.
Sovizi MR. Thermal behavior of drugs: investigation on decomposition kinetic of naproxen and celecoxib. J Therm Anal Calorim. 2010;102(1):285–9.
Gomes APB, Correia LP, da Silva Simões MO, Macêdo RO. Development of termogravimetric method for quantitative determination of ketoconazole. J Therm Anal Calorim. 2008;91(1):317–21.
Ludovic User’s Manual - v6.2. :210.
Bhujbal SV, Mitra B, Jain U, Gong Y, Agrawal A, Karki S, et al. Pharmaceutical amorphous solid dispersion: a review of manufacturing strategies. Acta Pharm Sin B. 2021;11(8):2505–36.
Struble LJ, Jiang Q. Effects of air entrainment on rheology. ACI Mater J. 2004;101:448–56.
Eitzlmayr A, Khinast J, Hörl G, Koscher G, Reynolds G, Huang Z, et al. Experimental characterization and modeling of twin-screw extruder elements for pharmaceutical hot melt extrusion. AIChE J. 2013;59(11):4440–50.
Bochmann ES, Steffens KE, Gryczke A, Wagner KG. Numerical simulation of hot-melt extrusion processes for amorphous solid dispersions using model-based melt viscosity. Eur J Pharm Biopharm. 2018;124:34–42.
Messaâdi A, Dhouibi N, Hamda H, Belgacem FBM, Adbelkader YH, Ouerfelli N, et al. A new equation relating the viscosity Arrhenius temperature and the activation energy for some Newtonian classical solvents. J Chem. 2015;2015:1.
Acknowledgements
We kindly thank Evonik and Shin-Etsu for providing samples. We further thank Sebastian Kappes, Institute of Pharmacy, University of Bonn, Germany, for reviewing the manuscript.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
Conceptualization: K.K. and K.G.W.; methodology: K.K.; investigation: K.K.; data curation: K.K.; resources: K.G.W.; writing—original draft preparation: K.K.; writing—review and editing: K.K., M.M. and K.G.W.; visualization: K.K.; supervision: K.G.W. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kayser, K., Monschke, M. & Wagner, K.G. ASD Formation Prior to Material Characterization as Key Parameter for Accurate Measurements and Subsequent Process Simulation for Hot-Melt Extrusion. AAPS PharmSciTech 23, 176 (2022). https://doi.org/10.1208/s12249-022-02331-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12249-022-02331-8