
ABSTRACT
The objective of the study was to identify the extragranular component requirements (level and type of excipients) to develop an immediate release tablet of solid dispersions prepared by hot melt extrusion (HME) process using commonly used HME polymers. Solid dispersions of compound X were prepared using polyvinyl pyrrolidone co-vinyl acetate 64 (PVP VA64), Soluplus, and hypromellose acetate succinate (HPMCAS-LF) polymers in 1:2 ratio by HME through 18 mm extruder. A mixture design was employed to study effect of type of polymer, filler (microcrystalline cellulose (MCC), lactose, and dicalcium phosphate anhydrous (DCPA)), and disintegrant (Crospovidone, croscarmellose sodium, and sodium starch glycolate (SSG)) as well as level of extrudates, filler, and disintegrant on tablet properties such as disintegration time (DT), tensile strength (TS), compactibility, and dissolution. Higher extrudate level resulted in longer DT and lower TS so 60–70% was the maximum amount of acceptable extrudate level in tablets. Fast disintegration was achieved with HPMCAS-containing tablets, whereas Soluplus- and PVP VA64-containing tablets had higher TS. Crospovidone and croscarmellose sodium were more suitable disintegrant than SSG to achieve short DT, and MCC was a suitable filler to prepare tablets with acceptable TS for each studied HME polymer. The influence of extragranular components on dissolution from tablets should be carefully evaluated while finalizing tablet composition, as it varies for each HME polymer. The developed statistical models identified suitable level of fillers and disintegrants for each studied HME polymer to achieve tablets with rapid DT (<15 min) and acceptable TS (≥1 MPa at 10–15% tablet porosity), and their predictivity was confirmed by conducting internal and external validation studies.
Similar content being viewed by others

INTRODUCTION
Solid dispersions represent a promising formulation approach for overcoming today’s major challenge in pharmaceutical industry of developing bioavailable solid dosage form for more than 50% of drug candidates that are poorly water soluble (1,2). Solid dispersions can be obtained from various techniques such as hot melt extrusion (HME), spray drying, and co-precipitation (3–5). However, HME is extremely suitable to prepare solid dispersion as this processing technique does not require any solvent or water, which avoids residual amounts of solvent and the accompanying stability risks during the shelf life of solid dispersion dosage form (6–8).
HME process involves transferring and melting of materials inside a heated barrel by rotating screws. The polymer and drug substance melt is then passed through a die opening to obtain extrudates, which can be further processed into granules, tablets, or beads. HME is a continuous, simple, and efficient process. No solvent or water is required, since the molten polymer can function as thermal binder. The intense mixing and agitation by screws during HME de-aggregates particles, reduces the particle size of drug substance, or allows miscible drug substance to dissolve in the molten polymer to form solid dispersion with improved content uniformity and/or bioavailability (6–11).
HME has been successfully applied to enhance solubility of poorly soluble drug substance through formation of an amorphous solid dispersion of drug substance in a polymeric (or lipid) carrier matrix (10,11). Proper selection of polymeric carrier, thorough evaluation of physicochemical properties, stability, and performance assessment of an amorphous solid dispersion are key aspects that should be evaluated to ensure successful development of bioavailable dispersion by HME process (12,13). However, to develop a commercial dosage form the solid dispersion needs to be converted to a final dosage form such as tablets or capsules. It has been observed that when solid dispersions are filled into capsules or compressed into tablets, the disintegration time might be too long (>30 min) due to formation of non-dispersible plug, which results in incomplete or non-optimal drug release (14–16). Polymers are major component in most of the amorphous solid dispersions as the typical drug loading is in the range of 10–50%. The polymers that are commonly used to form solid dispersions such as polyvinyl pyrrolidone (PVP), polyvinyl pyrrolidone co-vinyl acetate 64 (PVP VA64), hydroxypropyl methylcellulose (HPMC), polyethylene oxide (PEO), and hydroxyl propyl cellulose (HPC) have high binding and also gelling properties; hence, their presence in high level in the solid dispersion can result in very long disintegration time of tablets or capsules. It has also been observed that solid dispersions could be less compactible compared to the physical mixture of the blend, which could be attributed to the changes in mechanical properties of the polymer when the blend is passed through the extruder at high temperature and pressure (17,18). During extrusion, the reduced free volume retards molecular mobility, which can prevent further densification during tableting thereby reducing the tensile strength of extrudates (19). Hence, to develop a tablet of an amorphous solid dispersion of sufficient tensile strength and short disintegration time (<30 min), extragranular components with good compactibility and disintegration properties are required to be included in the formulation. However, limited published reports have conducted systematic studies to identify formulation composition requirements to develop immediate release solid dispersion tablets with acceptable drug product properties (20–23). To develop orally disintegrating tablets of ibuprofen solid dispersion prepared by HME process, Gryczke et al. evaluated various grades and level of crospovidone and croscarmellose sodium, which resulted in development of tablets with short disintegration time and acceptable hardness (21). They observed that HME tablets containing crospovidone (Kollidon CL-SF grade, particle size 10–30 μm) exhibited faster disintegration at each studied concentration and compaction force compared to Kollidon CL (particle size 110–130 μm). It was suggested that Kollidon CL-SF possessed a higher water uptake capacity (~7.5–8.5 g water/g polymer) than Kollidon CL (~3.5–5.5 g water/g polymer), which might have resulted in faster disintegration of ibuprofen HME tablets. Liu et al. studied effect of type of fillers (microcrystalline cellulose (MCC), lactose, and dicalcium phosphate dihydrate) to develop lipophilic hot melt-extruded matrix tablets (22). Tablets containing MCC or lactose exhibited increased dissolution rates and higher hardness values compared to dicalcium phosphate dihydrate.
The objective of the current study was to identify suitable type and level of extragranular excipients that are required to develop immediate release tablets of an amorphous solid dispersion prepared by HME process with short disintegration time (<15 min) and adequate tensile strength (≥1 MPa at 10–15% porosity) using a quality by design approach. Commonly used HME polymers such as PVP VA64, Soluplus, and HPMCAS were selected for evaluation in this study. Solid dispersions were prepared at 33.3% drug loading and 66.6% polymer loading using each studied HME polymer. It is hypothesized that polymer properties will have major influence on tablet properties as HME polymer is major component in these solid dispersions. The findings from this study may guide other scientists to develop HME tablets with adequate properties (fast disintegration and good tablet tensile strength) for the studied HME polymers.
MATERIALS AND METHODS
Materials
Compound X was supplied by the Chemical Development Department of Boehringer Ingelheim Pharmaceuticals, Inc. (Ridgefield, CT, USA). The copovidone also known as PVP VA 64 (Kollidon® VA64), Soluplus ®, and crospovidone (Kollidon® CL) were obtained from BASF (Ludwigshafen, Germany). Hypromellose acetate succinate (HPMCAS-LF) was obtained from Shin Etsu (Somerset, NJ). Microcrystalline cellulose (Avicel® PH 200) and croscarmellose sodium (Ac-Di-Sol® SD-71) were purchased from FMC BioPolymer (Philadelphia, PA). Lactose (Tablettose® 80) was purchased from Meggle (Wasserburg, Germany). Dicalcium phosphate anhydrous (DCPA) was purchased from Innophos (Cranbury, NJ). Sodium starch glycolate (SSG) was purchased from Roquette (Lestrem, France). Colloidal silicon dioxide (Aerosil® 200 P) was purchased from Evonik Degussa Corporation (Parsippany, NJ). Magnesium stearate was purchased from Peter Greven (Bad Münstereifel, Germany). Other pharmaceutical excipients of compendial grade were used as received.
Design of Experiments for HME Tablet Formulations
To systematically evaluate the extragranular component requirements for manufacture of immediate release HME tablets, a design of experiment (DOE) approach was employed. The DOE consisted of six factors—three categorial (type of polymer, filler, and disintegrant) and three continuous (levels of polymer, filler, and disintegrant) factors. The factors are described in Table I. Since the factors are both categorial and continuous, a mixture design was chosen based on Scheffe model. The magnesium stearate and colloidal silicon dioxide (CSD) level were kept constant at 1% each in the HME tablet.
Design-Expert® software (Stat-Ease Inc., Minneapolis, MN) was used for creating level IV optimal coordinate exchange design type. Level IV optimal design was selected as it provides lower average prediction variance across the region of experimentation. Design model was reduced quadratic and main effects with a total of 34 runs (Table II). A full factorial design would have resulted in 110 batches. The three-way and four-way interactions are rare hence excluded from the DOE design model, which resulted in reduction in total number of batches from 110 to 34. The design consisted of center point experiments and repeated runs to allow for evaluation of curvature in the chosen models. The effect of main factors and two-way interactions were assessed by this design. The response (dependent) variables were tablet disintegration time, tensile strength, compression force, and dissolution.
Preparation of Solid Dispersion by Hot Melt Extrusion
The compound X was blended with each of the studied HME polymer (PVP VA64, Soluplus®, HPMCAS-LF) in the ratio of 1 to 2, which resulted in drug concentration of 33.3%. For a 3-kg batch, blending was carried out using a 17-l Turbula blender (Glen Mills, Clifton, NJ) for 10 min at 34 rpm. The hot melt extrusion process was conducted using 18 mm extruder (Leistritz, Somerville, NJ), equipped with intermeshing co-rotating twin screws.
The 18-mm extruder consisted of seven heating zones, co-rotating twin screws, and a die plate with 3-mm round die opening. All heating zones were maintained at 180°C during the extrusion run for each studied HME polymer batch. The screw profile consisted of various types of conveying elements and a set of kneading elements of 30°, 60°, and 90° angle to provide adequate mechanical shear energy for formation of an amorphous solid dispersion. The 18-mm extruder was equipped with a twin screw gravimetric dosing feeder (K-Tron, Pitman, NJ). For each studied HME polymer batch, the extrusion was conducted at feed rate of 30 g/min and screw speed of 300 rpm. The extrusion process was monitored by recording melt temperature, melt pressure, and torque values throughout the extrusion run.
The cooled extrudates were milled using a quadro co-mill U10 (Waterloo, Canada) in two stages. First the extrudates were milled through eight mesh screen to break long extrudate into pieces followed by milling through 18 mesh screen using 17 Hz frequency at 1026 rpm for each studied HME polymer batch. Milled extrudates were stored in a sealed aluminum pouch until processed further to keep them moisture free.
Manufacture of HME Tablets
The final blend for each DOE run was prepared using the following procedure:
-
Step 1—The milled extrudates, filler, and disintegrant were weighed according to the DOE run formula and mixed in a Turbula blender (Glen Mills, Clifton, NJ) for 5 min at 34 rpm.
-
Step 2—Colloidal silicon dioxide and magnesium stearate were mixed by hand with a small amount of the blend from the step 1.
-
Step 3—The blend from step 2 was added to the remaining blend from step 1 and mixed in a Turbula blender (Glen Mills, Clifton, NJ) for 10 min at 34 rpm.
The final blend of each DOE run was compressed into tablets of varying porosity by changing the compression force on Instron-5867 system (Universal Testing System, Norwood, MA) using 30 kN load cell and 9 mm round flat-faced tablet punch set. The platen travel speed was maintained at 0.05 mm/s. Tablet weight, diameter, hardness, and thickness were measured to determine the tensile strength and porosity of the compact. The diametral compression test defined by Fell and Newton (24) was used to determine the tensile strength T, using the formula:
where P (N) is the diametrical tablet break force, D (cm) is the diameter of the tablet, and t (cm) is the tablet thickness.
The true density of final blend of each DOE run was measured in grams per cubic centimeter by helium pycnometer using a Micromeritics Accupyc 1330 helium pycnometer (GA, USA). The solid fraction (SF) and porosity (ε) were calculated based on the true density (ρ true), tablet volume (v), and tablet weight (W) as shown below (25):
For each DOE run, from the compaction profile, the compression force required to achieve target tablet porosity of 12 ± 3% was determined. Additional tablets were compressed at targeted compression force to conduct hardness, dissolution, and disintegration testing.
Characterization of Milled Extrudates and Tablets
X-Ray Powder Diffraction (XRPD)
XRPD analysis was performed at ambient temperature using a Bruker AXS X-Ray Powder Diffractometer Model D8 Advance (Karlsruhe, Germany), at 40 mA and 40 kV with CuKα radiation (1.54 Å) in parallel beam mode utilizing a scintillation detector. Samples were scanned over a range of 2θ values from 3° to 35° with a step size of 0.05° (2θ) and a counting time of 4 or 0.6 s. A 1-mm divergence slit was used with the incident beam along with 0.12-mm soller slits in the diffracted beam path. A sodium iodide scintillation detector was used.
Polarized Light Microscopy (PLM)
Polarized light microscopy was performed using an Olympus BX51 Polarizing Microscope (Olympus Optical Company, Westmont, IL) with objective of ×20 and ocular magnification of ×10. Small amount of sample was placed on the glass slide followed by addition of a drop of oil, which was covered with cover slip and then examined for birefringence. Sample analysis was done by using SPOT advanced software.
Thermal Analysis (mDSC)
Modulated differential scanning calorimetry (mDSC) was performed on a DSC Q1000 (TA Instruments, New Castle, DE). Data analysis was done using Universal Analysis 2000 thermal analysis software (TA instruments, New Castle, DE). Calibration was performed using sapphire as per the standard procedure recommended by TA Instruments. For mDSC testing, samples of 2–4 mg were weighed and placed in aluminum crimped pans. The samples were equilibrated at 20°C and then heating-cooling-heating cycle was performed. During heating cycle, the mDSC parameters were modulated at 0.636°C every 60 s with heating rates of 2°C/min from 20°C to 230°C followed by holding the sample isothermally for 1 min. During cooling cycle, the samples were cooled at 2°C/min to −10°C followed by holding the sample isothermally for 1 min and then the samples were subjected to heating cycle. All measurements were performed under nitrogen purge.
Particle Size distribution
Milled extrudates of each HME polymer batch were analyzed with the Sympatec QICPIC (Clausthal-Zellerfeld, Germany) system using the Aspiros attachment. The analysis was performed using from 1 to 2 bar pressure with a feed rate of 15 to 30 mm/s. Multiple runs were performed using sample amounts from 50 up to 350 mg of material. The quality of the resulting data was determined by the total particle count and the percent obscuration of the system.
The sieve analysis of milled extrudates was performed using an ATM Arrow Sonic Sifter (Model L3P, Advantech Manufacturing, New Berlin, WI). A sample size of about 5 g was shifted though 1000, 500, 250, 125, and 63 μm sieves with the sift amplitude set at 6, pulse enabled, and the timer set for 2 min.
Hardness, Disintegration, and Dissolution Testing of HME Tablets
The hardness testing was performed on tablets using the Schleuniger Pharmatron hardness tester (Solothum, Switzerland) where a single tablet is place between the moving metal platen and the diametrical force required to break the tablet was measured.
The disintegration testing was done in this study using USP disintegration apparatus (Erweka ZT 71, Doylestown, PA). The medium used was 0.1 N HCl at 37°C.
Dissolution of milled extrudates and tablets was performed using Leap OD Lite UV Fiber optic system (North Brunswick, NJ). A two-step non-sink dissolution method was developed to assess the performance of solid dispersions and HME tablets. The samples were suspended in 40 mesh basket. In the first step, dissolution testing was conducted in 300 ml of simulated gastric fluid at pH = 2 for 30 min and in the second step 600 ml phosphate buffer containing 0.15% sodium dodecyl sulfate (SDS) was added to make a final 900-ml volume of pH = 6.5 media containing 0.1% SDS. The amount of drug released was then monitored in the combined media at 330 nm.
RESULTS AND DISCUSSIONS
Hot Melt Extrusion Process and Extrudate Characterization
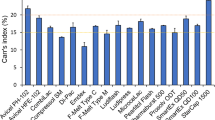
In this study, a weakly basic drug substance (referred in this article as compound X), which belongs to BCS class II category was used. The compound X has poor aqueous solubility (intrinsic solubility of 18 μg/ml), moderate hydrophobicity (log P of 2.6), and pK a of 2.5 (shows pH-dependent solubility). The compound X has melting point of 207°C, glass transition temperature (T g) of 77°C, and decomposes above 275°C. The anyhydrous free base form of compound X was used in the development of drug product. The average particle size (D 50 ) of the used lot was 14.3 μm, D 10 was 5.5 μm, and D 90 was 26.3 μm. The average bulk density of compound X was 0.226 g/ml, and tap density was 0.416 g/ml with resultant Carr index value of 45.6, which suggested poor flow characteristics of compound X.
The extrusion of compound X with each HME polymer (PVP VA64, Soluplus, and HPMCAS-LF) was conducted at 180°C in 18 mm extruder, which resulted in formation of transparent extrudates. For Soluplus and PVP VA64-containing batches, light yellow clear extrudates were obtained, whereas for HPMCAS-LF-containing batch, the extrudates were dark yellowish brown in color and less transparent. The degradation temperature of PVP VA64 (tdeg > 220°C) and Soluplus (tdeg > 250°C) is much higher than the extrusion temperature of 180°C; hence, no significant change in color of extrudates was observed during extrusion of compound X blend with PVP VA64 or Soluplus. Whereas, the degradation temperature of HPMCAS is around 190°C so at extrusion temperature of 180°C some changes in polymer structure might have occurred, which resulted in darker color of extrudates. During extrusion, the torque value was in the range of 50–55% for each polymer batch suggesting that selected extrusion conditions were adequate and did not result in generation of excessive pressure inside the extruder. For Soluplus- and PVP VA64-containing batches, the die melt pressure during extrusion was in the range of 145–155 PSI, whereas for HPMCAS-LF batch, the die melt pressure was higher in the range of 180–200 PSI, which could be due to high viscosity of the HPMCAS-LF polymer compared to Soluplus and PVP VA 64 polymers. The cleaning of extruder was challenging after the extrusion of HPMCAS-LF batch compared to Soluplus and PVP VA64 batch. The HPMCAS-LF-containing extrudates did not break into smaller pieces easily; hence, two-stage milling was required and also frequent blinding of the screen was observed during milling. Comparatively, it was easy to mill the Soluplus- or PVP VA64-containing extrudates. But to keep the milling procedure consistent, two-stage milling was employed during milling of each dispersion system.
Milled extrudates of each studied HME polymer batch were x-ray amorphous (Fig. 1). Polarized light microscopy did not reveal any presence of birefringence material for solid dispersion of compound X with PVP VA64 (Fig. 1). For compound X-Soluplus, solid dispersion slight birefringence was observed, which was more pronounced for compound X-HPMCAS-LF solid dispersion (Fig. 1). These results indicate that compound X has better miscibility in PVP VA64 polymer compared to Soluplus and HPMCAS-LF polymer at 1:2 ratio.

X-ray powder diffractograms and PLM images of compound X solid dispersion extrudates containing Soluplus, PVP VA64, and HPMCAS-LF polymer
The mDSC analysis of milled extrudates of compound X-PVP VA64 batch showed presence of only single T g of ~88.3°C and no melting endotherm, suggesting formation of an amorphous solid dispersion. For compound X solid dispersion with HPMCAS and Soluplus polymers, a small broad melting endotherm was observed by mDSC (Fig. 2), suggesting formation of a partially amorphous solid dispersion. However, HPMCAS- and Soluplus-containing solid dispersions were x-ray amorphous suggesting that these solid dispersions are primarily amorphous with low level of crystallinity, which could be detected only my more sensitive techniques such as PLM and mDSC.

DSC thermograms of compound X and solid dispersion extrudates of compound X containing Soluplus, PVP VA64, and HPMCAS polymer
Results of the sieve analysis on the milled extrudates are summarized in Fig. 3. The average particle size of the milled extrudates at 33.3% drug load for all three HME polymeric system was similar around 500 μm. For each studied HME polymer batch low percentage of fines below 63 μm and low percentage of large particles above 500 μm were observed. Even though there was a difference in the brittle property of the glassy extrudates of three studied HME polymeric systems, which required some adjustment to milling conditions while milling the extrudates, the particle size distribution of the milled extrudates of each HME polymer batch was similar. The number and volume distribution particle size curves that were obtained by QICPIC method (data not shown) for milled extrudates of each HME polymer batch were also similar. Hence, the milled extrudates from each HME polymer batch were considered to be suitable to be included in HME DOE study.

Sieve analysis results of compound X-milled extrudates containing Soluplus, PVP VA64, and HPMCAS-LF polymer
The compactibility profile of compound X, each studied HME polymer (PVP VA64, Soluplus, and HPMCAS) prior to extrusion, and milled extrudates of each studied HME polymer after extrusion is shown in Fig. 4. As shown in Fig. 4, the compactibility of milled extrudates of each studied polymer was lower compared to compactibility of “as is” HME polymers prior to extrusion. The compactibility of HPMCAS-containing extrudates decreased the most, whereas the compactibility of Soluplus-containing extrudates was not much lower compared to “as is” Soluplus (Fig. 4). Iyer et al. (19) noted that for HPMCAS polymer, an increase in pressure, heat, and shear stress during extrusion could result in significant increase in strength of milled HME extrudates with diminished ability to form strong bonds at points of contact during compaction; thus, HPMCAS extrudates may have reduced ability to form strong compacts upon compression.

Compactibility profiles of “as is” compound X and HME polymers and milled extrudates of each studied HME polymer after extrusion
HME Tablet Disintegration—Statistical Analysis
The tablet disintegration time range for PVP VA64-containing HME tablets was 202–2218 s, Soluplus-containing HME tablets was 16–1597 s, and HPMCAS-containing tablets was 7–64 s indicating strong influence of polymer type on disintegration time. On an average, the longest disintegration time was observed with PVP VA64 polymer, followed by Soluplus, and the shortest disintegration time was achieved with HPMCAS-LF polymer (less than 1.5 min). In combination with different type of disintegrants, the longest disintegration time was observed for PVP VA64-containing HME tablets with SSG followed by croscarmellose sodium and crospovidone, whereas for HPMCAS-LF-containing HME tablets, minimal influence of disintegrant type or other factors was observed on disintegration time.
The statistical analysis of tablet disintegration time results was performed, and results are presented in Table III. The disintegration time data fitted the log transform model well, and the lack of fit was not significant suggesting that the chosen model is valid. The "predicted R-squared" of 0.9346 was in reasonable agreement with the "adjusted R-squared" of 0.9441 (difference < 0.20), which indicated good predictability of the selected model. The residuals were randomly distributed, and a good correlation was observed between actual versus predicted values. The significant main factors were polymer type (D), disintegrant type (E), and filler type (F) (Table III). The interaction between filler level (B) and each categorial factor was significant (Table III). Also, interactions between extrudate/intragranular level (C) and disintegrant type (E) or polymer type (F) were found to be significant (Table III).
The effect of polymer type, disintegrant type, and disintegrant level on tablet disintegration time is depicted in Fig. 5. Statistical analysis results indicated that use of PVP VA64 polymer in combination with SSG or croscarmellose sodium as disintegrant would result in long tablet disintegration time, whereas use of crospovidone as disintegrant in PVP VA64-containing solid dispersion tablets would result in a much faster disintegration time. The use of Soluplus as HME polymer resulted in significant reduction in disintegration time of HME tablets. However, shortest disintegration time was attained with HPMCAS-LF polymer with any of the three studied disintegrants. Strong influence of type of polymer on tablet disintegration time was observed in the current study. PVP VA64 is a vinylpyrrolidone-vinylacetate copolymer with good water solubility, binding, and compactibility properties. Soluplus is a polyvinyl caprolactam-polyvinyl acetate-polyethylene glycol graft copolymer. It is a polymeric solubilizer with an amphiphilic chemical structure and is soluble in water. The PVP VA64 polymer is almost two times lower in molecular weight compared to Soluplus and it has high binding capacity. HPMCAS is a cellulosic polymer with four types of substituents (methoxy, hydroxypropyl, acetate, and succinate) semirandomly substituted on hydroxyl groups. It is an acidic polymer and dissolves above pH 5.5. Due to presence of relatively hydrophobic methoxy and acetate substituents, HPMCAS is water insoluble when un-ionized (below pH 5.5) hence does not swell or forms gel in low pH medium. In the present study, swelling to some extent and gel layer formation for PVP VA64-containing tablets were observed by image analysis (data not shown) compared to Soluplus- and HPMCAS-containing tablets in 0.1 N HCl medium, which might have resulted in long disintegration time for PVP VA64-containing HME tablets. Differences in chemical nature of the studied polymer, and their water solubility as well as binding capacity might have resulted in differences in HME tablet disintegration time as these polymers are present in high amount (40–53.3%) in HME tablets. Similar behavior was observed for other compounds in our lab when solid dispersion tablets were prepared using PVP VA64, Soluplus, and HPMCAS polymers.

Interaction plots for HME tablet disintegration time: effect of polymer type, disintegrant type, and disintegrant level
In general, use of crospovidone as disintegrant with any of the three studied HME polymers resulted in shortest disintegration time of HME tablets when average level of disintegrant was 7.5%, MCC was used as filler at 20.5%, and the intragranular milled extrudate level was 70.0% (Fig. 5). Decreasing the disintegrant level to 5.0% appears to impact mostly SSG and crospovidone with PVP VA64-containing tablets, by increasing the total disintegration time. Similarly increasing the disintegrant level to 10.0% appears to impact mostly SSG and crospovidone with PVP VA64-containing tablets, by decreasing the total disintegration time (Fig. 5). While developing orally disintegrating tablets of Ibuprofen solid dispersion using Eudragit® EPO polymer by hot melt extrusion process, Gryczke et al. (21) observed that 5–20% level of various grades of crospovidone (polyplasdone XL, polyplasdone XL10, and Kollidon CL-SF) and croscarmellose sodium (Vivasol) as well as 2–5% level of Kollidon CL grade of crospovidone results in short disintegration time. Laitinen et al. (23) achieved fast disintegration time of ~37 s for perphenazine solid dispersion tablets by including 15% crospovidone in the formulation. Similarly, Akbuga et al. (17) studied effect of three disintegrants such as corn starch, SSG, and crospovidone (Kollidon CL grade) on disintegration time of furosemide-PVP solid dispersion tablets and it was observed that fast disintegration was achieved with crospovidone-containing tablets due to faster water penetration rate compared to other disintegrants. Disintegrants are essential components of tablet formulations to achieve fast drug release. The ability to interact strongly with water is essential to disintegrant function. Combinations of swelling and/or wicking and/or deformation are the mechanisms of disintegrant action. Water penetration rate and rate of disintegration force development are generally positively related to the disintegrant efficiency in non-soluble matrices (26). The SSG imparts its disintegrating effect by rapidly swelling in contact with water (7–12-fold), which results in rupture of interparticulate bond and break up of tablet (26,27). The croscarmellose sodium acts as a super disintegrant by swelling to moderate level (4–8-fold) and wicking through capillary action to rupture the tablets. Whereas, crospovidone swells to some extent and primarily act as disintegrant through wicking action to generate the volume expansion and hydrostatic pressures necessary to provide rapid disintegration of tablets (26). In the present study, it was observed that crospovidone was more effective disintegrant compared to croscarmellose sodium and SSG for PVP VA64-containing HME tablets. Crospovidone has porous particle morphology, which facilitates rapid penetration of liquid into the tablet, as noted by Akbuga et al. (17). In addition, due to its high crosslink density crospovidone swells rapidly in water without gelling. Whereas, other disintegrants such as SSG and croscarmellose sodium have lower crosslink density, hence have a tendency to form gel when fully hydrated, particularly at higher use levels, which could result in slower disintegration time compared to crospovidone as observed in the present study and by several other researchers (26,28–30).
In the present study, the disintegrant level did not impact disintegration time significantly for each of the three studied disintegrants when HME tablets contained Soluplus or HPMCAS-LF polymer (Fig. 5), suggesting that other formulation components in addition to disintegrant type can influence tablet disintegration time and in this study it is polymer type. The intragranular level did not show any major impact on the HME tablet disintegration time in the presence of various combinations of disintegrants and polymers (data not shown).
The statistical analysis showed that when the MCC filler was at lowest (8%) or highest (33%) level, it resulted in corresponding decrease and increase in disintegration time. Holding all other factors constant but using lactose instead of MCC as the filler appears to shorten the disintegration of HME tablets (Fig. 6). Similar relationship between polymer and disintegrant type and resulting disintegration prevails with lactose as a filler in HME tablets instead of MCC. Holding all other factors constant but using DCPA as the filler appears to increase the disintegration time compared to lactose, similar to MCC (Fig. 6). Similar relationship between polymer and disintegrant type and resulting disintegration prevails when DCPA is used as a filler in HME tablets instead of MCC. Liu et al. (22) studied effect of type of fillers (MCC, lactose, and dicalcium phosphate dihydrate) to develop lipophilic hot melt-extruded matrix tablets. The tablets containing MCC or lactose exhibited increased dissolution rates and higher hardness values compared to dicalcium phosphate dihydrate. The observed differences in the disintegration and dissolution properties of the tablets were due to the differences in the solubility, swellability, and density of the filler excipients. Dicalcium phosphate is AN insoluble, non-swelling, and high density filler, which might have resulted in slower disintegration time in the present study compared to lactose, which is a soluble filler. MCC has a very high intra-particle porosity, which generally promotes swelling and disintegration of MCC-containing tablets by capillary action (31). However, MCC is highly compressible filler, which generally results in harder tablets hence might have resulted in slightly longer disintegration time compared to lactose especially at high MCC level in HME tablets.

Summary plots for HME tablet disintegration time: effect of filler type, disintegrant type, and polymer type
HME Tablet Compression Force—Statistical Analysis
The statistical analysis of compression force at 12 ± 3% porosity of HME tablets was conducted using ANOVA, which resulted in selection of linear mixture model. The adjusted R 2 (0.9487) and predicted R 2 (0.9397) values were within 0.20 of each other suggesting adequacy of the selected model to observe effect of extragranular components on compression force required to compact HME tablet at 12 ± 3% porosity. The significant main factors were D (polymer type) and E (disintegrant type) as the P value was less than 0.05. Significant interactions were observed between factors—filler level (B) in combination with D (polymer type), E (disintegrant type), and F (filler type). Also, interactions between the intragranular level (C) in combination with polymer type (D) and disintegrant type (E) were found to be significant.
Statistical analysis results showed that highest compression force was required for HPMCAS-containing tablets, followed by Soluplus and PVP VA64. Statistical analysis results were in line with the compactibility profile results obtained for milled extrudates of each studied HME polymer (Figs. 4 and 7). Similarly, Iyer et al. (19) observed in their study that upon hot melt extrusion, HPMCAS looses its compactibility and much higher compression force is required to form tablets of adequate tensile strength compared to PVP VA64. An increase in particle density and loss of voids can occur due to heat and shear stress during hot melt extrusion, which can reduce the ductility of the extruded material. The reduced ductility and bonding capacity were more significant for HPMCAS compared to other HME polymers such as Soluplus and PVP VA64, which resulted in higher compression force as noted in current study and by Iyer et al. (19). For HPMCAS-containing tablets, significant effect of disintegrant type on required compression force was observed at 60% intragranular level of extrudates (Fig. 7). When crospovidone was used as a disintegrant in the HPMCAS-containing tablets, lower compression force was required compared to other disintegrants at 60% intragranular level of extrudates (Fig. 7). As noted by Mohanchandran et al. (26), crospovidone is highly compressible material due to its unique particle morphology; hence, lower compression force is generally required to make tablets with desired tensile strength and porosity compared to other disintegrants. For PVP VA64- and Soluplus-containing tablets, disintegrant type did not affect the compression force to significant extent at 70% and 80% intragranular level of extrudates. The decrease in intragranular level to 60% mostly impacted HME tablet formulations containing SSG and croscarmellose Na by resulting in an increase in compression force for PVP VA64 and Soluplus polymer system. A higher compression force was required for DCPA-containing HME tablet formulation compared to MCC and lactose filler to achieve tablets with 12 ± 3% porosity (data not shown). DCPA is an inorganic material with high density, high dynamic hardness, and low bonding capacity; hence, to form tablets of adequate tensile strength, higher compression force is generally required for formulations containing DCPA compared to other fillers such as MCC and lactose (19).

Interaction plots for HME tablet compression force: effect of polymer type, disintegrant type, and intragranular level
HME Tablet Tensile Strength—Statistical Analysis
The statistical analysis of tablet tensile strength as one of the response variables was conducted using ANOVA, which resulted in selection of liner mixture model. The adjusted R 2 (0.920) and predicted R 2 (0.907) values were within 0.20 of each other, and lack of fit was not significant (P = 0.999) suggesting adequacy of the selected model to observe effect of extragranular components on tablet tensile strength at 12 ± 3% porosity of the HME tablet. The disintegrant type (E) and filler type (F) were significant main factors. Statistical analysis result showed that filler level (B) in combination with D (polymer type), E (disintegrant type), and F (filler type) was having a significant effect on tensile strength of the HME tablet (P < 0.05). The intragranular level (C) in combination with polymer type (D) and disintegrant type (E) was also having a significant effect (P < 0.05). The P value was close to 0.05 (P = 0.047) for the intragranular level (C) in combination with filler type (F) in HME tablet, suggesting that the effect was marginally significant.
Results of statistical analysis of HME tablet tensile strength indicated that for the given level of fillers and disintegrants, highest tensile strength was observed with Soluplus® and PVP VA64 polymer-containing tablets, whereas use of HPMCAS-LF resulted in low tablet tensile strength (Fig. 8). For HPMCAS, an increase in particle density and changes in polymer chain orientation during hot melt extrusion might have resulted in higher strength of extrudates, which might have resulted in diminished ability to form strong bonds at the point of contacts thereby resulting in tablets of lower tensile strength, as noted by Iyer et al. (19). The authors did not observe significant changes in material density for PVP VA64; hence, the extrudates were not very hard upon extrusion. Moreover, their study results indicate that strain index, a measure of elastic recovery after compression, is high for HPMCAS compared to PVP VA64, which might have resulted in lower tensile strength of tablets. The molecular weight and viscosity of HPMCAS-LF polymer are higher compared to Soluplus and PVP VA64, which could result in less ductile and more elastic extrudates with reduced bonding capacity compared to Soluplus and PVP VA64 as evident by milling challenges during milling of HPMCAS extrudates and higher compression force required during compaction in the current study.

Interaction plots for HME tablet tensile strength: effect of polymer type, disintegrant type, and disintegrant level
The HME tablet tensile strength with crospovidone or croscarmellose Na as disintegrant was significantly higher across all three polymeric system as compared to the HME tablet containing SSG (Fig. 8). Similarly, Gryczke et al. (21) observed that use of crospovidone or croscarmellose Na as disintegrant resulted in high tablet tensile strength while developing hot melt-extruded orally disintegrating tablets. Crospovidone is a porous material and acts as both binder and disintegrant in the tablet, which could result in stronger tablets compared to croscarmellose Na and mainly SSG (32). SSG is more elastic in nature; hence, it can negatively impact tablet tensile strength compared to crospovidone or croscarmellose Na (26). An increase in an intragranular level of extrudates from 60% to 80% resulted in a decrease in the tablet tensile strength for each type of studied polymer and disintegrant (Fig. 9). Inclusion of lactose in HME tablet resulted in a decrease in the tablet tensile strength significantly when compared with MCC (Fig. 10). MCC is a plastic filler, whereas lactose is a brittle filler; hence, better deformation under compaction pressure and bonding capacity can be achieved with MCC, which can result in higher tablet tensile strength as noted by other researchers (19,22). In the present study, inclusion of DCPA resulted in an increase in the tablet tensile strength the most, when compared to lactose or MCC (Fig. 10). Although DCPA requires high compression force to form compacts due to its high density and high dynamic hardness, but it has very low elasticity compared to other fillers such as MCC and lactose hence tablets of high tensile strength can be achieved using DCPA (19).

Interaction plots for HME tablet tensile strength: effect of polymer type, disintegrant type, and intragranular level

Summary plots for HME tablet tensile strength: effect of filler type, disintegrant type, and polymer type
HME Tablet Dissolution—Statistical Analysis
The dissolution testing of milled extrudates and HME tablets of each DOE run was conducted using two-step non-sink dissolution method. The overall dissolution profile for DOE batches of each studied HME polymer is presented in Figs. 11, 12, and 13.

Dissolution profiles of compound X and Soluplus HME DOE batches and milled extrudates

Dissolution profiles of compound X and PVP VA64 HME DOE batches and milled extrudates

Dissolution profiles of compound X and HPMCAS-LF HME DOE batches and milled extrudates
The Soluplus-containing milled extrudates and HME tablets showed slow dissolution rate with slow increase in release throughout the dissolution testing up to 120 min (Fig. 13). It is important to note that Soluplus-containing solid dispersion was not completely amorphous and HME tablets of each DOE run showed complete disintegration during the dissolution testing. Compared to milled extrudates, the drug release from HME tablet was higher for most of the DOE batches suggesting that extragranular components are influencing the drug release (Fig. 13). The HME tablets from DOE run no. 31 and 27 had very long disintegration time (>20 min), which could have resulted in slow drug release during first stage of dissolution testing. However, very low drug release from HME tablet of DOE run no. 16 cannot be explained by disintegration time as the tablet disintegration time was short (~2 min), suggesting that other factors are influencing drug release. In general, the slow dissolution rate obtained from Soluplus dispersions is similar to previous preliminary experimental results (data not shown) and appears to be inherent characteristic of compound X/Soluplus solid dispersion. In a study conducted by Pudlas et al. (33), the impact of different excipients on drug release behavior in HME formulations was analyzed. Their results indicate slow drug release from Soluplus-containing extrudates compared to PVP VA64-containing extrudates, which was attributed to stronger interaction between ibuprofen and Soluplus than that of water and Soluplus, thus making it difficult for water molecules to displace drug molecules H-bonded to the polymer and facilitate drug release. It was also noted that hydrophilicity of PVP VA64 is higher than Soluplus, which can result in faster drug release from PVP VA64-containing HME formulations (33).
The dissolution from PVP VA64-containing milled extrudates and HME tablets of various DOE runs was very different suggesting strong influence of tablet properties and extragranular components on drug release (Fig. 13). It is important to note that PVP VA64-containing solid dispersions were completely amorphous suggesting higher miscibility of compound X in PVP VA64 polymers compared to Soluplus and HPMCAS. The HME tablets of varying DOE runs showed complete disintegration during the dissolution testing. The HME tablets from DOE runs with greater than 15 min disintegration time (DOE run nos. 11, 18, 25, 28, and 33) showed very slow and suppressed drug release throughout the dissolution testing (Fig. 13). The DOE runs with less than 15 min disintegration time did show fast drug release during first stage of dissolution testing, but the dissolution profile was different compared to milled extrudate dissolution profile, suggesting that extragranular components type and level are affecting the drug release. It was interesting to note that drug release from fast disintegrating HME tablets was decreasing during the SIF phase of dissolution (second stage) compared to milled extrudates, suggesting that extragranular components are negatively impacting the supersaturation maintenance of the solid dispersion (Fig. 13). These dissolution results of PVP VA64-containing HME tablets clearly suggest that not only the solid dispersion components (intragranular excipients) but the extragranular components as well can positively or negatively impact the dissolution performance; hence, selection of these components requires careful evaluation while developing solid dispersion dosage form for any given compound. Similarly, other researchers have also observed differences in dissolution properties of the HME tablets when different types of extragranular excipients such as fillers and disintegrants are used (17,22,34).
The HPMCAS-LF polymer dissolves only above pH 5.5; hence, less than 10% drug release was observed for most of the DOE batches during first stage of dissolution testing, which is pH = 2 SGF media, for first 30 min (Fig. 13). The HPMCAS-containing HME tablets disintegrated rapidly during dissolution testing as the disintegration time of each DOE batch was less than 2 min. Compared to PVP VA64- and Soluplus-containing extrudates, the release from HPMCAS-containing extrudates was higher, which could be due to stronger interaction between compound X and HPMCAS compared to other polymers. Sarode et al. (35) observed high dissolution rate and supersaturation of various poorly soluble drugs due to strong drug-polymer interactions when Eudragit EPO, HPMCAS-LF, and Eudragit L-100-55 were used as HME polymers in comparison with PVP VA64. The drug release from HME tablets was lower than milled extrudates for most of the DOE runs, suggesting that presence of extragranular components in HPMCAS-containing HME tablet is negatively impacting the drug release (Fig. 13).
The statistical analysis was conducted at multiple dissolution time points (22, 38, 60, and 120 min) to understand the effect of DOE factors on various phases of dissolution testing. Statistical analysis results of these dissolution time points are summarized in Table IV. As expected, the polymer type had a significant impact on dissolution, which is mainly related to differences in polymer chemistry and interaction potential between drug substance and polymer (33,35). At early dissolution time points (<40 min), PVP VA64-containing dispersion and tablets showed higher drug release, whereas at later time points (>40 min), HPMCAS-containing dispersion and HME tablets showed higher release due to better maintenance of supersaturation. In the case of PVP VA64-containing tablets, some of the extragranular components had negative effect; hence, supersaturation could not be maintained in second stage of dissolution (Table IV). In general, an increase in intragranular level/extrudate level resulted in a decrease in drug release suggesting that an optimum balance between intragranular and extragranular level is required to achieve desired drug release. Use of croscarmellose Na or crospovidone resulted in higher drug release compared to SSG, which could be due to faster tablet disintegration when croscarmellose Na or crospovidone were used as disintegrants. Positive effect of crospovidone on enhancing dissolution from solid dispersion tablet was also observed by Akbuga et al. (17). The impact of disintegrant type on dissolution was more significant for PVP VA64-containing HME tablets (Table IV). In terms of filler, presence of lactose or MCC resulted in higher drug release compared to DCPA, which could be related to higher solubility of these fillers compared to DCPA. In addition, DCPA-containing tablets had higher tensile strength and longer disintegration time, which might have resulted in a negative impact on drug release. Liu et al. (22) observed that drug release from HME tablets decreased in the order of using MCC, lactose, and dicalcium phosphate dihydrate as the filler excipient, which was attributed to the differences in the solubility, swellability, and density of these filler excipients.
Optimization of HME Tablet Composition
Based on statistical models identified for each response variables (compression force, disintegration time, and tensile strength), the optimization analysis was performed to identify suitable combinations of extragranular excipients to achieve HME tablets with low compression force (optimization criteria set at <20 kN to achieve 12 ± 3% tablet porosity), high tablet tensile strength (optimization criteria set at ≥1 MPa), and short disintegration time (optimization criteria set at <15 min) for each studied HME polymer. These optimization criteria were mainly met when crospovidone was used as disintegrant and MCC was used as a filler for each studied HME polymer (PVP VA64, Soluplus, and HPMCAS-LF). The requirement for level of disintegrant and filler varied based on the type of studied HME polymer. Croscarmellose Na-containing formulations also met the optimization criteria at the slightly higher compression force, whereas SSG offered very limited opportunities to meet the requirements as its use resulted in long disintegration time, low tablet tensile strength, and also negatively affected the dissolution for each studied HME polymer. Lactose-containing formulations also met the optimization criteria mostly for the PVP VA64 and Soluplus polymer SYSTEM. Whereas, MCC was required as filler for HPMCAS-LF HME tablets to meet the optimization criteria due to loss of compactibility and compressibility during extrusion of HPMCAS-LF-containing blends. Adequate design space for each studied HME polymer was identified by optimization analysis based on the upper and lower limits chosen for each response variable. Internal and external validation studies were conducted to confirm the predictability of identified statistical models for each response variable by selecting one of the optimization solution/run for each studied HME polymer.
The internal validation was conducted using the same compound as used in HME DOE, i.e., compound X, using the optimization solutions obtained from statistical models. The PVP VA64-containing HME tablets had very long disintegration time for most of the DOE batches. Hence, compound X-PVP VA64 formulation was selected for validation study to test the predictivity of the statistical models to provide optimization solutions with acceptable disintegration time of <15 min. The HPMCAS-containing HME tablets showed low tensile strength for most of the DOE batches. Hence, compound X-HPMCAS formulation was also selected for validation study to evaluate the predictivity of the statistical models to provide optimization solutions with acceptable tablet tensile strength. Table V shows the formulation composition of these internal validation batches. The intragranular level/extrudate level was 60% for both validation batches. The compound X-to-polymer ratio of 1 to 2 was kept similar to HME DOE runs. For each validation batch, round tablets of 9 mm were compressed using Instron at the predicted compression forces obtained from optimization solutions. The PVP VA64-containing tablets were compressed at predicted compression force of 12 kN, which resulted in the HME tablet porosity of 10.6 ± 0.3%. The HPMCAS-containing tablets were compressed at predicted compression force of 16 kN, which resulted in the HME tablet porosity of 11.3 ± 0.4%. For each internal validation batch, the optimization criteria of less than 20 kN compression force to produce HME tablets with 12 ± 3% porosity were met. Figure 14a, b shows the comparison between optimization output and experimental data for tensile strength and disintegration time, respectively. It was observed that for tensile strength and disintegration time, experimental data followed similar trend as predicted by the optimization output for both dispersions. For PVP VA64 dispersion, disintegration time was higher than predicted value but met an optimization criterion of less than 15 min (Fig. 14b). The actual tablet tensile strength was higher than predicted value for each polymer system, but for both formulations, an optimization criterion of ≥1 MPa tensile strength was met (Fig. 14a), indicating good predictivity of developed statistical models for PVP VA64- and HPMCAS-containing HME tablet formulations.

Comparison of HME tablet properties of optimization output and experimental data of compound X internal validation study batches. a Tablet tensile strength. b Tablet disintegration time
An external validation study using different compound was also conducted to test if the findings from the HME DOE are generally applicable for each studied HME polymer even if a compound with different physicochemical characteristics than studied compound is used to prepare HME tablets. The compound A was of neutral form hence non-ionizable. The monohydrate crystalline form was selected for development. The compound A had poor aqueous solubility (40–50 μg/ml at pH 2–7.4), moderate hydrophobicity (Log P of 2.6), and a broad melting peak around 110–120°C. The compound A was selected for the external validation study as previous work in our lab had showed that solid dispersion tablets of compound a prepared by hot melt extrusion process had very long disintegration time (30–60 min).
To conduct external validation study, the compound A HME batches of all three polymeric system (compound A/Soluplus, compound A/PVP VA64, compound A/HPMCAS-LF) were extruded using 18-mm twin screw extruder at ratio of 1 to 2 with resulting dispersion drug loading concentration of 33.3%. The compound A had low melting point, which resulted in low extrusion temperature of 145°C for Soluplus, 150°C for PVP VA64, and 165°C for HPMCAS-containing batches. HME tablets of compound A dispersion were prepared based on optimization solution identified for each studied HME polymer. Table VI lists the formulation composition of these external validation study batches.
Similar to the internal validation results, the optimization solutions for each studied HME polymer predicted well on tested response factors for external validation batches. The external validation batches were compressed at the predicted compression force from optimization output, which resulted in the desired HME tablet porosity of 12 ± 3% (Table VI). Figure 15a, b shows comparison between optimization output and experimental data for tablet disintegration time and tensile strength, respectively. The results show that even though experimental data was not exactly same as predicted optimization output, the trends were similar. The tablet disintegration time was less than 15 min for external validation batch of each studied HME polymer (meets optimization criterion of <15 min) (Fig. 15a). Similarly, the tablet tensile strength was in acceptable range for each studied HME polymer formulation and met the optimization criterion of ≥1 MPa for tablet tensile strength (Fig. 15b).

Comparison of HME tablet properties of optimization output and experimental data of compound A external validation study batches. a Tablet disintegration time. b Tablet tensile strength
Overall, the results from internal and external validation study demonstrated good predictivity of the developed statistical models for solid dispersions of each studied HME polymer (PVP VA64, HPMCAS, and Soluplus) to achieve HME tablets with rapid disintegration time (less than 15 min) and acceptable tensile strength (≥1 MPa at 12 ± 3% tablet porosity). The validation study results support the hypothesis that polymer type has strong influence on HME tablet quality attributes as polymer is one of the major components of solid dispersions. In the current study, the extragranular component requirements to develop an immediate release HME tablet of required strength could be identified for each studied HME polymer by conducting systematic evaluation using quality by design approach.
CONCLUSIONS
The effect of intragranular and extragranular components on HME tablet properties could be identified by statistical analysis of HME DOE using mixture design. This systematic evaluation based on quality by design approach indicated that higher intragranular (extrudate) level in the HME tablets results in longer disintegration time and lower tensile strength. In general, 60–70% was the maximum amount of acceptable intragranular level in HME tablets. Fast disintegration of HME tablets was achieved with HPMCAS-containing solid dispersion with minimal influence of other formulation components on tablet disintegration. Soluplus- and PVP VA64-containing solid dispersion showed better extrudability, lower compression force, and higher tablet tensile strength compared to HPMCAS-containing solid dispersion. Use of crospovidone or croscarmellose sodium resulted in faster disintegration compared to SSG, hence considered as more suitable disintegrants to prepare immediate release HME tablets for PVP VA64 or Soluplus-containing solid dispersions. MCC was a suitable filler to prepare HME tablets with acceptable tensile strength for each studied polymer. When the disintegration time of HME tablet is too long (more than 15 min), then lactose can be considered as alternate filler but it can result in lower tablet tensile strength so appropriate level should be selected accordingly. Results of this study indicated that the influence of extragranular components on dissolution from tablets should be carefully evaluated while finalizing the HME tablet composition, as it varies for each polymer system and also depends on type as well as level of interactions between drug substance and polymer. Results of internal and external validation studies confirmed that the developed statistical models can predict optimization solutions for each studied HME polymer (PVP VA64, Soluplus, and HPMCAS) to achieve HME tablets with rapid disintegration time (less than 15 min) and acceptable tensile strength (≥1 MPa at 12 ± 3% tablet porosity). The findings from current study could be used as a starting point to prepare immediate release solid dispersion tablets for a new drug substance using PVP VA64, Soluplus, or HPMCAS-LF polymer.
REFERENCES
Leuner C, Dressman J. Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm Biopharm. 2000;50:47–60.
Vasconcelos T, Sarmento B, Costa P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov Today. 2007;12:1068–75.
Shah S, Maddineni S, Lu J, Repka MA. Melt extrusion with poorly soluble drugs. Int J Pharm. 2013;453:233–52.
Paudel A, Worku ZA, Meeus J, Guns S, van den Mooter G. Manufacturing of solid dispersions of poorly water soluble drugs by spray drying: formulation and process considerations. Int J Pharm. 2013;453:253–84.
Shah N, Iyer RM, Mair HJ, Choi DS, Tian H, Diodone R, et al. Improved human bioavailability of Vermurafenib, a practically insoluble drug, using an amorphous polymer stabilized solid dispersion prepared by a solvent-controlled coprecipitation process. J Pharm Sci. 2013;102:967–81.
Serajuddin AT. Solid dispersion of poorly water-soluble drugs: early promises, subsequent problems, and recent breakthroughs. J Pharm Sci. 1999;88:1058–66.
Agrawal A, Dudhedia MS, Patel AD, Raikes MS. Characterization and performance assessment of solid dispersions prepared by hot melt extrusion and spray drying process. Int J Pharm Sci. 2013;457:71–81.
Horspool K, Agrawal A, Mujumdar S, Bhugra C, Qiang D. Enabling formulation approaches: considerations to advance solid dispersions into late stage development. BioPharma Asia. 2013;2:4–13.
Breitenbach J. Melt extrusion: from process to drug delivery technology. Eur J Pharm Biopharm. 2002;54:107–17.
Repka MA, Majumdar S, Battu SK, Srirangam R, Upadhye SB. Applications of hot melt extrusion for drug delivery. Expert Opin Drug Deliv. 2008;5:1357–76.
Crowley MM, Zhang F, Repka MA, Thumma S, Upadhye SB, Battu SK, et al. Pharmaceutical applications of hot melt extrusion: part I. Drug Dev Ind Pharm. 2007;33:909–26.
Horspool K, Bhugra C, Agrawal A, Mujumdar S, Telang C, Qiang D. Enabling formulation approaches for poorly soluble compounds in early development. Biopharma Asia. 2012;1:26–35.
Agrawal AM, Dudhedia MS, Zimny E. Hot melt extrusion: development of an amorphous solid dispersion for an insoluble drug from mini-scale to clinical scale. AAPS PharmSci Tech. 2015. doi:10.1208/s12249-015-0425-7.
Puri V, Dantuluri AK, Bansal AK. Investigation of atypical dissolution behaviour of an encapsulated amorphous solid dispersion. J Pharm Sci. 2010;1-9.doi:10.1002/jps22462.
Drooge DJ, Hinrichs WL, Frijlink HW. Anomalous dissolution behaviour of tablets prepared from sugar glass based solid dispersions. J Control Release. 2004;97:441–52.
Craig DQ. The mechanisms of drug release from solid dispersions in water-soluble matrix. Int J Pharm. 2002;231:131–44.
Akbuga J, Gursoy A, Yetimoglu F. Preparation and properties of tablets prepared from furosemide-PVP solid dispersion systems. Drug Dev Ind Pharm. 1988;14:2091–108.
Ford JL, Rubinstein MH. Formulation and aging of tablets prepared from indomethacin-poly(ethylene glycol) 6000 solid dispersions. Pharm Acta Helv. 1980;55:1–7.
Iyer R, Hegde S, Zhang Y, Dinunzio J, Singhal D, Malick A, et al. The impact of hot melt extrusion and spray drying on mechanical properties and tableting indices of materials used in pharmaceutical development. J Pharm Sci. 2013;102(102):3604–13.
Demuth B, Nagy ZK, Balogh A, Vigh T, Marosi G, Verreck G, et al. Downstream processing of polymer-based amorphous solid dispersions to generate tablet formulations. Int J Pharm. 2015;486:268–86.
Gryczke A, Schminke S, Maniruzzaman M, Beck J, Douroumis D. Development and evaluation of orally disintegrating tablets (ODTs) containing ibuprofen granules prepared by hot melt extrusion. Colloids Surf B: Biointerfaces. 2011;86:275–84.
Liu J, Zhang F, McGinity JW. Properties of lipophilic matrix tablets containing phenylpropanolamine hydrochloride prepared by hot melt extrusion. Eur J Pharm BioPharm. 2001;52:181–90.
Laitinen R, Suihko E, Bjorkqvist M, Riikonen J, Lehto V, Jarvinen K, et al. Perphenazine solid dispersions for orally fast-disintegrating tablets: physical stability and formulation. Drug Dev Ind Pharm. 2010;35(5):601–13.
Fell JT, Newton JM. Determination of tablet strength by diametral compression test. J Pharm Sci. 1970;59:688–91.
Tye CK, Sun CC, Amidon GE. Evaluation of the effects of tableting speed on the relationships between compaction pressure, tablet tensile strength, and tablet solid fraction. J Pharm Sci. 2005;94:465–7.
Mohanachandran PS, Sindhumol PG, Kiran TS. Superdisintegrants: an overview. Int J Pharm Sci Rev Res. 2011;6:105–9.
Delalonde M, Fitouri R, Ruiz E, Bataille B, Sharkawi T. Impact of physicochemical environment on the super disintegrant functionality of cross-linked carboxymethyl sodium starch: insight on formulation precautions. AAPS PharnSci Tech. 2014;16:407–12.
Iyer SR, Sivakumar R, Siva P, Sajeeth CI. Formulation and evaluation of fast dissolving tablets of risperidone solid dispersion. Int J Pharm Chem Bio Sci. 2013;3:388–97.
Jain S, Jain S, Mishra A, Garg G, Modi RK. Formulation and characterization of fast disintegrating tablets containing cefdinir solid dispersion. Int J Pharm Life Sci. 2012;3:2190–9.
Raviteja P, Muralidhar S, Ramesh R, Narayana TV, Vasantha Kumar P, Vijay KG. Formulation and evaluation of valsartan fast disintegrating tablets using solid dispersion technique. Int J Inn Pharm Res. 2013;4:274–80.
Chamsai B, Sriamornsak P. Novel disintegrating microcrystalline cellulose pellets with improved drug dissolution performance. Powder Technol. 2013;233:278–85.
Mehta S, De Beer T, Remon JP, Vervaet C. Effect of disintegrants on the properties of multiparticulate tablets comprising starch pellets and excipients granules. Int J Pharm. 2012;422:310–7.
Pudlas M, Kyeremateng SO, Williams LA, Kimber JA, Lishaut HV, Kazarian SG, et al. Analyzing the impact of different excipients on drug release behaviour in hot-melt extrusion formulations using FTIR spectroscopic imaging. Eur J Pharm Sci. 2015;67:21–31.
Zhang YE, Schwartz JB. Effect of diluents on tablet integrity and controlled drug release. Drug Dev Ind Pharm. 2000;26:761–5.
Sarode AL, Sandhu H, Shah N, Malick W, Zia H. Hot melt extrusion (HME) for amorphous solid dispersions: predictive tools for processing and impact of drug-polymer interactions on supersaturation. Eur J Pharm Sci. 2013;48:371–84.
Author information
Authors and Affiliations
Corresponding author
Additional information
Guest Editors: Feng Zhang and Michael A. Repka
Rights and permissions
About this article

Cite this article
Agrawal, A., Dudhedia, M., Deng, W. et al. Development of Tablet Formulation of Amorphous Solid Dispersions Prepared by Hot Melt Extrusion Using Quality by Design Approach. AAPS PharmSciTech 17, 214–232 (2016). https://doi.org/10.1208/s12249-015-0472-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12249-015-0472-0