
Abstract
Na+,K+-ATPase activity was determined in erythrocyte membranes from 12 phenylketonuric patients of both sexes, aged 8.8 ± 5.0 y, with plasma phenylalanine levels of 0.64 ± 0.31 mM. The in vitro effects of phenylalanine and alanine on the enzyme activity in erythrocyte membranes from healthy individuals were also investigated. We observed that Na+,K+-ATPase activity was decreased by 31% in erythrocytes from phenylketonuric patients compared with normal age-matched individuals (p < 0.01). We also observed a significant negative correlation between erythrocyte Na+,K+-ATPase activity and plasma phenylalanine levels (r = −0.65;p < 0.05). All PKU patients with plasma phenylalanine levels higher than 0.3 mM had erythrocyte Na+,K+-ATPase activity below the normal range. Phenylalanine inhibited in vitro erythrocyte Na+,K+-ATPase activity by 22 to 34%, whereas alanine had no effect on this activity. However, when combined with phenylalanine, alanine prevented Na+,K+-ATPase inhibition. Considering that reduction of Na+,K+-ATPase activity occurs in various neurodegenerative disorders leading to neuronal loss , our previous observations showing a significant reduction of Na+,K+-ATPase activity in brain cortex of rats subjected to experimental phenylketonuria and the present results, it is proposed that determination of Na+,K+-ATPase activity in erythrocytes may be a useful peripheral marker for the neurotoxic effect of phenylalanine in phenylketonuria.
Similar content being viewed by others

Main
PKU is an inherited metabolic disease caused by a severe deficiency of Phe hydroxylase activity. As a consequence, Phe and its deaminated metabolites accumulate in urine, blood, and tissues. Neurologic dysfunction is the clinical hallmark of PKU, but the mechanisms by which brain damage occur are complex and poorly understood (1).
Na+,K+-ATPase (EC 3.6.1.3), the membrane-bound enzyme responsible for the active transport of Na+ and K+ across the cell membrane, utilizes 30 to 60% of the ATP synthesized in the brain (2). Considering that Na+,K+-ATPase activity is essential for synaptic and cellular functions, it is possible that reduction in the activity of this enzyme due either to inhibitors or to ATP depletion may disrupt normal brain development (3). In this context, it has been demonstrated that injection of ouabain, a potent Na+,K+-ATPase inhibitor, into the rat striatum and substantia nigra causes selective neuronal loss in up to 70% of the damaged area with glial cell and macrophage proliferation in the core of the lesion (4). In addition, it has been demonstrated that the extent of neuronal loss observed after injection of specific and nonspecific Na+,K+-ATPase inhibitors into the dorsal hippocampus of rats roughly paralleled their potency as inhibitors of the enzyme (5).
We have previously reported that Na+,K+-ATPase activity is reduced in synaptic plasma membrane from brain cortex of rats subjected to chronic hyperphenylalaninemia (6) and that this reduction is prevented by the simultaneous administration of Ala (7). We have also observed that Phe inhibits in vitro Na+,K+-ATPase activity in membranes from normal rats, and Ala reverses this inhibition by competition at the same binding site (8).
Because human brain tissue is not available for direct measurement of Na+,K+-ATPase activity, it is important to identify peripheral markers that might reflect the effects of Phe on this enzyme activity in the CNS. Therefore, considering that α1 is the main Na+,K+-ATPase isozyme in both brain and erythrocyte membranes (9) and that the enzymes from brain and erythrocyte membranes have similar pharmacologic and biochemical characteristics (10), the aim of the present study was to investigate whether Na+,K+-ATPase activity in erythrocytes is altered in PKU patients, as well as whether this measurement could be used as a possible marker for the effects of Phe on the enzyme activity in the brain. First, we compared Na+,K+-ATPase activity in erythrocyte membranes from PKU patients and from age-matched controls. Next, we investigated the in vitro effects of Phe or Ala, alone or combined, on the enzyme activity in erythrocyte membranes from healthy children.
METHODS
Subjects.
Blood specimens from 12 fasting PKU children of both sexes (seven boys and five girls) aged 8.8 ± 5.0 y on a low Phe diet with poor acceptance and from 15 normal age-matched individuals (controls) were used for the experiments. All patients were followed up by the same physician at the Medical Genetic Service of the Clinical Hospital of Porto Alegre and were under the same protocol for phenylketonuric patient management. Besides being affected by PKU, no other disorder was identified in the patients. Erythrocytes were obtained after centrifugation (400 ×g for 10 min) of peripheral venous blood collected into heparinized syringe for routine plasma Phe determination. Controls had normal plasma Phe levels and no clinical and laboratory evidence of any metabolic disease, whereas PKU patients had plasma Phe levels of 0.64 ± 0.31 mM and moderate to severe mental retardation. At the time of blood sample collection, nine patients had plasma Phe levels higher than 0.3 mM and three had plasma Phe levels of 0.3 mM or lower. Plasma Ala levels did not differ significantly between patients (0.41 ± 0.15 mM) and controls (0.47 ± 0.17 mM) (t(25) = 0.64;p > 0.5). Plasma Phe and Ala levels were measured according to McCaman and Robins (11) and Joseph and Marsden (12), respectively. This work was approved by the ethical committee of the university and was conducted according to the principles expressed in the Declaration of Helsinki. All studies were conducted with informed consent.
Preparation of erythrocyte membranes.
Erythrocyte membranes were prepared by the method of Matteucci et al. (13) with some modifications. The membrane preparation was carried out at 4°C. Briefly, erythrocytes from 2 mL of fresh venous blood were separated by centrifugation at 400 ×g for 10 min. Packed erythrocytes were washed twice in 20 mL of an isotonic medium, pH 7.4, containing 0.25 M mannitol, 20 mM Tris, and 1 mM EDTA. The pellet was resuspended in 2 mL of the above medium. After the addition of 18 mL of deionized water, the suspension was stirred on ice for 15 min and centrifuged at 20,000 ×g for 20 min. The pellet was resuspended in 2 mL of mannitol-Tris-EDTA buffer, stirred for 15 min after the addition of 18 mL of deionized water, and centrifuged at 20,000 ×g for 20 min. The last procedure was repeated twice. Ghost membranes were suspended in 0.5 mL of 40 mM Tris-HCl buffer, pH 7.4, to a final protein concentration of 0.8 to 1.5 mg/mL. Membranes stored at −20°C retained the enzyme activity for at least 2 wk. The Hb that remained attached to the membrane surface was measured with the kit 527-A (Sigma Chemical Co., St. Louis, MO, U.S.A.), and the value was subtracted from the total protein concentration.
Enzyme assay.
Mg2+-ATPase and Na+,K+-ATPase activities were assayed according to Tsakiris and Deliconstantinos (14). Ten microliters of erythrocyte membranes was added to the reaction mixture for the Mg2+-ATPase and Na+,K+-ATPase assay containing 5.0 mM MgCl2, 80.0 mM NaCl, 20.0 mM KCl, 40.0 mM Tris-HCl buffer, pH 7.4, in a final volume of 0.2 mL. The reaction was started by the addition of ATP (vanadium-free disodium salt) to a final concentration of 3.0 mM. The reaction was stopped after 10 min by the addition of 0.2 mL of 0.66 M trichloroacetic acid (TCA). Mg2+-ATPase was assayed under the same conditions but with the addition of 1.0 mM ouabain. Na+,K+-ATPase activity was calculated by the difference between the two values. For the in vitro studies, Phe or Ala was dissolved in 40 mM Tris-HCl buffer, pH 7.4, and added to the incubation mixture to a final concentration of 0.3 to 1.2 mM. Released inorganic phosphate (Pi) was measured by the method of Chan et al.(15). Results were expressed as nmol Pi/min·mg protein. Protein was measured by the method of Bradford (16), using BSA as standard. All assays were run in triplicate. All chemicals were purchased from Sigma Chemical Co., St. Louis, MO, U.S.A., and were of analytical grade.
Data were analyzed by t test or by 1-way ANOVA, followed by the Duncan multiple range test when the F test was significant. Pearson linear correlation was used to compare plasma Phe levels and erythrocyte Na+,K+-ATPase activities. All analyses were performed with a PC compatible computer using the Statistical Package for the Social Sciences (SPSS) software.
RESULTS
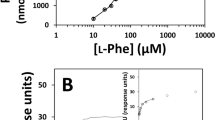
The activities of membrane-bound Na+,K+-ATPase and Mg2+-ATPase in erythrocytes from controls were 10.7 ± 1.05 nmol Pi/min·mg protein and 9.7 ± 1.50 nmol Pi/min·mg protein, respectively. These values are similar to those obtained by others (13, 17). Erythrocyte Na+,K+-ATPase specific activity (7.4 ± 1.60) was significantly decreased by 31% in PKU patients (t(25) = 6.02;p < 0.001), whereas Mg2+-ATPase activity (8.83 ± 1.48) was not significantly affected (t(25) = 1.87;p > 0.05). Figure 1 shows a significant negative correlation between plasma Phe levels and erythrocyte Na+,K+-ATPase activity in PKU patients (r = −0.65;p < 0.05). It can also be seen that all PKU patients with plasma Phe levels higher than 0.3 mM had erythrocyte Na+,K+-ATPase activity below the normal range.

Correlation between erythrocyte Na+,K+-ATPase activity and plasma Phe levels in patients with PKU. Horizontal lines are mean and 95% confidence interval for 15 age-matched healthy controls. r = −0.65;p < 0.05 (Pearson correlation).
Next, we determined the in vitro effects of Phe, Ala, or Ala plus Phe on the enzyme activities in erythrocyte membranes from normal individuals (Figs. 2,3,and 4). Phe had no effect on Mg2+-ATPase activity [F (4, 35) = 0.25;p > 0.9] at any concentration tested but significantly inhibited Na+,K+-ATPase activity by 22 to 34% [F (4, 35) = 3.18;p < 0.02] (Fig. 2). Ala had no effect on Na+,K+-ATPase [F (4, 35) = 1.76;p > 0.15] or Mg2+-ATPase activity [F (4, 35) = 0.30;p > 0.87] (Fig. 3). When Ala to a final concentration of 0.6 mM was added to the incubation medium containing Phe (0.3–1.2 mM), no effect was observed on Na+,K+-ATPase [F (4, 25) = 0.22;p > 0.94] or Mg2+-ATPase activity [F (4, 25) = 0.28;p > 0.91] (Fig. 4).

In vitro effect of Phe on Na+,K+-ATPase and Mg2+-ATPase activities in human erythrocyte membranes. Data are mean ± SEM for eight independent experiments performed in triplicate. [ ▪] Na+,K+-ATPase;[ □] Mg2+-ATPase. *Different from control, p < 0.01 (Duncan's multiple range test).

In vitro effect of Ala on Na+,K+-ATPase and Mg2+-ATPase in human erythrocyte membranes. Data are mean ± SEM for eight independent experiments performed in triplicate. [ ▪] Na+,K+-ATPase;[ □] Mg2+-ATPase.

In vitro effect of Ala plus Phe on Na+,K+-ATPase and Mg2+-ATPase activities in human erythrocyte membranes. 0.6 mM Ala was added to all experiments except the control. Data are mean ± SEM for six independent experiments performed in triplicate. [ ▪] Na+,K+-ATPase;[ □] Mg2+-ATPase.
We also tested the effect of Ala on the Na+,K+-ATPase activity in erythrocyte membranes from PKU patients. Ala did not alter the activity of Na+,K+-ATPase [PKU 7.26 ± 1.68 nmol Pi/min·mg protein; PKU plus Ala 7.61 ± 1.82 nmol Pi/min·mg protein (t9 = 0.92;p > 0.35)] or Mg2+-ATPase [PKU 8.63 ± 1.35 nmol Pi/min·mg protein; PKU plus Ala 8.27 ± 2.24 nmol Pi/min·mg protein (t9 = 0.63;p > 0.57)].
DISCUSSION
Although severe mental retardation is the clinical hallmark of untreated PKU patients, the mechanisms underlying the neurologic dysfunction in this disorder are probably multiple and not fully known. We have recently demonstrated that rats subjected to chemically induced hyperphenylalaninemia have a significant decrease of Na+,K+-ATPase activity in synaptic plasma membrane from brain cortex (6). Therefore, considering the importance of Na+,K+-ATPase activity for brain development and function, it would appear desirable to measure the activity of this enzyme in the brain of PKU children. However, because human brain specimens are not usually available for direct biochemical evaluation, it seems worthwhile to identify peripheral markers that could reflect neurotoxicity by noninvasive means (18). Several parameters of neurotransmission have been identified in platelets, lymphocytes, and erythrocytes whose pharmacologic and biochemical characteristics are similar to those of the CNS (19). So, Na+,K+-ATPase activity in red blood cells has been considered a reliable peripheral neurotransmitter parameter (10). On the other hand, reduction of Na+,K+-ATPase activity in erythrocytes from patients with bipolar illness has been considered evidence that a primary or secondary dysfunction of the Na+,K+-ATPase plays a predisposing or direct etiologic role in this disease (20). In addition, reduced Na+,K+-ATPase activity was found in the brain of uremic rats with neurologic dysfunction (21) as well as in erythrocytes of uremic patients (22). In these situations, the reduction in this enzyme activity was caused by the accumulation of endogenous ouabain-like factors, a fact indicative that erythrocytes are effective peripheral markers of neurologic dysfunction.
Because α1 is the Na+,K+-ATPase isozyme predominant in both brain and erythrocyte membranes (10), the main objective of the present study was to determine Na+,K+-ATPase activity in erythrocyte membranes of PKU patients in the hope to verify whether the erythrocyte enzyme activity could serve as a peripheral marker by which Phe neurotoxicity could be monitored in PKU.
We observed that Na+,K+-ATPase activity was decreased by 31% in erythrocyte membranes from PKU patients, whereas Mg2+-ATPase was not affected, indicating a specific effect on Na+,K+-ATPase. We also observed a significant negative correlation between plasma Phe levels and erythrocyte Na+,K+-ATPase activity and that only PKU patients presenting plasma Phe levels higher than 0.3 mM had erythrocyte Na+,K+-ATPase activity below normal levels. These results suggest that determination of erythrocyte Na+,K+-ATPase activity in PKU patients may reflect Phe toxicity.
We have previously reported that Ala prevents by competition the in vitro Na+,K+-ATPase activity inhibition provoked by Phe in synaptic plasma membrane of cerebral cortex from normal rats (6, 8). We have also demonstrated that Ala administration to rats subjected to chemically induced PKU prevents the reduction of Na+,K+-ATPase activity in synaptic plasma membrane of rat brain cortex. Taking into consideration these results, we decided to add Ala to the enzyme assay in erythrocyte membranes from PKU patients and observed no modification of the Na+,K+-ATPase activity. Considering that Phe is present at insignificant amounts in the isolated washed erythrocyte membranes from PKU patients, it was not surprising that Ala could not reverse this inhibition, because reversion occurs by competition with Phe. Therefore, it is possible that the reduced enzyme activity found in the erythrocyte membrane from PKU patients was not due to a reversible enzymatic inhibition by Phe as observed in vitro but to a diminution of the number of active enzyme molecules in the membrane due to diminished synthesis and/or increased degradation, a mechanism to be elucidated. However, we cannot rule out an in vivo inhibition by a direct effect of Phe on Na+,K+-ATPase activity.
Next, we studied the in vitro effects of Phe, Ala, or Ala plus Phe on Na+,K+-ATPase activity in erythrocyte membranes from healthy controls. We observed that Phe significantly inhibited Na+,K+-ATPase activity in the range of 0.3–1.2 mM. Because 0.3 mM of Phe already caused an inhibition of erythrocyte Na+,K+-ATPase activity in vitro different from what is observed in vivo in PKU in which plasma Phe levels of 0.3 mM do not provoke reduction of this enzyme activity (Fig. 1), it is possible that the mechanism involved in the in vivo and in vitro inhibition is distinct. It should be also considered that the intracellular Phe concentration in erythrocytes is possibly lower than that of plasma because the uptake of this amino acid by human red blood cells seems to be not so efficient (23, 24). If that is the case, plasma Phe concentrations in PKU patients do not necessarily reflect erythrocyte Phe concentrations. In this context, it has been demonstrated that Phe concentrations in the brain of PKU patients are 3–4-fold lower than those of plasma (25).
We also observed that the in vitro enzyme activity inhibition caused by the presence of Phe in the incubation medium and the reduction of Na+,K+-ATPase activity shown in the PKU patients are specific for Na+,K+-ATPase, because Mg2+-ATPase activity was not affected by this amino acid. In addition, we demonstrated that Ala alone had no effect on the in vitro Na+,K+-ATPase activity but prevented the inhibitory effect of Phe on the enzyme when added simultaneously to the incubation medium.
Taken together, these results indicate that the biochemical characteristics of Na+,K+-ATPase from rat brain cortex and from human erythrocytes are similar with respect to the effects of Phe and Ala on Na+,K+-ATPase activity. Furthermore, considering that α1 is the predominant isoform of Na+,K+-ATPase in brain and in erythrocytes, it is possible that the reduction of erythrocyte Na+,K+-ATPase activity detected in the present study also occurs in the CNS of PKU children.
A great body of evidence associates neurotoxicity with a reduction of Na+,K+-ATPase activity, suggesting that reduction in Na+,K+-ATPase activity may be a link between several common neurotoxic mechanisms (26). In this context, it has been demonstrated that 0.05–0.5-μM ouabain concentrations that inhibit Na+,K+-ATPase activity by 30%(27) provoke an increase of Na+ uptake and cytosolic-free Ca2+ concentration and a decrease of the membrane potential of synaptosomes from cerebral cortex of rats (28). In addition, spongiform encephalopathy has been associated with a 40% decrease of Na+,K+-ATPase activity in brain cortex of a neonate (29). The neurotoxicity provoked by Na+,K+-ATPase inhibition was also associated with glutamate release and excitotoxicity (4, 30). Therefore, if the 31% reduction of Na+,K+-ATPase activity identified in the present study in the erythrocytes of PKU patients also occurs in their brain, it is tempting to speculate that reduction of Na+,K+-ATPase activity may be one of the mechanisms by which Phe is toxic to the brain. If this is the case, determination of Na+,K+-ATPase activity in human red blood cells may be a useful peripheral marker for Phe neurotoxicity in PKU. We should, however, rule out other disorders affecting PKU patients known to reduce erythrocyte Na+,K+-ATPase activity, such as uremia (22), rheumatoid arthritis (31), diabetes mellitus (32), and essential hypertension (33). It should be emphasized that more work will be needed to support these preliminary observations that Na+,K+-ATPase activity in red blood cells is useful as a marker for neurotoxicity in PKU.
Abbreviations
- Phe:
-
phenylalanine
- Ala:
-
alanine
- PKU:
-
phenylketonuria
References
Scriver CR 1995 Whatever happened to PKU?. Clin Biochem 28: 137–144
Erecinska M, Silver IA 1994 Ions and energy in mammalian brain. Prog Neurobiol 43: 37–71
Lees GJ 1991 Inhibition of sodium-potassium-ATPase: a potentially ubiquitous mechanism contributing to central nervous system neuropathology. Brain Res Rev 16: 283–300
Lees GJ, Leong W 1995 The sodium-potassium-ATPase inhibitor ouabain is neurotoxic in the rat substantia nigra and striatum. Neurosci Lett 188: 113–116
Lees GJ, Leong W 1994 Brain lesions induced by specific and non-specific inhibitors of sodium-potassium ATPase. Brain Res 649: 225–233
Wyse ATS, Bolognesi G, Brusque AM, Wajner M, Wannmacher CMD 1995 Na+,K+-ATPase activity in the synaptic plasma membrane from the cerebral cortex of rats subjected to chemically induced phenylketonuria. Med Sci Res 23: 261–262
Wyse ATS, Noriler ME, Borges LF, Floriano PJ, Silva CG, Wajner M, Wannmacher CMD 1999 Alanine prevents the decrease of Na+,K+-ATPase activity in experimental phenylketonuria. Metab Brain Dis 14: 95–101
Wyse ATS, Wajner M, Wannmacher CMD 1998 Kinetics of alanine reversal on the inhibition of Na+,K+-ATPase activity by phenylalanine and phenylactate in the synaptic plasma membrane from the cerebral cortex of rats. Med Sci Res 26: 141–143
Sweadner KJ 1992 Overlapping and diverse distribution of Na+,K+-ATPase isozymes in neurons and glia. Can J Physiol Pharmacol 70: S255–S259
Hoffmann JF, Kennedy BG, Lunn G 1981 Modulators of red cell Na/K pump rates. Prog Clin Biol Res 56: 5–9
McCaman MW, Robins E 1962 Fluorimetric method for the determination of phenylalanine in serum. J Lab Clin Med 59: 885–890
Joseph MH, Marsden CA 1986 Amino acids and small peptides. In: Lim CK (ed) HPLC for Small Peptides. IRL Press, Oxford, 13–17.
Matteucci E, Cocci F, Pellegrini L, Gregori G, Giampietro O, 1994– 95. Measurement of ATPases in red cells: setting up and validation of a highly reproducible method. Enzyme Protein 48: 105–119
Tsakiris S, Deliconstantinos G 1984 Influence of phosphatidylserine on (Na+,K+)-stimulated ATPase and acetylcholinesterase activities of dog brain synaptosomal plasma membranes. Biochem J 220: 301–307
Chan K, Delfert D, Junger KD 1986 A direct colorimetric assay for Ca2+-stimulated ATPase activity. Anal Biochem 157: 375–380
Bradford MM 1976 A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72: 248–254
Sigström L, Waldeström J, Karlberg P 1981 Characteristics of active sodium and potassium transport in erythrocytes of healthy infants and children. Acta Pediatr Scand 70: 347–352
Maier WE, Costa LG 1990 Na+/K+-ATPase in rat brain and erythrocytes as a possible target and marker, respectively, for neurotoxicity: studies with chlordecone, organotins, and mercury compounds. Toxicol Lett 51: 175–189
Stahl SM 1985 Peripheral models for the study of neurotransmitter receptors in man. Psychopharmacol Bull 21: 663–671
El-Mallakh RS, Wyatt RJ 1995 The Na,K-ATPase hypothesis for bipolar illness. Biol Psychiatry 37: 235–244
Fraser CL, Sarnacki P, Arieff AI 1985 Abnormal sodium transport in synaptosomes from brain of uremic rats. J Clin Invest 75: 2014–2023
Vásárhelyi B, Sallay P, Balog E, Reusz G, Tulassay T 1996 Altered Na+,K+-ATPase activity in uraemic adolescents. Acta Paediatr 85: 919–922
Guidotti GG, Gazzola GC, Borghetti AF, Franchi-Gazzola R 1975 Adaptive regulation of amino acid transport across the cell membrane in avian and mammalian tissues. Biochim Biophys Acta 406: 264–279
Hagenfeldt L, Arvidsson A 1980 The distribution of amino acids between plasma and erythrocytes. Clin Chim Acta 100: 133–141
Novotny EJ, Avison MJ, Herschkowitz N, Petroff OAC, Prichard JW, Seashore MR, Rothman DL 1995 In vivo measurement of phenylalanine in human brain by proton nuclear magnetic resonance spectroscopy. Pediatr Res 37: 244–249
Lees GJ 1993 Contributory mechanisms in the causation of neurodegenerative disorders. Neuroscience 54: 287–322
Sweadner KJ 1979 Two molecular forms of (Na+,K+)-stimulated ATPase in brain. J Biol Chem 254: 6060–6067
Satoh E, Nakazato Y 1992 On the mechanism of ouabain-induced release of acetylcholine from synaptosomes. J Neurochem 58: 1038–1044
Renkawek K, Renier WO, de Pont JJHHM, Vogels OJM, Gabreels FJM 1992 Neonatal status convulsivus, spongiform encephalopathy, and low activity of Na+,K+-ATPase in the brain. Epilepsy 33: 58–64
Brines ML, Robbins RJ 1992 Inhibition of α2-α3-sodium pump isoforms potentiates glutamate neurotoxicity. Brain Res 591: 94–102
Testa I, Rabini RA, Corvetta A, Danieli G 1987 Decreased Na+,K+-ATPase activity in erythrocyte membrane from rheumatoid arthritis patients. Scand J Rheumatol 16: 301–305
Finotti P, Palatini P 1986 Reduction of erythrocyte (Na-K+)ATPase activity in type I (insulin dependent) diabetic subjects and its activation by homologous plasma. Diabetologia 29: 623–628
Ringel RE, Hamlyn JM, Hamilton BP, Pinkas GA, Chalew SA, Berman MA 1987 Red blood cell Na+,K+-ATPase in men with newly diagnosed or previously treated essential hypertension. Hypertension 9: 437–443
Acknowledgements
The authors thank Dr. Ricardo Flores Pires from the Medical Genetic Service of the Clinical Hospital, Porto Alegre, RS, Brazil, for supplying red blood cells from patients and controls.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported in part by grants from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq-Brazil), Programa de núcleos de Excelência (PRONEX), Fundação de Amparo à Pesquisa do Rio Grande do Sul (FAPERGS-Brazil), and Pro-Reitoria de Pesquisa da Universidade Federal do Rio Grande do Sul (PROPESQ/UFRGS).
Rights and permissions
About this article
Cite this article
Bedin, M., Estrella, C., Ponzi, D. et al. Reduced Na+,K+-ATPase Activity in Erythrocyte Membranes from Patients with Phenylketonuria. Pediatr Res 50, 56–60 (2001). https://doi.org/10.1203/00006450-200107000-00012
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-200107000-00012
