Article Text

Statistics from Altmetric.com
Poster presentations
Clinical Trials (In Progress)
P218 KEYNOTE-585: randomized, phase 3 study of chemotherapy + pembrolizumab vs chemotherapy + placebo as neoadjuvant/adjuvant treatment for patients with gastric or gastroesophageal junction (G/GEJ) cancer
Yung-Jue Bang1, Eric Van Cutsem2, Charles Fuchs3, Atsushi Ohtsu4, Josep Tabernero5, David Ilson6, Woo Jin Hyung7, Vivian Strong6, Thorsten Goetze8, Takaki Yoshikawa9, Laura Tang6, Linda Sun10, Aisha Hasan10, Minori Koshiji11, Kohei Shitara4
1Seoul National University Hospital, Seoul, Republic of Korea; 2University Hospitals Gasthuisberg Leuven and KU Leuven, Leuven, Belgium; 3Yale Cancer Center, New Haven, CT, USA; 4National Cancer Hospital East, Chiba, Japan; 5Vall d'Hebron University Hospital and Institute of Oncology (VHIO), Barcelona, Spain; 6Memorial Sloan Kettering Cancer Center, New York, NY, USA; 7Yonsei Cancer Hospital, Yonsei University Health System, Seoul, Republic of Korea; 8Institute of Clinical Cancer Research, UCT University Cancer Center, Frankfurt, Germany; 9Kanagawa Cancer Center, Kanagawa, Japan; 10Merck & Co., Inc., Kenilworth, NJ, USA; 11Merck & Co., Inc., New York, NY, USA
Correspondence: Yung-Jue Bang (bangyj@snu.ac.kr)
Background
In KEYNOTE-012 (NCT01848834) and KEYNOTE-059 (NCT02335411), pembrolizumab demonstrated manageable safety and promising antitumor activity alone or in combination with chemotherapy in patients with advanced G/GEJ cancer. Compared with chemotherapy alone, chemotherapy combined with pembrolizumab in the neoadjuvant/adjuvant setting can provide additional benefit to patients with locally advanced, resectable G/GEJ cancer. KEYNOTE-585 is a phase 3, randomized, double-blind study of chemotherapy combined with pembrolizumab versus chemotherapy combined with placebo as neoadjuvant/adjuvant treatment for locally advanced resectable G/GEJ cancer.
Methods
Key eligibility criteria in KEYNOTE-585 (NCT03221426) include age ≥18 years; previously untreated G/GEJ adenocarcinoma (Siewert type 2 or 3 tumors; Siewert type 1 tumor eligibility limited to those for whom planned treatment is perioperative chemotherapy and resection), with no evidence of metastatic disease; planning to undergo surgery after preoperative chemotherapy; Eastern Cooperative Oncology Group performance status 0-1; adequate organ function; no active autoimmune disease. Patients will be randomly assigned 1:1 to receive chemotherapy + pembrolizumab (arm 1) or chemotherapy + placebo (arm 2). Stratification factors are geographic region (Asia vs Non-Asia), primary tumor location (stomach vs GEJ), and tumor stage (II/III vs IVa). All patients will receive neoadjuvant (preoperative) chemotherapy + pembrolizumab every 3 weeks (Q3W) for 3 cycles or chemotherapy + placebo Q3W for 3 cycles followed by surgery and then adjuvant chemotherapy + pembrolizumab Q3W for 3 cycles or chemotherapy + placebo Q3W for 3 cycles followed by monotherapy with pembrolizumab or placebo Q3W for 11 cycles; treatment will continue for up to 17 cycles overall. Chemotherapy consists of cisplatin 80 mg/m2 intravenously + either capecitabine 1000 mg/m2 twice daily orally or 5-fluorouracil 800 mg/m2 intravenously (investigator’s choice). Pembrolizumab 200 mg was administered intravenously. Adjuvant monotherapy consists of pembrolizumab (arm 1) or placebo (arm 2). Primary end points are overall survival (OS), event-free survival, and rate of pathologic complete response (defined as no invasive disease and histologically negative nodes) per central review. Adverse events (AEs) are graded per National Cancer Institute Common Terminology Criteria for Adverse Events v4.0 and will be monitored for 30 days after treatment end (90 days for serious AEs). Patients are followed up for survival every 12 weeks until death, withdrawal from study, or study termination. Planned enrolment is approximately 800 patients.
Trial Registration
ClinicalTrials.gov, NCT03221426
P219 Phase 1/1b, first-in-human study of the PI3K-gamma inhibitor IPI-549 as monotherapy and combined with nivolumab in patients with advanced solid tumors
Antoni Ribas1, David Hong2, Anthony Tolcher3, Ryan Sullivan4, Geoffrey Shapiro5, Bartosz Chmielowski1, Les Brail6, Lucy Lee6, Suresh Mahabhashyam6, Claudio Dansky Ullmann6, Michael Postow7, Jedd Wolchok7
1University of California, Los Angeles, Los Angeles, CA, USA; 2MD Anderson Cancer Center, Houston, TX, USA; 3South Texas Accelerated Research Therapeutics (START), San Antonio, TX, USA; 4Massachusetts General Hospital, Boston, MA, USA; 5Dana-Farber Cancer Institute, Boston, MA, USA; 6Infinity Pharmaceuticals, Inc., Cambridge, MA, USA; 7Memorial Sloan Kettering Cancer Center, New York, NY, USA
Correspondence: Les Brail (Les.Brail@infi.com)
Background
IPI-549 is a potentially first-in-class, oral, potent, selective PI3K-gamma inhibitor being developed as an immuno-oncology therapeutic in multiple cancer indications. Preclinical solid tumor research has shown that PI3K-gamma blockade in tumor-associated macrophages by IPI-549 results in transcriptional reprogramming of the pro-tumor macrophage phenotype (M2) to its anti-tumor counterpart (M1). Within this setting, IPI-549 has demonstrated activity as monotherapy and still greater activity when combined with checkpoint inhibitor therapies. Importantly, the latter approach has been found to overcome checkpoint inhibitor resistance in specific resistant models.
Methods
This study is being conducted to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of IPI-549 to ultimately determine its recommended Phase 2 dose and activity, both as monotherapy and in combination with nivolumab, in patients with advanced solid tumors. As shown in Fig. 1, the design includes four parts: 1) dose escalation (DE) of IPI-549 monotherapy; 2) DE of IPI-549 with fixed-dose nivolumab; 3) monotherapy expansion; and 4) combination expansion in specific tumor types, including non-small cell lung cancer, melanoma, and squamous cell carcinoma of the head and neck, with de novo or acquired resistance to checkpoint inhibitors (Fig. 1).
Results
This trial is currently in progress. No results are available.
Progress update: Monotherapy DE enrollment is complete for once-daily (QD) IPI-549 doses up to 60 mg, while monotherapy expansion enrollment has been initiated at 60 mg QD. Combination DE enrollment is complete for QD doses of 20 mg and 30 mg IPI-549 combined with fixed-dose nivolumab (240 mg once every 2 weeks).
Conclusions
The collective preclinical data highlight the key role of PI3K-gamma in the immuno-suppressive tumor microenvironment and provide a strong rationale for the ongoing clinical study of IPI-549.
Trial Registration
ClinicalTrials.gov: NCT02637531

See text for description
P220 A phase 2, multicenter study to evaluate the efficacy and safety using autologous tumor infiltrating lymphocytes (LN-145) in patients with recurrent, metastatic, or persistent cervical carcinoma
Amir Jazaeri1, Robert Edwards2, Emese Zsiros3, Robert Brown4, Igor Gorbatchevsky4, Sam Suzuki4, Maria Fardis4, Robert Wenham5
1University of Texas MD Anderson Cancer Center, Houston, TX, USA; 2Magee-Women's Hospital of University of Pittsburgh Medical Center, Pittsburgh, PA, USA; 3Roswell Park Cancer Institute, Buffalo, NY, USA; 4Iovance Biotherapeutics, San Carlos, CA, USA; 5Moffitt Cancer Center, Tampa, FL, USA
Correspondence: Robert Brown (robert.brown@iovance.com)
Background
Adoptive cell therapy (ACT) may be effective in treating immunogenic tumors with high mutational load such as melanoma and virally-associated tumors like cervical cancer with several patients in studies performed by other institutions achieving durable complete response for years. HPV infection increases mutational load, thus providing additional neoantigen targets ideal for the polyclonal nature of ACT. As outcomes for patients with recurrent, metastatic or persistent cervical cancer remain extremely poor, there is an enormous need for novel immunotherapeutic approaches with curative potential such as ACT.
Methods
Clinical trial C-145-04 (NCT03108495) is a prospective, phase 2 multicenter, open-label study evaluating the efficacy of a single autologous tumor infiltrating lymphocyte infusion (LN-145) followed by IL-2 after a non-myeloablative lymphodepletion (NMA-LD) regimen in patients with recurrent, metastatic, or persistent cervical cancer who have failed at least one prior systemic therapy. The clinical trial protocol requires resection of a tumor lesion which is then shipped to a central GMP manufacturing facility for TIL extraction, expansion, and preparation of the final infusion product (LN-145). One week prior to LN-145 shipment and infusion, patients undergo NMA-LD consisting of cyclophosphamide (60 mg/kg) daily x 2 days followed by fludarabine (25 mg/m2) daily x 5 days. LN-145 is infused 24 hours after the last dose of fludarabine followed by up to 6 doses of IL-2 (600,000 IU/kg) every 8-12 hours. Simon’s two-stage optimal design with one-sided alpha level=0.025 and 80% power will be used to compare an objective response rate (ORR) of 5% vs. 20% in the first stage (n=15 subjects). If two or more ORR are observed, trial will expand to Stage 2 (n=47). The primary endpoint is the ORR per RECIST v1.1. Secondary endpoints include complete response, duration of response, disease control rate, progression free- and overall survival; and the safety summarization of treatment-emergent adverse events (AEs) including serious AEs, AEs leading to discontinuation, and clinical laboratory tests. Patients must have been treated with at least 1 systemic chemotherapy or immunotherapy treatment for recurrent, metastatic, or persistent cervical cancer and, in addition to the tumor targeted for excision and TIL manufacture, must have an additional measurable lesion for assessment of response. Other major eligibility criteria include amongst others: adequate bone marrow, liver, pulmonary, cardiac and renal function; ECOG performance status of 0 or 1. Systemic steroids greater than 10 mg/day prednisone equivalents are prohibited as are a history of serious immunotherapy-related adverse events.
Trial Registration
ClinicalTrials.gov identifier: NCT03108495
P221 A phase 2 study to evaluate the safety and efficacy using autologous tumor infiltrating lymphocytes (LN-145) in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck
Rom Leidner1, James Ohr2, Robert Brown3, Sam Suzuki3, Igor Gorbatchevsky3, Maris Fardis3, Robert Ferris2
1Earle A. Chiles Research Institute – Providence Cancer Center, Portland, OR, USA; 2Hillman Cancer Center at UPMC, Pittsburgh, PA, USA; 3Iovance Biotherapeutics, San Carlos, CA, USA
Correspondence: Rom Leidner; Robert Brown (robert.brown@iovance.com)
Background
Adoptive cell therapy (ACT) may be effective in treating immunogenic tumors with high mutational load such as melanoma and virally-associated tumors like cervical cancer with several patients in studies performed by other institutions achieving durable complete response for years. Despite the heterogeneity of squamous cell carcinomas of the head and neck (HNSCC), most tumors are either virally-associated (e.g., HPV in oropharyngeal) or carry high mutational load (e.g., tobacco-related) providing an increased diversity of potential targets ideal for the polyclonal nature of ACT. Furthermore, outcomes for patients with recurrent and/or metastatic HNSCC remain poor. Therefore, a clear rationale exists for the potential application of ACT in patients with HNSCC.
Methods
Clinical trial C-145-03 (NCT03083873) is a prospective phase 2 multicenter, open-label study evaluating the efficacy of a single autologous tumor infiltrating lymphocyte infusion (LN-145) followed by IL-2 after a non-myeloablative lymphodepletion (NMA-LD) regimen in patients with recurrent and/or metastatic HNSCC. Study-related therapy begins with resection of a tumor lesion that is then shipped to a central GMP manufacturing facility where TIL are extracted, expanded, packaged, and shipped for administration (LN-145). One week prior to LN-145 infusion, patients undergo NMA-LD consisting of cyclophosphamide (60 mg/kg) daily x 2 days followed by fludarabine (25 mg/m2) daily x 5 days. LN-145 is infused 24 hours after the last dose of fludarabine followed by up to 6 doses of IL-2 (600,000 IU/kg) every 8-12 hours. Simon’s two-stage optimal design with one-sided alpha level=0.025 and 80% power will be used to compare an objective response rate (ORR) of 5% vs. 20% in the first stage (n=15 subjects). If two or more ORR are observed, trial will expand to Stage 2 (n=47). The primary efficacy endpoints are the objective response rate per RECIST v1.1 and the safety summarization of treatment-emergent adverse events (AEs) including serious AEs, AEs leading to discontinuation, and clinical laboratory tests. Secondary efficacy endpoints include CR, DOR, PFS, and OS. Patients must have been treated with at least one systemic chemotherapy or immunotherapy treatment for recurrent and/or metastatic HNSCC and, in addition to the tumor targeted for excision and TIL manufacture, must have an additional measurable lesion for assessment of response. Additional eligibility criteria include amongst others: adequate bone marrow, liver, pulmonary, cardiac, and renal function; ECOG performance status of 0 or 1. Systemic steroids greater than 10 mg/day prednisone equivalents are prohibited as are a history of serious immunotherapy-related adverse events.
Trial Registration
ClinicalTrials.gov identifier:NCT03083873
P222 Clinical trial in progress: A phase 1b trial of talimogene laherparepvec (T-VEC) in combination with dabrafenib and trametinib in advanced melanoma with an activating BRAF mutation
Kenneth Byrd1, Nibedita Chakraborty2, Mehmet Kocak1, Alisa Harber2, Ari Vanderwalde2
1University of Tennessee Health Science Center, Memphis, TN, USA, Memphis, TN, USA; 2West Cancer Center, Germantown, TN, USA, Germantown, TN, USA
Correspondence: Kenneth Byrd (kbyrd@westclinic.com)
Background
Patients with BRAF-mutant advanced melanoma have been shown to respond at high rates to combination BRAF plus MEK inhibition. Response rates to single-agent immune therapies tend to be modest, but are often durable, while response rates to BRAF plus MEK inhibition tend to be high but transient. While early attempts at combining targeted and immune therapy resulted in dose-limiting hepatic toxicity, more recent attempts with newer agents have shown significant promise. T-VEC is an oncolytic viral immunotherapy which was designed to selectively replicate in tumors resulting in lytic cell death, antigen release, and production of GM-CSF to enhance systemic immune response. In a prior randomized, phase 3 study of T-VEC versus GM-CSF, durable response rate was statistically improved with T-VEC, with a strong trend towards improved overall survival. Dabrafenib and trametinib, inhibitors of BRAF and MEK, respectively, have been shown in combination to result in response rates of up to 75% and improvement in progression-free and overall survival compared to single-agent targeted therapy and chemotherapy. Combining T-VEC with dabrafenib and trametinib may further enhance antitumor immune responses in addition to preserving the targeted effect.
Methods
This is a Phase Ib, prospective, single arm study of T-VEC given in combination with standard doses of dabrafenib and trametinib in advanced melanoma with an activating BRAF mutation. The primary endpoint of the study is tolerability as measured by dose-limiting toxicities seen in the first 5 weeks of treatment. Key secondary endpoints include progression-free survival, objective response rate, change in tumor burden, time to response, and duration of response among responders. Tumor-level responses in injected and uninjected tumors, and characterization of immune markers in pre-study and on-study biopsies will be exploratory endpoints. Key eligibility criteria include unresectable stage IIIB-IV BRAF mutant melanoma, presence of measurable and injectable disease, no active cerebral metastases or autoimmune diseases, and any number of prior lines of therapy but no prior receipt of T-VEC. T-VEC (106 PFU/mL first dose, 108 PFU/mL subsequent doses) will be administered by intralesional injection into cutaneous, subcutaneous, or nodal lesions on week 1 day 1, week 4 day 1, and every 2 weeks thereafter until disappearance of injectable lesions, complete response, progressive disease, intolerance of study treatment, or 24 months after starting therapy, whichever occurs first. Dabrafenib and trametinib will be given until progression or intolerance. Twenty subjects are to be enrolled at a single U.S. institution.
Trial Registration
NCT03088176
P223 KEYNOTE-199: Phase 2 nonrandomized study of pembrolizumab in patients with PD-L1+ and PD-L1– metastatic castration-resistant prostate cancer
Johann de Bono1, Josep Piulats2, Marine Gross-Goupil3, Jeffrey Goh4, Kristiina Ojamaa5, Christopher Hoimes6, Ulka Vaishampayan7, Raanan Berger8, Ahmet Sezer9, Ronald De Wit10, Charles Drake11, Haiyan Wu12, Christian Poehlein12, Emmanuel S. Antonarakis13
1Royal Marsden Hospital and the Institute of Cancer Research, London, United Kingdom; 2Instituto Catalan de Oncologia, Hospital Duran i Reynals, Hospitalet de Llobregat, Barcelona, Spain; 3Institut Bergonie, Bordeaux, France; 4Royal Brisbane & Women’s Hospital, Brisbane, Australia; 5East Tallinn Central Hospital, Tallinn, Estonia; 6University Hospitals Seidman Cancer Center, Cleveland, OH, USA; 7Karmanos Cancer Institute, Detroit, MI, USA; 8Chaim Sheba Medical Center, Ramat-Gan, Israel; 9Baskent Üniversitesi Adana Uyg. ve Arast, Adana, Turkey; 10Erasmus MC Cancer Institute, Rotterdam, Netherlands; 11Columbia University, New York, NY, USA; 12Merck & Co., Inc., Kenilworth, NJ, USA; 13Johns Hopkins Kimmel Cancer Center, Baltimore, MD, USA
Correspondence: Johann de Bono (Johann.DeBono@icr.ac.uk)
Background
Metastatic castration-resistant prostate cancer (mCRPC) treatment has included suppression of androgen receptor signaling, palliative radiation therapy, and chemotherapy. As expression of the programmed death 1 (PD-1) receptor and its ligand PD-L1 is present in a subset of mCRPC lesions, targeting this pathway may be an attractive treatment option. KEYNOTE-199 (NCT02787005) is a nonrandomized, multinational, multicohort open-label phase 2 study to evaluate the anti–PD-1 antibody pembrolizumab in patients with mCRPC.
Methods
Patients must be ≥18 years old with histologically or cytologically confirmed prostate adenocarcinoma without small-cell histology, measurable disease (RECIST v1.1) or detectable bone metastases by whole-body bone scintigraphy and no RECIST v1.1 measurable tumors, supplied tumor sample for PD-L1 expression (new or archived), disease progression within 6 months before screening, and ECOG performance status 0-2. As of June 2017, 2 additional cohorts were added called cohorts 4 and 5. For these cohorts patients had to fail or show signs of failure on current prechemotherapy enzalutamide; patients could fail abiraterone treatment before enzalutamide. For cohort 4, patients will be enrolled with RECIST v1.1-measurable disease (n=80) and for cohort 5 patients will be enrolled with bone metastases only or bone-predominant disease (n=40). For both cohorts patients will receive pembrolizumab 200 mg every 3 weeks (Q3W) plus current enzalutamide regimen. For the original cohorts (cohorts 1-3), patients must have been treated with ≥1 targeted endocrine therapy (abiraterone or enzalutamide) and ≤2 chemotherapy regimens; one must have contained docetaxel. Patients also must be undergoing androgen deprivation with serum testosterone <50 ng/dL. Patients will be enrolled based on PD-L1 status and RECIST v1.1 measurability and will receive pembrolizumab 200 mg Q3W: PD-L1–positive, RECIST v1.1 measurable disease (cohort 1), PD-L1–negative, RECIST v1.1 measurable disease (cohort 2; cohorts 1 and 2 combined, n=200), and bone metastases and RECIST v1.1 nonmeasurable disease (cohort 3, n=50). All patients will continue until documented confirmed disease progression, unacceptable adverse events (AEs), or illness that prevents further treatment. Imaging response will be assessed every 9 weeks for approximately 1 year and every 12 weeks thereafter, per central imaging vendor review (RECIST v1.1) and the Prostate Cancer Clinical Trials Working Group 3 guidelines. AEs will be monitored throughout the study. Primary end points are overall response rate for cohorts 1 and 2 combined and for cohorts 1, 2, and 4. Key secondary end points include safety and tolerability, duration of response, disease control rate, radiographic progression-free survival, and overall survival.
Trial Registration
ClinicalTrials.gov, NCT0278700
P224 Immunological correlates observed in an Interim analysis of the Phase 3 ADAPT Trial evaluating Rocapuldencel-T (AGS-003), for the treatment of patients with metastatic renal cell carcinoma (mRCC)
Mark DeBenedette, Alicia Gamble, Ana Plachco, Marcus Norris, Charles Nicolette
Argos Therapeutics Inc, Durham, NC, USA
Correspondence: Mark DeBenedette (mdebenedette@argostherapeutics.com)
Background
Rocapuldencel-T is an investigational patient-specific immunotherapeutic comprised of autologous dendritic cells programmed with RNA from the patient’s tumor to express tumor-specific antigens and thereby induce a memory T cell response. Rocapuldencel-T is engineered to secrete IL-12, a critical cytokine for memory-T cell formation, to induce an antigen-specific memory T cell response. We previously reported results from a Phase 2 study showing a correlation between the magnitude of the induced memory-T cell response and overall survival (OS) in advanced RCC patients. We therefore measured the antigen-specific memory T cell response and the level of secreted IL-12 in patients receiving Rocapuldencel-T to assess the relationship of these parameters to each other and to overall survival (OS).
Methods
The ADAPT trial is designed to evaluate OS in subjects with newly diagnosed mRCC receiving Rocapuldencel-T in combination with standard-of-care (SOC) versus SOC alone. As part of this interim analysis, immune monitoring was performed on patients (N=146) enrolled in the United States and randomized to the combination arm. The change in the number of CD28+/CD45RA- antigen-specific memory T cells present after in vitro stimulation of PBMCs with Rocapuldencel-T was measured with multi-color flow cytometry. The amount of IL-12 secreted by each patient’s immunotherapeutic was measured by a cytokine bead capture method (N=179). The increase in the number of antigen-specific memory T cells after administration of Rocapuldencel-T and the concentration of IL-12 secreted by each patient’s immunotherapeutic where correlated with OS.
Results
Data from this interim analysis revealed a statistically significant increase in the number of CD28+/CD45RA- memory T cells after as few as three doses of Rocapuldencel-T administered three weeks apart. The increase above baseline became a statistically significant correlate with OS after seven doses. Furthermore, the amount of IL-12 each patient’s immunotherapeutic produced showed a statistically significant correlation with both the magnitude of the memory T cell response and OS.
Conclusions
This interim analysis demonstrated that administration of Rocapuldencel-T resulted in an increase in antigen-specific memory T cells and that the magnitude of the induced memory T cell response after seven doses correlated with OS. Additionally, the amount of IL-12 secreted by each patient’s immunotherapeutic correlated with both the magnitude of the induced memory T cell response and OS. Therefore, the level of IL-12 secretion may serve as a predictive biomarker for T cell response to Rocapuldencel-T and favorable clinical outcome, warranting further investigation.
Trial Registration
ClinicalTrials.gov identifier-NCT01582672
P225 Functional reversal of Foxp3+ T regulatory activity in patients enrolled in the Phase 3 ADAPT Trial evaluating Rocapuldencel-T for the treatment of patients with metastatic renal cell carcinoma (mRCC)
Mark DeBenedette, Joe Horvatinovich, Elizabeth Grogan, Larissa Benavente, Irina Tcherepanova, Charles Nicolette
Argos Therapeutics Inc, Durham, NC, USA
Correspondence: Mark DeBenedette (mdebenedette@argostherapeutics.com); Joe Horvatinovich
Background
Rocapuldencel-T is an investigational patient-specific immunotherapeutic comprised of autologous dendritic cells programmed with RNA from the patient’s tumor to express tumor-specific antigens and induce a memory-T cell response. The Phase 3 ADAPT trial is designed to evaluate overall survival (OS) in subjects with newly diagnosed mRCC receiving Rocapuldencel-T in combination with standard-of-care (SOC) versus SOC alone. Current SOC first-line treatment with sunitinib is shown to decrease regulatory T cells (Tregs), thus potentially modulating anti-tumor activity. Therefore, FoxP3+ Treg cell counts in patients receiving Rocapuldencel-T were measured ex vivo in whole blood samples and the relationship to OS was assessed for each arm of the study.
Methods
For this interim analysis, whole blood samples collected at intervals during the course of the ADAPT trial were stained immediately ex vivo by multi-color flow cytometry to determine the number of CD4+/CD25+/CD127-/Foxp3+ Treg cells. The correlation between the number of Tregs present and overall survival was determined for the combination arm subjects (N=177) and the SOC arm subjects (N=80) for whom data was available. We further studied the impact of Rocapuldencel-T on Treg phenotype and function after in vitro stimulation of PBMCs from treated subjects.
Results
As reported elsewhere, data from this interim analysis showed a decline in the number of Tregs after the first cycle of sunitinib. Surprisingly, the number of Tregs at baseline or at any time point measured, positively correlated with OS in the combination arm, but negatively correlated with OS in the SOC arm. Furthermore, in vitro stimulation of autologous PBMCs with Rocapuldencel-T resulted in conversion of Tregs to a proinflammatory cell phenotype that proliferates in culture, thus providing a possible explanation for the positive correlation between the number of Tregs and OS in the combination arm.
Conclusions
In this interim analysis, baseline Tregs positively correlated with survival only in the Rocapudencel-T arm and not in the SOC arm. These data suggest that Rocapudencel-T exerts a biologic effect catalyzed by the presence of Tregs, potentially by conversion to T effector cells, resulting in favorable clinical outcome. This is consistent with the in vitro observation that Rocapudencel-T can convert Tregs to T effector cells. Therefore, the level of Tregs in whole blood samples may serve as a useful baseline biomarker predictive of favorable long-term clinical outcome in patients treated with Rocapudencel-T while also predicting poor outcome in patients receiving conventional SOC.
Trial Registration
ClinicalTrials.gov identifier-NCT01582672.
P226 FRACTION-RCC: a randomized, open-label, adaptive, phase 2 study of nivolumab in combination with other immuno-oncology agents in patients with advanced RCC
Corina Candiani Taitt1, Robert J. Motzer2, Toni K. Choueiri3, Bernard J. Escudier4, Timothy Kuzel5, Michael A. Carducci6, Suresh Nair7, Scott S. Tykodi8, Sarah Tannenbaum-Dvir1, Megan Wind-Rotolo1, Katy L. Simonsen1, Paula M. Fracasso1
1Bristol-Myers Squibb, Princeton, NJ, USA; 2Memorial Sloan Kettering Cancer Center, New York, NY, USA; 3Dana-Farber Cancer Institute, Boston, MA, USA; 4Gustave Roussy Cancer Centre, Villejuif Cedex, France; 5Rush University Medical Center, Chicago, IL, USA; 6Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University, Baltimore, MD, USA; 7Lehigh Valley Health Network, Allentown, PA, USA; 8University of Washington and Fred Hutchinson Cancer Research Center, Seattle, WA, USA
Correspondence: Corina Candiani Taitt (ea@chrysalismedical.com)
Background
Nivolumab, a fully human IgG4 monoclonal antibody (mAb) that targets the PD-1 receptor, is approved for patients with advanced renal cell carcinoma (RCC) after prior antiangiogenic therapy based on superior overall survival vs everolimus (CheckMate 025; Motzer RJ, et al. N Engl J Med. 2015). Nivolumab has also shown promising antitumor activity in combination with ipilimumab (a fully human IgG1 mAb that targets cytotoxic T-lymphocyte antigen 4) in patients with metastatic RCC, supporting the rationale that nivolumab in combination with other immuno-oncology (IO) agents or targeted therapies may improve outcomes in patients with advanced RCC. Given the rapid development of novel IO agents, traditional study designs may not efficiently evaluate all possible IO-IO and IO-targeted therapy combinations. Fast Real-time Assessment of Combination Therapies in Immuno-ONcology (FRACTION) is an innovative clinical trial program with a rolling, adaptive platform design that allows for the addition of new regimens as well as the withdrawal of ineffective regimens. Here we describe the study concept, key design components, and first IO treatment combinations of FRACTION-RCC, a phase 2, randomized, open-label, adaptive study in advanced RCC (NCT02996110).
Methods
FRACTION-RCC is envisioned to accelerate the development of the next generation of IO combinations for patients with metastatic RCC. Patients with advanced RCC with a clear-cell component will be enrolled based on prior IO treatment and randomized to receive nivolumab plus BMS-986016 (a fully human IgG4 mAb that targets lymphocyte activation gene 3) or nivolumab plus ipilimumab. Enrollment is continuous and may offer patients consecutive treatment options based on treatment exposure and response. Primary endpoints include objective response rate, duration of response, and progression-free survival rate at 24 weeks. The secondary endpoint is safety. Biomarker analyses will also be performed. New treatment combinations will be added over time to explore their potential benefits and provide a continuous flow of treatment options for patients whose cancer progresses on existing treatments.
Trial Registration
ClinicalTrials.gov, NCT02996110
P227 A trial to evaluate the safety, immunogenicity and clinical activity of a helper peptide vaccine plus PD-1 blockade
Craig Slingluff, Jr, Elizabeth Gaughan, William Grosh, Gina Petroni, Kimberly Bullock
University of Virginia, Charlottesville, VA, USA
Correspondence: Elizabeth Gaughan (egaughan@virginia.edu)
Background
PD-1 blocking antibodies are standard first line therapy for management of advanced melanoma. Monotherapy response rates to these agents are reported between 30-40%; therefore, the majority of patients require additional therapy [1, 2]. Strategies to improve the benefit of these agents are currently focused on combination with other systemic agents and adjunctive local treatment measures. We are evaluating the combination of pembrolizumab with a vaccine incorporating 6 peptides that induces CD4+ helper T cell (TH) responses (6MHP) and has proven activity in patients with advanced melanoma [3]. The primary end point is determination of the safety and tolerability of the combination of 6MHP vaccine plus Pembrolizumab and estimation of the CD4+ T cell response rate to 6MHP in the blood and sentinel immunized node. Secondary endpoints include evaluation of the induction of epitope-spreading and the T cell infiltration of the tumor microenvironment.
Methods
This is an open-label, phase I/II study to evaluate the safety, immunogenicity and clinical activity of the 6MHP vaccine and pembrolizumab (MEL64, NCT02515227). Candidates may be resistant or naïve to systemic immunotherapy agents. Patients with prior PD-1 antibody exposure are included if the patient failed to experience a clinical response after 12 weeks or progressed on treatment. All subjects will receive 6MHP on days 1, 8, 15, 43, 64, and 85. Pembrolizumab will be administered intravenously (IV) every 3 weeks for up to 2 years. Biopsy specimens will be collected from the tumor (days 1 and 22) and a sentinel immunized node (SIN; day 22). Biopsy and peripheral blood specimens will be used in the immunologic analyses. Overall target sample size is based upon having sufficient information to assess whether combined treatment with 6MHP vaccine plus pembrolizumab increases the immunogenicity of 6MHP alone in the entire study population. Maximum target sample size is 40 patients. Trial is currently enrolling patients.
Results
Trial in Progress.
Conclusions
Trial in Progress.
Trial Registration
NCT02515227
References
1. Robert C, Schachter J, Long GV, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med. 2015;372(26):2521-32.
2. Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015;373(1):23-34.
3. Slingluff CL Jr, Petroni GR, Olson W, et al. Helper T cell responses and clinical activity of a melanoma vaccine with multiple peptides from MAGE and melanocytic differentiation antigens. J Clin Oncol. 2008;26(30):4973-80.
P228 Pembrolizumab and decitabine for relapsed and refractory acute myeloid leukemia (PD-AML)
Catherine Lai, Oetjen Karolyn, Katherine Lindblad, Christin Destefano, Meghali Goswami, Hong Yuen Wong, Therese Intrater, Debbie Draper, Laura Dillon, Christopher Hourigan
National Heart, Lung and Blood Institute, National Institutes of Health, Bethesda, MD, USA
Correspondence: Christopher Hourigan (hourigan@nih.gov)
Background
While a variety of different treatment regimens have been studied for patients with relapsed/refractory acute myeloid leukemia (AML) clinical outcomes unfortunately remain dismal. There appears to be no single superior therapeutic approach and the current standard of care is referral to an appropriate clinical trial. The unique combination of pembrolizumab and decitabine used in this trial was selected for investigation based on several factors: 1) Largely non-overlapping adverse reaction profiles for these agents. 2) Both agents already FDA approved potentially allowing for rapid translation/adoption. 3) Both agents have previously evaluated dosing schedules that are compatible with one another. 4) Theoretical possibility of synergy given respective mechanisms of action.
Methods
PD-AML (17-H-0026, NCT02996474) is an investigator sponsored, single-institution, single-arm open-label ten subject pilot study to evaluate the feasibility of a novel combination of pembrolizumab and decitabine in adults with relapsed/refractory AML. Secondary objectives will explore efficacy and determine time to first response, best response and duration of best response. Laboratory objectives include investigation of changes in AML clonal composition and disease burden during therapy, and measurement of changes in immune parameters associated with clinical efficacy and/or toxicity. Up to eight 21 day cycles of pembrolizumab are given with decitabine given on days 8-12 and 15-19 on alternative cycles (ie: cycles 1, 3, 5 and 7) (Fig. 1).
Results
This clinical trial opened in February 2017 and is currently in progress. Currently seven patients have reached response and toxicity assessment time-points. Updated results will be presented at the meeting.
Conclusions
Acute myeloid leukemia is a heterogeneous group of diseases with distinct molecular and phenotypic characteristics. Even within a single patient AML may be polyclonal at any examined time-point, and this clonal composition can change over time with the clone predominant at presentation not necessarily the one responsible for relapse and death. We hypothesize that effective pembrolizumab therapy for refractory/relapsed AML may be associated with changes in the leukemic clonal composition due to differences in immunogenicity between clones. The oligoclonal nature of AML biology, together with a blood and bone marrow distribution highly amenable to repeated sampling of the sites of disease burden, provides a near unique opportunity to investigate fundamental mechanisms underpinning treatment efficacy of this new combination of immunotherapeutic drugs.
Trial Registration
[https://clinicaltrials.gov/ct2/show/NCT02996474]
FDA-IND: 131826

See text for description
P229 Withdrawn
P230 Phase I/II safety and efficacy study of image guided intratumoral CD40 agonistic monoclonal antibody APX005M in combination with systemic pembrolizumab in metastatic melanoma patients
Daniel Johnson H Johnson, Srisuda Lecagoonporn, Chantale Bernatchez, Cara Haymaker, Salah Bentebibel, Marc Uemura, Cassian Yee, Rodabe Amaria, Sapna Patel, Hussein Tawbi, Isabella Glitza, Michael A. Davies, Michael K. Wong, Wen-Jen Hwu, Patrick Hwu, Willem Overwijk, Adi Diab
UT-MD Anderson Cancer Center, Houston, TX, USA
Correspondence: Daniel Johnson H Johnson (dhjohnson@mdanderson.org); Adi Diab
Background
Checkpoint blockade has become a major modality in the treatment of metastatic melanoma (MM). However, long-term survival and durable remission rates remain low and new treatment options are needed. CD40 activation on antigen presenting cells (APCs) initiates their maturation and ability to prime and activate CD8+ T cells through upregulation of co-stimulatory molecules (CD80, CD86, CD70, 4-1BBL, OX40L, and GITR-L) as well as expression of cytokines such as IL-12. Furthermore, CD40 activation cause macrophages to develop a more tumoricidal phenotype and tumor cells to increase MHC I expression.
Direct intratumoral (IT) immune modulation utilizes the tumor as a “vaccine site” to generate a tumor specific immune response. We hypothesize that (IT) injection of a CD40 agonist such as APX005M, will “immunize” patients against melanoma neoantigens through “licensing” of tumor infiltrating APCs for tumor specific T cell priming and activation. In preclinical mouse models, we have shown that IT administration of the recombinant adenovirus encoding the dendritic cell-activating CD40L induces CD8+ T cell-mediated systemic activity against B16 melanoma. Importantly, IT rAdCD40L also augmented the activity of anti-PD-1.
Methods
This phase I/II trial (NCT02706353) evaluates the safety, efficacy, and immunological impact of IT administration of APX005M (CD40 agonistic mAb) in combination with systemic pembrolizumab in pts with MM. An accelerated 3+3 design was used for the phase 1 dose escalation portion of this study. Pts will receive IT APX005M at escalating doses every 3 weeks for a total of 4 doses. Image guidance will allow for injection of visceral, nodal, and soft tissue metastases. The single-arm phase 2 expansion will evaluate the overall response rate (ORR) of this regimen 12 weeks after initiation of treatment. Some key inclusion criteria: confirmed cutaneous or mucosal melanoma; measurable, unresectable stage-III or stage-IV disease, and at least 2 injectable lesions. Key exclusion criteria include: prior immunotherapy, uveal melanoma, active autoimmune disease, or active immunodeficiency. A sample size of 26 patients will have 75% power to detect an improvement from a null ORR of 33% to 55%, using a one group chi-square test and assuming a one-sided α–level of 5%. Immune analysis will be performed on pre and on-treatment tumor/liquid biopsies including but not limited to quantification of dendritic cells and T cells both in injected and non-injected tumors.
Trial Registration
NCT02706353
P231 KEYNOTE-590: randomized, phase 3 study of chemotherapy + pembrolizumab vs chemotherapy + placebo as first-line therapy for patients with advanced esophageal or esophagogastric junction (E/EGJ) cancer
Ken Kato1, Manish Shah2, Peter Enzinger3, Jaafar Bennouna4, Lin Shen5, Antoine Adenis6, Ying Zhu7, Pooja Bhagia7, Minori Koshiji7, Toshihiko Doi8
1National Cancer Center Hospital, Tokyo, Japan; 2Weill Cornell Medical College, New York Presbyterian Hospital, New York, NY, USA; 3Dana Farber Cancer Institute, Boston, MA, USA; 4CHU de Nantes, Nantes, France; 5Beijing Cancer Hospital, Beijing, China; 6Institut du Cancer de Montpellier, Montpellier, France; 7Merck & Co., Inc., Kenilworth, NJ, USA; 8National Cancer Center East, Chiba, Japan
Correspondence: Ken Kato (kenkato@ncc.go.jp)
Background
No chemotherapeutic regimens or targeted agents are approved specifically for esophageal cancer; available options have limited benefit and substantial toxicity. In the phase 1b KEYNOTE-028 study (NCT02054806), pembrolizumab monotherapy demonstrated manageable safety and durable antitumor activity in heavily pretreated patients with PD-L1–positive advanced esophageal carcinoma. In KEYNOTE-059 (NCT02335411), combining chemotherapy (cisplatin and 5-fluorouracil) and pembrolizumab as first-line treatment for patients with advanced gastric or gastroesophageal junction cancer resulted in encouraging efficacy and manageable safety. This suggests chemotherapy plus pembrolizumab as a potential therapeutic strategy for esophageal cancer. KEYNOTE-590 is a randomized, double-blind, multicenter phase 3 study of cisplatin and 5-fluorouracil plus pembrolizumab versus cisplatin and 5-fluorouracil plus placebo in patients with advanced E/EGJ carcinoma.
Methods
Eligible patients are ≥18 years of age; have locally advanced unresectable or metastatic adenocarcinoma or squamous cell carcinoma of esophagus or metastatic Siewert type 1 adenocarcinoma of EGJ, measurable disease per RECIST 1.1, ECOG performance status 0-1, adequate organ function; have had no prior therapy for advanced disease, no autoimmune disease, no active infection; and can provide a newly obtained or archival tissue sample. Patients will be randomly assigned 1:1 to cisplatin 80 mg/m2 IV every 3 weeks (Q3W) for 6 cycles plus 5-fluorouracil 800 mg/m2 continuous IV on days 1-5 Q3W IV plus pembrolizumab 200 mg IV Q3W or cisplatin 80 mg/m2 IV Q3W for 6 cycles plus 5-fluorouracil 800 mg/m2 continuous IV on days 1-5 Q3W plus placebo Q3W IV. Treatment will continue up to 2 years. Response will be assessed using CT (preferred) or MRI every 9 weeks by central imaging per RECIST v1.1. Adverse events will be graded per NCI CTCAE v4.0 and up to at least 30 days after the end of treatment. Primary end points are PFS per RECIST v1.1 and OS in all patients and in patients with PD-L1 positive or negative tumor expression (combined positive score ≥10% or <10% using immunohistochemistry). Secondary end points include ORR per RECIST v 1.1, duration of response, safety, and health-related quality of life. PFS and OS will be compared between groups using a stratified log-rank test; hazard ratios will be estimated using a Cox regression model. The Kaplan-Meier method will be used to estimate event rates within groups. Enrollment is planned for approximately 700 patients.
Trial Registration
ClinicalTrials.gov, NCT03189719
P232 A pilot study to evaluate the clinical and immunological effects of incorporating a CD40-agonistic antibody into the multimodality treatment of resectable esophageal and GE junction cancers
Andrew H. Ko, Lawrence Fong
University of California San Francisco, San Francisco, CA, USA
Correspondence: Andrew H. Ko (andrew.ko@ucsf.edu
Background
Targeting CD40, a member of the TNF receptor superfamily found on antigen presenting cells (APCs), represents a promising cancer immunotherapeutic strategy. Activation of this costimulatory molecule results in improved antigen processing and presentation and cytokine release from activated APCs, enhancing T cell responses. Additionally, CD40 is expressed on many tumor cells that, when activated, results in tumor cell apoptosis and inhibition of tumor growth. APX005M (Apexigen, San Carlos, CA) is a humanized IgG1 anti-CD40 agonistic antibody that binds to CD40 with high affinity. In a FIH phase I clinical trial in patients with advanced solid tumors, APX005M was relatively well tolerated, with cytokine release syndrome (CRS) as the DLT at doses above the RP2D. Importantly, correlative studies show that APX005M produces dose-dependent activation of APCs, T cell activation, and increases in circulating cytokine levels.
Methods
This pilot trial represents the first to evaluate a CD40-agonistic antibody in esophageal/GE junction cancer, a disease in which IO agents (particularly PD-1 mAbs) have demonstrated promising activity. The study is also the first to explore combining IO with chemoradiation in the neoadjuvant setting, as this multimodality approach represents the standard of care for patients with resectable esophageal/GE junction cancer and is optimally conducive for serial tumor tissue acquisition. A total of 16 patients with resectable (uT1-3N0-1) squamous cell or adenocarcinoma of the esophagus or GE junction will be enrolled. Chemoradiation consists of radiation (5040cGy in 28 daily fractions) and low-dose carboplatin plus paclitaxel weekly x 5, as per standard of care. APX005M 0.3 mg/kg (1 dose level below single-agent MTD) is given every 3 weeks for a total of 4 doses, with the first dose administered two weeks prior to the initiation of concurrent chemoradiation. APX005M administration is offset by 2-3 days from chemotherapy to avoid the steroid premedication administered with paclitaxel. Tumor tissue is acquired via endoscopic biopsy at baseline and following the single-dose “run-in” of APX005M; and then again at esophagectomy, which occurs 1-2 months following completion of chemoradiation. Serial blood collections are also performed at multiple pre-defined timepoints. In addition to assessing the feasibility, safety, and preliminary efficacy (as measured by pathologic complete remission rate) of this novel combination, analyses of tumor tissue and blood will be performed, including Tissue Multiplex Immunohistochemistry and flow cytometry for both APC and T cell activation, as well as T cell receptor sequencing for T cell repertoire diversity.
P233 A phase I study to evaluate the safety of multi-antigen stimulated tumor specific cellular therapy (MASCT-I) in patients with advanced solid tumors
Ruihua Xu1, Xiaoshuang Li2, Yanjun Kong2, Jianchuan Xia1, Desheng Weng1, Xiaoshi Zhang1, Xing Zhang1
1Sun Yat-sen university affiliated oncology hospital, Guangzhou, China; 2HRYZ Biotech Company, Shenzhen, China
Correspondence: Ruihua Xu (kyj8@sina.com)
Background
Tumor-specific immune responses are known to be initiated by tumor associated and/or specific antigen-sensitized dendritic cells (DCs), that can effectively process and present tumor antigens to CD4+ and CD8+ T cells. MASCT-I is a sequential immune cell therapy for solid tumor, which included multi-antigen loaded DC vaccines followed by the adoptive transfer of anti-tumor specific T cells. DC vaccines are produced from patients’ autologous PBMC-derived DC loaded with multiple tumor associated antigen peptides and are injected into patients to induce active anti-tumor immunity. Anti-tumor specific T cells are stimulated from the same patient by co-cultured with DC vaccines and Anti-PD1 antibody in vitro, are then infused into the patient to target tumor cells.
Methods
This is a single center, three stages phase I study. Stage1 and 2 will enroll (3+3 design) patients with advanced (unresectable) or recurrent bladder cancer or soft tissue sarcoma who have failed all standard therapies, Patients will be treated with MASCT-I. If the dose limitation toxicity (DLT) in the first cycle of MASCT-I is <33.3%, the stage 2 will begin. Patients who have advanced recurrent or metastatic bladder cancer with Gemcitabine and Cisplatin (GP) chemotherapy achieving clinical benefit (group 1) will be treated with MASCT-I as maintenance therapy. Also, patients who have advanced recurrent or metastatic sarcoma with achieving clinical benefit after MAID or CAV/IE (predominant Doxorubicin regimens) (group 2) will be treated with MASCT-I combined with Ifosfamide as maintenance therapy too. During the stage 2, if, in the first cycle of MASCT-I treatment, the DLT is < 33.3%, stage 2 will extend to stage 3. Approximately additional 24 patients will be enrolled in group 1 and 2. The primary objective is safety and tolerability. Secondary objectives include DCR, PFS, TTP, OS. As of 17 May 2017, Three patients were enrolled and completed Stage 1 without any DLT and treatment related SAEs. Five AEs are related with treatment, they are pain on the injection site, fatigue, pruritus and arthralgia, which were all grade 1. Recruitment is ongoing for dose expansion (stage 2). Clinical trial information: NCT030343
P234 AST-VAC2: An allogeneic dendritic cell cancer immunotherapy entering clinical trials in patients with lung cancer in the advanced and adjuvant setting
Christian Ottensmeier1, Hayley Farmer-Hall2, Gary Acton2, Heike Lentfer3, Kevin Nishimoto4, Uzma Shoukat-Mumtaz4, Erik Whiteley4, Rob Allen4, Jane Lebkowski4
1University of Southampton, Southampton, United Kingdom; 2Cancer Research UK, London, United Kingdom; 3Cancer Research UK, Potters Bar, United Kingdom; 4Asterias Biotherapeutics, Fremont, CA, USA
Correspondence: Jane Lebkowski (rallen@asteriasbio.com)
Background
Primary lung cancer is the most common malignancy after non-melanocytic skin cancer with deaths exceeding those from any other type of malignancy worldwide. In advanced (TNM stage IIIA, IIIB and IV) disease, only palliative therapy is available which carries a high risk of toxicity and limited potential to extend life. Intradermal delivery of dendritic cells (DCs) carrying an immunogenic cargo offers a novel approach to address malignancy mediated through upregulated telomerase expression and decreased cell death. AST-VAC2 is a mature, allogeneic DC vaccine derived by differentiating H1 human embryonic stem cells (hESCs) into mature DCs, transfected to express the tumor associated antigen human telomerase reverse transcriptase (hTERT) and lysosomal associated membrane protein 1 (LAMP-1) [1,2]. LAMP-1 fusion proteins enable target hTERT peptides to be directed to HLA (Human Leukocyte Antigen) II and HLA I receptors on the surface of endogenous DCs, invoking dual, antigen-specific CD4+ and CD8+ responses [3]. Preclinical investigations have shown that AST-VAC2 phagocytose, process, and present antigen upon maturation. Furthermore they produce immunostimulatory cytokines, migrate in response to cytokines, and activate antigen specific T cell responses.
Methods
The first-in-human trial will evaluate safety, tolerability, immunogenicity and therapeutic potential in adult NSCLC patients in advanced (metastatic or locally advanced disease) and adjuvant (currently radiologically disease free) settings. Patients who are positive for the HLA-A2 allele (expressed by AST-VAC2) will receive 6 weekly doses of 1 x 107 AST-VAC2 cells and will be monitored for one year as the primary follow-up point and subsequently for up to five years for long-term follow-up. Safety assessments as well as immunological monitoring for the generation and maintenance of hTERT specific T cells will be key endpoints. Results will be used to expand the clinical indications for assessment and support future combination immunotherapy approaches.
References
1. Tseng SY, Nishimoto KP, Silk KM, Dawes GN, Waldmann H, Fairchild PJ, Lebkowski JS, Reddy A. Generation of immunogenic dendritic cells from human embryonic stem cells without serum or feeder cells. Regen. Med. 2009; 4(4):513-526.
2. Nishimoto KP, Tseng S-Y, Lebkowski JS, Reddy A. Modification of human embryonic stem cell-derived dendritic cells with mRNA for efficient antigen presentation and enhanced potency. Regen. Med. 2011;6(3):303–318.
3. Su Z, Dannull J, Yang B, Dahm P, Coleman D, Yancey D,Sichi S, Niedzwiecki D, Boczkowski D, Gilboa E, Vieweg J. Telomerase mRNA-Transfected Dendritic Cells Stimulate Antigen-Specific CD8+and CD4+T Cell Responses in Patients with Metastatic Prostate Cancer. J Immunol. 2005.174: 3798–3807.
P235 Nivolumab in patients with advanced or metastatic non-small cell lung cancer (Stage IIIb/IV) who have received at least one prior systemic chemotherapeutic regimens
Sung Yong Lee, Sang Mi Chung, Ju Whan Choi, Young Seok Lee, Jong Hyun Choi, Jee Youn Oh, Kyung Hoon Min, Gyu Young Hur, Jae Jeong Shim, Kyung Ho Kang
Korea University Medical Center, Seoul, Republic of Korea
Correspondence: Sung Yong Lee (syl0801@korea.ac.kr)
Background
Nivolumab, a human programmed death 1 (PD-1) immune checkpoint inhibitor antibody, has been shown to increase overall survival in non-small cell lung cancer (NSCLC) patients. This immune checkpoint blockade has been approved in Korea, United States, the European Union, and other countries for the treatment of advanced NSCLC that has progressed after platinum-based chemotherapy. Nivolumab has demonstrated longer overall survival than docetaxel among the previously treated NSCLC patients. We have assessed the safety of nivolumab in 8 patients with previously treated, locally advanced or metastatic NSCLC who were enrolled in the NSCLC Expanded Access Program (EAP) in Korea University Guro Hospital.
Methods
This EAP program included subjects with histologically or cytologically documented NSCLC who have relapsed after systemic treatment with a minimum of 1 prior systemic treatment for stage IIIB/stage IV disease. Subjects were treated with 3 mg/kg of nivolumab IV every 2 weeks for a maximum of 24 months. Each 14-day dosing period constituted a single cycle. Patients included in the analysis had received ≥ 1 dose of nivolumab and were monitored for adverse events (AEs).
Results
As of June 30, 2017, eight patients participated. Median age was 63.0 years. All participants were male. Except for 1 (12.5%) current smoker and 1 (12.5%) never smoker, other 6 (75%) patients were former smokers. The average of smoking period was 27 pack-years. 2 had squamous and 6 had non-squamous histology. At the time of enrollment, 4 had bone, 3 had brain and 3 had brain metastases. Best response rates was 12.5% and disease control rate was 50%. Median progression free survival was 99.5 days (95% CI 50.1-174.7). During the nivolumab chemotherapy, 2 patients had pneumonia and 1 had stroke. Other 5 patients had no critical complication during the treatment.
Conclusions
Our study showed that EAP participants had some lower response rate and more prolonged PFS compared to previously reported in Checkmate-017 and in Checkmate-057. And nivolumab EAP showed good safety profiles. In conclusion, treatment with Nivolumab is safe and effective for patients who have previously received heavily chemotherapy.
P236 Innate immunotherapy of neuroblastoma and PD-1 checkpoint blockade
Holger Lode1, Maxi Zumpe1, Madlen Juettner1, Sascha Troschke-Meurer1, Evelyne Janzek2, Romana Schaefer2, Hans Loibner2, Nikolai Siebert1
1University Medicine Greifswald, Greifswald, Germany; 2Apeiron Biologics, Vienna, Austria
Correspondence: Holger Lode (lode@uni-greifswald.de)
Background
Passive immunotherapy of cancer is established for a variety of malignant diseases. Anti-GD2 antibody (Ab) ch14.18/CHO (dinutuximab beta) showed activity for the treatment high-risk neuroblastoma (NB) patients and received recently marketing approval in the EU. Here we demonstrate that one important mechanism of action in patients is antigen specific Ab-dependent cellular cytotoxicity (ADCC) and we report that ADCC impacts on PD-1/PD-L1 checkpoint regulation that can be targeted by co-treatment with an inhibitor.
Methods
53 patients received 100 mg/m2 ch14.18/CHO (d8-17), 6x106 IU/m2 sc IL-2 (d1-5; 8-12 and 160 mg/m2 oral 13-cis-RA (d19-32) in a closed single center program (53 pts). Polymorphisms in Fcγ-receptor genes 2A (H131R), -3A (V158F) and -3B (NA1/NA2) were determined by real-time PCR. Expression of PD-L1 and PD-1 was analyzed by RT-PCR and flow cytometry. Effect of PD-1/PD-L1 blockade and ch14.18/CHO-mediated anti-NB immune response was evaluated using anti-PD-1 Ab both in vitro (Nivolumab) and in the syngeneic GD2+ NB NXS2 mouse model (anti-mouse PD-1).
Results
We identified 33/53 patients with low affinity FCGR alleles (FCGR2A-H131R/R and/or FCGRA3A-V158 F/F). These patients showed lower PFS rates compared to 20/53 patients with high affinity polymorphisms (p < 0.01). ADCC levels on day 15 of cycle 1 in pts with high affinity polymorphisms showed an ADCC increase of 20±6% compared to 11±2% in the control. The correlation with functional immune parameter ADCC and clinical outcome confirm its role for clinical efficacy.
Interestingly, tumor specific ADCC in the presence of LA-N-1 neuroblastoma cells, leukocytes and sub-therapeutic ch14.18/CHO concentrations (10 ng/ml) results in a strong increase of the PD-L1 expression and incubation with IL-2 further enhanced this effect. Blockade with Nivolumab reversed the PD-L1-dependent inhibition of ADCC. Finally, mice treated with ch14.18/CHO in combination with PD-1 blockade showed strongest reduction of tumor growth, longest survival rate as well as the highest level of NB cell lysis mediated by serum and leukocytes of treated mice compared to controls.
Conclusions
Patient studies clearly reveal ADCC as mechanism of ch14.18/CHO against neuroblastoma, and this upregulates the inhibitory checkpoint PD-1/PD-L1. Combination of ch14.18/CHO with PD-1/PD-L1 blockade results in synergistic treatment effects. Similar upregulation of PD-L1 expression by suboptimal ADCC was also seen with the human GD2+ osteosarcoma cell line MG63. This suggests a broader applicability and to consider combinations of passive immunotherapy of cancer with PD-1/PD-L1 checkpoint inhibition.
P237 Phase I study of adoptive transfer of iNKT cells for treating patients with relapsed/advanced hepatocellular carcinoma
Jun Lu1, Xuli Bao1, Jia Guo1, Wenfeng Sun2, Hui Chen2, Yanpin Ma1, Xiongwei Cui1
1Beijing YouAn Hospital, Capital Medical University, Beijing, China; 2Beijing Gene Key Life Technology Co.Ltd., Beijing, China
Correspondence: Jun Lu (lujun98@ccmu.edu.cn)
Background
Invariant Natural Killer T (iNKT) cells represent a distinctive subset of T lymphocytes characterized by an invariant receptor Vα24/Jα18 in human, which play critical roles in regulating anti-tumor immunity by bridging innate and adaptive immune responses. We have previously demonstrated that the accumulation of circulating iNKT cells provide a better prognosis for hepatocellular carcinoma (HCC) patients, the adoptive transfer of ex vivo expanded human iNKT cells in HCC tumor-bearing NOD/SCID mice led to the decrease in tumor size, revealing the iNKT immunotherapy may bring clinical benefits for the HCC patients.
Methods
The phase I trial enrolls patients who have relapsed/advanced HCC tumor relapsed or metastasized through the body after standard treatment or the patients cannot receive standard treatment under current conditions. The purpose of this study is to find the biggest dose of iNKT cells that is safe and tolerance, to see how long they last in the body, to learn the immune-response, the side effects and if the iNKT cells will help people with relapsed/advanced HCC. Key eligibility criteria include age ≥18 years, with HCC (BCLC, stage C) proved by histopathology or proved by CT or MRI imaging system, relapsed after previous therapy and no effective therapies known at this time and life expectancy of ≥ 12 weeks. Three different dosing schedules will be evaluated. Three patients will be evaluated on each dosing schedule. The following dose levels will be evaluated: Loading Dose 1: 3x107/m2; Loading Dose 2: 6x107/m2; Loading Dose 3: 9x107/m2. The doses are calculated according to the actual number of iNKT cells. Human Interleukin-2 will be given at a dose of 25,000 IU/kg/day for 5-14 days. Tegafur will be given at a dose of 40~60 mg bis in die (BID) 2 weeks. immune responses were measured by Elispot and ELISA; flow cytometry assays were performed to evaluate the effects on immune cell subsets. Tumor biopsies were evaluated for iNKT cells by immunohistochemistry. Incidence of treatment-emergent adverse events were defined as signs/symptoms, laboratory toxicities, and clinical events that are possibly, likely, or definitely related to study treatment adverse events assessed according to NCI-CTCAE v4.0 criteria 2. HCC progression was evaluated by imaging according to the irRC standard.
Results
To date, one patient has been started on therapy and is in week 3 monitoring period, is tolerating treatment well, with no significant toxicities thus far. Data regarding the peripheral blood iNKT cells response will be presented.
Trial Registration
ClinicalTrials.gov identifier NCT03175679.
P238 Phase I study of adoptive transfer of specific hepatocellular carcinoma antigens CD8+ T cells for treating patients with relapsed/advanced HCC
Jun Lu1, Xuli Bao1, Yanpin Ma1, Hui Chen2, Wenfeng Sun2, Jia Guo1, Xiongwei Cui1
1Beijing YouAn Hospital, Capital Medical University, Beijing, China; 2Beijing Gene Key Life Technology Co.Ltd., Beijing, China
Correspondence: Jun Lu (lujun98@ccmu.edu.cn)
Background
Hepatocellular carcinoma (HCC) is one of the most prevalent cancers for neoplastic deaths and shows a high recurrence rate. Adoptive T cell therapy, involving the ex vivo selection and expansion of antigen-specific T cell clones by MHC/peptide tetramer sorting, provides a method of augmenting antigen-specific immunity against HCC. To generate of a high number of cytotoxic T lymphocytes (CTLs), kind of effective T cells that specific recognizing and killing antigen targeted cells through cloning amplification after receiving antigen information from antigen presented cells, is a promising strategy for adoptive therapy.
Methods
The phase I trial enrolls patients who have HCC tumor relapsed or metastasized through the body after standard treatment or the patients cannot receive standard treatment under current conditions. The purpose of this study is to evaluate the safety and tolerance as well as the potential clinical efficacy of an adoptive transfer of CD8+ T cells, sorted with human leukocyte antigen (HLA)-peptide multimers and specific for Glypican (GPC)-3 /New York Esophageal Squamous-1 (NY-ESO-1) /alpha-fetoprotein (AFP) antigens and cultured in vitro. Key eligibility criteria include age ≥18 years, with HCC (BCLC, stage C) proved by histopathology or proved by CT or MRI imaging system, relapsed after previous therapy and no effective therapies known at this time and life expectancy of ≥ 12 weeks. Three different dosing schedules will be evaluated. Three patients will be evaluated on each dosing schedule. The following dose levels will be evaluated: Loading Dose 1: 3x107/m2; Loading Dose 2: 6x107/m2; Loading Dose 3: 9x107/m2. The doses are calculated according to the actual number of GPC3/NY-ESO-1/AFP CTLs. Human Interleukin-2 will be given at a dose of 25,000 IU/kg/day for 5-14 days. Tegafur will be given at a dose of 40~60 mg bis in die (BID) for 2 weeks. Immune responses were measured by Elispot and ELISA, flow cytometry assays were performed to evaluate the effects on immune cell subsets. Tumor biopsies were evaluated for CTLs by immunohistochemistry. Incidence of treatment-emergent adverse events were defined as signs/symptoms, laboratory toxicities, and clinical events that are possibly, likely, or definitely related to study treatment adverse events assessed according to NCI-CTCAE v4.0. HCC progression was evaluated by imaging according to the irRC standard.
Results
To date, 5 patients have been enrolled and 2 of them are in week 2 monitoring period, with no significant toxicities thus far. Data regarding the peripheral blood antigen-specific CTL cells response will be presented.
Trial Registration
ClinicalTrials.gov identifier NCT03175705
P239 Phase 3 study of Pembrolizumab plus Chemoradiation (CRT) vs CRT alone for locally advanced head and neck squamous cell carcinoma (LA-HNSCC): KEYNOTE-412
Jean-Pascal Machiels1, Chia-Jui Yen2, Lisa Licitra3, Danny Rischin4, John Waldron5, Barbara Burtness6, Vincent Gregoire1, Sanjiv Agarwala7, Yun Gan Tao8, Jeffrey Yorio9, Sercan Aksoy10, Sadakatsu Ikeda11, Ruey-Long Hong12, Joy Yang Ge13, Holly Brown13, Behzad Bidadi13, Lillian Siu5
1Cliniques Universitaires Saint-Luc, Brussels, Belgium; 2National Cheng Kung University Hospital, Tainan City, Taiwan; 3Fondazione IRCCS Istituto Nazionale dei Tumori, Milan, Italy; 4Peter MacCallum Cancer Centre, East Melbourne, Australia; 5Princess Margaret Cancer Centre, Toronto, ON, Canada; 6Yale University School of Medicine, New Haven, CT, USA; 7St. Luke's Cancer Center–Anderson, Easton, PA, USA; 8Institut Gustave Roussy, Villejuif, France; 9Texas Oncology–Austin Central, Austin, TX, USA; 10Hacettepe Universitesi Tip Fakultesi, Ankara, Turkey; 11Medical Hospital, Tokyo Medical and Dental University, Tokyo, Japan; 12National Taiwan University Hospital, Taipei City, Taiwan; 13Merck & Co., Inc., Kenilworth, NJ, USA
Correspondence: Jean-Pascal Machiels (Jean-pascal.machiels@uclouvain.be)
Background
CRT with cisplatin is the standard of care for patients with LA-HNSCC not treated by surgery. Preclinical data in murine cancer models show improved tumor growth control and survival when RT is combined with a programmed death 1 (PD-1) inhibitor. Pembrolizumab has been found to be effective for treating recurrent/metastatic HNSCC, and initial results from a phase 1b study suggest that pembrolizumab plus CRT is tolerable in patients with LA-HNSCC. KEYNOTE-412 (NCT03040999) is a phase 3, randomized, placebo-controlled, double-blind trial to determine the efficacy and safety of pembrolizumab given concomitantly with CRT and as maintenance therapy versus placebo plus CRT in LA-HNSCC.
Methods
Eligibility includes patient age ≥18 years; newly diagnosed, treatment-naive, oropharyngeal p16 positive (any T4 or N3), oropharyngeal p16 negative (any T3-T4 or N2a-N3), or larynx/hypopharynx/oral cavity (any T3-T4 or N2a-N3) SCC; evaluable tumor burden (RECIST v1.1); ECOG performance status 0-1; results available from local testing of human papillomavirus status for oropharyngeal cancer; eligible for definitive CRT and not considered for primary surgery per investigator decision; tissue from a core or excisional biopsy for programmed death ligand 1 (PD-L1) biomarker analysis. Patients will be randomly assigned (1:1) to receive pembrolizumab 200 mg every 3 weeks plus CRT, including radiotherapy (RT; accelerated [70 Gy, six 2 Gy fractions/wk] or standard [70 Gy, five 2 Gy fractions/week] fractionation) plus cisplatin 100 mg/m2 Q3W for 3 cycles only, or placebo Q3W plus CRT. Treatment will be stratified by RT regimen (accelerated vs standard), tumor site/p16 status (oropharynx p16 positive vs p16 negative or larynx/hypopharynx/oral cavity), and disease stage (III vs IV). A priming dose of pembrolizumab or placebo will be given 1 week before CRT, followed by 2 doses during CRT, and an additional 14 doses after CRT, for a total of 17 pembrolizumab or placebo infusions. Treatment will be discontinued upon centrally confirmed disease progression, unacceptable toxicity, or patient/physician decision to withdraw. Disease status will be assessed by computed topography or magnetic resonance imaging 12 weeks after CRT, every 3 months for 3 years, then every 6 months for years 4 and 5. Safety will be monitored throughout the study and for 30 days after treatment. The primary end point is event-free survival. Secondary end points include overall survival, safety, and patient-reported outcomes. Recruitment will continue until ~780 patients are enrolled.
Trial Registration
ClinicalTrials.gov, NCT03040999.
P240 Phase 1b trial of cabozantinib in combination with atezolizumab in patients with locally advanced or metastatic urothelial carcinoma or renal cell carcinoma
Manuel Caitano Maia1, Neeraj Agarwal2, Bradley McGregor3, Ulka Vaishampayan4, Toni K. Choueiri3, Marjorie Green5, Colin Hessel6, Christian Scheffold6, Gisela Schwab6, Thomas Powles7, Sumanta Pal1
1City of Hope, Duarte, CA, USA; 2Huntsman Cancer Hospital, Salt Lake City, UT, USA; 3Dana-Farber Cancer Institute, Boston, MA, USA; 4Karmanos Cancer Center, Detroit, MI, USA; 5Genentech, South San Francisco, CA, USA; 6Exelixis Inc., South San Francisco, CA, USA; 7Barts Cancer Institute, London, United Kingdom
Correspondence: Manuel Caitano Maia (hannah.welz@fishawack.com)
Background
Cabozantinib is an oral receptor tyrosine kinase inhibitor targeting MET, VEGFR, and TAM family receptors (TYRO3, AXL, and MER). It is approved for use in patients with advanced renal cell carcinoma (RCC) after prior therapy with antiangiogenic/VEGFR-targeted therapy, and has demonstrated clinical activity in urothelial carcinoma (UC). In clinical studies, cabozantinib exposure resulted in an increase in circulating CD8+ T cells and reduction of immune-suppressive monocytes and Tregs. In preclinical tumor models, treatment with cabozantinib resulted in an increase of MHC class 1 expression on tumor cells and a reduction of myeloid-derived suppressor cells. These observations support that cabozantinib may facilitate an immune-permissive tumor environment and may enhance the response to immune checkpoint inhibitors. Atezolizumab, an anti-PD-L1 monoclonal antibody, is approved for use in locally advanced or metastatic UC for patients who are either cisplatin-ineligible or have disease progression during or following platinum-containing chemotherapy. It is also approved for use in patients with metastatic non–small cell lung cancer who have disease progression during or following platinum-containing chemotherapy. Here, we present the study design of an ongoing phase 1b study combining cabozantinib with atezolizumab in patients with locally advanced or metastatic UC or RCC.
Methods
This multicenter, phase 1b, open-label study aims to assess safety, tolerability, preliminary efficacy, and pharmacokinetics of cabozantinib in combination with atezolizumab (NCT03170960). The study will enroll patients with advanced UC (including bladder, renal pelvis, ureter, urethra) or RCC. It consists of two stages: a dose-escalation stage and an expansion-cohort stage. In the dose-escalation stage (3+3 design), a recommended cabozantinib dose for the combination will be established. In the expansion stage, four tumor-specific cohorts will be enrolled, and the primary objective is to determine the objective response rate in each cohort. The four expansion cohorts are (1) patients with UC who have progressed on or after platinum-containing chemotherapy; (2) chemotherapy-naïve patients with UC who are ineligible for cisplatin; (3) chemotherapy-naïve patients with UC who are eligible for cisplatin; and (4) previously untreated patients with RCC with clear cell histology. Exploratory objectives include correlation of tumor and plasma biomarkers, and changes in immune cell profiles with clinical outcome. The study has been initiated and enrollment target is up to 120 patients across the 4 cohorts in the expansion-cohort stage.
Trial Registration
ClinicalTrials.gov: NCT03170960.
P241 A first-in-human study of ALX148: CD47 blockade to enhance innate and adaptive immunity for advanced solid tumor malignancy and non hodgkin lymphoma
Nehal Lakhani1, Patricia LoRusso2, Anuradha Krishnamurthy3, Timothy O'Rourke1, Philip Fanning4, Yonggang Zhao5, Hong Wan4, Jaume Pons4, Sophia Randolph4, Wells Messersmith3
1START Midwest, Grand Rapids, MI, USA; 2Yale Cancer Center, New Haven, CT, USA; 3University of Colorado Cancer Center, Aurora, CO, USA; 4Alexo Therapeutics Inc., South San Francisco, CA, USA; 5Skyview Research, Norristown, PA, USA
Correspondence: Sophia Randolph (srandolph@alexotherapeutics.com)
Background
CD47, a marker of self, is upregulated by tumors to evade the immune system. Blocking the interaction between CD47 and SIRPα, its receptor on myeloid cells, disrupts a key immune checkpoint and may enhance innate and adaptive immunity against cancer. ALX148 is a high affinity, engineered fusion protein containing the N-terminal D1 domain of SIRPα, which binds and blocks CD47, and is genetically linked to an inactive human Fc domain to minimize toxicity. ALX148 enhanced activity of multiple anti-cancer targeted antibodies and checkpoint inhibitors with minimal effect on normal blood cells in nonclinical models. This phase 1 study evaluates the safety, tolerability, pharmacokinetic (PK) and pharmacodynamic profiles of ALX148 in patients with advanced malignancy.
Methods
The primary study objective is to characterize the safety profile of ALX148 first as a single agent and then in combination with established anti-cancer antibodies. Cohorts (3-6 pts) with advanced malignancy receive escalating doses of ALX148, intravenously, once weekly or once every other week. Tumor response, PK, and target occupancy (TO) are characterized as secondary objectives. Preliminary single agent data are reported from the data cutoff, July 21, 2017 and will be updated at the time of presentation.
Results
Ten patients received ALX148 (4 males/6 females; 0.3 mg per kilogram (mpk), 3; 1.0 mpk, 4; 3.0 mpk, 3) as of data cutoff. Median age was 63 (37-76) yrs and ECOG PS 0/1: 1/9. Four patients experienced treatment related adverse events (AEs) which were predominantly low grade and included 1 each at 0.3 mpk (G1 Headache, Rash, Fatigue); 1.0 mpk (G3 Anemia, G1 Dysgeusia); and at 3.0 mpk (G2 Decreased Appetite, Hypersensitivity). As of the data cut-off no pts have experienced a dose-limiting toxicity. One patient achieved SD (0.3 mpk; leiomyosarcoma) for 16 weeks. ALX148 initial PK showed increased exposure with increasing dose and noticeable accumulation with repeated dosing, likely driven by target saturation. Dose dependent TO on CD47 by ALX148 was observed on RBCs and T cells. The magnitude and duration of TO increased with repeat dosing.
Conclusions
ALX148 is well tolerated in patients with advanced solid tumors with favorable PK/TO characteristics and no significant hematologic toxicity at doses evaluated. Accrual is ongoing. When the maximum tolerated dose/optimal biological dose of single agent ALX148 is established, patients with advanced malignancy will be evaluated with ALX148 in combination with anticancer antibodies.
Trial Registration
ClinicalTrials.gov identifier NCT03013218.
References
1. Weiskopf K. Cancer immunotherapy targeting the CD47/SIRPα axis.Eur J Cancer. 2017;76:100-109.
P242 A phase 1 multicenter, dose escalation study of CBT-501, a novel anti-PD-1 inhibitor in subjects with select advanced or relapsed/recurrent solid tumors
Purvi Patel, Mamatha Reddy, Melissa Lopez, Neil Sankar, Sarath Kanekal, Mike Li, Sanjeev Redkar, Gavin Choy
CBT Pharmaceuticals, Inc., Pleasanton, CA, USA
Correspondence: Sanjeev Redkar (sanjeev.redkar@cbtpharma.com)
Background
Programmed death-1 (PD-1, CD279) is an inhibitory co-receptor expressed on antigen-activated and exhausted T and B cells. PD-1/PD-L1 axis inhibition by targeted-antibodies, increases the T cell proliferation and cytotoxicity. This represents a promising mechanism to stimulate the anti-tumor activity of the immune system. CBT-501, genolimzumab (GB226) is a novel humanized IgG4 monoclonal antibody targeting the PD-1 membrane receptor on T lymphocytes and other cells of the immune system. CBT-501 demonstrated highly specific binding to PD-1 of human (Kd=505 pM) and cynomolgus (Kd=7.2 nM). CBT-501 efficiently inhibited the binding of PD-L1/L2 to PD-1 for both human and monkey and enhanced human T cell activation in the Mixed Lymphocyte Reaction (MLR) assay. CBT-501 has demonstrated anti-tumor activity in the in vivo animal model and no abnormal drug-related toxicity has been observed in the GLP toxicology studies. Data from all pre-clinical pharmacodynamics and toxicology studies of CBT-501 indicate pharmacological activity at effective doses with a wide margin of safety. Based on these findings, a Phase 1 study has been initiated with CBT-501 in Australia.
Methods
CBT-501-01 is a Phase 1, multicenter, dose escalation study of CBT-501 in subjects in select advanced or relapsed/recurrent solid tumors. The primary study objective is to identify the overall safety and tolerability, including any dose limiting toxicities (DLT), and determine the recommended Phase 2 dose (RP2D) in subjects with advanced solid tumors. Secondary objectives include assessing efficacy by overall response rate (ORR), best overall response rate (BOR) per RECIST v1.1 and irRECIST, time to response, duration of response (DOR), disease control rate (DCR) by RECIST v1.1 and irRECIST, progression free survival (PFS), and determining the pharmacokinetic (PK) parameters. Exploratory objectives involve the assessment of PD-1 and PD-L1 expression, receptor occupancy and the host immune response (immune modulation) in blood peripheral-blood mononuclear cells (PBMCs) or formalin-fixed paraffin-embedded (FFPE) samples. This is a 2-part study with a dose-escalation segment and dose and disease expansion cohorts of CBT-501. In Part 1, dose escalation (3+3 design) will occur among 3 cohorts to determine the RP2D. The tumor type(s) with the most robust clinical signal relative to response rate and safety/tolerability will be selected for further evaluation in the expansion cohort (Part 2). Approximately 32 subjects will be enrolled in the dose and disease expansion and treated at the RP2D, as determined in Part 1.
Trial Registration
Clinical Trial Registry Number: NCT03053466.
P243 Withdrawn
P244 A phase 1, first-in-human, open-label, dose escalation study of MGD013, a bispecific DART® protein binding PD-1 and LAG-3 in patients with unresectable or metastatic neoplasms
Sadhna Shankar1, Manish Patel2, George Blumenschein3, Erika Hamilton4, Jason Luke5, Ross La-Motte Mohs1, Kalpana Shah1, Lisa Adali-Piston1, Syd Johnson1, Ezio Bonivini1, Paul Moore1, Jon Wigginton1, Jim Vasselli1
1MacroGenics Inc., Rockville, MD, USA; 2Florida Cancer Specialists, Sarah Canon Research Institute, Sarasota, FL, USA; 3MD Anderson Cancer Center, Houston, TX, USA; 4Sarah Cannon Research Institute, Nashville, TN, USA; 5University of Chicago, Chicago, IL, USA
Correspondence: Sadhna Shankar (shankars@macrogenics.com)
Background
Lymphocyte-activation gene 3 (LAG-3) is a membrane protein in the immunoglobulin superfamily that binds to major histocompatibility complex class II (MHC-II). LAG-3 engagement negatively regulates T cell proliferation and differentiation. Blockade of PD-1 and LAG-3 in animal tumor models enhanced antitumor immunity via distinct, non-redundant signaling pathways that fostered the accumulation of functionally competent CD8+ T cells in mice [1]. Dual targeting of PD-1 and LAG-3 may help reverse effector cell exhaustion and increase the response rates and/or effectiveness of immunotherapy beyond that observed with single agents alone. MGD013 is an Fc-bearing bispecific tetravalent (bivalent for each antigen) DART® protein engineered as a hinge-stabilized immunoglobulin G4 molecule and designed to concomitantly bind PD-1 and LAG-3, thereby contributing to sustain or restore the function of exhausted T cells. MGD013 may enhance T cell activation in a synergistic fashion beyond that observed with the anti–PD-1 and anti–LAG-3 monoclonal antibodies alone or in combination. A bispecific format for target engagement may confer biologic advantages that may translate to clinical advantages over antibody combinations.
Methods
This is an open-label, dose escalation / cohort expansion phase 1 study (NCT03219268) designed to characterize the safety, tolerability, pharmacokinetics, pharmacodynamics, and preliminary antitumor activity of MGD013. Patients with unresectable, locally advanced or metastatic solid tumors of any histology are enrolled in the dose escalation phase. Sequential escalating flat doses ranging from 1 mg to 1600 mg every 2 weeks are evaluated in successive cohorts of 1 to 6 patients each. A single patient dose escalation design is utilized in the lower dose cohorts. The escalation approach transitions to a conventional 3+3 design after the first three cohorts. Occurrence of a drug-related Grade 2 adverse event in a single patient cohort will lead to enrollment of 3 additional patients at that dose level. Occurrence of a DLT in a single patient cohort will trigger transition to a conventional 3+3 design. Response is first determined at 8 weeks. Patient management is guided by response assessment according to irRECIST. MGD013 dosing may continue up to 2 years based on response. Cohort expansion phase will start after maximum tolerated dose is determined and will be restricted to 5 tumor types, including solid tumors and hematological malignancies.
Trial Registration
Clinicaltrials.gov- NCT03219268.
References
1. Grosso JF, Kelleher CC, Harris TJ, Maris CH, Hipkiss EL, De Marzo A, et al.LAG-3 regulates CD8+ T cell accumulation and effector function in murine self- and tumor-tolerance systems. J Clin Invest. 2007;117(11):3383-92.
P245 CAPRA: A Phase 1b study of intratumoral Coxsackievirus A21 (CVA21) and systemic pembrolizumab in advanced melanoma patients
Ann Silk1, Howard Kaufman1, Nashat Gabrail2, Janice Mehnert1, Jennifer Bryan1, Jacqueline Norrell1, Azra Haider1, Daniel Medina1, Praveen Bommareddy3, Darren Shafren4, Mark Grose4, Andrew Zloza1
1Rutgers Cancer Institute of New Jersey, New Brunswick, NJ, USA; 2Gabrail Cancer Center, Canton, OH, USA; 3Rutgers University, Piccataway, NJ, USA; 4Viralytics Limited, Sydney, Australia
Correspondence: Ann Silk (ann.w.silk@rutgers.edu)
Background
Coxsackievirus A21 (CVA21) is a novel bio-selected oncolytic, immunotherapeutic agent. Intratumoral (i.t.) CVA21 injection can induce selective tumor-cell infection, immune-cell infiltration, IFN-g response gene up-regulation, increased PD-L1 expression, tumor cell lysis and systemic anti-tumor immune responses. A clinical trial evaluating combination CVA21 and pembrolizumab in patients with melanoma was initiated and preliminary data on a pre-established futility endpoint are presented here.
Methods
This is a single-arm, multi-institutional open-label phase Ib clinical trial of i.t. CVA21 and i.v. pembrolizumab for treated or untreated unresectable Stage IIIB-IVM1c melanoma. Subjects with injectable disease receive up to 3 x 108 TCID50 CVA21 i.t. on Days 1, 3, 5, 8, and then every 3 weeks for up to 19 injections. Subjects also receive pembrolizumab (2mg/kg) i.v. every 3 weeks starting on Day 8. The primary endpoint is safety/tolerability by incidence of dose-limiting toxicity. Secondary endpoints include best ORR by immune-related response criteria, progression-free survival, overall survival, quality of life.
Results
To date, 22 subjects have started on protocol therapy. Overall, the adverse events have been low-grade constitutional symptoms related to CVA21 and expected pembrolizumab-related side effects. One subject had Grade 3 increased hepatic enzymes that was considered related to pembrolizumab. No DLT’s have been reported. Currently, 19 patients are evaluable for investigator response assessment. Among the evaluable subjects (n=19), the ORR was 63% (12/19). The DCR (CR+PR+SD) is currently 84% (16/19). In subjects with stage IVM1c disease, the ORR is 78% (7/9). The study has met its primary statistical futility endpoint of achieving ≥2 confirmed objective responses (CR or PR) in the first 12 patients enrolled. One of the 12 responders displayed early pseudo-progression and later developed a partial response.
Conclusions
Based on these initial results, the sample size has now been expanded to enroll up to 50 patients including subjects refractory to anti-PD1 therapy. Combination therapy of CVA21 and pembrolizumab may represent a new approach for the treatment of patients with injectable advanced melanoma.
Trial Registration
NCT02565992.
Consent
Written informed consent was obtained from all of the patients for participation in the study and use of the data for publication.
P246 Phase 1/2 study of in situ vaccination with tremelimumab + intravenous (IV) durvalumab + poly-ICLC in patients with select relapsed, advanced cancers with measurable, biopsy-accessible tumors
Craig Slingluff, Jr.1, Sunita Hack2, Paul Schwarzenberger3, Toni Ricciardi3, Mary Macri3, Aileen Ryan3, Ralph Venhaus3, Nina Bhardwaj4
1University of Virginia, Charlottesville, VA, USA; 2Ludwig Institute for Cancer Research, New York, NY, USA; 3Ludwig Cancer Research, New York, NY, USA; 4Icahn School of Medicine at Mt Sinai, New York, NY, USA
Correspondence: Craig Slingluff, Jr. (cls8h@virginia.edu)
Background
Immunotherapy has demonstrated promising antitumor activity in various advanced cancers. Combined tumor targeting from multiple drugs with unique mechanisms may provide further improved outcomes. Tremelimumab (TRE) is a CTLA-4 antibody and durvalumab (DUR) blocks PD-L1. Poly-ICLC is a toll-like receptor 3 agonist. Intratumoral (intra-T) injection of poly-ICLC directly alters the tumor microenvironment (TME), and by creating an in situ vaccination, may trigger a clinically effective systemic anti-tumor response when also combined with DUR and TRE.
Methods
This is an ongoing Phase 1/2, open-label, multicenter study (NCT02643303). The study evaluates the use of intra-T administration of TRE and IV DUR + poly-ICLC (intra-T and intramuscular [IM]) to determine the safety, preliminary efficacy and immune activity of this regimen in patients with advanced, measurable, biopsy-accessible tumors: head and neck squamous cell carcinoma, breast cancer, sarcoma, merkel cell carcinoma, cutaneous T cell lymphoma, melanoma, genitourinary cancer, and other solid tumors. Phase 1 determines the recommended combination dosing (RCD) for the regimen with dose de-escalation based on dose limiting toxicities (DLTs) and standard 3 + 3 rules. Starting doses are: DUR, 1500 mg IV; TRE, 75 mg IV; TRE, 10 mg intra-T; poly-ICLC, 1 mg intra-T/IM. Phase 1 starts with Cohort 1A (DUR + poly-ICLC). Upon demonstration of tolerability, enrollment proceeds with Cohort 1B (DUR + IV TRE + poly-ICLC) and Cohort 1C (DUR + intra-T TRE + poly-ICLC). The RCD is the highest dose at which < 2/6 patients have DLTs. In Phase 2, up to 66 evaluable patients are treated using the RCD regimen, with enrollment of 6 patients per tumor type initially, and enrollment of 6 additional patients per 3 tumor types contingent upon at least 1 response among the initial 6 patients. Study endpoints are RCD and safety, objective response rate, progression-free survival, and overall survival. Exploratory endpoints are biological activity, including effects on the TME and immunological responses. Enrollment opened on 28 Dec 2016.
Results
Trial in Progress.
Conclusions
Trial in Progress.
Trial Registration
Clinicaltrials.gov: NCT02643303.
P247 Study Design: Phase 1 dose escalation, multi-tumor study to assess safety, tolerability and antitumor activity of genetically engineered MAGE-A4 SPEAR T cells in HLA-A2+ subjects with MAGE-A4+ tumors
David S Hong1, Melissa Johnson2, Anthony J Olszanski3, Marcus Butler4, Connie Erickson-Miller5, Malini Iyengar5, Trupti Trivedi5, Karen Chagin5, Rafael Amado5
1The University of Texas MD Anderson Cancer Center, Houston, TX, USA; 2Sarah Cannon, Nashville, TN, USA; 3Anthony.Olszanski@FCCC.edu, Philadelphia, PA, USA; 4Princess Margaret Cancer Centre, Toronto, ON, Canada; 5Adaptimmune, Philadelphia, PA, USA
Background
MAGE-A4 is a cancer/testis antigen that has been identified in 13-48% of non-small cell lung cancer (NSCLC), urothelial, melanoma, head and neck, ovarian, gastric and esophageal tumors. This study (NCT03132922) will evaluate the safety and tolerability of genetically engineered autologous specific peptide enhanced affinity receptor (SPEAR) T cells (MAGE-A4c1032T cells) directed towards a MAGE-A4 peptide expressed on tumors in the context of HLA-A *02. Antitumor activity will also be assessed.
Methods
This first-in-human T cell dose escalation study utilizes a modified 3+3 design to evaluate safety, including dose limiting toxicities (DLT). Secondary objectives include anti-tumor activity (overall response (per RECIST v1.1), duration of response, time to response, progression-free survival, overall survival) and translational research assessments. Patients are screened under a separate protocol (NCT03132922). Those who are HLA-A*02 positive (with the exception of A*02:05) and have inoperable or metastatic (advanced) NSCLC, urothelial cancer, melanoma, or squamous cell head and neck, ovarian, gastric or esophageal tumors with MAGE-A4 expression and meet all other entry criteria are eligible for treatment. Subjects must have prior treatments as described in the table (Table 1). Patients must have received standard of care therapies and have measurable disease.
Following leukapheresis, the T cells are isolated, transduced with a lentiviral vector containing the MAGE-A4c1032 TCR, and expanded with CD3/CD28 beads. Subjects are given lymphodepleting chemotherapy (fludarabine 30 mg/m2/day and cyclophosphamide 600 mg/m2/day, on days -7, -6 and -5) prior to infusion of transduced cells. Groups 1, 2 and 3 will consist of 3-6 subjects, and the transduced cell doses will be as follows, respectively: 0.1 × 109 (±20%), 1 × 109 (±20%), and 5 × 109 (range: >1.2 – 6 × 109). The DLT observation period will be during the first 30 days following the infusion of MAGE-A4 SPEAR T cells for each patient in all groups. Following the dose escalation, up to 10 patients will be enrolled at the target dose.
Disease assessments will be conducted at week 6, 12, 18 and 24, and then every 3 months until confirmation of disease progression or at 2 years post-infusion. On study tumor biopsies and blood samples will be evaluated to compare the pre- and post-T cell infusion immune profile for association with treatment outcome.
Trial Registration
NCT03132922.
Eligibility Criteria
P248 Study Design: An open-label randomized pilot study of NY-ESO-1 SPEAR T cells alone or in combination with pembrolizumab in HLA-A2+ subjects with relapsed and refractory multiple myeloma (NCT03168438)
Aaron P Rapoport1, James E Hoffman2, Myo Htut3, Taiga Nishihori4, Karen Chagin5, Thomas Faitg5, Elliot Norry5, Trupti Trivedi5, Rafael Amado5
1University of Maryland School of Medicine, Baltimore, MD, USA; 2Sylvester Cancer Center at the University of Miami, Miami, FL, USA; 3City of Hope, Duarte, CA, USA; 4Moffit Cancer Center, Tampa, FL, USA; 5Adaptimmune, Philadelphia, PA, USA
Background
NY-ESO-1 and LAGE-1a are cancer/testis antigens that are expressed frequently in multiple myeloma (MM) and are often associated with poor prognosis. This two-arm randomized study will evaluate the safety and efficacy of genetically engineered autologous specific peptide enhanced affinity receptor (SPEAR) T cells (NY-ESO-1c259T cells) directed towards a NY-ESO-1/LAGE-1a peptide expressed on tumor cells in the context of HLA-A*02, alone and in combination with pembrolizumab.
Methods
This open label randomized pilot study will evaluate safety, efficacy (using the International Myeloma Working Group Uniform Response Criteria), and translational research endpoints. Patients must meet these criteria: ≥ 18 yrs old; HLA-A*02:01, A*02:05 or A*02:06 positive; have histologically confirmed diagnosis of MM with either primary refractory or relapsed/refractory disease expressing NY-ESO-1 and/or LAGE-1a; prior therapies including IMiD and a proteasome inhibitor as separate lines or a combined line of therapy; and adequate organ function. Subjects who have relapsed after autologous hematopoietic cell transplantation (HCT) or are unable to receive autologous HCT are eligible. Target enrollment for this study is 20 subjects, with 10 in each arm; patients will be randomly assigned to a treatment arm. Eligible subjects who do not receive the T cell infusion may be replaced.
Following apheresis, the T cells are isolated and expanded with CD3/CD28 beads, transduced with a lentiviral vector containing the NY-ESO-1c259 TCR, and 1– 8 × 109 transduced T cells are infused intravenously on day 1 after lymphodepletion with fludarabine 30 mg/m2/day and cyclophosphamide 600 mg/m2/day on days -7 to -5 and granulocyte-colony stimulating factor support starting on day -4. In Arm 1, SPEAR T cell infusion is the only investigational product administered. Subjects in Arm 2 will receive SPEAR T cell infusion followed by an initial 200 mg dose of pembrolizumab on day 22 (week 3). If toxicities preclude week 3 treatment, the first dose may be given at week 6. Subsequent doses of pembrolizumab will be given every 3 weeks up to week 108 post T cell infusion.
In both arms, safety will be assessed at each clinic visit. Disease response is assessed at weeks 1 and 3, every 3 weeks until week 24, every 6 weeks until week 72, and then every 12 weeks until confirmed progression of disease. On study biopsies and blood samples will be evaluated to compare the pre- and post-T cell infusion immune profile for association with treatment outcome.
Trial Registration
NCT03168438.
P249 A phase 1 study of the safety, tolerability, and pharmacokinetics (PK) of MGA012 (anti-PD-1 antibody) in patients with advanced solid tumors
Nehal Lakhani1, Janice M. Mehnert2, Drew Rasco3, Michael Gordon4, Joanna Lohr5, Pepi Pencheva5, Sharad Sharma5, Hua Li5, Ross LaMotte-Mohs5, Paul Moore5, Jichao Sun5, Bradley Sumrow5, Jon Wigginton5, John Powderly6
1START Midwest - South Texas Accelerated Research Therapeutics, LLC, Grand Rapids, MI, USA; 2Rutgers Cancer Institute of New Jersey, New Brunswick, NJ, USA; 3START - South Texas Accelerated Research Therapeutics, LLC, San Antonio, TX, USA; 4Honor Health Research Institute, Scottsdale, AZ, USA; 5MacroGenics, Inc., Rockville, MD, USA; 6Carolina BioOncology Institute, Huntersville, NC, USA
Correspondence: Bradley Sumrow (sumrowb@macrogenics.com)
Background
MGA012 is a humanized, IgG4κ monoclonal antibody (mAb) that recognizes human programmed cell death protein 1 (PD-1). MGA012 binds to PD-1 expressing T cells, inhibits PD-1 and PD-L1/PD-L2 interactions, and disrupts the negative signaling axis to restore T cell function. The biological activity of MGA012 is comparable to replicas of approved anti-PD-1 mAbs when assessed in vitro, including blockade of PD-1 and PD-L1/PD-L2 interactions, inhibition of PD-1 signaling, and enhancement of T cell effector function.
Methods
This phase 1, dose escalation study will characterize the safety, tolerability, PK/PD, immunogenicity, and preliminary anti-tumor activity of MGA012 administered IV every two or four weeks in patients with advanced solid tumors. MGA012 has been evaluated in sequential dose escalation cohorts (1-10 mg/kg) of 3 to 6 patients each, using a 3+3 design. Four tumor-specific expansion cohorts will be treated at the maximum tolerated dose of MGA012. Selective cohort expansion was allowed during escalation to gather further safety and PK/PD data.
Results
At the data cutoff, 33 patients (12M/21F, median age 63 years) with diverse tumor types were treated at doses from 1-10 mg/kg, including 21 patients on treatment at the time of data cutoff. MGA012 has demonstrated acceptable tolerability with no dose-limiting toxicities (DLTs). Treatment-related adverse events (AEs) occurred in 20/33 (61%) patients, most commonly fatigue (n=9) and nausea (n=5). Treatment-related Grade ≥3 AEs occurred in 3/33 (9%) patients and include increased lipase (n=2) and vaginal ulceration/inflammation (n=1). A single treatment-related serious adverse event (SAE) has been reported (aphasia occurring in conjunction with the emergence of new brain metastases). Immune-related AEs (irAEs) were limited to rash (n=3), infusion-related reaction (n=1), and vaginal ulceration/inflammation (n=1). MGA012 has PK features consistent with other IgG4 monoclonal antibodies, as well as full and sustained receptor occupancy at all dosing levels tested, consistent with its known binding characteristics. Twenty-two patients were response evaluable at the data cutoff. Three patients have experienced unconfirmed partial responses (including ovarian, MSI-high colorectal and uterine papillary serous carcinoma), and 4 additional patients experienced stable disease as a best response. Others had radiographic progressive disease or clinical progression.
Conclusions
MGA012 has demonstrated an acceptable safety profile, predictable PK/PD, and early evidence of anti-tumor activity. Subsequent to dose escalation, patients will be enrolled on tumor-specific monotherapy expansion cohorts. Future trials also are planned for combination testing of MGA012 with T cell directed, CD3-based DART® molecules.
Trial Registration
NCT03059823.
P250 LTX-315, an oncolytic peptide converts “cold” tumors to “hot” in a majority of patients with advanced cancer: results from an ongoing phase I study
Aurelien Marabelle1, Jean-Francois Baurain2, Ahmad Awada3, Rebecca Kristeleit4, Dag-Erik Jøssang5, Nina-louise Jebsen5, Delphine Loirat6, Andrew Saunders7, Wenche Olsen7, Berit Nicolaisen7, Baldur Sveinbjornsson7, Vibeke Sundvold Gjerstad7, Øystein Rekdal7, Pal Brunsvig8, Jerome Galon9, Fabienne Hermitte10, Bjorn-Tore Gjertsen5, Anna Armstrong11, James Spicer12
1Gustave Roussy, Villejuif, France; 2Saint-Luc University Hospital, Brussel, Belgium; 3Institut Jules Bordet, Brussel, Belgium; 4Univerity College London Hospital, London, United Kingdom; 5Haukeland Univeristy Hospital, Bergen, Norway; 6Institut Curie, Paris, France; 7Lytix Biopharma, Oslo, Norway; 8Oslo University Hospital, Oslo, Norway; 9Laboratory of Integrative Cancer Immunology, INSERM, Paris, France; 10INSERM, Paris, France; 11Christie Hospital, Manchester UK, Manchester, United Kingdom; 12King’s College London, London, United Kingdom
Correspondence: Øystein Rekdal (baldur.sveinbjornsson@lytixbiopharma.com)
Background
LTX-315 disintegrates cytoplasmic organelles (e.g mitochondria) and induces immunogenic cancer cell death in preclinical in vivo models. Intratumoral LTX-315 increases tumor-infiltrating lymphocytes (TILs) and induces complete tumor regression in several rodent models. Systemic (abscopal) anti-tumor immune responses can be enhanced upon combination with immune checkpoint inhibitors (ICI). Here we report preliminary results of the phase 1 trial which evaluates intratumoral LTX-315 in monotherapy or in combination with ICI.
Methods
Patients with advanced solid tumors were immunologically primed with weekly intratumoral LTX-315 into a single accessible lesion over 6 weeks. Additional injections could be administered thereafter every 2 weeks. ICI combinations included ipilimumab (melanoma cohort) and pembrolizumab (TNBC cohort). Biopsies of injected lesions were taken at baseline and after LTX-315 treatment. Immunoprofiling was performed using immunohistochemistry and Nanostring analysis. Twenty nine patients have been treated with LTX-315 monotherapy. LTX-315 monotherapy was administered at doses of 2-7mg to a median of 1.8 tumor lesions (range 1-6) for a median of 9 weeks (range 1-33).
Results
In 29 patients, all LTX-315-related adverse events were CTC grade 1 or 2, most commonly local erythema, flushing, pruritis and hypotension, usually resolving within minutes of injection. Related grade 3 (3 patients) or 4 (1) allergic/anaphylaxis adverse events occurred and resolved without sequelae. Of 44 injected lesions in 20 evaluable LTX-315 monotherapy patients, 2 lesions regressed completely, > 50% regression was seen in 5 lesions, and 20 remained stable. Significant increases in CD8+ TILs occurred in 67% (14 of 21) patients with evaluable biopsies. The HalioDx Immune Gene Signature analysis of LTX-315-treated tumors showed upregulation of genes involved in immune-mediated tumor regression (effector T cells, TH1 orientation, chemokines and cytokines). Regression of distant non-injected tumors (irRC) criteria has been observed in 11 of 30 tumors in 9 patients. Stable disease (SD) of at least 7 weeks duration in non-injected tumors (median duration 11 weeks) by immune-related RECIST criteria (irRC in evaluable patients) occurred in 50% of LTX-315 monotherapy patients (4 melanoma, 3 sarcoma, and 1breast cancer patients).
Conclusions
This phase 1 study demonstrates that intratumoral LTX-315 is generally safe and tolerable. Intratumoral LTX-315 triggers an increase in TILs as assessed by IHC, and generates a transition from a cold to a hot tumor transcriptome as assessed by Immunosign® gene signature. Moreover, local and systemic clinical benefit (achievement of SD in non-injected sites per irRC criteria) was observed.
Trial Registration
Clinical trial information: NCT01986426.
P251 A study to evaluate the safety and efficacy of the CD40 agonistic antibody APX005M in combination with nivolumab in subjects with non-small cell lung cancer and subjects with metastatic melanoma
Linda Garland1, Ravi Salgia2, Melissa Johnson3, Amy Weise4, Gerald Linette5, Thomas Tremblay6, Ovid Trifan6, Martin Edelman7
1University of Arizona Cancer Center, Tucson, AZ, USA; 2City of Hope, Duarte, CA, USA; 3Sarah Cannon Research Institute, Tennessee Oncology, Nashville, TN, USA; 4Karmanos Cancer Center, Detroit, MI, USA; 5Abramson Cancer Center, University of Pennsylvania, Philadelphia, PA, USA; 6Apexigen Inc., San Carlos, CA, USA; 7Fox Chase Cancer Center, Philadelphia, PA, USA
Correspondence: Martin Edelman (otrifan@apexigen.com)
Background
Blocking immune checkpoint PD-1, PD-L1 and CTLA-4 function enhance antitumor immunity, leading to durable clinical responses for a subset of patients with melanoma, lung cancer and other tumor types. However, the majority of patients with melanoma or lung cancer continue to have short or no response to checkpoint blockade therapies and thus require novel approaches to stimulate the antitumor immune response such as immune stimulatory antibodies. Recently, Zippelius and co-authors [1] showed in preclinical models that CD40 engagement with an agonistic mAb leads to a T cell and IFN-γ dependent upregulation of PD-L1 on tumor infiltrating monocytes and macrophages, thereby promoting a negative feedback loop, which hampers CD40 induced T-cell responses. This resistance mechanism was successfully circumvented by co-administration of PD-1/PD-L1 blocking antibodies. To this end, Apexigen Inc., is developing APX005M a humanized monoclonal IgG1 CD40 agonistic antibody that stimulates both innate and adaptive immune response. APX005M recognizes a unique epitope that overlaps with the CD40 ligand binding sites and uses FcRγIIb to cluster CD40, thus mimicking CD40L engagement. As a result of antigen presenting cell (APC) activation, APX005M enhances T-cell response to tumor antigens. APX005M combined with antibodies against PD-1 or PD-L1 synergistically enhances T-cell responses. In a phase 1 trial, APX005M was administered IV every 21 days to adult subjects up to 1mg/kg with an acceptable safety profile and has demonstrated a dose-dependent activation of APCs and T cells and increases in circulating levels of cytokines.
Methods
Study APX005M-002 is a Phase 1b–2 study of APX005M administered in combination with nivolumab every 21 days to adult subjects with platinum pre-treated immunotherapy naïve non-small cell lung cancer (NSCLC) or metastatic melanoma after failure of anti-PD-1/PD-L1 therapy (MM). The Phase 1b portion will establish the maximum tolerated dose and the recommended Phase 2 dose of APX005M with nivolumab. The Phase 2 portion will evaluate safety and efficacy of the combination in each of the two distinct tumor types. Inclusion criteria include: age ≥ 18 years, histologically documented NSCLC or MM, measurable disease by RECIST 1.1, ECOG performance status 0-1, adequate organ function. Exclusion criteria include: concomitant anti-cancer therapy, history of bone marrow transplantation, active coagulopathy, previous immune mediated disorders, active infections or uncontrolled intercurrent illness. Recruitment is ongoing, with a target enrollment of approximately 100 subjects across 7 centers in the United States.
Trial Registration
Clinical trial information: NCT03123783.
References
1. Zippelius A, Schreiner J, Herzig P, Müller P. Cancer Immunol Res. 2015;3:236-44.
P252 Phase 1 study to evaluate the safety and tolerability of the CD40 agonistic monoclonal antibody APX005M in subjects with solid tumors
Marwan Fakih1, David Bajor2, Ronac Mamtani3, Thomas Tremblay4, Ovid Trifan4, Robert Vonderheide3
1City of Hope, Duarte, CA, USA; 2University Hospitals Cleveland Medical Center, Cleveland, OH, USA; 3Abramson Cancer Center, University of Pennsylvania, Philadelphia, PA, USA; 4Apexigen Inc., San Carlos, CA, USA
Correspondence: Ovid Trifan (otrifan@apexigen.com)
Background
APX005M is a humanized monoclonal IgG1 CD40 agonistic antibody developed by Apexigen, which mimics the natural ligand CD154. APX005M binds with high affinity to human CD40 leading to antigen presenting cell (APC) activation and subsequent T cell activation. APX005M enhances T cell proliferation, IFN-γ production and T-cell response to tumor antigens. In comparison with other anti-human CD40 agonistic antibodies, such as CP-870,893/RG7876, SGN-40, and ADC-1013/JNJ-64457107 analogs, APX005M is the most potent CD40 agonist and outperforms all others in many measures of immune activation. In a first in human clinical trial APX005M was administered IV every 21 days and has demonstrated a dose-dependent activation of APCs and T cells and increases in circulating levels of IL-12, IFN-γ, TNFα and IL-6. APX005M was escalated up to 1mg/kg with a good safety profile.
Methods
Study APX005M-001 was originally designed as a multicenter Phase 1 dose escalation study of APX005M administered every 3 weeks to subjects with solid tumors and was amended to introduce two new dosing schedules for APX005M (every 2 weeks and every 1 week). Currently, primary objectives of the study are to evaluate the safety of APX005M administered intravenously (IV) every 2 weeks and every week and to determine the maximum tolerated dose (MTD) and recommended Phase 2 dose (RP2D) of APX005M for the every 2 weeks and every 1 week schedules. Secondary objectives include determining the pharmacokinetics (PK) of APX005M and preliminary assessment of clinical response. Inclusion criteria include: age ≥ 18 years, histologically or cytologically documented diagnosis of urothelial carcinoma, melanoma, squamous cell carcinoma of the head and neck, non-small cell lung cancer, or any solid tumor with high microsatellite instability status (MSI-high), measurable disease by RECIST 1.1, ECOG performance status 0-1, adequate organ function. Exclusion criteria include: concomitant anti-cancer therapy, history of bone marrow transplantation, active coagulopathy, previous immune mediated disorders, active infections or uncontrolled intercurrent illness. Recruitment is ongoing, with a target enrollment of approximately 20 subjects across 3 centers in the United States.
Trial Registration
Clinical trial information: NCT02482168.
P253 Prioritizing tumor types for treatment with a novel immunotherapy: LYC-55716 a small-molecule RORγ agonist
Xiao Hu1, Xikui Liu1, Hongxiu Li1, Madhumita Bogdan1, Yilin Gao1, Brian Fox2, H. Jeffrey Wilkins3, Laura Carter1
1Lycera Corp., Ann Arbor, MI, USA; 2Celgene Corp., Seattle, WA, USA; 3Lycera Corp., Plymouth Meeting, PA, USA
Correspondence: H. Jeffrey Wilkins (autumn@ahkcommunications.com)
Background
RORγ is the master transcription factor for Type 17 effector T cell differentiation and function. RORγt expression is induced by cytokines such as IL-6, TGF-β, IL-1b, and IL-23 and is activated by endogenous ligands derived from the cholesterol biosynthetic pathway. Synthetic RORγ agonists augment the activity of this transcriptional regulator by modulating a gene expression program in immune cells, resulting in enhanced effector functions and decreased immunosuppression. LYC-55716 and other RORγ agonists have shown promise as monotherapy and combination therapy in syngeneic tumor models. During Phase 1 clinical testing of this compound, preclinical and bioinformatics assessments were undertaken to prioritize tumor types that may respond to RORγ agonist therapy, for possible inclusion in a Phase 2a trial.
Methods
An RORγ agonist signature was derived from transcriptional profiling of primary murine and human T cells treated with or without RORγ agonists. Using a panel of murine syngeneic models and The Cancer Genome Atlas (TCGA) dataset, a series of bioinformatic analyses were conducted to provide information across tumor types on (a) RORγ expression, (b) RORγ biology, including sterol mobilizing genes and correlations with prognosis, and (c) general immune parameters. Public data sets were also assessed for correlations of RORγ signature genes and prognosis. For each assessment, tumor types were prioritized, then compared across categories to determine a final ranking.
Results
Target expression: 15 tumors types were identified, with >20% of samples expressing RORγt. However, based on lack of correlation between baseline RORγt expression and efficacy of RORγ agonists in preclinical models, these criteria were extended to include additional tumors that express mRNA for factors known to induce RORγ expression. Target biology: As a surrogate for endogenous ligand levels, TCGA analysis revealed differentially expressed sterol synthesis and efflux genes across tumor types. RORγt expression correlated significantly with patient survival in 5 tumor types. Immune parameters: Consideration of tumors with immune infiltrates, high mutational burden, and reports of prior immunotherapy success highlighted 8 tumor types. After combining these findings, 7 tumor types met all selection criteria: non–small-cell lung; ovarian; stomach adenocarcinoma; and head and neck squamous cell, renal clear cell, hepatocellular, and esophageal carcinomas.
Conclusions
Preclinical data from syngeneic tumor models and bioinformatic analyses of TCGA database prioritized 7 tumors for LYC-55716 monotherapy. Our findings support the inclusion of these tumor types in the Phase 2a clinical trial.
P254 A phase 1b/2 study of CD40 agonistic monoclonal antibody (APX005M) together with gemcitabine and nab-paclitaxel with or without nivolumab in untreated metastatic pancreatic adenocarcinoma patients
Mark O’Hara1, Rosemarie Mick2, Jaclyn Lyman3, Jingying Xu3, Maryam Hosseini3, Theresa LaVallee3, Pier Federico Gherardini3, Barbara Vance1, Ovid Trifan4, Ute Dugan5, Aiman Shalabi6, Ramy Ibrahim3, Robert Vonderheide1
1University of Pennsylvania, Philadelphia, PA, USA; 2University of Pennsylvania Perelman, Philadelphia, PA, USA; 3Parker Institute for Cancer Immunotherapy, San Francisco, CA, USA; 4Apexigen, San Carlos, CA, USA; 5Bristol-Myers Squibb, New York City, NY, USA; 6Cancer Research Institute, New York, NY, USA
Correspondence: Ramy Ibrahim; Robert Vonderheide (jxu@parkerici.org)
Background
Pancreatic cancer is one of the most lethal malignancies of the gastrointestinal tract. While check point inhibitors such as αCTLA4 and αPD-1 have been effective in melanoma and lung cancer, clinical benefit of immunotherapy in the management in pancreatic cancer subjects has not been yet established. A recent study using a genetically engineered mouse model of pancreatic ductal adenocarcinoma (PDA) demonstrated that despite robust expression of PD-1 and PD-L1 in the tumor microenvironment, treatment with αPD-1 with or without αCTLA-4 failed to improve the survival of mice or slow the growth of PDA tumors. However, administration of αCD40, gemcitabine (Gem) and nab-paclitaxel (NP), induces T cell immunity in mice with PDA, controls tumor growth and significantly improves survival in a CD8+ T cell dependent manner. In particular, αCD40/Gem/NP plus αPD-1 nearly double the median overall survival in genetically engineered KPC mice with pre-established spontaneous pancreatic tumors. Moreover, the capability of treated mice to reject second and third subcutaneous tumor challenges in a CD8+ T cell-dependent fashion thereby rendering long-term survival suggests the establishment of antitumor immune memory with curative potential.
Methods
This is a multi-center, open label study evaluating the combination of APX005M with Nivolumab and standard chemotherapy (Gem and NP). The Phase 1b will define the recommended Phase 2 dose of APX005M when combined with the standard dose of Gem and NP, with or without Nivolumab. The second part of the study is a 3-arm randomized Phase 2 (35 subjects per treatment arm) aimed to evaluate the activity (overall survival) of APX005M combined with Gem and NP, with or without Nivolumab with Gem and NP. Main inclusion criteria include: age ≥ 18 years, documented diagnosis of pancreatic adenocarcinoma with metastatic disease, measurable disease by RECIST 1.1, ECOG performance status 0-1, normal organ function. Main exclusion criteria include: concomitant anti-cancer therapy, previous exposure to CD40, PD-1, PD-L1, CTLA-4 mAbs or any other immunomodulatory agents, previous immune mediated disorders, active infections or uncontrolled intercurrent illness. The study will be exploring different doses of APX005M in combination with approved doses of Nivolumab and chemotherapy. The main safety endpoints include the frequency of DLT and incidence of AEs. The main efficacy endpoints include overall survival, and 1-year OS rate in each treatment arm.
Results
This trial is open for enrollment.
Conclusions
Clinical trial information: NCT02482168.
Combination Therapy (IO/IO, IO/Standard of Care, IO/Other)
P255 Depleting blood arginine with AEB1102 (Pegzilarginase) exerts additive anti-tumor and synergistic survival benefits when combined with immunomodulators of the PD-1 pathway
Giulia Agnello, Mark Badeaux, Susan Alters, David Lowe, Scott Rowlinson
Aeglea BioTherapeutics, Inc., Austin, TX, USA
Correspondence: Giulia Agnello (gagnello@aegleabio.com); Scott Rowlinson
Background
Tumor dependence on specific amino acids for survival and proliferation is well recognized and has been exploited effectively in the clinic through the use of asparaginases for the treatment of acute lymphoblastic leukemia. Sensitivity of tumors to L-Arginine (L-Arg) deprivation results from an impaired ability to synthesize L-Arg, most commonly due to decreased functional expression of argininosuccinate synthase. Native human arginase 1 is not a viable drug candidate due to low activity and low stability in serum. We have developed a novel cobalt substituted, PEGylated human arginase 1 (AEB1102, Pegzilarginase) with enhanced pharmacological properties. We and others have successfully utilized arginase 1 to impart an anti-tumor effect through L-Arg starvation in multiple tumor types in vitro and in vivo (e.g. AEB1102 single agent efficacy in melanoma, SCLC, sarcoma, large cell NSCLC, Merkel cell carcinoma). Given that arginase 1 has been reported to be immune suppressive, immune neutral (PMID: 23717444), or immune promoting (PMID: 27043409) in different experimental settings and by different groups, we have investigated the impact of systemic depletion of L-Arg on the anti-tumor efficacy of immune checkpoint inhibitors.
Methods
Murine syngeneic models (e.g. CT26, MC38) were dosed with AEB1102 alone and in combination with immunomodulatory anti-PD-L1 monoclonal antibody (mAb).
Results
Combination therapy of AEB1102 with anti-PD-L1 resulted in an additive anti-tumor effect with improved survival benefit (increased life span (ILS) 55-129%) compared to AEB1102 (ILS 29-33%) and anti-PD-L1 (ILS 7-33%) monotherapies. In addition, in the CT26 model, complete tumor regression (non-palpable tumors) was observed in 37% of the mice; importantly, complete responses were observed only in the combination therapy group. When the complete responders were re-challenged with fresh CT26 cells, tumors failed to establish, suggesting the development of an immune memory response as a result of the previously administered combination therapy of AEB1102 and anti-PD-L1. Administration of AEB1102 as a monotherapy or in combination with anti-PD-L1 in the CT26 model was associated with an increase in tumor-infiltrated CD45+ cells, indicating that AEB1102 promotes T-cells accumulation in the tumor microenvironment.
Conclusions
Collectively, these results demonstrate that in addition to tumor growth inhibition, L-Arg depletion in the tumor microenvironment enhances the effectiveness of immunotherapy. AEB1102 is currently in Phase 1 (monotherapy) clinical trials. These data open the possibility of clinical combination of AEB1102 with immunomodulators of the PD-1 pathway to further improve outcomes in cancer patients.
P256 G100 and ZVex®-based combination immunotherapy induces near complete regression of established glioma tumors in mice
Tina C. Albershardt, Jordan E. Krull, Reice D. James, Peter Berglund, Jan ter Meulen
Immune Design, Seattle, WA, USA
Correspondence: Tina C. Albershardt (tina.albershardt@immunedesign.com)
Background
Glioblastoma (GBM) is a malignant brain tumor with an overall survival of < 3.3% at 5 years. Novel immunotherapies are being explored but face limitations due to low infiltration of activated T cells. ZVex is an integration-deficient lentiviral vector-based platform that targets dendritic cells in vivo to generate tumor-specific T cells. Intratumoral injections of G100, which contains glucopyransosyl lipid A (GLA, a synthetic TLR4 agonist), modulate the tumor microenvironment through induction of proinflammatory responses. We have previously shown in the B16 model that G100 induces T cell homing chemokines CXCL9 and CXCL10, increases number of T cells in the tumor, enhances intratumoral antigen presentation, and results in antigen spreading of the T cell response. Combined systemic and in situ immunization with ZVex/OVA and G100, respectively, completely eradicated established B16/OVA melanomas in mice. Here, we report that, in an orthotopic GBM model, the combined regimen with ZVex/OVA and G100 induced near complete regression of established GL261 syngeneic gliomas in C57BL/6 mice.
Methods
Female C57BL/6 mice were stereotactically cannulated in the left striatum. At study initiation, 2 x 105 GL261 glioma cells expressing ovalbumin (GL261/OVA) were inoculated through the cannula. Tumor growth was monitored via imaging of luciferase activity, twice weekly. On Day 7, mice with tumors (6.8 x 105 avg. radiance) were immunized by a single subcutaneous injection of ZVex/OVA. G100 (2 μg GLA) was administered through the cannula, also on Day 7, and then once weekly for the duration of the study, for a total of three injections. Animals were sacrificed when displaying signs of cachexia and > 20% weight loss, usually around 25 days after tumor inoculation.
Results
GL261/OVA-bearing mice treated with G100 or ZVex/OVA alone exhibited delayed tumor growth or modest tumor regression, respectively (mean reduction of 17% and 72%, respectively, ranging from none to 83%), while mice treated with ZVex/OVA and G100 combination exhibited significantly greater reduction in tumor size, averaging 98% (74%-99.8%).
Conclusions
This study demonstrates that ZVex/OVA and G100 combination therapy was very successful in controlling growth of GL261 gliomas, presumably in part by chemotactically directing peripheral T cells to the brain, and suggest the potential for treatment of patients with GBM. The precise mechanism of synergy in this GBM model is currently under investigation. Both ZVex-based therapies and G100 are being evaluated in multiple Phase 1 and 2 clinical trials, and their combination is being investigated in a Phase 1 trial in soft tissue sarcoma patients.
P257 Phase II basket study of olaparib and durvalumab: Biomarker analysis in germline BRCA-mutated (gBRCAm) HER2-negative metastatic breast cancer (MBC) and relapsed small-cell lung cancer (SCLC) patients
Helen Angell1, Vidalba Rocher Ros1, Nathan Standifer2, Zhongwu Lai1, Christopher Gresty1, Jean-Pierre Delord3, Maja De Jonge4, Sophie Postel-Vinay5, Antoine Italiano6, Matthew G Krebs7, Bella Kaufman8, Yeon Hee Park9, Susan Domchek10, Pia Herbolsheimer11, Darren Hodgson1
1AstraZeneca, Cambridge, United Kingdom; 2MedImmune, Mountain View, CA, USA; 3Institut Claudius Regaud-Oncopole, Toulouse, France; 4Erasmus MC Cancer Institute, Rotterdam, Netherlands; 5Gustave Roussy Cancer Campus, Villejuif, France; 6Institut Bergonié, Bordeaux, France; 7The University of Manchester and The Christie NHS Foundation Trust, Manchester, United Kingdom; 8Sheba Medical Centre, Ramat Gan, Israel; 9Samsung Medical Centre, Sungkyunkwan University School of Medicine, Seoul, Republic of Korea; 10Hospital of the University of Pennsylvania, Philadelphia, PA, USA; 11AstraZeneca, Gaithersburg, MD, USA
Correspondence: Helen Angell (Helen.Angell@astrazeneca.com)
Background
Poly(ADP-ribose) polymerase inhibitors (PARPi) trap PARP at sites of single-strand DNA breaks, and generate genomic instability and cell death in homologous recombination deficient tumour cells. Both intrinsic and PARPi-induced DNA repair defects may attract tumour-infiltrating lymphocytes (TILs), upregulate programmed cell death ligand-1 (PD-L1) and release tumour neo-antigens upon cell death. The MEDIOLA study (NCT02734004) assessed the effect of the PARPi, olaparib, alone and in combination with durvalumab (anti-PD-L1), on immune cell pharmacodynamics, and the correlation of candidate predictive markers on disease control rate (DCR).
Methods
Patients with HER2-negative, gBRCAm MBC (n=25), or relapsed SCLC (n=38) received olaparib (300 mg PO BID) for 4 weeks, followed by a combination of olaparib and durvalumab (1.5 g IV every 4 weeks) until progression. At 4 weeks after initial olaparib, paired biopsies were conducted for immuno-oncology profiling. Primary endpoints were DCR at 12 weeks, safety and tolerability. Biomarker endpoints included archival (Fig. 1) and paired biopsy analysis of tumour cell (TC) and immune cell (IC) PD-L1 expression (SP263), densities of CD3 and CD8 T-cells (cells/mm2), tumour mutation status and peripheral blood immunophenotyping.
Results
Olaparib caused small reductions in circulating T, B and Natural Killer (NK) cells on day 29 and thereafter in 86–93% of MBC and 81–91% of SCLC patients, followed by evidence of post-durvalumab 2-fold or greater increases in Ki67+CD8+ T-cells in 46% of MBC and 50% of SCLC patients on day 57. In MBC (n=25) 5/5 (100%) PD-L1 TC≥1% patients had controlled disease at 12 weeks, compared with 9/14 (64.3%) PD-L1 TC<1% patients (no data for 6/25 patients). In SCLC (n=38) PD-L1 TC≥1% patients had a DCR of 1/6 (16.7%) compared with a PD-L1 TC<1% DCR of 9/27 (33.3%) at 12 weeks (no data for 5/38 patients). At the time of analysis, 1/2 MBC with paired biopsy data had a partial response (PR) and demonstrated dramatic increases in CD3/CD8 densities and PD-L1 TC positivity 4 weeks post-olaparib monotherapy. 1/3 SCLC paired biopsies had PRs and showed increases in CD3/CD8 densities, but no PD-L1 TC expression, compared with no change in 2/3 patients with stable disease. Updated results will be presented.
Conclusions
Olaparib results in modest reductions in circulating T, B and NK cells: however, this effect does not disrupt durvalumab-based increases in circulating, proliferating cytotoxic T-cells. Preliminary data suggest that olaparib treatment may increase levels of TILs, but more data are required. Data collection and analysis is ongoing.

Biomarker analysis summary
P258 Resource use and cost implications associated with treatment-free interval experienced on immunotherapies
Michael Atkins1, Ahmad Tarhini2, Apoorva Ambavane3, David McDermott4, Komal Gupte-Singh5, Josh Weng3, Corey Ritchings5, Meredith Regan7, Agnes Benedict6, Sumati Rao5
1Georgetown-Lombardi Comprehensive Cancer Center, Washington, DC, USA; 2University of Pittsburgh School of Medicine and Cancer Institute, Pittsburgh, PA, USA; 3Evidera, Inc., Bethesda, MD, USA; 4Beth Israel Deaconess Medical Center, Boston, MA, USA; 5Bristol-Myers Squibb, Princeton, NJ, USA; 6Evidera, Inc., Budapest, Hungary; 7Dana-Farber Cancer Institute, Boston, MA, USA
Correspondence: Michael Atkins (mba41@georgetown.edu)
Background
Immunotherapies, including ipilimumab, nivolumab, and nivolumab+ipilimumab, are recommended for the treatment of advanced melanoma. Patients treated with immunotherapies often experience clinical benefit beyond treatment discontinuation, resulting in a significant treatment-free interval (TFI) until subsequent treatment is needed. However, resource use during TFI is not well understood. The objective of this study was to estimate the resource use and costs associated with the TFI experienced after first-line immunotherapies.
Methods
Patient-level data (N=882) from the phase 3 CheckMate 067 and phase 2 CheckMate 069 trials were analyzed to estimate the healthcare resource use associated with the management of advanced melanoma. TFI is defined as time from first-line discontinuation to second-line initiation. Concomitant medications, laboratory tests, procedures, consultations, hospitalizations, and surgeries were analyzed. The annual rate of resource use was estimated for ipilimumab, nivolumab, and nivolumab+ipilimumab, and stratified by disease phase (progression-free or progressed defined by RECIST v.1.1). Mean annual total costs and 95% confidence intervals (CIs) were estimated by applying unit costs from publicly available sources in the United States.
Results
Annual costs per patient during the TFI on various immunotherapies are presented below (Table 1) Although confidence intervals are wide, ipilimumab is associated with higher total cost/year compared to nivolumab+ipilimumab and nivolumab in the progression-free phase (difference in costs $5,624-$6,481), and compared to nivolumab+ipilimumab and nivolumab in the progressed phase (difference in costs $354-$3,834). Progression increases the costs of melanoma management across all treatments by $12,486-$16,823. Key resources utilized across all treatments were hospitalizations (34-62% of total costs) in the progression-free phase, due to management of adverse events, and hospitalizations and surgeries (>94% of total costs) in the progressed phase, due to disease worsening.
Conclusions
Immunotherapies for advanced melanoma, particularly nivolumab+ipilimumab, may lead to a long TFI before starting second-line therapy. The estimated annual cost of managing advanced melanoma is low in the TFI prior to disease progression, especially with nivolumab and nivolumab+ipilimumab. These results require comparison with real-world evidence to measure the impact of bias due to the controlled clinical trial setting.
See text for description
P259 Pushing the accelerator and releasing the brake: testing the soluble LAG-3 protein (IMP321), an antigen presenting cell activator, together with pembrolizumab in unresectable or metastatic melanoma
Victoria Atkinson1, Andrew Haydon2, Melissa Eastgate3, Amitesh Roy4, Adnan Khattak5, Christian Mueller6, Tina Dunkelmann6, Chrystelle Brignone7, Frederic Triebel7
1Princess Alexandra Hospital, 4102 Woolloongabba, Australia; 2Alfred Hopital, 3004 Melbourne, Australia; 3Royal Brisbane and Women's Hospital, 429 Herston, Australia; 4Flinders Medical Centre, Bedfork Park, Australia; 5Fiona Stanley Hospital, Murdoch, Australia; 6Prima BioMed GmbH, Berlin, Germany; 7Immutep S.A.S., Châtenay-Malabry, France
Correspondence: Victoria Atkinson (Victoria.Atkinson@health.qld.gov.au)
Background
IMP321 is a recombinant soluble LAG-3Ig fusion protein binding to MHC class II molecules and mediating antigen presenting cell (APC) activation followed by CD8 T-cell activation. The activation of the dendritic cell network and the subsequent T cell recruitment at the tumor site with IMP321 may lead to stronger anti-tumor CD8 T cell responses than observed with pembrolizumab alone. This combination of an APC activator with an immune checkpoint inhibitor (ICI) is aiming to increase efficacy without additional toxicity. We report initial results of the first 2 cohorts of a dose escalation phase I trial (TACTI-mel, NCT02676869) with pembrolizumab and IMP321 at different dose levels.
Methods
In this study, melanoma patients treated with pembrolizumab being without a complete response or fast progression after 3 cycles received pembrolizumab per standard dosing plus either 1 mg (n=6; cohort 1) or 6 mg (n=6, cohort 2) IMP321 injections s.c. (every 2 weeks for 6 months) from cycle 5 onwards. Patients without progressive disease (PD) at the end of the 6 months combination therapy continue on pembrolizumab monotherapy.
Results
In total, 12 patients (11 male, 1 female) with a median age of 62 years (range 48-85) were enrolled between Apr 2016 and Feb 2017. Four patients completed the 6 months combination treatment. One patient discontinued due to serious adverse event (SAE), unrelated to both study drugs. Two patients withdrew consent and five discontinued prematurely due to PD. No dose limiting toxicities for the combination have been reported. No SAE were found related to IMP321. All patients were evaluable according to irRC and 6 showed a reduction of ~50 % after IMP321 initiation. This includes one patient with a confirmed complete response while having PD on pembrolizumab monotherapy before study start.
Conclusions
One (1) and 6 mg IMP321 given s.c. every 2 weeks in combination with pembrolizumab are safe and testing of the highest dose (30 mg) is underway. The 6 late tumor responses seen after the combination was started may point out to the benefit of adding a systemic APC activator to an ICI.
P260 Glembatumumab vedotin (GV), an anti-gpNMB antibody-drug conjugate (ADC), in combination with varlilumab (V), an anti-CD27 antibody, in advanced melanoma
Omid Hamid1, Anna C. Pavlick2, C. Lance Cowey3, Lowell Hart4, Douglas B. Johnson5, Jose Lutzky6, Aaron Alizadeh7, David Spigel8, Neal Rothschild9, April Salama10, Robert Weber11, Jason J. Luke12, Ying Wang13, Michael Yellin13, Yi He13, Rebecca G. Bagley13, Patrick Ott14
1The Angeles Clinic and Research Institute, Los Angeles, CA, USA; 2New York University School of Medicine, New York, NY, USA; 3Baylor University Medical Center, Dallas, TX, USA; 4Florida Cancer Specialists, Fort Myers, FL, USA; 5Vanderbilt University Medical Center and Vanderbilt Ingram Cancer Center, Nashville, TN, USA; 6Mount Sinai Comprehensive Cancer Center, Miami Beach, FL, USA; 7Northside Hospital, Atlanta, GA, USA; 8Tennessee Oncology, PLLC, Nashville, TN, USA; 9Florida Cancer Specialists, West Palm Beach, FL, USA; 10Duke University, Durham, NC, USA; 11St. Mary's Medical Center, San Francisco, CA, USA; 12University of Chicago, Chicago, IL, USA; 13Celldex Therapeutics, Inc., Hampton, NJ, USA; 14Dana-Farber Cancer Institute, Boston, MA, USA
Correspondence: Omid Hamid (rbagley@celldex.com)
Background
gpNMB is an internalizable transmembrane glycoprotein expressed in multiple tumor types. The ADC GV delivers the potent cytotoxin MMAE to gpNMB+ cells and showed promising activity as monotherapy in advanced melanoma refractory to checkpoint inhibitors (CPI). Preclinical results showed synergistic antitumor activity between ADC with CPI [1, 2], via dendritic cell (DC) activation; ADC-MMAE augmented the immune response induced by V, an agonist monoclonal antibody against the T cell costimulatory molecule CD27. This Phase 2 study was conducted to evaluate the activity and safety of the GV/V combination in advanced melanoma.
Methods
Patients with advanced melanoma, progressive after ≤1 chemotherapy, ≥1 checkpoint inhibitor (CPI), and if BRAF mutation ≥1 BRAF or MEK + BRAF inhibitor, were treated with GV (1.9 mg/kg q3w until progression/intolerance) in combination with V (3.0 mg/kg) on day 1 of weeks 1, 3, 9, 15, 21, and 27. Retrospective analysis on pre-study tumors and skin biopsies include gpNMB expression and infiltrating lymphocytes by immunohistochemistry plus gene expression profiling. Primary objective is to evaluate objective response rate (ORR) (RECIST 1.1).
Results
Thirty-four patients enrolled: median age of 61 years; 59% male; 18% BRAFV600 mutated; 59% ≥3 lines prior therapy; 76% prior anti-CTLA-4; 100% prior anti-PD-1/PD-L1 inhibitor; 97% Stage IV; 71% M1c. Of 30 response evaluable patients, emerging tumor response data shows 2 confirmed partial responses (PR) (ORR = 7%, CI: 0.8, 22.1), and 1 single time point PR before patient withdrew consent. 48% patients had tumor shrinkage. Median PFS = 2.6 months and median OS = 4.4 months; 3 patients remain on treatment and 18 patients in survival follow up. Most common treatment related toxicities include alopecia, rash, fatigue, neuropathy, nausea, and vomiting. Tumor cells in 84% of patients with samples (n=25) tested to date are 100% gpNMB+.
Conclusions
The GV/V combination was well tolerated without evidence of additive toxicity. There was no apparent enhanced clinical benefit of GV/V over GV alone in this patient population, perhaps because immune checkpoint molecules remained unblocked and/or of a dearth of antigen presenting cells in tumors. Correlative biomarker analyses are ongoing and will be presented. For further insights into the synergy of ADC and immunotherapy, a cohort evaluating GV with anti-PD-1 in CPI-refractory melanoma is enrolling and a cohort investigating GV with the DC growth factor FLT3L (CDX-301) is planned.
References
1. Muller P, et al.Cancer Immunol Res. 2014; 2:741-755.
2. Muller P, et al. Sci Transl Med. 2015; 7:315rfa188.
P261 Molecular signatures of combination immunotherapy of prostate cancer using a Listeria-based PSA vaccine and radiation
Emily Bongiorno1, Trevor Baybutt1, Carla Portocarrero1, Adam Snook1, Adam Dicker1, Sandra Hayes2, Ulrich Rodeck1
1Thomas Jefferson University, Philadelphia, PA, USA; 2Advaxis Immunotherapeutics, Inc., Princeton, NJ, USA
Correspondence: Emily Bongiorno (emily.bongiorno@jefferson.edu)
Background
Radiotherapy (RT) has the potential to amplify immune responses triggered by tumor vaccines, including ADXS-PSA, a live-attenuated Listeria monocytogenes (Lm)-based vector expressing human PSA. Earlier observations suggest that the two treatment modalities cooperatively induce regression of syngeneic mouse prostate cancer cells expressing human PSA (TPSA23), though immune correlates of efficacy and tumor recurrence are poorly understood.
Methods
We compared efficacy of different sequencing regimens of combination RT/vaccine treatments on TPSA23 tumor growth in syngeneic mice. Using the optimal sequencing protocol, tumors were collected post-implantation to assess immune infiltrate and function during initial tumor regression (day 20) and upon resumption of tumor growth (day 38). Correlates of treatment efficacy were determined by transcriptome analysis, phenotypic analyses of infiltrates and TCR sequencing.
Results
We confirmed that combination RT/ADXS-PSA is superior to single modality treatments. Concurrent administration of RT/vaccine was the most effective treatment schedule and was associated with enhanced T cell activation and robust IFNg signatures in the tumor microenvironment. This was reflected in increased intratumoral CD4 and CD8 T cell infiltration in mice receiving RT/vaccine. TCRb chain sequencing revealed elevated and sustained T cell diversity in tumor tissues of RT/vaccine-treated mice, when compared with mice receiving single modality treatments. In these residual tumors resident and/or memory T cell phenotypic markers were increased. Transcriptome analysis of recurring tumors further revealed induction of PD-L1 as a function of treatment. Targeting of the PD1/PD-L1 axis via a PD1 blocking antibody administered in addition to radiation and ADXS-PSA (triple combination) further amplified tumor growth inhibition in mice receiving dual RT/vaccine therapy.
Conclusions
Combining RT with the ADXS-PSA vaccine leads to effective tumor growth inhibition and induces robust, persistent antitumor immunity within the tumor environment. Transcriptome analysis during treatment revealed increased PD1 expression as a potential resistance mechanism and a PD1 blocking antibody provided further therapeutic benefit. These results support the rationale for combining Listeria-based vaccines with radiation in the clinic.
P262 Phosphatidylserine targeting antibody in combination with tumor radiation and immune checkpoint blockade promotes anti-tumor activity in mouse B16 melanoma
Sadna Budhu1, Rachel Giese1, Olivier De Henau1, Roberta Zappasodi1, Luis Felipe Campesato1, Aditi Gupta1, Christopher Barker1, Bruce Freimark2, Jedd D. Wolchok1, Taha Merghoub1
1Memorial Sloan Kettering Cancer Center, New York, NY, USA; 2Peregrine Pharmaceuticals, Inc., Tustin, CA, USA
Correspondence: Jedd D. Wolchok (budhus@mskcc.org); Taha Merghoub
Background
Phosphatidylserine (PS) is a phospholipid that is exposed on surface of apoptotic cells, viable tumor cells, tumor endothelium and activated immune cells. It has been shown to promote immunosuppressive signals in the tumor microenvironment. In a mouse B16 melanoma model, targeting PS in combination with immune checkpoint blockade promoted greater anti-tumor activity than either agent alone. This combination was shown to enhance CD4+ and CD8+ T cell infiltration and activation in the tumors of treated animals. Radiation therapy (RT) is an effective focal treatment of primary solid tumors, but is less effective in treating metastatic solid tumors as a monotherapy. RT induces immunogenic tumor cell death and enhances tumor-specific T cell infiltration in treated tumors. The abscopal effect, a phenomenon in which tumor regression occurs outside the site of RT, has been observed in both preclinical and clinical trials when RT is combined with immunotherapy.
Methods
Mice were injected intradermally on the hind limb with 105 B16F10 melanoma cells. 7-10 days after implantation, tumors were treated locally with 15 Gy RT. 1 day after RT, mice were given antibodies to PS (mch1N11) and PD-1 (RMP 1-10) intraperitoneally every 3 days. Tumor surface area and overall survival of mice were used to determine efficacy of the combinations. For FACS analysis, tissues were collected between 1-10 days after RT.
Results
In this study, we show that irradiation of B16 melanoma causes an increase in PS expression on the surface of viable tumor and immune infiltrates. We subsequently examined the effects of combining RT with an antibody that targets PS and anti-PD-1. We found that treatment with mch1N11 synergizes with RT to improve anti-tumor activity and overall survival in tumor bearing mice. In addition, the triple combination of mch1N11, RT and anti-PD-1 treatment displayed even greater anti-tumor and survival benefit. Analysis of the immune response in the tumors of treated animals revealed an increase in M1-like macrophages in the tumors after treatment with RT and mch1N11. In addition, analysis of the systemic immune responses revealed an increase in antigen-specific CD8 T cell infiltration in the tumors as well as increased activation, effector function and differentiation in the triple combination therapy.
Conclusions
This finding highlights the potential of combining these agents to improve outcome in patients with advanced-stage melanoma and may inform the design of future clinical trials with PS targeting in multiple cancers.
P263 Preclinical development of a Vaccine-Based Immunotherapy Regimen (VBIR) that breaks immunological tolerance and induces high titer and long lived T-cell responses to a tumor-associated self-antigen
Helen Cho1, Risini Weeratna2, Joe Binder1, Bassel Akache2, Rajeev Nepal3, Paul Cockle4, Marianne Martinic1, Michael Dermyer1, Stanley Dai1, James Merson1, Karin Jooss4
1Pfizer Inc., San Diego, CA, USA; 2National Research Council of Canada, Ottawa, ON, Canada; 3Pfizer Canada Inc., Kirkland, QC, Canada; 4Gritstone Oncology Inc., Emeryville, CA, USA
Correspondence: Helen Cho (Helen.k.cho@pfizer.com)
Background
A successful therapeutic cancer vaccine will activate the immune system to rapidly induce potent, balanced and durable CD8 and CD4 T-cell responses to selected tumor antigens that can reduce tumor growth, lead to tumor regression or prevent/delay the onset of new lesions.
Methods
The magnitude and quality of T-cell responses against self (rhesus Prostate Specific Membrane Antigen; rhPSMA) and non-self (human PSMA; hPSMA) antigens delivered by various vaccine platforms in non-human primates (NHPs) given with and without a check point inhibitor, anti-CTLA4 monoclonal antibody (tremelimumab; anti-CTLA4), were evaluated.
Results
Assessment of T-cell responses against self (rhPSMA) and non-self (hPSMA) antigens delivered by various vaccine platforms in NHPs, demonstrated that the AdV vector was the most potent delivery platform for breaking of immune tolerance to a self-antigen.
Many humans have immunity to adenovirus that could blunt the ability of an adenovirus vaccine to effectively prime an immune response. This limitation can be overcome by utilizing an AdV of a different serotype for which humans do not have pre-existing immunity. However, even with a low/no seroprevalent AdV, an AdV boost vaccination following an AdV prime vaccination can only be effective when the neutralizing immunity to the vector wanes in between the vaccinations, making the regimen less suitable for patients with high tumor burden or fast progressive disease. Therefore, plasmid DNA boost vaccinations delivered intramuscularly by electroporation were evaluated for durable expansion of AdV primed T-cell responses. Administration of anti-CTLA4 delivered subcutaneously to achieve high local concentrations in the vaccine draining lymph nodes, given concurrently with the AdV prime and DNA boost vaccinations was shown to induce and expand durable and polyfunctional (IFNγ+, TNFα+ and/or IL-2+) T-cell responses to a tumor-associated self-antigen in NHPs.
Conclusions
In summary, we have developed VBIR that consists of a heterologous prime-boost vaccine approach given in combination with local delivery of tremelimumab to maximize vaccine potency while minimizing the negative impact of neutralizing antibodies to the AdV vector. These results have encouraged clinical development of this unique immunotherapeutic regimen.
P264 Immune modulation by low dose sunitinib combined with a cancer vaccine based immunotherapeutic regimen provides therapeutic benefit to tumor bearing mice
Helen Cho1, Cindy Li1, Antonio Boccia1, Steve Burgess1, Terri Harder1, Paul Cockle2, Jim Eyles1, Marianne Martinic1, Bryan Clay1, Kam Chan1, Stanley Dai1, Michael Dermyer1, Joe Binder1, Steve Kurzyniec3, Terrina Bayone1, Joan Guo4, Robert Hollingsworth1, James Merson1, Karin Jooss2
1Pfizer Inc., San Diego, CA, USA; 2Gritstone Oncology Inc., Emeryville, CA, USA; 3Shimadzu Scientific Instruments, San Diego, CA, USA; 4Moderna Therapeutics, Cambridge, MA, USA
Correspondence: Helen Cho (Helen.k.cho@pfizer.com)
Background
Sunitinib is a receptor tyrosine kinase (RTK) inhibitor approved as a monotherapy for the treatment of advanced renal cell carcinoma (RCC), gastrointestinal stromal tumors and advanced pancreatic neuroendocrine tumors. It inhibits angiogenesis and targets RTKs (PDGFR, VEGFR, KIT, FLT3, and RET) involved in tumor cell growth. Sunitinib also has been shown, in preclinical studies and patients, to have immunomodulatory properties that reduce myeloid derived suppressor cells (MDSCs) and restore a Th1 cytokine profile. The approved dosing regimens for RCC have a safety profile that is overall manageable with the most commonly reported sunitinib-related grade 3 adverse events including hypertension, fatigue, diarrhea and hand-foot syndrome. Although the tolerability of sunitinib as monotherapy was acceptable in advanced stage, metastatic cancer patients, the drug’s side effect profile could potentially pose challenges for use in patients with earlier stage cancer and/or when combined with other oncology therapies. Thus, lowering the dose of sunitinib might improve the safety of combinations, including combinations with cancer vaccines, thus enabling treatment of cancers of all stages.
Methods
The anti-tumor efficacy of lowered doses of sunitinib as a monotherapy, and in combination with a cancer vaccine, was investigated in immune-competent mouse tumor models. To understand the mechanism of such efficacy, the impact of the treatments on MDSC profiles in the periphery and tumors, T cell IFNγ production, and antigen-specific immune responses was evaluated. Additionally, adoptive immune cell transfer experiments were conducted to monitor survival of MDSCs and T cells in mice treated with sunitinib.
Results
At doses below the maximum biologic efficacy dose in mice (< 40 mg/kg q.d.), sunitinib selectively and significantly reduced the survival and frequency of MDSCs in the periphery and tumors, which resulted in increased IFNg production by T cells. Importantly, when low dose sunitinib was given concurrently with a rat HER2 (rHER2)-based heterologous prime-boost vaccine, it significantly reduced the growth of subcutaneous tumors and prolonged survival of tumor-bearing rHER2 transgenic mice compared to mice receiving only monotherapies. Addition of an anti-CTLA4 monoclonal antibody (anti-CTLA4) significantly enhanced the vaccine-induced immune response and prolonged survival.
Conclusions
The combination of the vaccine, sunitinib, and anti-CTLA4 further increased efficacy and survival. These results indicate that low dose sunitinib, alone or together with either a cancer vaccine or a cancer vaccine plus anti-CTLA4, maintains its immune modulatory properties and may be a valuable component of a safe and efficacious vaccine-based immunotherapy regimen.
P265 Optimizing targeted therapy and immune checkpoint blockade therapy in Kras mutant lung cancer
Hyejin Choi1, Jiehui Deng2, Tarik Silk1, Ann Powers1, Jonathan Boiarsky1, Taha Merghoub3, Kwok-Kin Wong2, Jedd Wolchok1
1MSKCC, New York, NY, USA; 2New York University Langone Medical Center, New York, NY, USA; 3Medicine, New York, NY, USA
Correspondence: Taha Merghoub (choih3@mskcc.org); Kwok-Kin Wong; Jedd Wolchok
Background
KRAS is the most commonly identified driver oncogene in lung cancer. However, to date there is no effective therapy available for KRAS mutant lung cancers. To identify the most effective therapy, we studied the impact of Kras signaling targeted therapy (MEK inhibition) on the immune microenvironment, in order to formulate a combinatorial therapy using targeted therapy and immunotherapy, with a goal of optimally enhancing tumor apoptosis and promoting long-term immune response simultaneously. MEK signaling is a downstream of Kras signaling pathway and critical for tumor growth and T cell activation. Conventionally, MEK inhibitor is treated daily/continuously. However, to enhance tumor apoptosis and promote T cell activation on MEK inhibition, we hypothesized intermittent administration of MEK inhibitor will confer T cells temporal release from MEK signaling inhibition allowing T cells activated, while tumor growth is suppressed and immunotherapy can be combined based on T cell activation/inhibitory markers change on this treatment schedule.
Methods
We have treated T cells from Kras lung cancer bearing mice or tumor bearing mice with Selumetinib or Trametinib in a pulsatile/intermittent or continuous way, then analyzed T cell phenotype changes, tumor progression, and survival.
Results
Ex vivo T cell study showed that pulsatile treatment of MEK inhibitor, Selumetinib and Trametinib, showed highly increased CTLA4 expression and mild increase of PD1 in CD8+ T cells and CD4+Foxp3- T cells, compared to continuously treated group which is a standard regimen. This result was confirmed in intermittently treated KrasG12D/+; p53-/- transplantable mouse model and GEMM as well. Moreover, CD8+ T cells from pulsatile group showed increase of Ki67 and 4-1BB expression, suggesting that CD8+ T cells are more activated in pulsatile group. In vivo tumor study using Kras mutant lung cancer animal model showed intermittent treatment suppressed tumor growth better than continuous treatment. Better response, more activated phenotype of CD8+ T cells including increased CTLA4 expression in intermittent group lead us to test combination of intermittent MEK inhibitor treatment with anti-CTLA4 antibody to maximize anti-tumor T cell activity and it is currently under investigation.
Conclusions
In summary, we found that pulsatile/intermittent treatment with MEK inhibitor showed better response and more activated phenotype of T cells including increased CTLA4 expression compared to continuous treatment. This study suggests that optimized intermittent schedule of MEK inhibitor treatment is essential to maximize T cell mediated anti-tumor activity in combination with anti-CTLA4 therapy and this will benefit Kras mutant lung cancer patients.
P266 Significant enhancement of expanded natural killer cells against GD2 pediatric solid tumors (ST) in combination with ALT-803 (IL-15 superagonist) and dinutuximab
Yaya Chu1, Nang Kham Su1, Jeremy Rosenblum1, Hing C. Wong2, Dean A. Lee3, Mitchell S Cairo1
1New York Medical College, Valhalla, NY, USA; 2Altor Bioscience, Miramar, FL, USA; 3Nationwide Children’s Hospital, Columbus, OH, USA
Correspondence: Yaya Chu (yaya_chu@nymc.edu)
Background
Children with recurrent and/or metastatic osteosarcoma (OS), neuroblastoma (NB) and glioblastoma (GBM) have a dismal even-free survival (EFS) (<25%). Dinutuximab is an anti-GD2 monoclonal antibody that has significantly increased EFS in children with GD2+ neuroblastoma [1]. ALT-803 is a superagonist of an IL-15 variant bound to an IL-15RαSu-Fc fusion with enhanced biological activity [2]. Our group has successfully expanded peripheral blood Natural Killer cells (exPBNK) with irradiated feeder cells [3].
In this research, we aim to determine if the combination of ALT-803 and dinutuximab significantly enhances exPBNK cell in vitro cytotoxicity against GD2+ OS, NB and GBM.
Methods
PBMCs were expanded with lethally irradiated K562-mbIL21-41BBL cells [4]. ExPBNK cells were isolated using Miltenyi NK cell isolation kits as we previously described.3 ALT-803 was generously provided by Altor BioScience Corporation. NK proliferation, NK receptors expression and cytotoxicity were assessed as we previously described [3]. Dinutuximab (generously provided by United Therapeutics) was used for antibody-dependent cellular cytotoxicity (ADCC) assays. IFN-γ and perforin levels were evaluated by ELISA assays. GD2+ OS, NB and GBM cell lines were used as target cells.
Results
ALT-803 significantly promoted exPBNK in-vitro proliferation by increasing the phosphorylation of Akt, Stat3/5 and p38 MAPK. ALT-803 increased NK activating receptors expression: NKG2D, NKp30, NKp44, and NKp46.
ALT-803 significantly enhanced exPBNK mediated ADCC with dinutuximab in a E:T dependent manner (p<0.001) against OS, NB and GBM cells (Fig. 1). ALT-803 significantly enhanced IFN-γ (p<0.001) and perforin (p<0.001) release from exPBNK when it was combined with dinutuximab against OS, NB and GBM cells compared to exPBNK, ALT-803+exPBNK, or dinutuximab+exPBNK.
Conclusions
ALT-803 significantly enhanced exPBNK ADCC and IFN-γ and perforin release with dinutuximab against GD2+ OS, NB and GBM cells. In vivo studies using NOD/SCID human solid tumor xenografts are under investigation.
References
1. Yu AL, Gilman AL, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. The New England journal of medicine 2010; 363:1324-34.
2. Xu W, Jones M, et al. Efficacy and mechanism-of-action of a novel superagonist interleukin-15: interleukin-15 receptor alphaSu/Fc fusion complex in syngeneic murine models of multiple myeloma. Cancer Res 2013; 73:3075-86.
3. Chu Y, Hochberg J, et al. Targeting CD20+ Aggressive B-cell Non-Hodgkin Lymphoma by Anti-CD20 CAR mRNA-Modified Expanded Natural Killer Cells In Vitro and in NSG Mice. Cancer Immunol Res 2015; 3:333-44.
4. Denman CJ, Senyukov VV, et al. Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. PLoS One 2012; 7:e3026.

See text for description
P267 Withdrawn
P268 Targeting the tumor microenvironment with first-in-class Semaphorin4D MAb for combination immunotherapy
Elizabeth Evans1, Holm Bussler1, Crystal Mallow1, Christine Reilly1, Sebold Torno1, Maria Scrivens1, Cathie Foster1, Alan Howell1, Leslie Balch1, John E. Leonard1, Terrence L. Fisher1, David Jenkins2, Clint Allen3, Paul Clavijo3, Siwen Hu-Lieskovan4, Antoni Ribas5, Ernest Smith1, Maurice Zauderer1
1Vaccinex, Rochester, NY, USA; 2Tesaro, Waltham, MA, USA; 3NIH/NIDCD, Bethesda, MD, USA; 4UCLA, Los Angeles, CA, USA; 5Vaccinex, Los Angeles, CA, USA
Correspondence: Elizabeth Evans (eevans@vaccinex.com)
Background
Mechanistic findings in preclinical studies demonstrate that antibody blockade of Semaphorin 4D (SEMA4D, CD100) reduces expansion of MDSC and shifts the balance of immune cells within the TME to facilitate tumor rejection. Efficacy is further enhanced when combined with various immunotherapies.
Methods
Anti-SEMA4D antibodies were evaluated in combination with other immunotherapies in preclinical models. Anti-tumor activity and immune response was characterized by immunohistochemistry, flow cytometry, functional assays, and cytokine, chemokine and gene expression analysis. These data support clinical combination trials of VX15/2503, a humanized IgG4 antibody targeting SEMA4D, with immune checkpoint inhibition.
Results
SEMA4D restricts migration of monocytes and promotes expansion of suppressive myeloid cells in vitro. Strong expression of SEMA4D at the invasive margins of growing tumors in vivo restricts the infiltration and modulates polarization of leukocytes in the TME. Antibody blockade of SEMA4D in preclinical models facilitated recruitment of activated DCs and T lymphocytes with concurrent reduction in M2 macrophage and Treg within TME [1]. MDSCs were significantly reduced in tumor and blood following treatment and new data characterizing MDSC function will be described. This significant shift in the immune contexture is associated with durable tumor rejection and immunologic memory in murine colon, breast, HNSCC, and melanoma models. New translational data characterizing expression of SEMA4D and its receptors in human tumors will be shown. Importantly, anti-SEMA4D treatment can further enhance activity of immunotherapies and chemotherapy. For example, combinations with immune checkpoint inhibitor anti-LAG3 or anti-CTLA-4 cause complete tumor regression in 90% or 100% of mice, as compared to ~20% with monotherapy (p<0.01). New preclinical data include synergistic activity of combinations of anti-SEMA4D with anti-LAG3 and additional studies combining with epigenetic modulators, including treatment of established tumors.
Conclusions
SEMA4D blockade represents a novel mechanism to promote functional immune infiltration into the tumor and enhance immunotherapy. Treatment with VX15/2503 was well tolerated in a Phase I trial in patients with advanced refractory solid tumors [2]. Plans for several clinical trials will be presented, including a Phase 1b/2 of combination therapy with avelumab in immunotherapy naïve NSCLC, and combination with anti-PD-1 or Ipilimumab in various indications. A neoadjuvant trial of VX15/2503 with anti-PD-1 in patients with metastatic colorectal and pancreatic cancers will be described, as well as introduction of a Phase 1/2 trial of VX15/2503 in pediatric and osteosarcoma patients.
Trial Registration
NCT01313065.
References
1. Evans, EE et al. Cancer Immunol Res. 2015 Jun;3(6):689-701.
2. Patnaik A et al. Clin Cancer Res. 2016 Feb 15;22(4):827-36.
P269 Epigenetic reprogramming of the tumor microenvironment increases tumor sensitivity to multivalent immunotherapy combinations with an IL-15 superagonist plus vaccine or immune checkpoint blockade
Kristin C. Hicks1, Karin M. Knudson1, Anthony S. Malamas1, Frank R. Jones2, Peter Ordentlich3, Shahrooz Rabizadeh4, Hing C. Wong5, James W. Hodge1, Jeffrey Schlom1, Sofia R. Gameiro1
1National Cancer Institute, National Institutes of Health, Bethesda, MD, USA; 2Etubics Corporation, Seattle, WA, USA; 3Syndax Pharmaceuticals, Inc., Waltham, MA, USA; 4NantCell, LLC, Culver City, CA, USA; 5Altor BioScience Corporation, Miramar, FL, USA
Background
The clinical promise of cancer immunotherapy relies on the immune system recognizing and eliminating tumor cells identified as non-self. However, solid malignancies evade host immune surveillance by multiple mechanisms, including epigenetic deregulation and promoting a tumor microenvironment (TME) that suppresses infiltration and function of immune effector cells. The TME hampers T and NK cell maturation, recruitment, and function, ultimately causing their functional exhaustion via numerous immunosuppressive pathways, including upregulation of immune checkpoints such as PD-L1. Epigenetic silencing of genes involved in antigen processing and tumor immune recognition has been associated with worse prognosis in a wide spectrum of malignancies. Hence, there is an unmet clinical need to develop effective therapeutic strategies that can reprogram the TME to restore tumor immune recognition and reverse immune evasion. We recently demonstrated that entinostat, a class I histone deacetylase (HDAC) inhibitor reverses carcinoma immune escape to T cell-mediated lysis.
Methods
We hypothesize that the immune-mediated tumor elimination promoted by the IL-15/IL-15Ra superagonist ALT803 in combination with PD-L1 checkpoint blockade or a therapeutic adenoviral vaccine targeting CEA (Ad-CEA) will be augmented by the epigenetic reprograming of the TME induced by entinostat.
Results
In preclinical studies, ALT803 has been shown to exhibit potent antitumor activity in multiple murine models of cancer through the expansion of NK and CD8+ T cells with high effector function. Here, we demonstrate that entinostat modulates the cell-surface phenotype of murine colon and breast carcinoma cells to become more amenable to immune-mediated elimination. In the MC38-CEA murine model of colon carcinoma, proper scheduling of entinostat administration significantly augmented the antitumor activity promoted by ALT803 plus Ad-CEA, resulting in increased survival. Further, in the 4T1 murine model of triple-negative breast cancer, the combination of entinostat with ALT803 and a monoclonal antibody (mAb) targeting PD-L1 significantly reduced primary tumor weight relative to ALT803 plus anti-PD-L1 therapy. Treatment with the triple therapy in the neoadjuvant setting resulted in significant reduction of the number of 4T1 tumor-forming cells in the lung, with over 85% of animals cured. Current studies aim to elucidate the immune mechanisms associated with the antitumor effects observed with these therapeutic interventions.
Conclusions
Overall, these studies examine the rationale for combining entinostat with multivalent immunotherapy combinations, including cytokines, mAbs targeting PD-L1, and therapeutic cancer vaccines.
P270 Simultaneous PD-1 blockade is detrimental to the anti-tumor effects mediated by the agonist OX40 antibody
Seema Gupta1, Rajeev Shrimali2, Shamim Ahmad3, Vivek Verma1, Peng Zeng1, Sudha Ananth1, Pankaj Gaur1, Rachel Gittelman4, Erik Yusko4, Catherine Sanders4, Harlan Robins4, Scott Hammond5, John Janik1, Mikayel Mkrtichyan3, Samir Khleif1
1Georgia Cancer Center, Augusta University, Augusta, GA, USA; 2Georgia Cancer Center, Augusta University, Dallas, TX, USA; 3Georgia Cancer Center, Augusta University, South San Francisco, CA, USA; 4Adaptive Biotechnologies, Seattle, WA, USA; 5MedImmune LLC, Gaithersburg, MD, USA
Correspondence: Samir Khleif (segupta@augusta.edu)
Background
The checkpoint inhibitor antibodies (Abs) have improved the anti-tumor response although in a limited number of patients [1]. The combination of these Abs has enhanced the efficacy but with increased adverse events [1]. Since immune-stimulatory agonist Abs like anti-OX40 significantly increase the immune response [2], their combination with the anti-PD-1 Abs is being tested in clinical trials [3] with at least one clinical trial demonstrating lack of benefit [4] and the rest are not yet reported. Moreover, the potential immune outcome of such a combination are currently lacking. Therefore, purpose of this study was to determine the therapeutic and immune outcome of combining anti-PD-1 with anti-OX40 in an immune-primed environment.
Methods
Effects of adding PD-1 blockade to anti-OX40/vaccine treatment on tumor growth and survival were evaluated in a TC-1 tumor mouse model using different treatment schedules and immune responses were assessed. In vitro mechanistic studies were carried out in pMel-1 CD8+ T-cells.
Results
Anti-OX40 treatment resulted in significant enhancement of antigen-specific CD8+ T-cells tumor-infiltration leading to strong anti-tumor response and prolonged survival of mice. Interestingly, we found that simultaneous addition of anti-PD-1 to anti-OX40 completely abrogated these effects. Despite an increase in IFNγ-producing E7-specifc CD8+ T-cells in the spleens of mice treated with the combination, these cells underwent significant apoptosis in both the periphery and the tumor. Consistent with increased apoptosis, immunoSequencing analysis of the tumor and spleen T-cell population showed reduction in T-cell fraction and clonality following the addition of anti-PD-1. We further showed that delay in anti-PD-1 addition resulted in no significant change in T-cell apoptosis, however, it did not add to the anti-tumor response of the anti-OX40 treatment.
Conclusions
These results indicate that anti-PD-1 added at the initiation of therapy exhibits a detrimental effect on the positive outcome of anti-OX40. This may provide an important insight into why some of the immune combination clinical trials are not producing the intended outcome, demonstrating the need to rigorously test the combination partners and sequencing before employing them in the clinic.
References
1. Page DB, et al. Immune modulation in cancer with antibodies. Annu Rev Med. 2014;65:185-202.
2. Linch SN et al: OX40 Agonists and Combination Immunotherapy: Putting the Pedal to the Metal. Front Oncol. 2015;5:34.
3. “ClinicalTrials.gov.”April 15, 2017 from https://clinicaltrials.gov/ct2/results?term=ox40&Search=Search">https://clinicaltrials.gov/ct2/results?term=ox40&Search=Search.
4. Infante, et al. A phase Ib dose escalation study of the OX40 agonist MOXR0916 and the PD-L1 inhibitor atezolizumab in patients with advanced solid tumors. Presentation in ASCO Annual Meeting, 2016.
P271 Phase 1b/2 dose-escalation study of ARRY-382, an oral inhibitor of colony-stimulating factor-1 receptor (CSF1R), in combination with pembrolizumab for treatment of patients with advanced solid tumors
Wael A. Harb1, Melissa L. Johnson2, Jonathan W. Goldman3, Amy Weise4, Justin A. Call5, Arkadiusz Z. Dudek6, Rene Gonzalez7, C. Lance Cowey8, Sybil Zildjian9, Kati Maharry9, Ashwin Gollerkeri9, Justin F. Gainor10
1Horizon Oncology Research, Inc., Lafayette, IN, USA; 2Sarah Cannon Research Institute, Nashville, TN, USA; 3UCLA Medical Center, Santa Monica, CA, USA; 4Karmanos Cancer Institute, Detroit, MI, USA; 5Utah Cancer Specialists, Salt Lake City, UT, USA; 6Regions Cancer Care Center, St. Paul, MN, USA; 7University of Colorado Cancer Center, Aurora, CO, USA; 8Baylor Health Care System, Dallas, TX, USA; 9Array BioPharma Inc., Boulder, CO, USA; 10Massachusetts General Hospital, Boston, MA, USA
Correspondence: Wael A. Harb (drharb@me.com)
Background
CSF1 regulates tumor-associated macrophages and myeloid-derived suppressor cells, which are critical tumor microenvironment modulators of immune response. Combining a programmed death 1 (PD-1) inhibitor with a CSF1R inhibitor in preclinical models shows enhanced antitumor activity. ARRY-382 is a highly selective oral inhibitor of CSF1R. Studies of ARRY-382 monotherapy identified the maximum tolerated dose (MTD) as 400 mg once daily (QD), with biologic activity at doses ≥200 mg QD. This 3-part, phase 1b/2 study evaluates ARRY-382 in combination with pembrolizumab, a humanized monoclonal antibody targeting PD-1.
Methods
Phase 1b (part A) determined the MTD and recommended phase 2 dose (RP2D) of ARRY-382 plus pembrolizumab in patients with selected advanced solid tumors. Patients were enrolled in 3 successive cohorts of ARRY-382 at doses of 200, 400, or 300 mg QD plus pembrolizumab 2 mg/kg every 3 weeks (Q3W). The primary endpoint was the incidence of dose-limiting toxicities (DLTs). Secondary endpoints included safety, pharmacokinetics of ARRY-382, and objective response rate.
Results
Twenty patients enrolled in part A; 19 were treated: 200 mg QD, n=6; 400 mg QD, n=7; 300 mg QD, n=6. Median age was 59 years; tumor types were pancreatic (n=6), colorectal (n=5), ovarian (n=3), gastric and melanoma (n=2 each), and triple-negative breast (n=1). The mean number of prior therapies was 2.9 (range, 1–5). DLTs of grade 3 increased aspartate aminotransferase (AST)/alanine aminotransferase/bilirubin and grade 3 increased creatine phosphokinase (n=1 each, 400-mg dose) and grade 3 pancreatitis (n=1, 300-mg dose) were observed. The most common (>2 patients across doses) grade 3/4 adverse events (AEs) by dose cohort (200, 300, and 400 mg) were increased AST (1, 1, and 2 patients), increased lipase (0, 3, 0), and rash (1, 1, 1), respectively. One patient in the 200-mg cohort discontinued treatment because of an AE of pneumonitis in cycle 5. ARRY-382 pharmacokinetics was unaffected by pembrolizumab. Preliminary efficacy data will be presented.
Conclusions
ARRY-382 plus pembrolizumab has a manageable safety profile. The RP2D of ARRY-382 plus pembrolizumab 2 mg/kg Q3W is 300 mg QD. This combination is currently being evaluated in the study in patients with melanoma and non–small-cell lung cancer.
Trial Registration
ClinicalTrials.gov: NCT02880371.
P272 The anti-tumor effect of radiation therapy is enhanced with the addition of TTI-621 (SIRPαFc), an immune checkpoint inhibitor blocking the CD47 “do not eat” signal
Lei Cui, Hui Chen, Alison O'Connor, Debbie Jin, Jeffrey Winston, Robert Uger, Lisa Johnson
Therapeutics Inc., Mississauga, ON, Canada
Correspondence: Lisa Johnson (lisa@trilliumtherapeutics.com)
Background
CD47 is an immune checkpoint that binds to signal regulatory protein alpha (SIRPα) and delivers a “do not eat” signal to suppress macrophage phagocytosis. Tumor cells frequently overexpress CD47 to evade macrophage-mediated destruction. TTI-621 (SIRPαFc) is an immune checkpoint inhibitor consisting of the CD47 binding domain of human SIRPα linked to the Fc region of human IgG1 designed to both: 1) block the CD47 “do not eat” signal, and 2) engage macrophage Fcγ receptors with IgG1 Fc to enhance phagocytosis and antitumor activity.
Radiation therapy (RT), a primary mode of cancer treatment, induces immunogenic cell death, and is associated with release of tumor antigens, induction of pro-inflammatory cytokines, and enhanced migration and infiltration of immune cells, including macrophages, to tumor sites. Thus, the combination of RT and TTI-621 may improve the therapeutic effect over either treatment modality alone. Herein, we report the efficacy of the combination of RT and the CD47-blocking agent TTI-621 in xenograft tumor models.
Methods
The in vivo efficacy of RT, TTI-621, and RT+TTI-621 was evaluated in B cell lymphoma (SU-DHL-6) and solid tumor xenografts, including the radio-insensitive A549 lung adenocarcinoma. TTI-621 (10 mg/kg) or vehicle were administered intratumorally 30 min prior to RT, 3 times per week for 4 weeks. Tumors were locally irradiated using an image-guided small animal irradiator (225 kVp, 13 mA) at a dose of 4-6 Gy for 3 fractions. Tumor volumes were monitored using standard caliper measurement. Systemic toxicity of the treatments was evaluated by body weight change. Tumor-associated macrophages were quantitatively assessed using flow cytometry and immunohistochemistry.
Results
RT+TTI-621 had a more profound effect on tumor control in both lymphoma and solid tumor models than RT alone. In SU-DHL-6, 88% (7/8) of mice treated with both TTI-621 and radiation were tumor-free at the end of the study whereas none were tumor-free when treated with RT or TTI-621 alone, although there was tumor growth delay with each individual therapy compared to vehicle. RT+TTI-621 led to an increased infiltration of macrophages at the tumor sites in SU-DHL-6. Significant tumor control was also observed in A549 tumor bearing mice treated with RT+TTI-621 compared to either treatment alone. No toxicity was observed for any of the treatments.
Conclusions
The current study demonstrates the combination of TTI-621 and radiation therapy is superior to either treatment alone and provides supportive evidence for dual modality therapy in a variety of tumors.
P273 Pembrolizumab and afatinib for recurrent or metastatic head and neck squamous cell carcinoma
Hsiang-Fong Kao, Huai-Cheng Huang, Bin-Chi Liao, Ruey-Long Hong
National Taiwan University Hospital, Taipei, Taiwan
Correspondence: Hsiang-Fong Kao (hfkao.tw@gmail.com); Ruey-Long Hong
Background
Head and neck squamous cell carcinoma (HNSCC) is an important malignancy in Taiwan. Anti-PD-1, including nivolumab or pembrolizumab, have shown the efficacies against head and neck squamous cell carcinoma. Afatinib, an irreversible EGFR tyrosine kinase inhibitor (TKI), had showed its activity against HNSCC. The role of afatinib for cancer immunotherapy have not been explored. In animal model, afatinib can suppress the carcinogenesis by inhibiting the function of macrophage [1]. EGFR targeted therapy can increase the MHC expression, enhance dendritic cell function, and increase the infiltrating T cell in the tumor [2]. In syngeneic mice cancer models, EGFR TKI can suppress the glycosylation of PD-L1 and sensitize the mice to anti-PD-1 therapy [3]. We hypothesized that adding afatinib with pembrolizumab may improve the treatment efficacy for patients with recurrent or metastatic HNSCC.
Methods
Patients with locally advanced or metastatic HNSCC were retrospectively reviewed in a university hospital in Taiwan. For patients taking pembrolizumab, the combination with afatinib will be discussed between the physician and the patient. For patients taking pembrolizumab and afatinib (P+A), the medical records were reviewed.
Results
From Nov 2016 to Jul 2017, there were 35 patients taking P+A. The median age was 59 years, and 32/35 pts were men. Oral cavity cancer is the most common type of HNSCC in the cohort (29/35 pts). The treatment were: pembrolizumab 200mg: 30pts; 2mg/kg: 5pts; afatinib 40mg QD: 34pts; 30mg QD: 1 pt. The maximal cycles of pembrolizumab per patient is four cycles. Sixteen patients took P+A as first line therapy. Until Jul 30, 2017, 29 pts had evaluable outcomes. The ORR (CR+PR) is 48.2% (14/29), DCR (CR+PR+SD) was 69.0% (20/29), and the median PFS: 184 days (32-332). The common toxicities were: diarrhea 40%; skin rash 37%; mucositis 29%; hand-foot-skin reaction 23%; weight loss 17%; and elevated AST/ALT 9%. Grade >= 3 toxicities happened in 8.6% patients. There was no treatment related pneumonitis in the cohort.
Conclusions
The addition of afatinib with pembrolizumab showed good efficacy (ORR: 48.2%, DCR: 69.0%, PFS: 6.0 months) and tolerable toxicities. Further confirmatory prospective trial is indicated.
References
1. Lanaya, Hanane, et al. EGFR has a tumor-promoting role in liver macrophages during hepatocellular carcinoma formation. Nature cell biology. 2014; 16.10:972.
2. Concha-Benavente, Fernando, and Robert L. Ferris. Oncogenic growth factor signaling mediating tumor escape from cellular immunity. Current Opinion in Immunology. 2017;45:52-59.
3. Li, Chia-Wei, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nature Communication. 2016;7:12632.
P274 Combination of NKTR-214 and radiotherapy (RT) to reverse anergy and expand tumor-specific CD8 T Cells
Joshua Walker1, Melissa Kasiewicz2, Michael McNamara2, Ian Hilgart-Martiszus2, Ute Hock3, Deborah Charych3, William Redmond2
1Oregon Health & Science University, Portland, OR, USA; 2Providence Portland Medical Center, Portland, OR, USA; 3Nektar Therapeutics, San Francisco, CA, USA
Correspondence: William Redmond (melissa.kasiewicz@providence.org)
Background
We investigated therapeutic and mechanistic synergy between single-dose RT and systemic administration of NKTR-214.The effects of NKTR-214 with and without RT on anergic, tumor-specific CD8 T cells were characterized. NKTR-214 is a CD122-biased cytokine agonist conjugated with releasable chains of polyethylene glycol. NKTR-214 provides sustained signaling through the IL2 receptor pathway (IL2Rβγ) to preferentially activate and expand effector CD8 T and NK cells over regulatory T cells. Preclinical models demonstrated NKTR-214 preferentially expands effector CD8 T and NK cells in the tumor resulting in marked tumor growth suppression as a single-agent and in combination with checkpoint inhibitors. RT can induce antigen-release and epitope spreading, while NKTR-214 activates and expands antigen-specific effector populations. We hypothesized the combination of systemic NKTR-214 and RT would generate better therapeutic responses than either treatment alone. A phase I/II trial is in progress to evaluate NKTR-214 safety and efficacy in an outpatient setting as monotherapy and in combination with nivolumab.
Methods
NKTR-214 was dosed 0.8 mg/kg alone or together with high-dose RT (20 Gy x 1) in multiple murine models, including an established CD8 T cell anergy model using Nur77-GFP reporter CD8 T cells. Activation markers on CD4, CD8 and NK cells in blood, lymph and tumor were evaluated by flow cytometry and gene expression (mRNA) profiling of the irradiated and non-irradiated tumors. In addition, immunohistochemistry was utilized to visualize immune infiltration.
Results
Consistent with prior observations, NKTR-214 induced activation marker expression by CD4, CD8 T and NK cells in the blood, lymph nodes and tumor. The combination of RT and NKTR-214 resulted in unique effects including increased absolute lymphocyte counts, increased expression of CD8 activation markers (CD25, PD1) in the blood and tumor, increased intratumoral NK cells, and a significant survival benefit over monotherapy. Evaluation of tumor infiltrating lymphocytes (TIL) indicates combination therapy reverses tumor-associated T cell anergy and results in a higher frequency of recently activated CD8 TIL.
Conclusions
The combination of NKTR-214 and radiotherapy is synergistic, provides significantly better anti-tumor responses and reverses T cell anergy.
P275 Harnessing the innate and adaptive immune system to eradicate treated and distant untreted solid tumors
Saul Kivimae, Werner Rubas, Rhoneil Pena, Marlene Hennessy, Yolanda Kirksey, Wildaliz Nieves, Myong Lee, Clive Law, Kavitha Bhasi, Phi Quach, Janet Cetz, John L. Langowski, Christie Fanton, Jode Zandro Aquino, Zhongxu Ren, Haiying Cai, BoLiang Deng, Wen Zhang, Neel K. Anand, Jennifer Riggs-Sauthier, Steve Doberstein, Jonathan Zalevsky
Nektar Therapeutics, San Francisco, CA, USA
Correspondence: Saul Kivimae (skivimae@nektar.com); Jonathan Zalevsky
Background
NKTR-262 is a novel therapeutic, which delivers sustained intratumoral engagement of the TLR 7/8 pathway, promoting an immune stimulatory environment and tumor antigen release. When NKTR-262 is administered in combination with NKTR-214, a CD122-biased cytokine agonist currently in clinical trials as a monotherapy and in combination with nivolumab, the combined effect of innate immune stimulation and enhanced antigen presentation with sustained T cell activation leads to systemic tumor immunity.
Methods
Balb/c mice were implanted s.c. with bilateral CT26 colon carcinoma tumors. Once established, one tumor was treated with a single peritumoral dose of NKTR-262, while NKTR-214 was administered i.v. on q9dx3 schedule. Regression of treated tumors and the abscopal effect in contralateral tumors was assessed by tumor size measurements. Immune cell activation in both tumors was assessed by flow cytometry. Cytokine induction was measured in plasma, blood cells and treated tumors by MSD and qRT-PCR.
Results
Combination treatment with NKTR-262 and NKTR-214 eliminated both tumors in up to ≥95% of mice. Single agent NKTR-262 or NKTR-214 treatment led to complete responses in ≤30% of mice. Combination treatment of NKTR-262 plus NKTR-214 induced a two-step immune response in treated and untreated tumors. At early timepoints, accumulation of activated neutrophils correlated with tumor cell death and dendritic cell activation. The innate response was followed by increased CD8+ T cells and a reduction of immunorepressive cells. Single agent treatment showed only a subset of the cellular changes observed in the combination.
Conclusions
We present a designed combination therapy that mimics a natural immune response by activating a broad immune cell network. Combining NKTR-262 and NKTR-214 engages the entire immune activation cascade required for systemic tumor clearance from local tumor antigen production to a sustained systemic T cell response. Unlike treatments that stimulate downstream components of select immune pathways without eliciting systemic tumor immunity, a comprehensive anti-tumor immune activation by coordinated engagement of innate and adaptive immune cells may increase the success of immune therapy for patients
P276 Immunoswitch particles target activation of anti-tumor CD8+ T cells to inhibit tumor growth
Alyssa Kosmides, John-William Sidhom, Andrew Fraser, Catherine Bessell, Jonathan Schneck
Johns Hopkins School of Medicine, Baltimore, MD, USA
Correspondence: Alyssa Kosmides (akosmides@gmail.com)
Background
Recent advances in cancer immunotherapy have led to the advent of treatments that boost an anti-tumor response, such as checkpoint blockade and co-stimulatory molecules. However, these approaches are effective for only a subset of patients and target non-specific molecules, thus the large doses required result in off-target toxicities. Here, we have developed a nanoparticle that synergizes checkpoint blockade and T cell costimulation on a single injectable therapeutic. Nanoparticles are conjugated with antibodies against 4-1BB, expressed by T cells, and anti-PD-L1, expressed by tumor cells and target these pathways while also physically linking the effector and target cell to increase efficacy. These dual targeting particles, termed “immunoswitch particles”, localize treatment to the tumor site and are effective at 10-100X lower doses than systemically injected drug and thus have the potential to reduce cost and off-target toxicities.
Methods
Immunoswitch particles were synthesized by conjugating 80nm iron-dextran nanoparticles with agonistic anti-4-1BB and antagonistic anti-PD-L1 antibodies. In vitro efficacy was assessed by co-culturing PD-L1hi tumor cells with 4-1BB/PD-1hi CD8+ T cells and measuring cytotoxicity, cytokine secretion, and effector-target cell conjugation. In vivo efficacy was measured by comparing intratumoral immunoswitch and soluble antibody treatment in multiple murine tumor models.
Results
Immunoswitch particles were demonstrated to increase CD8+ T cell cytokine secretion when co-incubated with T cells and cognate B16 tumor cells compared to an equivalent amount of soluble antibody in vitro. Immunoswitch particles also increased effector-target cell conjugation both in vitro (Fig. 1) and in vivo. In vivo, immunoswitch treatment, but not an equivalent amount of soluble antibody, delayed/reversed tumor growth in both a B16-SIY and MC38-OVA tumor treatment model in the absence of adoptively transferred cells (Fig. 2). This was caused by a change in biodistribution, altered CD8+ T cell repertoire, and an increase in cytokine secretion by tumor-specific CD8+ T cells (Fig. 3A). Despite local intra-tumoral treatment, new data shows that MC38-OVA cured mice have systemic immunity against the OVA antigen. These mice have a systemically circulating repertoire of tumor-specific cells, demonstrated by increased in vivo killing of intravenously injected OVA-expressing cells (Fig. 3B) and protection against a re-inoculation of MC38-OVA.
Conclusions
Here, we have demonstrated the efficacy of a new dual-targeting immunoswitch particle for cancer immunotherapy. Immunoswitch particles synergize checkpoint blockade with co-stimulation by confining the antibodies to a rigid nanoparticle surface. This new approach can be extended to different disease models and new checkpoint or co-stimulatory target molecules.

. See text for description

See text for description
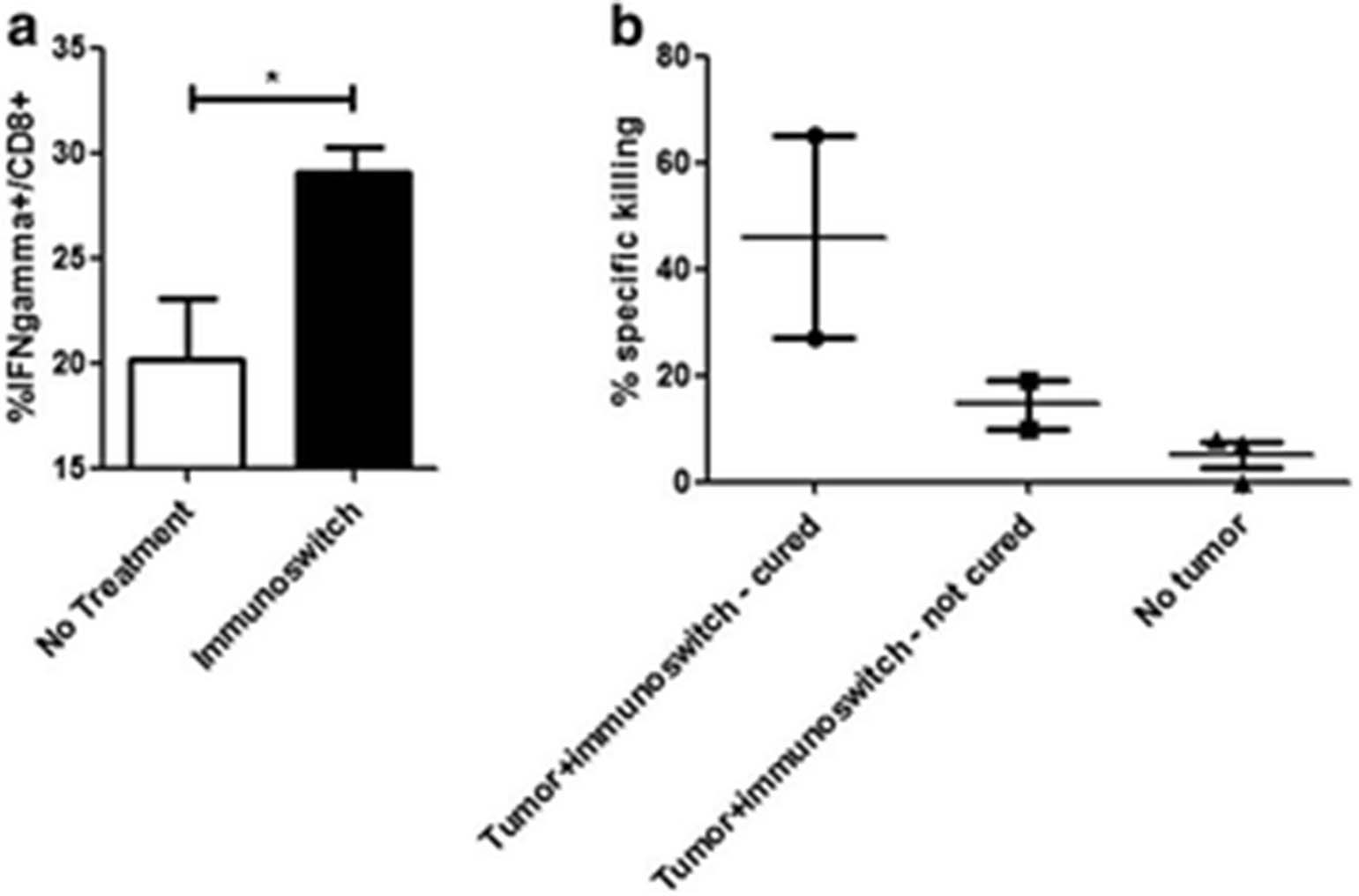
See text for description
P277 STING agonist treatment increases response to chemotherapy and immune checkpoint blockade therapy in a syngeneic murine model of high-grade serous ovarian cancer
Abdi Ghaffari1, Nichole Peterson1, Kasra Khalaj1, Andrew Robinson2, Julie-Ann Francis2, Madhuri Koti1
1Queen's University, Kingston, ON, Canada; 2Kingston Health Sciences Center, Kingston, ON, Canada
Correspondence: Madhuri Koti (kotim@queensu.ca)
Background
Background: High Grade Serous Carcinoma of the ovary is mostly diagnosed at late stages and primarily treated with surgery followed by platinum/taxane chemotherapy. Unfortunately, majority of the patients exhibit resistance to chemotherapy and ultimately succumb to the disease. We previously reported that tumours from patients with early recurrence show an immunosuppressed pre-existing tumour immune microenvironment with decreased expression of genes involved in Type I Interferon (IFN1) and T helper type 1 response [1, 2, 3]. We thus hypothesized that response to chemotherapy and overall survival of HGSC patients can be improved by stimulating the IFN1 response in the TME post chemotherapy.
Methods
In this pre-clinical study we tested the efficacy of a novel “Stimulator of Interferon Genes” agonist in immunocompetent mice implanted with ID8-TRP53-/- mouse ovarian cancer cells. Post treatment tumour immune cell profiles were measured using a combination of flow cytometry and CyTOF based profiling. Tumour immune transcriptomic alterations were measured using the NanoString mouse pan cancer immune profiling panel. Log-rank test based survival analysis was performed to determine significant differences in overall survival post treatment.
Results
Flow cytometry and NanoString based analysis of tumours collected at endpoint showed higher intra-tumoural PD-1+ and CD69+CD62L-, CD8+T cells, increased expression of IFN response, antigen presentation and MHCII, genes, respectively, in tumours from STING agonist treated mice compared to those from vehicle treated mice. In addition to significantly decreased ascites accumulation and decreased tumour burden, survival of mice treated with a combination of Carboplatin + STING agonist + anti-PD-1 therapy was significantly longer compared to Carboplatin + STING agonist, Carboplatin only, STING only and vehicle treated mice.
Conclusions
Findings from this study are foundational to future clinical trials aimed at combinatorial immunomodulatory therapies to improve chemotherapy response and overall survival of HGSC patients.
References
1. Koti M, SiuA, Clément I, Bidarimath M, Turashvili G, EdwardsA, et al. A distinct pre-existing inflammatory tumour microenvironment is associated with chemotherapy resistance in high-grade serous epithelial ovarian cancer. Br J Cancer [Internet]. 2015;112(7):1215–22.
2. Au KK, Le Page C, Ren R, Meunier L, Clément I, Tryshkin K, et al. STAT1-associated intratumoural T H 1 immunity predicts chemotherapy resistance in high-grade serous ovarian cancer. J Pathol Clin Res. 2016.
3. Au KK, Peterson N, Truesdell P, Reid-schachter G, Khalaj K, Ren R, et al. Gynecologic Oncology CXCL10 alters the tumour immune microenvironment and disease progression in a syngeneic murine model of high-grade serous ovarian cancer. Gynecol Oncol [Internet]. Elsevier Inc.; 2017;4–13.P278.
P278 A Rational Combination of Standard of Care and Immunotherapy Increases Survival Against Glioblastoma
Erik Ladomersky, Derek Wainwright
Northwestern University, Chicago, IL, USA
Correspondence: Derek Wainwright (erik.ladomersky@northwestern.edu)
Background
Glioblastoma (GBM) is a fatal primary brain tumor that, even after maximum surgical resection, radiotherapy (RT) and chemotherapy, is associated with a median overall survival (OS) of 14.6 months. Combinatorial treatment approaches that simultaneously address tumor growth, as well as the immunosuppressive microenvironment, may prove to be more effective than current clinical strategies. This work aims to determine the therapeutic efficacy of combining the novel, pharmaceutical-grade IDO1 enzyme inhibitor, BGB-5777, with PD-1 blockade and/or whole brain radiation, in a syngeneic, immunocompetent mouse glioblastoma model.
Methods
Mice were intracranially-engrafted with 2x105 GL261 or CT-2A murine GBM cells, then treated, beginning day 14 post-tumor cell implantation, with 500μg loading dose IgG control mAb followed by three 200μg doses, administered every third day (n=7), or 2 Gy WBRT for 5 days, PD-1 mAb (J43) administered the same as IgG control mAb, and 100mg/kg BGB-5777 BID for 4 weeks (n=9).
Results
Mice engrafted with the GL261 tumors and treated with the combination therapy showed a significant increase in overall survival (53 days) compared to IgG treated mice (25 days). This therapy regimen also significantly increased durable survival (>120 days) (P<0.0001) in mice ic. unmodified GL261 or CT-2A cells. (Fig. 1A) Paradoxically, mice with GBM cells overexpressing mouse IDO1 cDNA and treated with triple therapy showed a similar proportion of durable survivors (40%) vs. mice with similar tumors treated with IgG control (P<0.0001). (Fig. 1B) Interestingly, the survival effects seen in engrafted with unmodified GL261 tumors correlated to a significant decrease in GBM-infiltrating Treg levels (P<0.01), however this same effect was not seen in mice with GL261 mIDO1 O/E tumors. (Fig. 1C) These results suggest that effectiveness of triple therapy is independent of tumor IDO1 expression level and Treg accumulation. Results from the analysis of WT and IDO1-/- mice with intracranial tumor indicate that in the presence of non-tumor IDO1, IDO1 inhibition is necessary to confer survival benefit (P<0.01). (Fig. 2)
Conclusions
Ultimately, the data suggest that using radiation to induce potential immunogenicity and/or inflammation in GBM, while co-inhibiting immunosuppression, is a rational and potentially clinically-beneficial pursuit.

. See text for description

See text for description
P279 An oral small molecule combination therapy targeting PD-L1, VISTA and Tim-3 immune inhibitory checkpoints exhibits enhanced anti-tumor efficacy in pre-clinical models of cancer
Adam Lazorchak1, Troy Patterson1, Yueyun Ding1, Pottayil G. Sasikumar2, Naremaddepalli S. Sudarshan2, Nagaraj M. Gowda2, Raghuveer K. Ramachandra2, Dodheri S. Samiulla2, Mohammed Rafi2, Nagesh Gowda2, Sreenivas Adurthi2, Jiju Mani2, Rashmi Nair2, Murali Ramachandra2, David Tuck1, Timothy Wyant1
1Curis, Inc., Lexington, MA, USA; 2Aurigene Discovery Technologies Limited, Bengaluru, India
Correspondence:Adam Lazorchak (adam.lazorchak@icloud.com)
Background
Antibodies targeting immune inhibitory checkpoint pathways have transformed cancer therapy. However, the majority of patients fail to respond to therapies targeting single immune checkpoints due, in part, to the compensatory activity of alternative immune suppressive pathways. A therapeutic strategy that targets multiple immune checkpoints may significantly improve the anti-tumor response rate, however this strategy may also increase the risk of severe treatment related immune mediated side effects. Orally bioavailable, small molecule immune checkpoint antagonists with short in vivo half-lives are ideal candidates for combination cancer immune therapy due to a short drug washout time which may improve safety and flexibility in scheduling which may increase efficacy.
Methods
The anti-tumor efficacy of orally administered CA-170 (a PD-L1/2 and VISTA/PD-1H antagonist), CA-327 (a PD-L1/2 and Tim-3 antagonist) or the combination of CA-170 plus CA-327 was studied in the mouse syngeneic CT26 colon carcinoma tumor model. Compound dosing was initiated within 1-3 days of tumor implantation or in mice bearing established tumors (90-170 mm3). Populations of immune cells were measured by flow cytometric analysis in tumor or blood. Ex vivo functional assays were performed on tumor derived CD8+ T cells to evaluate effector cell function and gene expression profiling was performed on tumors.
Results
Significant anti-tumor efficacy was observed in CT26 tumor bearing mice treated with either CA-170 or CA-327 compared to vehicle treated animals. Flow cytometric analysis of the tumors taken from these mice revealed that both compounds significantly increased the total number of CD45+ immune cells within the tumor relative to vehicle treated animals. Tumor growth inhibition positively correlated with increased numbers of activated CD8+ T cells and IFN-γ+ CD8+ effector T cells within the tumor. A significant increase in anti-tumor efficacy was observed when CA-170 and CA-327 were administered together as an oral combination therapy, when compared to animals treated with either compound alone.
Conclusions
These non-clinical data demonstrate a proof-of-concept showing that a combination therapy consisting of oral small molecules that antagonize the PD-L1/2, VISTA and Tim-3 pathways significantly enhance anti-tumor efficacy. CA-170 is currently undergoing Phase I clinical testing and CA-327 is in pre-clinical development. The results of our study provide a strong rational for the continued development of combination therapies using small molecule immune checkpoint antagonists for the treatment of advanced cancers.
P280 Combination lymphoma immunotherapy using intratumoral virus-like particles containing CpG TLR9 agonist combined with checkpoint blockade
Caitlin Lemke-Miltner1, Sue Blackwell1, Arthur Krieg2, George Weiner1
1University of Iowa, Iowa City, IA, USA; 2Checkmate Pharmaceuticals, Cambridge, MA, USA
Correspondence: Caitlin Lemke-Miltner (caitlin-lemke@uiowa.edu)
Background
Checkpoint blockade of immune inhibitory pathways is an exciting approach to cancer immunotherapy, however there remains considerable room for improvement through the use of unique immunotherapeutic combinations. We explored the combination of intratumoral injection (in situ immunization) with a virus-like particle (VLP) combined with anti-PD1 antibody therapy. The VLP is designated CMP-001 and is composed of an immunostimulatory CpG oligodeoxynucleotide TLR9 agonist encapsulated in the Qb bacteriophage protein. CMP-001 was previously evaluated clinically in over 700 subjects in non-cancer trials where it stimulated a strong Th1 cytokine response. The current studies were based on the hypothesis that intratumoral CMP-001 can augment the development of a tumor-specific T cell response that can be maintained by anti-PD-1 therapy.
Methods
The impact of CMP-001 with and without anti-PD1 was assessed in both human (in vitro) and mouse (in vitro and in vivo) systems. In vivo therapy studies in mice included the B16F0 melanoma and A20 B cell lymphoma models. CMP-001, or saline control, was delivered intratumorally starting one week after tumor challenge. Anti-PD-1, or isotype control, was administered systemically starting one week after tumor challenge. Tumor growth and survival was followed. In some experiments, tumor inoculation was done bilaterally while a unilateral tumor was injected with CMP-001 to allow for assessment of response of both the treated and untreated tumor.
Results
In vitro, CMP-001 stimulated IFNα production, as well as other pro-inflammatory cytokines, from human and mouse mononuclear cells (from the peripheral blood and spleen respectively). This was only seen when anti-Qβ antibody was present.
Intratumoral CMP-001 enhanced survival, and reduced tumor growth of treated tumors in the B16F0 model, and of both treated and untreated tumors in the A20 lymphoma model. Anti-PD-1 enhanced this effect in the A20 model. Depletion of T cells in mice eliminated the anti-tumor effect in both the treated and untreated tumors.
Conclusions
We conclude CMP-001 can induce a robust Th1 response in both murine and human systems in a manner that is dependent on opsonization of the VLP with anti-Qb antibody. The combination of intratumoral CMP-001 and systemic anti-PD-1 leads to development of a systemic anti-tumor T cell response in two murine tumor models and is a promising immunotherapy combination worthy of clinical evaluation. A Phase 1 clinical trial of the combination of anti-PD-1 and CMP-001 is underway in advanced melanoma. These results will be presented separately.
P281 Treatment with a VEGFR-2 antibody results in intra-tumor immune modulation and enhances antitumor efficacy of PD-L1 blockade in syngeneic murine tumor models
yanxia li1, David Schaer1, Marguerita O’Mahony1, Ivan Inigo1, Qi Li1, Nelusha Amaladas1, Erik Rasmussen1, Thompson Doman2, Jason Manro2, Mary Murphy1, Macrina Francisco1, Gerald Hall1, Michael Kalos1, Ruslan Novosiadly1, Bronislaw Pytowski1
1Eli Lilly and Company, New York, NY, USA; 2Eli Lilly and Company, Indianapolis, IN, USA
Correspondence: David Schaer (yanxia.li@lilly.com)
Background
The activation of VEGFR-2 by its primary cognate ligand VEGF has been principally implicated in tumor angiogenesis. However, emerging data has suggested that the VEGF/VEGFR-2 axis also mediates a suppressive effect on the anti-tumor immune response. Inhibition of this pathway has demonstrated the potential to facilitate T cell migration into the tumor and to reduce the direct immune inhibitory activity promoted by tumor endothelial cells. Clinical studies that combine anti-angiogenic antibodies to immune checkpoint inhibitors have shown promising results in a number of solid tumors. However, the mechanisms underlying immunomodulatory activity of agents that target the VEGF/VEGFR-2 axis remain incompletely understood.
Methods
Ramucirumab is an approved VEGFR-2 targeted antibody currently undergoing clinical trials in combination with PD-1/PD-L1 inhibitory antibodies. The emerging data from these early trials has prompted us to undertake non-clinical studies with potential to uncover mechanisms that mediate the enhancement of anti-tumor immune response by the blockade of VEGFR-2. Using murine EMT6 breast and MC38 colon carcinoma models we investigated the immunomodulatory effects of DC101, a surrogate antibody that blocks mouse VEGFR-2, and its ability to increase the anti-tumor efficacy of checkpoint inhibition. Intra-tumor immune-related changes were evaluated by flow cytometry, immunohistochemistry (IHC) and nCounter gene expression analysis (NanoString).
Results
Anti-VEGFR2 monotherapy resulted in increased T cell infiltration into the tumors. Combination of anti-VEGFR-2 with anti-PD-L1 resulted in greater anti-tumor efficacy compared to anti-PD-L1 monotherapy in both MC38 and EMT6 tumor models. Mice achieving complete tumor regressions after the combination treatment resisted tumor rechallenge demonstrating the development of immunologic memory. Analysis of changes in the tumor microenvironment by flow cytometry during combination therapy showed increased myeloid and T cell infiltration. nCounter gene expression analysis confirmed that anti-VEGFR-2 treatment enhanced inflammation and immune activation gene expression signature in monotherapy, and this effect was much more pronounced after the combination treatment. Pathway analysis highlighted that the combination effect could be attributable to enhanced innate immune response (e.g. dendritic cell maturation, antigen presentation) and T cell activation. These results were corroborated by flow cytometry, showing increased MHCI and MHCII expression on DCs and macrophages, along with increased PD-L1 expression during anti-VEGFR-2 monotherapy.
Conclusions
Taken together, these results highlight the potential of anti-VEGFR-2 antibodies to partially ameliorate intra-tumor immune suppression, providing insights into the mechanisms by which combined VEGFR-2/PD-L1 antibody therapy leads to increased anti-tumor efficacy.
P282 Immunostimulatory CD40L/4-1BBL Gene Therapy Enhances aPD-1 Antibody Therapy in Experimental Models
Jessica Wenthe, Emma Eriksson, Ann-Charlotte Hellström, Angelica Loskog
Uppsala University, Uppsala, Sweden
Correspondence: Angelica Loskog (angelica.loskog@igp.uu.se)
Background
Lung cancer is a deadly disease responding poorly to conventional therapy. Checkpoint blockade antibodies inhibiting PD-1/PD-L1 have shown clinical benefit. Since the success of PD-1 blockade therapy prerequisites the presence of tumor-reactive T-cells, we hypothesized that an activating immunotherapy with high potency to stimulate T-cell responses would be an ideal candidate to combine with anti-PD-1 antibody treatment. LOAd703 is a replication-restricted adenovirus serotype 5/35. It is armed with two immunostimulatory transgenes, 4-1BB ligand (4-1BBL) and a trimerized human CD40 ligand (TMZ-CD40L). Both molecules are co-stimulatory molecules in the dendritic cell (DC)/T-cell synapse. This study aims to evaluate if immunostimulatory gene therapy, such as LOAd703, is a good enhancer of checkpoint blockade therapy.
Methods
The capacity of LOAd703 was tested using the lung cancer cell lines A549 and H727. Tumor viability was evaluated in vitro using MTS viability assay. Co-culture experiments were analyzed in real time using Incucyte technology. Cells were phenotyped with multicolor flow cytometry and cytokine patterns investigated by MesoScale. A20 cells that can be infected with LOAd703 virus were used in a syngeneic model in BalbC mice.
Results
Tumor cells were killed by oncolysis post infection with high dose virus (100MOI) in vitro and in vivo in multiple xenograft mouse models (lung, ovary, pancreas, colon, bladder). In tumor cell/PBMC co-culture,addition of anti-PD-1 antibody did not increase tumor cell apoptosis. If OKT3/IL2 was added apoptosis was confirmed in the cultures peaking at day 5. Combination of OKT3/IL2 with anti-PD-1 antibody significantly enhanced the effect. LOAd703 was added to tumor cell/PBMC co-cultures at a low MOI to delay oncolysis. LOAd703 induced tumor cell death, starting already at day 2 and the activity was continuously rising throughout the experiment. Combining LOAd703 with anti-PD-1 antibody significantly increased apoptosis induction of tumor cells while a control virus was less effective. At endpoint, LOAd703 plus anti-PD-1 was twice as effective as OKT3/IL2 plus anti-PD-1. The T cells were increased in the same groups as tumor cell killing was previously noted. IFNg, TNF, IL2 were all enhanced by LOAd703 and OKT3/IL2. In line with in vitro data, LOAd703 could enhance in vivo tumor control of anti-PD-1 therapy.
Conclusions
In conclusion, LOAd703 is a potent stimulator of T cell responses and potentiates the effect of aPD-1 therapy.
P283 Differential impact of chemotherapy on tumor-associated antigen-specific immunogenicity in cynomolgus macaques
Maria Josic, Aaron Longworth, Joe Binder, Helen Cho, Erick Gamelin, Siradanahalli Guru, Eugenia Kraynov, Steve Burgess, Bryan Clay, Charlie Huang, Jannine Landry, Mark Lesch, Cindy Wei Li, Shangjin Li, Pavinder Kaur, Sophie Muscat-King, Peter Weady, Dan Xu, James Merson, Robert Hollingsworth, Marianne Martinic
Pfizer, San Diego, CA, USA
Correspondence: Marianne Martinic (maria.josic@pfizer.com)
Background
Pfizer’s vaccine-based immunotherapy regimen (VBIR) is a cancer treatment which combines a tumor-associated antigen (TAA)-specific vaccine, delivered via an AdC68 adenovirus and DNA regimen, with the immunomodulatory agent tremelimumab (anti-CTLA4 antibody) for T cell expansion. Various tumor types that are targets for VBIR, including non-small cell lung, triple negative breast, ovarian, and pancreatic cancer, are treated with chemotherapy as the standard of care (SOC). Accordingly, VBIR clinical administration may take place in patients who are currently undergoing chemotherapy. Synergistic effect has been reported between immune therapies and chemotherapy, either due to immunogenic cell death or a direct effect on immunosuppressive cells. However, limited information is known about the impact of chemotherapy on a vaccine-induced de novo immune response.
Methods
A nonhuman primate model was developed to most precisely mimic clinical therapy conditions, including pharmacokinetics and dynamics, dosing regimens, and supplemental treatments, allowing evaluation of potential chemotherapy impact on the VBIR-induced immune response. Macaques were dosed with a VBIR targeting a self-version of a human TAA with or without the following SOC treatments: paclitaxel/gemcitabine, paclitaxel/carboplatin, docetaxel/carboplatin, or doxorubicin. Complete blood counts and immunophenotyping were performed to track changes in immune cell profile as a biomarker of response. TAA-specific IFN-γ+ CD8+ and CD4+ T cell responses and humoral responses were measured throughout the study.
Results
In non-tumor bearing macaques, different chemotherapies impacted VBIR-induced immune responses with varying degrees of severity. TAA-specific IFN-γ+ CD8+ T cell responses ranged from unaffected with doxorubicin, to moderately reduced with carboplatin-based doublet chemotherapies, to almost entirely ablated with paclitaxel/gemcitabine. TAA-specific IFN-γ+ CD4+ T cell responses, however, were generally unimpaired by chemotherapy: indeed, in many instances chemotherapy boosted CD4+ T cell responses. TAA-specific humoral responses were superior in animals treated with carboplatin-based doublet chemotherapies, while paclitaxel/gemcitabine continued to preclude an effective immune response. Doxorubicin treated animals demonstrated a strongly diminished humoral response, attributed to B cell depletion. The impact of chemotherapy on the VBIR-induced immune response was also dependent on antigenic strength, with responses to a weaker antigen more dramatically reduced than were responses to a more immunogenic antigen.
Conclusions
Careful clinical trial planning is critical for vaccine-based immunotherapy regimen and chemotherapy combinations that will not only permit robust immune response activation and maintenance but also provide the adequate conditions for optimal synergistic anti-tumor effect.
P284 T cell priming by Toca 511 and 5-FC coupled with T regulatory cell depletion by αCTLA-4 synergistically enhances anti-tumor immune memory in a mouse model of glioma
Leah Mitchell, Kader Yagiz, Anthony Munday, Fernando Lopez, Daniel Mendoza, Douglas Jolly
Tocagen, San Diego, CA, USA
Correspondence: Leah Mitchell (lmitchell@tocagen.com)
Background
Toca 511 (vocimagene amiretrorepvec) is a gamma retroviral replicating vector that selectively infects cancer cells in vivo and encodes cytosine deaminase. In combination with the prodrug, 5-fluorocytosine (5-FC), Toca 511 produces 5-fluorouracil (5-FU) locally in the tumor microenvironment. Prior work has demonstrated a reduction in immunosuppressive myeloid cells and an increase in CD4 and CD8 T cells in tumors while T regulatory cells remain unchanged with treatment with Toca 511 and 5-FC.
Methods
This work, in a mouse model of glioma, aimed to determine if the addition of a checkpoint inhibitor, αCTLA-4, would provide therapeutic benefit to Toca 511 and 5-FC.
Results
Initially, we noted that Toca 511 and 5-FC was highly efficacious and that it provided little room for further improvement and therefore combination with αCTLA-4 did not show additive benefit against the primary cancer. However tumor associated Regulatory T cells were significantly reduced with αCTLA-4 treatment and long term memory was significantly improved with the combination as shown in adoptive transfer studies. Adoptive transfer of immune cells from animals that cleared their primary tumor through Toca 511, 5-FC, and αCTLA-4 showed 100% survival benefit to animals bearing orthotopic gliomas, significantly greater than the ~50% survival seen with transfer from animals that cleared primary tumor through Toca 511 and 5-FC alone. Further, αCTLA-4 treatment during clearance of primary tumors resulted in a marked reduction of memory T regulatory cells in secondary tumors after adoptive transfer. To determine if αCTLA-4 in combination with Toca 511 and 5-FC could reduce primary tumor burden we developed a model using a submaximal infection level of Toca 511. Specifically, restricting Toca 511 infection to only 2% of tumor cells limited the tumor growth arrest activity of 5-FC and the loss of efficacy with 2% infection was rescued when 5-FC treatment was combined with αCTLA-4.
Conclusions
These data suggest that αCTLA-4, and other compounds that target T regulatory T cells, should be evaluated in patients receiving Toca 511 and Toca FC to determine if the combination confers additional clinical benefit.
P285 Antibody and T cell response profiling of pancreatic cancer patients before and after chemotherapy reveals increased recognition of antigens suitable for immunotherapy
Giorgia Mandili1, Moitza Principe1, Emanuela Mazza1, Sara Bulfamante1, Laura Follia1, Giulio Ferrero1, Andrea Evangelista2, Daniele Giordano1, Paola Cappello1, Francesco Novelli1
1University of Turin, Torino, Italy; 2Hospital Città della Salute e della Scienza di Torino, Torino, Italy
Correspondence: Francesco Novelli (franco.novelli@unito.it)
Background
Pancreatic ductal adenocarcinoma (PDA) is one of the most lethal cancer, both for lack of effective screening method and for resistance to chemotherapy (CTX) and radiotherapy. At present surgical resection is the only potentially curative option. Once diagnosed, CTX, radiation or combination therapy regimens are used to treat patients, but responses remain poor. However, some chemotherapeutic agents have an immune modulating effect and a combination of CTX and immunotherapy could increase therapeutic efficacy. Thus, more immunogenic antigens can be induced by CTX and targeted by passive or active immunotherapy. To discover TAAs that might be selected for immunotherapy, antibody response in PDA patients’ sera were analyzed before and after CTX. TAAs selected based on their increased recognition after CTX were used to evaluate whether PDA patient autologous T cells have an increased TAAs specific response after CTX.
Methods
Antibody response in sera of 29 PDA patients, before and after CTX treatments, has been analyzed by Serological Proteome Analysis (SERPA). The production of IFN-g and IL-10 by PBMC stimulated in vitro with recombinant TAAs was evaluated by ELISA.
Results
CTX increases the number of TAAs recognized by PDA patients. Specifically, the set of TAAs we identified are proteins whose mRNA expression have been found up-regulated in PDA patients. We observed a positive correlation between patients survival and the increased antibodies production against alpha-enolase (ENO1), glyceraldehyde-3-phosphate dehydrogenase (G3P), tubulin (TUBB5), and keratin, type II cytoskeletal 8 (K2C8). Of note, 48% of PDA patients sera after CTX treatment showed an increase of Complement Dependent Cytotoxicity (CDC) against human cell lines. Moreover in about 50% of the same PDA patient cohort the in vitro specific T cell response to recombinant TAAs that were mostly recognized by antibodies after CTX (ENO1, G3P, K2C8 and Far Upstream Binding Protein 1 (FUBP1) switched from a pro-tumor regulatory (low IFN-g/IL10 ratio) to anti-tumor effector (high IFN-g/IL10 ratio) phenotype after one or two rounds of CTX.
Conclusions
Data indicated that in PDA patients CTX induces an increase of IgG antibody to ENO1, G3P, TUBB5 and K2C8 whose expression is upregulated in PDA that may have a prognostic role. CTX also increases the ability of IgG kill PDA cells by CDC. Finally , CTX switches the T cell response to ENO1, G3P, K2C8 or FUBP1 from regulatory to effector and thus renders these TAAs promising targets for the design of new immunotherapeutic approach to improve the efficacy of CTX.
P286 Efficacy of interleukin 2 and interleukin 15 for in situ vaccination in a mouse melanoma model
Alexander Rakhmilevich1, Anna Hoefges1, Jacob Slowinski1, Kayla Rasmussen1, Mackenzie Heck1, Michael Meagher2, Alan Korman3, Paul Sondel1
1University of Wisconsin-Madison, Madison, WI, USA; 2St. Jude Children’s Research Hospital/Children’s GMP, LLC, Memphis, TN, USA; 3Bristol-Myers Squibb Company, Redwood City, CA, USA
Correspondence: Alexander Rakhmilevich (rakhmil@humonc.wisc.edu)
Background
We have previously shown that a direct intratumoral (IT) injection of immunocytokine (IC), an anti-GD2 antibody linked to interleukin 2 (IL-2), can serve as an in situ vaccine and synergize with anti-CTLA-4 antibody to induce T cell-mediated antitumor effects. We have also shown a synergy of this approach with activation of innate immunity with an agonistic anti-CD40 monoclonal antibody (anti-CD40) and CpG (Rakhmilevich et.al. Journal of Immunology 2017). We used these two immunotherapeutic approaches to test whether IT treatment with a mixture of IL-2 and anti-GD2 antibody, hu14.18K322A, will be effective against subcutaneous melanoma. In addition, we hypothesized that IL-15, a cytokine having several activities similar to those of IL-2 except induction of T regulatory cells (Tregs), may be as or more effective than IL-2 for in situ vaccination.
Methods
GD2+ B78 mouse melanoma cells were injected subcutaneously in C57BL/6 mice. Anti-CD40 and anti-CTLA-4 were given intraperitoneally, and CpG, IL-2, IL-15 and K322A were given IT. IL-2 and IL-15 were given at the dose of 7.5x104 units per injection daily for 5 days. Tumor growth and survival were followed. In some experiments, tumors were analyzed by flow cytometry.
Results
Flow cytometric analyses showed that IL-2 treatment increased Tregs within a tumor whereas IL-15 did not. When the treatments were started on day 6 post tumor cell implantation, IL-2 and IL-15 combined with K322A induced similar reductions of B78 tumor growth, and complete tumor regression with the addition of anti-CTLA-4. When the treatments were started on day 22 post tumor cell implantation, IL-2 and IL-15, combined with K322A, anti-CTLA-4, anti-CD40 and CpG, also induced comparable antitumor effects resulting in survival of 40-60% of mice.
Conclusions
In two therapeutic approaches, IL-2 and IL-15, combined with other immunotherapeutic agents, induced similar antitumor effects.
P287 Immunological effects of checkpoint blockade plus galectin-3 inhibition with GR-MD-02 in a first-in-human phase I clinical trial
William Redmond1, Yoshinobu Koguchi1, Christopher Fountain1, Peter Traber2, Brendan Curti1
1Providence Portland Medical Center, Portland, OR, USA; 2Galectin Therapeutics, Norcross, GA, USA
Correspondence: William Redmond (william.redmond@providence.org)
Background
Immunosuppression of tumor-infiltrating lymphocytes (TIL) is a major obstacle to creating effective therapies for patients with metastatic cancer. Galectin-3 (Gal3), a lectin family member, is expressed by numerous cancers and immune cell subsets. Serum Gal3 expression is higher in patients with metastatic disease and is associated with reduced survival in patients with metastatic melanoma. Furthermore, Gal3 has been implicated in disease progression via the promotion of angiogenesis and metastasis. Interestingly, extracellular Gal3 induces immune suppression via inhibiting TIL function, promoting M2 macrophage polarization and mobilizing myeloid cells from the bone marrow to promote a metastatic niche within the tumor. We hypothesized that Gal3 inhibition in conjunction checkpoint blockade immunotherapy would improve TIL function while inhibiting tumor growth and metastasis. Preclinical studies revealed that Gal3 blockade with GR-MD-02 and agonist anti-OX40, anti-CTLA-4 or anti-PD-1 mAb therapy enhanced tumor-specific immunity and improved survival in tumor-bearing mice.
Methods
We initiated 2 phase I clinical trials at our Institute to evaluate the safety and immunological effects of GR-MD-02 plus checkpoint blockade immunotherapy. These studies are investigating dose escalations of GR-MD-02 with the standard therapeutic dose of anti-CTLA-4 (ipilimumab/ipi) or anti-PD-1 (pembrolizumab/pembro) in patients (pts) with advanced melanoma (ipi; pembro), HNSCC (pembro), or NSCLC (pembro) (NCT02117362, NCT02575404).
Results
The GR-MD-02 + ipi study has completed enrollment. Three patients received GR-MD-02 at 1 mg/kg, 3 at 2 mg/kg and 2 at 4 mg/kg. There were no DLTs for GR-MD-02; however there was 1 grade 3 AE from ipilimumab (diarrhea) that occurred after dose 4 of ipi. The GR-MD-02 + pembro study is ongoing. Six patients were enrolled at the 2 mg/kg dose level to gain more data from the immunological monitoring assays. Three patients were treated at 4 mg/kg. Eight of the 9 enrolled patients have melanoma and 1 has HNSCC. There have been no DLTs related to GR-MD-02 or pembro, but there one patient experienced transient grade 3 tumor-related pain. Two pts in cohort 1 (2 mg/kg GR-MD-02) had objective responses (1 PR, 1 CR) and two pts in cohort 2 (4 mg/kg) have PR at the first response assessment. All responses have been observed in patients with melanoma.
Conclusions
These data demonstrate that the Gal3 inhibitor GR-MD-02 can be combined safely with checkpoint blockade in patients with metastatic disease and that melanoma regression was observed in multiple patients following combination therapy. Comprehensive immunological monitoring is being conducted to provide insight into potential mechanisms of action.
Trial Registration
NCT02117362, NCT02575404
P288 KY1044, a novel anti-ICOS antibody, elicits long term in vivo anti-tumour efficacy as monotherapy or in combination with immune checkpoint inhibitors
Richard C.A. Sainson, Anil Thotakura, Nahida Parveen, Gwenoline Bohris, Robert Rowlands, Miha Kosmac, Jamie Campbell, Ian Kirby, Volker Germaschewski, Matthew McCourt
Kymab Ltd, Cambridge, United Kingdom
Correspondence: Richard C.A. Sainson (richard.sainson@kymab.com); Matthew McCourt
Background
The inducible co-stimulator molecule (ICOS/CD278) is a member of the CD28/CTLA-4 family that is up-regulated upon T cell activation. In the tumour microenvironment, ICOS expression levels vary in different immune cell subtypes, with expression on highly immunosuppressive regulatory T cells (Treg: CD4+/FOXP3+) significantly higher than that on effector CD8+ T cells. The high expression levels on Tregs highlights the potential of targeting ICOS to deplete these cells and enhance the anti-tumour immune response when used in combination with immune checkpoint blockers.
Methods
Using the Kymouse™ platform, we have identified a novel, fully human antibody called KY1044. KY1044 is an anti-ICOS subclass G1 kappa monoclonal antibody that selectively binds to dimeric ICOS (Fc fusion) with an affinity of less than 2nM as measured by SPR using Fabs of KY1044. This antibody, which binds ICOS from human, cynomolgus monkey, rat and mouse with similar affinity, was used in several in vitro and in vivo assays to refine its mechanism of action and assess the anti-tumour efficacy in pre-clinical models.
Results
Using in vitro reporter and primary cell assays, we have demonstrated that KY1044 can affect the immune context via a dual mechanism of action. On the one hand, KY1044 is extremely potent (low pM EC50) at depleting ICOShigh Tregs and on the other hand KY1044 was shown to stimulate ICOSInt effector T-cells by increasing their IFNγ and TNFα production. Since KY1044 is cross reactive to the rodent orthologue of ICOS, a mouse effector enabled version of KY1044 (mIgG2a) was generated and tested in syngeneic tumour models. Using this antibody, we have confirmed strong anti-tumour efficacy as monotherapy or in combination with surrogates of “approved” immune checkpoint blockers. Noteworthy, and confirming the in vitro data, pharmacodynamic studies demonstrated a long-term depletion of Tregs and a significant increase in the effector T cell to Treg ratio in response to KY1044.
Conclusions
Altogether, the in vitro and in vivo properties of this novel, fully human anti-ICOS antibody support the continued development of KY1044 as a treatment option to activate an anti-tumour immune response.
P289 Dual cIAP1/XIAP inhibitor ASTX660 synergizes with radiation therapy and PD-1 blockade to enhance anti-tumor immunity
Roy Xiao1,2, Clint Allen1,3, Linda Tran1, So-Jin Park1, Zhong Chen1, Carter Van Waes1, Nicole Schmitt1,3
1NIDCD, NIH, Bethesda, MD, USA
Correspondence: Nicole Schmitt (nicole.schmitt@nih.gov)
Background
Head and neck squamous cell carcinomas (HNSCCs) frequently harbor genomic mutations in cell death pathways. Nearly 30% of HNSCC overexpress Fas-Associated Death Domain (FADD), with or without BIRC2/3 genes encoding cellular Inhibitor of Apoptosis Proteins 1/2 (cIAP1/2), critical components of the Tumor Necrosis Factor (TNF) Receptor signaling pathways. ASTX660 is a novel dual cIAP1/XIAP antagonist in clinical trials for advanced solid tumors and lymphomas.
Methods
Murine oral cancer 1 (MOC1) cells were used in vitro and in vivo to investigate the anti-tumor activity of ASTX660 alone and in combination with tumor necrosis factor receptor (TNFR) superfamily ligands, radiation, cisplatin chemotherapy, and anti-PD-1 checkpoint blockade. OT-1 T cells were used to investigate the effects of ASTX660 on antigen-specific T cell killing of ovalbumin-expressing MOC1 (MOC1ova) cells.
Results
ASTX660, at nanomolar concentrations, sensitized MOC1 cells to TNFα and stimulated cytotoxic T lymphocyte (CTL) killing of MOC1ova. CTL killing was found to be predominantly mediated by perforin/granzyme B during the earliest stages of killing and release of death ligands TNFα, TRAIL, and FasL as a sustained mechanism of killing. Using MOC1 cells in vivo, ASTX660 synergized with radiation therapy (XRT), cisplatin chemotherapy, and PD-1 blockade to significantly delay or eradicate MOC1 tumors. These combination therapies significantly increased CD8+ T cells and dendritic cells, as well as T cell activity. Depletion of CD8+ T cells and NK cells in vivo revealed both to be important components of the anti-tumor response enhanced by ASTX660+XRT.
Conclusions
These findings serve to inform future studies of IAP inhibitors and support the potential for future clinical trials investigating ASTX660 with XRT and immunotherapies such as PD-1/PD-L1 blockade in HNSCC.
P290 Agonist redirected checkpoint (ARC), TIM3-Fc-OX40L, for cancer immunotherapy
George Fromm, Suresh de Silva, Kellsey Johannes, Arpita Patel, Josiah Hornblower, Taylor Schreiber
Shattuck Labs, Inc., Research Triangle Park, NC, USA
Correspondence: Taylor Schreiber (tschreiber@shattucklabs.com)
Background
Current attempts at combination immunotherapy with bispecific antibodies, linked scFv’s or T cell engagers have not demonstrated that both checkpoint blockade and TNF receptor activation (agonism) can be achieved with a single molecule. This is likely due to the fact that these molecules lose target avidity when engineered to bind multiple targets with monovalent antigen binding arms. Fusion proteins incorporating the extracellular domain (ECD) of type I membrane proteins (eg. Enbrel, Orencia) or type II membrane proteins (eg. OX40L-Fc, GITRL-Fc), linked to the hinge-CH2-CH3 domain of antibodies are both functional, despite the fact that the ECDs are in opposite orientation. Here we report the generation of a two-sided fusion protein incorporating the ECD of TIM3 and the ECD of OX40L, adjoined by a central Fc domain.
Methods
Shattuck synthesizes both murine and human versions of ARC proteins, and assesses them using a litany of biochemical assays to determine molecular weight, subunit composition & binding affinity; molecular assays to characterize in vitro/ex vivo binding, in vitro functional activity; and anti-tumor efficacy in multiple syngeneic tumor model systems. The human TIM3-Fc-OX40L has advanced into cell line development and early manufacturing.
Results
The TIM3 end of the fusion protein binds GAL9 and phosphtidylserine (PS) on the surface of human tumor cells. The OX40L end of the fusion protein binds OX40 on the surface of primary T cells. TIM3-Fc-OX40L activates NFkB signaling in cells engineered to overexpress OX40 and an NFkB-luciferase reporter. Additionally, the TIM3-Fc-OX40L ARC added to primary human PBMCs along with the super-antigen Staphylococcal enterotoxin B, induced robust secretion of the cytokines IL2 and TNFa. In vivo, TIM3-Fc-OX40L stimulates significant expansion of antigen-specific CD4 and CD8 T cells in mice adoptively transferred with OT-I/OT-II cells and vaccinated with ova/alum. Finally, the therapeutic activity of TIM3-Fc-OX40L in established murine MC38 and CT26 tumors was significantly superior to either TIM3 blocking antibody, OX40 agonist antibody or combination antibody therapy. Importantly, a pharmacodynamic biomarker of tumor rejection was identified by coordinated elevations in serum IFNγ, IL-2, IL-4, IL-5, IL-6 and IL-17A.
Conclusions
These data demonstrate feasibility and functional activity of a novel chimeric fusion protein platform, providing checkpoint blockade and TNF superfamily costimulation in a single molecule, which is uniquely advantageous because the construct links those two signals in the same microenvironmental context, at the time in which T cells are engaging cognate tumor antigen.
P291 Agonist redirected checkpoint (ARC), SIRPα-Fc-CD40L, for cancer immunotherapy
Suresh de Silva, George Fromm, Arpita Patel, Kellsey Johannes, Josiah Hornblower, Taylor Schreiber
Shattuck Labs, Inc., Research Triangle Park, NC, USA
Correspondence: Taylor Schreiber (tschreiber@shattucklabs.com)
Background
Current attempts at combination immunotherapy with bispecific antibodies, linked scFv’s or T cell engagers have not demonstrated that both checkpoint blockade and TNF receptor activation (agonism) can be achieved with a single molecule. This is likely because these molecules lose target avidity when engineered to bind multiple targets with monovalent antigen binding arms. Fusion proteins incorporating the extracellular domain (ECD) of type I membrane proteins (eg. Enbrel, Orencia) or type II membrane proteins (eg. SIRPα-Fc, GITRL-Fc), linked to the hinge-CH2-CH3 domain of antibodies are both functional, despite the ECDs being in opposite orientation. We report the generation of a two-sided fusion protein incorporating the ECD of SIRPα (CD172a) and the ECD of CD40L, adjoined by a central Fc domain.
Methods
Shattuck synthesizes both murine and human versions of ARC proteins, and assesses them using a litany of biochemical assays to determine MW, subunit composition & binding affinity; molecular assays to characterize in vitro/ex vivo binding & functional activity; and anti-tumor efficacy in syngeneic tumor models. The human SIRPa-Fc-CD40L has completed cell line development and single cell cloning and is in late-stage manufacturing.
Results
The SIRPα end of the ARC binds immobilized CD47 at 3.59 nM affinity and binds CD47 on the surface of human tumor cells both in vitro and in vivo, but does not bind human platelets or RBCs. Importantly, no hemolytic activity has been observed with the human SIRPa-Fc-CD40L ARC; where significant hemolysis has been reported with comparative CD47 mAbs. The CD40L end of the ARC binds immobilized CD40 at 756 pM affinity and binds CD40 on primary macrophages. The SIRPa-Fc-CD40L ARC stimulates functional activity in NFkB-luciferase reporter cells (CD40 driven activation of NFkB) and when added ex vivo to human PBMCs primed with the super-antigen SEB; increases secretion of IL2 and TNFa. Furthermore, when activated human macrophages were co-cultured with CD47 positive human tumor cells, SIRPα -Fc-CD40L was shown to enhance phagocytosis of human tumor cells. Finally, the therapeutic activity of SIRPα-Fc-CD40L in established murine MC38 and CT26 tumors was superior to either CD47 blocking antibody, CD40 agonist antibody or combination antibody therapy.
Conclusions
These data demonstrate feasibility and functional activity of a novel chimeric fusion protein platform, providing checkpoint blockade and TNF superfamily costimulation in a single molecule. Signal replacement of CD47 by CD40L may uniquely poise macrophages in the tumor microenvironment for activation and cross-presentation of tumor antigens following enhanced tumor cell phagocytosis.
P292 Pre-clinical activity of a novel immunotherapy combination of CAVATAK (Coxsackievirus A21), anti-PD1 blockade and an IDO inhibitor in melanoma
Gough Au, Min Yuan, Yvonne Wong, Darren Shafren
Viralytics Limited, Sydney, Australia
Correspondence: Darren Shafren (darren.shafren@viralytics.com)
Background
Coxsackievirus A21 (CAVATAKTM) is a bio-selected oncolytic immunotherapy virus. A Phase Ib trial of i.v. CAVATAK (NCT01227551) in advanced cancer patients has displayed viral tumor targeting and initial indications of antitumor activity in some lesions. Intratumoral CAVATAK injection of melanoma lesions can induce selective tumor-cell infection, immune-cell infiltration, IFN-g response gene up-regulation, increased PD-L1 and IDO (indoleamine-pyrrole 2,3-dioxygenase) expression, tumor cell lysis and systemic anti-tumor immune responses. Blockade of programmed death protein-1 (PD1) and/or IDO inhibition in many cancer patients has resulted in substantial tumor responses. We investigated anti-tumor activity in a B16-ICAM-1 melanoma immune competent mouse model of a novel combination of CAVATAK, an anti-PD1 mAb and an IDO inhibitor.
Methods
Palpable flank tumor of murine melanoma B16-cells expressing human ICAM-1 were propagated to assess the antitumor activity of CAVATAK, an anti-mouse PD1 (mPD1) mAb and an murine IDO inhibitor in an immune competent mouse model. CAVATAK was administered i.v, while anti mPD1 mAb was delivered via the i.p route and the IDO inhibitor in drinking water.
Results
Notable single agent antitumor activity against the B16-ICAM-1 tumors was only observed in mice treated with anti-PD1 blockade relative to saline controls. Significant survival benefits were only observed in mice treated with the CVA21-anti-mPD1 doublet or the CVA21, anti-PD1 and IDO inhibitor triplet combinations. While not significant, we observed a positive trend in both reduction of overall tumor burden and survival in mice treated with the CVA21, anti-mPD1 doublet and the CVA21, anti-mPD1 and IDO inhibitor triplet compared with mice receiving anti-mPD1 blockade and IDO-inhibition. All combinations of CVA21, anti-mPD1 and IDO blockade appeared to be generally well tolerated.
Conclusions
The notable anti-tumor activity and survival benefit mediated by the combination of CAVATAK, PD1 blockade and IDO inhibition observed in the presented melanoma model supports potential clinical evaluation of such a novel immunotherapeutic combination treatment regimen.
P293 Talimogene Laherparepvec combined with anti-PD-1 based immunotherapy for unresectable stage III-IV melanoma: a case series
Lillian Sun1, Pauline Funchain2, Jung-Min Song2, Michael McNamara2, Brian Gastman3
1Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH, USA; 2Cleveland Clinic Taussig Cancer Institute, Cleveland, OH, USA; 3Cleveland Clinic, Cleveland, OH, USA
Correspondence: Brian Gastman (sunl2@ccf.org)
Background
Checkpoint inhibitors have become standard of care for treating advanced melanoma. By blocking key inhibitory pathways, checkpoint inhibitors reinvigorate effector T cells to overcome an immunosuppressed microenvironment. A key preceding step to effective tumor killing is the release and presentation of tumor antigen. Talimogene Laherparepvec (T-VEC) is an oncolytic virus recently approved as an intratumoral therapy for treating unresectable stage IIIB-IV metastatic melanoma. As monotherapy, it confers modest efficacy in controlling disease progression in patients with locoregional lesions. T-VEC is used to directly lyse tumor cells and promote anti-tumor immunity via the release of tumor antigens and virus-encoded GM-CSF. The mechanisms of action for T-VEC and checkpoint inhibitors are highly complementary, raising the possibility of synergistic benefit from combining these two therapies.
Methods
We reviewed 10 consecutive cases of stage IIIC to stage IVM1b melanoma patients that received T-VEC plus either pembrolizumab or ipilimumab/nivolumab, treated between January 2016 and July 2017 at the Cleveland Clinic with a median follow-up of 7 months (range: 4 to 13 months). Responses of injected (on-target) and uninjected (off-target) lesions were evaluated according to RECIST 2.0.
Results
The overall response rate for on-target lesions was 90%, with 5 patients experiencing a complete response of injected lesions. Two patients had off-target lesions, both of whom experienced complete response of their uninjected distant metastases. Overall survival of this cohort was 80%. Of the two patients who died, one died of causes unrelated to melanoma. Checkpoint inhibitor therapy was interrupted for 2 patients due to adverse events, with one patient experiencing Grade 3 nephritis and the other patient experiencing Grade 3 diarrhea. There were 2 patients who experienced progression of disease. Taking this into account with the 2 patients who died, overall progression-free survival for this cohort was 60%.
Conclusions
There may be potential synergistic benefit in combining checkpoint inhibitors with T-VEC injection in patients with unresectable melanoma. Although this study is limited by the lack of randomization and short follow-up time, the data revealed positive signals in the responses of both on-target and off-target lesions to the combination of T-VEC and checkpoint inhibitor treatment. In addition, the results show a better response rate than previous publications using T-VEC. Ongoing clinical trials will elucidate the true clinical benefit of this combinational therapy.
P294 Cost of adverse events associated with immunotherapy monotherapy versus targeted therapy in elderly metastatic melanoma patients
Jackson Tang1, Zhiyi Li1, Syed Mahmood2, Sameer Ghate2
1Asclepius Analytics, New York, NY, USA; 2Novartis Pharmaceuticals Corporation, East Hanover, NJ, USA
Correspondence: Jackson Tang (jtang@asclepiusanalytics.com)
Background
The National Comprehensive Cancer Network guidelines recommend the use of immunotherapy (IO) or targeted therapy (TT) (if BRAF-mutated) for the first-line treatment of metastatic melanoma (MM). Both IO and TT can lead to treatment-related adverse events (AEs). The objective of the study is to estimate and compare the cost of treatment-related AEs between elderly MM patients receiving IO monotherapy and those receiving TT.
Methods
A retrospective cohort study was conducted using Medicare research identifiable files from 2006 to 2014. Eligible patients had ≥1 MM diagnosis and ≥1 prescription for an IO (ipilimumab or pembrolizumab) or TT (dabrafenib, trametinib, vemurafenib, or dabrafenib+trametinib) that was FDA-approved during the study period. Patients were assigned to the IO or TT cohort based on the most recent therapy used, and had a minimum of 3 months follow-up within the study period. 10 categories of AEs were identified based on a review of IO or TT package inserts, as well as AEs that were common among MM patients. Cost per AE was calculated as the average per-episode cost (inpatient + outpatient) incurred in the 30 days after the first occurrence of the AE. Costs were inflated to 2017 USD and compared between the cohorts using the Wilcoxon rank-sum test, with a significance level of 0.05.
Results
The study included 266 IO patients and 159 TT patients. The TT cohort was younger (72.8±14.3 vs. 73.7±12.6 years; p=0.04) and less likely to be male (57.2% vs. 63.2%; p=0.01) compared with the IO cohort. The 30-day AE costs for each cohort are summarized in Table 1. The average costs associated with gastrointestinal, respiratory, and pain-related AEs were significantly higher in the IO cohort than the TT cohort (difference: $9270, $2654, and $2155, respectively; p<0.01). In contrast, the average costs associated with pyrexia and/or chills, and other AEs (including decreased appetite/anorexia, fatigue, or infections) were significantly lower in the IO cohort (difference: -$5761, -$598 respectively; p<0.05). There was no significant difference in the costs associated with hematologic/lymphatic, central nervous system/psychiatric, metabolic/nutritional, skin/subcutaneous tissue, or cardiovascular AEs.
Conclusions
The costs associated with treatment-related AEs among elderly MM patients were substantial. Patients treated with IO incurred higher costs after gastrointestinal, respiratory, and pain-related AEs, while patients treated with TT incurred higher costs after pyrexia/chills, decreased appetite/anorexia, fatigue, or infections.
P295 Economic burden of adverse events associated with immunotherapy and targeted therapy for metastatic melanoma in the US elderly population
Jackson Tang1, Zhiyi Li1, Syed Mahmood2, Sameer Ghate2
1Asclepius Analytics, New York, NY, USA; 2Novartis Pharmaceuticals Corporation, East Hanover, NJ, USA
Correspondence: Jackson Tang (jtang@asclepiusanalytics.com)
Background
The National Comprehensive Cancer Network guidelines for the first-line treatment of metastatic melanoma (MM) recommend the use of immunotherapy (IO) or targeted therapy (TT) (if BRAF-mutated), both of which are associated with treatment-related adverse events (AEs). The occurrence of AEs is associated with increased healthcare resource use and costs. The objective of this study is to estimate the incremental costs of experiencing treatment-related AEs among elderly MM patients receiving IO monotherapy or TT.
Methods
A retrospective cohort study was conducted using Medicare research identifiable files from 2006 to 2014. Eligible patients had ≥1 MM diagnosis, ≥1 prescription for an IO (ipilimumab or pembrolizumab) or TT (dabrafenib, trametinib, vemurafenib, or dabrafenib+trametinib) that was FDA-approved during the study period, and a minimum of 3 months of follow-up. 10 categories of AEs were identified based on a review of IO or TT package inserts, as well as AEs that were common among MM. The incremental cost for each AE category was determined by comparing the 30-day expenditures (inpatient + outpatient) between patients with the AE versus patients without the AE using a generalized linear model with a log-link function and gamma distribution, adjusting for baseline covariates. All costs were inflated to 2017 USD.
Results
A total of 425 patients were included. The mean age was 73.4 years (SD±13.8), 61% were male, 94% were white, and the mean baseline Charlson comorbidity index was 8.5 (SD±2.3). Table 1 summarizes the adjusted 30-day incremental cost of each AE. The adjusted 30-day incremental cost was highest for respiratory AEs ($24,150; 95% confidence interval [CI] $17,630–30,671), followed by central nervous system/psychiatric disorders ($21,932; 95% CI $16,011–27,854), metabolic/nutritional disorders ($19,776; 95% CI $14,239–25,314), skin/subcutaneous tissue AEs ($19,183; 95% CI $14,195–24,170), fever (pyrexia) and/or chills ($18,976; 95% CI $13,663–24,289), pain ($18,406; 95% CI $13,805–23,008), cardiovascular AEs ($16,393; 95% CI $12,131–20,655), hematologic/lymphatic AEs ($15,850; 95% CI $11,888–19,813), gastrointestinal AEs ($13,699; 95% CI $10,138–17,261), and other AEs (e.g. decreased appetite/anorexia fatigue, infections (including folliculitis)) ($9,754; 95% CI $7,315–12,192).
Conclusions
Among elderly MM patients in the United States, the incremental costs of treatment-related AEs are substantial. These findings may help to inform comparisons between treatments, and thereby aid in clinical and budgetary decision-making in this population.
P296 Exosomes shuttle TREX1-sensitive IFN-stimulatory dsDNA from irradiated cancer cells to dendritic cells
Claire Vanpouille-Box1, Julie Diamond1, Nils Rudqvist1, Karsten Pilones1, Yasmeen Sarfraz1, Silvia Formenti2, Sandra Demaria1
1Weill Cornell Medicine, New York, NY, USA; 2Weill Cornell, New York, NY, USA
Correspondence: Sandra Demaria (claire.vanpouille.box@gmail.com)
Background
Radiotherapy (RT) used at immunogenic doses leads to accumulation of cytosolic dsDNA in the cancer cells, which activates interferon type I (IFN-I) via the cGAS/STING pathway. Cancer cell-derived IFN-I is required to recruit BATF3-dependent dendritic cells (DCs) to poorly immunogenic tumors and trigger anti-tumor immune responses in combination with immunotherapies. Importantly, we have recently demonstrated that TREX1 regulates radiation immunogenicity by degrading cytosolic dsDNA (Vanpouille-Box et al., 2017 Nat Commun). Tumor-derived dsDNA has also been shown to be critical for cGAS/STING-mediated IFN-I by tumor-infiltrated DCs (Woo et al., 2014 Immunity). Here we hypothesized that activation of DCs by tumor-derived dsDNA is modulated by RT and regulated by TREX1. Exosomes are secreted by cancer cells and can carry dsDNA. Thus, we also tested whether tumor-derived exosomes (TEX) can deliver dsDNA to DCs.
Methods
TEX were purified from supernatants of mouse carcinoma TSA or TSA knock-in for Trex1 (TSAKI Trex1) cells that were mock-treated (UT-TEX), or irradiated with 3 doses of 8Gy (RT-TEX). Protein composition and content of dsDNA was analyzed. TEX were incubated in vitro with primary DCs, or used to vaccinate BALB/c mice (n=6) by 3 s.c. injections followed by challenge with TSA cells to evaluate the development of protective anti-tumor immunity. Tumor-specific CD8+ T cells were identified using H2-Ld/AH1 peptide pentamers.
Results
Double-stranded DNA content of RT-TEX was significantly higher than that of UT-TEX. In vitro, RT-TEX but not UT-TEX induced the upregulation CD40, CD80 and CD86 on DCs, and the production of IFNb, which was dependent on STING expression by DCs. When TREX1 is upregulated in TSAKI Trex1 cells, dsDNA amount of RT-TEX was markedly reduced, indicating that it is largely derived from cytosolic dsDNA present in the irradiated parent cells. Most importantly, enforced TREX1 expression abrogated the ability to RT-TEX to induce IFN-I in recipient DCs. In vivo, vaccination with UT-TEX led to 100% tumor outgrowth, while 2/6 mice vaccinated with RT-TEX were protected from tumor development. Remaining tumors grew significantly slower compared to UT-TEX treated animals. Tumor-specific CD8+ T cells were significantly increased in the tumors and spleen of RT-TEX vaccinated mice.
Conclusions
Overall these results identify RT-TEX as a mechanism whereby IFN-stimulatory dsDNA is transferred from cancer cells to DCs. Importantly, they also demonstrate that TREX1 in the irradiated cancer cells regulate the production of IFN-I by DCs. Findings further support the use of RT doses that do not induce TREX1 to achieve in situ vaccination by RT.
P297 Predicting the efficacy of combination immunotherapy in animal models using tumor microenvironment immune cell profiling
Ava Vila-Leahey, Genevieve Weir, Alecia MacKay, Valarmathy Kaliaperumal, Cynthia Tram, Marianne Stanford
Immunovaccine, Halifax, NS, Canada
Correspondence: Ava Vila-Leahey (avilaleahey@imvaccine.com)
Background
Due to the complex nature of the tumor microenvironment, combinations of immune modulating compounds are likely required for maximal clinical benefit in advanced cancer patients. In our previous work, we have demonstrated that a therapeutic cancer vaccine combined with metronomic cyclophosphamide (mCPA) can enhance T cell infiltration of the tumor. This treatment can be added to treatments such as anti-PD-1 to provide enhanced immune infiltration and better tumor control in murine models. A part of this increased efficacy was due to the increased levels of these molecules in the tumors of treated animals. In this study, we have conducted a systematic flow cytometry survey of checkpoint molecules expressed on T cells within the tumor microenvironment after immune therapy, and utilized this to predict the potential of combining vaccine therapy with other clinical stage antibodies targeting the checkpoint system.
Methods
HPV16 E7-transformed C3 cells were implanted subcutaneously into C57BL/6 mice, and then treated with mCPA (20mg/kg/day PO), followed by vaccination with HPV16 E749-57 peptide in a DepoVaxTM platform (DPX-R9F). Additional mice were also treated with anti-PD-1 (200 mg) or anti-OX40 (100 mg). Tumors were collected for analysis 10 days post-vaccine treatment, and digested for staining for flow cytometry to observe differences in infiltrating immune cells and markers on tumor infiltrating immune cells.
Results
We found that expression of the inhibitory checkpoint markers such as PD-1 and TIM-3 were increased, but activating checkpoint molecules GITR and OX40 were not increased, on T cells after treatment with DPX/mCPA, and no checkpoint molecules are altered further with either anti-PD-1 or OX40 antibody. We performed a tumor challenge study with the C3 model and antibodies targeting either PD-1, GITR or OX40 to determine whether targeting a molecule upregulated by the vaccine would have greater effect than targeting a molecule that is not altered. Antibodies targeting PD-1, GITR or OX40 had significant synergistic activity with DPX-R9F and mCPA treatment, but anti-PD-1 antibody was most potent in tumor growth inhibition. (Fig. 1)
Conclusions
In conclusion, we have found that an extensive tumor immune profile of treatment regimes in animal models can accurately predict the efficacy of combination immunotherapy, and activating checkpoint molecules do not necessarily enhance the efficacy of treatment in our tested models. Current studies are exploring additional checkpoint molecules in this model that were shown to be elevated in tumor infiltrating CD8+ T cells with vaccine treatment to maximize combination immunotherapy potential.
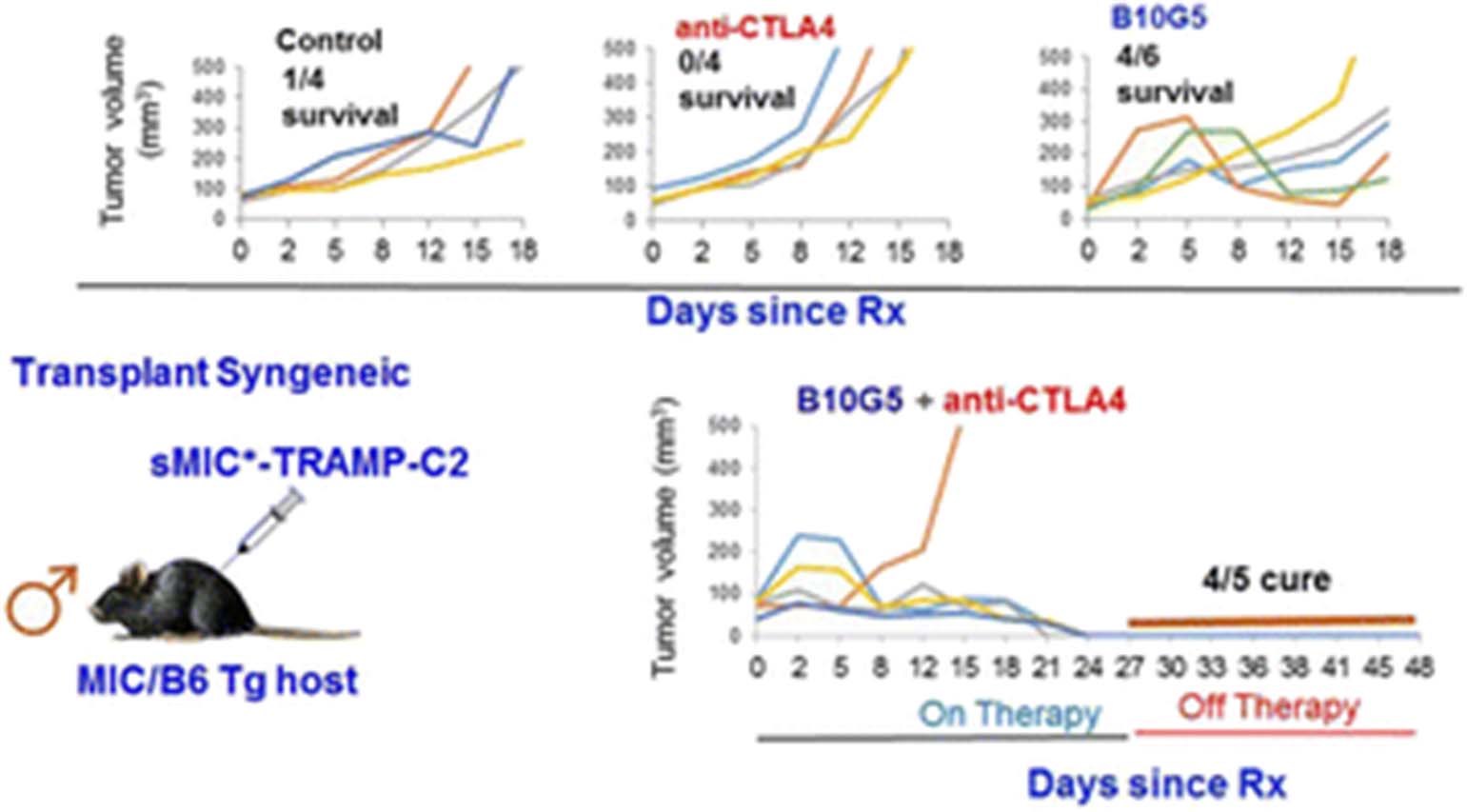
See text for description
Emerging Models and Imaging
P298 Utilization of murine breast cancer models in preclinical immuno-oncology pharmacology
Dylan Daniel, Sumithra Urs, Kevin Guley, David Draper, Alden Wong, Dan Saims, Scott Wise, Maryland Rosenfeld Franklin
MI Bioresearch, Ann Arbor, MI, USA
Correspondence: Dylan Daniel (ddaniel@molecularimaging.com)
Background
The field of immuno-oncology is persistently challenged by the need for more syngeneic mouse models in any given tissue type. The most prevalently used syngeneic model for breast cancer is the 4T1 mammary cancer cell line. The 4T1 cell line has useful traits for immuno-oncology research including a highly metastatic phenotype that leads to extensive lymph node and lung metastasis. The tumors have a highly immunosuppressed microenvironment with regulatory T cells (Tregs) and large numbers of granulocytic myeloid derived suppressor cells (G-MDSCs). Radiation can induce changes in an immunosuppressive microenvironment, and focal beam radiotherapy remains an important therapeutic strategy for the treatment of breast cancer. For this reason, we established a radiation dose response on established 4T1 tumors for the purpose of guiding future immunotherapy combinations.
Nonetheless, there are some important challenges with using the 4T1 model, in particular with studying the activity of T cell checkpoint inhibitors and costimulatory antibodies. Mice bearing 4T1 tumors develop a fatal pulmonary hypersensitivity upon repeated treatment with rat antibodies to PD-1, PD-L1 or OX40. 4T1 tumors are completely resistant to PD-1 pathway blockade and the hypersensitivity reaction makes it challenging to perform combination studies with novel test agents.
Methods
We have characterized two mammary cancer alternatives to 4T1, EMT-6 and E0771. The in vivo growth characteristics and baseline immune cell profiling have been completed for these models with some notable immune profile differences. Further, we have completed an efficacy study of E0771 using an array of immunotherapies.
Results
All three models have a similar proportion of CD4+ T cells and Tregs. In contrast, 4T1 has a much larger proportion of G-MDSCs while E0771 is almost entirely lacking in G-MDSCs with EMT-6 in between the two. The content of monocytic MDCSs (M-MDSCs) is nearly reciprocal with E0771 having a proportionately high population and 4T1 having a minimal number. These immune profile characteristics can be factored into model selection decisions.
An efficacy study in the E0771 model showed that it is highly sensitive to several checkpoint inhibitors, including anti-PD-1, anti-PD-L1, and anti-CTLA-4. E0771 shows a more modest response to anti-LAG3. Costimulatory agonist antibodies to OX40, CD137 (4-1BB) and glucocorticoid induced TNFR-related protein (GITR) were highly active. The indoleamine 2,3-dioxygenase 1 (IDO1) inhibitor epacadostat had no impact on the growth of E0771.
Conclusions
These data enable rational combination strategies and provide alternatives to breast cancer studies in 4T1.
P299 Cell therapy - TRAcking, Circulation, & Safety (CT-TRACS): The Health and Environmental Sciences Institute (HESI)’s new collaborative effort to address the challenges of cell therapies translation
Brooke Helfer1, Charles O'Hanlon1, Patricia Murray2, Keiji Yamamoto3, Gregory Mullen4, Carla Herberts5, William Shingleton6, Lucilia Mouriès7
1Celsense, Inc, Pittsburgh, PA, USA; 2University of Liverpool, Liverpool L69 3BX, United Kingdom; 3Takeda Pharmaceutical Company Limited, Fujisawa, Kanagawa, Japan; 4King's College London, London, United Kingdom; 5Medicines Evaluation Board, Netherlands, 3531 AH Utrecht, Netherlands; 6GE Healthcare, Amersham, United Kingdom; 7ILSI Health and Environmental Sciences Institute, Washington, DC, USA
Correspondence: Brooke Helfer (brooke@celsense.com)
Background
Cell therapies show great therapeutic promise in the field of immunotherapy. To realize their full clinical potential there is a need for greater understanding of their mode of action, migration after administration, delivery, persistence at sites of action, and whether their localization or distribution may cause safety issues. There are several existing and emerging tools available to develop pharmacokinetics data on cell-derived therapies to improve our understanding but adoption by investigators has been limited. Furthermore, the regulatory landscape is not clearly defined for these emerging therapeutics.
Methods
The Health and Environmental Sciences Institute’s Emerging Issues Committee recently launched a multi-sector collaborative sub-committee to identify key needs for assessing the safety of cell therapies and identify opportunities to meet these needs. This program, the Cell Therapy - TRAcking, Circulation, & Safety (CT-TRACS) sub-committee, provides a platform for developers, researchers, regulators, imaging specialists and other stakeholders to interact, discuss challenges and identify best practices to ensure that therapies are safe and effective. The sub-committee aims to bring awareness on how existing cell tracking technologies, methods, and best practices can benefit the clinical translation of these new therapies.
Results
Since its inception in December 2015, CT-TRACS gathered more than 60 members from 25 organizations across the United States, Europe and Japan. The sub-committee has convened monthly and the focus of the group has been narrowed to Cell Fate, i.e., distribution, survival/engraftment and phenotype, post-administration, in vivo, as well as evaluating the tumorigenic potential of cell-based therapies. Our initial goals have been to: 1. evaluate current cell-based therapies safety assessment practice and tools; 2. develop best practices for application of available tools for safety assessment of cell therapies and/or identify gaps in safety assessment; 3. organize a workshop to present findings of the sub-teams and develop recommendations for next steps; and 4. initiate a manuscript describing the needs and gaps identified, to build confidence in safety assessment approaches for clinical applications. To date, we have held our first scientific session at an international cell therapy meeting and are compiling the results of a survey of therapeutic stakeholders’ needs and wishes in the imaging of cellular therapeutics, survey shared herein.
Conclusions
The CT-TRACS project is open to all current HESI members as well as new participants with relevant technical expertise. The program encourages inquiries by those with interest in providing support for these innovative efforts.
P300 PSMA-associated PET imaging of CAR T cells
IL Minn1, David Huss2, Hye-Hyun Ahn1, Tamara Chinn2, Andrew Park1, Jon Jones2, Mary Brummet1, Steven Rowe1, Polina Sysa-Shah1, Yong Du1, Hyam Levitsky2, Martin Pomper1
1Johns Hopkins Medical Institutions, Baltimore, MD, USA; 2Juno Therapeutics, Seattle, WA, USA
Correspondence: Hyam Levitsky (david.huss@junotherapeutics.com); Martin Pomper1
Background
Chimeric antigen receptor (CAR) T cells have demonstrated clinical benefit in numerous hematological malignancies and hold promise for application in solid tumors. As a “living drug”, CAR T cells traffic throughout the body with dynamic expansion and contraction kinetics. This dynamic nature limits the utility of standard blood pharmacokinetic analyses where the temporal and spatial distribution of CAR T cells is only partially captured. Therefore, a non-invasive imaging technique that allows tracking of CAR T cells may enhance the understanding of in vivo CAR-T behavior and inform associations with safety and efficacy. Previous attempts to track gene modified T cell therapies with PET imaging have been limited by immunogenicity of PET reporters, poor tissue penetration of large molecular weight PET ligands and/or limited sensitivity.
Methods
To improve the ability to track CAR T cells, we engineered CD19-directed CAR T cells to express a truncated form of human prostate-specific membrane antigen (tPSMA) as a reporter. We then used the PSMA-directed small molecule PET ligand [18F]DCFPyL for in vitro and in vivo CAR T visualization.
Results
We demonstrate high level surface co-expression of tPSMA and the CD19-directed CAR. The addition of tPSMA to the CAR T cells did not impact in vitro or in vivo anti-tumor functionality. Phantom imaging studies demonstrated that PET with [18F]DCFPyL could reliably detect as few as 4000 CD19-tPSMA CAR T cells in a 50uL volume. We developed a spontaneous metastatic acute lymphoblastic leukemia model by subcutaneously injecting CD19+ Nalm6-ffLuc-eGFP tumor cells into immunodeficient NSG mice. Metastatic lesions were observed in liver, spleen and bone marrow, providing an opportunity to evaluate CAR T infiltration into primary subcutaneous and secondary disseminated tumor sites. CD19-tPSMA CAR T cell treatment eradicated Nalm6 tumors from mice up to 90 days post-treatment. PET/CT with [18F]DCFPyL visualized infiltration of CAR T cells at primary and metastatic tumor sites, which was confirmed by immunohistochemical analyses.
Conclusions
These preclinical results establish a new technology for whole body, non-invasive tracking of CAR T cells and support translation into the clinic.
P301 A dual in vivo and in silico system to model tertiary lymphoid structure formation and anti-tumor immune response in the murine tumor microenvironment
Rana Falahat, Yohsuke Yagawa, Mark Robertson-Tessi, Susan L. Zhou, Alexander Anderson, James J. Mulé, Adam Mailloux
H. Lee Moffitt Cancer Center, Tampa, FL, USA
Correspondence: Adam Mailloux (adam.mailloux@moffitt.org)
Background
Tertiary Lymphoid Structures (TLS) are highly organized foci of lymphocytes and antigen-presenting cells that predict increased survival across multiple solid tumors, and are associated with a previously identified gene expression signature composed of twelve chemokines (12-CK-GES) [1]. These chemokines likely result from chronic lymphotoxin-beta receptor activation on resident stromal cells or by paracrine production involving subsequent infiltrates. The exact nature and timing of this induction is unclear due in part to insufficient models of TLS formation. Here, we build upon insights gained from the 12-CK-GES using an implantable three-dimensional bioscaffold to study the interaction of developing murine tumor, endogenous infiltrate, and implanted stroma. Concurrently, we use this system to parameterize an integrated mathematical model of the microenvironment, which can then re-inform our three-dimensional bioscaffold model.
Methods
Injectable bioscaffolds were prepared using primary lymph node reticular fibroblast cells implanted in either Matrigel or chitosan hydrogels loaded with recombinant Lymphotoxin-α1β2 (LT) or individual chemokines from the 12-CK-GES, with or without murine MC-38 colon carcinoma cells. In some experiments, recombinant factors were loaded in lipid-coated silica microparticles to delay release. Preparations were then injected subcutaneously in C57BL/6 mice. Implants were resected at later time points and dissociated for flow cytometry or cryosectioned for histology and immunofluorescent staining. Resulting data, along with those from in vitro chemotaxis assays, were used to parameterize an in silico model of TLS formation simulated in two dimensions using fixed stromal cells, three types of immune cells, and discrete chemokine fields.
Results
LT, when implanted with stromal cells, can induce organized aggregates of endogenous lymphocytes, significantly increase infiltration of T cells, B cells, and dendritic cells (all p<0.05), and prevent the growth of MC-38 tumors in resected implants (p<0.001). In silico model runs predict polarized activation of stromal cells and subsequent production of CCL19, CCL21, and CXCL13 is sufficient to compartmentalize lymphoid aggregates into discrete B and T cell zones and promote anti-tumor activity.
Conclusions
This dual-model system has identified an important role for LT activation of stromal cells in the induction of TLSs and suggests components of the 12-CK-GES are vital for TLS organization.
References
1. Messina JL, Fenstermacher DA, Eschrich S, et al. 12-Chemokine gene signature identifies lymph node-like structures in melanoma: potential for patient selection for immunotherapy? Scientific Reports 2012; 2:765-770.
P302 Characterizing immunotherapy-induced lymphocyte infiltration at the single patient level using CANscriptTM, an ex-vivo human tumor model
Munisha Smalley, Basavaraja Shanthappa, Hans Gertje, Mark Lawson, Baraneedharan Ulaganathan, Allen Thayakumar, Laura Maciejko, Padhma Radhakrishnan, Aaron Goldman
Mitra Biotech, Woburn, MA, USA
Correspondence: Aaron Goldman (msmalley@mitrabiotech.com)
Background
The presence and activity of lymphocytes within the tumor is critical for clinical response to cancer immunotherapy, such as immune checkpoint blockade. Poor lymphocyte infiltration into the tumor, known as a ‘cold’ phenotype, is associated with modest clinical response. High baseline infiltration of effector lymphocytes is considered ‘hot’, and patients are predicted to respond more favorably to treatment. Despite these fundamental predictive biomarkers, patient-to-patient response and durability remains highly variable. There is an urgent gap in available methods to study lymphocyte infiltration, trafficking and spatial heterogeneity induced by different cancer immunotherapies in individual patients.
Methods
Here, we used CANscriptTM, an ex-vivo human tumor model that recapitulates and preserves the native, patient-autologous tumor microenvironment, including peripheral blood mononucleated cells (PBNC). Utilizing tissue from breast cancer patients classified as either ‘cold’ (N=5) or ‘hot’ (N=5), we studied lymphocyte infiltration under pressure of immune checkpoint blockade for 72h using pembrolizumab (a-PD-1) and avelumab (a-PDL-1). Using flow cytometric analysis, we characterized infiltrating lymphocytes, and coupled these data with multiplex immunohistochemistry (CD3+, CD4+, CD8+, CD56+, CD25+) to map proximity of tumor cells to lymphocytes before and after treatment, ex-vivo.
Results
We determined that immune checkpoint blockade induced unique patterns of migration and infiltration of effector T-cells (Teff), T-regulatory (Treg) cells and natural killer (NK)-cells in ‘hot’ vs ‘cold’ tumors. Furthermore, we determined that, in some instances, ‘cold’ tumors can be driven towards a ‘hot’ phenotype characterized by trafficking of active immune lymphocytes following treatment, which corresponded to differential ratio of Teff to Treg compared to baseline.
Conclusions
Taken together, these data demonstrate the utility of CANscriptTM as a platform to characterize response to immunotherapy in a spatial context. Such an advance in our preclinical methods to study immuno-modulators at the individual patient level can help guide treatment decisions for clinicians while simultaneously functioning as a platform to study and discover mechanisms of clinical efficacy for emerging drugs.
References
1. Brijwani N, Jain M, Dhandapani M. Rationally co-targeting divergent pathways in KRAS wild-type colorectal cancers by CANscript technology reveals tumor dependence on Notch and Erbb2, Sci. Rep. 2017;7:1502-1513.
2. Majumder B, Baraneedharan U, Thiyagarajan S. Predicting clinical response to anticancer drugs using an ex vivo platform that captures tumour heterogeneity, Nat. Comm. 2015;6:6169-6183.
P303 Immune checkpoint inhibitor responses in humanized mouse melanoma models using patient-derived xenografts
Rajasekharan Somasundaram, Marilda Bequiri, Ling Li, Meenhard Herlyn, Meaghan Kiernan, Kar Muthumani, Hyeree Choi
The Wistar Institute, Philadelphia, PA, USA
Correspondence: Rajasekharan Somasundaram (shyam@wistar.org)
Background
Melanoma patients develop resistance to both chemo- and targeted-therapy drugs. Promising pre-clinical and clinical results with immune checkpoint inhibitors using antibodies directed against CTLA-4 and PD-1 have re-energized the field of immune-based therapies in melanoma. However, only a third of melanoma patients respond to immune checkpoint blockade. Currently available mouse xenograft and transgenic mouse melanoma models have several short comings and are unable to address the basis of drug resistance and immune non-responsiveness that are frequently observed in melanoma patients. Thus, there is an urgent need to establish an in vivo model with a human immune microenvironment that can address issues of therapy resistance.
Methods
For this, our laboratory has developed a humanized mouse melanoma model using patient-derived xenografts (PDX). Immunodeficient NSG mice are reconstituted with human CD34+ cells and after 7-9 weeks, mature human CD45+ cells are observed in circulating blood. Humanized mice were then challenged with HLA-matched melanoma PDX and the functional ability of human immune cells to restrict tumor growth is monitored.
Results
Delayed tumor growth was observed in humanized mice indicating in-vivo sensitization of human immune cells to melanoma. This was confirmed by in-vitro demonstration of human lymphocytes from tumor-bearing mice showing enhanced cytokine expression after stimulation with melanoma antigen peptides. Further, cytotoxic T-cells derived from melanoma peptide stimulation could functionally lyse tumor cells in vitro. In preliminary therapy studies, most tumor-bearing humanized mice treated with anti-PD-1 antibody showed restricted tumor growth. Anti-PD-1 antibody therapy resulted in enhanced infiltration of CD4+ and CD8+ T-cells that correlated with tumor response.
Conclusions
Our results suggest that humanized mouse melanoma model can be explored further to understand the causes of therapy resistance and immune non-responsiveness.
P304 Predicting pre-clinical tumour response to anti-PD-1 immunotherapy with computational modelling
Damijan Valentinuzzi1,2, Urban Simončič1,2, Katja Uršič3, Martina Vrankar3, Maruša Turk2, Robert Jeraj1,2,4
1Jožef Stefan Institute, Ljubljana, Slovenia; 2Faculty of Mathematics and Physics – University of Ljubljana, Ljubljana, Slovenia; 3Institute of Oncology Ljubljana, Ljubljana, Slovenia, Ljubljana, Slovenia; 4University of Wisconsin – Madison (WI)
Correspondence: Damijan Valentinuzzi (damijan.valentinuzzi@ijs.si)
Background
Majority of patients fail to respond to anti-programmed death-1 (anti-PD-1) immunotherapy and the reasons for this remain largely unknown. The aim of this study was to develop a computational model, able to predict antitumour response to anti-PD-1 on the basis of key biological properties that might serve as predictive biomarkers of response.
Methods
Interplay between tumour cells and tumour infiltrating lymphocytes (TILs) is described with deterministic population model. It incorporates intrinsic tumour parameters (growth rate (k)), as well as other biological parameters, such as major histocompatibility complex (MHC) class I and PD-1 ligand (PD-L1) appearance on tumour cells, PD-1 appearance on TILs, anti-PD-1 pharmacodynamics, dosing and scheduling regimen, etc. Most of the parameters were taken from literature. The remaining free parameters (k, TILs infiltration rate, tumour cells-TILs interaction rate) were fitted to experimental data, where B16-F10 melanoma was treated with anti-PD-1 [1]. Predictive ability of the model was tested on independent experiment from literature, namely B16-OVA treated with anti-PD-1 [2]. Simulated tumour growth curves were compared to experimental data and sensitivity study of key parameters was performed.
Results
Simulated tumour growth curves are in good agreement with experimental data. Maximum deviation of simulated control tumour volume (solid line – blue) to experimental data (blue squares) is −25% (day 13). On the other hand, the model slightly underestimates the effect of treatment with anti-PD-1. Maximum deviation of simulated anti-PD-1-treated tumour volume (solid line – red) to experimental data (red squares) is +40% (day 17). Sensitivity study reveals that dosing and scheduling regimen does not importantly affect treatment outcome (data not shown). The most sensitive parameter of the model is MHC class I appearance. By modulating MHC class I appearance from 1% to 100%, while keeping other parameters fixed, we are able to simulate responders as well as non-responders to anti-PD-1.
Conclusions
Model predictions of antitumour response to anti-PD-1 are within expectations. The predictions might be even more reliable if model parameters were, due to their inter-patient variability and presumably dynamic nature, actually measured for every specific experiment/patient. The emphasis should be on MHC class I appearance as it might be one of the predictive biomarkers of response to anti-PD-1.
References
1. Kleffel, Sonja, et al. Melanoma cell-intrinsic PD-1 receptor functions promote tumor growth. Cell. 2015; 162.6:1242-1256.
2. Sánchez-Paulete, Alfonso R., et al. Cancer immunotherapy with immunomodulatory anti-CD137 and anti–PD-1 monoclonal antibodies requires BATF3-dependent dendritic cells. Cancer discovery. 2016; 6.1:71-79.
P305 Unravelling the immune contexture of pre-invasive lesions of the lung by multispectral imaging
Angela Vasaturo1, Celine Mascaux2, Mihaela Angelova1, Benedict Buttard1, Jean-Paul Sculier3, Jerome Galon1
1Centre de Recherche des Cordeliers, Université Pierre et Marie Curie, Sorbonne Universités, Paris, France; 2Aix Marseille University, APHM, Marseille, France; 3Institut Jules Bordet, Centre des Tumeurs de l'Université Libre de Bruxelles, B-1000 Brussels, Belgium, Bruxelles, Belgium
Correspondence: Jerome Galon (angelavasaturo@gmail.com)
Background
Lung cancer is the leading cause of cancer deaths world- wide and despite advances in therapy, the overall survival rate for lung cancer patients remains only 15%.
As most of sporadic cancers, lung cancer emerges from pre-neoplastic lesions characterized by morphological and molecular changes. If morphological changes of pre-invasive bronchial lesions are well characterized, the cause and effect relationship between those changes and the immune response is still un- known. Though, we identified gene expression alterations that suggest a role of the innate and adaptive immunity in the transformation towards carcinoma.
Methods
In order to characterize the evolution of the immune response in pre-invasive bronchial lesions, we have optimized different multispectral 7 colors immunofluorescence panels, by using the Tyramide Signal Amplification (TSA) technology.
Results
We have performed multiplex staining on FFPE human bronchial biopsies (N=114) at 8 successive morphological stages of lung squamous carcinogenesis, from normal, to low grades dysplasia, high grades, to carcinoma. Images of each biopsy have been acquired multispectrally and digitally analyzed to identify and quantify the density and the tissue distribution of different immune cell types. We aimed to characterize the immune infiltrates at different stages of carcinogenesis and elucidate the role of different cell subtypes in tumor development and progression and the possible causal relationship between the immune phenotypes in pre-neoplastic lesions and tumor progression and patient prognosis.
Conclusions
Evaluation of the immune contexture and prognostic assessment of precancerous lesions of the lung may identify promising new biomarkers for early detection and targets of novel therapeutic strategies for lung cancer.
Immune Modulation, Cytokines, and Antibodies
P306 A protein extract from fermented wheat germ promotes NK cell-mediated lymphoma eradication in mouse xenografts
Gustavo Barisone1, Yunpeng Ma1, Mastewal Abuhay1, Robert O'Donnell1, Kathleen Lundeberg1, Sonia Gowda1, William Murphy2, Joseph Tuscano1
1University of California Davis, Sacramento, CA, USA; 2University of California, Sacramento, CA, USA
Correspondence: Joseph Tuscano (gabarisone@ucdavis.edu)
Background
Proteomic and genomic data has allowed for the development of promising targeted agents for NHL [1]. Most have acute and chronic toxicities that limit efficacy. The use of complementary and alternative medicines has increased during the last decade. However, scientific evidence of their efficacy is scarce. Fermented wheat germ extract (FWGE) has been claimed to have anti-cancer properties in many tumor types. FWGE therapeutic activity has been attributed to its content of benzoquinones [2].
Methods
A protein fraction (FWGP) was isolated by FPLC and proteins identified by mass spectrometry. Direct cytotoxic was studied in vitro using NHL cell lines. Immunomodulatory properties were evaluated ex vivo by measure immune cell activation in human PBMCs and isolated NK cells. In vivo experiments used nu/nu NHL xenografts with or without NK cell depletion; endpoints were tumor volume and toxicity. In vivo immunomodulatory effects were evaluated by treating tumor-free BALB/c mice with FWGP and measuring NK cell killing activity and degranulation.
Results
FWGP was cytotoxic in 17 cancer cell lines (IC50 = 20-171 μg/ml in NHL, 12-27 μg/ml in colon and 70-144 μg/ml in lung) and induced apoptosis by increasing levels of caspase-3, PARP, BAK, BAD and p53, while reducing levels of AKT. FWGP increased % NK cells, production of IFg and GrB, and NK-mediated killing. In vivo efficacy was confirmed, with no toxicity, in pre-emptive and established models. In vivo treatment with FWGP+rituximab was as effective as R-CHOP, with 90% complete remission. NK depletion resulted in no response to FWGP. These results support the hypotheses that FWGP augments NK-mediated tumor killing. Proteomic profiling identified 844 proteins. An active fraction consisted of 169 proteins.
Conclusions
FWGP represents a promising immunomodulatory agent with anti-tumor activity, minimal toxicity and low cost. Our results suggest FWGP has direct lymphomacidal activity by inducing apoptosis and indirect anti-tumor efficacy by enhancing NK-mediated tumor eradication. Further experimental validation will allow translation of al “alternative” product into mainstream medicine.
References
1. Abuhay, M., et al. The HB22.7-vcMMAE antibody-drug conjugate has efficacy against non-Hodgkin lymphoma mouse xenografts with minimal systemic toxicity. Cancer Immunol Immunother. 2016; 65(10):1169-75.
2. Hidvegi, M., et al. MSC, a new benzoquinone-containing natural product with antimetastatic effect. Cancer Biother Radiopharm. 1999. 14(4):277-89.
P307 Expression and function of PD-1 and TIM-3 in non-small cell lung cancer (NSCLC)
Jonathan Travers, Krtisten McEachern, Srimoyee Ghosh, Sridhar Ramaswamy, David Jenkins
1TESARO Inc., Waltham, MA, USA
Correspondence: David Jenkins (dianna.bartel@ashfieldhealthcare.com)
Background
The use of anti-programmed cell death protein 1 (PD-1) and PD-Ligand 1 agents in the treatment of non-small cell lung cancer (NSCLC) has been well established but many patients are either intrinsically resistant or become refractory during therapy. One potential resistance mechanism is the upregulated expression of additional checkpoint receptors such as T cell immunoglobulin and mucin domain 3 (TIM-3), a transmembrane receptor that binds multiple putative ligands, and that has been shown to negatively regulate the function of T cells that co-express PD-1 [1].
Methods
We examined the immunophenotype and checkpoint receptor expression of over 100 NSCLC samples from primary surgical resections. From a subset of these samples, we evaluated T cell functional status by gene expression analysis on sorted PD-1+ and TIM-3+ CD8+ T cells as well as ex vivo stimulation assays to evaluate cytokine production. Furthermore, we used ex vivo and in vivo studies to assess the effect of blockade of PD-1 and TIM-3 alone and in combination on T cell activation and anti-tumor activity.
Results
We showed that primary NSCLC samples display heterogeneity in both their baseline immune infiltrate and also PD-1 and TIM-3 checkpoint receptor expression. We examined mRNA expression of multiple immune genes on sorted PD-1+ and TIM-3+ CD8+ T cells, and found that PD-1/TIM-3 double positive cells express reduced interleukin-2 (IL-2) and tumor necrosis factor alpha (TNFα), but similar mRNA levels of interferon gamma (IFNγ) when compared to double negative cells. This phenotype is recapitulated in CD8+ T cells derived from patient samples stimulated with PMA and ionomycin, where we found PD-1 and TIM-3 double positive cells to be significantly deficient in IL-2, but not IFNγ production. Importantly, in addition to their expression being associated with T-cell dysfunction, we also found that blockade of PD-1 and TIM-3 was associated with increased T cell activation and anti-tumor activity in ex vivo and in vivo models, suggesting a potential functional role for the inhibition of TIM-3, in addition to PD-1, in the enhancement of anti-tumor immunity.
Conclusions
Taken together, these data provide further evidence that TIM-3 may play a role in intrinsic resistance to single agent anti-PD1 therapy in NSCLC and support evaluating the combination of anti-PD-1 and anti-TIM-3 agents in the clinic.
References
1. Koyama, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7:10501.
P308 A PD-1 x CTLA-4 bispecific DART® protein with optimal dual checkpoint blockade and favorable tolerability in non-human primates
Alexey Berezhnoy1, Kurt Stahl1, Kalpana Shah1, Tim Gaynutdinov1, Gurunadh Chichili1, Daorong Liu1, Rebecca Johnson1, Ross La Motte-Mohs1, Jessica Hill1, Jonathan Li2, Sergey Gorlatov1, Valentina Ciccarone1, Ralf Alderson1, Hua Li1, James Tamura1, Jennifer Brown1, Jon Wigginton1, Ezio Bonvini1, Paul Moore1, Syd Johnson1
1Macrogenics, INC, Rockville, MD, USA; 2Macrogenics, INC, South San Fancisco, CA, USA
Correspondence: Alexey Berezhnoy (berezhnoya@macrogenics.com)
Background
Immunotherapy with the combination of monoclonal antibodies that block PD-1 and CTLA-4 has shown clinical benefit beyond that observed with either mAb alone. A PD-1xCTLA-4 bispecific DART protein was designed to induce antitumor immunity through simultaneous targeting of both checkpoint pathways via administration of a single molecule. The DART protein increases checkpoint blocking activity on PD-1/CTLA-4 dually expressing cells, while displaying distinct immunological effects of CTLA-4 blockade in vivo absent evidence of toxicity.
Methods
A PD-1xCTLA-4 DART protein was engineered as a tetravalent bispecific molecule from humanized anti-PD-1 and anti-CTLA-4 mAb sequences in a human hinge-stabilized IgG4 backbone. PK, PD and toxicology studies were performed in cynomolgus monkeys.
Results
The PD-1xCTLA-4 DART molecule demonstrated binding to immobilized PD-1 protein and PD-1-expressing cells lines, inhibition of PD-1 interaction with PD-L1 or PD-L2, as well as reversal of PD-1-mediated T-cell signal inhibition in gene-reporter assays comparable to that supported by a replica of nivolumab. Similarly, binding, ligand blocking and rescue of CTLA-4-mediated T-cell suppression was comparable to that supported by a replica of ipilimumab. The DART molecule demonstrated activation properties comparable to the combination of ipilimumab and nivolumab replicas in a variety of human primary T-cell assays and showed enhanced B7-ligand binding blockade over that mediated by the ipilimumab replica on PD-1/CTLA-4 double-positive cells. In the cynomolgus monkey, the PD-1xCTLA-4 DART molecule exhibited a PK profile consistent with that of an IgG4 and was well tolerated, with no mortality or significant adverse findings up to 75 mg/kg QWx3, the highest dose tested. T-cell expansion in the peripheral blood and lymphoid organs was observed, which was attributable to the CTLA-4 blocking arm, since no such finding was observed with similar or higher doses of the anti-PD-1 constituent of the bispecific molecule.
Conclusions
PD-1xCTLA-4 DART protein binds and blocks its targets, with increased activity on dual PD-1/CTLA-4-expressing cells.
The DART molecule enhances T-cell responses in vitro to the level achieved by a combination of nivolumab and ipilimumab replicas.
PD-1xCTLA-4 DART protein was well tolerated in cynomolgus monkeys, with a safety profile similar to that observed with PD-1 blockade alone, while demonstrating biological effects of CTLA-4 antagonism.
The favorable safety and tolerability profile of the PD-1xCTLA-4 DART molecule combined with its enhanced activity on PD-1/CTLA-4 double-positive cells suggest a potential for an improved therapeutic window for PD-1/CTLA-4 co-blockade strategies, with the administration of a single molecule providing dosing convenience and ease of incorporation into additional therapeutic regimens.
P309 Treatment with heterodimeric IL-15 promotes effector T cell infiltration into tumors
Cristina Bergamaschi1, Konstantinos Dimas1, Dimitrios Stellas1, Bethany Nagy1, Shawn Jensen2, Bernard Fox2, Barbara Felber1, George Pavlakis1
1National Cancer Institute at Frederick, Frederick, MD, USA; 2Earle A Chiles Research Institute, Portland Providence Cancer Center, Portland, OR, USA
Correspondence: Cristina Bergamaschi (cristina.bergamaschi@nih.gov)
Background
The presence of tumor-infiltrating effector T cells is considered the most predictive biomarker for clinical benefit in response to immunotherapies. IL-15 is a cytokine important for the proliferation, activation and mobilization of lymphocytes, including natural killer and CD8+T cells. We have previously shown that bioactive IL-15 in vivo comprises a complex of the IL-15 chain with the IL-15 receptor alpha chain that are together termed heterodimeric IL-15 (hetIL-15). Several preclinical models have indicated the ability of IL-15 to enhance the response of the immune system against cancer, and based on these results hetIL-15 has advanced to clinical trials.
Methods
We have produced hetIL-15 and tested its anti-tumor activity in several murine cancer models. Analysis of lymphocytes in lymphoid organs and in tumors was performed by flow cytometry and multi-color immunohistochemistry. Chemokine and cytokine levels were determined using electrochemiluminescence (MSD) and ELISA assays.
Results
Repeated injections of hetIL-15 in mice were effective in delaying tumor growth in the MC38 colon carcinoma, TC-1 cervical carcinoma and B16 melanoma models. The combination of hetIL-15 and adoptive cell transfer of melanoma specific Pmel-1 cells showed anti-tumor efficacy in B16-bearing mice in absence of lymphodepletion. The E0771 orthotopic breast cancer model showed delay in tumor progression and significantly reduced lung metastasis upon hetIL-15 treatment. A significantly reduced onset of lung metastasis was also observed in 4T1 breast cancer-bearing mice. Flow cytometry and multi-color immunohistochemistry assays showed increased trafficking and persistence of CD8+ T cells, including tumor specific T cells, into the tumors and an increased CD8+/Treg ratio, upon hetIL-15 administration. Importantly, hetIL-15 treatment led to preferential enrichment of adoptively transferred tumor-specific CD8+T cells in the B16 tumor in an antigen-dependent manner. Tumor infiltration by CD8+T cells was accompanied by increased plasma levels of CXCL10. Tumor-resident CD8+ T cells showed features of activated effector cells with enhanced proliferation (Ki67+) and high cytotoxic potential (Granzyme B+). Upon ex-vivo stimulation, an increased frequency of both CD8+ and CD4+ T cells producing IFNg was observed in the tumors of mice treated with hetIL-15.
Conclusions
Our results show that hetIL-15 administration may be a general method to enhance T cell entry in non-inflamed tumors, increasing the success rate of immunotherapy interventions. Preclinical cancer studies support the use of hetIL-15 in tumor immunotherapy approaches to promote the development of anti-tumor responses by favoring effector over regulatory cells. The effect of hetIL-15 on metastasis establishment in orthotopic models may provide synergies against metastatic disease.
P310 CSF1/CSF1R signaling blockade triggers release of matrix-degrading proteases in mouse models
Stefan Bissinger1, Martina Schmittnaegel2, Ioanna Keklikoglou2, Michele De Palma2, Sabine Hoves1, Carola Ries1
1Roche Innovation Center Munich, 82377 Penzberg, Germany; 2The Swiss Institute for Experimental Cancer Research (ISREC), School of Life Sciences Ecole Polytechnique Fédérale de Lausanne (EPFL), Lausanne, Switzerland
Correspondence: Stefan Bissinger (stefan.bissinger@roche.com)
Background
The heterotypic interplay between cancer cells and their microenvironment provides an opportunity for therapeutic targeting. The abundant tumor-associated macrophage (TAM) infiltrate can be substantially reduced in mouse tumor models and cancer patients by colony-stimulating factor 1 receptor (CSF1R) signaling blockade [1]. Notably, TAM depletion provides marked clinical benefits in diffuse-type tenosynovial giant cell tumors [2,3]. However, facial edema is reported as the most common adverse event of TAM elimination in patients [2,4]. We here sought to gain insight into the molecular mechanisms mediating edema formation. To this aim, we characterized antibody exposure, CSF1 levels and an array of extracellular matrix-degrading and restructuring metalloproteinases (MMPs) in tumor-bearing and tumor-free mice treated with an anti-CSF1R antibody.
Methods
Western Blotting and multiplex assays of tumors and sera of anti-mouse CSF1R mAb (clone 2G2)-treated mice showed an association of TAM elimination with an early intratumoral and systemic release of a specific set of MMPs, including MMP-2, -3 and -8, in multiple transplant (MC38, E0771, KPL-4 and PyMT) and de novo (MMTV-PyMT) tumor models.
Results
In addition, we found that the early increase of MMPs in the face of CSF1/CSF1R pathway blockade was independent of tumor burden and was accompanied by a significant increase of body weight in tumor-free mice following long-term exposure to the antibody. The body weight gain may therefore indicate the enhanced retention of body fluids. Discontinuation of the CSF1R antibody reinstated unaltered body weight and MMP levels. We excluded platelets and neutrophils as sources of MMP release, even if they accumulated in tumor-bearing mice during CSF1R inhibition. We also examined the effects of blocking the CSF1R ligand CSF1, which is one of the two known ligands of the CSF1R [2,4]. CSF1 antibodies had no impact on the binding of IL-34 (the second CSF1R ligand) to CSF1R. Similar to CSF1R blockade, an anti-mouse CSF1 antibody (clone 5A1) provoked an early systemic surge of a subset of MMPs.
Conclusions
Collectively, our data suggest that CSF1 and CSF1R blocking antibodies induce the release of a distinct set of matrix-degrading proteases (MMP-2,-3 and -8), but not metastasis-promoting proteases (MMP-9 and -12), which may be potentially causative of edema formation. Further studies may inform optimized dosing schedules of CSF1/CSF1R targeting regimens for cancer patients.
References
1. Ries CH, et al. Cancer Cell 2014;25(6):846-59.
2. Cassier PA, et al. Lancet Oncol 2015;16(8):949-56.
3. Tap WD, et al. N Engl J Med 2015;373(5):428-37.
4. Papadopoulos KP, et al. Clin Cancer Res. 2017.
P311 Clinical outcomes of PD-1 inhibition by PD-L1 expression level across malignancies in 204 consecutive patients in a real world oncology setting
Kenneth Byrd1, Tristan Hayes1, Mike Martin2, Lee Schwartzberg2, Ari Vanderwalde2
1University of Tennessee Health Sciences Center, Memphis, TN, USA; 2West Cancer Center, Germantown, TN, USA
Correspondence: Kenneth Byrd (kbyrd@westclinic.com)
Background
The utility of the PD-L1 biomarker in predicting response to anti-PD-1 agents has been inconsistent across malignancies. In this study, we describe outcomes of patients treated with anti-PD-1 agents by PD-L1 expression level.
Methods
Molecular profiling of tumors in patients with advanced cancer was performed at West Cancer Center using Caris Molecular Intelligence Profile testing, which includes PD-L1 percentage assessed by immunohistochemical staining. Patients were included in this retrospective analysis if they were treated with a PD-1 inhibitor and had available PD-L1 results between November 2014 and May 2017. Patients were assessed by PD-L1 expression level, defined as negative (0% expression) or positive (>1% expression), regardless of staining intensity. PD-L1 positive samples were further subclassified into PD-L1 low (1-4%), intermediate (5-49%) and high (≥50%). Best overall response using RECIST 1.1 criteria was retrospectively assessed using 2-physician review of radiologic data. Progression free survival (PFS) and overall survival (OS) were assessed using the Kaplan-Meier method.
Results
204 patients with quantifiable PD-L1 expression were treated with PD-1 inhibitors. Primary tumors included 125 non-small cell lung cancers, 31 melanomas, 12 renal cell carcinomas, and 36 others. 110 (54%) tumors were PD-L1 negative and 94 (46%) were PD-L1 positive (22 [11%] low, 37 [18%] intermediate, and 35 [17%] high). ORR was 39% for PD-L1 positive versus 17% for PD-L1 negative (p=<0.001). Best response for each PD-L1 level is shown in Table 1. The estimated median PFS was 6.4 months for PD-L1 positive versus 3.0 months for PD-L1 negative (HR 0.59; p=0.001; 95% CI, 0.43 to 0.81). The estimated median OS was 17.3 months for PD-L1 positive versus 6.9 months for PD-L1 negative (Fig. 1; HR 0.64; p=0.021, 95% CI, 0.44 to 0.94). Increasing PD-L1 expression was associated with a statistically significant improvement in PFS and OS. There was a statistically significant difference among PFS and OS with increasing PD-L1 levels (PFS p = 0.002; OS p = 0.026). Multivariate analysis did not identify tumor type as a predictor of response.
Conclusions
PD-L1 staining of any level predicted for improvements in ORR, PFS, and OS with PD-1 inhibitors across multiple malignancies. The higher the PD-L1 staining, the greater the likelihood of benefit. These data provide important real world confirmation for the potential utility of global PD-L1 testing in clinical practice, regardless of malignancy.
P312 AGEN2034, a novel anti-PD-1 antibody that combines effectively with CTLA-4 pathway blockade to enhance T cell activity
Dhan Chand1, David Savitsky1, Ana Gonzalez1, Christopher Clarke1, Andrea Schuster2, Elise E. Drouin1, Jeremy D. Waight1, Cornelia Mundt2, Gerd Ritter1, Taha Merghoub4, David Schaer4, Rikke B Homlgaard4, Roberta Zappasodi4, Marc van Dijk5, Jennifer S. Buell1, Jean-Marie Cuillerot1, Robert Stein1, Nicholas S Wilson1
1Agenus Inc., Lexington, MA, USA; 2Former employee of Agenus Switzerland Inc., Basel, Switzerland; 4Memorial Sloan Kettering Cancer Center, New York, NY, USA; 5Agenus Switzerland Inc., Basel, Switzerland
Correspondence: Dhan Chand (dhan.chand@agenusbio.com)
Background
PD-1 (or CD279) is a co-inhibitory receptor that suppresses T cell function upon binding to its ligands, PD-L1 or PD-L2. PD-1 signaling functions cooperatively with CTLA-4 to limit T cell activation during priming by antigen presenting cells, leading to reduced proliferation, cytokine and chemokine production and cell survival. Anti-PD-1 antibody therapies that block the interaction between PD-1 and its ligands have shown durable clinical benefit both as single agents, but particularly in combination with antibodies that antagonize CTLA-4.
Methods
AGEN2034 (anti-PD-1; IgG4) was discovered using a proprietary mammalian display technology, Retrocyte Display™. Binding kinetics and affinity to PD-1 were characterized by surface plasmon resonance. Cell-based potency was determined in a Jurkat PD-1+ NFAT reporter-based assay where luciferase activity was measured as an endpoint of PD-1/PD-L1 antagonism. The pharmacological effects of AGEN2034 alone or in combination with CTLA-4 pathway blockade using a novel high affinity anti-CTLA-4 IgG1 antibody, AGEN1884, was assessed in vitro using peripheral blood mononuclear cells (PBMC) from healthy donors. Prior to human clinical trials, pharmacokinetic and pharmacodynamic profiling of AGEN2034 were performed in cynomolgus monkeys both alone and in combination with CTLA-4 blockade.
Results
AGEN2034 selectively binds to human and cynomolgus PD-1 with high affinity (Kd<1nM) and sub-nanomolar EC50. The Fc interactions of AGEN2034 are minimized via selection of a human IgG4 Fc region. In primary human immune cell assays, AGEN2034 showed a dose-dependent increase in T cell cytokine and proliferative responses. Notably, AGEN2034 combined effectively with AGEN1884 in a dose-dependent manner to further enhance T cell responsiveness. AGEN2034 was well tolerated, and no-observed-adverse-effect level (NOAEL) could be established up to 40 mg/kg in non-human primates. Furthermore, the combination of AGEN2034 and anti-CTLA-4 blockade promoted a dynamic pharmacodynamic response in cynomolgus monkeys, including a transient increase in proliferation and ICOS (inducible co-stimulator molecule) expression in a subset of central memory and effector memory T cells.
Conclusions
The functional attributes of AGEN2034 combined with the favorable pharmacokinetic and pharmacodynamic profile in cynomolgus monkeys are ideally suited for clinical development. Moreover, AGEN2034 combined effectively with CTLA-4 blockade in a range of preclinical assays to enhance antigen-specific T cell responsiveness and produced a dynamic pharmacodynamic response in non-human primates. AGEN2034 is currently under evaluation in a Phase 1/2 study in subjects with advanced tumors and cervical cancer (NCT03104699) and clinical studies to evaluate AGEN2034 in combination with AGEN1884 are planned.
P313 In vivo effect of albumin binding domains attached to immune modulators
Haomin Huang, Keneshia Haenssen, Anil Bhate, Supriya Sanglikar, John Baradei, Shan Liu, Senthil Kumar, Zihao Cui, Richard Hampton, Robert Kramer, John Cini
Sonnet BioTherapeutics, Cranbury, NJ, USA
Correspondence: Haomin Huang (johncini@sonnetbio.com); Keneshia Haenssen; Anil Bhate; Supriya Sanglikar; John Baradei; Shan Liu; Senthil Kumar; Zihao Cui; Richard Hampton
Background
Recombinant therapeutic proteins < 50Kd (eg., receptor ligands, cytokines) exhibit short circulation half-lives (mins/hour vs. days for IgGs) which limit their therapeutic utility. A specific way to increase the pharmacokinetic half-life of these agents is via conjugation to circulating albumin. Here we describe the creation of an albumin binding single chain fragment antibody (ScFv-ABD) that binds albumin in circulation, is recycled by binding to the FcRn (similar to IgG’s) and then recycled after cellular uptake resulting in increased half-life of the appended therapeutic protein. A second advantage to linking a therapeutic protein to an ScFv-ABD is improved tumor target delivery as numerous studies have shown that albumin accumulates in tumors and inflamed tissues.
Methods
Sonnet BioTherapeutics, using a XOMA phage library, has developed scFv ABD fusion constructs with several different small therapeutic proteins (recombinant interleukin proteins and scFvs targeting relevant immune-oncology receptors). These various ABD constructs have high binding affinity to mouse, human & cyno circulating serum albumin thereby preventing renal clearance and retaining benefits of FcRn mediated recycling of albumin for extended PK. Early studies to investigate improved tumor accumulation translates into anti-tumor efficacy in vivo and have shown that ScFv-ABD enhances tumor targeting.
Results
Our characterized scFV-ABD constructs have demonstrated that;
biologic activity is retained when the therapeutic protein is attached via the N- or C-terminus suggesting utility for delivering more than one therapeutic protein/scFv;
half-life in mouse serum in vivo was extended from minutes to hours/days for three different recombinant proteins and scFv with MWs of 10-80Kd;
in an established B6F10 melanoma model these ABD constructs have demonstrated markedly superior reductions in tumor growth and improved overall survival compared to the same constructs without the ABD. Superior efficacy was observed with lower doses and with a single dose of the ScFv-ABD constructs vs free recombinant protein
Conclusions
We will describe several examples of improved half-life, tumor accumulation and efficacy using our albumin linkage approach that is leading to the selection of drug candidates for clinical development.
P314 Glioblastoma stem-like cell targeting antibodies identified using yeast display biopanning
Paul Clark, Michael Zorniak, Benjamin Umlauf, Yongku Cho, Eric Shusta, John Kuo
University of Wisconsin - Madison, Madison, WI, USA
Correspondence: John Kuo (clark@neurosurgery.wisc.edu)
Background
BACKGROUND: Glioblastoma stem-like cells (GSCs) are hypothesized to evade current therapies and cause tumor recurrence, contributing to poor patient survival. Existing cell surface markers for GSCs are developed from embryonic or neural stem cell systems; however, currently available GSC markers are suboptimal in sensitivity and specificity. We hypothesized that the GSC surface proteome could be mined with a yeast display antibody library to reveal novel immunophenotypes.
Methods
MATERIALS AND METHODS: A naïve yeast expression library of single-chain human antibodies (scFv) was mined using biopanning against patient-derived GSCs. Discovered unique clones were characterized for qualitative binding affinity against 5 patient-derived GSC lines, 5 matched non-GSC/GBM lines, and 2 normal cell lines. Presumptive GSC-specific antibodies were purified from yeast, and GSC specificity and affinity determined by confocal microscopy and flow cytometry. GSC targeting in vitro was evaluated using flow cytometry, and in vivo after intravenous administration of near infrared fluorescent tagged scFv to immunodeficient mice harboring orthotopic GBM xenografts.
Results
RESULTS: Nine rounds of positive selection against patient-derived GSCs enriched for GSC-binding scFv, with selected pools also negatively screened against normal human astrocytes, neural stem cells, and serum-cultured GBM (i.e. non-GSC). We identified 62 unique scFv clones from ~600 candidates by differential PCR and restriction analysis. Clone 9.7 (heavy chain only, termed VH-9.7) demonstrated specificity against 5 patient-derived GSC lines, with minimal binding to non-GSC and normal controls. Purified VH-9.7, produced predominantly in a monomeric form, had a GSC binding affinity (Kd) of 74.3 ± 9.85 nM. Flow cytometry using 5 GSC lines verified VH-9.7 specific GSC labeling: 17-115-fold higher compared to normal astrocytes and 10-65-fold higher compared to non-GSC/GBM lines. After intravenous injection, VH-9.7 significantly localized to GSC-derived GBM xenografts in mice [92 ± 11 relative fluorescent units (RFI), n=3, p<0.05], compared to control non-targeting scFv (11 ± 7.8 RFI).
Conclusions
CONCLUSIONS: Rapid screening via yeast antibody library biopanning identified human-specific antibodies that demonstrated GSC specificity compared to both non-GSC (i.e. bulk of GBM) and normal neural cells. Identified antibodies could potentially be developed into immunotargeted diagnostics and therapeutics in brain cancer.
P315 A bispecific fusion protein, ACDClx, can selectively activate T cells against glioblastoma cells in vitro
Rebecca Cook1, Sierra Bichler1, Andrew Diamos1, Braeden Schaefer1, Andrew Niemann1, Hugh Mason1, Tsafrir Mor1, Rachael Sirianni2, Joseph Blattman1
1Arizona State University, Tempe, AZ, USA; 2Barrow Neurological Institute, Phoenix, AZ, USA
Correspondence: Rebecca Cook (Rebecca.McCall@asu.edu)
Background
Glioblastoma (GBM) is a highly invasive and fatal form of brain tumor with a median survival of approximately 15 months. Treatment for GBM is hampered by the blood-brain barrier (BBB) and small populations of cells that resist conventional therapies. There is an urgent need for innovative new therapies to target all GBM cells within a patient. Chlorotoxin is a small peptide derived from the venom of the deathstalker scorpion that has been shown to be highly selective for all GBM cells, does not bind healthy tissue, is non-toxic to humans, and has been demonstrated to cross the BBB. To this end, we have designed a T cell engaging molecule, anti-CD3/chlorotoxin (ACDClx), composed of the variable heavy (VH) and light (VL) fragments of an anti-CD3 antibody (2C11) tethered to chlorotoxin. Here, we show that T cells can be selectively activated against GBM cells, only in the presence of ACDClx.
Methods
The gene sequence encoding for His-tagged ACDClx was cloned into a deconstructed and improved geminiviral vector and introduced into agrobacteria for needleless infiltration into leaf tissue of Nicotiana benthamiana. ACDClx was extracted from tissue 4 days post-infiltration and purified via nickel affinity chromatography. T cell activation was evaluated via calcium flux assay (Fluo-4) using ionomycin as a positive control and CD69 expression using full length anti-CD3 antibody as a positive control. Activity was measured using freshly isolated mouse splenocytes and mouse GBM cells (GL261-LucNeo). For flow cytometry, lymphocytes were gated from the splenocyte population and further gated based on CD4 and CD8 markers.
Results
Expression and purification from N. benthamiana resulted in a yield of 614ug ACDClx per gram of leaf tissue with greater than 99% purity (Figure 1A). Expression of CD69 was observed when splenocytes and GBM cells were incubated with ACDClx or anti-CD3 antibody, but not with media alone (Figure 1B). T cell activation as measured by an increase in calcium flux over baseline was observed when ACDClx was added to a mixture of splenocytes and GBM cells (Figure 1C). Flux was not observed when mock isolated protein from mock-infiltrated plants was added to splenocytes and GBM, nor with splenocytes and ACDClx alone.
Conclusions
Our results indicate that ACDClx can be expressed to high levels in plant tissue and has the capacity to selectively activate T cells in vitro. These results provide further motivation for our current studies of ACDClx in vitro and in an immunocompetent model of GBM in vivo.
P316 Impact of anti- PD1 on TIL phenotype and function
Caitlin Creasy1, Cara Haymaker2, Marie-Andrée Forget2, Gopal Singh2, Coya Tapia2, Jeane Marie Painter2, Funda Meric-Bernstam2, Chantale Bernatchez2, Aung Naing2
1MD Anderson Cancer Center University of Texas Health Graduate School of Biomedical Sciences, Houston, TX, USA; 2University of Texas MD Anderson Cancer Center, Houston, TX, USA
Correspondence: Chantale Bernatchez (cacreasy@mdanderson.org); Aung Naing
Background
Therapeutic antibodies targeting PD-1 have demonstrated efficacy in several solid tumor types with durable responses in a subset of patients. However most patients will either not respond to therapy or progress after an initial response. It is not fully understood how this treatment may alter the phenotype or function of Tumor-infiltrating Lymphocytes (TIL). In this study, methods have been optimized to derive TIL from a single core biopsy and applied to growing TIL from pre-treatment and on-treatment biopsies in patients receiving anti-PD-1 to investigate drug-induced changes in TIL phenotype and function.
Methods
Tumor samples are obtained from an ongoing Phase II clinical trial of anti-PD-1 in cohorts of patients with rare solid tumor types (NCT02721732). Mandatory core biopsies are taken at baseline and on day 15-21 after the first cycle of anti-PD1 (Pembrolizumab, 200 mg). TIL are propagated ex vivo utilizing IL-2 and an agonistic anti-4-1BB antibody (Urelumab, BMS), with or without anti-CD3 (clone OKT3). TIL phenotype and function are evaluated after 2 or 3 weeks of culture.
Results
TIL cultures were first initiated by mincing one core biopsy into 3-6 tumor fragments and cultured in media containing IL-2 and the anti-4-1BB mAb. Observed TIL growth was poor with only 11% of all samples yielding over 2 million TIL per fragment after 3 weeks (2 /17 samples). The addition of anti-CD3 to the regimen dramatically improved TIL outgrowth, with 78% and 80% of the baseline biopsy and on-treatment biopsy samples, respectively, growing over 108 million TIL (n=38/49 baseline; 24/30 treatment, range 10 million to 357.7 million). Phenotypic analysis of the expanded TIL showed an effector memory differentiation status at both time points. However TIL grown from samples obtained after anti-PD-1 dosing showed enhanced proportion of CD8+ TIL and enriched CTLA-4 expression in both CD4 and CD8 TIL subsets. PD-1 expression on expanded CD8+ and CD4+ TIL was maintained or elevated after therapy. Analysis of 4 patients’ paired baseline and on-treatment TIL show that CD4+ and CD8+ TIL grown post anti-PD-1 treatment secrete significantly more of the effector cytokines IFN-γ, IL-13, and TNF-α following anti-CD3 re-stimulation.
Conclusions
Our study highlights phenotypical and functional differences of TIL after a single dose of anti-PD-1. Additionally we demonstrate that it is possible to grow TIL in numbers that would be sufficient to proceed with rapid expansion and adoptive cell transfer from one core biopsy.
Trial Registration
NCT02721732
P317 In vivo efficacy and mechanism of action of anti-TIGIT monoclonal antibody CASC-674
Peter de Vries, Robert Rosler, Janelle Taylor, Scott Peterson
Cascadian Therapeutics, Inc., Seattle, WA, USA
Correspondence: Peter de Vries (pdevries@cascadianrx.com)
Background
TIGIT is a coinhibitory immune checkpoint receptor expressed on regulatory T cells (Tregs), cytotoxic T cells and NK cells. TIGIT ligands include CD155 and CD112, which are expressed on antigen presenting cells and a variety of tumors. These ligands also bind the activating receptor CD226, often co-expressed with TIGIT, creating a network that modulates adaptive and innate immune response in a manner analogous to the CD28-CTLA4-CD80-CD86 network. We previously reported on the characterization of a panel of fully human antagonistic monoclonal antibodies that bind with subnanomolar affinity to mouse, NHP and human TIGIT and block ligand-receptor interactions and signaling in T cells (AACR, 2017). Here we report on the in vivo mechanism of action and efficacy of CASC-674 in a number of syngeneic tumor models.
Methods
A lead candidate antibody (CASC-674) was selected and produced as mouse IgG2a and tested in syngeneic in vivo mouse tumor models. To explore the mechanism of action in vivo, CT26 tumor bearing mice were treated with CASC-674 alone or in combination with anti-PD1/anti-PD-L1 and their tumors and spleens isolated and evaluated by flow cytometry for various lymphoid and myeloid cell subsets.
Results
CASC-674 as single agent has significant anti-tumor activity in the anti-PD1/anti-PD-L1 insensitive CT26 colorectal tumor model, resulting in 8/10 complete regressions (CR). Combination treatment with anti-PD-1/PDL-1 did not enhance the anti-tumor effect compared to CASC-674 alone. Tumor free mice from this study were re-challenged 8 weeks after the last treatment with CT26 cells in the opposite flank of the original inoculum and no CT26 tumor growth was observed in any of the mice after 30 days, demonstrating that previous treatments resulted in an immune memory response. In addition to the CT-26 model, CASC-674 demonstrated significant anti-tumor effects in EMT6, H22 and MBT-2 syngeneic mouse tumor models.
In CT26 tumors, treatment with CASC-674 but not an IgG1 version of the same variable domains, shifted the immunosuppressive phenotype to an inflammatory anti-tumor phenotype. CASC-674 alone and in combination with anti-PD1 significantly reduced the % of Tregs and exhausted (PD1+ Ki67+) CD8+ cells and increased the % of cytotoxic CD8+ cells (PD-1- Ki67+) and anti-tumor (CD155+) M1 macrophages.
Conclusions
These results combined with the antitumor effect observed suggest Fc functionality is critical for the effect of CASC-674. The observed single agent anti-tumor effect in several different tumor models of CASC-674 and its unique human, non-human primate, and murine cross-reactivity supports consideration of CASC-674 as a therapeutic development candidate.
P318 Anti-tumor efficacy and enhancement of T cell effector functions by EOS084448, an antagonist anti-TIGIT antibody
Gregory Driessens1, Julia Cuende1, Sofie Denies1, Chaterine Hoofd1, Florence Lambolez1, Shruthi Prassad1, Virginie Rabolli1, Anthony Cooper2, Christophe Quéva1
1iTeos Therapeutics, Gosselies, Belgium; 2Adimab, Lebanon, NH, USA
Correspondence: Gregory Driessens (gregory.driessens@iteostherapeutics.com)
Background
T cell Immunoreceptor with Ig and ITIM domains (TIGIT) is an ITIM domain- containing co-inhibitory receptor preferentially expressed by NK, CD8+ and CD4+ T cells as well as by regulatory T cells (Treg). Several ligands are described to bind to TIGIT with PVR (CD155) showing the highest affinity. CD226 (DNAM-1), a co-stimulatory receptor also expressed on NK and T cells compete with TIGIT for PVR binding but with a lower affinity. Co-expression of TIGIT and CD226 receptors on T and NK effector cells suggests a role in the fine control of their activation
Methods
Antagonistic anti-TIGIT antibodies were selected by Adimab, LLC. using a synthetic library of human antibodies presented on the surface of yeast. Anti-TIGIT mAbs were characterized for affinity to recombinant human TIGIT (Biacore), for binding to human primary T cells, for competition to CD155 binding as well as for functional activity on cells engineered to express TIGIT or on human primary T cells. An anti-TIGIT mAb cross-reactive to mouse TIGIT was used to evaluate the anti-tumor efficacy of anti-TIGIT in the CT26 syngeneic model.
Results
Anti-TIGIT mAb EOS084448 affinity for human TIGIT was 0.25nM, translating into potent binding to human primary T cells with an average EC50 of 0.09nM. EOS084448 inhibited CD155 binding to TIGIT at the surface of TIGIT-expressing cells with an EC50 of 0.16nM. EOS084448 antagonistic activity was evaluated in a TIGIT:CD155 reporter bioassay that resulted in activation of the IL-2 promoter and by measuring IFNg secretion by minimally stimulated human primary CD8+ T cells; EOS084448 potency in these 2 assays was 8nM and 0.4nM. A surrogate anti-TIGIT mAb with a 26,7nM Kd for mouse TIGIT was evaluated in established CT26 mouse tumors. Anti-TIGIT monotherapy delayed CT26 tumor growth and achieved complete response in few mice. Complete tumor regression occurred in most of the animals receiving the anti-TiGIT mAb combined with anti-PD-1 mAb. Anti-tumor efficacy was associated with an increased CD8+ T cell : Treg ratio, an increase TH1/TH2 cytokine ratio and a transcriptional signature indicating an increased cytolytic T cell activity.
Conclusions
In vitro and in vivo data demonstrate the potential for EOS084448 to promote antitumor immunity and efficacy and supports the rationale for its clinical evaluation.
P319 Long-term disease-free survival (DFS) of metastatic melanoma (mM) and renal cell cancer (mRCC) patients following high-dose interleukin-2 (HD IL2)
Joseph Clark1, Brendan Curti2, Elizabeth Davis3, Howard Kaufman4, Asim amin5, Ajjai Alva6, Theodore Logan7, Ralph Hauke8, Gerald Miletello9, Ulka Vaishampayan10, Douglas Johnson3, Richard White5, Peter Wiernik11, Janice Dutcher12
1Loyola University, Maywood, IL, USA; 2Providence Cancer Center, Portland, OR, USA; 3Vanderbilt University, Nashville, TN, USA; 4Rutgers University, New Brunswick, NJ, USA; 5Carolinas Health Care System, Charlotte, NC, USA; 6University of Michigan, Ann Arbor, MI, USA; 7University of Indiana, Indianapolis, IN, USA; 8Midwest Cancer Center, Omaha, NE, USA; 9Hematology/Oncology Clinic, LLP, Baton Rouge, LA, USA; 10Wayne State University, Detroit, MI, USA; 11Cancer Research Foundation of New York, Chappaqua, NY, USA; 12Cancer Research Foundation of New York, Bronx, NY, USA
Correspondence: Janice Dutcher (jpd4401@aol.com)
Background
HD IL2 treatment produces durable complete responses (CRs) and surgical CRs. Patients achieving partial response (PR) and stable disease (SD) demonstrate improved survival compared with patients who progress.
Methods
Eleven HD IL2 treatment centers identified patients with survival > 5 years after HD IL2. DFS was from end of IL2 to June 2017. Treatment courses generally consisted of 2 1-week cycles of HD IL2, 600,000-720,000 U/kg IV every 8 hours. We collected data on patients treated with HD IL2 alone, or HD IL2 plus local therapy (surgery or radiation (SRS) leading to CR) with survival > 5 years after HD IL2.
Results
Ninety-nine patients are reported: 46 mRCC (male 32, female 10, unknown 4) and 53 mM (male 31, female 22). Median age at HD IL2 treatment of mRCC patients is 54 years (range, 39-73 years) and of mM patients is 53 years (range, 24-76 years). Sites of metastatic disease for mM patients were lymph nodes (LN), lungs, bone, liver, brain, and other organs, and for mRCC patients were lung, LN, adrenal, bone, and other organs. The majority of patients received 2-3 courses of IL2 (63 of 99 patients) and 18 received 1 course, with the overall number of IL2 courses ranging from 0.5 to 4 courses. Among the 46 mRCC patients, there are 38 CRs, 5 surgical CRs and 3 PRs with no further treatment. Among 53 mM patients, there are 42 CRs, 2 near CRs, and 9 Surgical/SRS CRs without further treatment. DFS in these patients after HD IL2 ranges from 5+ years to 30+ years, median 10+ years. 27 mRCC and 31 mM are alive > 10 years after IL2. Long-term toxicity among these 99 patients includes hypothyroidism-5 patients, arthralgias/arthritis-6 patients, vitiligo-3 patients, and 1 patient each: neuropathy, PVCs, and normal pressure hydrocephalus. Additional patients may be added as records become available.
Conclusions
We document long-term DFS (>5 years) after CR or PR from HD IL2 alone. Surgical or SRS conversion of PR to CR can produce durable CRs. Long-term DFS was observed in patients with visceral and bone metastases, not only those with LN or lung sites of metastases. Neither age, sex, nor number of courses of IL2 predicted long-term DFS. Chronic toxicity due to IL2 is uncommon in long-term survivors.
P320 Targeting CD38 beyond haematological malignancies: a panel of anti-CD38 antibodies with unique functional properties
Nina Eissler1, Pascal Merchiers1, Simone Filosto1, Rahul Chaitanya Khanolkar1, Beatriz Goyenechea1, Dominic Smethurst1, Kevin Moulder1, Sergio A. Quezada2, Anne Goubier1
1Tusk Therapeutics, Stevenage, United Kingdom; 2UCL Cancer Institute, London, United Kingdom
Correspondence: Anne Goubier (nina.eissler@tusktherapeutics.com)
Background
Because of its high level of expression on haematological cancers, depleting antibodies targeting CD38, an ectoenzyme with hydrolase and cyclase activity, have been generated and showed clinical benefits in particular against multiple myeloma. Interestingly, CD38 is not restricted to haematological cancer cells but also expressed on many different immune subsets including NK and effector T cells, exhausted PD-1+ T cells, suppressive myeloid cells, and regulatory T and B cells. Anti-CD38 antibodies will therefore not only impact CD38-expressing tumour cells but also both effector and suppressive immune cells, as illustrated by the increased interest for CD38 as a target in Immuno-Oncology.
Methods
An antibody production and screening campaign has been initiated resulting in a panel of fully human CD38-binding antibodies. All antibodies have been screened for their potential to induce ADCC, apoptosis, ADCP, and CDC. The potential to deplete CD38-expressing cells has been verified in vivo in lymphoma engrafted mice. Additionally, all antibodies have been evaluated for their ability to influence effector T cell and NK cell activation.
Results
We have produced a portfolio of antibodies targeting distinct epitopes of CD38. These antibodies exert ADCC when directed against CD38-overexpressing targets, while showing differential capacity to induce ADCP and CDC. This translated into differential inhibition of in vivo tumour growth of human lymphoma tumours in SCID mice. Most interestingly, some of these antibodies augment TCR-induced proliferation and activation of human T cells in vitro, with an activity ranging from strong T cell activators that increase proinflammatory cytokine release, to medium or weak activators resulting in low or no cytokine release. In addition, in tumour coculture models, anti-CD38 antibodies increased NK cell activation and proliferation. Regarding CD38 enzymatic function, i.e. hydrolase and cyclase activity, our antibodies display differential profiles in terms of blockade or augmentation of both activities. Effects of anti-CD38 antibodies on immune effector cells, suppressive immune cells as well as tumour cells will be further explored in patient-derived ex vivo tumour models.
Conclusions
We present a portfolio of CD38-targeting antibodies with distinct activity profile. The broad expression and multiple functions described for CD38 underline the importance of being able to choose from a range of antibodies that can address the different functionalities depending on the most prominent role of CD38 in each disease setting. The new class of anti-CD38 antibodies presented here will be further explored for their potential to improve response rates, especially in solid tumours.
P321 Ibrutinib in combination with agonist αOX40 mAb and CTLA-4 blockade induces Eomeshi CD8 T cells and promotes tumor regression
Dana Emerson1, Michael Mcnamara2, Ian Hilgart-Martiszus2, Mohammad Farhad2, William Redmond2
1Oregon Health & Science University, Portland, OR, USA; 2Earle A. Chiles Research Institute, Portland, OR, USA
Correspondence: William Redmond (emersoda@ohsu.edu)
Background
Antagonist monoclonal antibodies (mAb) targeting T cell checkpoints such as CTLA-4 or PD-1 have shown efficacy in treating a subset of patients with metastatic disease. In preclinical models, CTLA-4 blockade synergized with an agonist anti-OX40 mAb to enhance the expansion and effector function of tumor-specific T cells. Previous studies demonstrated that combined aOX40/aCTLA-4 therapy also induced the generation of CD8 T cells expressing high levels of Eomesodermin (Eomes), a transcription factor known to regulate CD8 T cell differentiation and memory. Eomes expression is negatively regulated in T cells by the T cell signaling kinase ITK (interleukin-2 inducible T cell kinase). Importantly, the FDA-approved Bruton’s tyrosine kinase inhibitor ibrutinib also blocks ITK, thus providing a potential means of modulating Eomes expression. We sought to characterize these Eomeshi CD8 T cells and investigate the mechanisms regulating the generation of this novel subset.
Methods
Wild-type C57BL/6 mice were challenged by 1x106 TRAMP-C1 tumor cells in the right flank. Wild-type BALB/C mice were challenged by 5x104 4T1 tumor cells orthotopically in the mammary fat pad. Mice were treated starting day 7 with 200ug aCTLA-4 (clone 9D9), 200ug aOX40 (Clone OX86), or 150ug Ibrutinib. All treatments were injected i.p. Tumor growth (area) was assessed with microcallipers every 2 to 3 days. Mice were killed when tumors exceeded 175mm2 for experiments tracking tumor growth and survival. Lymph nodes and tumors were harvested on day 14 to assess and characterize T cell responses by flow cytometry.
Results
Our data revealed that these Eomeshi CD8 T cells expressed significantly less PD-1 on their surface compared to Eomeslo CD8 T cells (97% vs. 57%, respectively), while maintaining high levels of IFN-g production within the tumor. Additionally, we confirmed that clinical concentrations of ibrutinib do not inhibit T cell receptor signaling in CD8 T cells. Next, demonstrated that the combination of ibrutinib/aOX40/aCTLA-4 therapy enhanced the frequency of Eomeshi CD8 T cells in 4T1 tumor-bearing mice, a model of triple negative breast cancer. Additionally, this triple therapy significantly enhanced IFN-g and TNF-a expression by CD4 and CD8 T cells in the tumor and draining lymph nodes, which was associated with tumor regression and enhanced survival in TRAMP-C1 and Myc-CaP models of prostate adenocarcinoma.
Conclusions
Taken together, these data demonstrate that combined ibrutinib/aOX40/aCTLA-4 therapy induced a robust population of Eomeshi CD8 T cells with enhanced effector function capable of mediating tumor regression in multiple pre-clinical tumor models.
P322 Antitumor effects of radiation combined with intratumoral anti-Tem8 mAb and IL2
Amy K. Erbe1, Kathryn Komro1, Arika Feils1, Mackenzie Heck1, Sabrina VandenHeuvel1, Manasi Mohan1, Peter M. Carlson1, Jacquelyn A. Hank1, Alexander Rakhmilevich1, Zachary S. Morris1, Amit Chaudhary2, Brad St. Croix3, Paul M. Sondel1
1University of Wisconsin-Madison, Madison, WI, USA; 2Meso Scale Diagnostics, Rockville, MD, USA; 3National Cancer Institute, Frederick, MD, USA
Correspondence: Paul M. Sondel (aerbe@wisc.edu)
Background
Tumor Endothelial Marker 8 (Tem8) is overexpressed on endothelial cells that line the tumor vasculature. Chaudhary et al. [Cancer Cell, 2012] showed in several tumor models that the anti-Tem8 monoclonal antibody, L2mAb, augmented the activity of anti-cancer agents leading to slowed tumor growth. Tem8 is also overexpressed on tumor cells themselves in several tumor types, including melanoma. We have recently shown that radiation (RT) given in combination with intra-tumoral injection (IT) of a tumor-specific antibody and interleukin-2 (IL2) can elicit an in situ vaccine effect for mice bearing a B78 syngeneic melanoma. Here, we show that RT+L2mAb+IL2 treatment resulted in improved outcome for mice bearing a Tem8+ B78 melanoma.
Methods
B78 melanoma cells were transduced to overexpress Tem8 and injected into C57BL/6 mice. Mice bearing a ~200mm3 tumor received external beam RT (12Gy), followed by IT injection of L2mAb (2mg/kg) and/or IL2 (150,000U) on days 5-9 post-RT. Mice were monitored for tumor growth and overall survival. Statistical differences in tumor volume were determined by Two-Way ANOVA followed by Tukey’s test for multiple comparisons, and Log-rank/Mantel-Cox test was used for overall survival. Chi-Square test was used to compare response rates.
Results
Treatment with RT+L2mAb resulted in slower tumor growth as compared to RT alone (p<0.001). By including IL2 to RT+L2 (i.e. RT+L2mAb+IL2), tumor growth was significantly reduced as compared to mice treated with either RT alone (p<0.001) or with RT+L2 (p<0.001). In our preliminary study, only the combination of RT+L2mAb+IL2 yielded any mice that were tumor free (2 tumor-free mice out of 5); treatment with RT+L2mAb, or RT+IL2 did not generate any tumor-free mice.
Conclusions
Tem8-targeted therapy given in conjunction with other anti-cancer agents has shown promising effects. Prior to this study, anti-Tem8 antibody given in combination with RT or IL2 had not been investigated. Here, we show that RT followed by Tem8-specific antibody significantly improved outcome, and that addition of IL2 further improved outcome. Inhibition of tumor-angiogenesis via anti-Tem8 antibodies may block the function of the Tem8 that is expressed on tumor endothelial cells, slowing tumor growth. However, since RT+L2mAb treatment resulted in improved outcome when given in combination with IL2, antibody-dependent-cell-mediated cytotoxicity may also contribute to the mechanism of action of this therapy.
P323 Novel treatment of cutaneous T cell lymphoma: Targeting TNFR2, an oncogene and marker of potent Tregs, with anti-TNFR2 antibodies
Heather Torrey1, Audrey Defusco1, Danielle Baum1, Ziba Rahbar2, Michael Khodadoust2, Youn H. Kim2, Denise Faustman1
1Massachusetts General Hospital/Harvard Medical School, Charlestown, MA, USA; 2Stanford University School of Medicine/Cancer Institute, Stanford, CA, USA
Correspondence: Denise Faustman (faustman@helix.mgh.harvard.edu)
Background
Tumor necrosis factor receptor 2 (TNFR2) is a lymphoid marker of the most potent regulatory T cell (Treg) subtype, which is enriched in the tumor microenvironment, and a commonly expressed oncogene in human tumors. Anti-TNFR2 antibodies can inhibit both Tregs and tumors with specificity for the tumor microenvironment [1], and recent data shows that TNFR2 is a candidate oncogene in cutaneous T cell lymphoma (CTCL) with recurrent point mutations and gain of function alteration of TNFR2, resulting in abnormal expression of TNFR2 on CD4+CD26- tumor cells [2].
Methods
We screened novel TNFR2-directed antagonistic antibodies for their ability to kill leukemic cells in Stage IV CTCL (Sézary syndrome) subjects with failure on diverse drug regimens, as well as their ability to induce killing of tumor-associated Tregs and unleash effector T cell (Teffector) proliferation. Studies were performed in vitro on sorted CD4+CD26- Sézary cells or V-beta specific populations when a tumor was typed.
Results
At baseline, CTCL blood samples showed significant burdens of tumor cells within the CD26- subset of CD4 cells (averaging 45-95% of this subfraction), vs control blood (18% CD26- cells on average). In CTCL subjects, Treg numbers (CD4+CD25hiFoxp3) were elevated at baseline (11% vs 7% control, p< 0.05), Teffectors were depressed (3% vs 8% control, p< 0.05), and Treg/Teffector ratios were abnormally elevated (8% vs 1% control, p< 0.05).
Regardless of underlying therapy used in vivo, TNFR2 antagonism showed dose responsive killing of tumor cells within the CD26- fraction of peripheral CD4 T cells. TNFR2 antagonism also had specificity for tumor cells vs CD26- cells from paired controls. In vivo treatment of CTCL subjects with anti-proliferative agents such as methotrexate hindered TNFR2 antagonism driven killing, demonstrating the specificity of TNFR2 antagonism for rapidly proliferating cells.
Dose response experiments in vitro showed TNFR2 antagonism also had the desired dual effect of Treg killing combined with unleashing of Teffector proliferation at 48-72 hours.
Conclusions
TNFR2 is a recurrent genomic gain alteration in CTCL that can potentially be targeted to directly stop tumor cell growth by antibody-induced cell death, eliminate Tregs of the tumor microenvironment and unleash Teffector proliferation.
References
1. Torrey H, Butterworth J, Mera T, et al. Targeting TNFR2 with antagonistic antibodies inhibits proliferation of ovarian cancer cells and tumor-associated Tregs. Sci Signal. 2017;10(462):pii: eaaf8608.
2. Ungewickell A, Bhaduri A, Rios E, et al. Genomic analysis of mycosis fungoides and Sézary syndrome identifies recurrent alterations in TNFR2. Nat Genet. 2015;47(9):1056-60.
P324 Co-targeting of mesothelin and CD47 with bispecific antibodies for efficient elimination of solid tumors
Krzysztof Masternak, Limin Shang, Vanessa Buatois, Stefano Majocchi, Eric Hatterer, Xavier Chauchet, Valéry Moine, Lucile Broyer, Marie H. Kosco-Vilbois, Nicolas Fischer, Walter G. Ferlin
Novimmune S.A., Geneva, Switzerland
Correspondence: Walter G. Ferlin (wferlin@novimmune.com)
Background
Mesothelin (MSLN) is a cell surface glycoprotein overexpressed in several human cancers, including mesothelioma, pancreatic-, ovarian-, lung- and gastric cancer. MSLN overexpression is associated with poor prognosis, with serum levels of soluble MSLN a biomarker of disease severity in mesothelioma patients. While a promising target in cancer, monoclonal antibodies (mAbs) targeting MSLN have demonstrated limited efficacy in clinical trials. Current MSLN-targeted approaches in development have thus incorporated novel modalities to enhance tumor-killing potential (e.g., Antibody Drug Conjugates and CAR-T cells). CD47, an immune check-point, interacts with SIRPα providing a ‘don’t eat me’ signal that allows healthy cells to limit elimination by immune cells, in particular macrophages. CD47 upregulation in solid cancers is correlated with poor clinical prognosis, almost certainly by allowing tumor cells to escape immune surveillance by phagocytes. Clinical development of mAbs to CD47 is hindered by the ubiquitous expression of CD47 leading to rapid drug elimination and significant hematological toxicity including anemia. To address these concerns, we have employed a bispecific antibody (biAb) approach that pairs a high affinity anti-MSLN targeting arm to an anti-CD47 arm of an optimized affinity that drives the efficacious binding only on MSLN-positive cells. This MSLN/CD47 biAb approach, therefore, is designed to target the CD47-SIRPα pathway in the tumor microenvironment to more efficiently harness the immune system for tumor eradication.
Methods
Fully human biAbs targeting both MSLN and CD47 were generated. An array of biAbs coupling MSLN-targeting arms binding to different epitopes on MSLN with a common CD47-targeting arm have been tested in antibody dependent cellular phagocytosis (ADCP) and antibody dependent cellular cytotoxicity (ADCC) assays in vitro plus in mouse xenograft experiments. The following human cell lines were used: NCI-N87 (gastric), HPAC (pancreatic), OVCAR3 (ovarian), Caov-3 (ovarian), NCI-H226 (mesothelioma) and MSLN-transfected HepG2 (hepatic).
Results
The MSLN/CD47 biAbs significantly enhanced macrophage-mediated ADCP of NCI-N87, HPAC, OVCAR3 and Caov-3 as compared to the anti-MSLN mAb, Amatuximab. In addition, the biAbs also demonstrated superior ADCC of NCI-N87 cells and NCI-H226. When tested in a xenograft tumor model using MSLN-transfected HepG2 cells, therapeutic treatment with MSLN/CD47 biAbs significantly prevented tumor development, while Amatuximab and B6H12, an anti-CD47 mAb, only slightly delayed tumor growth.
Conclusions
Using a novel biAb approach that focuses the blockade of the innate immune checkpoint receptor CD47 to MSLN-expressing tumors substantially enhances their elimination in vitro and in vivo. Thus, MSLN/CD47 targeting biAbs are a potentially superior strategy in managing MSLN-positive solid tumors.
P325 Characterization of the anti-CTLA-4 antibody AGEN1884, including toxicology and pharmacology assessments in non-human primates
Randi Gombos1, Ana Gonzalez1, Mariana Manrique1, Dhan Chand1, David Savitsky1, Benjamin Morin1, Ekaterina Breous-Nystrom2, Christopher Dupont1, Rebecca Ward1, Cornelia Mundt3, Benjamin Duckless4, Hao Tang4, Mark Findeis1, Andrea Schuster3, Jeremy Waight1, Dennis Underwood1, Christopher Clarke1, Gerd Ritter5, Taha Merghoub6, David Schaer6, Marc van Dijk2, Jennifer Buell1, Jean-Marie Cuillerot1, Robert Stein1, Elise Drouin1, Nicholas WIlson1
1Agenus Inc., Lexington, MA, USA; 2Agenus Switzerland Inc., Basel, Switzerland; 3Former employee of Agenus Switzerland, Inc., Basel, Switzerland; 4Former employee of Agenus Inc., Lexington, MA, USA; 5Ludwig Institute for Cancer Research, New York, NY, USA; 6Memorial Sloan Kettering Cancer Center, New York, NY, USA
Correspondence: Randi Gombos (randi.gombos@agenusbio.com); Nicholas WIlson
Background
Cytotoxic T lymphocyte antigen-4 (CTLA-4) is an important negative regulator of T cell function. Together with CD28, these receptors exemplify a co-inhibitory and co-stimulatory signaling axis that dynamically sculpts the interaction of antigen-specific T cells with antigen presenting cells (APCs). Preclinical studies have demonstrated that anti-CTLA-4 antibodies can enhance tumor-specific immunity through a variety of mechanisms including: i) blockade of CD80 or CD86 binding to CTLA-4; ii) preventing CTLA-4-expressing regulatory T cells from physically removing CD80 and CD86 from the surface of APCs; and iii) selective elimination of CTLA-4-expressing intratumoral regulatory T cells by an Fcg receptor-dependent mechanism.
Methods
Here we describe the pharmacological and toxicological characterization of a novel human IgG1 anti-CTLA-4 antagonist antibody, AGEN1884. Binding, blocking, T cell activation as well as Fcgamma receptor-mediated activity of AGEN1884 were evaluated in vitro. The activity and tolerability of AGEN1884 was further assessed in vivo using a non-human primate model. Our in vitro and in vivo assessments extended to a direct comparison of AGEN1884 with an IgG2 Fc variant, AGEN2041.
Results
AGEN1884 potently enhanced T cell responsiveness in vitro, and combined effectively with other immunomodulatory antibodies targeting co-inhibitory and co-stimulatory receptors on T cells. AGEN1884 was well-tolerated in non-human primates and was confirmed to modulate cellular and humoral immune responses to co-administered reporter vaccines. In addition to the activity of AGEN1884 as a monotherapy, a memory T cell proliferative response was observed in peripheral blood of animals when co-administered with an anti-PD-1 antibody. Finally, we provide a comparison of the in vitro and in vivo functional properties of an IgG2 variant of AGEN1884, revealing important antibody isotype differences that may have an impact on the design of optimal dosing regimens in patients.
Conclusions
Taken together, the pharmacologic properties of AGEN1884 support its clinical investigation as both a single therapeutic agent and in combination therapies.
P326 Isoform specific TGF-β inhibition in combination with radiation therapy as a novel immune therapeutic approach to cancer therapy
Aditi Gupta1, Sadna Budhu1, Rachel Giese1, Jacques van Snick2, Catherine Uyttenhove2, Gerd Ritter3, Jedd Wolchok1, Taha Merghoub1
1Memorial Sloan Kettering, New York, NY, USA; 2Ludwig Institute for Cancer Research Ltd Brussels Branch, Brussels, Belgium; 3Ludwig Institute for Cancer Research, Ltd, New York, NY, USA
Correspondence: Taha Merghoub (guptaa@mskcc.org); Jedd Wolchok
Background
TGF-β is a pleotropic cytokine, which has emerged as a potential target in cancer treatment due to its dual role in tumorigenesis and homeostasis. There are three isoforms of TGF-β (TGF-β1, TGF-β2 and TGF-β3), which are secreted by immune and non-immune cells as a latent complex. Depending on the local context, TGF-β adopts opposing roles in carcinogensis and in modulating the immune system. These dueling roles of TGF-β are dependent on its secretion and activation. Local radiation therapy (RT) can activate TGF-β via reactive oxygen species. Such TGF-β expression is linked to radioresistance and dose-limiting toxicities, reducing the effectiveness of RT. In these studies, we aim to characterize the effect of RT on the temporal and cell-specific expression patterns of TGF-β isoforms in mouse tumor models. This will inform treatment regimens combining isoform specific anti-TGF-β therapy with RT.
Methods
Fluorescence-activated cell sorting (FACS): C57BL/6 mice were implanted on the hind limb with B16-F10 melanoma cells. On day 10, tumors were irradiated locally with 15 Gy. Expression of TGF-β isoforms was measured at 1, 3 and 5 days post-RT by FACS.
In-vivo: C57BL/6 mice were implanted with tumors and irradiated as described. Mice were treated (10/group) with anti-TGF-β1, anti-TGF-β3 or a pan-TGF-β antibody beginning 1 day after RT given intraperitoneally (200 ug/mouse) every other day for 8 doses. Tumor growth and overall survival were monitored. A similar experiment was conducted in the 4T1 breast cancer model, in which mice were treated 1 day prior to radiation.
Results
FACS data indicated that TGF-β1 and TGF-β3 expression increases on most immune cells in the tumor 1 day after RT, decreases 3 days after RT and reaches a peak 5 days after RT. Preliminary in-vivo studies demonstrate that both αTGF-β1 and αTGF-β3 as monotherapies have activity against B16 melanoma. In combination with RT, αTGF-β3 trends towards inhibiting tumor growth. Similar observations were obtained in a 4T1 breast model; however, αTGF-β3 alone and in combination with RT as well as αTGF-β1 + RT showed a significant delay against tumor growth. No significant differences in survival were seen in either tumor model.
Conclusions
TGF-β1 and TGF-β3 are expressed on numerous lymphoid and myeloid cells in B16 tumors and spleens. TGF-β isoform expression peaks 5 days post-RT. Anti-TGF-β therapy is effective in delaying tumor growth and may synergize with RT in certain cancers. This demonstrates rationale for the use of anti-TGF-β therapy to enhance the effectiveness of RT in cancer.
P327 Intratumor injection of tumor-specific antibody and IL2 triggers in situ vaccination following local radiation therapy
Emily I. Guy1, Sara Busche1, Peter Carlson1, Clinton Heinze1, Ciara N. Schwarz1, Raghava N. Srirameneni1, Jasdeep Kler1, Michael M. Meagher2, Amy K. Erbe1, Jacquelyn A. Hank1, Alexander L. Rakhmilevich1, Paul M. Sondel1, Zachary S. Morris1
1University of Wisconsin School of Medicine and Public Health, Madison, WI, USA; 2St. Jude Children's Research Hospital/Children's GMP, LLC, Memphis, TN, Memphis, TN, USA
Correspondence: Emily I. Guy (eguy@wisc.edu)
Background
In murine models of GD2+ melanoma, GD2+ neuroblastoma, and EGFR+ head and neck cancer, we have reported a cooperative interaction between radiation and intratumor (IT) injection of tumor-specific antibody (anti-GD2 hu14.18K322A or anti-EGFR cetuximab). Consistent with a process mediated by antibody-dependent cell-mediated cytotoxicity, this interaction required the Fc portion of the antibody, host Fcʏ receptor, NK cells, and tumor expression of the antibody-targeted antigen. In this GD2+ melanoma model, combined treatment with RT + IT-hu14.18-IL2 immunocytokine (a fusion protein of hu14.18 antibody and IL2) markedly enhanced response compared to radiation or IT-IC alone, radiation + IT-hu14.18 antibody, or radiation + intravenous-IC. In those studies, radiation + IT-IC induced an in situ vaccine effect, resulting in a tumor-specific memory T cell response. Here we test whether IT administration of non-fused tumor-specific antibody and IL2 may elicit an in situ vaccination response following local radiation.
Methods
C57BL/6 mice were flank engrafted with syngeneic GD2+ B78 melanoma and 5-week tumors (~200 mm3) were treated with single fraction 12 Gy radiation, IT-IL2 (150,000U), and/or IT-hu14.18K322A (50μg). IT injections were given daily on days 6-10 after radiation. Outcomes included tumor response and rates of complete regression, overall survival, tumor-specific memory (tested in disease-free mice by contralateral flank injection with B78 melanoma >90 days after radiation), and immunohistochemistry on tumors resected at day 12 after radiation.
Results
The combination of local radiation + IT-hu14.18K322A + IT-IL2 resulted in greater tumor regression compared to radiation + IT-hu14.18K322A or radiation + IT-IL2 [50% (19/38) aggregate complete tumor regression vs 0% (0/11) and 25% (3/12), respectively, p<0.001]. No mice were rendered disease-free with these IT-treatments in the absence of radiation. Kaplan-Meier analysis demonstrated improved survival with radiation + IT-hu14.18K322A + IT-IL2 compared to radiation + IT-hu14.18K322A and radiation + IT-IL2 (log-rank p<0.0001; 80% alive at day 65 vs 0% and 50%, respectively). Thus far,100% (6/6) of mice rendered disease-free by combined radiation + IT-hu14.18K322A + IT-IL2 have rejected subcutaneous B78 re-implantation at >90 days after radiation, compared to 0/10 naïve control mice. Preliminary immunohistochemistry analyses suggest that IT-IL2 may increase tumor infiltrating CD8+ T cells in this preclinical model.
Conclusions
We present evidence of a cooperative anti-tumor effect with the combination of local radiation and IT injection of both tumor-specific antibody and IL2. Given the widespread availability of tumor-specific antibodies, this may offer a viable approach to pursuing in situ tumor vaccination in many diverse types of cancer using off-the-shelf reagents.
P328 Therapeutic application of radiation-induced anti-tumor antibody
Tsuguhide Takeshima, Raquibul Hannan
UT Southwestern Medical Center, Dallas, TX, USA
Correspondence: Raquibul Hannan (Raquibul.Hannan@utsouthwestern.edu)
Background
Radiation therapy (RT) has significant immune modulatory properties that, when strategically integrated with immunotherapy, may improve cancer treatment outcomes. It has been previously reported that RT induces tumor-specific antibodies [1]; however, this response needs to be better characterized and its therapeutic potential evaluated. Antibodies binding to the Fc receptor of dendritic cells (DC) have been shown to direct DC to the tumor site [2]. We hypothesize that radiation-induced anti-tumor antibodies (RT-Ab) increase dendritic cell (DC) trafficking to the irradiated tumor to increase tumor antigen presentation. Here, we explore the therapeutic application of RT-Ab.
Methods
Multiple syngeneic murine cell lines were used (EG7, B16, LLC, 4T1), along with their respective murine strains. Fluorescence tagged anti-mouse antibodies (eBiosciences) and anti-CD11c antibodies (eBiosciences) were used for flow cytometry.
Results
Multiple syngeneic murine tumor grafts (B16, LLC, 4T1) were grown in the hind legs of respective murine strains and focally irradiated with a single 15Gy RT dose, with post-RT sera (pRTs) obtained at different time intervals. Flow cytometric analysis using anti-mouse antibodies of the pRTs incubated with the respective tumor cell showed a significant increase in RT-Ab that peaked between days 7-13. This increase was more pronounced for a single fraction of 15Gy when compared to 4.5Gy on 5 consecutive days while the tumor growth delay was equal between the two groups. Next, flow cytometry was performed on bone marrow-derived dendritic cells (BMDC) from the respective murine strains co-cultured with pRTs, which showed an increase in the binding of RT-Ab to DCs when compared to normal or non-irradiated tumor bearing mice sera. When an Fc receptor blocking agent was used, this binding was significantly inhibited, which suggested Fc receptor-mediated binding of RT-Ab to the BMDC. Lastly, we demonstrated that RT-Ab-bound-BMDC (RT-DC), when administered as an autologous DC therapy in conjunction with focal RT, increased the therapeutic effect of focal tumor RT significantly compared to multiple control conditions.
Conclusions
RT induces a tumor-specific antibody response that increases DC trafficking to the tumor via Fc receptor-mediated targeting. This likely contributes to RT-mediated tumor antigen presentation and adaptive anti-tumor immunity. Amplification of the RT-Ab response may have therapeutic applications.
References
1. Nesslinger NJ et. al. Standard treatments induce antigen-specific immune responses in prostate cancer. Clin Cancer Res. 2007 Mar 1;13(5):1493-502. PubMed PMID: 17332294.
2. Franki SN et. al. Dendritic cells loaded with apoptotic antibody-coated tumor cells provide protective immunity against B-cell lymphoma in vivo. Blood. 2008 Feb 1;111(3):1504-11.
P329 Dual blockade of PD1 and CTLA4 with bispecific antibody XmAb20717 promotes human T cell activation and proliferation
Michael Hedvat1, Matthew Bernett1, Gregory Moore1, Christine Bonzon1, Rumana Rashid1, Seung Chu2, Kendra Avery1, Alex Nisthal1, Umesh Muchhal1, John Desjarlais1
1Xencor, Inc., Monrovia, CA, USA; 2schu@xencor.com, Monrovia, CA, USA
Correspondence: John Desjarlais (mhedvat@xencor.com)
Background
Treatment of melanoma patients with nivolumab plus ipilimumab increases progression-free-survival compared to each monotherapy. The increase in efficacy of the combination regimen is accompanied by an increase in adverse events. Since PD1+CTLA4+ tumor-infiltrating-lymphocytes are dysfunctional in the tumor microenvironment, we developed XmAb20717 to selectively target PD1+CTLA4+ double-positive T cells in an effort to recapitulate efficacy of the combination regimen while reducing toxicity.
Methods
XmAb20717 component antibodies binding to PD1 and CTLA4 with favorable stability and functionality were assembled in a bispecific antibody platform with substitutions in the Fc domain to suppress effector function. XmAb20717 was evaluated in vitro by measuring antibody binding and de-repression of super-antigen stimulated peripheral blood lymphocytes (PBMCs) and in vivo by monitoring the engraftment of human PBMCs in NSG mice (huPBMC-NSG) by flow cytometry. To evaluate anti-tumor efficacy we monitored the growth of established cancer cells in huPBMC-NSG following treatment.
Results
Optimized candidate single-chain Fvs were confirmed to bind PD1 and functionally block PDL1 and PDL2 binding to PD1. We also generated optimized anti-CTLA4 Fabs. Anti-PD1 and anti-CTLA4 targeting component antibodies were assembled into XmAb20717, which displayed favorable biophysical and manufacturing properties. XmAb20717 enhanced IL2 secretion in vitro 4.1-fold relative to a negative control antibody (p < 0.01, n = 15 donors) in response to antigenic challenge of previously stimulated T cells, with 1.6-fold superior activity compared to an anti-PD1 bivalent antibody (p < 0.01, n = 15 donors). XmAb20717 enhanced T cell engraftment (9.6-fold) and IFNγ secretion (2.9-fold) compared to vehicle controls in huPBMC-NSG mice (p < 0.001, n = 10/group), with equivalent activity to a combination regimen of anti-PD1 and anti-CTLA4 bivalent antibodies. Blockade of PD1 and CTLA4 with an equimolar mixture of monovalent component antibodies that compose XmAb20717 lead to inferior activity compared to XmAb20717, suggesting that binding selectivity of the bispecific antibody contributes to function. XmAb20717 also exhibited anti-tumor activity in preliminary anti-tumor studies in mice.
Conclusions
Dual blockade of PD1 and CTLA4 with XmAb20717 resulted in T cell activation that is comparable to a combination of bivalent antibodies targeting PD1 and CTLA4. Specific targeting of human lymphocytes positive for both PD1 and CTLA4 with XmAb20717 may promote similar efficacy compared to a combination of bivalent antibodies while reducing adverse events. These data suggest that clinical development of XmAb20717 is warranted for the treatment of human malignancies.
P330 T cell immunotherapies trigger innate immunity and aseptic inflammation leading to potent anti-tumor and off-targets effects
Daniel Hirschhorn1, Jacob Ricca1, Bilel Gasmi1, Levi Mark Mangarin1, Olivier De Henau1, Sadna Budhu1, Yanyun Li1, Czrina Cortez1, Yang Xia1, Hong Zhong1, Cailian Liu1, Roberta Zappasodi1, Travis Hollmann1, Mario Lacouture1, Jean Albrengues2, Mikala Egeblad2, Wolchok Jedd1, Taha Merghoub1
1Memorial Sloan Kettering Cancer Center, New York, NY, USA; 2Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, USA
Correspondence: Wolchok Jedd (hirschhd@mskcc.org); Taha Merghoub
Background
Mobilizing the immune system to treat advanced cancers is now a clinical reality. Successful immune-based therapies that treat tumors are often accompanied by immune-related adverse events (irAE) including toxicities that can occasionally present with severe and lethal symptoms. The primary immunotherapies currently in clinic include agents that activate T cell responses such as checkpoint blockade of inhibitory pathways and infusion of ex-vivo tumor-derived or T cell receptor (TCR)-transgenic or chimeric antigen receptor (CAR)-modified T cells. While the beneficial and toxic effects of T cell-based immunotherapies in the clinic are being extensively explored, the precise mechanisms of tumor elimination and irAE remain the subject of intense investigation.
Methods
In the present study, we treated established tumors with melanoma-specific adoptive CD4+ T cell transfer and costimulation via OX40 or checkpoint blockade with anti-CTLA-4.
Results
We found that, in spite of co-opting the adaptive immune response to treat cancer, acute local inflammation, resembling delayed-type hypersensitivity, plays a fundamental role in tumor elimination and related toxicities in a model of irAE. While OX40 or CTLA-4 antibodies stimulated T cells are necessary for initiating a therapeutic response, activation of endogenous neutrophils constitutes an important and necessary effector mechanism of tumor destruction and irAE. Upon closer examination, we found extensive neutrophil extracellular traps (NETs) in ear pinnae of treated mice and in melanoma patients suffering from immunotherapy-induced irAE.
Conclusions
Our results illustrate the involvement of innate immunity in promoting tumor elimination and subsequent side effects with immunotherapies that engage T cells.
P331 Involvement of estrogen and progesterone in the modulation of indoleamine 2,3 dioxygenase - IDO – expression in cultured mammary carcinoma cells of female dog
Jose Roberto Kfoury Jr, Pedro Bianchi, Flavia Ponce, Rafael Leandro, Tulio Yoshinaga
School of Veterinary Medicine and Animal Sciences, University of Sao Paulo, Sao Paulo, Brazil
Correspondence: Jose Roberto Kfoury Jr (jrobertok@usp.br)
Background
Indoleamine 2,3 dioxygenase - IDO is an enzyme that prevents the establishment of an immune response in the microenvironment in which it is expressed by catabolizing the amino acid tryptophan. The deprivation of tryptophan and the generation of its metabolites, mainly kynurenine, impairs effector T- cells proliferation leading them to apoptosis [1] and, by its interaction with the aryl-hydrocarbon receptor (AhR) in CD4+ T-cells, favors the expansion of T-regulatory cells [2]. It is known that several cancer cells and leucocytes in the tumor microenvironment express IDO and are sensitive to hormones. Steroidal hormones, such as estrogen and progesterone, are capable of altering immune functions in cells and may influence IDO expression; nonetheless, the mechanisms involved are still poorly understood. Our group previous data have shown that the progesterone was directly involved in IDO expression modulation via its receptor in dendritic and CD4+ T cells from the maternal-fetal interface of Wistar rats [3]. Therefore, this study aims to investigate whether this mechanism is present in the female dog mammary carcinoma microenvironment and if it occurs in a similar way with estrogen.
Methods
Cells of mammary carcinoma from bitches were treated with exogenous progesterone and estrogen and their respective receptor antagonists tamoxifen and mifepristone. IDO expression was analyzed by immunohistochemistry, flow cytometry and the mRNA by real-time PCR. IDO quantification was obtained by western blot technique.
Results
IDO quantification exhibited the same pattern as mRNA expression. There was an increase of the enzyme expression and mRNA in the estrogen treated group, in contrast to the decrease observed in the progesterone group. When the cells were subjected to the hormonal inhibitors, an evident decrease of IDO expression percentage and the respective mRNA was verified following the supplementation of tamoxifen and a restoration of IDO expression values and the mRNA after the addition of the progesterone inhibitor, mifepristone.
Conclusions
These findings strongly suggest that progesterone and estrogen participate indirectly in the modulation of IDO through their membrane receptors.
References
1. Munn DH., Mellor AL: IDO in the Tumor Microenvironment Inflammation, Counter-Regulation, and Tolerance. Trends in Immunology. 2016; 37(3):193-207.
2. Harden JL, Egilmez NK: Indoleamine 2,3-Dioxygenase and Dendritic Cell Tolerogenicity. Immunol Invest. 2012; 41:738-764.
3. Bianchi PKF, Leandro RM, Poscai AN, Yoshinaga T, Gonçalez PO, Kfoury Junior JR: Progesterone Decreases in vitro Indoleamine 2, 3-dioxygenase Expression in Dendritic and CD4+ Cells from Maternal-Fetal Interface of Rats. Immunol Invest. 2017; 46(5):1–13.
P332 Pre-clinical efficacy and tolerability of NKTR-255, a polymer-conjugated IL-15 for immuno-oncology
Peter Kirk, Peiwen Kuo, Murali Addepalli, Takahiro Miyazaki, Jane Gunther, Laurie VanderVeen, Ping Zhang, Palakshi Obalapur, Mekhala Maiti, Amol Murkar, Arunasree Lanka, Werner Rubas, Jonathan Zalevsky
Nektar Therapeutics, San Francisco, CA, USA
Correspondence: Peter Kirk (pkirk@nektar.com)
Background
IL-15 is a cytokine that activates T-cells and NK cells and has long been recognized for its potential as an immunotherapeutic agent for the treatment of cancer. Exploiting this potential has been challenging due to unfavorable pharmacokinetic properties. NKTR-255 is a polymer-conjugated IL-15 that shows improved plasma exposure while retaining potency and high affinity for IL-15Rα. Here we investigate the pharmacodynamics, pre-clinical efficacy and tolerability of NKTR-255.
Methods
To assess the pharmacodynamic effects of NKTR-255 in mice and non-human primates NKTR-255 was delivered intravenously and whole blood was collected at the indicated timepoints; flow cytometry was used to measure signaling activity (STAT5 phosphorylation), proliferative status (Ki-67 expression) and absolute frequency of various lymphocyte subpopulations. Cytotoxicity assays were performed by incubating NK cells purified from spleens of NKTR-255-treated mice with dye-labelled YAC-1 target cells and assessing extent of cell-killing by flow cytometry. Efficacy was studied using the CT-26 lung metastasis model; briefly, 105 CT-26 cells were injected into the tail-vein of mice, NKTR-255 treatment was initiated the following day, lungs were harvested on day 13 and metastases were counted.
Results
NKTR-255 induces rapid and sustained signaling in lymphocytes following intravenous administration in mice and non-human primates. This sustained signal results in proliferation of CD8 T-cells and a preferential expansion of the CD8 central memory population. NK cells increase in number and in Granzyme B expression, concomitant with an increase in cytotoxic potential. The robust induction of CD8 and NK cell proliferation is maintained upon repeat dosing. In a mouse model of tumor metastasis to the lungs, NKTR-255 treatment results in an 85% reduction in the number of metastases. This efficacy is dependent on NK cells but not CD8 T-cells, as demonstrated in cell-depletion studies. Toxicology assessments demonstrate that NKTR-255 is well tolerated at efficacious dose levels.
Conclusions
NKTR-255 provides a sustained IL-15 signal, resulting in profound and sustained immune activation and anti-tumor activity. NKTR-255 is well tolerated and its pharmacokinetic properties and pharmacodynamic effects translate well from rodents to non-human primates, supporting further development.
P333 MPL-5821, an ESM™-p38 MAPK inhibitor, enhances tumor immune response and M1 macrophage polarization in a 3D Ex Vivo system utilizing fresh tumor microspheroids of lung cancer patients
Melanie Mediavilla-Varela1, Melba Marie Page1, Jenny Kreahling1, Martin Perry2, David Moffat2, Scott Antonia1, Soner Altiok1
1Nilogen Oncosystems, Tampa, FL, USA; 2Macrophage Pharma Limited, Windsor, United Kingdom
Correspondence: Soner Altiok (jenny@nilogen.com)
Background
Lung cancer is one of the most common causes of death worldwide. Immunotherapy has demonstrated durable responses and tolerability in subsets of lung cancer patients. Macrophages are a significant component of the tumor microenvironment and the predominant phenotype (M2-like) is frequently associated with one supportive of tumor immune evasion. However, the plasticity of macrophages offers the opportunity for therapeutic intervention to repolarize the phenotype to one that is non-immunosuppressive and supportive of an anti-tumor immune response (M1-like). MPL-5821 is a p38 MAPK inhibitor employing Esterase Sensitive Motif (ESM™) technology [1] to principally target myelomonocytic cells. This study evaluates the immunomodulatory effect of MPL-5821 in a 3D ex vivo assay of lung cancer.
Methods
Fresh tumor tissues obtained from consented patients with non-small cell lung cancer at the time of surgical resection were utilized in a proprietary 3D ex vivo tumor microsphere assay with intact tumor immune microenvironment. Tumor microspheres were treated with MPL-5821 for 36 hours. At the end of the treatment, flow cytometry analysis was performed to assess M1/M2 plasticity and TIL proliferation (CD3+/Ki-67+). Additionally, multiplex human cytokine assay was used to simultaneously analyze the differential release of cytokines in culture media and gene expression analysis was performed using the NanoString PanCancer Immune Profiling panel which contains probes to quantitate 770 immune function genes.
Results
MPL-5821 at 10nM and 50nM effectively inhibited the p38 signaling pathway in the tumor samples and led to profound inhibition of the immunosuppressive cytokine IL-10 and pro-inflammatory cytokines TNFα and IL-6. Evidence of enhanced T-cell proliferation and activation was observed in several of the tumor samples. The most statistical significant up-regulated genes were associated with antigen processing/presentation and MHC class II protein binding and included CD4, CR1, CD74 and multiple HLA genes. CD40 and ICOSLG genes were up-regulated in several tumors as were CXCL9 and 10. Chemokine genes CXCL3, 5, 7 and 8, associated with a pro-angiogenic macrophage phenotype, were the most statistically down regulated genes along with the M2 phenotype associated genes CCL13, 23 and 24. Down-regulation of PD-L1 and IDO gene expression was also seen in several tumors.
Conclusions
This lung patient derived ex vivo approach indicates that MPL-5821 may alleviate myelomonocytic cell induced immunosuppression and enhance antigen responsiveness suggesting potential clinical implications in the treatment of lung cancer.
References
1. Needham LA, et al, J Pharmacol Exp Ther. (2011), 339 :132-42.
P334 Inhibition of IDO activity by epacadostat (INCB024360) activates tumor infiltrating lymphocytes in a patient-derived 3D ex vivo system of lung cancer and alleviates stromal immunosuppression
Melanie Mediavilla-Varela1, Melba Marie Page1, Jenny Kreahling1, Peggy Scherle2, Scott Antonia1, Soner Altiok1
1Nilogen Oncosystems, Tampa, FL, USA; 2Incyte Corporation, Wilmington, DE, USA
Correspondence: Soner Altiok (jenny@nilogen.com)
Background
Immune evasion is one of the major hallmarks of cancer and identifying mechanisms by which cancer cells evade the immune system have become a major strategy against cancer. IDO (indoleamine 2,3-dioxygenase) is a tryptophan catabolizing enzyme expressed constitutively by tumor cells and different components of immune cells present within the tumor microenvironment. It has been shown that high expression of IDO increases the number of Tregs and blocks the proliferation of effector T cells. Thus, inhibiting the IDO pathway is a promising strategy to restore immune system responses to more easily identify and destroy cancer cells. This study evaluates the immunomodulatory effect of an IDO inhibitor epacadostat (INCB024360) on the immunosuppressive effect of cancer-associated fibroblasts and activation of tumor infiltrating lymphocytes in a 3D ex vivo assay utilizing fresh patient tumor samples
Methods
3D ex vivo studies were performed with fresh tumor tissue obtained from consented NSCLC patients. Tumor samples were treated with IDO inhibitor at 1μM for 48 hours. HPLC analysis on kynurenine and tryptophan was performed to verify target inhibition in the ex vivo model. A multiplex human cytokine assay was used to simultaneously analyze the differential release of cytokines in culture media. Additionally, NanoString PanCancer Immune Profiling platform containing probes to quantitate 770 immune function genes was used to determine positive and negative associations between expression of immune function genes and TIL activation by ex vivo treatment. Furthermore, autologous patient-derived cell lines (CAF and TILs) were utilized in an in vitro assay to determine the role of IDO inhibition on CAF-mediated immunosuppression.
Results
3D ex vivo studies showed a significant decrease in kynurenine demonstrating that epacadostat effectively inhibited the enzymatic activity of IDO in the tumor microenvironment accompanied by increased release of pro-inflammatory cytokines such as IFNγ. Treatment with epacadostat demonstrated decreased expression of genes involved in tumor growth (CCL25) and increased expression of antitumor immune response genes (CXCL14, CCL19 and CCL21). These studies showed epacadostat at an effective concentration of 1μM induced specific changes in the microenvironment and increased immune response. Furthermore, the autologous patient derived cell line in vitro assay determined that epacadostat overcame CAF induced inhibition of TIL activity.
Conclusions
This patient-derived 3D ex vivo approach demonstrated the immunomodulatory activity of epacadostat in NSCLC and indicates that inhibition of IDO activity may overcome stroma-induced immunosuppression in lung cancer. Studies on the effects of epacadostat in combination with anti-PD1 in the same culture systems are currently ongoing.
P335 Characterization of a novel differentiated anti-CTLA-4 antibody (ADU-1604) in vitro and in vivo
Maaike Hendriks1, Joost Kreijtz1, Paul Vink1, David Lutje Hulsik1, Imke Lodewijks1, Astrid Bertens1, Jos van de Crommert1, Maurice Habraken1, Wout Janssen1, Judith Stammen-Vogelzang1, Weiwen Deng2, Laura Hix Glickman2, Meredith Leong2, Sarah McWhirter2, Thomas Dubensky2, Hans van Eenennaam1, Andrea van Elsas1
1Aduro Biotech Europe, Oss, Netherlands; 2Aduro Biotech, Inc., Berkeley, CA, USA
Correspondence: Joost Kreijtz (jkreijtz@aduro.com)
Background
Targeting the CTLA-4 immune checkpoint with antibodies, either alone or in combination with PD-1/PD-L1 inhibitors shows clinical activity and durable responses in advanced cancer. The use of anti-CTLA-4 may augment other immunotherapies. Indeed, in syngeneic mouse tumor models anti-CTLA-4 strongly enhanced anti-tumor efficacy of live attenuated double-deleted Listeria monocytogenes (LADD) immunotherapy and of the stimulator of interferon genes (STING) pathway activator ADU-S100, indicating combination potential. Activity of anti-CTLA-4 is enhanced by Fc receptor engagement and tumor-selective depletion of regulatory T cells in mice, but this is unproven as of yet for cancer patients. Here we present a novel, humanized anti-CTLA-4 antibody originating from Aduro’s proprietary B-Select platform.
Methods
Among a panel of mouse anti-human CTLA-4 antibodies, 27A was humanized and functionally characterized on an IgG1 and IgG4 backbone. Both variants, named hCTLA4.27H6L1.i1 and .i4 were characterized in vitro for binding to human and cynomolgus CTLA-4, blocking of CD80/CD86 and functional activity. Both antibodies are compared for their capacity to induce tumor rejection in patient-derived tumor graft models in humanized mice.
Results
The IgG1 and IgG4 variants bound recombinant human CTLA-4 with a Kd of 2.7 ± 2.9 nM and 1.3 ± 0.5 nM as measured by Bio-Layer Interferometry, and cell-expressed CTLA-4 with an EC50 of 0.042 ± 0.000 nM and 0.048 ± 0.002 nM, respectively. Binding to endogenous CTLA-4 was demonstrated by flow cytometry using primary activated human and non-human primate lymphocytes. Both IgG1 and IgG4 variants potently blocked the interaction of CD80 and CD86 with human CTLA-4 (IC50 ranged from 1-2 nM). Using swap mutants of mouse and human CTLA-4 and cross-competition assays by Bio-Layer Interferometry, these antibodies showed a unique binding profile indicating a previously undiscovered epitope on CTLA-4. Functional characterization demonstrated that both antibodies enhanced activation and IL-2 production by human primary T cells or peripheral blood mononuclear cells (PBMCs) co-stimulated by Raji cells or Staphylococcal enterotoxin B, in a dose-dependent manner. The IgG1 variant (in contrast to IgG4) bound to low and high-affinity Fcγ receptors inducing antibody-dependent cell-mediated cytotoxicity (ADCC) mediated by natural killer (NK) cells and CD16+ monocytes towards CTLA-4+ cells. Similarly, complement-dependent cytotoxicity was differentially induced in vitro.
Conclusions
Overall, both hCTLA4.27 derivatives compared favorably to benchmarks in their biophysical and functional characteristics. The IgG1 variant, designated ADU-1604, is progressing through preclinical development.
P336 Preclinical characterization of MGA012, a novel clinical-stage PD-1 monoclonal antibody
Ross La Motte-Mohs1, Kalpana Shah1, Jennifer G. Brown1, Ralph F. Alderson1, Doug Smith2, Sergey Gorlatov1, Valentina Ciccarone1, James K. Tamura1, Hua Li1, Leilei He1, Farha Vasanwala1, Jill R. Rillema2, Monica Licea2, Jessica Hill1, Christina Wolff1, Massimiliano Pascuccio1, Francine Z. Chen2, Yan Chen2, Anushka De Costa2, Ann Easton2, Alexey Berezhnoy1, Jonathan Li2, Jeffrey Nordstrom1, Scott Koenig1, Ezio Bonvini1, Syd Johnson1, Paul A. Moore1
1MacroGenics, Inc., Rockville, MD, USA; 2MacroGenics, Inc., South San Francisco, CA, USA
Correspondence: Ross La Motte-Mohs (lamottemohsr@macrogenics.com)
Background
Monoclonal antibodies (mAbs) that target immune checkpoint pathways, such as the cytotoxic T lymphocyte-associated antigen-4 and the programmed cell death protein 1 (PD-1) pathways, have demonstrated broad clinical efficacy against a variety of malignancies as monotherapy or in a combination. MGA012 is a novel anti-PD-1 mAb developed to disrupt the PD-1 interaction with PD-L1/PD-L2 to restore or improve T-cell function as stand-alone therapy or in combination with other immune approaches.
Methods
Murine PD-1 mAbs were generated and benchmarked against replicas of the approved mAbs, nivolumab and pembrolizumab. Several mAbs with favorable characteristics were further chimerized or humanized. MGA012, a humanized, hinge-stabilized IgG4κ mAb, was selected based on binding and biophysical properties as well as a functional characterization inclusive of enhanced T-cell activation following superantigen re-stimulation.
Results
MGA012 bound human PD-1 with an affinity equal to or exceeding those of replicas of nivolumab or pembrolizumab. MGA012 bound PD-1-expressing cell lines and chronically-activated T cells, blocked PD-1 interactions with PD-L1/PD-L2, resulting in inhibition of PD-1 signaling and enhanced antigen-driven cytokine secretion to levels comparable to those observed with nivolumab or pembrolizumab replicas. Furthermore, characterization of MGA012 in ex-vivo tumor microenvironment immune models showed activation profiles recapitulating the benchmark PD-1 mAbs. MGA012 showed combinatorial activity in vitro when added to anti-CTLA-4 or anti-LAG-3 mAbs and enhanced the activity of a T-cell redirecting molecule in a mouse tumor model. MGA012 showed no unexpected cross-reactivity in human tissues, with staining observed primarily in lymphocytes and lymphoid organs. In a repeat-dose (10-150 mg/kg QWx4) study in cynomolgus monkeys, PK was linear with a beta half-life of 11.2 days (±4.6 SD) and full PD-1 occupancy on circulating T cells at all doses tested. Occupancy of ≥80%, persisting for 4-7 weeks, was also observed in monkeys receiving a single 10 mg/kg dose. MGA012 was well tolerated in cynomolgus monkeys and demonstrated a favorable safety profile with a no-adverse effect level (NOAEL) of 150 mg/kg.
Conclusions
MGA012 is a novel anti-PD-1 mAb with favorable preclinical characteristics, including PD-1 binding and biophysical properties, PD-1 pathway blockade, the ability to enhance T-cell responses in vitro and in vivo, and a favorable PK and safety profile in cynomolgus monkeys. Clinical trials are ongoing [NCT03059823] or in planning stage to ascertain the safety and preliminary activity of MGA012 alone or in combination with other immune oncology agents, including T-cell redirecting bispecific DART® molecules.
Trial Registration
ClinicalTrials.gov NCT03059823.
P337 Preclinical characterization of MGD013, a PD-1 x LAG-3 bispecific DART® molecule
Ross La Motte-Mohs1, Kalpana Shah1, Jennifer G. Brown1, Doug Smith2, Sergey Gorlatov1, Valentina Ciccarone3, James K. Tamura1, Hua Li1, Leilei He1, Farha Vasanwala1, Christine Shoemaker1, Jonathan Li2, Shereen Saini2, Jill R Rillema2, Monica Licea2, Jessica Hill1, Arin Whiddon1, Massimiliano Pascuccio1, Francine Z. Chen2, Anushka De Costa2, Ann Easton2, Peter Lung2, Kurt Stahl1, Jeffrey Nordstrom1, Scott Koenig1, Ezio Bonvini1, Syd Johnson1, Paul A. Moore1
1MacroGenics, Inc., Rockville, MD, USA; 2MacroGenics, Inc., South San Francisco, CA, USA; 3MacroGenics, Inc., Rockville, CA, USA
Correspondence: Ross La Motte-Mohs (lamottemohsr@macrogenics.com)
Background
Monoclonal antibodies (mAbs) that target the immune checkpoints, cytotoxic T lymphocyte-associated antigen-4 (CTLA-4) and programmed cell death protein-1 (PD-1), have shown enhanced clinical anti-tumor activity when given in combination, triggering interest in determining whether additional checkpoint inhibitor combinations may afford enhanced clinical benefit. Lymphocyte activation gene-3 (LAG-3) is another immune checkpoint expressed on activated T cells and tumor infiltrating lymphocytes. Recognizing the therapeutic potential of dual checkpoint blockade, we have engineered MGD013, a IgG4κ bispecific DART molecule to bind PD-1 and LAG-3 concomitantly or independently and disrupt these non-redundant inhibitory pathways to further restore exhausted T-cell function.
Methods
Proprietary PD-1 and LAG-3 mAbs were generated and selected based on binding characteristics, biophysical properties, the ability to block their respective receptor/ligand axes and to synergize in T-cell stimulation assays. Humanized sequences were incorporated into a tetravalent bispecific DART format and benchmarked against combinations of replicas of the approved PD-1 mAb, nivolumab, and BMS-986016 anti-LAG-3 mAb, which is currently under clinical evaluation. MGD013 biological activity was evaluated in various primary cell-based immune assays. Safety was assessed in cynomolgus monkey toxicology studies performed at MPI (Mattawan, MI) under Institutional Animal Care and Use Committee-approved protocols.
Results
MGD013 bound with high affinity to human and cynomolgus monkey PD-1- and LAG-3-expressing cells and blocked PD-1/PD-L1, PD-1/PD-L2 and LAG-3/HLA (MHC-II) interactions, with resultant signaling blockade. Functional characterization revealed enhanced cytokine secretion in response to antigen stimulation that was greater than that of the combination of individual equimolar amounts of PD-1 and LAG-3 mAbs. MGD013 was well-tolerated in a repeated-dose (Q1Wx4) cynomolgus monkey toxicology study. Except for the occurrence of watery feces in a few animals, no MGD013-related adverse findings were noted, including hematological or clinical chemistry changes, serum cytokine levels or gross and microscopic histological findings, establishing a no-observed adverse-effect level (NOAEL) of 100 mg/kg.
Conclusions
MGD013 is a bispecific DART molecule capable of simultaneously blocking the PD-1 and LAG-3 pathways, resulting in enhanced T-cell activation compared to single or combination mAb blockade. MGD013 has demonstrated a favorable pre-clinical safety and toxicological profile and is currently initiating clinical testing [NCT03219268].
Trial Registration
ClinicalTrials.gov NCT03219268
P338 Combined IL-6 and CTLA4 blockade enhances CD8+ T cell infiltration via CXCR3 and limits growth of pancreatic cancer in murine models
Christopher McQuinn1, Matthew Farren2, Thomas Mace1, Jacob Bowers3, Reena Shakya1, A. Brad Ferris2, Gregory Young1, William Carson, III1, Chrystal Paulos3, Bassel El-Rayes2, Gregory Lesinski2
1The Ohio State University, Columbus, OH, USA; 2Winship Cancer Institute of Emory University, Atlanta, GA, USA; 3Medical University of South Carolina, Charleston, SC, USA
Correspondence: Gregory Lesinski (gregory.b.lesinski@emory.edu)
Background
Immune checkpoint blockade has not shown efficacy in pancreatic cancer. We hypothesized the cytokine interleukin-6 (IL-6) enhances immune suppression in this disease, and represents a logical target to augment immunotherapy. Prior publications from our group and others demonstrate IL-6 is derived from pancreatic stroma, is inversely associated with overall survival in metastatic patients and enhances efficacy of anti-PD-L1 antibodies in pre-clinical models. This report addresses the efficacy of combined IL-6 and CTLA4 blockade, and identifies a mechanism by which this combination enhances effector T cell infiltration into tumors.
Methods
In vivo efficacy of Ab targeting IL-6 and CTLA4 was tested in mice bearing subcutaneous MT-5 murine pancreatic tumor cells with KrasG12D and TP53R172H mutations. CXCR3 blocking antibodies were used for in vivo mechanistic studies, while immune biomarkers were analyzed using flow cytometry or immunohistochemistry.
Results
Combined blockade of IL-6 and CTLA4 inhibited tumor growth rate as compared to controls (p’s <0.05). IHC analysis revealed increased T cells in tumors from combination treated mice (p=0.035 vs. anti-IL-6; p=0.038 vs. anti-CTLA4; p<0.0001 vs. iso control). Analysis of systemic immune biomarkers was conducted via flow cytometric analysis of splenocytes. Despite the role for IL-6 in expanding MDSC, no change in monocytic (CD11b+Ly6G-Ly6C+) or granulocytic (CD11b+Ly6G+Ly6Clow) cells were observed. However, analysis of T cell subsets revealed both anti-CTLA4 alone or the combination increased cells with Th1 or Th17, but not Th2 phenotypes (p<0.05). Cells with a CD4+CD25+FoxP3+ phenotype were increased in combination treated mice (p<0.05). We are studying if these cells are suppressive or a byproduct of enhanced activation marker expression. Blockade of IL-6, CTLA4 or the combination altered circulating cells with phenotypic properties of naïve, effector and central memory T cells (based on CD44/CD62L). Notably, circulating CD4+CD44+CD62L+ T cells were significantly higher in the combination (p<0.05). Consistent with T cell infiltration data, sequencing of TCRβ within n=3 representative tumors from each group of mice revealed a trend toward increased number of both productive templates and rearrangements. Finally, in vivo administration of Ab targeting CXCR3 limited the efficacy of the treatment combination, and reduced CD8+ T cell infiltration into tumors.
Conclusions
IL-6 blockade enhances efficacy of antibodies targeting CTLA4 in pre-clinical models of pancreatic cancer. Mechanistically, this involves CXCR3-mediated intratumoral infiltration of CD8+ T cells. These data suggest that clinically-available IL-6/IL-6R targeted agents could be repurposed to enhance immune checkpoint blockade as a novel combination therapy.
P339 Critical role of hematopoietic progenitor kinase 1 in anti-tumor immune responses
Dan You1, Stephen Hillerman1, Jesse Swanson1, Ching-Ping Ho1, Anwar Murtaza1, Rukiye Eraslan1, Miguel Sanjuan1, John Hunt1, Luisa Salter-Cid1, Joshua Curtin2
1Bristol-Myers Squibb, Lawrence Township, NJ, USA; 2Johnson & Johnson Pharmaceutical, Spring House, PA, USA
Correspondence: Jinqi Liu (jinqi.liu1@gmail.com)
Background
Hematopoietic progenitor kinase 1 (HPK1) was previously reported as a negative regulator of T cell responses due to its ability to attenuate T cell receptor (TCR) signaling. T cells from HPK1 knockout (HPK1-/-) mice demonstrated elevated proliferation and cytokine production in response to TCR engagement and are resistant to prostaglandin E2 (PGE2)-mediated suppression. Additionally, HPK1-/- bone marrow derived dendritic cells (BMDCs) eliminate established Lewis Lung Carcinoma more effectively compared with HPK1 +/+ BMDCs, suggesting HPK1 is a critical negative regulatory component in key immune cell types involved in anti-tumor immunity. To further investigate the potential value of targeting HPK1 pharmacologically in cancer immunotherapy, we generated HPK1 kinase dead mice (HPK1 KD). Extensive characterization of these mice was conducted via employing syngeneic tumor models and ex vivo studies. We report here our findings on the blockade of HPK1 kinase activity in modulating anti-tumor immune responses.
Methods
HPK1 KD mice were generated and characterized, including immune cell phenotyping in peripheral blood and secondary lymphoid organs. These mice were then subjected to in vivo stimulation with agonistic anti-CD3 followed by measurement of plasma cytokines. HPK1 WT and KD mice were further interrogated in syngeneic tumor models in combination with a cyclooxygenase inhibitor (celecoxib) or anti-PD1. Anti-tumor efficacy was monitored, immune phenotyping was conducted for draining lymph nodes and tumor tissues, and ex vivo studies were performed to evaluate the enhancement of immune responses.
Results
HPK1 KD mice were grossly normal and immune phenotyping revealed no difference compared with WT mice. HPK1 KD mice demonstrated superior anti-tumor efficacy in a sarcoma model. Significantly better anti-tumor efficacy with 50-70% tumor free was observed in HPK1 KD mice compared to WT mice treated with either celecoxib or anti-PD1, in sarcoma and colorectal tumor models, respectively. Further immune phenotyping and ex vivo studies indicated enhanced CD4+ and CD8+ T cell populations as well as IFNγ and TNFα secretion along with reduced ratios of CD8/Tregs. Cytolytic activities against syngeneic tumor cells were also augmented with HPK1 KD CD8+ T cells compared with WT CD8+ cells.
Conclusions
Genetic blockade of HPK1 kinase activity revealed a significant enhancement of immune responses and resulted in improved anti-tumor efficacy in combination with inhibition of PGE2 or PD-1 pathways.
P340 In vitro effects and mechanism of hu14.18-IL2 immunocytokine against GD2 positive pediatric malignancies
Holger Lode1, Maxi Zumpe1, Madlen Juettner1, Sascha Troschke-Meurer1, Evelyne Janzek2, Romana Schaefer2, Bettina Neuchrist2, Nikolai Siebert1, Hans Loibner2
1University Medicine Greifswald, Greifswald, Germany; 2Apeiron Biologics, Vienna, Austria
Correspondence: Holger Lode (lode@uni-greifswald.de)
Background
Passive immunotherapy with anti-GD2 antibody (Ab) ch14.18/CHO (dinutuximab beta) showed activity for the treatment high-risk neuroblastoma (NB) patients and received recently marketing approval in the EU. Immunocytokines are antibody-cytokine fusion proteins that combine the targeting properties of monoclonal antibodies with the immune stimulatory function of cytokines. Here we demonstrate activity and mechanism of hu14.18-IL2 against a variety of GD2 positive pediatric tumor cell lines (neuroblastoma, osteosarcoma, Ewing’s sarcoma, retinoblastoma) in preclinical models.
Methods
Expression of the target antigen GD2 on LAN-1 (neuroblastoma), MG63 (osteosarcoma), TC-71 (Ewing’s sarcoma) and Y79 (retinoblastoma), and PD-1/PD-L1 checkpoint on LAN-1 was analyzed by flow cytometry. Hu14.18-IL2 mediated ADCC, CDC and whole blood cytotoxicity (WBT) was determined by 51Cr release assays.
Results
We found expression of antigen GD2 on all cell lines derived of major neuro-ectodermal malignancies in childhood. The highest level was found in neuroblastoma but all other cell lines were clearly GD2 antigen positive. Importantly, hu14.18-IL2 was effective in mediating ADCC, CDC and WBT against all cell lines in vitro, and potency was found higher than that of ch14.18/CHO in the osteosarcoma and retinoblastoma models. The effects were antigen specific as addition of an anti-idiotypic antibody abrogated the cytolytic activity.
Interestingly, tumor specific ADCC in the presence of LA-N-1 neuroblastoma cells, leukocytes and sub-therapeutic hu14.18-IL2 concentrations (10-100 ng/ml) resulted in a strong increase of PD-L1 expression both on effector and target cells. This effect required cell-cell contact, since separation of effector cells from target cells using a membrane abrogated the activity. This provides a rationale to explore combinatorial approaches with agents that inhibit the PD-1/PD-L1 checkpoint.
Conclusions
Immunocytokine hu14.18-IL2 is effective against GD2 positive pediatric malignancies derived of neuroectodermal origin. Engaged immune effector functions by hu14.18-IL2 result in a concomitant upregulation of immune checkpoints that suggests a combinatorial approach with checkpoint inhibitors early during clinical development.
P341 Peripheral immunomodulatory changes in metastatic renal cell carcinoma (mRCC) patients treated with nivolumab
Lorena Almeida, Manuel Maia, Haejung Won, Dayson Moreira, Paulo Bergerot, Marcin Kortylewski, Sumanta Pal
City of Hope Comprehensive Cancer Center, Duarte, CA, USA
Correspondence: Manuel Maia (mcaitano@coh.org)
Background
CheckMate-009 was a biomarker-based study in which serial biopsies were conducted for patients receiving nivolumab [1]. Lesser data is available for fluxes in circulating biomarkers during the course of treatment. We hypothesized that immunomodulatory changes in blood would mirror previous observations in tissue.
Methods
Patients with mRCC receiving nivolumab as standard of care were prospectively enrolled. Blood samples were drawn at baseline, week 4 and week 12 during treatment. Analysis of samples for relevant white blood subsets was conducted by immersing peripheral blood mononuclear cells in a mixture of PBS, 2% FCS and 0.1% sodium azide with FcIII/IIR-specific antibody to block nonspecific binding and stain the cells with different combinations of fluorochrome-coupled antibodies. We further collected fluorescence data on Fortessa (Beckton Dickinson) and analyzed them using FlowJo software (Tree Star) [2]. Plasmas were analyzed for human cytokines using Luminex. Changes in cells and cytokines overtime in the overall population were interrogated. We also compared baseline levels of cytokines in responders (R; including partial/complete response and stable disease) and non-responders (P; primary progression).
Results
A total of 10 patients were included, with a total of 29 samples collected. During treatment, we observed a significant decrease in the proportion of CD3+CD4+ T cells and an increase in the CD3+CD8+ T cells in the overall population (p<0.05). In addition, the expression of PD-1 was significantly decreased in both CD4+ and CD8+ T cells overtime, while serum level of IL2R and CXCL9 significantly increased (p<0.05). Compared to P, R group showed a trend towards higher baseline levels of IFN-alpha and IFN-gamma (p=0.05 and 0.11, respectively) as well as VISTA expression on CD4+ T cells (p=0.15)
Conclusions
Our results emphasize the immunomodulatory changes in the peripheral blood occurring during treatment with nivolumab in mRCC pts. Some dynamics contrast with previous observations in tissue [3], reflecting temporal and spatial heterogeneity during immune system modulation by immunotherapies. Differential baseline levels of cytokines and immune checkpoint expression in the R group may be used as a predictive biomarker for nivolumab response if confirmed in larger series.
References
1. Choueiri, T. K., et al. Immunomodulatory Activity of Nivolumab in Metastatic Renal Cell Carcinoma. Clinical Cancer Research.2016;22(22): 546.
2. Chalmin F, et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human MDSC. J Clin Invest. 2010;120:457-71.
3. Gao, J., et al. VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat Med.2017;23(5): 551-555.
P342 Blood in circulation, with intact cascade systems, as a tool to predict first-infusion reactions and mechanism-of-action of immunotherapeutics
Erika Fletcher1, Mohamed Elthahir1, Frida Lindqvist2, Jonas Rieth1, Gunilla Törnqvist2, Justyna Leja-Jarblad1,2, Sara Mangsbo1,2
1Uppsala University, Uppsala, Sweden; 2Immuneed AB, Uppsala, Sweden
Correspondence: Sara Mangsbo (sara.mangsbo@farmbio.uu.se)
Background
Since the TGN1412 disaster in London, where an anti-CD28 superagonistic antibody induced a life-threatening cytokine storm in six healthy men during a clinical trial [1], risks of first-infusion reactions are important to study prior clinical trial initiation. These assays can constructed in different ways and depending on the setup, the sensitivity of the assay depends on the antibody tested. It is therefore of great need to validate a cytokine release assay with the potential to detect first-infusion reactions regardless of the nature of the antibody tested.
Methods
As the assays used today to predict cytokine release syndrome (CRS) are devoid of one or several blood components we setout to investigate if a modified chandler loop model could be used to predict CRS. The assay makes use of extracorporeal fresh whole blood in circulation, thus it contains blood immune cells, immunoglobulins along with intact cascade systems [2]. The challenge with such systems is to avoid clotting during assay performance, but correctly performed, will allow for both monitoring of cytokine release along with studies of ADCC and CDC.
Results
Uniquely the assessed agonistic antibodies were scored to induce CRS in blood from all tested donors (n=22) after only 4 hours of incubation in the loop assay, whereas non-agonistic antibodies associated with no or low infusion reactions in the clinic neither induce cytokine release nor cause false positive responses. Additionally, the value of an intact complement system in the assay was highlighted by the possibility to dissect out the mechanism-of-action of alemtuzumab and rituximab.
Conclusions
A modified chandler loop system can complement lymph node-like assays or be used as an excellent stand-alone test to investigate drug/blood interactions during preclinical development, or for individual safety screening prior a first-in-man clinical trial.
References
1. Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, Panoskaltsis N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 2006;355(10):1018-28.
2. Mangsbo SM, Sanchez J, Anger K, Lambris JD, Ekdahl KN, Loskog AS, Nilsson B, Totterman TH. Complement activation by CpG in a human whole blood loop system: mechanisms and immunomodulatory effects. J Immunol. 2009;183(10):6724-32.
P343 Novel immuno-oncology biologics derived via directed evolution of IgSF domains
Mark Maurer, Ryan Swanson, Michael Kornacker, Steve Levin, Chris Navas, Chelsea Gudgeon, Lawrence Evans, Martin Wolfson, Dan Ardourel, Daniel DeMonte, Joseph Kuijper, Sean MacNeil, Janhavi Bhandari, Mark Rixon, Stanford Peng
Alpine Immune Sciences, Inc., Seattle, WA, USA
Correspondence: Mark Maurer (mark.maurer@alpineimmunesciences.com)
Background
Our variant Ig domain (vIgD™) platform creates novel, therapeutically-applicable protein domains with tailored specificity and affinity. These vIgDs are created through directed evolution of immunoglobulin superfamily (IgSF) proteins and have unique biochemical properties including small size, single domain structure, and the capacity to interact with multiple counter-structures. Because many IgSF family members and their counter-structures are widely expressed on immune cells and tumors, the vIgD platform is well positioned for the development of immuno-oncology biologics with potential first-in-class mechanisms of action. Here, multiple therapeutic formats for vIgDs were developed and characterized.
Methods
Novel vIgDs were created with tailored affinities and modulatory activities against PD-1, TIGIT, PD-L1, CTLA-4, CD28, and/or ICOS. These domains were successfully developed into multiple therapeutic formats, including single and multiple domain Fc fusion proteins and vIgD-monoclonal antibody (V-mAb) fusion proteins. In addition to ligand binding and specificity assays, in vitro functional activity was characterized in several T cell-based assays including cell-based reporter systems for pathway agonism or antagonism, primary human mixed lymphocyte reactions (MLRs), and costimulation assays utilizing artificial APCs (assessed by proliferation and IFNg production).
Results
Several functionally active therapeutic vIgD-based molecules were created successfully. (1) Single-domain vIgD-Fc fusion proteins with tailored binding to CD28, CTLA-4, and PD-L1 demonstrated differential activity in T cell activation assays and, depending on their ligand binding profile, resulted in greater or reduced IFNγ production and T cell proliferation in human T cell activation assays. (2) Multi-domain vIgD-Fc fusion proteins demonstrated promising targeting of immunomodulatory pathways in cell-based reporter assays and MLRs as assessed by IL-2 signaling and IFNγ production. Efficacy was comparable to monoclonal antibodies against the individual vIgD targets. (3) V-mAbs demonstrated target-specific T cell proliferation and IFNγ production in vitro, using both recombinant target proteins or target-specific cell lines.
Conclusions
The vIgD platform has successfully generated multiple immuno-oncology therapeutic candidates, in various formats including single- and multiple-domain Fc fusion proteins as well as V-mAbs. These varied formats confer, from a single molecule, multiple advantages including the multi-target modulation capability of evolved IgSFs, and, where applicable, tumor localizing capability of partner molecules or domains. This platform may contribute to the next generation of immunotherapeutic proteins in an oncology setting and efforts are ongoing to develop these candidates for human therapeutic use.
P344 Development and functional characterization of COM902, a novel therapeutic antibody targeting the immune checkpoint TIGIT
Sandeep Kumar1, Radhika Desai1, Hsin-Yuan Cheng1, Kyle Hansen1, Andy Drake1, Patrick Wall1, Kathryn Logronio1, Gady Cojocaru2, John Hunter1, Mark White1, Spencer Liang1, Maya Kotturi1
1Compugen USA, Inc., South San Francisco, CA, USA; 2Compugen Ltd., Holon, Israel
Correspondence: Maya Kotturi (mayak@cgen.com)
Background
TIGIT is a coinhibitory receptor that is expressed on lymphocytes, including effector and regulatory CD4+ T cells (Tregs), effector CD8+ T cells, and NK cells, that infiltrate different types of tumors. Engagement of TIGIT with its reported ligands, poliovirus receptor (PVR) and PVR-like proteins (PVRL2 and PVRL3) directly suppresses lymphocyte activation. PVR is also broadly expressed in tumors, suggesting that the TIGIT-PVR signaling axis may be a dominant immune escape mechanism for cancer. We report here the biophysical and functional characterization of COM902, a therapeutic antibody targeting TIGIT. We demonstrate that co-blockade of TIGIT and a new checkpoint inhibitor, PVRIG, augments T cell responses.
Methods
Human phage display and mouse hybridoma antibody discovery campaigns were conducted to generate therapeutic anti-TIGIT antibodies. The resulting antibodies were evaluated for their ability to bind to recombinant and cell surface-expressed human TIGIT with high affinity. Cross-reactivity of the antibodies to cynomolgus and mouse TIGIT was also examined. A subset of antibodies that bound with high affinity to human TIGIT, and cross-reactive to cynomolgus TIGIT were further characterized for their ability to block the interaction between TIGIT and PVR. Blocking antibodies were screened for their ability to enhance antigen-specific T cell activation in vitro either alone, or in combination with an anti-PVRIG antibody, COM701.
Results
We identified a lead antibody, COM902, that binds to human TIGIT with high femtomolar affinity. This antibody bound to TIGIT endogenously expressed on human CD8+ T cells with higher affinity than tested benchmark antibodies, and was also cross-reactive to both cynomolgus and mouse TIGIT. When tested for in vitro activity, COM902 augmented cytokine secretion and tumor cell killing by CMV-specific CD8+ T cells with superior or equivalent potency to the tested benchmark antibodies. Combination of COM902 with an anti-PD1 antibody or COM701 resulted in enhanced CMV-specific CD8+ T cell activity. Furthermore, we demonstrated that TIGIT is predominantly expressed on Tregs and effector CD8+ T cells from solid tumors compared to peripheral blood, suggesting that these populations will likely be preferentially targeted by COM902.
Conclusions
We describe the development of a high affinity antagonistic TIGIT antibody, COM902, that is currently in preclinical development. We postulate that the femtomolar affinity of COM902 could result in lower and less frequent dosing in patients. COM902 can enhance human T cell activation either alone or in combination with other checkpoint antibodies. Thus, our data demonstrates the utility of targeting TIGIT, PD1, and PVRIG for the treatment of cancer.
P345 Tumor protective effect of anti-MUC1.IgE in pancreatic cancer
Kamiya Mehla1, James A Grunkemeyer1, Ragupathy Madiyalakan2, Christopher F Nicodemus3, Michael A Hollingsworth1
1University of Nebraska Medical Centre, Omaha, NE, USA; 2Oncoquest, Edmonton,, AB, Canada; 3AIT Strategies, Franconia, NH, USA
Correspondence: Michael A Hollingswort (kamiya.mehla@unmc.edu)
Background
Pancreatic adenocarcinoma remains a highly lethal disease, that has thus far proven to be highly refractory to immunotherapeutic strategies. Hence, new approaches are needed. There is epidemiological evidence that individuals with allergies have a lower incidence of pancreatic cancer [1]. This suggests that distinct immune surveillance (Th2 based) might underlie protection against pancreatic cancer observed in allergic individuals. We hypothesized that therapeutic targeting with IgE to enhance FcεRI signaling would trigger an effective anti-tumor immune response against pancreatic cancer. We posited that tumor antigen directed IgE/FcεRI cross-linking on mast cell and controlled release of histamine near intra-tumor blood vessels could be exploited to improve drug delivery in pancreatic cancer.
Methods
We therefore explored the efficacy of human tumor antigen targeted IgE antibody (humanized anti-MUC1.IgE) in combination of anti-PD-L1 (for relieving T cell exhaustion [2]) and PolyICLC (for effective dendritic cell maturation [2]) in a pre-clinical model of pancreatic cancer using mice transgenic for human MUC1 and FcεRI (hMUC1/hFcεRI).
Results
We observed that a combination of anti-MUC1.IgE+anti-PD-L1+PolyICLC induced antigen specific rejection of two different MUC1 expressing pancreatic tumors cell lines (Panc02.MUC1, KPC.MUC1) and prolonged the overall survival of mice challenged with subcutaneous and orthotopic tumors as compared to mice treated with an isotype control antibody (anti-PSA.IgE). Furthermore, anti-MUC1.IgE+anti-PD-L1+PolyICLC combination induced MUC1 specific memory responses as evidenced by antigen specific rejection/delays of tumors in mice re-challenged with Panc02.MUC1 tumors. Importantly, we observed that NK and CD8 T cells were required for the cell mediated anti-tumor responses, as in vivo depletion of these subtypes but not CD4 abrogated the tumor protective in mice bearing orthotopic tumors. Additionally, our study demonstrated a time dependent increase in intra-tumor vascular permeability (increased dextran-647 influx) post anti-MUC1.IgE treatment (iv) in subcutaneous tumor (Panc02.MUC1) bearing dTg mice.
Conclusions
Taken together, this is the first study to show that cellular immune responses induced by antigen specific stimulation of the IgE/FcεRI axis in combination with PolyICLC and anti-PD-L1provided tumor protective benefits against pancreatic cancer. Our study provides evidence for the clinical applicability and rapid translation of anti-MUC1.IgE based combination therapy against pancreatic tumors.
References
1. Gandini S, Lowenfel AB, Jaffee EM, Armstrong TD. Allergies and risk of pancreatic cancer: a meta-analysis with review of epidemiology and biological mechanisms. Cancer Epidemio Biomarker Prev. 2005;14(8):1908-16.
2. Nagato T, Celis E. A novel combinatorial cancer immunotherapy:poly-IC and blockade of the PD-1/PD-L1 pathway. Oncoimmunology. 2014;15:3:e28440.
P346 Inhibition of MMP9 yields improved effector T cell responses in a PD1-Axis refractory model
Amanda Mikels-Vigdal, Vladi Juric, Chris O'Sullivan, Erin Stefanutti, Andrew Greenstein, Maria Kovalenko, Jeremiah Degenhardt, Peng Yue, Victoria Smith
Gilead Sciences, Inc, Foster City, CA, USA
Correspondence: Amanda Mikels-Vigdal (amanda.mikels-vigdal@gilead.com)
Background
Matrix metalloproteinase 9 (MMP9) acts via diverse mechanisms to promote tumor growth and metastasis, and is a key component of the immune-suppressive myeloid inflammatory milieu. We developed a monoclonal antibody (AB0046) that inhibits murine MMP9 and assessed its mechanism of action in immunocompetent mice as a single agent or in combination with a murine anti-PDL1 antibody.
Methods
An orthotopic, syngeneic tumor model of Her2-driven breast cancer was utilized for both efficacy and pharmacodynamic studies involving RNA and T cell receptor (TCR) sequencing, flow cytometry, and protein analysis. Enzymatic analyses were performed on T cell chemoattractant CXCR3 ligands (CXCL9, CXCL10, and CXCL11) which were subsequently evaluated in chemotaxis assays.
Results
Anti-MMP9 treatment alone or in combination with an anti-PDL1 antibody decreased primary tumor growth as compared to IgG control-treated animals (56% vs 335% tumor growth increase, p=0.0005) or anti-PDL1 alone. Profiling of tumors by RNA sequencing revealed that inhibition of MMP9 resulted in elevated expression of genes associated with immune cell activation pathways (Hallmark Interferon Gamma Response, FDR p<0.001). Treatment with anti-MMP9 and anti-PDL1 antibodies decreased TCR clonality, with evidence of a more diverse TCR repertoire (p=0.005). Immunophenotyping of tumor-associated T cells by flow cytometry showed that anti-MMP9 and anti-PDL1 co-treatment promoted a 2.8-fold increase in CD3+ cells in tumors (p=0.01), which was associated with an increase in CD4+ T cells (3.2-fold increase; p=0.006) and CD8+ T cells (2.8-fold increase; p=0.013). In contrast, anti-MMP9 and combination treatment resulted in a decrease in tumor-associated regulatory T cells (CD25+ FoxP3+ cells, p = 0.04). MMP9 cleavage of T cell chemoattractant ligands in vitro rendered them functionally inactive for recruitment of activated primary human effector T cells. Luminex analysis of tumor protein lysates showed elevated levels of key immune cytokines IL-12p70, IL-18, and CXCL10.
Conclusions
Inhibition of MMP9 reduces tumor burden and promotes cytotoxic T cell infiltration in a PD1-axis refractory mouse model. The combination of nivolumab and GS-5745, a humanized anti-MMP9 inhibitory antibody, is being evaluated in gastric cancer (NCT02864381).
Trial Registration
ClinicalTrials.gov: NCT02864381.
P347 Anti-PD1 x anti-ICOS bispecific antibody XmAb23104 brings together PD1 blockade and ICOS costimulation to promote human T cell activation and proliferation
Gregory Moore, Michael Hedvat, Matthew Bernett, Christine Bonzon, Rajat Varma, Suzanne Schubbert, Sung-Hyung Lee, Kendra Avery, Rumana Rashid, Alex Nisthal, Liz Bogaert, Irene Leung, Seung Chu, Umesh Muchhal, John Desjarlais
Xencor, Inc., Monrovia, CA, USA
Correspondence: John Desjarlais (gmoore@xencor.com)
Background
Tumor infiltrating lymphocytes (TILs) often express multiple immune checkpoints and costimulatory receptors. Checkpoint blockade has demonstrated increased clinical response rates relative to other treatment options; however, many patients fail to achieve a response to checkpoint blockade. We sought to identify an additional therapeutic modality to stack with checkpoint blockade that could increase patient response rate. We hypothesized that engagement of T cell costimulatory receptors in concert with checkpoint blockade could further increase T cell activation and proliferation. The combination of checkpoint blockade with costimulation could be accomplished using a bispecific antibody format, with the potential benefits of reduced cost and more selective targeting of TILs to improve safety.
Methods
XmAb23104, which binds to immune checkpoint PD1 and T cell costimulatory receptor ICOS, was assembled in a bispecific antibody platform with substitutions in the Fc domain to suppress effector function. XmAb23104 was evaluated in vitro by measuring antibody binding to and cytokine release from Staphylococcal enterotoxin B (SEB) stimulated PBMCs. In vivo activity was evaluated using a mouse model in which human PBMCs are engrafted into NSG mice (huPBMC-NSG) and the extent of T cell engraftment monitored by flow cytometry.
Results
We produced XmAb23104, a PD1 x ICOS bispecific antibody that binds both PD1 and ICOS monovalently. XmAb23104 selectively targeted PD1+ICOS+ T cells more than monovalent controls, indicating that a single bispecific molecule was capable of avid co-engagement of both PD1 and ICOS. Surprisingly, despite monovalent engagement of ICOS, XmAb23104 promoted strong T cell activation above that expected of PD1 blockade alone, both in vitro and in vivo. In vitro, XmAb23104 enhanced IL2 production in an SEB stimulation assay (n = 16 donors) relative to control, anti-PD1 alone, and anti-ICOS alone (p < 0.001 for all comparisons), indicating productive combination of PD1 blockade plus ICOS costimulation. Analysis of XmAb23104-treated cells revealed hallmarks of ICOS signaling, including AKT phosphorylation and a multi-gene expression signature consistent with ICOS costimulation. In vivo, treatment of huPBMC-NSG mice with XmAb23104 promoted superior T cell engraftment to that found for anti-PD1 treatment alone. In a representative study, XmAb23104 induced a 14-fold increase in human CD45+ cells, while anti-PD1 treatment alone only promoted a 2-fold increase.
Conclusions
XmAb23104, a PD1 x ICOS bispecific antibody, brings together PD1 blockade and ICOS costimulation and promotes strong T cell activation in vitro and in vivo. Compelling activity suggests clinical development is warranted for the treatment of human malignancies.
P348 Dual blockade of PD-L1 and LAG-3 with FS118, a unique bispecific antibody, induces T-cell activation with the potential to drive potent anti-tumour immune responses
Matthew Kraman, Natalie Fosh, Katarzyna Kmiecik, Katy Everett, Carlo Zimarino, Mustapha Faroudi, Mateusz Wydro, Alexander Koers, Lesley Young, Michelle Morrow, Jacqueline Doody, Mihriban Tuna, Neil Brewis
F-star, Cambridge, United Kingdom
Correspondence: Michelle Morrow (michelle.morrow@f-star.com)
Background
Despite advances with therapies targeting the PD-1/PD-L1 pathway, many patients are refractory or relapse following treatment. LAG-3 expression on exhausted T cells and T-regulatory cells (Tregs) in the tumour may be responsible for this resistance and provides a rationale for co-treatment with antibodies targeting LAG-3 and PD-L1. An alternative approach is the development of a bispecific antibody encompassing binding sites for two antigens. FS118 is a bispecific antibody targeting LAG-3 and PD-L1 that provides dual pathway blockade with the potential to drive unique biology via co-binding of PD-L1 and LAG-3.
Methods
A LAG-3/PD-L1 mAb2 bispecific antibody, termed FS118, was engineered by introducing a distinct LAG-3 binding capability into the constant region of a human IgG1 molecule and assembled into a bispecific format with anti-PD-L1 Fabs. Additional mutations introduced into the Fc region suppress effector function. FS118 was evaluated in vitro for antigen binding and de-repression of LAG-3 and PD-L1 function in both a D011.10 T-cell activation system and a super-antigen stimulated peripheral blood mononuclear cells (PBMC) assay. Anti-tumour activity of a murine-specific molecule, mLAG-3/PD-L1 mAb2, was assessed in vivo in the MC38 mouse tumour model and associated immunophenotypic changes were evaluated using flow cytometry.
Results
FS118 is bivalent for both LAG-3 and PD-L1. It is capable of binding to both targets simultaneously and can de-repress the inhibitory function of human PD-L1 and human LAG-3 in an engineered murine T-cell system. FS118 is a potent activator of immune cell function generating sub-nanomolar EC50 values in a human PBMC assay as measured by cytokine with at least equivalent activity to a combination of anti-LAG-3 and anti-PD-L1.
In murine in vitro assay systems, mLAG3/PD-L1 mAb2 recapitulates the function of FS118 in human systems. MC38 tumour growth studies indicated that mLAG-3/PD-L1 could result in significant anti-tumour activity equivalent to a combination of antibodies targeting LAG-3 and PD-L1. Pharmacodynamic assessment demonstrated changes in the immunophenotype of tumour-infiltrating lymphocytes in the tumour of mLAG3/PD-L1 mAb2 treated mice.
Conclusions
Dual blockade of LAG-3 and PD-L1 with a bispecific antibody results in T-cell activation at least comparable to a combination of antibodies targeting LAG-3 and PD-L1 in primary T-cell assays and murine tumour models. These data provide evidence to support the rationale for clinical development of FS118, a LAG-3/PD-L1 mAb2, for the treatment of human cancer.
P349 Molecular-targeted radiotherapy with an alkyl-phosphocholine analog leads toimmunomodulation in a syngeneic murine melanoma model
Suresh Veerankutty, Peter Carlson, Dana Baiu, Clinton Heinze, Alexander Boruch, Amy Erbe, Joseph Grudzinski, Reinier Hernandez, Bryan Bednarz, Jamey Weichert, Paul Sondel, Zachary Morris, Mario Otto
University of Wisconsin, Madison, WI, USA
Correspondence: Mario Otto (motto@pediatrics.wisc.edu); Zachary Morris
Background
Ionizing radiation can influence the immune response to tumors, and the combination of external beam radiotherapy with immunotherapy has increasingly garnered attention in clinical trials. Little is known about the immunomodulatory effects of molecular-targeted radiopharmaceuticals. 18-(p-iodophenyl) octadecyl phosphocholine (NM404) is a phospholipid ether analog with selective sequestration in cancer cells [1]. Here, we report on the immunomodulatory properties of the radioactive isostere, 131I-NM404 in a syngeneic murine melanoma model.
Methods
In vitro cell uptake of NM404 was evaluated by flow cytometry using a fluorescent-labeled NM404 analog. Mice, bearing syngeneic B78 melanoma tumors, were injected with a subtherapeutic dose of 2.2 MBq 131I-NM404 via lateral tail vein. Mice were euthanized and tumor tissue was harvested at 4 consecutive 1-week intervals post injection; the half-life of iodine-131 is ~1 week. To evaluate markers of immunomodulation over time, quantitative PCR and immunohistochemistry (IHC) were performed on the serially collected tumor tissue samples.
Results
In vitro, murine B78 melanoma cells sequestered 3-4 times more NM404 than normal murine splenocytes, and 6-10 times more than primary human fibroblasts over an 18 hour incubation period. The single, sub-therapeutic dose of 131I-NM404 did not significantly reduce the growth rate of B78 tumors in the treated mice compared to a control cohort injected with equivalent mass dose of nonradioactive NM404 (excipient). Gene expression analysis revealed a marked modulation of various immune and tumor-associated markers during the course of radiotherapy, such as IL-1β, IFNγ, CXCL1, IL-18, TGF-β2, B7-H3, PD-L1 and PD-L2. Our preliminary data suggest time-dependent patterns. IHC demonstrated that the number of tumor-infiltrating CD4 and CD8 T cells did not change significantly over time.
Conclusions
Our preliminary results suggest time-dependent immunomodulation induced by 131I-NM404 in B78 flank tumors. Further research should address the influence of variables such as radioactive dose, choice of radionuclide, tumor type and location on immunomodulation. This information has the potential to provide guidance in the design of more effective therapeutic strategies combining molecular-targeted radiotherapy and immunotherapy for cancer patients.
References
1. Pinchuk AN et al. Synthesis and structure-activity relationship effects on the tumor avidity of radioiodinated phospholipid ether analogues. J Med Chem. 2006;49(7):2155-65.
P350 Breast tumor suppression mediated by therapeutic expression of chemerin, an innate leukocyte chemoattractant
Russell Pachynski1, Ping Wang1, Brian Zabel2, Eugene Butcher2
1Washington University School of Medicine, St Louis, MO, USA; 2Stanford University, Palo Alto, CA, USA
Correspondence: Russell Pachynski (rkpachynski@wustl.edu)
Background
The infiltration of immune cells into the tumor microenvironment can regulate growth and survival of neoplastic cells. Several studies have shown a correlation between increases in the number of effector immune cells present in a tumor and clinical outcomes in many human tumors, including breast. Current strategies employing checkpoint inhibitors aim to stimulate effector immune cells, however lack of adequate effector cell numbers within the tumor microenvironment can result in suboptimal responses to these agents. Here, we describe a novel strategy employing therapeutic overexpression of chemerin to recruit immune cells into the tumor microenvironment (TME) and suppress tumor growth.
Methods
Publically available whole genome expression datasets were analyzed for RARRES2 expression, comparing normal breast tissue to invasive breast cancers. Both the JC and EMT6 tumor lines were used in fully immune competent BALB/c mice. Mammary fat pads were used for inoculation, and chemerin-expressing tumors were compared to control tumors. Tumors were measured using standard caliper measurements, and infiltrating leukocytes measured by flow cytometry. Leukocyte depletion studies were performed using specific depleting antibodies.
Results
Our analyses of both TCGA and other public whole genome expression datasets show that RARRES2 (the gene for chemerin), a widely expressed endogenous chemoattractant protein for innate immune cells, is downregulated in several studies of human breast cancer. Significant downregulation of RARRES2 is seen in both invasive ductal and lobular breast cancers, compared to normal breast tissue. In mouse models using both the JC and EMT6 tumor lines, we have found that forced overexpression of chemerin within the TME significantly suppressed tumor growth with increased numbers of infiltrating leukocytes compared to controls. Systemic depletion of NK cells using anti-GM1 antibody treatment resulted in significant abrogation of chemerin’s anti-tumor effect, suggesting – at least in part – a reliance on the innate response.
Conclusions
We have shown, for the first time, the use of therapeutic overexpression of chemerin is effective at suppressing breast tumor growth in in vivo mouse models. This approach has been used successfully in our melanoma models, and may be a broadly applicable approach to increase the number of immune effector cells within the TME. Ongoing studies are looking at the combination of therapeutic chemerin modulation in combination with available checkpoint inhibitors for determination of additive or synergistic effects.
P351 A novel, individualized xenograft model of cancer immunotherapy and tumor growth inhibition by ALKS 4230
Lukas Pfannenstiel1, Jared Lopes2, Heather Losey2, Juan Alvarez2, Brian Gastman1
1Cleveland Clinic, Cleveland, OH, USA; 2Alkermes, Inc., Waltham, MA, USA
Correspondence: Lukas Pfannenstiel (pfannel@ccf.org)
Background
Recent successes in tumor immunotherapy highlight the curative potential of modulating patient anti-tumor immune responses. However, preclinical in vivo modeling of immune/tumor interactions often depend on a limited number of well-established cell lines. Of available treatment strategies, the use of cytokine therapy offers the advantage of using the patient’s own immune cells as anti-tumor effectors. ALKS 4230 is a fusion protein of circularly permuted IL-2 and IL-2 receptor α that is selective for the intermediate-affinity IL-2 receptor expressed on NK cells and subsets of memory and effector T cells. ALKS 4230 is currently in a phase 1 trial to evaluate safety and tolerability in the treatment of patients with refractory solid tumors.
Methods
In order to evaluate the ability of ALKS 4230 to promote and enhance anti-human tumor immune responses preclinically, individual xenograft tumor models were established in NOD-scid IL2Rgammanull mice (NSG) using tumor cells derived from metastatic melanoma patients following surgical resection. Upon tumor implantation and palpable growth, mice received an adoptive transfer of autologous, unexpanded PBMC from the same patient and treatment with ALKS 4230.
Results
We found that autologous T cells successfully engrafted NSG recipient mice after ALKS 4230 treatment similar to IL-2 and that both treatments induced cellular expansion over vehicle controls. Following treatment with ALKS 4230 and adoptive transfer of autologous PBMCs, PDX tumor-bearing mice consistently displayed increased numbers of both CD8 and CD4 T cells migrating into tumor tissue, preferential expansion of non-regulatory T cell subsets, and significant delays in tumor growth as compared to vehicle-treated controls.
Conclusions
Together these data support the rationale for ALKS 4230 as a novel immunotherapeutic for the treatment of melanoma and potentially other solid cancers, as well as the strategy of screening individual, patient-specific xenograft models to assess potential treatment efficacy.
P352 A novel CD73 blocking antibody restores T cell function and augments efficacy of Adenosine 2A Receptor (A2AR) inhibitor CPI-444
Emily Piccione, Jenny Rudnick, Barbara Daine-Matsuoka, Glen Mikesell, Richard Miller, Ian McCaffery
Corvus Pharmaceuticals, Burlingame, CA, USA
Correspondence: Emily Piccione (epiccione@corvuspharma.com)
Background
Adenosine accumulates in the tumor microenvironment (TME) through degradation of extracellular ATP by ectonucleotidases CD39 and CD73 and suppresses T cell activity through activation of A2AR. CD73 levels are elevated and are prognostic in certain tumors suggesting CD73 is an important immune-suppressive mechanism. Multiple CD73 antibodies are in clinical development that are allosteric CD73 inhibitors that stimulate internalization (type 2 mechanism), leading to incomplete inhibition of cell surface CD73 activity. We describe CPI-006, a novel CD73 antibody that directly inhibits CD73 enzymatic activity (type 1 mechanism).
Methods
CD73 activity was assayed by Malachite Green. Internalization was measured by flow cytometry. T cell proliferation was assayed by flow cytometry and cytokine levels by ELISA. Tumor CD73 was assessed by immunohistochemistry. Gene expression was determined by Nanostring.
Results
CPI-006 (nM affinity) was compared to CPX-016 (pM affinity), a potent type 2 CD73 antibody. CPI-006 completely inhibited CD73 catalytic activity across a broad range of CD73 expression. In contrast, CPX-016 incompletely inhibited CD73 activity (~60%) and was not effective towards cell lines with high CD73 expression.
CPI-006 was significantly more effective than CPX-016 at blocking suppression of T cell proliferation and cytokine secretion by adenosine (80% restoration compared to 45%). CPI-006 was effective across a broad range of CD73 expression while CPX-016 efficacy was restricted to samples with lower CD73 levels.
CD73 expression was evaluated by IHC in renal cell, melanoma, non-small cell lung and breast cancers (N=70 each). CD73 expression was heterogeneous in the tumor and stromal compartments; however, when expressed, CD73 levels were high, exceeding expression in the high expressing CD73 cell lines that were incompletely inhibited by CPX-016. These data suggest that type 2 antibodies will have limited efficacy in CD73 high expressing tumors.
Tumors with high CD73 expression are more responsive to A2AR inhibition [1] therefore we investigated the combined effect of CPI-006 with A2AR inhibitor, CPI-444. The combination significantly restored T cell proliferation and interferon-gamma production and suppressed adenosine responsive gene expression to levels greater than either agent alone.
Conclusions
CPI-006 is a novel CD73 blocking antibody that directly and completely inhibits CD73 activity, in contrast to type 2 antibodies in development. CPI-006 blocks CD73 activity and improves T cell function independent of CD73 expression levels and augments the effect of CPI-444.
References
1. Beavis, P.A., et al., Blockade of A2A receptors potently suppresses the metastasis of CD73+ tumors. PNAS. 2013;110(36):14711-6.
P353 Discovery and pre-clinical development of an orally available small molecule antagonist targeting the CD47/SIRPα pathway for cancer immunotherapy
Pottayil Sasikumar, Chennakrishna Gundala, Wesley Balasubramanian, Sudarshan Naremaddepalli, Archana Bhumireddy, Sandeep Patil, Amit Dhudashiya, Vijaysai Rayavarapu, Anirudha Lakshminarasimhan, Samiulla Dodheri, Kiran Aithal, Girish Daginakatte, Murali Ramachandra
Aurigene Discovery Technologies Limited, Bangalore, India
Correspondence: Murali Ramachandra (sasikumar_g@aurigene.com)
Background
The CD47/signal regulatory protein alpha (SIRPα) axis is a critical regulator of myeloid cell activation and serves as an immune checkpoint for macrophage mediated phagocytosis. Because of its frequent upregualtion in several cancers, CD47 contributes to immune evasion and cancer progression. Most of the current approaches in the immunotherapy focus on T-cell axis. Macrophages and other myeloid immune cells offer much promise as effectors of cancer immunotherapy, hence efforts to modulate them for therapeutic benefit are gaining momentum. In this regard disruption of CD47-SIRPα interaction is now being evaluated as a therapeutic strategy for cancer to stimulating macrophage mediated anti-tumor immune response. In view of the requirement for the intravenous dosing for all reported CD47 targeting agents, we sought to discover and develop an orally available small molecule CD47 antagonist. An oral CD47 agent potentially offers the convenience, flexibility to adjust the dose and schedule to address any emergent adverse events and ease of combination therapy.
Methods
Series of peptide disrupting CD47-SIRPα interactions were identified by rational design strategy based on CD47/SIRPα interacting interface. An FITC probe based cellular binding assay was established to identify high peptide fragments. The elements of this pharmacophore were incorporated into non-peptidic small molecule scaffolds resulting in lead compounds disrupting CD47/SIRPα interaction. The shortlisted compounds were further screened in a phagocytosis assay. In vivo efficacy was evaluated in A20 B Cell lymphoma syngeneic tumor model.
Results
Initial hits identified in FITC probe based cellular binding assay was further optimised for their activity in binding assay. Lead CD47 antagonists induced phagocytototic activity of human macrophages to a similar extent as commercially available anti-CD47 antibodies. Further optimization of these leads resulted in compounds with desirable physico-chemical properties and good oral bioavailability. An advanced lead CD47 antagonist inhibited primary tumor growth (~90%TGI) in a mouse syngeneic model of B-cell lymphoma upon twice a day oral dosing. Biomarker characterization and efficacy studies in additional tumor models are ongoing.
Conclusions
Rational design based on CD47/SIRPα interacting interface led to the identification of a novel and selective CD-47 antagonist with potent activity in cellular binding assay and phagocytosis assay. The lead compound exhibited desirable metabolic stability, solubility and oral bioavailability. In in-vivo studies, lead compound demonstrated significant anti-tumor efficacy in A20 B-cell lymphoma tumor model upon oral dosing. The above findings support further development of these orally bioavailable agents for use in the clinic.
P354 An orally bioavailable small molecule antagonist of VISTA and VSIG8 signaling pathways shows potent anti-tumor activity
Pottayil Sasikumar, Sudarshan Naremaddepalli, Raghuveer Ramachandra, Nagesh Gowda, Govardhan Alluri, Manikyalarao Yerramsetti, Sreenivas Adurthi, Jiju Mani, Rashmi Nair, Amit Dhudashiya, Samiulla- Dodheri, Nagaraj Gowda, Murali Ramachandra
Aurigene Discovery Technologies Limited, Bangalore, India
Correspondence: Murali Ramachandra (sasikumar_g@aurigene.com)
Background
Antibody-mediated blockade of immune checkpoint pathways have transformed the outlook for cancer therapy. While PD-1/PD-L1 antibodies primarily focus on T-cells to achieve anti-tumor efficacy, other cells in the tumor microenvironment such as myeloid cells including MDSCs also play a role in immune evasion. To overcome the immune resistance induced by MDSCs, V-domain Ig suppressor of T-cell activation (VISTA) expressed predominantly on myeloid cells and tumor-infiltrating lymphocytes is considered as an ideal target. Recent findings also support the role of VISTA pathway in clearance of apoptotic bodies and prevention of autoimmunity. VISTA is reported to mediate immune suppression through homophilic interaction as well as interaction with V-Set and immunoglobulin domain containing 8 (VSIG8). We sought to discover and develop an orally available small molecule VISTA antagonist targeting both VISTA and VSIG8 pathways. Unlike antibodies an oral gent potentially offers the convenience, flexibility to adjust dose and schedule to address any emergent adverse events and ease of combination therapy.
Methods
VISTA belongs to B7 family and shares sequence homology with the B7 family ligands PD-L1, a focused library of compounds mimicking the interaction of checkpoint proteins of B7 family was designed and synthesized. These compounds were tested in VISTA and VSIG8 specific functional assays. In-vivo efficacy was evaluated in mouse syngeneic B16F10 melanoma and CT26 colon carcinoma models. Impact of test agents on various immune populations was measured by flow cytometric analysis in tumor–bearing animals upon repeated dosing.
Results
Screening and analysis of the focused library of compounds led to the identification of hits capable of functional disruption of the checkpoint protein(s) signaling. Further optimization resulted in lead compounds targeting both VISTA and VSIG8 signaling pathways with desirable drug-like properties. Potent functional activity comparable to that obtained with an anti-VISTA or anti-VSIG8 antibody in rescuing effector functions was observed with the lead compound along with selectivity against other immune checkpoint proteins. An advanced lead compound exhibited sustained immune PD in tumor-bearing animals including desirable impact on myeloid and T-cells in both circulation and tumor. The advanced lead compound also exhibited significant efficacy in syngeneic pre-clinical tumor models of melanoma and colon cancers upon once a day oral dosing with excellent tolerability.
Conclusions
Our efforts have resulted in the identification of novel oral antagonists of VISTA and VSIG8 signaling. Desirable drug-like properties and anti-tumor efficacy at well-tolerated doses by the advanced lead compound in pre-clinical models supports its further development towards advancing to the clinic.
P355 Epigenetic modulation promotes tumor suppression and improves survival when combined with checkpoint inhibition in murine models of breast cancer
Evanthia Roussos Torres, Hayley Ma, Christine Rafie, Brian Christmas, Todd Armstrong, Elizabeth Jaffee
Johns Hopkins University, Baltimore, MD, USA
Correspondence: Evanthia Roussos Torres (ertorres@jhu.edu); Elizabeth Jaffee
Background
Checkpoint inhibition is a very successful treatment strategy in cancers that are naturally immunogenic by attracting T cells into the tumor microenvironment (TME) and promoting cytotoxic signaling pathways. Most breast cancers are not highly immunogenic likely due to an immunosuppressive microenvironment. One strategy to transform this TME is to use epigenetic modulation to affect activation and trafficking of myeloid derived suppressor cells (MDSCs), known to alter the immunogenicity of the TME and sensitize tumors to checkpoint modulation. We hypothesize that combinatorial therapy primes the TME by altering infiltration and function of MDSCs leading to a more robust T cell response.
Methods
We use the HER-2/neu transgenic mouse model with challenge of syngeneic cell lines. This model enables us to study the efficacy of different combinations of an anti-HER2 antibody (a-Her2), an epigenetic agent, the histone deacetylase inhibitor entinostat, and checkpoint inhibitors, anti -programmed cell death protein (PD-1) and anti-cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) antibodies, on tumor growth and to help identify co-stimulatory and inhibitory factors regulating T cell and MDSC responses. Characterization of tumor infiltrating lymphocytes (TIL) and their functional capabilities are being investigated in primary tumors using fluorescence-activated cell sorting, nanostring gene expression profiling, and immunohistochemistry.
Results
We found significant improvement in survival and delay in tumor growth in tumor bearing mice- treated with entinostat, anti-PD1, and anti-CTLA-4 and with a-Her2, entinostat and anti-PD1 or anti-CTLA4. We show that addition of entinostat to checkpoint inhibition leads to significantly increased infiltration of granulocytic-MDSCs and CD8+ T effector cells into the TME. Flow cytometric evaluation suggests increased T cell activation, exhaustion, and altered MDSC function. Functional assays of isolated MDSCs from mice treated with combination therapy demonstrates reduced suppressive ability of these cells. Gene expression profiling of isolated MDSCs and TIL is underway to help determine significant changes in immune related pathways that have lead to our observed outcomes.
Conclusions
Addition of entinostat and checkpoint inhibition to a-Her2 therapy significantly increases infiltration of innate and adaptive immune cells into the highly tolerant breast tumors and leads to improved survival and decreased tumor burden. Results suggest that this combinatorial treatment alters the function of the infiltrating cell types that lead to the observed phenotype. We aim to delineate genetic alterations responsible for these observations. It is our hope that our novel findings will provide further rationale for combination therapy and improve the response rate to immunotherapy in patients with breast cancer.
P356 Efficacy and Mechanism of Action of CXCR4 Inhibition in B16-OVA Melanoma Model
Ruchi Saxena1, Yan Wang2, James Mier1
1Beth Israel Deaconess Medical Center, Boston, MA, USA; 2X4 Pharmaceuticals, Cambridge, MA, USA
Correspondence: James Mier (rsaxena@bidmc.harvard.edu)
Background
In our previous studies, we demonstrated that combination of a CXCR4 antagonist with an anti-angiogenesis agent axitinib relieved myeloid-derived suppressor cells (MDSCs) mediated immunosuppression and suppressed HIF-2a expression, which resulted in synergistic antitumor effects in 786-0 and A498 RCC xenografts. To further study the CXCR4 mechanism of action in an immune-proficient background, we investigated the activity of a CXCR4 antagonist in a syngeneic mouse tumor model. As the current murine models for RCC do not share the same genetic alterations with the human disease, the B16-OVA model was selected for this study. The CXCR4 antagonist was tested in combination with axitinib or with checkpoint inhibitors which are part of the SOC in managing melanoma in clinic.
Methods
B16-OVA cells were implanted into C57BL/6 mice. Seven-days post implantation, mice were treated with axitinib, X4-136 (CXCR4 inhibitor) or anti-PD-L1/anti-CTLA4, or the combinations for 16 days. At sacrifice, tumors were excised and flash frozen in liquid nitrogen for Western Blot analysis or treated with collagenase for the analysis of subsets of T-cells by FACS.
Results
Axitinib alone modestly inhibited growth of B16-OVA tumors. X4-136, the CXCR4 inhibitor, alone demonstrated more robust activity than the combination treatment of anti-PD-L1 and anti-CTLA4. The anti-tumor activity of the anti-PD-L1 and anti-CTLA4 regimen was further enhanced in combination with X4-136.
Preliminary analysis of infiltrating immune cells by flow cytometry showed that axitinib treatment led to an increase in immunosuppressive Treg cell population in tumors, while treatment with X4-136 alone or the anti-PD-L1/anti-CTLA4 combination led to a decrease in Tregs. The decrease was more pronounced when X4-136 was used in combination with axitinib. Additional analysis of other immune cell subsets will also be presented.
This combination also led to a decrease in the expression of HIF-2α and cyclin D1 in comparison to vehicle treated mice as demonstrated by western blot analysis of tumors, suggesting an anti-proliferative effect. PARP cleavage was apparent when X4-136 was used in combination with axitinib indicating the role of apoptosis in anti-tumor effect. Additional analysis of tumors after treatment in earlier time points will also be presented.
Conclusions
X4-136 alone exhibited potent anti-tumor activity in the B16-OVA murine melanoma model. Added benefit was observed when X4-136 was added to either axitinib or to anti-PD-L1/anti-CTLA-4 treatments. Treatment benefits were associated with the reduction of Treg population in the tumor microenvironment. Induction of apoptosis was observed in combination treatment of axitinib and X4-136.
P357 The Tt-cell growth factor cocktail IL-2/IL-15/IL-21 enhances expansion and effector function of tumor-infiltrating T cells in a novel process developed by iovance
Ian Frank, Michelle Simpson-Abelson, Michael Lotze, Krit Ritthipichai, Christopher Mosychuk
Iovance Biotherapeutics, Tampa Bay, FL, USA
Correspondence: Michael Lotze (michelle.abelson@iovance.com)
Background
Adoptive T cell therapy with autologous tumor infiltrating lymphocytes (TIL) has demonstrated clinical efficacy in patients with metastatic melanoma and cervical carcinoma. In some studies, clinical outcomes have positively correlated with the total number of cells infused and/or percentage of CD8+ T cells. Most current production regimens solely utilize IL-2 to promote TIL growth, although enhanced lymphocyte expansion has been reported using IL-15 and IL-21-containing regimens. This study describes the positive effects of adding IL-15 and IL-21 to the standard IL-2-alone TIL generation protocol.
Methods
The process for generating TIL includes a pre-Rapid Expansion Protocol (pre-REP), in which TIL emigrate out of tumor fragments of 1-3 mm3 in media containing IL-2. To further stimulate TIL growth, TIL are expanded using a secondary culture period termed the Rapid Expansion Protocol (REP) that includes irradiated PBMC feeders, IL-2 and anti-CD3. In this study, a shortened pre-REP and REP expansion protocol was developed to expand TIL while maintaining the phenotypic and functional attributes of the final TIL product. This shortened TIL-generation protocol was used to assess the impact of IL-2 alone versus a combination of IL-2/IL-15/IL-21. These two culture regimens were compared for the generation of TIL grown from colorectal, melanoma, cervical, triple negative breast, lung and renal tumors. At the completion of the pre-REP, cultured TIL were assessed for expansion, phenotype, function (CD107a+ expression and IFNg release) and TCR Vβ repertoire.
Results
Enhanced TIL expansion (>20%), in both CD4+ and CD8+ cells in the IL-2/IL-15/IL-21 culture cocktail was observed across multiple tumor histologies. Preliminary analysis demonstrated a shift towards a predominantly CD8+ TIL population with a skewed TCR Vβ repertoire in TIL cultured with the IL-2/IL-15/IL-21, versus IL-2 alone. IFNg release and CD107a expression were also elevated in TIL cultured in the presence of IL-2/IL-15/IL-21 versus those cultured using IL-2 alone.
Conclusions
Rapidly expanding TIL ex vivo for adoptive cell therapy is essential in treating patients with cancer. We report an increased TIL-product yield, in addition to potentially beneficial phenotypic and functional differences, when TIL are cultured with IL-2/IL15/IL-21 as compared to IL-2 alone. We suggest a more robust process encompassing the use of IL-2/IL-15/IL-21 in TIL culture may provide a means to promote TIL expansion particularly in tumors with poor T cell infiltration.
P358 Development of CDX-1140, an agonist CD40 antibody for cancer immunotherapy
Lawrence Thomas1, Laura Vitale2, Thomas O'Neill2, Jenifer Widger2, Andrea Crocker2, Li-Zhen He2, Jeffrey Weidlick2, Karuna Sundarapandiyan2, James Storey1, Eric Forsberg1, Catherine Pilsmaker1, Lauren Gergel1, Elizabeth Do1, Joel Goldstein2, Henry Marsh1, Tibor Keler2
1Celldex Therapeutics, Needham, MA, USA; 2Celldex Therapeutics, Hampton, NJ, USA
Correspondence: Lawrence Thomas (Lthomas@celldex.com)
Background
Current limitations for immunotherapy approaches include poorly functioning events early in the immune response cycle, such as efficient antigen presentation and T cell priming. CD40 signaling in dendritic cells (DCs) leads to upregulation of cell surface costimulatory and MHC molecules and the generation of cytokines, which promotes effective priming of CD8+ effector T cells while minimizing T cell anergy and the generation of regulatory T cells. This naturally occurs through interaction with CD40 ligand (CD40L) expressed on CD4+ T-helper cells. CD40 signaling can also be achieved using specific monoclonal antibodies (mAbs), leading to the development of several agonist CD40 antibodies that have initiated clinical development.
Methods
Our approach for the development of a CD40 agonist antibody was to define a balanced profile between sufficiently strong immune stimulation and the untoward effects of systemic immune activation.
Results
CDX-1140 is a human IgG2 antibody derived from human Ig transgenic mice, that activates DCs as demonstrated by upregulation of costimulatory molecules and increased production of cytokines. This activity is Fc independent as it is maintained using an F(ab’)2 fragment of the antibody. CDX-1140 does not block the binding of CD40L and the addition of soluble CD40L greatly enhances DC activation by CDX-1140, suggesting that CDX-1140 may act synergistically with naturally expressed CD40L. CDX-1140 also activates B cells, and has direct anti-lymphoma activity in xenograft models. However, the antibody does not promote cytokine production in whole blood assays. Importantly, CDX-1140 has shown good pharmacodynamic and safety profiles in a cynomolgus macaque study investigating doses from 0.01 to 10 mg/kg. This study showed a clear dose response with respect to changes in hematologic and circulating cytokine values (IL-12p40) that were expected to result from CD40 activation.
Conclusions
These data support the potential of CDX-1140 as part of a cancer therapy regimen, and the phase 1 dose-escalation clinical study is anticipated to begin this year.
P359 Effect of targeted anti-GD2/-CD16 Bispecific NK cell Engager (BiKE) with or without IL-15 super agonist ALT803 against GD2+ Neuroblastoma (NB) and Ewing Sarcoma (ES)
Aradhana Tiwari1, Dina Edani1, Janet Ayello1, Hing C. Wong2, Mitchell S Cairo1
1New York Medical College, Valhalla, NY, USA; 2Altor BioscienceWebsiteDirections, Miramar, FL, USA
Correspondence: Aradhana Tiwari (aradhana_tiwari@nymc.edu)
Background
GD2 is a surface disialoganglioside that is a well-characterized immunotherapeutic target in NB and Sarcomas [1]. The efficacy of anti- GD2 antibodies depends on engaging functional NK cells to kill GD2-positive targets through antibody dependent cellular cytotoxicity (ADCC). However, NK cell number and function are decreased in most cancer patients at diagnosis, are further reduced by radiation chemotherapy, antigen loss and immunosuppressive tumor microenvironment (TME) which contribute to treatment failure. One of the new approaches to overcome TME resistance and improve NK cell mediated ADCC against tumor cells is using a BiKE [2]. ALT-803 is a superagonist of an IL-15 variant bound to an IL-15RaSu-Fc fusion with enhanced IL-15 biological activity.
To investigate the in-vitro activity of hu-anti-GD2/-CD16 BiKE with or without ALT803 against GD2 expressing NB/ES
Methods
Anti-GD2/-CD16 BiKE was constructed in pBudCE4.1 mammalian expression vector and transfected in to HEK293-EBNA Cells. Stable clone were selected by Zeocin for the secretion and purification of anti-GD2/-CD16 plasmid by ProBond™ Ni column and validated by Western blot. Cytotoxicity was examined against NB/ES cells with or without ALT803 (generously supplied by Altor Biosciences) with K562-mbIL21-41BBL expanded NK cells by DELFIA cytotoxicity assay at 10:1 E:T ratio.
Results
GD2 expressing NB (54.4± 10.61%) and ES (71.93±8.33%) cells were used for functional assays. Anti-GD2/-CD16 BiKE was purified from the transfected culture supernatant and validated by western blot (Fig. 1A). BiKE+NK compared to Medium+NK significantly increased NK mediated cytotoxicity against NB: SKNF1 (64.5±5.9% vs. 20.1±1.3%, p=0.002), SKNBE2 (67.4±4.02% vs. 15.1±0.9%, p=0.004), SHS5Y5 (68.9±0.9% vs. 30.2±0.85%, p=0.005) and ES: EWS502 (68.05±3.06% vs. 28.1±4.05%, p=0.004), A673 (66.02±4.05% vs. 20.3±0.8%, p=0.005) respectively. Further, BiKE+NK+ALT803 compared to BiKE+NK+rhuIL15 improved NK mediated cytotoxicity against NB: SKNF1 (73.9±9.3% vs. 56.5±8.5%, p=0.007) and SKNBE2 (63.9±0.6% vs. 40.5±0.49%, p=0.002) respectively, against NB/ES cell lines (Fig. 1B).
Conclusions
Our preliminary results demonstrated that the BiKE with ALT803 significantly enhanced NK cytotoxicity against NB and ES. Future studies will investigate the efficacy of this BiKE with ALT803 against GD2 expressing solid tumor in humanized NSG xenografted mouse model.
References
1. Perkins S M, Shinohara ET, DeWees T, Frangoul H. Outcome for children with metastatic solid tumors over the last four decades. PLoS One. 2014;9.
2. Gleason MK, et al. Bispecific and trispecific killer cell engagers directly activate human NK cells through CD16 signaling and induce cytotoxicity and cytokine production. Mol Cancer Ther. 2012;11:2674-2684.
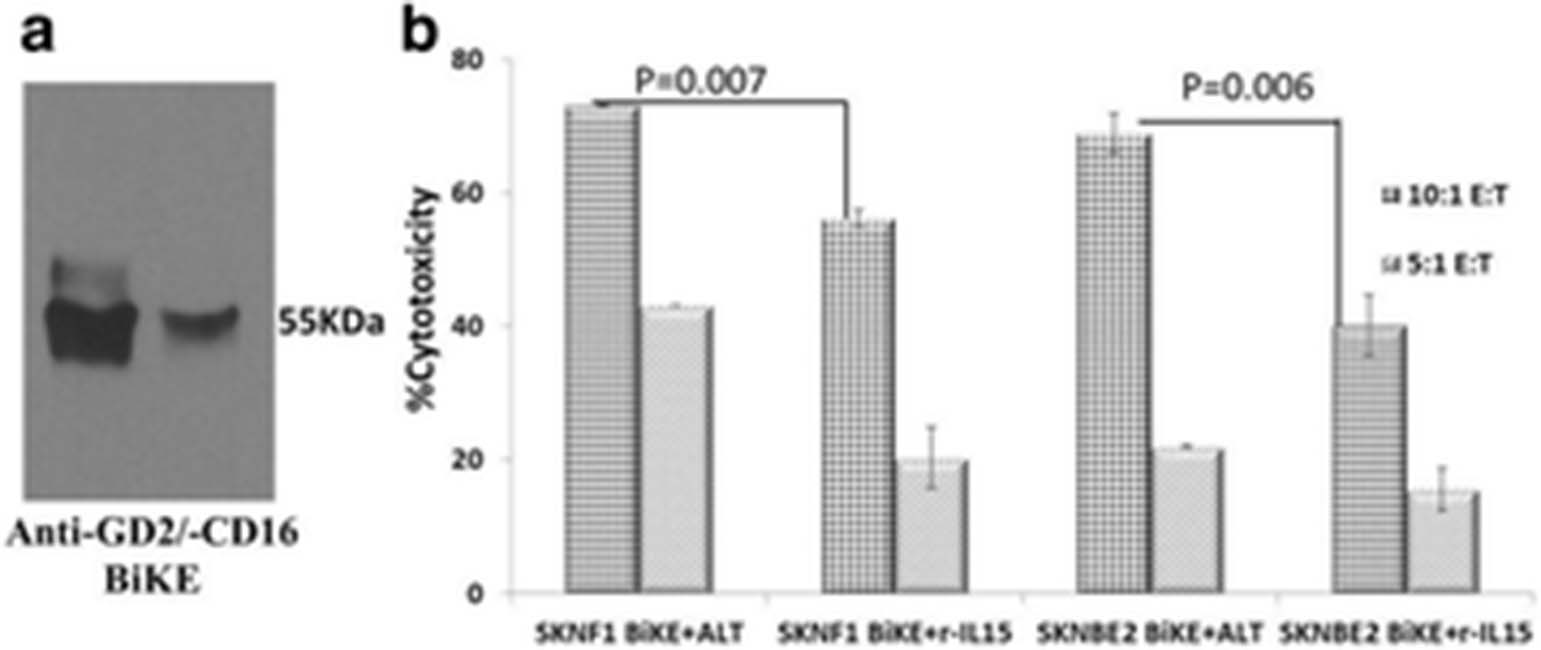
See text for description
P360 APX005M is a potent CD40 agonistic antibody capable of stimulating both innate and adaptive immune responses against cancer
Pia Björck, Erin Filbert, Ovid Trifan, Xiaodong Yang
Apexigen Inc., San Carlos, CA, USA
Correspondence: Xiaodong Yang (otrifan@apexigen.com)
Background
CD40 plays an important role in activation and regulation of both innate and adaptive immunity. Using the proprietary APXiMAB™ discovery platform, Apexigen discovered APX005M, a potent immune stimulatory CD40 agonistic antibody currently being developed for cancer immunotherapy. APX005M is a high-affinity humanized IgG1 antibody targeting human CD40. APX005M was engineered to carry an S267E mutation in its Fc region to enhance CD40 agonistic activity via cross-linking to the FcRγIIb.
Methods
PBMCs were obtained from healthy human subjects. Isolated cell subsets were cultured in various conditions with APX005M. The potential synergistic effect of APX005M with immune checkpoint inhibitors (ICI) was assessed by in vitro culture of human dendritic cells (DC) and allogeneic T cells with APX005M and anti-PD-1 or anti-PD-L1 antibodies. The duration of APC activation following exposure to APX005M was determined by culturing PBMCs with APX005M for 24 hours. Cell activation was measured at various times post washout.
Results
APX005M binds with high-affinity to human CD40 (Kd=0.12 nM) and recognizes a unique epitope that overlaps with the CD40 ligand (CD40L) binding domain thus mimicking CD40L-induced activation. APX005M is capable of activating human B cells (EC50=12 pM) and DC (EC50=0.49 nM) and also promotes proliferative responses of tumor-infiltrating T cells. APX005M’s CD40 agonistic activity depends on cross-linking of Fc-gamma receptors. Receptor occupancy studies revealed that 10% of CD40 receptor occupancy is sufficient to produce maximum APC and T cell activation. Short-term exposure (24 hours) of PBMC to APX005M induced long-lasting activation of B cells, monocytes and T cells that remained activated 2 weeks after removal of APX005M. In vitro co-culture of human DC and allogeneic T cells showed that APX005M induces a dose-dependent increase of CD4 and CD8 T cell proliferation and IFN-γ secretion, and the T-cell responses were further enhanced by anti-PD-1 or anti-PD-L1 antibodies suggesting that APX005M combined with ICI can synergistically stimulate T-cell responses.
Conclusions
The immune stimulatory activity of APX005M results from its unique epitope and is dependent on cross-linking by Fc-gamma receptors. The properties of APX005M make it an optimal CD40 agonist for stimulating anti-tumor immune responses while maintaining a good safety profile. Combination of APX005M with ICI enhances T-cell responses providing a rationale for the ongoing clinical studies combining APX005M with anti-PD-1 antibodies in NSCLC and melanoma patients (trials NCT03123783 and NCT02706353).
P361 A novel T-cell engaging bispecific antibody platform: Efficient In vivo tumor clearance with minimal cytokine release
Nathan Trinklein, Andrew Boudreau, Ben Buelow, Starlynn Clarke, Kevin Dang, Laura Davison, Shelley Force Aldred, Katherine Harris, Suhasini Iyer, Brett Jorgensen, Heather Ogana, Duy Pham, Payal Pratap, Udaya Rangaswamy, Ute Schellenberger, Harshad Ugamraj, Omid Vafa, Wim van Schooten
Teneobio, Menlo Park, CA, USA
Correspondence: Nathan Trinklein (ntrinklein@teneobio.com)
Background
Using transgenic rats and a unique sequence-based discovery approach, we have created a large collection of fully human anti-CD3 antibodies with diverse T-cell agonist activities. We have used these anti-CD3 antibodies to create a unique multi-specific platform for T-cell redirected tumor cell killing.
Methods
Our novel discovery platform combines antibody repertoire deep sequencing, high-throughput gene assembly, and recombinant expression and generates a much larger diversity of antibodies than traditional approaches. The CD3 antibodies identified by our platform show diverse in vitro T-cell activation profiles measured by CD69 upregulation, IL2, and IFNg production. Using our discovery platform, we have also generated human domain antibodies targeting tumor antigens that may be combined with our unique CD3 antibodies to create multi-specific antibodies.
Results
As one example, we have created a CD3xBCMA bispecific antibody (TNB-383B) for the treatment of multiple myeloma. TNB-383B kills multiple myeloma cells in vitro and in vivo in a BCMA-dependent manner, and kills primary patient myeloma cells ex vivo. The EC50 for cytotoxicity was in the single-digit nanomolar range for TNB-383B against MM cell lines in vitro. TNB-383B showed much reduced (ie IFN-ɣ) or absent (ie IL-2) cytokine release compared to other anti-CD3 antibodies. In vitro results were consistent across 10 healthy huPBMC donors. Ex vivo, TNB-383B efficiently lysed primary MM cells in the presence and absence of supplementary T-cells. In vivo, TNB-383B mediated clearance of MM tumors from NSG mice at doses as low as 10ng of bispecific antibody.
Conclusions
In summary, we have created a T-cell engaging bispecific antibody platform with tunable T-cell agonism that can be used to optimize the therapeutic index for a variety of tumor antigens.
P362 A novel assay using RNA aptamers to quantitate the fraction of IL2Ra (CD25) receptors occupied by IL2
Suresh Veeramani, Sue Blackwell, William Thiel, Paloma Giangrande, George Weiner
University of Iowa, Iowa City, IA, USA
Correspondence: George Weiner (suresh-veeramani@uiowa.edu)
Background
RNA aptamers bind to antigens in a manner analogous to antibodies and their binding can be quantitated using simple RT-PCR. Using IL2-IL2Ra as a model, we report a novel assay using IL2-IL2Ra-binding RNA aptamers that allows for quantitation of receptor occupancy by its ligand.
Methods
A modification of whole cell Systematic Evolution of Ligands by EXponential enrichment (SELEX) was used to select T regulatory (Treg) cell-specific RNA aptamers. Aptamers specific for common T cell antigens were precleared using normal donor CD4+IL2Ra- T cells, followed by enrichment for aptamers specific for CD4+IL2Ra+ Tregs obtained from the same donor. The process was repeated for eight rounds with each round using T cells from a different normal donor. High-throughput sequencing and bioinformatics analysis was used to select top Treg-binding aptamers. Binding of aptamers to unoccupied IL2Ra and IL2Ra occupied by IL2 was determined by RT-qPCR. Some aptamers bound preferentially to unoccupied IL2Ra while others bound preferentially to IL2Ra occupied by IL2. To determine the fraction of IL2Ra occupied by IL2, aptamers that bound preferentially to unoccupied receptor or to IL2-occupied receptor were mixed in equimolar quantities. Aptamer mix was added to IL2Ra-coated Dynabeads that were pre-incubated with various concentrations of IL2 to create various IL2-occupied receptor fractions. Aptamer binding was quantified by RT-qPCR using a set of primers specific for both aptamers. Fluorescent probes specific for the variable region for each aptamer were used to quantify each of the aptamers.
Results
Five of the top 12 Treg-binding aptamers recognized IL2Ra. Some IL2Ra-specific aptamers (e.g. Tr-8) bound preferentially to unoccupied IL2Ra and others (e.g. Tr-7) to the IL2-IL2Ra complex (Fig. 1). Standard curves were developed which allowed for determination of the fraction of IL2Ra occupied by IL2 (Fig. 2).
Conclusions
A Treg cell-based SELEX was used to select Treg-binding aptamers specific for IL2Ra. Aptamer pairs showing differential binding preferences towards unoccupied IL2Ra versus the IL2-IL2Ra complex were identified. Because the selected aptamer pairs share the same primers, RT-qPCR amplification using a single set of primers allowed for quantitation of the ratio of bound aptamers. The resulting ratio reflects the occupancy of the IL2Ra receptor by its ligand IL2. This technique could be valuable for studying the role of IL2-IL2Ra ligand-receptor complex in anti-tumor immunity. Importantly, modifications of this assay could be used to quantitate other receptor-ligand complexes relevant in the field of cancer immunotherapy.

See text for description

See text for description
P363 The OX40-CTLA-4 bispecific antibody, ATOR-1015, induces immune activation and anti-tumor effect
Niina Veitonmäki, Karin Hägerbrand, Mia Thagesson, Doreen Werchau, Karin Enell-Smith, Anna Rosén, Sara Frizell, Anne Månsson-Kvarhammar, Per Norlén, Furebring Tina, Peter Ellmark
Alligator Bioscience, Lund, Sweden
Correspondence: Niina Veitonmäki (niv@alligatorbioscience.com)
Background
ATOR-1015 is an OX40-CTLA-4 bispecific immune activating antibody developed for tumor-directed immunotherapy. The compound was generated by fusing a high affinity CTLA-4 binder, derived by FIND® optimization of CD86, to an agonistic OX40 antibody derived from the human antibody library ALLIGATOR-GOLD®.
ATOR-1015 binds both targets simultaneously resulting in cell-cell interactions expected to enhance the immuno-stimulating effect of the compound. The mode of action of ATOR-1015 is thought to be a combination of effector T cell activation and regulatory T cell (Treg) depletion.
Methods
Human primary cells used in in vitro assays were isolated from leukocyte concentrates from healthy donors. The in vitro effects were measured by using either standard cytokine release assays or Promega reporter assays.
Human OX40 transgenic (knock-in) mice were generated by genOway SA and female mice were used for the anti-tumor effect studies using syngeneic mouse models. Tumors and spleens from treated mice were analyzed for Treg and T cell populations by flow cytometry.
Results
ATOR-1015 dependent T cell activation and Treg depletion is supported by cell-based in vitro studies and in vivo syngeneic tumor models.
The ability to induce ADCC of human Treg was investigated using a FcγR expressing reporter assay demonstrating superior effect of ATOR-1015 compared to the monospecific antibodies. Further, ATOR-1015 has been shown to induce activation of T cells in the presence of CTLA-4 expressing cells.
Treatment with ATOR-1015 reduces tumor growth and prolongs survival in syngeneic tumor models in vivo using human OX40 transgenic mice. Further, ATOR-1015 treatment demonstrates an increase in the intratumoral CD8+ T cell/Treg ratio which is superior compared to the monospecific counterparts, without affecting systemic T cells.
Conclusions
ATOR-1015 suppresses/depletes Tregs and activates CD8+ T cells in vivo and is currently in the second phase of production, and process development. ATOR-1015 is planned to enter clinical trials in 2018.
P364 Discovery and pharmacological characterization of JNJ-63723283, an anti-programmed cell death protein-1 (PD-1) antibody that blocks PD-1 function
Catherine Ferrante1, Nikki DeAngelis1, Gordon Powers1, Jocelyn Sendecki1, Hillary Millar2, Bethany Mattson1, Darlene Pizutti1, Christina Lourdes Mayer1, Weirong Wang1, Raymond Brittingham1, John Wheeler1, Linda Barone1, Rupesh Nanjunda1, Eilyn R. Lacy1, Sheng-Jiun Wu1, Jinquan Luo3, Enrique Zudaire1, Kathryn Packman1, Brian Geist1, Kevin Trouba1, Matthew V. Lorenzi1, Raluca I. Verona1
1Janssen Research & Development, Spring House, PA, USA; 2Janssen Research & Development, Beerse, Belgium; 3Janssen Resaerch & Development, Spring House, PA, USA
Correspondence: Raluca I. Verona (RVerona@its.jnj.com)
Background
Programmed cell death protein-1 (PD-1), a negative immune checkpoint receptor, suppresses T cell function when bound to its ligands PD-L1 and PD-L2. Tumor cells exploit this pathway to evade immune surveillance; therefore, inhibiting the PD-1 pathway can restore T cell function, stimulating their anti-tumor response. Here we report the characterization of JNJ-63723283 (JNJ-283) to support new combination approaches in the future.
Methods
JNJ-283 was generated by phage panning against human and cynomolgus monkey (cyno) PD-1 extracellular domain followed by affinity maturation. In vitro activity was evaluated using cytomegalovirus (CMV) recall, mixed lymphocyte reaction (MLR), and Jurkat-PD-1 nuclear factor of activated T cells (NFAT) reporter assays. In vivo activity was assessed using human PD-1 knock-in mice implanted with MC38 tumors and a lung patient-derived xenograft (PDX) model (LG1306) using CD34+ cord blood humanized NSG mice. Pharmacodynamic, toxicokinetic, and safety assessments were performed in cyno following single (0.1, 1, 10 mg/kg) and/or repeat (10, 30, 100 mg/kg/wk up to 5 weeks) intravenous (IV) dosing.
Results
JNJ-283 displayed high affinity for human (1.72 ± 0.99 nM) and cyno (0.90 ± 0.08 nM) PD-1, blocked PD-1 binding to PD-L1 (IC50=111.7 ± 22.0 ng/mL) and PD-L2 (IC50=138.6 ± 12.4 ng/mL), and cross-competed with nivolumab and pembrolizumab analogs.
JNJ-283 dose-dependently increased T cell-mediated cytokine production in CMV recall and MLR assays and enhanced NFAT activity in a Jurkat-PD-1 reporter assay, indicating functional disruption of the PD-1/PD-L1 interaction. JNJ-283 in vitro activity was comparable to that of competitor molecule analogs.
Following intraperitoneal injection with 10 mg/kg JNJ-283 or nivolumab analog (2x weekly; day 7 after tumor implantation), mean MC38 tumor volume in PD-1 knock-in mice was significantly lower at days 17 (p<0.001) and 21 (p<0.0001) compared to control (Fig. 1). In the lung PDX model, 10 mg/kg JNJ-283 or pembrolizumab (q5d x 6) reduced (p<0.01) mean tumor volume compared to control.
JNJ-283 was tolerated in cyno following single and/or repeat dosing; mean drug exposures increased in a dose dependent manner. Primary findings attributed to JNJ-283 included proliferation of T-lymphocyte subsets, stimulation-related cytokine expression (in vitro), increased IgM and IgG titer secondary to antigen challenge, and decreased cellularity (lymphocytes) in the thymus.
Conclusions
JNJ-283, a high affinity anti-PD-1 antibody, was well-tolerated in toxicology studies and demonstrated robust activity in vitro as well as displayed anti-tumor efficacy in vivo comparable to that observed with other agents targeting PD-1. These data support the ongoing clinical study of JNJ-283 and combination potential with other modalities.

In vivo anti-tumor activity of JNJ-283 in human PD-1 knock-in mice implanted with MC38 tumors
P365 Development of JNJ-64158120, an anti-TIM-3 antibody to overcome innate and acquired mechanisms of resistance to PD-1 therapy
Nikki DeAngelis1, Gordon Powers1, Keri Dorn1, Matthew Chomo1, Douglas Dolfi1, Karla Wiehagen1, Douglas Marvel1, Bethany Mattson1, Darlene Pizutti1, Liam Campion1, Caitlin Morgan1, Brian Tomkowicz1, Nina Sabins1, Sandra Santulli-Marotto2, Jocelyn Sendecki1, John Wheeler1, Brian Whitaker1, Annmarie Winkis1, Linda Barone1, Deborah Kwok1, Eilyn R. Lacy1, Edmund Moon3, Steve Albelda3, Kathryn Packman1, Enrique Zudaire1, Matthew V. Lorenzi1, Raluca I. Verona1
1Janssen Research & Development, Spring House, PA, USA; 2Alios BioPharma/Janssen Research & Development, South San Francisco, PA, USA; 3University of Pennsylvania, Philadelphia, PA, USA
Correspondence: Raluca I. Verona (RVerona@its.jnj.com)
Background
Programmed cell death protein-1 (PD-1) pathway blockade has revolutionized the treatment of a variety of malignancies but has only demonstrated clinical benefit in a subset of patients. Emerging preclinical and clinical data have proposed T cell immunoglobulin and mucin domain-3 (TIM-3) as an inhibitory mechanism that is co-regulated with PD-1 to restrict T cell-mediated immune responses. Dynamic upregulation of TIM-3 has been observed in patients in response to anti-PD-1/PD-L1 therapy, implicating TIM-3 as a potential mechanism of immune resistance. These observations suggest that dual blockade of the TIM-3 and PD-1 pathways can broaden the effectiveness of anti-PD-1/PD-L1 therapy as a new immunotherapy for PD-1 responsive malignancies. Here we describe the characterization and functional activity of JNJ-64158120 (JNJ-120), an antibody targeting human TIM-3.
Methods
JNJ-120 in vitro activity was assessed by CMV, MART-1, and NK cell activation assays; in vivo activity was evaluated using a NY-ESO TCR transgenic model of T cell exhaustion and a human TIM-3 knock-in mouse model bearing MB49 murine bladder carcinoma tumors.
Results
JNJ-120 is a fully human, phage-derived, IgG2σ monoclonal antibody that binds to human (Kd=2.5 nM) and cynomolgus monkey (Kd=0.75 nM) TIM-3. JNJ-120 inhibited the binding of TIM-3 to two putative TIM-3 ligands, phosphatidyl serine and galectin-9 and enhanced T cell function in primary antigen-specific T cell assays (CMV and MART-1). In CMV stimulated PBMCs, JNJ-120 triggered expression of IFN-γ and TNF-α and enhanced CD137 expression on CD8+ and CD4+ T cells. Furthermore, in MART-1 assays, JNJ-120 enhanced IFN-γ, TNF-α, IL-2, and IL-8 production by CD8+ T cells. JNJ-120 also enhanced T cell function in combination with anti-PD-1 antibodies in peripheral blood samples derived from melanoma patients. JNJ-120 led to higher NK cell activation, as measured by CD69 expression and cytokine secretion, in IL-2-stimulated PBMCs derived from normal donors and cancer patients.
In vivo, JNJ-120 treatment enhanced the anti-tumor activity of PD-1 blockade in tumor-bearing TIM-3 knock-in mice and in a NY-ESO TCR transgenic model of T cell exhaustion. In the NY-ESO TCR model, JNJ-120 increased T cell infiltration in NY-ESO-expressing A549 tumors and, in combination with anti-PD-1 antibodies, enhanced the ability of infiltrating T cells to kill tumor cells ex-vivo.
Conclusions
These data demonstrated that targeting TIM-3 in combination with PD-1 leads to increased tumor T cell infiltration and superior anti-tumor efficacy compared to PD-1 blockade alone. These data support the clinical testing of PD-1 and TIM-3 in tumor types not broadly responsive to current immunotherapy regimens.
P366 Anti–PD-L1 mAb treatment combined with cisplatin modulates intratumoral immune responses and promotes antitumor effects
Daiko Wakita, Toshiki Iwai, Masamichi Sugimoto, Suguru Harada, Miho Suzuki, Kaname Yamamoto
Chugai pharmaceutical CO., LTD., Kamakura, Japan
Correspondence: Kaname Yamamoto (wakita.daiko59@chugai-pharm.co.jp)
Background
Immune checkpoint inhibitors such as anti–PD-L1/PD-1 agents have shown marked antitumor effect in a range of cancer types, and to further expand their effects, combination treatments with chemotherapeutic drugs have been actively investigated. Although cisplatin is widely used as a standard chemotherapy, the antitumor activity of cisplatin combined with immune checkpoint inhibitors remains largely unknown. Here, we investigated if anti–PD-L1 plus cisplatin combination can augment antitumor immunity in a syngeneic mouse tumor model.
Methods
E.G7-OVA cells, expressing ovalbumin (OVA) gene as a model tumor antigen, were subcutaneously inoculated into syngeneic C57BL/6 mouse. After tumor mass was established, anti-mouse PD-L1 mAb (anti–PD-L1; 10 mg/kg, three times a week) and cisplatin (1 mg/kg, once) were administered intraperitoneally on the day of treatment initiation (Day1). The frequency and activation status of immune cells in tumor tissues were evaluated by flow cytometry. For detection of IFNg-producing cells, the immune cells were stimulated with anti-CD3/anti-CD28 mAbs in vitro before staining.
Results
We found that anti–PD-L1 plus cisplatin combination resulted in profound effect leading to tumor shrinkage vs anti–PD-L1 alone or cisplatin alone even though each monotherapy showed significant inhibition of the tumor growth than the control group at Day17. In parallel with the clinical effect, the combination therapy significantly increased tumor-infiltrating CD8+ T cells vs each monotherapy at Day7 even though more tumor-infiltrating CD8+ T cells were observed in mice treated with anti–PD-L1 or cisplatin alone than control. Further, anti–PD-L1 plus cisplatin combination activated tumor-infiltrating CD8+ T cells, characterized by higher frequency of granzyme B-expressing cells than each monotherapy. We also observed increased OVA-specific CD8+ T cells with combination vs monotherapies. Additional analysis of CD4+ T cells showed that anti–PD-L1 plus cisplatin combination or anti–PD-L1 alone, but not cisplatin alone, induced IFNg-producing CD4+ T cells in the tumor tissues.
Conclusions
Anti–PD-L1 plus cisplatin combination therapy demonstrated marked antitumor effect in this mouse tumor model. This robust therapeutic effects may at least partly be due to the increased tumor-specific CD8+ T cells in tumor tissues. Cisplatin or anti–PD-L1 alone increased tumor-infiltrating CD8+ T cells, however anti–PD-L1 but not cisplatin induced IFNg+ CD4+ T cells at tumor sites. These differences in immune status in response to anti–PD-L1 or cisplatin may lead to the combined therapeutic effect and provide a rationale for the combination.
P367 X4P-001, an orally bioavailable CXCR4 antagonist, increases T cell infiltration in human metastatic melanoma
Robert Andtbacka1, Melinda Yushak2, Merrick Ross3, Kenneth Grossman1, Eleni Tsiroyanni4, Sarah Blanchette4, Lu Gan4, Yan Wang4, Mohammed Milhem5
1Huntsman Cancer Institute, Salt Lake City, UT, USA; 2Winship Cancer Institute, Atlanta, GA, USA; 3MD Anderson Cancer Center, Houston, TX, USA; 4X4 Pharmaceuticals, Cambridge, MA, USA; 5University of Iowa, Iowa City, IA, USA
Correspondence: Robert Andtbacka (jennifer@theyatesnetwork.com)
Background
The CXCR4/CXCL12 axis plays a central role in the trafficking of key immune cells in the tumor microenvironment. Enhanced survival is reported in multiple syngeneic mouse models when a CXCR4 antagonist is combined with a check-point inhibitor. X4P-001 is an oral, selective, allosteric inhibitor of CXCR4, and X4P-001 alone demonstrated robust inhibition of murine B16-OVA melanoma growth. We hypothesize that disruption of CXCR4/CXCL12 signaling will result in modulation of the immune cell profile within the tumor microenvironment and ultimately lead to increased CD8+ T-cell infiltration, favoring an improved response to checkpoint inhibitors in metastatic melanoma.
Methods
The primary objectives for this ongoing biomarker-driven Phase Ib trial of X4P-001 alone and with pembrolizumab are to evaluate the safety and tolerability in patients (pts) with metastatic melanoma, and to characterize the effects of X4P-001 alone and with pembrolizumab on tumor immune cell infiltrates. Serial biopsies of cutaneous or subcutaneous metastatic melanoma lesions and peripheral blood mononuclear cells (PBMCs) are collected at pre-dose, after 3 weeks of X4P-001 treatment and after 6 weeks of combination treatment. Biopsies are assessed by immunohistochemistry (IHC) for multiple markers including CD3, CD8, FoxP3 and CXCL12, and PBMCs are analyzed by flow cytometry for both lymphoid and myeloid-cells.
Results
As of June 30, 2017, 13 pts have been enrolled and 4 of the 13 pts have completed the study. The median age was 74 years (range 53-91). Most pts (12/13) had not received prior systemic therapy. X4P-001 was well tolerated. Treatment related AEs (related to either X4P-001 or pembrolizumab; >5%) of any grade were: diarrhea (15%), chill, fatigue, headache, ocular hyperemia, photophobia, pruritus, rash, and vomiting (7.7%). Two treatment related G3 SAEs were reported: acute diarrhea and immune-mediated drug-reaction. Three of the 4 pts who completed the study had IHC evaluable tumor samples. All showed an increase in T-cell infiltration in the central region of the tumors following both single agent and combination treatment. The percentage of Ki67 positive CD8+ T cells in PBMC were increased post treatment in all 4 pts. These data along with additional biomarker readouts in all enrolled patients will be presented.
Conclusions
Treatment with X4P-001 as a single agent, and in combination with pembrolizumab, is well tolerated with preliminary evidence of enhanced immune cell infiltration and activation. The enrollment of the study is near completion and further biomarker analysis is on-going.
P368 eFT508, a potent and highly selective inhibitor of MNK1 and MNK2, is an activator of anti-tumor immune response
Kevin Webster1, Rajesh Sharma1, Vikas Goel1, Craig Stumpf1, Jocelyn Stauton1, Peggy Thompson1, Gary Chiang1, Yichen Xu2, Hyun Yong Jin2, Davide Ruggero2
1eFFECTOR Therapeutics, San Diego, CA, USA; 2Helen Diller Family Comprehensive Cancer Center, San Francisco, CA, USA
Correspondence: Kevin Webster (kwebster@effector.com)
Background
eFT508 is a potent and highly selective inhibitor of MNK1 and MNK2, kinases that function to mediate tumor immune evasion downstream of MEK and MAPK signaling. eFT508 treatment establishes a regulatory program that promotes multiple steps in the cancer immunity cycle including antigen presentation and T cell priming, expansion of memory T cells, and prevention of T cell exhaustion.
Methods
The immunological effects of eFT508 have been evaluated in the context of normal human immune cells in vitro and in immunocompetent syngeneic and genetically engineered mouse models in vivo.
Results
eFT508 treatment of normal donor T cells has no deleterious effect on αCD3/αCD28 stimulated T cell proliferation or T cell viability in contrast to inhibitors acting upstream of MAPK signaling. eFT508 selectively down regulates key immune checkpoint proteins and the production of a subset of proinflammatory and immunosuppressive cytokines. In vitro mechanism of action studies have demonstrated that MNK selectively regulates gene expression at the level of mRNA translation via specific sequence elements in the 5’- and 3’-untranslated regions. In addition, eFT508 activated antigen presenting cells leading to more effective T cell priming. eFT508 also affected T cell memory formation, both in the context of specific peptide antigen stimulation and in a mixed lymphocyte reaction, shifting the distribution of T cells towards a CD62L+CD44+ central memory T cell population. eFT508 also enhanced the cytotoxic function of T cells from OT-I mice stimulated with SIINFEKL peptide demonstrating a dose-dependent increase of cell killing. Consistent with the mechanisms elaborated upon in vitro, eFT508 showed significant anti-tumor activity mediated through tumor infiltrating lymphocytes in the CT26 syngeneic tumor model as well as genetically engineered mouse models of NSCLC and HCC.
Conclusions
eFT508 treatment establishes a regulatory program that promotes anti-tumor immunity. eFT508 is currently under evaluation as a single agent in two phase 1/2 clinical trials for patients with advanced solid tumors and patients with advanced lymphoma. A biomarker driven proof of concept study, including mandatory pre- and on-treatment biopsies, to evaluate the immunological mechanism of action of the drug is planned to be initiated later this year. In addition, a phase 2 study evaluating eFT508, alone or in combination with avelumab, a PD-L1 immune checkpoint inhibitor, in microsatellite stable relapsed or refractory CRC patients is planned.
P369 Analysis of the TIGIT/PVRIG axis in human cancers to support indication selection and biomarkers for COM701 and COM902
Sarah Whelan, Ling Leung, David Bernardos, John Hunter, Mark White, Spencer Liang
Compugen USA, Inc., South San Francisco, CA, USA
Correspondence: Sarah Whelan (sarahw@cgen.com)
Background
PVRIG and TIGIT were identified by Compugen’s Predictive Discovery Platform as immune inhibitory receptors and have been reported to inhibit anti-tumor activity. We are pursuing clinical development of antagonistic antibodies to PVRIG (COM701) and to TIGIT (COM902). Here, we analyzed primary human cancer tissues and immune cells to characterize expression in the TIGIT/PVRIG axis to support indication selection and combination strategies for COM701 and COM902.
Methods
COM701 and COM902 were identified based on ability to block the interaction of PVRIG and TIGIT with their cognate ligands (PVRL2 and PVR respectively) and were screened for their ability to enhance antigen-specific CD8 T cell activation in a co-culture with tumor cell lines. Immunohistochemistry and Flow cytometry were performed to assess receptor/ligand expression in dissociated bladder, breast, colorectal, head and neck, lung, kidney, ovarian, prostate, and stomach tumors.
Results
Among the cancers examined, PVRIG and PVRL2 expression was highest in endometrial, lung, kidney, ovarian, and head and neck cancers compared to normal adjacent tissue. From dissociated tumors, PVRIG expression was detected on T and NK TILs whereas PVRL2 expression was detected on CD45- cells and myeloid cells. A co-expression analysis of PVRIG, TIGIT, and PD1 demonstrated that PVRIG was co-expressed with both TIGIT and PD1 and that PVRIG+TIGIT+PD1+ cells comprised a major proportion of CD8 TILs. In comparison to PD-L1, PVRL2 expression was more prevalent across several cancer types and expression of PVRL2 was detected in PD-L1 negative samples. In vitro, combination of COM701 with PD1 inhibitors or COM902 enhanced CD8 cytokine production and cytotoxic activity, with the triple combination of COM701, COM902, and PD-1 antibody yielding the greatest increase in functional activity. Several immune receptors were induced in response of PVRIG blockade by COM701 on CD8 T cells. Taken together, these data support indication selection and combination strategies for COM701 and COM902 and potential biomarkers that could be indicators of response.
Conclusions
In summary, we demonstrate that PVRIG and PVRL2 are induced in the tumor microenvironment of human cancers, and the potential of COM701 as a cancer therapeutic, either as a monotherapy or as a dual- or triple-combination therapy with antibodies targeting TIGIT, and PD-1. These data highlight the potential of this combination approach to expand the immune checkpoint inhibitor responsive cancer patient population, including those who are non-responsive to PD-1 inhibitors.
P370 Enhancement of tumor specific immunity by activation of CD40 through a bispecific molecule targeting CD40 and a tumor surface antigen
Shiming Ye1, Diane Yu1, Nicole Belmar1, Donghee Choi1, Debra Chao1, Mien Sho1, Han Kim1, Jean Cabel2, Danying Song2, Kelley Hiser2, John Harlan2, Catherine Zhang1, Yuni Fang1, Steve Keller1, Alan Wahl3, Patricia Culp4, Diane Hollenbaugh1
1AbbVie Biotherapeutics Inc., Redwood City, CA, USA; 2AbbVie Inc., North Chicago, IL, USA; 3Former AbbVie Employee, Del Mar, CA, USA; 4Former AbbVie Employee (currently working at Alector Inc.), South San Francisco, CA, USA
Correspondence: Shiming Ye (shiming.ye@abbvie.com)
Background
CD40 activation can bridge the innate and adaptive immune systems during immune activation. Agonistic anti-CD40 monoclonal antibodies (mAbs) have demonstrated anti-tumor activity in clinical studies. Due to the dose-limiting toxicity observed, an alternative approach to developing an anti-CD40 therapy, and potentially reducing systemic toxicity, is to achieve immune activation via a bispecific molecule that is maximally active in the presence of a tumor antigen (TA), but with limited activity in its absence.
Methods
Different bispecific formats targeting both CD40 and a TA were designed and constructed. Bispecific molecules with desired properties were screened and assayed for activation of primary B cells or monocyte-derived dendritic cells (moDC) co-cultured with TA positive or negative cells. Selected bispecific molecules were tested for anti-tumor activity using PC3 tumor cells with or without TA expression mixed with autologous moDC and T cells, cultured in vitro or inoculated in NSG mice. Syngeneic mouse models of TA positive tumors were used to test bispecific molecules for immune activation, anti-tumor potency, and tolerability.
Results
ABBV-428 is a bispecific molecule with single chain Fv (scFv) domains targeting human CD40 and a TA. ABBV-428 was less potent than a CD40 mAb in stimulating B cells and moDCs when cultured alone or in the presence of cells that do not express TA. However, ABBV-428 exhibited enhanced B cell and moDC activation when cultured with cells expressing TA. T cells were also activated by ABBV-428 when mixed with moDC in the presence of cell-surface TA, as evidenced by a reduction in growth of PC3 cells, both in vitro and in NSG mice. Although expression of the cell surface TA is necessary for immune-mediated anti-tumor activity, ABBV-428 inhibited the growth of both TA-positive and TA-negative PC3 cells as long as the TA was expressed within the tumor environment. This phenomenon was confirmed in a mouse model carrying syngeneic 4T1 tumors expressing the cell surface TA. A surrogate molecule of ABBV-428 elicited T cell responses against both TA-expressing and non-expressing 4T1 cells, where the anti-tumor activity was similar to anti-CD40 mAb, but did not elicit increases in serum cytokines and liver enzyme levels observed in anti-CD40 mAb treated mice.
Conclusions
ABBV-428 exhibits enhanced CD40 activation upon binding to TA-expressing cells, and provides tumor-specific immune stimulation with systemic administration. The enhanced tumor-specific immune activation is hypothesized to maximize anti-tumor potency while limiting systemic toxicity in clinical studies.
Impact of Diet, Exercise, and/or Stress on Antitumor Immunity
P371 Featuring adipocytes secretion as pharmacological target for adjuvant immunotherapy against breast cancer
Luis Henrique Correa, LÍVIA PIMENTEL SANT’ANA DOURADO, Kelly Grace Magalhaes
University of Brasilia, Brasilia, Brazil
Correspondence: Kelly Grace Magalhaes (kellymagalhaes@gmail.com)
Background
Obesity and adipose tissue have been shown to be associated with low grade inflammation resulting in cellular and humoral inflammatory factors that contribute to carcinogenesis1. It has been known that inflammation exerts an important role in carcinogenesis and tumor progression. Inflammatory molecules can potentially be used as adjuvant in immunotherapy against cancer. However, little is known about the differential role of brown and white adipocytes against breast cancer. In the present work, we aimed to characterize the differential function of brown and white adipocytes in breast cancer cell death in vitro and investigate the role of NLRP3 and caspase-1/11 in this process.
Methods
Brown and white adipose tissue were isolated from wild type and caspase-1/11 and NLRP3 knockout mice. Conditioned medium from these brown and white adipocytes were used to stimulate breast cancer cells 4T1. Breast cancer cells (I) viability was analyzed by MTT assay; (II) cell death was investigated by annexin V/PI staining and flow cytometry analysis (III) membrane pore formation was observed by PI staining and spectrophotometry analysis and (IV) lipid droplet biogenesis was analyzed by Bodipy staining and flow cytometry analysis.
Results
Our data showed that brown adipocytes conditioned medium triggered significant higher levels of breast cancer cell death and pore membrane formation and lower levels of lipid droplet biogenesis and cell viability compared to white adipocytes conditioned medium.
Conclusions
The absence of caspase-1/11 in brown adipocytes, but not NLRP3, enhanced these cell death and carcinogenic parameters in breast cancer cells. Identification of molecules from these adipocytes secretion is ongoing and can be potentially used as adjuvant immunotherapy agains breast cancer.
References
1. Alderton G. The supersized tumour microenvironment. Nature Reviews Cancer. 2015;15:575.
Mechanisms of Efficacy or Toxicity
P372 A syngeneic mouse model of CAR-T mediated toxicity and neuroinflammation
Eric Chadwick, Alyssa Noll, Yue Jiang, Ronald Hause, Rafael Ponce, Hyam Levitsky, Ruth Salmon
Juno Therapeutics, Seattle, WA, USA
Correspondence: Ruth Salmon (eric.chadwick@junotherapeutics.com)
Background
Interrogating the spectrum of systemic effects of CAR-T cell therapy, especially in the context of adverse events (i.e. systemic cytokine release syndrome (sCRS) and neurotoxicity) that have occurred in clinical trials, may provide important insights into factors contributing to the cause and/or progression of such events and potential interventions. Preclinical CAR-T cell efficacy models in immune-compromised mice do not capture the potential role of the host immune system in sCRS and central nervous system (CNS) pathologies that, in combination with other patient factors, may lead to severe neurotoxicity. We have developed a novel model system where conditioning immune-competent mice with cyclophosphamide (CPA) followed by transfer of murine CD19-directed CAR-T cells induces acute symptoms including systemic cytokine release in concordance with what is observed in patients that develop CRS and/or neurotoxicity. This systemic toxicity is accompanied by the alteration of gene expression levels in the mouse brain indicating a possible neuro-inflammatory response.
Methods
We examined the effects of murine CD19-directed CAR-T cell therapy in a syngeneic BALB/c model. Following dose optimization for both CPA conditioning and the intravenous transfer of CAR-T cells, mice were evaluated for acute toxicity and systemic and brain-related pathologies including serum cytokine levels, brain and peripheral organ histopathology by microscopy of hematoxylin and eosin-stained sections, evaluation of blood brain barrier (BBB) integrity by extravasation of fluorescently-labeled low-molecular weight dextrans, wet-dry brain weights indicative of cerebral edema, and flow cytometry and gene expression analysis of transcardially-perfused brain tissues.
Results
Conditioning with CPA, followed by administration of murine CD19-directed CAR T cells, but not control CAR-T cells, induced rapid weight loss, peripheral organ pathologies and elevated serum cytokine levels (including a spectrum of cytokines similar to those observed in clinical CRS). In the brain, we observed significant changes in gene expression indicative of neuro-inflammation including genes associated with interferon response pathways, vascular endothelial activation and oxidative stress, accompanied by CAR-T cell infiltrate into the brain. No evidence for overt brain histopathology was observed, nor increased BBB permeability or cerebral edema.
Conclusions
These findings describe a new animal model and highlight its potential use to elucidate the mechanisms underlying CAR-T-cell mediated toxicities and test proposed interventions to reduce neuro-inflammation that may arise from CD19-directed CAR-T cell therapies. Ongoing work seeks to identify and evaluate various pharmacological interventions with the potential to ameliorate systemic and neuro-inflammation in this model, with the goal to translate these learnings to the clinic.
P373 Optimizing anti-OX40 mediated immunotherapy: preclinical exploration of the relationship between antitumor activity and isotype choice, ligand blocking capacity, dose and schedule
Chan Gao1, John Engelhardt2, Gennaro Dito1, Susan Glick1, Megan Raymond1, Marie-Claude Gaudreau1, Alan Korman2, Michael Quigley1
1Bristol-Myers Squibb, Princeton, NJ, USA; 2Bristol-Myers Squibb, Redwood City, CA, USA
Correspondence: Michael Quigley (gyrase@gmail.com); Alan Korman
Background
Following the clinical success of checkpoint blockade, the field of cancer immunotherapy is rapidly expanding. Extensive preclinical data have demonstrated that treatment with an agonist OX40 antibody can result in anti-tumor immune responses both alone and in combination with other immune-targeting agents including CTLA-4 and PD-1 blockers. The reported mechanism of action for OX40 includes costimulation of effector T cells as well as reduction in regulatory T cell (Treg) suppression either through depletion or receptor engagement. However, the vast majority of published work utilizes a single, ligand non-blocking antibody to define the role of anti-OX40 in enhancing tumor immunity in vivo. We generated an alternative agonistic, ligand-blocking mouse OX40 antibody as well as series of isotype variants in order to better define the role of isotype, ligand blocking vs non-blocking epitopes, dose and schedule on the anti-tumor activity of anti-OX40 alone and in combination with PD-1 blockade in vivo.
Methods
The activity of OX40 agonist antibodies, OX86 (ligand non-blocker) and OX40.23 (ligand blocker), were tested across a dose range from 0.03 mg/kg to 10mg/kg in the CT26 tumor model. Anti-PD-1 (10 mg/kg) was dosed either concurrently or 5 days after initial OX40 treatment. On-treatment immune monitoring of peripheral and tumor-infiltrating lymphocytes was analyzed by flow cytometry.
Results
Fc-competent agonist OX86 antibodies display potent in vivo anti-tumor activity at 10 mg/kg, whereas an Fc inert antibody at the same dose had no effect on CT26 tumor growth. Relative anti-tumor activity was related to their ability to preferentially bind activating FcγR and deplete intratumoral Tregs. Maximal activity of OX40.23 was achieved at 3 mg/kg and 0.3 mg/kg as monotherapy and in combination with anti-PD-1, respectively. Interestingly, administration of OX40.23 at 10 mg/kg in both monotherapy and combination displayed diminished activity, accompanied by a reduction in peripheral T cell activation. Evaluation of OX40 RO demonstrated that peripheral and intratumoral RO was similar as was RO between monotherapy and combination treatment. Maximal anti-tumor activity of the combination was achieved well below 100% OX40 RO. Furthermore, for combination treatment, concurrent dosing resulted in greater anti-tumor activity than a staggered regimen.
Conclusions
Our results demonstrate the importance of isotype choice, ligand blockade capacity, dose and schedule on the in vivo anti-tumor activity of mouse OX40 agonist antibodies alone and in combination with PD-1 blockade. These data provide valuable insight relevant in biomarker, dose and schedule selection for agonist OX40 antibody-containing regimens currently in clinical development.
P374 Activation of 4-1BB on liver myeloid cells triggers hepatitis via an interleukin-27 dependent pathway
Ashvin R. Jaiswal1, Todd Bartkowiak1, Casey R. Ager1, Renee Chin1, Chao-Hsien Chen1, Pratha Budhani1, Midan Ai1, Matthew J. Reilley1, Manu M. Sebastian2, David S Hong1, Michael A Curran1
1The University of Texas MD Anderson Cancer Center, Houston, TX, USA; 2The University of Texas MD Anderson Cancer Center, Smithville, TX, USA
Correspondence: Ashvin R. Jaiswal (arjaiswal@mdanderson.org); Michael A Curran
Background
Agonist antibodies targeting the T cell co-stimulatory receptor 4-1BB (CD137) are among the most effective immunotherapeutic agents across multiple pre-clinical models of cancer. In the clinic, however, development of these agents has been stymied by dose-limiting liver toxicity. Lack of knowledge of the mechanisms underlying this toxicity has limited the potential to separate 4-1BB agonist driven anti-tumor immunity from hepatotoxicity.
Methods
The capacity of 4-1BB agonist antibodies to induce liver toxicity was investigated in immune competent mice, with or without co-administration of checkpoint blockade, via measurement of serum transaminase levels, through imaging of liver immune infiltrates, and via qualitative and quantitative assessment of liver myeloid and T cells via flow cytometry. Knockout mice were used to clarify the contribution of specific cell subsets, cytokines and chemokines.
Results
We find that activation of 4-1BB on liver myeloid cells is essential to initiate hepatitis. Once activated, these cells produce interleukin-27 which is required for liver toxicity. CD8 T cells infiltrate the liver in response to this myeloid activation and mediate tissue damage triggering transaminase elevation. FoxP3+ regulatory T cells limit liver damage and their removal dramatically exacerbates 4-1BB agonist hepatitis. Co-administration of CTLA-4 blockade ameliorates transaminase elevation, whereas PD-1 blockade exacerbates it. Loss of the chemokine receptor CCR2 blocks 4-1BB agonist hepatitis without diminishing tumor-specific immunity against B16 melanoma.
Conclusions
4-1BB agonist antibodies trigger hepatitis via activation of myeloid cells to produce Interleukin-27. Co-administration of CTLA-4 and/or CCR2 blockade may minimizing hepatitis but yield equal or greater antitumor immunity.
P375 Evaluation of surrogate endpoints for overall survival in patients treated with immunotherapies
Howard Kaufman1, Lawrence Schwartz2, William William3, Mario Sznol4, Michael del Aguila5, Craig Whittington5, Kyle Fahrbach6, Yingxin Xu6, Eric Masson7, Andrea Vergara-Silva8
1Rutgers Cancer Institute of New Jersey, New Brunswick, NJ, USA; 2Columbia University Medical Center, New York, NY, USA; 3MD Anderson Cancer Center, Houston, TX, USA; 4Yale School of Medicine, New Haven, CT, USA; 5Doctor Evidence, Santa Monica, CA, USA; 6Evidera, Bethesda, MD, USA; 7AstraZeneca, Waltham, MA, USA; 8AstraZeneca, Gaithersburg, MD, USA
Correspondence: Howard Kaufman (hk553@cinj.rutgers.edu)
Background
Surrogate endpoints have not been clearly determined in oncology immunotherapy (IT). Using both arm- and comparison-level data, this study explored the relationship between overall survival (OS) and clinical endpoints (objective response rate [ORR], disease control rate [DCR], and progression-free survival [PFS]) in patients treated with IT. The aim was to assess whether any of these endpoints could function as surrogates for OS.
Methods
A systematic review was conducted in MEDLINE™ and Embase (January 2005–March 2017), and supplemented with conference proceedings (2014–2017). Eligible studies were randomized controlled trials (RCTs) that investigated blocking antibodies targeting programmed cell death-1 (PD-1)/programmed cell death ligand-1 (PD-L1) or cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4). Studies were included in the arm-level analyses if the treatment arm’s absolute effects could be obtained for ORR, DCR, 6- and 9-month PFS, median PFS, median OS, or OS at 12 or 18 months. They were included in comparison-level analyses if the treatment’s relative effects (odds ratios [ORs] on ORR and DCR or hazard ratios [HR] on PFS and OS) were reported/could be derived.
Weighted linear regression models were fitted and adjusted R2 values estimated, with analyses stratified by treatment regimen (IT mono or IT plus chemotherapy), by type of IT (PD-1/PD-L1 or CTLA-4), and by indication, as data permitted.
Results
Twenty-nine RCTs involving 66 treatment arms (11,797 patients) were included. In the arm-level analyses, higher PFS rates at 6 months predicted better OS rates at 12 and 18 months, with similar trends across subgroups (Fig. 1A–D); results by type of IT revealed stronger correlations for PD-1/PD-L1 than CTLA-4 as potential surrogates for OS (Fig. 1A, Fig. 1B). Similarly, the comparison-level analyses only found the PFS HR to be moderately correlated with the OS HR (R2=0.372; P=0.002; Fig. 1e); this finding remained consistent when restricted to studies assessing IT monotherapy only (R2=0.460; P=0.002; Fig. 1F) and in subgroups stratified by treatment type or indication.
Conclusions
Among anti–PD-1/PD-L1 studies, PFS was an imperfect surrogate (low to moderate correlation) for OS, whereas other surrogate clinical endpoints were not correlated with OS. Thus, a new surrogate, such as a biomarker, is needed to better predict the OS benefit for IT.

Abstract Summary
P376 Evaluating CD38 as a therapeutic target in non-small cell lung cancer (NSCLC)
Michelle Kinder, Mark Mendonca, Khaja Syed, Melissa Parker, Patrick Wilkinson, Edward Thompson, Gerald Chu, Christopher Chiu, Natalie Hutnick
Janssen Research and Development, LLC, Spring House, PA, USA
Correspondence: Michelle Kinder (mkinder@its.jnj.com)
Background
Daratumumab (DARA), a human CD38-targeted monoclonal antibody, is approved as monotherapy and in combination with standard of care regimens for patients with relapsed or refractory multiple myeloma. DARA demonstrates direct on-tumor and immunomodulatory mechanisms of action. In multiple myeloma patients treated with DARA, CD38+ immunosuppressive cells including regulatory T-cells (Treg), myeloid derived suppressor cells (MDSC) and regulatory B-cells (Breg) are depleted. DARA also results in increased CD8+ T-cells and T-cell clonality. We hypothesized that DARA could be effective in solid tumors that have elevated levels of CD38 expression on the surface of tumor and/or immunosuppressive cells. Here, we investigated CD38 expression on lung cancer and intra-tumoral immune cells to determine whether NSCLC may be susceptible to the immunomodulatory effects of DARA.
Methods
CD38 RNA expression in lung cancer cell lines and tumor sections from The Cancer Genome Atlas (TCGA) database was investigated using RNA-sequencing. Protein expression on lung cancer cell lines, tumor-infiltrating immune cells, and peripheral blood (PB) immune cells was analyzed by flow cytometry. Primary tumor samples were evaluated by immunohistochemistry. The effect of DARA on lung cancer cell viability was measured in an in vitro assay. DARA-mediated depletion of immune cells in the PB of lung cancer patients was assessed ex vivo.
Results
CD38 RNA was highly expressed in lung tumor specimens from the TCGA database. Of the lung cancer cell lines investigated, 3/12 (A549, NCI-H2023, and NCI-H2073) showed detectable CD38 receptor. These cell lines demonstrated moderate receptor density (8,830, 20,900, and 58,100 average receptors/cell, respectively) compared to a multiple myeloma cell line (MM.1R; 75,590 average receptors/cell). Detectable CD38 receptor expression was observed on tumor cells from 5/21 primary NSCLC tumor samples and was highly expressed on intratumoral immunosuppressive cells including: monocytic MDSC, tumor-associated macrophages and Treg. In the PB of lung cancer patients, CD38 receptor was expressed on >50% of NK cells, B-cells, monocytic MDSC, immature MDSC, and monocytes. Significantly, DARA induced cell-mediated killing of CD38+ lung cancer cell lines in vitro, and depleted monocytic MDSC from the PB of lung cancer patients ex vivo.
Conclusions
Tumor cell CD38 expression is observed in a subset of lung cancers and is highly expressed on patient tumor-infiltrating immune cells and immunosuppressive cells in PB samples. Because DARA has demonstrated in vitro activity in depleting these cells, DARA may represent a novel therapeutic approach to targeting the immune microenvironment in solid tumors. Clinical trials in NSCLC are underway (NCT03023423) and/or planned.
P377 Cisplatin induces immunogenic cell death in preclinical models of head and neck squamous cell carcinoma
So-Jin Park1, Linda Tran1, Roy Xiao1,2, Carter Van Waes1, Nicole Schmitt1,3
1NIDCD, NIH, Bethesda, MD, USA; 2Cleveland Clinic Lerner College of Medicine, Bethesda, MD, USA; 3Johns Hopkins University, Bethesda, MD, USA
Correspondence: Nicole Schmitt (nicole.schmitt@nih.gov)
Background
Immunogenic cell death (ICD) is the process by which stressed cells exhibit translocation of markers including calreticulin and heat shock proteins (HSPs) to the cell surface along with release of HMGB1 and ATP. This activates toll-like receptors and adaptive immunity. Studies in several cancer types suggest that oxaliplatin induces superior ICD compared to cisplatin. This has never been studied in head and neck squamous cell carcinoma (HNSCC).
Methods
Three HNSCC cell lines were treated with sublethal doses of the platinum chemotherapy drugs cisplatin, oxaliplatin, and carboplatin for 48 hours. Cell surface levels of HSP70 and calreticulin were then assayed by flow-cytometry. Intracellular HMGB1, an indirect measurement of HMGB1 release, was also quantified. Release of HMGB1 was measured in the cell culture supernatants. A syngeneic mouse model was then used to compare the effects of cisplatin vs. oxaliplatin, alone or in combination with anti-PD-1 immunotherapy, on tumor growth and survival. A subset of tumors were analyzed for immune cell infiltrates by flow cytometry.
Results
All three platinum drugs induced translocation of HSP70 and calreticulin to the cell surface, as well as release of HMGB1 in multiple cell lines. Cisplatin was the superior ICD inducer in these cell lines. Cisplatin and oxaliplatin induced similar tumor growth delay when combined with anti-PD-1 immunotherapy in tumor-bearing, immunocompetent mice.
Conclusions
Treatment of HNSCC cells with platinum chemotherapy drugs appears to induce ICD, which may enhance anti-tumor immunity. Cisplatin, which is the standard chemotherapy drug for treatment of HNSCC, appears to be at least as effective as oxaliplatin as an ICD inducer in these preclinical models.
P378 Improving the efficacy of cancer immunotherapy: An intimate play of CD8 T and NK cells
Roman Uzhachenko1, J Shawn Goodwin1, William Hofmeister2, Anil Shanker1,3
1Meharry Medical College School of Medicine, Nashville, TN, USA; 2University of Tennessee Space Institute, Tullahoma, TN, USA; 3Vanderbilt-Ingram Cancer Center, Nashville, TN, USA
Correspondence: Anil Shanker (ashanker@mmc.edu)
Background
The interaction between the innate and adaptive immune components is fundamental for an effective antitumor immunity. Our studies in murine models of mastocytoma, kidney, breast and lung solid tumors showed that productive antitumor effector response relies on functional crosstalk between innate immune effectors—natural killer (NK) cells, and adaptive immune effectors—cytolytic CD8+T lymphocytes. We found that this lymphocyte cooperativity between CD8+T and NK cells can prevent the development of antigen-escape tumor variants.
Methods
Using a nanofiber matrix engineered to provide lymphocytes a controlled 3D interaction, we found that activated CD8+T cells (CD69highCD25high) formed multiple intercellular contacts with several naïve NK cells, while naïve CD8+T cells made single or no contact with NK cells.
Results
In lymphocyte coculture (physical contact possible), activated CD8+T and NK cells cross-regulated each other’s phenotype wherein NK cells polarized activated CD8+T cells towards T central memory phenotype and activated CD8+T lymphocytes induced acquisition of effector/regulatory phenotype by naïve NK cells. This cross-regulation of lymphocytes disappeared in a trans-well system (no physical contact) indicating the necessity of cell-to-cell physical interaction during CD8+T—NK crosstalk. Notably, intercellular physical interaction led to cross-regulation of mitoCa2+ oscillations in both activated CD8+T and NK cells. Inhibition of mitochondrial Ca2+ uptake or Na+/Ca2+ exchanger with Ru360 and CGP37157, respectively, mimicked observed alterations in both lymphocytes. Further, NK cells displayed increased oxidative signaling, Tyk2, Jak 1 and 3, Stat2 and Stat6 phosphorylation while inhibiting TCR- and various cytokine receptor-mediated signaling. In turn, NK cells selectively restrained IL-2 signaling in CD8+T cells by dampening activation-induced up-regulation of CD25, Stat5 phosphorylation, IL-2 synthesis and elevation in IL-2 uptake.
Conclusions
These data suggest a model, where mitochondrial Ca2+ flux acts as a key biological controller to guide cellular crosstalk allowing acquisition of NK cell effector/regulatory and T cell central-memory phenotypes upon their interaction. Further understanding of the characteristics and regulatory factors involved in this NK—CD8+T cell physical play in the tumor microenvironment will provide new insights on controlling immune escape variants of tumor. This could lead to novel strategies for effective cancer immunotherapies, with a potential of relapse-free survival in cancer patients.
P379 Distinct cellular mechanisms underlie anti-CTLA-4 and anti-PD-1 checkpoint blockade
Spencer Wei1, Jacob Levine2, Alexandria Cogdill1, Yang Zhao1, Nana-Ama Anang1, Miles Andrews1, Padmanee Sharma1, Jing Wang1, Jennifer Wargo1, Dana Pe'er2, James Allison1
1The University of Texas MD Anderson Cancer Center, Houston, TX, USA; 2Sloan Kettering Institute, New York, NY, USA
Correspondence: Spencer Wei (scwei@mdanderson.org); James Allison
Background
Immune checkpoint blockade is able to achieve durable responses in a subset of patients, however despite such significant clinical progress we still lack a fundamental understanding of the mechanisms of anti-CTLA-4 and anti-PD-1 induced tumor immune rejection. CTLA-4 and PD-1 regulate T cell activation through different molecular and cellular mechanisms and act at different stages of T cell activation. As such, we hypothesized that anti-CTLA-4 and anti-PD-1 checkpoint blockade induce tumor rejection through distinct cellular mechanisms.
Methods
To address this hypothesis, we utilized mass cytometry which enables characterization of more than 40 parameters at single cell resolution and unsupervised cellular classification. Using this approach, we comprehensively profiled the immune infiltrates of MC38 and B16BL6 murine tumors from mice treated with anti-CTLA-4, anti-PD-1, or control antibodies. We also performed similar analyses of surgically resected melanomas from patients being treated with checkpoint blockade therapy.
Results
In both tumor models, more than 13 distinct tumor infiltrating T cell populations were identified. Both anti-CTLA-4 and anti-PD-1 checkpoint blockade modulated the frequencies of only a subset of these tumor infiltrating T cell populations. Furthermore, of multiple exhausted-like CD8 T cell populations identified, the frequencies of only two subsets correlate with outcome, suggestive of functional heterogeneity within phenotypically exhausted T cells. Most notably, we find that anti-CTLA-4, but not anti-PD-1, modulates the CD4 effector compartment by inducing the expansion of Th1-like CD4 effector T cells. Observations from mass cytometry analyses of surgically resected melanoma tumors from patients being treated with anti-CTLA-4 or anti-PD-1 therapy were consistent with these preclinical findings.
Next we utilized similar methodologies to investigate the cellular mechanism of combination anti-CTLA-4 and anti-PD-1 therapy. Although combination therapy largely enhanced effects observed in monotherapies, combination therapy differentially modulated specific exhausted-like CD8 T cell populations. These data suggest that combination therapy modulates T cell function and mediates tumor rejection through mechanisms that are in part distinct from either monotherapy.
Conclusions
Our findings indicate that anti-CTLA-4 and anti-PD-1 modulate specific tumor infiltrating T cell populations and utilize distinct cellular mechanisms to induce tumor rejection. Anti-CTLA-4, but not anti-PD-1, induces expansion of CD4 effector T cells. Furthermore, these data suggest that combination therapy modulates T cell function differently than monotherapies. These findings have implications for the rational design of combinatorial therapeutic approaches and expand our understanding of the mechanisms that regulate T cell activity.
P380 The role of radiotherapy in driving anti-tumor CD8 T cell responses
Lauren Zebertavage1, Alejandro Alice2, Marka Crittenden2, Michael Gough2
1Oregon Health and Science University, Portland, OR, USA; 2Earle A Chiles Research Institute, Portland, OR, USA
Correspondence: Michael Gough (uhde@ohsu.edu)
Background
Radiotherapy (RT) is one of the three main arms of traditional cancer therapy, and has been show to synergize well with immunotherapies in preclinical models. Consistent with the findings of other investigators, in immunogenic murine models of radiation therapy and immunotherapy we have observed CD8+ T cell-dependent clearance of tumors given single, high-dose radiation (20Gy) and anti-PD1 checkpoint blockade. In this setting, RT has been proposed to generate tumor antigen-specific T cells following the release of antigens by dying cancer cells, and PD1 blockade allows T cell control of residual cancer cells in the suppressive tumor environment. We hypothesized that the role of radiation is to boost the number of existing tumor antigen-specific CD8 T cells and drive an influx of tumor-antigen reactive cytotoxic T cells into the tumor. If this hypothesis is true, then providing T cells through other means should be sufficient to replace radiation therapy in this therapeutic combination.
Methods
In order to deliver targeted beams of radiation, we use the Xstrahl Small Animal Radiation Research Platform (SARRP). Tumor-reactive CD8 T cell numbers were boosted using live-attenuated Listeria monocytogenes vaccines against potent tumor-specific CD8 model antigens. Nur77-GFP transgenic mice were used to measure antigen recognition by T cells in vitro and in vivo.
Results
We demonstrate that RT generates marginal increases in tumor-antigen specific T cells in the peripheral circulation. In order to determine whether higher numbers of tumor-responsive CD8s were driving RT plus checkpoint blockade efficacy, we vaccinated mice with attenuated Listeria expressing tumor-specific antigens. Using immunohistology and flow cytometry we demonstrate that these antigen-specific cells were present at high levels in the tumor. We demonstrate that antigen-specific T cells generated by vaccination kill antigen-pulsed targets in in vivo cytotoxicity assays, recognize cancer cells ex vivo, and respond to tumor-associated antigen within the tumor. However, mice treated with Listeria vaccination and PD1 blockade showed no tumor growth control advantage when compared to controls, despite the high level of antigen-specific T cells in the tumor.
Conclusions
These data demonstrate that while radiotherapy generates marginal increases in the number of antigen-specific T cells, which are log-fold fewer than generated by Listeria vaccination, RT is a superior partner for combination with checkpoint blockade. We find that generating large numbers of tumor antigen-specific T cells cannot substitute for the efficacy of radiotherapy in combination with anti-PD1 checkpoint blockade, and question whether the main driver of radiotherapeutic efficacy is augmenting CD8 T cell numbers.
Mechanisms of Resistance to Immunotherapy
P381 The BET bromodomain inhibitor ZEN-3694 modulates the expression of checkpoint receptors and immune suppressive factors in the blood of mCRPC patients
Eric Campeau1, Henrik Hansen1, Sanjay Lakhotia2, Karen Norek1, Laura Tsujikawa1, Sarah Attwell1
1Zenith Epigenetics, Calgary, AB, Canada; 2Zenith Epigenetics, San Francisco, CA, USA
Correspondence: Eric Campeau (sattwell@zenithepigenetics.com)
Background
Epigenetic regulation of the immune system plays a significant role in the response to immunotherapies. The potential for epigenetic modulators to prime the immune system and increase the duration and frequency of response to checkpoint inhibitors has been supported by both pre-clinical and clinical evidence. ZEN-3694 is an orally available inhibitor of the bromodomain and extra-terminal (BET) domain family of proteins currently in phase I clinical trials in metastatic castration-resistant prostate cancer (mCRPC) (NCT02705469 and NCT02711956). Previously, we have shown that ZEN-3694 modulates multiple checkpoint receptors, immune suppressive factors and cytokines in vitro, and acts synergistically with a PD-1 mAb to inhibit tumor growth in a MC-38 syngeneic mouse model. To follow up with these findings, we examined the ability of ZEN-3694 to modulate the expression of Immuno-Oncology target genes in mCRPC patients at multiple doses in our current phase 1 clinical trial.
Methods
The Nanostring nCounter® PanCancer Immune Profiling Panel was used to measure immune marker expression in patient whole blood RNA taken at 0, 4 and 24 h post dosing with ZEN-3694.
Results
ZEN-3694 modulates multiple checkpoints, suppressive factors and cytokines in peripheral immune cells 4 hours after a single dose. Most of these changes return to baseline at 24 hours, following clearance of the drug. Significant effects were detected at all doses tested, including at well-tolerated doses below the maximum tolerated dose. Multiple checkpoint receptors, including TIM3 and PD-L1, chemokines CCL2/CCR2 and IL-8, and the suppressive factors IDO1 and ARG1, were significantly inhibited 4 h post-dose across 16 patients. These results were also confirmed by real-time PCR for several of these markers. Furthermore, mRNA levels of multiple co-stimulatory markers, including ICOSLG and CD28, were maintained or induced. Several of these markers show a strong dose/exposure response, and Ingenuity® Pathway Analysis suggests that lower doses may be superior to higher doses for cancer immune response modulation.
Conclusions
Taken together, these data suggest that ZEN-3694 has the potential to modulate multiple factors involved in adaptive resistance to therapeutic PD-1 blockade, and therefore may improve response rate and duration in combination with a checkpoint inhibitor. This is the first presented clinical evidence that a BET bromodomain inhibitor can modulate PD-L1 and other relevant immuno-oncology targets in patients at therapeutically well-tolerated doses. Follow up studies to correlate these changes with protein expression, immune cell activation, and tumor-specific targets in patients are underway.
P382 ATR inhibition sequenced with radiation therapy abrogates immune exhaustion
David Clump1, Christopher Bakkenist1, Frank Vindetti1, Greg Delgoffe2, Robert Ferris1
1UPMC Hillman Cancer Center, Pittsburgh, PA, USA; 2University of Pittsburgh School of Medicine, Pittsburgh, PA, USA
Correspondence: David Clump (clumpda2@upmc.edu)
Background
Immunotherapy that targets the immunosuppressive interaction between the T cell activation-induced programmed cell death receptor 1 (PD-1) and its ligand PD-L1 and radiation therapy are used in the management of a majority of metastatic non-small cell lung cancer (NSCLC) patients. ATR is a DNA damage signaling kinase activated at damaged replication forks and ATR kinase inhibitors potentiate the cytotoxicity of DNA damaging chemotherapies and radiation
Methods
Here we show that ATR kinase inhibitors potentiate radiation-induced tumor immune responses in genetically engineered and syngeneic mouse models of Kras-mutant lung adenocarcinoma and colorectal carcinoma, respectively.
Results
ATR kinase inhibitors attenuate radiation-induced PD-L1 upregulation on tumor cells and dramatically decrease the number of tumor infiltrating Tregs. ATR kinase inhibitors further attenuate radiation-induced T cell exhaustion and increase T cell activity in the tumor microenvironment.
Conclusions
Our work raises the exciting possibility that a single pharmacologic agent may enhance the cytotoxic effects of DNA damaging chemotherapy and radiation while concurrently potentiating radiation-induced tumor immunity.
P383 Molecular and immune characterization of melanoma metastases with heterogeneous PTEN expression
Mariana Petaccia de Macedo1, Feng Wang2, Diego Marzese3, Courtney Hudgens2, Meredith McKean2, Khalida Wani2, Lauren Haydu2, Patrick Danaher4, Christopher Merritt4, Giang Ong4, Sarah Warren4, Joseph Beechem4, Dave Hoon3, Weiyi Peng2, Lawrence Kwong2, Michael Tetzlaff2, Alexander Lazar2, Michael Davies2
1AC Camargo Cancer Center, São Paulo, Brazil; 2MD Anderson Cancer Center, Houston, TX, USA; 3John Wayne Cancer Institute, Santa Monica, CA, USA; 4NanoString Technologies, Seattle, WA, USA
Correspondence: Michael Davies (mdavies@mdanderson.org)
Background
There is growing evidence that oncogenic pathways in tumor cells can impact the anti-tumor immune response and the efficacy of immunotherapy. Previously we showed that loss of PTEN inhibits immune infiltration and promotes resistance to immune-mediated tumor killing in melanoma [1]. Although PTEN expression is generally uniform in melanoma, we identified a cohort of melanoma metastases with distinct areas with (+) and without (-) PTEN protein expression. Comparative molecular and immune analyses were performed on the PTEN (+) and (-) regions of these tumors to improve our understanding of the mechanisms, pathogenesis, and significance of PTEN loss.
Methods
Core biopsies were performed on formalin-fixed paraffin-embedded FFPE blocks to isolate analytes from tumor-matched regions with (+) and (-) PTEN expression by immunohistochemistry (IHC). DNA was analyzed for somatic mutations and copy number variations (CNVs) in 200 cancer-related genes by next generation sequencing (NGS), and globally for DNA methylation patterns by Illumina Infinium Human Methylation450 arrays. Expression of proteins involved in signaling pathways (n=42) and immune populations/regulators (n=40) in distinct PTEN (+) and (-) regions within the tumors was quantified by Nanostring Digital Spatial Profiling.
Results
NGS showed that PTEN (+) and (-) regions did not differ in overall mutation patterns. PTEN (-) regions had increased rates of chromosomal loss affecting PTEN, EMB and ZNF567; gain of LETM1, WDR17 and SPATA4 was detected in PTEN (+) regions. Hierarchical clustering of DNA methylation patterns demonstrated overall clustering of samples by tumor. Loss of PTEN expression did not correlate with increased methylation of the Pten locus, but differential methylation of several potential regulatory transcription factors (Pax3, Gata2, Ikzf1, Pax5, and Gata5) was detected. Proteomic analysis identified multiple signaling and immune related proteins with significantly different expression between PTEN (+) and (-) regions. Loss of PTEN correlated with significantly increased P-AKT and P-P70S6K, consistent with increased activation of PI3K-AKT-mTOR signaling, and significantly decreased VISTA, CD8A, and CD3. PD-L1 expression did not correspond with PTEN status.
Conclusions
Intratumoral loss of PTEN in melanoma metastases corresponded with genetic alterations in the Pten gene but not its methylation status. Proteomic analysis supports that PTEN status corresponds locally with PI3K-AKT pathway activation and with heterogeneity of components of the anti-tumor immune response. Together the findings further support the biological significance of PTEN loss in melanoma.
Consent to publish
All patients provided informed consent.
References
1.Peng W, et al. Loss of PTEN promotes resistance to T-cell mediated immunotherapy. Cancer Discovery. 2016; 6 (2):201-216.
P384 Mechanisms of efficacy during TGFbR1 inhibition/cytotoxic combination therapy for rectal cancer
Andrew Gunderson, Kayla McCarty, Michaela Phillips, Emily Hodel, Michael Gough, Marka Crittenden, Kristina Young
Earle A. Chiles Cancer Research Institute, Portland, OR, USA
Correspondence: Andrew Gunderson (agun1107@gmail.com); Kristina Young
Background
Neoadjuvant chemoradiation is the standard of care for patients with locally advanced rectal cancer with the intention of downstaging and sphincter preservation prior to surgical intervention. Responses may be limited, however, in part due to TGFb1 regulated resistance to these cytotoxic modalities. We have shown previously that blocking ALK5 signaling through use of a small molecule inhibitor sensitizes CT26 tumor-bearing mice to the effects of radiation in part by boosting CD8+ T cell mediated cytotoxicity. The mechanism for this efficacy is currently unknown.
Methods
By using LY2157299 prior to 5-FU and hypofractionated radiation, tumor-bearing animals elicit suboptimal T cell responses capable of controlling tumor growth in a significant percentage of animals but fail to cure.
Results
Early results from a clinical trial of neoadjuvant TGFbR1 inhibition with chemoradiation in 3 rectal cancer patients demonstrate objective clinical responses and markers of enhanced anti-tumor immunity but indicators of compensatory immunosuppression are also observed in surgically excised tissue. One visually prominent aspect in a proportion of these patients is the infiltration of plasma cells into tumor parenchyma. Typically these fully differentiated B cells would be a hallmark of immune mediated rejection through secretion of high affinity immunoglobulin, but previous reports suggest they may also function to suppress cytotoxic T cells through TGFb1, IL-10 and PDL1. Correspondingly, we also observe increases in B regulatory populations and IL-10 expression in CT26 tumor draining lymph nodes from treated animals.
Conclusions
As B cell subsets express apoptotic cellular receptors similar to macrophages, we propose B cells may be one mechanism of resistance to this combination therapy and the direct blockade of their activity may optimize T cell effectiveness for patients with rectal cancer.
P385 A HSP-TLR-Wnt5a paracrine signaling axis drives CXCR2 ligand recruitment of myeloid-derived suppressor cells and represents a novel adaptive resistance mechanism to anti-PD-1 antibody therapy
Bala Theivanthiran1, Nicholas C. DeVito1, Kathy Evans1, Fei Zhao1, Christine Xiao1, Benjamin S. Goldschmidt2, Rob Edgar2, Alisha H. Holtzhausen3, April K.S. Salama1, John Lewicki4, John H. Strickler1, John A. Viator2, Brent A. Hanks5
1Duke University, Durham, NC, USA; 2Duquesne University, Pittsburgh, PA, USA; 3University of North Carolina, Chapel Hill, NC, USA; 4OncoMed Pharmaceuticals, Inc., Redwood City, CA, USA; 5Duke University, Durham, NC, USA
Correspondence: Brent A. Hanks (hanks004@mc.duke.edu)
Background
Despite the impressive impact generated by the anti-PD-1/PD-L1 antibody (ab) therapies in oncology, a significant percentage of patients exhibit either primary or secondary resistance to these treatment modalities. The evolution of these resistance mechanisms to cancer immunotherapy remains poorly understood. Myeloid-derived suppressor cells (MDSCs) have been demonstrated to suppress anti-tumor immunity and previous work has shown the granulocytic subset of MDSCs (Gr-MDSDCs) to migrate in response to CXCR2 chemokine gradients.
Methods
We utilized RNAseq differential gene expression analysis, serial tissue biopsy qrt-PCR, and SILAC-AHA metabolic labeling mass spectrometry-based differential secretome analysis to identify those genes and secreted proteins upregulated in melanomas escaping anti-PD-1 ab therapy in an autochthonous BRAFV600EPTEN-/- melanoma model. Multi-parameter flow cytometry and immunohistochemistry were utilized to investigate MDSC recruitment to tumor tissues. Lentiviral shRNA gene silencing and in vivo CD8+ T cell ablation assays were conducted to define the underlying signaling mechanisms driving MDSC recruitment to anti-PD-1 ab-treated melanomas. Circulating melanoma cells (CMCs) were derived from patients undergoing active anti-PD-1/PD-L1 ab therapy using a photoacoustic cytometry technique for single cell qrt-PCR validation of pre-clinical findings.
Results
Autochthonous BRAFV600EPTEN-/- melanomas upregulate the expression of several CXCR2 ligands, including CXCL5, following escape from anti-PD-1 ab therapy (Fig. 1A-B). This is associated with the rapid recruitment of Gr-MDSCs and an inhibition of local CD8+ T cell responses (Fig. 1C-D). We determined that melanoma CXCL5 expression is induced by a Wnt5a-YAP pathway and that Wnt5a is, in turn, regulated by local CD8+ T cell killing and paracrine heat shock protein (HSP)-TLR2 signaling (Fig. 2). The individual genetic silencing of Wnt5a and CXCL5 reverses Gr-MDSC recruitment in response to anti-PD-1 ab therapy and sensitizes these BRAFV600EPTEN-/- melanomas to checkpoint inhibition (Fig. 3). Indeed, blockade of Wnt-mediated signaling with a OMP-54F28 'Wnt trap' diminishes Gr-MDSC recruitment and augments the efficacy of anti-PD-1 ab therapy in this autochthonous melanoma model (Fig. 4). Plasma levels of CXCL5 correlate with progression and anti-PD-1 ab escape in the transgenic melanoma model while patient-derived CMCs also demonstrate CXCR2 ligand upregulation following progression on anti-PD-1/PD-L1 ab therapy (Fig. 5).
Conclusions
A HSP-TLR-Wnt5a paracrine signaling axis mediates the recruitment of Gr-MDSCs in response to anti-PD-1 ab therapy-mediated CD8+ T cell killing and represents a novel adaptive resistance mechanism to cancer immunotherapy. Additional studies are ongoing to exploit this pathway as a strategy for augmenting the therapeutic efficacy of anti-PD-1 ab therapy and for monitoring the development of immunotherapy resistance in advanced melanoma patients.

Escape from anti-PD-1 Ab Immunotherapy in a Transgenic Melanoma Model Involves the Recruitment of Gr-MDSCs. A. RNAseq differential gene expression analysis showing an upregulation of several CXCR2 ligands following anti-PD-1 ab escape in a transgenic BRAF(V600)-PTEN-/- melanoma model. B. RNAseq differential gene expression analysis demonstrating increased MDSC markers in BRAF(V600E)-PTEN-/- melanomas following escape from anti-PD-1 ab therapy. C. Immunohistochemistry and flow cytometry analysis demonstrating increased levels of Gr-MDSCs in anti-PD-1 ab-treated versus IgG isotype control-treated BRAF(V600E)-PTEN-/- melanomas. Representative IHC images and flow cytometry dot plots are provided. D. Serial tissue biopsy and qrt-PCR analysis of anti-PD-1 ab-treated versus IgG isotype control-treated BRAF(V600E)-PTEN-/- melanomas. All data is mean +/- SEM. *P<0.05
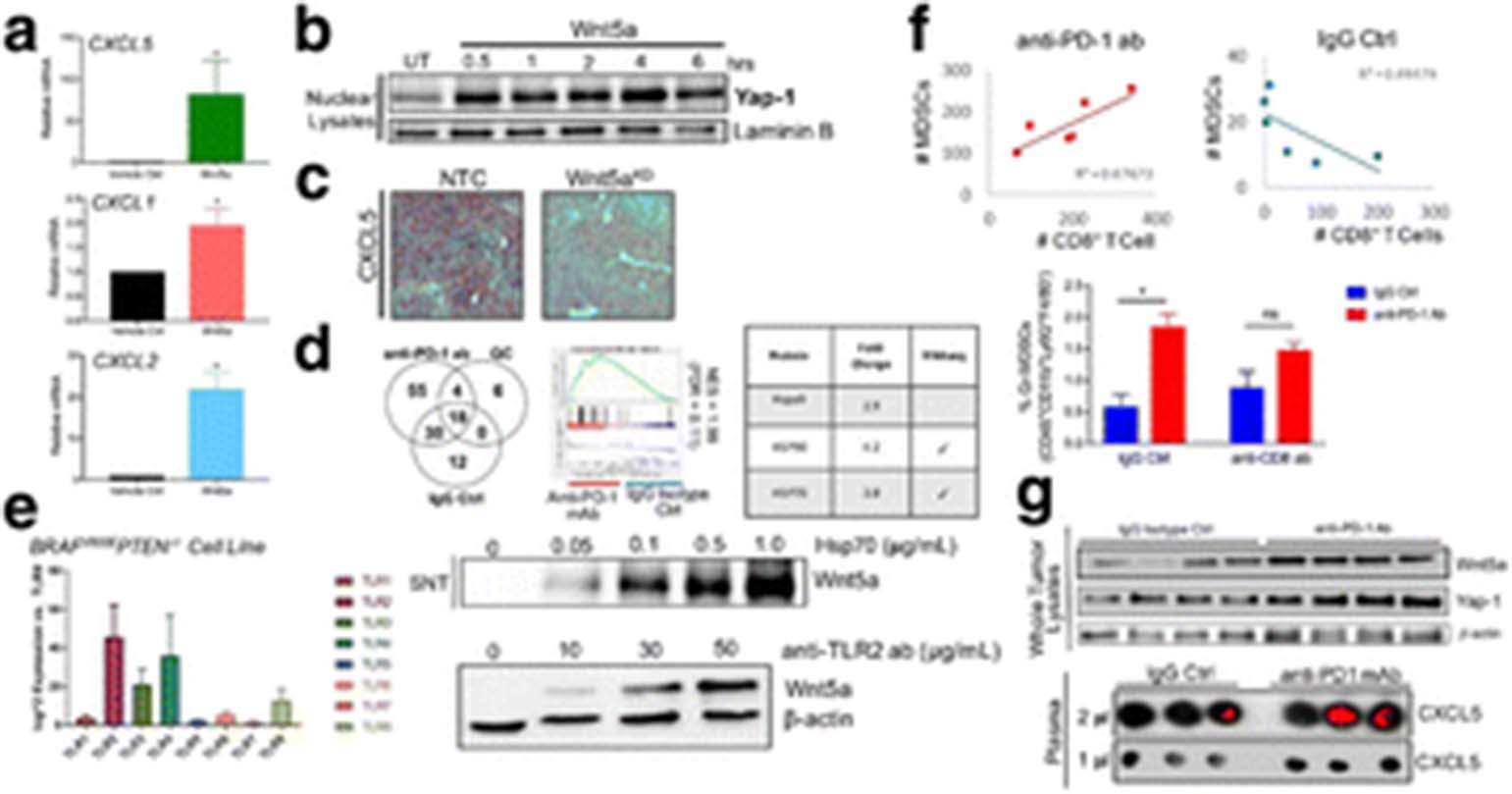
Induction of CXCL5 in Melanomas Undergoing Treatment with Anti-PD-1 Ab Therapy is Dependent Upon a Paracrine HSP-TLR-Wnt5a Signaling Pathway. A. Qrt-PCR analysis shows that Wnt5a stimulates the expression of several CXCR2 ligands in a BRAF(V600E)-PTEN-/- melanoma cell line. B. Western blot analysis of nuclear YAP1 levels in response to Wnt5a stimulation in a BRAF(V600E)-PTEN-/- melanoma cell line. C. Immunohistochemistry studies show that CXCL5 expression is significantly suppressed in Wnt5a-silenced BRAF(V600E)-PTEN-/- melanomas. D. SILAC-AHA metabolic labeling mass spectrometry-based differential secretome analysis and RNAseq differential gene expression analysis of BRAF(V600E)-PTEN-/- melanomas following treatment with anti-PD-1 ab vs IgG isotype control ab. Several cellular stress pathways identified as being upregulated in those melanomas undergoing anti-PD-1 ab therapy. Several heat shock proteins including HSP70 noted to be induced in anti-PD-1 ab-treated melanomas. E. BRAF(V600E)-PTEN-/- melanomas express elevated levels of TLR2 and TLR4 based on qrt-PCR. Western blot demonstrates that HSP70 and agonistic anti-TLR2 ab stimulates Wnt5a expression in the BRAF(V600E)-PTEN-/- melanoma cell line. F. top, CD8+ T cell levels and Gr-MDSC levels correlate significantly in BRAF(V600E)-PTEN-/- melanomas only when treated with anti-PD-1 ab therapy (p = 0.044). bottom, Ablation of CD8+ T cells diminishes the recruitment of Gr-MDSCs in response to anti-PD-1 ab therapy. G. top, Whole tissue Western blot analysis shows that anti-PD-1 ab therapy induces the upregulation of Wnt5a and YAP1 in BRAF(V600E)-PTEN-/- melanomas. bottom, Dot blot analysis shows that CXCL5 expression is upregulated in the serum of transgenic BRAF(V600E)-PTEN-/- mice undergoing treatment with anti-PD-1 ab therapy (red, blot density analysis). Representative blots shown. All data is mean +/- SEM. *P<0.05

The Wnt5a-CXCL5 Pathway Drives Gr-MDSC Recruitment in Response to anti-PD-1 Ab Therapy and Promotes the Development of anti-PD-1 Ab Adaptive Resistance. A. Flow cytometry analysis of Gr-MDSCs in Wnt5a-silenced BRAF(V600E)-PTEN-/- melanomas. B. Immunohistochemistry of Gr-MDSCS in Wnt5a-silenced BRAF(V600E)-PTEN-/- melanomas. C. Flow cytometry analysis demonstrates that Gr-MDSC recruitment in response to anti-PD-1 ab therapy is ablated in Wnt5a-silenced BRAF(V600E)-PTEN-/- melanomas. Representative flow cytometry dot plots provided. D. IFNg ELISPOT and tissue immunofluorescence show that melanoma Wnt5a silencing enhances the activation of TRP2-specific CD8+ T cells and promotes the expression of PD-L1. Representative images provided on the left. E. Wnt5a silencing sensitizes BRAF(V600E)-PTEN-/- melanomas to anti-PD-1 ab therapy. F. CXCL5 silencing suppresses Gr-MDSC recruitment and sensitizes BRAF(V600E)-PTEN-/- melanomas to anti-PD-1 ab therapy. Representative flow cytometry dot plots shown. All data is mean +/- SEM. *P<0.05

Inhibition of Wnt Signaling Suppresses Gr-MDSC Recruitment in Response to anti-PD-1 Ab Therapy and Augments the Efficacy of this Checkpoint Inhibitor in a Transgenic BRAF(V600E)-PTEN-/- Melanoma Model. A. Immunohistochemistry of CXCL5 and Gr-1 in BRAF(V600E)-PTEN-/- melanoma tissue undergoing treatment with anti-PD-1 ab therapy in the absence and presence of OMP-18R5 (vantictumab, anti-Fzd7 ab) or OMP-54F28 (ipafricept, Fzd8-Fc). B. Histologic analysis of resected lung tissues to enumerate lung metastases in the transgenic BRAF(V600E)-PTEN-/- melanoma model following the indicated treatment regimens. C. Flow cytometry analysis of BRAF(V600E)-PTEN-/- melanoma tissue following the indicated treatment regimens. CD45+CD3+CD8+ T cells and CD45+CD4+FoxP3+ regulatory T cell (Treg) populations quantitated and expressed as a CD8+ T cell : Treg ratio. D. TRP2 IFNg ELISPOT analysis performed using harvested splenocytes from transgenic BRAF(V600E)-PTEN-/- mice after undergoing the indicated therapy. Representative wells shown on the right. All data is mean +/- SEM. *P<0.05
Circulating Melanoma Cell (CMC) Qrt-PCR Analysis Demonstrates Enhanced CXCR2 Ligand Expression in Progressing Melanoma Patients Following anti-PD-1/anti-PD-L1 Ab Therapy. A. Schematic showing clinical specimen acquisition and analysis work flow. Samples are harvested from patients before and during checkpoint inhibitor immunotherapy. CMCs are enriched through centrifugation, analyzed by the photoacoustic flow cytometer, and subjected to integrated fluidic chip single cell qrt-PCR analysis of select target genes. Samples positive for CD45 or negative for both MART1 and S100 are excluded from analysis. Target gene expression normalized to Ubb expression levels. 6 CMCs analyzed per patient time point. B. Spider plot of target lesion tumor volume versus time for an advanced melanoma patient undergoing anti-PD-L1 ab therapy. Blood samples taken at week 0 prior to therapy and week 24. C. MART1 immunofluoresence analysis of captured CMCs. Brightfield images shown on left. D. CMC single-cell qrt-PCR analysis and quantitation of CXCR2 ligands and other target genes of interest in the same patient described in B. Data has been normalized to week 0 expression levels of respective target genes
P386 Tracking the dynamic response to vaccine prime/oncolytic boost immunotherapy identifies key mechanisms of immune resistance in metastatic ovarian cancer
AJ Robert McGray1, Raya Huang1, Sebastiano Battaglia1, Cheryl Eppolito1, Anthony Miliotto1, Mukund Seshadri1, Kyle Stephenson2, Brian Lichty2, Kunle Odunsi1
1Roswell Park Cancer Institute, Buffalo, NY, USA; 2McMaster University/Turnstone Biologics, Hamilton & Ottawa, Canada
Correspondence: Kunle Odunsi (bobmcgray@gmail.com)
Background
While spectacular responses to cancer immunotherapy have been observed, most responses are short-lived with ultimate tumor relapse. Using a vaccine prime/oncolytic boost immunotherapy, we sought to define robust biomarkers indicative of therapeutic response, response durability, as well as changes leading to tumor relapse in a pre-clinical mouse model of intraperitoneal metastatic ovarian cancer.
Methods
We utilized a heterologous prime/boost vaccine to treat ovarian cancer, combining an adjuvant-based vaccine with a novel oncolytic Maraba viral vector to generate robust anti-tumor T cell responses. To investigate treatment impact on both anti-tumor immunity and local events within the tumor, we combined analyses of immune cells within the tumor microenvironment with transcriptional profiling of solid tumors, and pre-clinical MRI to directly monitor changes in intraperitoneal tumors in response to therapy. This approach led to the identification/testing of candidate combination treatments with the potential to improve prime/boost vaccination.
Results
Heterologous prime/boost vaccination elicited robust tumor-specific CD8+ T cell responses, with high numbers of therapy-induced CD8+ T cells effectively trafficking to the tumor microenvironment. This approach greatly improved tumor control and long-term survival compared to vaccine priming alone, however the combination therapy was not curative and cellular analysis suggested that T cells within the tumor microenvironment were functionally suppressed. Tumor transcriptional profiling revealed that prime/boost vaccination led to induction of numerous inflammatory processes, with gene signatures consistent with CD8+ T cell infiltration, upregulation of multiple co-stimulatory/ immune checkpoint receptors, and induction of chemokine networks associated with lymphoid and myeloid cell trafficking. Based on these signatures, we tested candidate combination therapies to further enhance the impact of prime/boost vaccination, of which checkpoint blockade using αPD-1 produced the greatest benefit. Analysis of the response to prime/boost vaccination + αPD-1 by MRI revealed significant intratumoral inflammation after Maraba boosting followed by dramatic tumor regression, consistent with pseudo-progression. Tumors were effectively controlled, but ultimately relapsed. Using MRI guided assessment, treated tumors were subsequently isolated during all phases of this response and subjected to RNAseq analysis. The findings from these studies will be presented.
Conclusions
Our data suggest that interrogating the tumor microenvironment during the course of vaccine prime/oncolytic boost immunotherapy can identify candidate therapeutic targets, as well as key biomarkers of therapeutic response and/or intratumoral changes leading to tumor progression.
P387 IFNγ signaling in Tregs acts as a barrier to immunotherapeutic response
Abigail Overacre-Delgoffe1, Maria Chikina1, Rebecca Dadey1, Hiroshi Yano1, Erin Brunazzi1, Gulidanna Shayan2, William Horne1, Jessica Moskovitz2, Jay Kolls1, Cindy Sander2, Yongli Shuai1, Daniel Normolle1, John Kirkwood2, Robert Ferris2, Greg Delgoffe2, Tullia Bruno2, Creg Workman1, Dario Vignali1
1University of Pittsburgh, Pittsburgh, PA, USA; 2University of Pittsburgh Cancer Institute, Pittsburgh, PA, USA
Correspondence: Abigail Overacre-Delgoffe (overacre@pitt.edu); Dario Vignali
Background
Checkpoint blockade (anti-PD1/PDL1) has displayed striking, durable responses in cancer patients; however, many still succumb to disease. The underlying reasons for this lack of response remain obscure. Therefore, identifying resistance mechanisms to current immunotherapies is critical. While regulatory T cells (Tregs) are required to maintain immune homeostasis, they are also a major barrier to the anti-tumor immune response, and as such, are an attractive therapeutic target. We have previously shown that intratumoral Treg stability is reliant on the Neuropilin-1 (Nrp1):Semaphorin-4a axis. In its absence, Tregs become fragile and secrete IFNγ while maintaining Foxp3 expression [1,2]. However, 1) whether Treg fragility is required for response to immunotherapy, and 2) the underlying Treg-mediated mechanisms of immunotherapeutic resistance in mice and patients remain unclear.
Methods
We utilized syngeneic tumor models, including MC38 (anti-PD-1 sensitive) and B16.F10 (anti-PD-1 resistant) in both Foxp3Cre-YFP and Ifngr1L/LFoxp3Cre-YFP mice. Mice were treated with various immunotherapies to assess the role of IFNγ signaling by Tregs in mediating resistance. In addition, we are assessing the direct effect of IFNγ in vivo through the use of novel delivery mechanisms. Lastly, we are determining the impact of Treg fragility on patient response rate after PD-1 blockade in metastatic melanoma and head and neck squamous cell carcinoma.
Results
Using Foxp3Cre-YFP and Ifngr1L/LFoxp3Cre-YFP mice, we found that response to PD-1 blockade required IFNγ-induced Treg fragility, as Ifngr1L/LFoxp3Cre-YFP mice treated with anti-PD1 were completely resistant to therapy. In addition, PD-1 blockade led to increased IFNγ secretion by Tregs, an effect that was absent in Ifngr1L/LFoxp3Cre-YFP mice. Furthermore, pre-treatment of tumor-derived Tregs with IFNγ led to significantly reduced suppressive function in both mouse and human Tregs. We are currently extending our studies to patients treated with anti-PD1 in order to determine whether Treg fragility is associated with immunotherapeutic response.
Conclusions
Overall, we have shown that IFNγ-induced Treg fragility is required for anti-PD1 response and that IFNγ leads to reduced Treg suppressive capacity while maintaining Foxp3 expression. These studies uncover a novel potential resistance mechanism to immunotherapy, and provides an avenue to target Tregs selectively in the tumor microenvironment to bolster current immunotherapies.
References
1. Delgoffe GM, et al. Stability and function of regulatory T cells is maintained by a neuropilin-1-semaphorin-4a axis. Nature. 2013; 501: 252-256.
2. Overacre-Delgoffe AE, et al. Interferon-γ Drives Treg Fragility to Promote Anti-tumor Immunity. Cell. 2017. 169. 1130-1141.
P388 Neutrophil-rich inflammation and PD1/PD-L1 inhibitor resistance in advanced non-small cell lung cancer patients
Wungki Park1, Sandra Algaze2, Vaia Florou1, Diana Saravia1, Deukwoo Kwon1, Gilberto Lopes1
1University of Miami, Miller School of Medicine, Sylvester Comprehensive Cancer Center, Miami, FL, USA; 2University of Miami, Miller School of Medicine, Miami, FL, USA
Correspondence: Gilberto Lopes (wungkipark@gmail.com)
Background
The pro-tumoral role of neutrophils in chronic inflammation in carcinogenesis and metastasis has been well-reported in the animal models in the literature.[1] However, the role of neutrophil-to-lymphocyte ratio (NLR) and the acquired resistance to PD1/PD-L1 inhibitors (PD1/PD-L1i) in advanced non-small cell lung cancer patients (NSCLC) has not been described.
Methods
We retrospectively evaluated 115 advanced NSCLC patients who received PD-1 and PD-L1 inhibitors as their second-line therapy after platinum-based chemotherapy. Two groups were defined: Progressors (P) as patients who had radiographic and clinical progression after at least 2 months of treatment and Clinical Durable Responders (CDR) as patients with at least treatment duration of 6 months and ongoing response. Patients with baseline NLR > 10 were excluded. We performed paired-samples T test to analyze the difference of each patient’s NLR at baseline and at progression in P group. In CDR group, the difference of each patient’s NLR at baseline and at censored time while receiving ongoing PD-1/PD-L1i above 6 months were compared. Mann-Whitney test was performed to compare individual patient’s NLR at baseline and at progression with the presence of bone metastasis.
Results
There were 72 patients in P group and 43 patients in the CDR group. Total 82 patients received nivolumab, 25 patients received pembrolizumab, and 8 patients received atezolizumab. The duration of clinical response in P ranged from 2 to 17.4 months and 6.5 to 23.8 months in CDR group. The mean NLR at baseline in P group was 4.89 and 6.59 at progression. Patients in P group had statistically significant increases of NLR at progression with mean increase of 1.69. The 95% confidence interval was 3.04 to 0.35 (p-value = 0.01). However, there was no statistical difference between the NLR at baseline and censored time while receiving ongoing treatment in CDR group (95% C.I.: -1.90 - 0.822, p-value=0.428). Bone metastasis did not correlate with the degree of NLR. (p=0.56) Patients who received corticosteroid at the time of progression were not included in the study.
Conclusions
Persistent increase of NLR may represent unfavorable pro-tumoral inflammation and can represent unfavorable peripheral balance of PD-1 and PD-L1 interplay in myeloid-effector T cell immune synapse. This may serve as acquired resistance mechanism of PD-1/PD-L1i and warrants further investigation.
References
1. Fridlender ZG, Sun J, Kim S, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: "N1" versus "N2" TAN. Cancer cell. 2009;16:183-194.
P389 Aurora kinase inhibition enhances the efficacy of immunotherapy
Simone Punt1, Shruti Malu2, Rina Mbofung1, Jodi McKenzie1, Zhe Wang3, Leila Williams1, Jie Qing Chen1, Sourindra Maiti1, Trang Tieu1, Weiyi Peng1, Chengwen Liu1, Chunyu Xu1, Marie-Andrée Forget1, Cara Haymaker1, Jahan Khalili4, Nikunj Satani1, Florian Muller1, Laurence Cooper5, Jason Roszik1, Rodabe Amaria1, Chantale Bernatchez1, Timothy Heffernan1, Patrick Hwu1
1The University of Texas MD Anderson Cancer Center, Houston, TX, USA; 2The University of Texas MD Anderson Cancer Center (current: Eli Lilly and Company), Indianapolis, IN, USA; 3The University of Texas MD Anderson Cancer Center (current: The Journal of Experimental Medicine), New York, NY, USA; 4The University of MD Anderson Cancer Center (current: Personal Peptides LLC), Seattle, WA, USA; 5The University of Texas MD Anderson Cancer Center (current: Ziopharm Oncology, Inc.), Houston, TX, USA
Correspondence: Simone Punt (s.vd.garde@outlook.com)
Background
Immunotherapy has improved clinical outcomes for a subpopulation of melanoma patients, but over half of treated patients develop primary or secondary resistance. Current efforts are thus focused on developing combinatorial strategies involving immunotherapy and other treatment modalities, such as targeted therapy, to improve clinical outcomes. We aimed to identify factors that regulate the tumor cell response to T cell mediated cytotoxicity, with the goal to develop more efficacious treatment combinations.
Methods
A high-throughput screen of 850 bioactive compounds was used to identify proteins/pathways that confer resistance to T cell mediated cytotoxicity (Fig. 1A). A comboscore was calculated for apoptosis (cleaved caspase-3) induced by combined compound and T cell treatment compared to either treatment alone (Fig. 1B). A library of 576 ORF was subsequently used to identify proteins that induce resistance to T cell mediated cytotoxicity. The genes with the lowest comboscore in the ORF screen were analyzed using Nanostring on 23 samples from adoptive cell therapy (ACT) treated melanoma patients. One of the hits was further validated using in vitro and in vivo assays.
Results
After challenging melanoma cell lines with the compounds and autologous TILs, Aurora kinase inhibitors (AURKi) obtained a high comboscore. Two Aurora kinase inhibitors were validated to synergize with T cell mediated cytotoxicity in multiple melanoma cell lines (example of enhanced cytotoxicity in one cell line shown in Fig. 1C). Complementary to this finding, Aurora kinase A (AURKA) and AURKB overexpression induced resistance to T cell cytotoxicity. Additionally, patients who did not respond to ACT expressed higher levels of AURKA compared to responding patients (p=0.04), further demonstrating the relevance of Aurora kinase in immune suppression. The combination of AURKi AZD1152 and anti-CTLA4 significantly reduced tumor growth (p<0.05) and enhanced survival (p<0.01) as compared to either therapy alone in an MC38/gp100 in vivo model. Ongoing studies are now aimed at identifying the molecular mechanisms by which AURKi enhance T cell mediated cytotoxicity.
Conclusions
Aurora kinase inhibition may not be cytotoxic per se, but we showed that AURKi treatment sensitizes tumor cells to T cell mediated cytotoxicity and enhances the efficacy of immunotherapy. The combination of AURKi treatment and immunotherapy may thus induce an effective and durable response in melanoma patients, making Aurora kinase inhibitors a promising combination strategy with immunotherapy, especially for patients who do not respond to immunotherapy alone.

Aurora kinase inhibitors were identified to enhance T cell mediated cytotoxicity. (A) Patient derived melanoma tumor cells were incubated with one of 850 different compounds per well in triplicate in a high-throughput screen. After 24 hours, the compound was washed off and the tumor cells were co-cultured with autologous TILs for three hours, followed by intracellular staining for cleaved caspase-3. The percentage caspase-3 positive tumor cells is used as a read-out for the amount of apoptosis among DDAO stained tumor cells. (B) The comboscore is calculated to assess the enhanced cytotoxicity of combined compound and TIL treatment over either treatment alone. (C) Mel2549 cells were treated with 2uM AZD1152 or DMSO, TIL at a 3:1 E:T ratio or the combination of AZD1152 and TIL. Four other patient derived melanoma cell lines showed similar enhanced cytotoxicity for the combination treatment
P390 Combination radiotherapy and αOX40/αCTLA-4 immunotherapy reverses anergy and prevents development of functional exhaustion within tumor-specific CD8 T cells
Joshua Walker1, Melissa Kasiewicz2, William Redmond2
1Oregon Health & Science University, Portland, OR, USA; 2Providence Cancer Center, Portland, OR, USA
Correspondence: William Redmond (walkejos@gmail.com)
Background
The immunosuppressive tumor microenvironment and chronic T cell stimulation that occurs in the presence of cancer results in CD8 T cell dysfunction that has been difficult to reverse with immunotherapy (IT) alone. This dysfunction can be separated into two categories of anergy and exhaustion. Anergy being a poor proliferative response to stimulation after initial priming with inadequate costimulation or suboptimal T cell receptor ligation, and functional exhaustion being the inability of T cells to kill or produce effector cytokines in response to stimulation. We hypothesized the addition of ionizing radiotherapy (RT) to IT with agonist αOX40 & blocking αCTLA-4 antibodies would reverse tumor associated CD8 T cell anergy and exhaustion.
Methods
We utilized a model of CD8 T cell anergy in which transgenic, OVA-specific, OT-I CD8 T cells are adoptively transferred into centrally tolerant POET-1 mice bearing OVA expressing tumors. After development of anergy/exhaustion, 21 days after transfer, animals were treated with a single 20 Gy fraction of RT, αOX40/αCTLA-4, or combined IT/RT. To assess anergy/exhaustion in endogenous CD8 T cell responses, CT26 colon carcinoma bearing BALB/c mice were treated with IT, RT, or combined IT/RT either 10 or 17 days after tumor implantation. The frequency/function of tumor-specific (OT-I or AH1) CD8 T cells was monitored in blood, lymph node (LN), and tumor infiltrating lymphocytes (TIL). Reporter Nur77-GFP-OT-I CD8 T cells expressing green fluorescent protein in response to TCR ligation were used to measure TCR signaling following IT/RT.
Results
We demonstrate OT-I cells become anergic/exhausted and produce few effector cytokines 21 days after adoptive transfer. Combination therapy with IT/RT resulted in increased TCR stimulation (measured by GFP expression) and reversed anergy (measured by proliferation) within OT-I T cells in blood, LN, and TIL compared to IT or RT alone. Combination therapy was able to reverse functional exhaustion (measured by cytokine production) within LN but not TIL in this model. In CT26 tumor bearing mice combination IT/RT significantly increased (up to 4-fold) the frequency of tumor-specific endogenous AH1 CD8 TIL and prevented development of functional exhaustion within TIL compared to IT or RT alone.
Conclusions
Combined ablative RT and IT with αOX40/αCTLA-4 results in increased CD8 TCR signaling, reversal of T cell anergy, and can prevent or reverse functional exhaustion in some cases. These novel results suggest the addition of RT to IT can effectively reverse tumor-associated T cell dysfunction where IT alone is insufficient and provide rationale for early phase clinical trial design.
P391 Non-conventional inhibitory CD4+Foxp3-PD-1hi T cells as a biomarker of immune checkpoint blockade activity
Roberta Zappasodi, Sadna Budhu, Matthew Hellmann, Michael Postow, Yasin Senbabaoglu, Yanyun Li, Cailian Liu, Hong Zhong, Billel Gasmi, Daniel Hirschhorn-Cymerman, Katherine Panageas, Taha Merghoub, Jedd Wolchok
MSKCC, New York, NY, USA
Correspondence: Jedd Wolchok (zappasor@mskcc.org)
Background
A significant proportion of cancer patients do not respond to immune checkpoint blockade therapy. To deepen our understanding of the mechanisms of action of this strategy, we explored the role of a subset of CD4+Foxp3-negative T cells expressing PD-1 (4PD1hi), which we found to be up-regulated after CTLA-4 blockade in association with limited response to treatment.
Methods
4PD1hi frequency was monitored by flow cytometry. Their function was tested in standard in vitro suppression assays and 3D collagen-fibrin gel killing assays. RNAseq gene expression analyses were performed on a Proton sequencing system at the MSK Genomics Core Facility.
Results
We observed that 4PD1hi accumulate intratumorally as a function of tumor burden in untreated tumor-bearing hosts. In addition, mouse and human circulating and intra-tumor 4PD1hi inhibit T-cell functions in a PD-1/PD-L1 dependent fashion. Interestingly, CTLA-4 blockade promotes intratumoral and peripheral 4PD1hi increases in a dose-dependent manner, while combination with PD-1 blockade mitigates this effect and significantly improves anti-tumor activity. In keeping with these observations, we found that patients have a significantly higher risk of death if high 4PD1hi levels persist after PD-1 blockade. Mechanistically, we provide evidence that mouse and human 4PD1hi resemble follicular-helper-T-cell(TFH)-like cells and that CTLA-4 blockade increases B-cell co-stimulatory potential in vivo and in vitro, pointing to 4PD1hi as a possible downstream effect of enhanced T-cell priming during CTLA-4 blockade.
Conclusions
These findings broaden our understanding of the incremental activity of combination checkpoint blockade. They also provide a new pharmacodynamic and prognostic biomarker that may inform the design of optimal combination schedules and checkpoint blockade dosage.
Microbiome
P392 Microbiome analyses in patients with previously treated, deficient DNA mismatch repair/microsatellite instability-high metastatic colorectal cancer treated with nivolumab ± ipilimumab: CheckMate 142
Scott Kopetz1, Vittorina Zagonel2, Michael J. Overman1, Ray McDermott3, Michael A. Morse4, Franklin L. Chen5, James Lee6, Rebecca A. Moss7, Lilan Ling8, Alexander Greenfield7, Danielle Greenawalt7, Z. Alexander Cao7
1MD Anderson Cancer Center, Houston, TX, USA; 2Istituto Oncologico Veneto IOV-IRCSS, Padova, Italy; 3St. Vincent's University, Dublin, Ireland; 4Duke University Medical Center, Durham, NC, USA; 5Novant Health Oncology Specialists, Winston-Salem, NC, USA; 6University of Pittsburgh School of Medicine UPMC, Pittsburgh, PA, USA; 7Bristol-Myers Squibb, Princeton, NJ, USA; 8Memorial Sloan-Kettering Cancer Center, New York, NY, USA
Correspondence: Scott Kopetz (chris.geraci@chrysalismedical.com)
Background
Microbiota may have a role in the carcinogenesis of colorectal cancer (CRC) through chronic low-grade inflammation and the influence of microbiota-derived metabolites on host metabolism and immune function.[1,2] Microbiome diversity was significantly different between patients with melanoma who responded to anti–PD-1 therapy and those who did not respond.[3] Here, we explore the relationship between microbiota and response to nivolumab ± ipilimumab in patients with deficient DNA mismatch repair/microsatellite instability-high (dMMR/MSI-H) CRC enrolled in CheckMate 142 (NCT02060188).
Methods
Patients received nivolumab 3 mg/kg Q2W or nivolumab 3 mg/kg + ipilimumab 1 mg/kg Q3W × 4 doses followed by nivolumab 3 mg/kg Q2W until discontinuation. 16S ribosomal RNA sequencing was performed on baseline stool samples. Alpha diversity and differential measures of operational taxonomic units were evaluated at each taxa level. Microbiota differences were compared between patients with an investigator-assessed best overall response (BOR) of partial response (PR) vs those with a BOR of progressive disease (PD). Due to small patient numbers, those treated with nivolumab and nivolumab + ipilimumab were combined for the analysis.
Results
Baseline stool samples were collected from 72 patients. Among these patients, BORs of PR were achieved in 25, stable disease in 31, and PD in 14; 2 were not evaluable. Alpha diversity was not statistically different between patients with a BOR of PR vs those with PD. The abundance of Actinobacteria Micrococcaceae was 2.7-fold lower in patients with a BOR of PR compared with patients with a BOR of PD (P=0.0009; Fig. 1); significant differences were also observed in the genus Rothia (2.7-fold lower in patients with a PR; P=0.0007). These results remained significantly different (P=0.04) after corrections for false discovery rate. Analyses on additional microbiota will be presented.
Conclusions
Patients with dMMR/MSI-H CRC who achieved a BOR of PR with nivolumab ± ipilimumab treatment were found to have a lower abundance of Actinobacteria Micrococcaceae, and in particular, the genus Rothia, compared with those with a BOR of PD. These results suggest that there may be differences in the gut microbiota of patients with dMMR/MSI-H CRC who respond to treatment with checkpoint inhibitors vs those who progress. Additional analyses investigating correlations with tumor immune phenotype and microenvironment, and other clinical parameters, are warranted.
References
1. Oke S, Martin A. Therap Adv Gastroenterol. 2017;10:417-428.
2. Gao R et al. Eur J Clin Microbiol Infect Dis. 2017; 36 :757-769.
3. Sivan A et al. Science. 2015; 350:1084-1089.
See text for description
P393 Association of microbiome to nivolumab response in metastatic renal cell carcinoma (mRCC)
Manuel Maia1, Valeriy Poroyko1, Hae Won2, Paulo Bergerot1, Lorena Almeida1, Nazli Dizman1, JoAnn Hsu1, Jeremy Jones2, Ravi Salgia1, Sumanta Pal1
1City of Hope Comprehensive Cancer Center, Duarte, CA, USA; 2Beckman Research Institute, City of Hope Comprehensive Cancer Center, Duarte, CA, USA
Correspondence: Manuel Maia (mcaitano@coh.org)
Background
Checkpoint inhibitors (CPI) have improved clinical outcomes in various cancer types, including mRCC [1]. Predictive factors of benefit from CPIs are not well defined. Relationship of the microbiome to benefit from CPIs has been suggested in preclinical studies [2]. Here we provide the first evidence of association between microbiome composition and response to nivolumab in mRCC patients (pts).
Methods
Stool samples were prospectively collected from pts with mRCC at 3 time points relative to nivolumab treatment (baseline, week 4 and week 12) and used to assess gut microbiota composition in two different groups: responders (R; including complete/partial response and stable disease) and non-responders (P; primary progression). In short, microbial DNA was extracted, 16s rRNA gene tags (v4) were generated by PCR amplification and sequenced using MiSeq (Illumina). Sequence reads were processed by Mothur software, assembled in operational taxonomic units (OTUs), taxonomically annotated to the level of genus and used to construct Bray-Curtis dissimilarity matrices. The similarity of samples was visualized by principal coordinates analysis (PCoA) and further confirmed by k-means clustering (k=2) and ANOSIM tests. Differentially abundant taxa were determined by METASTATS and compared between R and P.
Results
Of 11 pts, 9 were evaluable for response. In this study, 25,304 OTUs were attributed to 165 genera from 8 phyla. PCoA analysis reveals that the first two principal coordinates can explain 49.2% of data set variation. Subsequent k-means clustering shows an almost complete separation of microbiota in R and P groups. The produced clusters almost perfectly aligned with R and P groups, although ANOSIM of this separation was not significant (p=0.07). The analysis of microbiota membership in P and R groups revealed 4 differentially abundant taxonomic units at the genus level (Fig. 1), with 2 present above 1% abundance. Namely, Roseburia spp and Faecalibacterium spp were significantly elevated in R as compared to P (p<0.05). Notably, no significant differences were noted in Bifidobacterium spp or Bacteriodes spp in R and P (P=0.15 for both).
Conclusions
Our results are the first to associate specific microbial genera to nivolumab response in mRCC and warrant confirmation in larger series. Furthermore, manipulation of the stool microbiome as a means of modulating nivolumab response should be investigated.
References
1. Motzer RJ, et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med.2015: 1803-1813.
2. Vetizou M,et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. 2015; 350: 1079-84.
Relative abundance of selected genera demonstrating significant differences between responders (R) and progressors (P). P: Primary progression group. R: Responders group
P394 Commensal microbiota associated with anti-PD-1 efficacy in metastatic melanoma patients
Vyara Matson, Jessica Fessler, Riyue Bao, Tara Chongsuwat, Yuanyuan Zha, Maria-Luisa Alegre, Jason Luke, Thomas Gajewski
University of Chicago, Chicago, IL, USA
Correspondence: Thomas Gajewski (vmatson@uchicago.edu)
Background
Anti-PD-1-based immunotherapy is having a major impact on cancer outcomes, but only a subset of patients respond favorably. To predict response and ultimately improve therapeutic efficacy, it is of interest to identify variables that cause this inter-patient heterogeneity. Pre-clinically, commensal bacterial species have been demonstrated to modulate spontaneous anti-tumor immunity and the outcome of immunotherapy and chemotherapy in mice (1-3). Here, we examine the association between gut microbiota composition and response to anti-PD-1 immunotherapy in patients with melanoma.
Methods
Stool samples were collected from 42 metastatic melanoma patients prior to immunotherapy and microbial composition was analyzed through an integration of 16S rRNA gene sequencing, metagenomic shotgun sequencing, and quantitative PCR for selected bacteria. Clinical response was determined using Response Evaluation Criteria In Solid Tumors (RECIST) version 1.1. Germ-free mice were colonized with stool from responding and non-responding patients, and tumor growth kinetics and anti-tumor immune responses were evaluated.
Results
There were 16 responders and 26 non-responders. There was a significant association between the commensal microbiota composition and clinical response. Sixty-two operational taxonomic units (OTUs) assigned from the 16S rRNA gene sequencing dataset were differentially abundant in responders relative to non-responders. Forty-three of these OTUs were assigned potential species-level identities, based on metagenomics sequencing of the same stool samples. Selected responder-associated species, including Bifidobacterium longum, Akkermansia muciniphila, Collinsella aerofaciens, and Enterococcus faecium were also confirmed by quantitative PCR. Reconstitution of germ-free mice with fecal material from a responding patient led to improved tumor control, augmented T cell responses, and a distinct microbiota compared to a non-responding patient.
Conclusions
Our results indicate a correlation between the abundance of certain gut bacteria and clinical response to anti-PD-1 immunotherapy in cancer patients, and suggest a mechanistic impact of the commensal microbiota on anti-tumor immunity
References
1. Sivan A, Corrales L, Hubert N, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science. 2015; 350: 1084-1089.
2. Daillere R, Vétizou M, Waldschmitt N, et al. Enterococcus hirae and Barnesiella intestinihominis Facilitate Cyclophosphamide-Induced Therapeutic Immunomodulatory Effects. Immunity. 2016; 45: 931-943.
3. Iida N, Dzutsev A, Stewart CA, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science. 2013; 342 : 967-970.
P395 DNA repair genetic aberrations and tumor mutation burden in biliary tract cancers
Reham Abdel-Wahab1,2, Rachna Shroff1, Jean Nicolas Vauthey1, Eugene Jon Koay1, Siraj Ali3, Lawrence Kwong1, Mingxin Zuo1, Asif Rashid1, Milind Javle1
1MD Anderson Cancer Center, Houston, TX, USA; 2Assiut University Hospital, Assiut Faculty of Medicine, Assiut, Egypt; 3Foundation Medicine, Cambridge, MA, USA
Correspondence: Milind Javle (raali@mdanderson.org)
Background
Immune checkpoint inhibitors (ICPI) has been approved for several cancers. The association between tumor mutational burden (TMB) and ICPI has been previously studied and high TMB is associated with prolonged progression free survival (PFS) after ICPI. DNA repair gene aberrations (GAs) including those leading to microsatellite instability (MSI) is associated with response to ICPI. Data regarding this in biliary tract cancers (BTC) are limited.[1]
Methods
A comprehensive genomic profiling of 422 formalin-fixed, paraffin-embedded tumor tissue from BTC patients, including 92 gall bladder cancer and 330 cholangiocarcinoma, was performed using next generation sequencing. We included 20 DNA repair genes that were previously described.[1] These included direct DNA repair genes (ATM, ATR, BRCA1, BRCA2, FANCA, FANCD2, MLH1, MSH2, MSH6, PALB2, POLD1, POLE, PRKDC, RAD50, SLX4) and caretaker genes that induce genomic instability (BAP1, CDK12, MLL3, TP53, BLM). TMB was calculated in 205 samples by counting the number of mutations across a 1.25Mb region and classified into three groups; low (< 6 mutation/Mb), intermediate (6 – 19 mutation/Mb), and high (≥20 mutation/Mb). We assessed MSI status by a computational algorithm examining 114 intronic homopolymer loci.
Results
Direct DNA repair genes were identified in 61 BTC tumors (14.5%) including GAs in ATM (5.5%) or BRCA2 (2.1%). Majority of the tumors had GAs in caretaker genes (42.9%), predominately in TP53 (33.4%), and BAP1 (9.5%). However, FANCD2, POLD1, POLE, PRKDC, RAD50, SLX4, BLM mutations were not identified in our cohort. TMB was evaluated in 205 patients. Among patients with direct DNA repair GAs, 20 out of 35 (57.2%) had intermediate or high TMB as compared with 47 out of 170 (27.7%) patients without direct DNA repair GAs (P < .0001). Patients with caretaker GAs also had significantly higher TMB [intermediate and high (48.4%)] than patients without these mutations (P < .0001). MSI was determined in 88 patients and classified as stable, ambiguous, and high. All patients with MSI high had direct DNA repair GAs (P < .0001).
Conclusions
DNA repair GAs occur at a relatively high frequency in BTC and are associated with higher TMB and MSI. Future clinical trials targeting this subgroup with ICPI are warranted.
References
1. Chae YK, Anker JF, Carneiro BA, Chandra S, Kaplan J, Kalyan A, Santa-Maria CA, Platanias LC, Giles FJ. Genomic landscape of DNA repair genes in cancer. Oncotarget. 2016;7(17):23312-21.
P396 Evolving immune response in lung squamous carcinogenesis
Celine Mascaux1, Mihaela Angelova2, Angela Vasaturo2, Jennifer Beane3, Kahkeshan Hijazi4, Geraldine Anthoine5, Karen Willard-Gallo5, Annick Haller6, Vincent Ninane7, Arsène Burny5, Jean-Paul Sculier8, Avrum Spira9, Jérôme Galon2
1Marseille Early Phases Cancer Trials Center CLIP, Marseille, France; 2Laboratory of Integrative Cancer Immunology, Paris, France; 3Boston Univ. School of Medicine, Boston, MA, USA; 4Research Center for Modeling and Simulation, Islamabad, Pakistan; 5Institut Jules Bordet, Université Libre de Bruxelles, Brussels, Belgium; 6University Chidren's Hospital Queen Fabiola, Brussels, Belgium; 7CHU Saint-Pierre - Service de Pneumologie, Brussels, Belgium; 8Institut Jules Bordet, Centre des Tumeurs de l'Université Libre de Bruxelles, Brussels, Belgium; 9Boston University Medical Center, Boston, MA, USA
Correspondence: Jérôme Galon (mihaela.angelova@crc.jussieu.fr)
Background
Molecular characterization of pre-invasive and invasive bronchial lesions can provide insights into the mechanisms behind genesis and development of squamous cell carcinoma, revealing potentially promising new biomarkers for early detection and treatment of lung cancer.
Methods
At 8 different morphological stages of lung squamous carcinogenesis, from normal, to low grades dysplasia, high grades dysplasia, to carcinoma, fresh frozen human bronchial biopsies (n=122) were subjected to gene expression profiling (Agilent microarrays) from 77 patients. A linear mixed-effects model was applied to infer modules of gene co-expression, adjusted for smoking status, gender, and cancer history as fixed effects, and patient as a random effect. Immune cell-specific gene signatures were used to characterize the immune response through the different development stages.
Results
The gene expression alterations could distinguish four groups of successive stages of development. There were eight modules of co-expressed genes with different expression patterns across the stages: up- or down-regulation among the early or late steps, linear increase or decrease in expression, and a biphasic pattern. Transitioning to early grade, there was a depletion of immune gene expression. However, at the transition to high-grade dysplasia, there was a significant up-regulation of immune/inflammatory response genes, representing T helper 1, myeloid cells, as well as co-inhibitory and co-stimulatory molecules.
Conclusions
At a critical stage of carcinogenesis there is a significant modification of the immune response. The up-regulation of immune genes in high-grade suggests a major role of the surrounding microenvironment thorough adaptive and innate immune response, but also a tumor versus host immunosuppressive response before tumor invasion across the basal membrane. This analysis may identify promising new immune signatures of carcinogenesis, and may provide novel therapeutic targets for lung cancer.
P397
Withdrawn
P398 Tumor mutation load assessment of FFPE samples using an NGS based assay
Ruchi Chaudhary1, Asha Kamat1, Warren Tom1, Iris Casuga1, Dinesh Cyanam2, Dumitru Brinza1, Janice Au-Young1, Fiona Hyland1
1Thermofisher Scientific, South San Francisco, CA, USA; 2Thermofisher Scientific, Ann Arbor, MI, USA
Correspondence: Asha Kamat (asha.kamat@thermofisher.com); Ruchi Chaudhary
Background
Background – Tumors with high mutation load respond to therapy with immune checkpoint inhibitors. Hence high mutation load of a tumor can act as a predictive biomarker to immuno-therapy. We developed an Ion S5 Sequencing System assay to determine the mutation status of solid tumors.
Methods
Methods: Our assay uses an FFPE sample, utilizing an Ampliseq based target panel that covers 1.7 Mb of genomic DNA including 409 genes. The assay does not require matched normal sample to estimate the mutation status of a tumor.
Results
Results: Our panel exhibits >90% sensitivity and >95% specificity in distinguishing between high and low mutation burden samples. Using a variety of public annotation sources, we have successfully eliminated approximately 98% of germline variants. Matched tumor-normal analyses on 9 lung and colorectal samples suggested that our single sample analysis on tumor samples detects mutation load with strong correlation (r=0.9358) with tumor-normal analysis. On engineered control from Acrometix, mutation load count consistently comprise true mutations with positive predictive value of 89%. To assess reproducibility, we compared the mutation load in library replicates for a cohort of 10 samples (FFPE and fresh frozen tumors, Acrometrix control, and NIST cell-lines) and observed high correlation (r=0.9948).
Conclusions
We have developed a single tumor-only assay for accurate estimation of tumor mutation burden, useful for cancer immuno-therapy research and development.
P399 Rationally improving T cell-mediated cancer immunotherapy using Sleeping Beauty mutagenesis to identify novel drug targets
Laura Rogers, Adam Dupuy, George Weiner
University of Iowa, Iowa City, IA, USA
Correspondence: Laura Rogers (laura-rogers@uiowa.edu)
Background
T cells have amazing potential to eliminate tumor cells throughout the body, evidenced by the clinical success of immunotherapies designed to enhance T cell function like immune checkpoint blockade and chimeric antigen receptor T cell (CAR-T) therapies. Expanding success to a broader number of patients is a top priority in the field. T cell infiltration into tumors appears to be an important prerequisite for the success of both immune checkpoint blockade and CAR-T therapies. Thus, increasing T cell infiltration into tumors could have beneficial therapeutic impact.
Methods
We have designed a forward genetic screen to identify T cell genes that contribute to intratumoral T cell accumulation using Sleeping Beauty (SB) transposon insertional mutagenesis in T cells. Our systematic screen approach allows us to evaluate both loss- and gain-of-function mutations across the entire genome in immunocompetent melanoma and lymphoma models that preserve the complexity of the tumor microenvironment. Importantly, the vast majority of genes we identified were not previously known to be involved in tumor-associated T cell biology, and likely would not have been considered as drug targets to enhance immunotherapy. These genes have the potential to modify important T cell functions including trafficking to the tumor, clonal expansion, and sustained viability once inside the tumor.
Results
We identified 312 tumor-enriched genes that were mutated in tumors, but not the spleen, of individual mice. Twenty of these were detected in more than one mouse, representing strong gene candidates for validation. We demonstrate that one gene candidate is functionally associated with T cell response to activation signals. Specifically, CRISPR-mediated knockout of this gene in a murine T cell line resulted in cytokine production defects after T cell receptor stimulation. Additional screening has begun using other tumor models and anti-PD-1 therapy. Preliminary data from these indicate that few genes are conserved across tumor models and treatment groups.
Conclusions
We are currently investigating the role of candidate genes in intratumoral T cell accumulation and immunotherapy enhancement using an in vivo approach. Together, these experiments have the potential to expand our understanding of T cell infiltration into tumors and may provide previously unexplored strategies to rationally enhance immunotherapy efficacy.
P400 Evaluation of tumor genetics and microenvironment through next-generation sequencing (NGS)
Alex So, Shannon Kaplan, Joyee Yao, Shile Zhang, Aaron Wise, Kristina Kruglyak, Naomi O'Grady, Jeremiah Mcdole, Marina Bibikova
Illumina, San Diego, CA, USA
Correspondence: Alex So (alexsoas@gmail.com)
Background
The effectiveness of cancer immunotherapy is determined by the genetic basis of the patient’s tumor as well as the state of the tumor microenvironment. For instance, tumor mutation burden (TMB) and microsatellite instability (MSI) status correlate with patient response to checkpoint inhibitors [1]. Also, the presence of immune cells in the tumor microenvironment correlates with immunotherapy responsiveness. Thus, this project aims to develop a next-generation sequencing (NGS) workflow that reports various biomarkers that can be used to guide cancer immunotherapy selection. Here we present our data on using whole exome sequencing (WES) and whole transcriptome sequencing (WTS) to report human leukocyte antigen (HLA) sequence, TMB load, prioritized neoantigens, and presence of immune cells.
Methods
WES and WTS libraries were generated using Illumina’s TruSeq® Exome and RNA Access library preparation kits, respectively. The samples were paired-end sequenced (2x75bp) using HiSeq® 2000 and 2500 instruments.
Results
Here we demonstrate a workflow using tumor/normal WES and tumor-only WTS to determine MSI status and expressed TMB load. We also show data that WES can be used to haplotype HLA Class I genes with 97% accuracy. Using the HLA haplotype sequence, we computationally predicted putative neoantigens and experimentally validated a subset, as true binders to their respective major histocompatibility complex. In addition, we applied FRICTION, a novel algorithm for cell deconvolution, to WTS data extracted from bulk tumor to quantify the fraction of immune cells present in the tumor microenvironment. Our titration experiments demonstrated our method’s linearity in quantifying CD4+ T cells, CD8+ T cells, and CD19+ B cells in a variety of tissue backgrounds (median R2 > 0.97). Finally, we applied FRICTION to WTS data from melanoma samples extracted from different subjects and showed that our workflow could also quantify the fraction of immune cells in these samples when compared to quantitation by flow cytometry.
Conclusions
In summary, we demonstrate the utility of a NGS workflow that uses WES and WTS to report biomarkers relevant to immunotherapy and characterize the inflammation status of the tumor microenvironment.
References
1. Rizvi, NA, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2016;348:1-13.
Oncolytic Viruses and Intratumoral Therapies
P401 Intratumoral expression of IL12 using the ZVex® dendritic cell-targeting lentiviral vector exerts potent anti-tumor effects via induction of multiple immune effectors, including CD8 T cell responses
Tina C. Albershardt, Jardin Leleux, Rebecca S. Reeves, Jan ter Meulen, Peter Berglund
Immune Design, Seattle, WA, USA
Correspondence: Tina C. Albershardt (tina.albershardt@immunedesign.com)
Background
Interleukin 12 (IL12), produced by antigen-presenting cells, plays a pivotal role in the interplay between the innate and adaptive arms of the immune system. IL12 treatment has been shown to augment cytotoxic T lymphocyte and TH 1 responses and anti-tumor effects. However, its use as a systemic therapeutic agent is limited due to toxicity. Intratumoral administration of IL12 is therefore being explored as an alternative route. We have previously reported on the strong anti-tumor effects of IL12 delivered intratumorally using Immune Design’s dendritic cell-targeting lentiviral platform ZVex in various murine tumor models. Here, we evaluated the mechanism of action mediating the local and systemic immune responses induced by intratumoral ZVex/IL12 in different animal models.
Methods
Tumor cell lines (B16 melanoma, CT26 colon carcinoma, 4T1 breast cancer, A20 lymphoma, P815 mastocytoma, GL261 glioma) were implanted subcutaneously into mice. ZVex was engineered to express murine IL12 and administered as a single intratumoral injection into palpable tumors (50-100 mm3), typically at 7 days post-tumor inoculation. Mice were monitored twice weekly for tumor growth and survival. In lymphocyte depletion studies, anti-CD8, anti-CD4 or anti-NK1.1 antibodies were administered twice weekly, and their effect on immediate and long-term anti-tumor response was studied.
Results
A single intratumoral injection of ZVex/mIL12 induced complete tumor regression in B16, CT26, A20, GL261, and P815 models, delayed tumor growth in the 4T1 model. In mice depleted of either CD8 T, CD4 T, or NK cells, complete tumor regression was still observed in 13-40% of mice. However, only depletion of CD8 T cells together with CD4 T and/or NK cells completely abrogated the anti-tumor response, suggesting that CD8 T cells are required but not sufficient effectors in ZVex/mIL12-mediated anti-tumor control. Furthermore, depletion of CD4 T cells resulted in very pronounced vitiligo, demonstrating that intratumoral ZVex/mIL12 profoundly breaks self-tolerance, likely in the absence of regulatory T cells.
Conclusions
Taken together, the results demonstrate that intratumoral administration of IL12 induces complex protective cellular immune responses, requiring CD8 effector T cells for maximum efficacy.
P402 Chemotherapy enhances oncolytic recombinant poliovirus efficacy
Jeffrey Bryant1, Michael Brown2, Matthias Gromeier2
1Duke University, Durham, NC, USA; 2Duke University Medical Center, Durham, NC, USA
Correspondence: Jeffrey Bryant (jeffrey.bryant@duke.edu); Matthias Gromeier
Background
Oncolytic viruses are capable of direct tumor lysis along with the ability to activated pro-inflammatory programs that may contribute to the recruitment of adaptive immune effector responses. We use a recombinant poliovirus:rhinovirus hybrid, PVSRIPO, which has tropism for neoplastic cells, due to the ectopic expression of the poliovirus receptor, CD155, and a reliance on oncogenic signaling to initiate viral protein translation. PVSRIPO is currently in Phase-I/II clinical trials against recurrent glioblastoma multiforme (GBM). The first patients treated with PVSRIPO are radiographically tumor-free and clinically normal >52 months after single intratumoral infusion of PVSRIPO. On May 10th, 2016, the FDA/CBER granted Breakthrough Therapy Designation to PVSRIPO. A co-incidental finding emerging from our ongoing trial is remarkable durable, complete responses in patients upon treatment with chemotherapy following tumor progression after PVSRIPO administration.
Methods
This study used syngeneic mouse tumor models (TRAMPC2 and CT2A) to test the same chemotherapy combination used in the clinic. Both the clinical trial and mouse study use chemotherapy doses that induce severe lymphotoxic side effects which have been suggested to augment antitumor immunity when combined with an immunotherapy regimen. To mirror what is done in the clinic the established mouse tumors receive a single injection of PVSRIPO followed a week later by a single round of the alkylating chemotherapeutic drug Temozolomide (TMZ). Tumor growth and end point immune cells populations were monitored.
Results
This study demonstrates that this PVSRIPO TMZ combination can be tested in syngeneic mouse models resulting in both an increase in survival and a delay in tumor growth over both PVSRIPO and TMZ treatment alone. Our data also suggests that this combination induces an increase in memory and infiltrating Treg cell populations.
Conclusions
Ongoing work is being done to further validate these finding in multiple mouse tumor models including intra-cerebral brain tumors models and to determine the mechanisms contributing to the synergy seen resulting from the PVSRIPO and chemotherapy combination. These intriguing results suggest that the memory T cells seen after PVSRIPO chemotherapy combination may be responsible for the new anti-tumor immune response. However, chemotherapy also induced an increased tumor infiltrating inhibitory Treg cells. Future work will look at ways to enhance this positive memory T cell population while preventing intratumoral Treg cell accumulation.
P403 Intratumoral administration of a multigene construct by electroporation can effectively modulate anti-tumor response in a murine B16.F10 model
Shawna Shirley1, Anandaroop Mukhopadhyay1, Christoph Burkart1, Jocelyn Wright1, Robert Pierce2, Jean Campbell2, David Canton1
1Oncosec Medical Incorporated, San Diego, CA, USA; 2Fred Hutchinson Cancer Research Center, Seattle, WA, USA
Correspondence: David Canton (dcanton@oncosec.com)
Background
Immunomodulatory cytokines, such as interleukin-12 (IL-12) are attractive candidates for cancer immunotherapy. IL-12 is a pro-inflammatory cytokine with potent anti-tumor effects; however, systemic administration of IL-12 shows limited clinical efficacy and dose-associated toxicity. In preclinical and clinical studies, intratumoral (IT) delivery of IL-12 plasmid DNA by electroporation (EP) can provide a safe and effective alternative for efficacious dosing. To augment the effects of IL-12, we developed a DNA plasmid platform that allows for delivery of agents that modulate multiple immune pathways as well as tumor- or patient-specific neoantigens. Polycistronic IL-12 Immune Modulator plasmid (PIIM) is a single plasmid encoding IL-12, and a fusion of Flt3L to an antigen. Flt3L is a ligand that stimulates dendritic cell (DC) maturation and enhances antigen processing and presentation. The encoded antigen can be a viral or shared antigen, or a patient-specific neoantigen, which enables customization to patient populations, as well as providing an aid to monitoring antigen-specific immune response(s) that can be correlated to patient outcomes. Here we demonstrate the first functional characterization of PIIM.
Methods
Two PIIM constructs were created for functional characterization: PIIM-OVA (IL12~Flt3L-OVA) for mouse experiments, and PIIM-NYESO1 (IL12~Flt3L-NYESO1) for testing in human cells. The expression and functional activity of PIIM components were determined. Treated tumors and spleens were assessed for transcriptional changes by NanoString® and phenotypic changes by flow cytometry. Systemic effects of PIIM were assessed using a syngeneic two-tumor model of B16.F10 in which only one tumor received IT-PIIM-EP while the other contralateral lesion remained untreated.
Results
PIIM-OVA and PIIM-NYESO1 secrete functional IL-12p70, Flt3L-OVA and Flt3L-NYESO1 fusion proteins as assessed by ELISA, flow and cell-based assays. PIIM promotes DC maturation and antigen-specific T cell proliferation both ex vivo and in vivo. Hydrodynamic-based gene delivery of PIIM-OVA lead to splenomegaly and significantly increased splenic CD11c+ DCs. Furthermore, IT-PIIM-EP lead to generation of splenic OVA-specific CD8s and increased APM gene expression. When introduced intratumorally in a mouse two-tumor model, IT-PIIM-EP delays B16.F10 tumor growth in both treated and contralateral tumors compared to untreated controls resulting in increased overall survival.
Conclusions
PIIM represents a novel approach to cancer immunotherapy. A combination of functional immune modulators can be expressed locally in the tumor microenvironment that increase inflammatory infiltrate, enhances antigen presentation and produces a systemic T cell response specific to the antigen encoded on the plasmid. This customizable approach has the potential to improve therapeutic outcome by enhancing adaptive-immunity and addressing patient-specific neoantigens needs.
P404 Targeting T-cells to human cancer associated fibroblasts using an oncolytic virus expressing a FAP-specific T-cell engager
Brian Champion1, Joshua Freedman2, Rebecca Ashfield2, Margaret Duffy2, Alice Brown1, Leonard Seymour2, Kerry Fisher2
1PsiOxus Therapeutics Ltd, Abingdon, United Kingdom; 2Oxford University, Oxford, United Kingdom
Correspondence: Brian Champion (brian.champion@psioxus.com)
Background
Enadenotucirev (EnAd) is a chimeric oncolytic group B adenovirus with potent and selective anti-tumor activity against a range of epithelial cancer cells, with a blood stability profile that enables systemic dosing and has been administered intravenously to over 100 cancer patients. As an approach to immunogene therapy targeting stromal rich tumors, we have created transgene-modified variants of EnAd expressing a bi-specific T-cell engager (BiTE) molecule recognizing human fibroblast activating protein (FAP) on cancer associated fibroblasts (CAFs) and CD3 on T-cells. CAFs play a pivotal role in the development of solid carcinomas by facilitating invasion, coordinating angiogenesis and establishing and maintaining an immune suppressive microenvironment in the tumor stromal tissue. We hypothesize that production of BiTE proteins by virus infected tumor cells could be an effective way of modifying the stromal microenvironment to drive effective anti-tumor immunity and would bypass delivery and safety issues related to systemic dosing of BiTE proteins.
Methods
Bi-specific T-cell engager constructs comprising linked ScFv antibodies specific for human FAP and CD3 were designed and used to generate EnAd viruses expressing the BiTE such that transgene expression was under the control of either a CMV promoter or the virus major late promoter to allow broad or tumor-selective expression, respectively. Tumor and fibroblast cell lines, together with control BiTE proteins and BiTE-encoding viruses, were used to evaluate initial activities of the FAP-BiTE viruses prior to testing with freshly isolated malignant peritoneal ascites from patients with advanced cancer.
Results
We have shown that the viruses selectively express and secrete functional FAP-specific BiTEs following infection of tumor cell lines. In co-culture with FAP+ fibroblasts and PBMC-derived T cells this leads to T cell-activation and cytotoxicity towards the fibroblasts. Similarly, infection of primary malignant cancer ascites with FAP-BiTE-encoding viruses led to polyclonal activation of endogenous T-cells and depletion of FAP+ cells from the cultures. Unlike activation of T-cells with anti-CD3/CD28, FAP-BiTE activation was also shown to be effective even in the presence of immunosuppressive (cell-free) ascitic fluid samples added to the cultures.
Conclusions
In this study, we have shown that CAFs can be effectively targeted for T-cell mediated destruction by a FAP-BiTE transgene-bearing oncolytic virus, with an associated strong activation of endogenous T-cells to kill endogenous CAFs even in the presence of an immunosuppressive microenvironment. Systemic dosing of such a virus to patients with stromal rich tumors may provide an effective approach for driving effective anti-tumor immunity.
P405 Harnessing pre-existing antiviral immunity to treat solid tumors
Nicolas Cuburu1, Rina Kim1, Sergio Pontejo2, Cynthia Thompson1, Douglas Lowy1, John Schiller1,3
1National Cancer Institute, NIH, Bethesda, MD, USA; 2National Institute of Allergy and Infectious Diseases, NIH, Bethesda, MD, USA; 3NCI/NIH, Bethesda, MD, USA
Correspondence: Nicolas Cuburu (cuburun@mail.nih.gov); John Schiller
Background
Cancer immunotherapy rely on adaptive immune responses against tumor-associated antigens. The diversity of tumor antigens makes the development of cancer vaccines a highly personalized endeavor. Epitope spreading results from the induction of de novo responses against tumor antigens not initially included in vaccines [[1]. Intratumoral therapies also require to induce de novo responses against tumor antigen for abscopal therapeutic effect.
Human cytomegalovirus is highly prevalent in humans with polyfunctional T cell responses expanding with age [2]. We questioned whether redirecting pre-existing anti-cytomegalovirus T cells into solid tumor could arrest tumor growth, induce epitope spreading, and confer long-term anti-tumor immunity?
Methods
Persistently infected mice with mouse cytomegalovirus (MCMV) were challenged with TC-1 tumor cells expressing human papillomavirus (HPV) E6 and E7 oncogenes. We generated HPV pseudovirons expressing MCMV antigens or peptides derived from minimal MCMV epitopes. These reagents were injected intratumorally together with the poly(I:C), as adjuvant. We monitored tumor growth, survival and measured anti-tumor CD8+ T cell responses against an E7 immunodominant epitope using MHC-I tetramer and intracellular cytokine stainings. In rechallenge experiments, we evaluated the induction of long-term anti-tumor immunity. Using PanCancerImmune panel (Nanostring), we analyzed the tumor immune microenvironment after treatment.
Results
Transduction of TC-1 tumors with HPV pseudovirions expressing MCMV antigens or peptidic MCMV epitopes caused the expansion of MCMV-specific CD4+ and CD8+ T cells. These treatments also caused broad modifications of the tumor immune environement. Intratumoral injection of MCMV CD8 epitopes provoked the arrest of tumor growth. Intratumoral injection of MCMV CD4 epitopes with poly(I:C) promoted induction of E7-specific CD8+ T cells. Sequential administration of CD4 and CD8 MCMV epitopes together with poly(I:C) was the best protocol to eradicate pre-existing tumor of 5 to 10mm3, and rechallenge experiments showed anti-tumor immunity up to 4 months after the last treatment.
Conclusions
Our results provide a proof of concept to design “antigen-agnostic” intratumoral therapies based on pre-existing antiviral T cells. Such approach change the tumor immune microenvironment, induce epitope spreading, and confer long term anti-tumor immunity. These findings prompt further evaluation in other spontaneous tumor models and provide a model to decipher the mechanisms of epitope spreading and the role of CD4 help.
References
1. Gulley JL, et al. Immune impact induced by PROSTVAC (PSA-TRICOM), a therapeutic vaccine for prostate cancer. Cancer Immunol Res. 2014;2(2):133-41.
2. O'Hara GA, et al. Memory T cell inflation: understanding cause and effect. Trends Immunol. 2012;33(2):84-90.
P406 Dissecting the mechanisms underlying antitumor effects of live oncolytic vaccinia and heat-inactivated vaccinia
Weiyi Wang, Peihong Dai, Ning Yang, Stewart Shuman, Taha Merghoub, Jedd Wolchok, Liang Deng
Memorial Sloan Kettering Cancer Center, New York, NY, USA
Correspondence: Liang Deng (dengl@mskcc.org)
Background
Preclinical and clinical studies have shown that viral-based immunotherapy has the potential to overcome resistance to immune checkpoint blockade and to fill the unmet needs of many cancer patients. Oncolytic viruses are defined as engineered or naturally occurring viruses that selectively replicate in and kill cancer cells. Oncolytic viruses also induce antitumor immunity. We recently showed that intratumoral (IT) delivery of inactivated modified vaccinia virus Ankara (iMVA) induces antitumor systemic immunity via the STING-mediated cytosolic DNA-sensing pathway and Batf3-dependent CD103+/CD8α+ dendritic cells (DCs). The combination of IT iMVA and systemic delivery of immune checkpoint blockade is highly effective in treating large established tumors and distant metastasis [1]. MVA is non-replicative in tumor cells, and therefore whether or not viral replication is necessary for antitumor effects of IT oncolytic DNA virus is unknown.
Methods
In the study, we engineered a replication-competent, attenuated vaccinia virus (VC) by removing the Z-DNA-binding domain of vaccinia virulence gene E3 and inserting murine GM-CSF gene into the thymidine kinase (TK) locus (E3L∆83N-TK--mGM-CSF). We compared antitumor effects of live VC and iVC in murine tumor models.
Results
We found that IT Heat-inactivated VC (iVC; by heating the live virus at 55°C for 1 h) is more effective in generating systemic antitumor immunity than live VC murine tumor models. The antitumor effects of live VC also requires Batf3-dependent DCs. IT Heat-iVC induces higher numbers of infiltrating activated CD8+ and CD4+ T cells in the non-injected tumors than live VC and is more potent in depleting tumor-associated macrophages than live VC in both injected and non-injected distant tumors. The surviving mice treated with Heat-iVC are more effective in rejecting tumor rechallenge than those treated with live-VC, indicating that IT Heat-iVC generates stronger antitumor memory T cell responses than live VC.
Conclusions
In conclusion, our results support the use the IT delivery of inactivated vaccinia virus as a safe and effective cancer immunotherapy either alone or in combination with immune checkpoint blockade antibodies.
References
1. Dai P, Wang W, Serna-Tamayo C, Ricca JM, Zamarin D, Shuman S., Merghoub T, Wolchok JD, and Deng L. Intratumoral delivery of inactivated modified vaccinia virus Ankara (iMVA) induces systemic antitumor immunity via STING and Batf3-dependent DC. Science Immunology. 2017;2: eaal1713.
P407 In situ vaccination for cancer immunotherapy: treat locally, respond systemically
Nicole Steinmetz1, P. Jack Hoopes2, Mee Ree Sheen2, Chenkai Mao2, Scott Sieg1, Steven Fiering2, Patrick :Lizotte2, Amy Wen3
1Case Western Reserve University, Cleveland, NH, USA; 2Geisel School of Medicine at Dartmouth, Lebanon, NH, USA; 3Case Western Reserve University, Cleveland, OH, USA
Correspondence: Steven Fiering (fiering@dartmouth.edu)
Background
Immunotherapy for cancer is making impressive impacts in the clinic. One strategy dating back to William Coley in 1900 is in situ vaccination. This approach puts immunostimulatory reagents into an identified tumor to break the local immunosuppression, stimulate a local anti-tumor response and most importantly stimulate systemic antitumor immune responses to eliminate metastatic disease. This is essentially an antitumor therapeutic vaccination, because the tumor provides the antigens and the adjuvants are the immunostimulatory reagents, thus “in situ vaccination”. The approach has been part of standard of care for superficial bladder cancer using BCG bacteria. There are many immunostimulatory reagents that can be used and each has different capabilities. We have done previous studies with attenuated microorganisms including Toxoplasma gondii, and Listeria monocytogenes.
Methods
Our recent focus has been on a plant virus that cannot infect animals, compea mosaic virus (CPMV). Complete CPMV or CPMV lacking nucleic acids (eCPMV) are grown in plants and biochemically purified.
Results
CPMV nanoparticles were used in mouse cancer models and community dogs with oral melanoma and other tumors. These particles are composed of assembled viral capsid proteins. eCPMV has no nucleic acids and no recognized immunostimulatory reagents. However, eCPMV is strongly immunostimulatory through unknown pathways and causes changes in the tumor microenvironment that lead to primary tumor reduction or elimination and potent resistance to metastatic tumors (1). The treatment is immune-mediated but response in the lungs requires different immune components than response in flank tumors of the same B16F10 melanoma cell line. Tumor reduction or elimination occurs in many anatomic locations with multiple tumor types and in multiple strains of inbred mice. Treatment of primary tumors by direct intratumoral injection mediates robust rejection of a rechallenge with the same tumor. Response in community dogs was highly effective at both eliminating the local tumor and resisting metastatic disease. The mechanisms and pathways of immunostimulation are under investigation.
Conclusions
In situ vaccination has considerable potential for clinical use. In addition to the inherent immunostimulatory adjuvant properties of CPMV, they are a versatile platform to which other reagents for immune modulation can be attached. This demonstration of the value of select viral-like nanoparticles for treatment of cancer opens a new avenue of cancer immunotherapy.
References
1. Lizotte PH, Wen AM, Sheen MR, Fields J, Rojanasopondist P, Steinmetz NF, Fiering S. In situ vaccination with cowpea mosaic virus nanoparticles suppresses metastatic cancer. Nat Nanotechnol. 2016;11:295-303.
P408 Tumor cell-intrinsic STING signaling and regulation of IFN-β gene expression
Blake Flood1, Leticia Corrales2, Thomas Gajewski1
1University of Chicago, Chicago, IL, USA; 2Aduro Biotech, Chicago, IL, USA
Correspondence: Blake Flood (blakeflood@uchicago.edu)
Background
Our laboratory has previously shown that immunogenic tumors spontaneously activate the innate immune system through the STING pathway. The STING pathway senses cytosolic DNA, which activates a signal transduction pathway culminating in phosphorylated IRF3 that translocates to the nucleus where it acts as a transcription factor to induce several genes including IFN-β. STING signaling and IFN-β receptor signaling in tumor-infiltrating immune cells, in turn, are required for optimal priming of CD8+ T cells against tumor antigens. Based on this notion, STING agonists were developed and tested as a pharmacologic approach to activate the pathway.
Methods
We stimulated various cell populations present in the tumor microenvironment as well as several tumor cell lines with STING agonists to test their ability produce IFN-β and analyze each step in STING pathway signaling.
Results
We observe that tumor cells themselves frequently fail to produce IFN-β in response to STING agonists or cytoplasmic DNA, arguing that loss of activation of this pathway might occur regularly as a component of oncogenesis. Surprisingly, we find that tumor cells retain expression of each gene in the STING pathway and STING signal transduction is intact up to and including nuclear translocation of IRF3. ChIP assays demonstrate IRF3 is unable to bind the IFN-β promoter but can still bind the promoters of other genes.
Conclusions
These data indicate tumor cells fail to express IFN-β following STING pathway activation due to a defect in transcription factor binding to the IFN locus. Based on ChIP experiments the defect in IRF3 DNA binding may be specific to the IFN-β locus. Sequencing of the locus shows no mutations in or around the binding site so we are currently investigating epigenetic mechanisms regulating IFN-β gene expression.
P409 Efficacious anti-melanoma immunity induced by OX40 ligand-expressing oncolytic adenovirus Delta-24-RGDOX
Hong Jiang, Andrew Dong, Caroline Carrillo, Verlene Henry, Xuejun Fan, Yisel Rivera-Molina, Francisco Puerta-Martinez, Teresa Nguyen, Karen Clise-Dwyer, Frederick Lang, Candelaria Gomez-Manzano, Juan Fueyo
The University of Texas MD Anderson Cancer Center, Houston, TX, USA
Correspondence: Hong Jiang (hjiang@mdanderson.org)
Background
Oncolytic adenoviruses are highly immunogenic and cancer-selective but show limited efficacy as a single agent. Immune checkpoint modulation has shown efficacy in a variety of cancers but is associated with nonspecific T-cell activation, and has had a limited effect in tumors with a nonimmunogenic microenvironment.Thus, combining these two strategies likely resulted in both efficacious and specific cancer therapy. To this end, we constructed the oncolytic adenovirus Delta-24-RGDOX which expressed the immune co-stimulator OX40L. This new virus induces a superior immunotherapeutic effect in immunocompetent mouse glioma models than its predecessor Delta-24-RGDOX.
Methods
In a s.c./s.c. melanoma mouse model with B16F10-Red-FLuc cells, Delta-24-RGDOX and/or anti-CD47 antibody were injected into the first implanted tumor. The tumor growth of the first and second implanted tumors was monitored with bioluninescence. The survival curves were plotted according to the Kaplan–Meier method. Survival rates in the different treatment groups were compared using the log-rank test. The immune cells in the tumor were isolated and analyzed with flow cytometry.
Results
Delta-24-RGDOX expressed OX40L effectively in mouse melanoma cells. Compared to treatment with PBS, four doses of intratumoral injection of the virus significantly inhibited the growth of both the injected tumors and the untreated distant tumors, resulting in prolonged survival of the mice with 40% long-term survival (P = 0.01). The surviving mice is resistant to rechallenging with the same tumor cells but is susceptible to lung cancer cells, suggesting the development of immune memory specific to the virus-injected tumor type. Through flow cytometry analysis of the tumor-infiltrating lymphocytes, we found the virus injection increased the presence of CD3+ T lymphocytes, CD3+CD4+ helper T cells and CD3+CD8+ cytotoxic T cells in the tumor, causing a decreased ratio of CD4+/CD8+ cells. To further increase the efficacy of Delta-24-RGDOX, we combined the virus with intratumoral injection of an antibody against CD47, a “don’t eat me” signal overexpressed on tumor cell surface to protect them from phagocytosis. We found high expression levels of CD47 in cultured B16 cells and the tumor cells from the xenografts. Intratumoral injection of Delta-24-RGDOX caused much more phagocytes presented at the tumor site than PBS treatment. Combination of anti-CD47 antibody with Delta-24-RGDOX mediated longer survival time than the virus alone (median survival: 56 days vs. 26 days).
Conclusions
Delta-24-RGDOX induced efficacious local and systemic anti-melanoma immunity in B16-C57BL/6 mouse model and combination of the virus with anti-CD47 antibody further increased the therapeutic efficacy.
P410 Generation of therapeutic RNAs to induce immunogenic cell death and interferon expression in cancer cells
Jaewoo Lee1, Youngju Lee2
1Duke University Medical Center, Durham, NC, USA; 2Duke University, Durham, NC, USA
Correspondence: Jaewoo Lee (jaewoo.lee@duke.edu)
Background
Pattern-recognition receptors (PRRs) are immunological sensors that initiate the host defense response against infections. They are located at the cell surface, within endosomal compartments and in the cytoplasm, where they are poised to recognize different molecular signatures associated with invading pathogens. In addition to anti-infectious immunity, the activation of RNA-sensing PRRs can mediate programmed cell death of infected cells, which allows the host to efficiently block viral replication by sacrificing infected cells. Transfection with certain types of RNA ligands that stimulate RIG-I-like receptor (RLR) family can induce type I interferon (IFN) and immunogenic cell death (ICD) in cancer cells, which together orchestrate anti-tumor immune responses that can prevent tumor recurrence and metastasis. Currently RNA-sensing PRR agonists have demonstrated little or no overall benefit to patients with cancers because of, mostly but not exclusively, toxicity driven by non-specific induction of immune reactions. In this study, we generate and screen multiple nuclease-resistant RNA molecules that can differentially induce immunogenic cancer cell death with or without concomitant expression of IFN-β, pro-inflammatory cytokines and pro-coagulation mediators.
Methods
Therapeutic RNAs are designed using mFold RNA structure prediction program and generated by in vitro transcription. All RNAs contain 5’triphosphate and 2’fluoro pyrimidine that are recognized by RLRs and resistant to nucleases. Immunogenic cell death in human melanoma, pancreatic cancer and prostate cancer cells and PBMCs was analyzed by western blot, ELISA and Flow cytometry. In vivo anti-tumor effects of the RNAs were induced by intratumoral transfection with RNAs into immunocompromised mice bearing human melanoma.
Results
We generated two novel and distinct ssRNA molecules (Immunogenic Cell- killing RNA (ICR)2 and ICR4). ICR2 and ICR4 differentially stimulated cell death and PRR signaling pathways and induced different patterns of cytokine expression in cancer and innate immune cells. Interestingly, damage-associated molecular patterns (DAMPs) released from ICR2- and ICR4-treated cancer cells had distinct patterns of stimulation of innate immune receptors and coagulation. Finally, ICR2 and ICR4 inhibited in vivo tumor growth as effectively as polyI:C. ICR2 and ICR4 are potential therapeutic agents that differentially induce cell death, immune stimulation and coagulation when introduced into tumors.
Conclusions
ICR2 and ICR4 are potent PRR-stimulating cytotoxic agents against human cancers. ICR2 and ICR4 have distinctive immune stimulatory and coagulating activities. Combination of dual checkpoint inhibitors and ICR4 and/or ICR2 would be a potent and effective anti-cancer therapies against advanced cancers.
P411 Immunotherapy with BO-112, a novel double-stranded RNA-based agent, promotes tumor cell death and boosts T cell immunity in preclinical mouse models
Lourdes Planelles1, Angela Aznar2, MªJosé Gómez-Sánchez1, Mercedes Pérez-Olivares1, Saray Garasa2, Mercedes Pozuelo1, Ivan Márquez-Rodas3, Marisol Quintero1, Ignacio Melero2
1BIoncotech Therapeutics, Valencia, Spain; 2Center for Applied Medical Research (CIMA) & University of Navarra, Pamplona, Spain; 3Hospital General Universitario Gregorio Marañón, Madrid, Spain
Correspondence: Lourdes Planelles (lplanelles@bioncotech.com)
Background
Unresponsive or refractory tumors are the main research objective in cancer immunotherapy. Strategies that promote immunogenic tumor cell death in the context of type I interferon (IFN) signaling can be combined effectively with existing therapies that target checkpoint inhibitors to boost anti-tumor immunity. BO-112 is a double-stranded synthetic RNA currently undergoing phase I clinical trials for intratumor injection. To enhance local antitumor immunity, BO-112 bears poly I:C chains that trigger endosomal Toll-like receptor 3 and cytosolic helicases.
Methods
B16F10 and B16F10-OVA melanoma, MC38 colon carcinoma or 4T1 breast carcinoma cells (5 x 105) were implanted subcutaneously in the right flank of C57BL/6J and BALB/c mice; a second tumor was engrafted in the left flank to evaluate distant antitumor effects. Beginning 7-10 days post-implant, mice were treated intratumorally with BO-112 only in the primary lesion. Therapeutic efficacy was evaluated by monitoring tumor growth and survival, and cellular responses were analyzed by multiparametric flow cytometry. Antigen-specific CD8+ T cell in vivo priming was addressed by H-2Kb tetramer analysis in B16F10-OVA tumors. The role of CD4+ or CD8+ T cells was studied by depletion and depletion/rechallenge experiments. Combined BO-112 and anti-PD-L1 (programmed death-ligand 1) antibody therapy was evaluated in established B16F10 melanomas.
Results
In vitro and in vivo studies showed that BO-112 triggered cell death in tumor cells of distinct tissue origins (melanoma, colon or breast carcinoma). The cytotoxic effect was also detected in engineered B16 cells that do not respond to type I IFN. Intratumor BO-112 administration in all three mouse tumor models showed therapeutic effects, with complete tumor regression in up to 30% of mice. FACS analyses of tumor-derived cell suspensions showed that BO-112 increased the tumor immune infiltrate and CD8+ T cell frequency. Moreover, the receptors PD1 and CD137, which denote activation, were upregulated on CD8+ T cells. BO-112 treatment enhanced intratumor OVA-specific CD8+ T cells, and depletion and rechallenge experiments confirmed CD8+ T cell involvement in its immunotherapeutic effects. A combination of BO-112 and anti-PD-L1 antibody significantly improved the poor response to PD-L1 blockade in B16F10 and B16F10-OVA tumor-bearing mice. Responses were observed in treated and in untreated distant tumors, which provides a rationale for clinical testing of this combination.
Conclusions
These data identify local BO-112 release as a new immunotherapy agent beneficial in the treatment of refractory and recurrent tumors, especially when combined with checkpoint inhibitors. A phase Ib clinical trial is being designed to address this question.
Trial Registration
http://ClinicalTrials.gov identifier, NCT02828098.
P412 Antitumor immunity in patients with locally advanced soft tissue sarcoma treated with hafnium oxide nanoparticles and radiation therapy
Jérôme Galon1, Marick Laé2, Juliette Thariat3, Sébastien Carrere4, Zsuzsanna Papai5, Martine Delannes6, Philippe Rochaix6, László Mangel7, Fabienne Hermitte8, Zoltán Sapi9, Tamas Tornoczky7, Vincent Servois2, Isabelle Birtwisle Peyrottes3, Raphaël Tetreau4, Marie-Christine Château4, Sébastien Paris10, Hervé Brisse2, Sylvie Bonvalot2
1INSERM, Paris, France; 2Institut Curie, Paris, France; 3Centre Antoine Lacassagne, Nice, France; 4Centre Régional De Lutte Contre Le Cancer Paul Lamarque, Montpellier, France; 5Medical Centre, Hungarian Defence Forces, Budapest, Hungary; 6Institut Universitaire du Cancer Toulouse - Oncopole, Toulouse, France; 7Pecs University, Pecs, Hungary; 8HalioDX, Marseille, France; 9Semmelweis University, Budapest, Hungary; 10Nanobiotix, Paris, France
Correspondence: Sébastien Paris (agnes.pottier@nanobiotix.com)
Background
Soft tissue sarcoma (STS) is a large and heterogeneous group of malignant mesenchymal neoplasms characterized by a strong tendency toward local recurrence and metastatic spreading. Consistently, the immune microenvironment in sarcomas is highly variable. A new class of material with high electron density, hafnium oxide, was designed at the nanoscale to efficiently absorb ionizing radiation from within the tumor cells and augment the dose deposited to a tumor. These nanoparticles (HfO2-NP) administered in a single intratumor injection and activated by fractionated radiotherapy are evaluated in a phase II/III trial in patients with locally advanced STS as neoadjuvant treatment. Besides, beyond the broadly cytotoxic effect of radiation therapy (RT), RT may promote the release of tumor neoantigens during cancer cell death and stimulate local immunological effects.
Here, we explore the effects of nanosized hafnium oxide exposed to RT in terms of tumor immune profile changes in patients with STS when compared to RT alone.
Methods
Tumor tissues pre- (biopsy) and post-treatment (resection) are collected from patients with locally advanced STS (NCT02379845), who received either HfO2-NP activated by RT or RT alone. Immunohistochemistry and Digital Pathology for immune biomarkers and Pan-Immune gene expression profiling are analyzed.
Results
A significant increase of CD8+ T cells and a marked increase of CD3+ and PD-1 T cells and CD103+ immune cell infiltration post- vs pre-treatment are observed for HfO2-NP + RT while no differences are seen for RT alone (more than 10 patients analyzed in each arm). Functional analysis of genes expression up-regulated in HfO2-NP + RT post- vs pre-treatment shows an enrichment of cytokine activity (IL7, IFNA, IL11, IFNG), adaptive immunity (RAG1, TAP1, TAP2, TBX21, IFNG, LTK, CD37, CD22) and T cell receptor signaling pathway (CD28, CTLA4, CD274, BTLA, TIGIT, CD5, ZAP70) when compared to RT.
Conclusions
Promising results are observed in patients who received HfO2-NP + RT in terms of immune cells infiltration post- vs pre-treatment when compared to RT. Moreover, HfO2-NP + RT induces a specific adaptive immune pattern. So far, nanosized hafnium oxide exposed to RT bring substantial changes to the tumor immune profile in patients with STS when compared to RT. As such, it may convert immunologically “cold” tumor into “hot” tumor and be effectively combined with immunotherapeutic agents across oncology. More tissue samples are under evaluation to reinforce these findings.
P413 Transforming immunologically “cold” tumor into “hot” tumor with hafnium oxide nanoparticles and radiation therapy
Sébastien Paris, Audrey Darmon, Ping Zhang, Maxime Bergère, Laurent Levy
Nanobiotix, Paris, France
Correspondence: Sébastien Paris (agnes.pottier@nanobiotix.com)
Background
Hafnium oxide, an electron-dense material, was designed at the nanoscale to increase the radiation dose deposited from within the cancer cells: “Hot spot” of energy deposit where the nanoparticles are when exposed to radiation therapy (RT). Preclinical studies have demonstrated increase of cancer cells killing in vitro and marked antitumor efficacy in vivo with presence of these nanoparticles (HfO2-NP) exposed to RT, when compared to RT alone. HfO2-NP is intended for a single intratumor injection and is currently evaluated in clinical trials including soft tissue sarcoma, head and neck, prostate, liver and rectum cancers.
Here, we explore the ability of nanosized hafnium oxide exposed to RT to bring substantial immune cells infiltrations in the tumors and convert immunologically “cold” tumor into “hot” tumor.
Methods
CT26 (murine colorectal cancer cells) were subcutaneously injected in the flank of BALB/c mice. Once the mean tumors volume reached 115±30 mm3, tumors were intratumor injected with HfO2-NP and irradiated with 2Gyx3 or 4Gyx3, or irradiated only. Tumors were collected 5 days after the last RT fraction and analyzed for immune cell infiltrates by immunohistochemistry (2Gyx3 and 4Gyx3) and cytokines content by flow cytometry (2Gyx3).
A second study evaluated HfO2-NP exposed to RT vs RT alone using the 4T1 murine breast cancer model. Cells treated or not with HfO2-NP were exposed to irradiation (40Gy). Irradiated cells (1.106) (or phosphate-buffered saline as control) were inoculated subcutaneously into the flank of BALB/c mice (vaccination phase). Seven days after, mice were challenged with untreated 4T1 cells (1.106) (challenge phase). Grown tumors (challenge site) were collected 19 days after the challenge phase and analyzed for immune cell infiltrates by immunohistochemistry.
Results
In mice bearing CT26 tumors, a marked increase of cytokines content and immune cell infiltrates was observed with HfO2-NP + 2Gyx3 when compared to RT alone. The tumor immune cell infiltrates were further enhanced with HfO2-NP + 4Gyx3.
In mice inoculated with 4T1 cells treated with HfO2-NP + 40Gy, a marked increase of immune cell infiltrate (CD8+) was observed in tumors when compared to tumors in mice inoculated with 4T1 cells treated with 40Gy and control.
Conclusions
These in vivo data generated from CT26 and 4T1 tumor models suggest that HfO2-NP + RT triggers immunogenic conversion of the tumor microenvironement when compared to RT alone. HfO2-NP treatment may represent a therapeutical approach for broad applications since it does not rely on any molecular characteristics of the tumor.
P414 PVSRIPO is an interferon-resistant, immunotherapeutic oncolytic virus
Ross Walton, Michael Brown, Eda Holl, David Boczkowski, Vidya Chandramohan, Smita Nair, Matthias Gromeier
Duke University, Durham, NC, USA
Correspondence: Ross Walton (rww7@duke.edu)
Background
Oncolytic viruses are attractive cancer therapies because they selectively lyse cancer cells, but show attenuation in normal cells. Many oncolytic virus are attenuated through susceptibility to innate antiviral immune responses, like Type I Interferon (IFN) responses. Until recently it was believed that cancer cells lack these responses. However, our lab and others have shown cancer cells can have intact IFN responses which inhibit IFN-susceptible viruses, but may activate potent anti-cancer immunity. Innate immunity in cancer is thus an area of extreme interest.
Our lab developed an oncolytic human rhinovirus:poliovirus chimera, PVSRIPO, which is in clinical trial against glioblastoma multiforme, an aggressive form for brain cancer. Using PVSRIPO, and the related picornavirus encephalomyocarditis virus (EMCV), we investigated MDA5 and IFNs in infection of cancer cells. MDA5 is a pattern recognition receptor, targeted to viral double-stranded RNA produced by picornaviruses. We also investigated PVSRIPO’s role in anti-tumor immune responses after treatment of cancer cells.
Methods
DM440, a human melanoma cell line, was engineered with stable MDA5 knock-down by lentivirus transduction containing MDA5-targeting small-hairpin RNA. PVSRIPO or EMCV was added at a multiplicity of infection (MOI) of 0.1 or 0.01 and samples collected for immunoblot, ELISA, or viral titer. Cells were also incubated with IFN-α2 for 24 hours, infected, and assayed as above. Human dendritic cells (DCs) were infected at 1, 10, 50, or 100 MOI and collected as above. DCs were also treated with oncolysate from PVSRIPO-infected SUM149 human breast cancer cells, non-infectious lysate, or virus-filtered lysate. DCs were analyzed by flow cytometry or used for a cytotoxic HLA-matched T-cell assay using europium-labeled target cells, including cancer cells, tumor antigen transfected DCs, and control antigen expressing DCs.
Results
DM440 cells produce and respond to IFNs in an MDA5-dependent manner after PVSRIPO and EMCV infection. MDA5 activation and IFN treatment inhibits EMCV replication significantly more than PVSRIPO replication. Human DCs can be productively infected and activated with PVSRIPO without toxicity. Oncolysate activates human DCs in a virus dependent manner, and DCs activated in this way promote cytotoxic T-cell activation against tumor-specific antigen.
Conclusions
PVSRIPO is an IFN-resistant oncolytic virus that provokes MDA5-dependent IFN responses in cancer cells, without significant viral inhibition. Further, PVSRIPO infects DCs, activating them without significant cytotoxicity, which can lead to tumor-specific cytotoxic T-cell responses, and may lead to antigen spreading. It is possible this potent DC activation phenotype is due to PVSRIPO’s resistance to IFNs.
P415 Upregulation of PD-L1 in tumor microenvironment is a resistance mechanism for oncolytic virus immunotherapy
Dmitriy Zamarin, Jacob Ricca, Anton Oseledchyk, Mathieu Gigoux, Taha Merghoub, Jedd Wolchok
Memorial Sloan Kettering Cancer Center, New York, NY, USA
Correspondence: Dmitriy Zamarin (zamarind@mskcc.org)
Background
Intralesional therapy with oncolytic viruses (OV) leads to activation of multiple immune pathways such as type I IFN. Both OV and OV-activated type I IFN pathway can exert a variety of pleiotropic effects on both immune and non-immune cells, activating resistance to the immune system on both local and abscopal level. Identification of such pathways could provide insights into the mechanisms of OV-mediated T cell activation as well as generate rationale for combinatorial strategies.
Methods
Using Newcastle Disease Virus (NDV) as a model OV, we explored the effect of intratumoral OV therapy on the microenvironment of the treated and distant tumors in syngeneic bilateral flank tumor models, and explored combination therapies using intratumoral NDV with systemic immune checkpoint blockade.
Results
Intratumoral therapy with NDV led to upregulation of a range of activating and inhibitory immune targets in the treated and distant tumors. While NDV therapy shifted the balance from exhausted to effector T cell phenotype in distant tumors, it was not sufficient for complete tumor rejection. We further demonstrate that infection with NDV leads to upregulation of PD-L1, an effect that is mediated early through paracrine action of type I IFN. Consistent with these findings, intratumoral therapy with NDV or IFNα results in upregulation of PD-L1 on both tumor cells and tumor-infiltrating lymphocytes in the treated tumors. In contrast to NDV, intratumoral IFNα therapy had no effect on late PD-L1 expression, immune cell infiltration, or growth of distant tumors, highlighting that late PD-L1 upregulation is likely a reflection of adaptive immune resistance to the increased tumor infiltrating lymphocytes. Systemic therapeutic targeting of PD-1 or PD-L1 in combination with intratumoral NDV resulted in rejection of both treated and distant tumors. Anti-tumor efficacy was fully dependent on CD8 cells and on early presence of NK cells.
Conclusions
These findings provide implications for timing of PD-1/PD-L1 blockade in conjunction with OV therapy and highlight that understanding of adaptive mechanisms of immune resistance to specific OV will be important for rational design of appropriate combinatorial approaches with these agents.
Other
P416 Anti-tumor effects and immunological response following immune stimulating interstitial laser thermotherapy
Jakob Axelsson, Cristina Pantaleone
Clinical Laserthermia Systems AB, Lund, Sweden
Correspondence: Jakob Axelsson (jakob@clinicallaser.se)
Background
Immune stimulating interstitial laser thermotherapy (imILT) is a local ablation modality designed for local destruction of tumor tissue as well as to optimize systemic immunologically mediated anti-cancer effects. Safety and feasibility in the clinic has been studied and previous rodent data show absopal effects. This study expands on the mechanisms behind these immunological effects.
Methods
An imILT treatment protocol was developed and a syngenic mouse tumor model was established. Mice were inoculated subcutaneously with tumor cells and tumor growth was monitored. When tumors reached adequate size imILT treatment was performed and subsequently another tumor was inoculated and tumor progression was measured. In another set of experiments, two tumors were inoculated simultaneously (twin tumor model) and one of them was treated at appropriate size. Tumor progression of both tumors were monitored. Immune infiltrate and morphology of all tumors were studied.
Results
Preliminary data show both abscopal effects in the twin tumor model as indicated as reduced growth of the non-treated tumor of the imILT treated mice as compared to the control mice. Memory effects can also be demonstrated in the first set of experiment as seen as reduced growth and take of tumor inoculation in mice which have previously been receiving imILT as compared to control mice.
Conclusions
Taken together these data suggest immunologically mediated effects are evoked following imILT treatment. This new information will be useful for deducing the mechanisms of action involved in these effects.
P417 Post-marketing safety of checkpoint inhibitors: analysis of the FDA adverse event reporting system
Rawad Elias1, Jennifer Rider2, Osama Rahma3
1Boston University Medical Center, Boston, MA, USA; 2Boston University School of Public Health, Boston, MA, USA; 3Dana-Farber Cancer Institute, Boston, MA, USA
Correspondence: Rawad Elias (rawadelias@hotmail.com)
Background
Information about the toxicity profile of immune checkpoint Inhibitors (ICIs) is limited. Available data primarily comes from clinical trials. However, subjects enrolled in clinical trials are usually fitter than patients typically seen in clinical practice and therefore may have a different toxicity profile.
Methods
We reviewed data from the FDA Adverse Event Reporting System (FAERS) summarizing adverse events (AEs) associated with the PD-1 inhibitors (nivolumab and pembrolizumab); PD-L1 inhibitor (atezolizumab); and CTLA-4 inhibitor (ipilimumab). We restricted our analysis to reports that included only an ICI as suspect agent. For each agent, we performed a descriptive analysis of hospitalization (HO) and death (DE) outcomes, as well as AEs of special interest (AESI). We compared the distribution of each outcome within age groups (<65 years; 67-75; >75) using Mantel-Haenszel chi -quare test for trend.
Results
Our analysis included a total of 21,588 safety reports. 415 for atezolizumab, 10,026 for nivolumab, 4,808 for pembrolizumab, and 6,339 for ipilimumab (Table 1). There was a statistically significant trend of more hospitalization with increasing age for all drugs. Prevalence of any of the AESI was higher as age increased for all drugs (p<0.0001) except for atezolizumab (p 0.12). However, no statistically significant trend by age was found for any individual AESI (data not shown). Proportion of older patients who experienced death was higher for pembrolizumab (p<0.001) and ipilimumab (p 0.002).
Conclusions
Our analysis suggests that older patients receiving pembrolizumab, Nivolumab or ipilimumab are more likely to develop immune related AEs, and to be hospitalized. This resulted in higher rate of deaths only in pembrolizumab and ipilimumab. The smaller sample size of the patients receiving atezolizumab may have contributed to the lack of statistical significance. Older patients treated with ICIs should be monitored carefully for treatment-related AEs. Toxicity of ICIs should be further evaluated prospectively in the setting of trials in older adults that would take in consideration impact of physiologic and immune aging.
See text for description
P418 Bromodomain and extraterminal region inhibitors slow melanoma tumor growth by altering the tumor microenvironment
Dan A. Erkes1, Claudia Capparelli1, Shea A. Heilman1, Timothy J. Purwin1, Adam C. Berger1, Michael A. Davies2, Andrew E. Aplin1
1Thomas Jefferson University, philadephia, PA, USA; 2MD Anderson Cancer Center, houston, TX, USA
Correspondence: Andrew E. Aplin (dan.erkes@jefferson.edu)
Background
Melanoma is the deadliest form of skin cancer, yielding a 2-16% survival rate for metastatic patients. Despite the onset of new successful therapies, i.e. targeted inhibitors and immunotherapy, there is a large need for drugs that alter the entire tumor microenvironment. To this end, we aim to study drugs that have widespread tumor microenvironment effects, such as epigenetic modifiers, i.e. bromodomain and extra terminal domain inhibitors (BETi), which slow tumor growth in a variety of cancers and effect inflammatory responses. Thus, BETis potentially represent a novel treatment for metastatic melanoma, as they can target tumor cell growth and anti-tumor immune mechanisms.
Methods
Several mouse and human BRAFV600E melanoma cell lines, immune competent mouse models, and a patient-dreived xenograft model were used to uncover the effects of BETi treatment on melanoma.
Results
Here we show that the BETi PLX51107 was able to differentially impact BRAFV600E melanoma tumor growth in a number of mouse and human melanoma cell lines in vitro and in vivo. PLX51107 induced cell cycle arrest and cell death of melanoma cell lines by increasing Bim and decreasing Cyclin B1 in mouse and human lines. Using immune competent mouse models, BETi treatment altered the immunogenicity of the microenvironment of responsive melanoma tumors by lowering the expression of anti-inflammatory PD-L1 and FasL, while increasing pro-inflammatory MHC-I. This was also seen in mouse and human melanoma cell lines and cancer-associated fibroblasts. PLX51107 treatment differentially altered tumor infiltrating lymphocyte (TIL) populations in mouse melanoma models, increasing activated, proliferating, and functional intratumoral CD8+ T cells and leading to a CD8+ T cell mediated tumor growth delay in highly responsive tumors. However, in moderately or poorly responsive tumors, these CD8+ T cell populations were unchanged or decreased after BETi treatment. As BETi treatment was a successful primary therapy, it was prudent to determine its efficacy as a second line therapy. In this vein, an immune compotent mouse model and patient-derived xenograft model were used to demonstrate BETi efficacy on anti-PD-1 resistant tumors. BETi treatment was able to delay the growth of anti-PD-1 resistant tumors, by increasing intratumoral CD8+ T cells and pushing tumors to a more responsive IPRES signature.
Conclusions
As hypothesized, BETi treatment had wide-ranging effects on melanoma tumors by altering the tumor cells themselves, the microenvironment, and anti-tumor immunity to induce robust tumor growth delay. From these findings, BETis represent a potential primary or secondary treatment for metastatic melanoma patients.
P419 Tobacco use, awareness and cessation among Malayali tribes, Yelagiri Hills, Tamil nadu, India
Delfin lovelina Francis (delfin_lovelina@yahoo.co.in)
Dr MGR Medical University, Chennai, India
Background
Health is a state of complete wellbeing free from any discomfort and pain. Despite remarkable world-wide progress in the field of diagnostic, curative and preventive medicine, still there are large populations of people living in isolation in natural and unpolluted surroundings far away from civilisation, maintaining their traditional values, customs, beliefs and myths. India has the second largest tribal population of the world next to the African countries. About half of the world’s autochthonous people live in India, thus making India home to many tribes which have an interesting and varied history of origins, customs and social practices. The present study was conducted to assess the tobacco use, awarness and its effect on health among Malayali tribes, Yelagiri Hills, Tamil nadu, India.
Methods
The inhabitants of the 14 villages of the Yelagiri hills, who have completed 18years and residing for more than 15years present on the day of examination and who were willing to participate in the study were included.
Data was collected from a cross-sectional survey, using a Survey Proforma, clinical examinationand a pre-tested questionnaire which included Demographic data, tobacco habits. An intra-oral examination was carried out by a single examiner to assess the Oral Health Status using WHO Oral Health Surveys – Basic Methods Proforma (1997).SPSS version15 was used for statistical analysis.
Results
Results showed that among 660 study population, 381(57.7%) had no formal education. Among the study population 75%) had the habit of alcohol consumption. Of those who had the habit of smoking, 26% smoked beedi, 10.9% smoked cigarette, 65% chewed raw tobacco, 18% chewed Hans and 28% had a combination of smoking and smokeless tobacco usage. The reason for practicing these habits were as a measure to combat the cold, relieving stress and body pain after work, and the lack of awareness of the hazards of the materials used. Prevalence of oral mucosal lesions in the study population was due to tobacco usage and alcohol consumption and lack of awareness regarding the deleterious effects of the products used.
Conclusions
From the results of this study it may be concluded that the Malayali tribes were characterized by a lack of awareness about oral health, deep rooted dental beliefs, high prevalence of tobacco use and limited access to health services.
P420 Selection of first-in-human starting dose of anti-OX40 agonist monoclonal antibody BMS-986178 using a pharmacokinetic/pharmacodynamic-based approach
Christine Huang1, Yan Feng1, Bryan Barnhart2, Michael Quigley1, John Huber1, Akintunde Bello1, Punit Marathe1, Praveen Aanur1, Timothy Reilly1, Zheng Yang1
1Bristol-Myers Squibb, Princeton, NJ, USA; 2Bristol-Myers Squibb, Redwood City, CA, USA
Correspondence: Christine Huang (christine.huang@bms.com)
Background
Following the clinical success of checkpoint blockade, the field of cancer immunotherapy rapidly expanded. Co-stimulatory molecules from the TNF receptor superfamily, including OX40, may be a promising approach to enhance the benefits of immunotherapy. BMS-986178 is a fully human agonist antibody of the immunoglobulin G1 isotype that binds with high affinity to the human OX40 receptor, currently being developed for the treatment of advanced solid tumors.
Methods
BMS-986178 does not bind to mouse OX40, necessitating antibody surrogates to assess in vivo activity of OX40 agonism. Two hamster anti-mouse OX40 agonist mAbs (reformatted as mouse antibodies: mIgG1 and mIgG2a) were studied in the mouse MC38 colon adenocarcinoma model. A pharmacokinetic/pharmacodynamic (PK/PD)-based approach was employed to integrate the preclinical data and guide the first-in-human (FIH) starting dose selection.
Results
In vitro, binding EC50 of mIgG1 and mIgG2a mAbs in activated mouse T-cells was similar to in vitro binding EC50 of BMS-986178 in activated human T-cells. PK/PD analysis of the anti-tumor efficacy data showed maximum efficacy at an AUC of 100 μg*h/mL with a Cmax of 25 μg/mL. Targeting the same AUC and Cmax in mice into a human PK/PD prediction model, the human efficacious dose was projected to be 1 mg/kg.
The proposed FIH starting dose was then selected to be 0.25 mg/kg, 4-fold below the projected efficacious dose. First, using preclinical GLP safety assessments, the proposed starting dose was 68- to 80-fold lower than 1/6th of the highest non-severely toxic dose (120 mg/kg/week) in monkeys. Second, the Cmax (6.25 μg/mL) at the starting dose was 5-fold below the highest no-effect concentration (33 μg/mL) evaluated in the in vitro cytokine release assay. Third, there was a >10-fold exposure margin between the minimal pharmacologic effect (antigen-specific T-cell or antibody responses) observed in monkeys at doses of 2-4 mg/kg and the FIH starting dose selected.
The FIH trial was successfully initiated at 20 mg (0.25 mg/kg with a BW of 80 kg). BMS-986178 was well tolerated without any adverse event at this dose. OX40 receptor occupancy observed in peripheral blood at the starting dose maintained ~80% in line with the prediction made from in vitro cellular binding affinity and clinical information observed from other anti-OX40 antibodies.
Conclusions
This PK/PD-based approach was successfully applied to select the FIH starting dose of BMS-986178. This strategy appropriately balanced the selection of a safe dose, while minimizing the number of cancer patients receiving sub-therapeutic doses.
P421 Co-administration of dexamethasone with checkpoint blockade therapy increases survival in brain tumor model
Marsha-Kay Hutchinson, Amber Giles, Heather Sonnemann, Jinkyu Jung, Caitlin Reid, Deric Park, Mark Gilbert
1National Institutes of Health, Bethesda, MD, USA
Correspondence: Amber Giles (hutchinsonmn@mail.nih.gov); Deric Park; Mark Gilbert
Background
The use of corticosteroids for therapeutic benefit must be weighed against risks of adverse consequences associated with these drugs. Brain tumor patients are routinely prescribed dexamethasone to reduce tumor-associated edema. Checkpoint blockade, a type of immune therapy, is currently being investigated as a potential treatment for brain tumors. Although glucocorticoid signaling has been shown to attenuate the immune response, the effect of glucocorticoids on the anti-tumor immune response during checkpoint blockade remains unclear. Here, we propose that dexamethasone’s ability to upregulate T-cell checkpoint molecules such as CTLA-4 might be an immunosuppressive mechanism, but can be countered in unison with checkpoint blockade therapy.
Methods
Healthy donor T cells were tested for response to dexamethasone. T cell proliferation, cell cycle analysis, apoptosis, glucose uptake, and protein expression were assessed with flow cytometry and Western blots. Transcriptional changes were assessed with qPCR. CTLA-4 was blocked using monoclonal antibodies in vitro on human PBMCs and in vivo with the GL261 syngeneic glioblastoma model.
Results
Checkpoint molecule CTLA-4 was increased by dexamethasone treatment upon stimulation. Unexpectedly, dexamethasone did not elicit a direct lymphotoxic effect on T cells but significantly reduced T cell entry into cell cycle. Dexamethasone also abated CD28 signaling as shown by reduced AKT phosphorylation and reduced glucose uptake. Blockade of CTLA-4 resulted in a substantial reversal of these effects. In addition, we discovered that antigen-experienced memory T cells were invulnerable to dexamethasone treatment. In vivo, dexamethasone and CTLA-4 blockade provided a survival benefit to tumor bearing mice.
Conclusions
These results suggest that mature T cells, which predominate in the tumor microenvironment, are resilient to the negative effects of corticosteroids. Thus, corticosteroids are unlikely to destroy anti-tumor T cells. For patients who have not responded to immune therapy, CTLA-4 blockade may provide protection from corticosteroids to the naïve T cell pool.
P422 Overall survival and treatment patterns among real-world patients with metastatic non-small cell lung cancer not previously treated with systemic therapy for advanced cancer
Jason C. Simeone1, Beth Nordstrom1, Ketan Patel2, Alyssa B. Klein3
1Evidera, Waltham, MA, USA; 2AstraZeneca, Milton, United Kingdom; 3AstraZeneca, Gaithersburg, MD, USA
Correspondence: Alyssa B. Klein (caroline.moreau@evidera.com)
Background
The majority of patients with non-small cell lung cancer (NSCLC) are diagnosed at an advanced stage of disease, typically with a poor prognosis. While treatment of metastatic NSCLC is focused on extending survival, real-world data on the effectiveness of existing therapies and patterns of treatment is limited.
Methods
Patients with metastatic NSCLC, that are EGFR and ALK WT and no prior systemic therapy for advanced disease were identified from the Flatiron Oncology electronic medical record database from 2013–2016. Treatment patterns, including regimens administered and time to treatment, were summarized. The median overall survival (OS) from the initial diagnosis of metastatic NSCLC was calculated from Kaplan-Meier curves, with associated 95% confidence intervals (CI).
Results
A total of 10,123 patients were eligible for analysis. The mean age of patients was 68.0 ± 10.0 years, and 54.0% were male. The most commonly observed regimens after the diagnosis of metastatic disease were nivolumab (17.4%), carboplatin + pemetrexed (16.0%), and carboplatin + paclitaxel (15.0%), while 25.4% did not receive treatment. The median time from diagnosis of metastatic disease to start of first-line therapy was 36 days (range: 1–1,770) and the median number of therapy lines was 1 (range: 1–8). The median OS was 11.2 months (95% CI: 10.9–11.6) from the initial diagnosis of metastatic disease (Fig. 1).
Conclusions
In this real-world study, overall survival of metastatic patients was less than 12 months, and one quarter of patients did not receive treatment after diagnosis. This evidence indicates a potential unmet need for additional treatment options, as more patient information such as biomarkers become available to physicians at initial diagnosis.

Overall Survival for Patients with Stage IV NSCLC, From Initial Diagnosis of Advanced or Metastatic NSCLC
P423 Overall survival and treatment patterns among real-world patients with stage IIIB non-small cell lung cancer treated with platinum-based chemotherapy
Jason C. Simeone1, Beth Nordstrom1, Ketan Patel2, Alyssa B. Klein3
1Evidera, Waltham, MA, USA; 2AstraZeneca, Milton, United Kingdom; 3AstraZeneca, Gaithersburg, MD, USA
Correspondence: Alyssa B. Klein (caroline.moreau@evidera.com)
Background
Platinum-based chemotherapy has been the standard of care in the management of patients with advanced non-small cell lung cancer (NSCLC), but evidence of effectiveness and patterns of treatment in a real-world setting is needed.
Methods
Electronic medical record data of patients from the Flatiron Oncology database with Stage IIIB NSCLC treated with at least two cycles of platinum-based chemotherapy during 2013–2016 were identified; patients with prior evidence of metastatic disease were excluded. Treatment patterns were examined, including the duration and number of lines received. The median overall survival (OS) from the start date of the third cycle of chemotherapy was calculated from Kaplan-Meier curves, with 95% confidence intervals (CI).
Results
A total of 1,226 patients met all criteria for study inclusion. The mean age of patients was 68.5 ± 9.0 years, and 51.9% were male. The most common regimens observed at any time during follow-up were carboplatin + paclitaxel (43.9%), nivolumab (23.7%), and carboplatin + pemetrexed (21.0%); patients received a median of 2 lines of therapy (range: 1–7). The median OS was 19.4 months (95% CI: 17.4–21.6) from the start of the third cycle of chemotherapy (Fig. 1).
Conclusions
Overall survival among Stage IIIB NSCLC patients who were treated with platinum-based chemotherapy in a real-world setting was less than 20 months. Previous literature indicates survival of 7-13 months among advanced NSCLC patients treated with chemotherapy. However, the present study excludes patients with metastatic disease. The difference in OS may also be related to the use of newer, more effective treatments that were not available in earlier studies. nt-->
Overall Survival of Patients with Stage IIIB NSCLC, From Start of Third Cycle of Chemotherapy
P424 Efficacy of pembrolizumab (MK-3475) in patients with adrenocortical carcinoma
Mohammed Amir Habra, Matthew Campbell, Camilo Jimenez, Daniel Karp, David Hong, Vivek Subbiah, Shubham Pant, Jeane Painter, Saria Khan, Chantale Bernatchez, Bettzy Stephen, Anas Alshawa, Coya Tapia, Tito Mendoza, Rivka Colen, Kenneth Hess, Funda Meric-Bernstam, Aung Naing
MD Anderson Cancer Center, Houston, TX, USA
Correspondence: Aung Naing (anaing@mdanderson.org)
Background
Adrenocortical carcinoma (ACC) is an orphan endocrine malignancy with poor prognosis and limited response to chemotherapy. ACC can express immune markers which could make it susceptible to immunotherapy. So far, there is no published clinical trial about using immunotherapy in ACC.
Methods
This is a phase II study of pembrolizumab in patients with rare tumors including a pre-specified ACC cohort (http://ClinicalTrials.gov identifier, NCT02721732). Patients received pembrolizumab 200 mg intravenously once every 3 weeks. Response was assessed every 9 weeks using RECIST1.1. The primary end point was progression-free survival at 27 weeks (PFS27wk). Mandatory biopsies are taken at baseline, on cycle 1 day 15 -21, and at the time of progression.
Results
A total of 11 patients were enrolled and treated. At the time of analysis, patients received a median of 6 treatment cycles (range 2-17). A PFS of greater than 27 weeks was seen in 3/11 patients (27%). The first patient reached 37% tumor reduction (partial response) while the second patient had a response of 41% tumor reduction (partial response) and the third patient had stable disease. The remaining 8 patients had evidence of disease progression within 27 weeks of starting the study (median time 18 weeks, range 6-27 weeks). Progression was seen in most patients with hormonally active ACC (6/7) and only in 2/4 patients without evidence of hormonal excess at the time of their initial diagnosis. The safety profile of pembrolizumab was very favorable and none of these patients had severe grade 3 or grade 4 adverse effects.
Conclusions
Single agent pembrolizumab may be an effective option in subset of ACCs without hormonal overproduction while it is associated with high failure rate among patients with hormonally active ACCs. Expanding the study to hormonally silent ACCs is needed to verify the initial observation from limited number of patients treated in this study. Translational data will be presented at this meeting.
Trial Registration
http://ClinicalTrials.gov identifier, NCT02721732.
P425 Efficacy of pembrolizumab in patients with cutaneous aquamous cell carcinoma
Renata Ferrarotto, Bonnie Glisson, George Blumenschein, David Hong, Sarina Piha-Paul, Dipti Jain, Anas Alshawa, Jeane Painter, Kenneth Hess, Rivka Colen, Chantale Bernatchez, Charles Lu, Bettzy Stephen, Coya Tapia, Tito Mendoza, Funda Meric-Bernstam, Aung Naing
The University of Texas MD Anderson Cancer Center, Houston, TX, USA
Correspondence: Aung Naing (anaing@mdanderson.org)
Background
Cutaneous squamous cell carcinoma (cSCC) is the 2nd most common malignancy in the US. While the majority of patients are cured with surgery, approximately 6,400 die annually as a consequence of disease. There is limited data on the role of chemotherapy in the treatment of metastatic cSCC. The high-mutational burden, the presence of tumor-infiltrating lymphocytes, immunosuppression as a risk factor, and evidence of direct immunosuppressive effects of UV radiation suggest that cSCC is appropriate for the clinical study of checkpoint inhibitors.
Methods
This is a phase II study of pembrolizumab in patients with rare tumors including a pre-specified cutaneous squamous cell carcinoma cohort (http://ClinicalTrials.gov identifier, NCT02721732). Patients received pembrolizumab 200 mg intravenously once every three weeks. The response was assessed every nine weeks using RECIST1.1. The primary end point was progression-free survival at 27 weeks (PFS27wk). Mandatory biopsies are taken at baseline, on cycle 1 day 15 -21, and at the time of progression.
Results
A total of 11 patients were enrolled and treated. At the date of analysis, patients received a median of 5.5 treatment cycles (range 1-15). Three out of eleven patients have been treated beyond 27 weeks. Four patients had a partial response and the tumor reduction from the baseline have reached 33%, 55%, 63%, and 70%. The fifth patient had stable disease while the other five patients had a progressive disease within 27 weeks of starting the study. The last patient has not had his first restaging scans yet. The safety profile of pembrolizumab was very favorable.
Conclusions
Single agent pembrolizumab may be effective in a patient with advanced cutaneous squamous cell carcinoma. Expanding the study is needed to verify the initial observation. Translational data will be presented at this meeting.
Trial Registration
http://ClinicalTrials.gov identifier, NCT02721732.
P426 Antibody-drug conjugate induced cytotoxicity of tumor cell lines by targeting the SAS1B N-terminus
Arabinda Mandal1, Mriganka Mandal2, Walter Olson1, Jagathpala Shetty1, Kiley Knapp3, Eusebio Pires1, Todd Bauer1, Timothy Bullock4, John Herr4, Craig Slingluff1
1University of Virginia, Charlottesville, VA, USA; 2Albemarle High School, Charlottesville, VA, USA; 3University, Charlottesville, VA, USA; 4University of Virginia.edu, Charlottesville, VA, USA
Correspondence: Craig Slingluff (wco3j@virginia.edu)
Background
SAS1B (ASTL, Ovastacin) is a zinc metalloproteinase found normally in growing oocytes that has been detected in uterine cancer cells, and its expression is restricted to oocytes, among adult tissues. Thus, it is classified as a cancer-oocyte neoantigen [1]. Treatment of uterine tumor cells with anti-SAS1B polyclonal antibodies and complement arrested growth, and a saporin-immunotoxin directed to SAS1B caused cell death. The goal of the present study was to develop immunotherapeutic monoclonal antibodies against the human SAS1B target, to reduce risks of off-target effects during therapy.
Methods
Monoclonal antibodies (mAbs) were generated to recombinant (r) human (h) SAS1B immunogen without its signal peptide.
Results
SAS1B transcript in human uterine and lung cancer cell lines was confirmed by PCR and sequencing. SB2 and SB5 mAbs demonstrated Western immunoreactivity to rhSAS1B expressed in E. coli or HEK293T cells, and recognition of SAS1B was confirmed by mass spectrometry on immunoprecipitated antigen. SAS1B deletion constructs mapped the epitopes recognized by the SB2 and SB5 mAbs to the N-terminal domain between residues 32-40. Surface expression of SAS1B in live uterine, lung and pancreatic tumor cell lines was detected by both mAbs. The expression of SAS1B N-terminus on the surface of live tumor cells and its cytoplasmic expression from uterus (SNU539, Fig. 1B), ovary, breast, melanoma (Malme3M, Fig. 1C) lung (H226, Fig. 1D), and were confirmed by flow cytometry and SAS1B peptide blocking. SAS1B expression was not evident on the surface of normal peripheral blood mononuclear cells. Monoclonal antibodies SB2 and SB5, complexed with a Fab-Duocarmycin DM, that incorporated a cathepsin-cleavable linker arm, were cytotoxic to SAS1B positive uterine (SNU539, Fig. 1A), pancreatic and lung tumor cells at 10 - 100 picomolar concentrations.
Conclusions
The SB2 and SB5 mAbs offer promise as candidate immunotherapeutic entities for targeting SAS1B positive cancer cells with defined epitopes located in the SAS1B N-terminal domain suitable for immunotherapeutic targeting.
References
1. Pires ES, et al. Membrane Associated Cancer-Oocyte Neoantigen SAS1B/Ovastacin is a Candidate Immunotherapeutic Target for Uterine Tumors. Oncotarget. 2015; 6:30194-30211.

Profile of SAS1B expression on tumor cell lines by ADC and flow cytometry
P427 Democratizing analysis of cancer data using the cancer genomics cloud
Anurag Sethi (anurag.sethi@sbgenomics.com)
Seven Bridges Genomics, Inc, Boston, MA, USA
Background
The advent of next generation sequencing has resulted in the generation of petabytes of multi-dimensional information, but the access and analyses of this data remains challenging. This difficulty is exemplified when we consider data generated by the efforts of The Cancer Genomics Atlas (TCGA) network. Historically, the only means for gaining insight from the TCGA data was to download the complete TCGA repository, which can require several weeks with a highly optimized network connection, followed by analyses in a very expensive high performance computational cluster.
Methods
The Cancer Genomics Cloud Pilots project seeks to democratize cancer data analysis by co-localizing data with the computational resources. Funded by the National Cancer Institute, the Cancer Genomics Cloud Pilot (www.cancergenomicscloud.org) project enables researchers to leverage the power of cloud computing to gain actionable insights on cancer biology and human genetics from massive public datasets including TCGA.
Results
The CGC removes the need to download data, enables easy querying, and much more.
Conclusions
We will highlight our approach to optimized computation, data mining, and visualization solutions that address the challenges associated with analyses of petabyte-scale datasets and beyond.
P428 PD-1 blockade activates CD4 T cells and the innate immune response for glioblastoma eradication
Sarah R. Klein, Maria C. Speranza, Prafulla C. Gokhale, Margaret K. Wilkens, Kristen L. Jones, Apoorvi Chaudhri, Paul T. Kirschmeier, David A. Reardon, Gordon J. Freeman
Dana-Farber Cancer Institute, Boston, MA, USA
Correspondence: Gordon J. Freeman (maria_speranza@dfci.harvard.edu)
Background
Cancer immunotherapy has come of age as a result of substantive advances in basic and translational tumor immunology [1]. In fact, multiple advanced clinical trials with human anti-PD-1 antibodies (i.e. nivolumab and pembrolizumab) have convincingly established the ability of anti-PD-1 antibodies to trigger clinically significant tumor destruction in multiple tumor types, reinforced by dramatic anti-cancer responses in animal models and encouraging durability in early clinical trials [2].
Methods
Through flow cytometry and in in vivo intracranial injection of mouse glioma tumor cells we recently demonstrated that PD-1 blockade elicits an anti-tumor immune response resulting in tumor rejection and long-term survival in approximately 50% of mice, despite the absence of accumulating CD8+ cytotoxic T cells in the tumor or draining lymph nodes [3].
Results
In this investigation, we provide evidence for the role of conventional CD4+ T cells and the innate immune response in PD-1 mediated anti-glioma immunity in this model. In response to anti-PD-1 monotherapy, intratumoral CD4+ T cells, but not CD8+ T cells, expressed significantly elevated levels of IFN-γ and TNF-α pro-inflammatory cytokines and the cytotoxic enzyme, granzyme B. Tbet, GATA3, and EOMES, transcription factors required for T cell proliferation, activation, and effector function, were also up-regulated in CD4+, but not CD8+ T cells in the brains of mice treated with PD-1 mAbs when compared to controls. We demonstrated that depletion of CD4+ or CD8+ T cells, but not NK, was sufficient to completely ablate anti-PD-1-mediated tumor eradication and long-term survival. CD4+ T cell activation was accompanied by the classical activation and M1 polarization of resident microglia and tumor-infiltrating macrophages. Moreover, CSF-1 inhibition with PLX3397 to deplete microglia and macrophages did not affect tumor growth, but did lead to a significant survival advantage when combined with anti-PD-1 mAb.
Conclusions
Together, these studies demonstrate for the first time a role for CD4+ T cells and the innate immune response in the eradication of glioblastoma by PD-1 blockade.
References
1. Mittal D, Gubin MM, Schreiber RD et al. New insights into cancer immunoediting and its three component phases--elimination, equilibrium and escape. Current opinion in immunology. 2014;27:16-25.
2. Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027-1034.
3. Checkpoint Blockade in an Orthotopic, Immunocompetent Model. Cancer Immunol Res. 2015;4(2): 124–35.
Personalized Vaccines and Technologies/Personalized Medicine
P429 Nanoparticle based enrichment and expansion of self and neo-epitope specific CD8+ T cells in murine melanoma
Catherine A Bessell, Joan Glick Bieler, John-William Sidhom, Jonathan P Schneck
Johns Hopkins University, Baltimore, MD, USA
Correspondence: Jonathan P Schneck (cbessel1@jhmi.edu)
Background
T cell responses against neo-antigens has created a new avenue to target and kill patients’ tumors while decreasing autoimmune side effects. Neo-antigen-specific CD8+ T cells have the potential benefit to be a source of T cells that are not limited by peripheral tolerance or deleted from the T cell repertoire by central tolerance. To understand the differences between T cell responses against neo- and self-antigens, these two types of T cell populations must be compared side-by-side for anti-tumor responses and phenotypic differences.
Methods
Following another group that identified mutated proteins in B16 murine melanoma [1], our current work has identified the short peptide epitopes that can stimulate neo-antigen T cell responses. Artificial antigen presenting cells (aAPCs) were used to access neo- and self-antigen tumor specific responses in the endogenous T cell population of naïve and B16 tumor bearing animals. In a B16-SIY melanoma model, neo-epitope Kb-SIY serves as a model to collect T cells for self and neo-epitopes and analyze the T cell receptor (TCR) repertoire of the separate populations.
Results
After exposure to tumor, neo-epitope T cell responses showed larger fold expansion, while T cell expansions for self-antigen Kb-TRP2 expanded similar in the naïve animals. TCR repertoire analysis of neo- and self-antigen reactive T cells showed a highly conserved V beta gene usage in the naive Kb-SIY TCR repertoire, while the naïve Kb-TRP2 TCR repertoire had a more broad V beta selection. The tumor did not alter the V beta usage for Kb-SIY repertoire, however the Kb-TRP2 repertoire changed to different V beta genes after tumor exposure.
Conclusions
The increased expansion ability of neo-antigens after tumor exposure demonstrates the potential anti-tumor ability of neo-epitope responses compared to self-epitope T cell responses. TCR repertoire analysis of neo- and self-antigen reactive T cells shows that the tumor shifts the TCR clonal diversity of self-antigen specific populations to but leaves the neo-epitope repertoire intact and similar to the naïve repertoire. From this work, we show proof of concept of a streamlined method to screen candidate neo-epitopes, identify rare endogenous T cell populations, and assess the benefit of T cell directed immunotherapies on different T cell populations.
References
1. Castle JC, Kreiter S, Diekmann J, Lower M, van de Roemer N, de Graaf J, Selmi A, Diken M, Boegel S, Paret C, Koslowski M, Kuhn AN, Britten CM, Huber C, Tureci O, Sahin U. Exploiting the Mutanome for Tumor Vaccination. Cancer Res. 2012; 72:1081-1091.
P430 Neoantigen identification using ATLASTM across multiple tumor types highlights limitations of prediction algorithms
Jason Dobson, Huilei Xu, Kyle Ferber, Johanna Kaufmann, Judy Jacques, Christine McCoy, Michael O'Keeffe, Yana Ostrovsky, Crystal Cabral, Pamela Carroll, Theresa Zhang, Jessica Flechtner, Wendy Broom
Genocea Biosciences, Cambridge, MA, USA
Correspondence: Wendy Broom (jason.dobson@genocea.com)
Background
Neoantigens arise from tumor-specific, somatic mutations and have the potential to be recognized by T cells that are associated with anti-tumor immune responses. Since they are non-self, they are hypothesized to provide an attractive therapeutic modality since T cells that can respond to those sequences have not undergone thymic selection. The ATLASTM platform enables identification of biologically relevant CD4+ and CD8+ T cell neoantigens in any subject in an unbiased manner.
Methods
Multiple patients with solid tumors were analyzed. CD14+ monocytes and T cell subsets were isolated from patient peripheral blood mononuclear cells. Monocytes were differentiated into dendritic cells (MDDC), and T cells were non-specifically expanded. Whole exome sequencing was performed on tumor biopsies and matched normal genomic DNA, and tumor specific changes (single nucleotide variants and insertion/deletions) were identified and cloned into E. coli expression vectors with and without co-expressed listeriolysin O to enable presentation via MHC class I or class II, respectively. For each patient, their unique clones were co-cultured with autologous MDDCs in an ordered array, then their CD4+ or CD8+ T cells were added and incubated overnight. T cell activation was determined by measurement of TNF-α and IFN-γ in the supernatants by meso-scale discovery. Cytokine concentrations were normalized against responses to negative control bacteria and neoantigens were defined as clones that elicited responses >3 median absolute deviations of the median of negative controls.
Results
ATLASTM identified CD4+ and CD8+ T cells responses to up to 15% of mutant polypeptide sequences, across a broad cohort of patients with different tumor types, including tumors with either low or high mutational burden. “Inhibitory” neoantigens, which shut off all cytokine production, were also identified in each subject. Many neoantigens were not predicted by algorithms. When exploring neoantigens by tumor type, no patterns in overall mutational burden, RNA expression level, or DNA mutant allele frequency have yet been identified.
Conclusions
The ATLASTM platform empirically defines which potential neoantigens created by somatic mutations elicit immune responses in individual patients independently of a patient’s HLA type and T cell receptor repertoire. The identification of activating and inhibitory neoantigens for CD8+ T cells, as well as for CD4+ T cells for which the algorithmic approaches do not perform nearly as well, provides the opportunity to identify better targets to include in a vaccine formulation. Genocea is currently developing personalized cancer vaccines with neoantigens prioritized by ATLASTM which we hypothesize will provide better therapeutic impact than algorithmic approaches.
P431 Identification of immunogenic breast cancer neoantigens exposed by radiation therapy
Claire Lhuillier, Nils Rudqvist, Takahiro Yamazaki, Tuo Zhang, Lorenzo Galluzzi, Sandra Demaria
Weill Cornell Medical College, New-York, NY, USA
Correspondence: Sandra Demaria (cfl2002@med.cornell.edu)
Background
Recent studies have highlighted the key role of mutation-generated neoantigens in tumor response to immunotherapy [1]. We have previously shown in the 4T1 mouse model (syngeneic with BALB/c mice) of immune-checkpoint blockade-resistant metastatic breast cancer that local radiation therapy (RT) combined with CTLA-4 blockade induces the CD8+ T cell-mediated regression of irradiated tumors and limits metastatic lung colonization [2]. Preliminary analysis of the T-cell receptor repertoire indicated that unique clonotypes are expanded in treated tumors, suggesting that tumor rejection involves T cells reactive to a set of tumor antigens that are made available to the immune system by RT. Therefore, we hypothesize that RT, by changing the transcriptional profile of cancer cells, may expose antigenic mutations not transcribed at sufficient levels in untreated tumors and hence promote priming of T cells to these unique mutated antigens.
Methods
We performed whole-exome sequencing and RNA sequencing (RNA-seq) of 4T1 cells irradiated (8GyX3) or not (0Gy) in vitro to identify tumor-specific mutations. We used several algorithms to predict MHC-I (H2d)-binding epitopes from these mutated genes, and we selected those with a predicted affinity <500 nM. Based on the RNA-seq data, we prioritized variants that were upregulated by radiation. These candidate peptides were synthesized and tested in vitro for binding to H2-Ld or H2-Kd in a MHC stabilization assay using RMA-S cells. Finally, the mutated peptides with the highest affinity were used to vaccinate BALB/c mice, followed by challenge with 4T1 cells to test for the induction of protective anti-tumor immunity.
Results
Out of 309 total mutations initially identified in the 4T1 cancer cells, we selected and tested in vitro 17 candidate neo-epitopes. Two of them bound H2-Ld with a high affinity and were used alone or in combination to vaccinate mice. Our preliminary data indicate that the combination of these two neoepitopes is immunogenic and delays the tumor growth after challenge with 4T1 cells.
Conclusions
In conclusion, these data provide initial proof-of-principle evidence that RT can expose existing neoantigens to the immune system.
References
1. Schumacher TN, and Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015; 348: 69-74.
2. Demaria S, Kawashima N, Yang AM, Devitt ML, Babb JS, Allison JP, and Formenti SC Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2005;11:728-734.
P432 Exploiting large scale HLA peptidomics to generate novel Immunotherapies; A data-driven approach to neoantigen prioritization
Alex S. Powlesland, Geert P.M. Mommen, Ricardo J. Carreira, David Lowne, Michael J. Cundell, Floriana Capuano, Bent K. Jakobsen
Immunocore Ltd, Abingdon, United Kingdom
Correspondence: Alex S. Powlesland (alex.powlesland@gmail.com)
Background
Peptides presented to the immune system on HLA complexes are valuable targets for immunotherapeutic treatments. Identifying the full complement of peptides derived from a particular protein that are presented on major class I HLA restrictions will provide a vital step toward increasing the speed and viability of many immunotherapeutic strategies. Advances in next generation sequencing and single cell technologies have enabled the accurate capture of somatic mutations accumulated by a tumour, yet a significant hurdle remains how this information can be utilised for immunotherapeutic benefit. In particular, identifying which somatic mutations produce neoantigens, (peptides that contain a somatic mutation and are presented to the immune system in complex with HLA), is crucial to linking genetic changes with immunological impact.
Methods
Our approach to understanding the targetable human HLA peptidome is based on three key principles; achieving full proteome coverage, maximising individual protein coverage, and focusing on dominant HLA restrictions. By integrating novel cell biology, mass spectrometry, and bioinformatic technologies in over 1000 individual experiments we have dramatically increased the depth of the HLA ligandome captured and achieved near total coverage of the protein-coding genome, with 90% of the proteome captured for HLA-A*02:01.
Our comprehensive genome coverage has enabled us to probe both directly and indirectly for the presence of neoantigens. Known somatic mutations within immortalised lines were used to generate bespoke reference databases that has led to direct identification of many hundreds of neoantigens.
Results
Proteins that were found to contain neoantigens appeared to follow the processing and presentation behaviours of their unmutated equivalent. We have therefore found our HLA peptide dataset is able to offer significant value in predicting the likelihood of a somatic mutation creating a neoantigen.
To test this, somatic mutations reported in 980 cell lines were probed against the database of HLA peptides. On average we find one peptide containing the mutated amino acid for every 5 somatic mutations reported. By incorporating the HLA background of the cell carrying the mutation we narrow this prediction to one high affinity HLA peptide for every 14 somatic mutations reported. Comparing the peptides predicted in this analysis with those directly identified by mass spectrometry, we are able to show that we can prioritise mutation data by accurately predicting the presence and relative abundance of neoantigens.
Conclusions
An integrative approach to HLA peptidomics has delivered a powerful reference database that can be used as a source for developing novel immunotherapies.
P433 Computational pipeline for the PGV-001 (personalized genomic vaccine) clinical trial
Alexander Rubinsteyn1, Julia Kodysh2, Nina Bhardwaj1, Jeffrey Hammerbacher1
1Icahn School of Medicine at Mount Sinai, New York, NY, USA; 2Icahn School of Medicine, New York, NY, USA
Correspondence: Alexander Rubinsteyn (alex.rubinsteyn@gmail.com)
Background
PGV-001 (NCT02721043) is a phase I clinical trial at Mount Sinai Hospital, studying the safety and immunogenicity of a multi-peptide personalized genomic vaccine for treatment of several different malignancies. Patients are treated with a TLR3 agonist (Poly-ICLC) and ten synthetic long peptides containing tumor mutations that are predicted to be abundantly expressed and to form mutated MHC ligands. The personalized vaccine is administered as an intramuscular injection and is given to each patient 10 times over a span of 6 months. Thus far, two patients (of an eventual twenty) have enrolled in the trial and one has been treated. The vaccine is administered in the adjuvant setting for patients who undergo a complete resection and have no evidence of residual disease.
Methods
Tumor DNA and RNA are extracted from fresh frozen tissue immediately following surgery. Normal DNA is extracted from a patient blood sample. Both DNA samples are enriched for coding regions using an exon capture kit. Normal DNA is sequenced to mean on-target coverage of 150x, whereas tumor DNA is sequenced to 300x coverage. RNA is enriched for mRNA using poly-A capture and then >100M paired-end reads are sequenced. Somatic variants are detected from the tumor and normal DNA sequencing data. The RNA sequencing data is then used to assemble the most abundant coding sequence for each variant, which naturally phases adjacent variants and allows for an allele-specific estimate of expression. Each coding sequence is then translated to a mutated protein fragment, which is ranked according to (1) expression and (2) predicted binding affinity to patient MHC I molecules. The window of wildtype amino acids around a mutation in each peptide is optimized to improve the odds of successful solid phase synthesis.
Results
Though only two patients have enrolled in PGV-001 so far, we have evaluated the pipeline on a variety of patient (and mouse) samples across different kinds of cancers. We demonstrate that even cancers with intermediate mutational burden still yield sufficient predicted neoantigens to potentially benefit from vaccination. Furthermore, even with our existing attempt to optimize peptide sequences for manufacturability, we have still found many peptide sequences cannot be synthesized or purified. Analysis of these peptide sequences hint at additional rules which may improve synthetic yield for peptide-based neoantigen vaccines.
Conclusions
We hope that discussion of the design and rationale for PGV-001's computational pipeline will help other groups start their own personalized vaccine programs.
Trial Registration
NCT02721043.
P434 Great Apes Adenoviral vaccine encoding neoantigens synergizes with immunomodulators to cure established tumors in mice
Anna Morena D'Alise1, Gabriella Cotugno1, Guido Leoni1, Francesca Langone1, Imma Fichera1, Maria De Lucia2, Rosa Vitale1, Adriano Leuzzi1, Elena Di Matteo1, Antonella Folgori1, Stefano Colloca1, Alfredo Nicosia1,2, Jonathan Zalevsky3, Elisa Scarselli1
1Nouscom srl, Rome, Italy; 2University of Naples Federico II, Naples, Italy; 3Nektar Therapeutics, San Francisco, CA, USA
Correspondence: Elisa Scarselli (g.napolitano@nouscom.com)
Background
Current immunotherapies based on checkpoint inhibitors (CPI) revealed the importance of neoantigens-specific T cells for tumor control. Here, we developed a novel neoantigens vaccine approach based on the use of a viral vector, Great Apes Adenovirus (GAd), encoding multiple cancer neoantigens in tandem. NKTR-214 is a CD122-biased cytokine agonist currently in clinical trials. NKTR-214 is designed to expand a specific population of cancer-killing T cells, known as tumor-infiltrating lymphocytes. Synergy between the two drugs was investigated.
Methods
Balb/c mice were implanted subcutaneously with CT26 colon carcinoma cells. Once tumors were established, mice were randomized and vaccinated with a single intramuscular injection of GAd encoding up to 20 different CT-26 neoantigens identified by NGS and selected with a NousCom proprietary pipeline. NKTR-214 was administered i.v. on q9dx3 schedule. Tumor growth in treated animals was monitored and neoantigen-specific T cells were measured both in the periphery and in tumors.
Results
Combination treatment of a GAd-CT26 neoantigens vaccine with NKTR-214 results in tumor regression in 90% of mice. Single agent NKTR-214 treatment led to tumor regression in < 40% of mice. Interestingly, all animals cured by the combo were resistant to a second tumor challenge. NKTR-214 treatment rescued CD4 T cell reactivity against at least one of the predicted neoantigens, likely induced by the tumor itself. GAd vaccination in the presence of NKTR-214 induced a greater breadth and depth of immune response. When compared to Gad vaccine alone, the Gad + NKTR-214 combination had immune reactivity detected against a larger number of vaccine-encoded neoantigens and there was a greater proportion of IFN-gamma+ CD4 and CD8 T cells against each vaccine-encoded neoantigens. Tumors in regression of mice treated by the combo were highly enriched in T cells reactive to vaccine-encoded neoantigens.
Conclusions
Vaccination with GAd encoding neoantigens is a very effective and safe approach to strengthen and broaden tumor-killing T cells. Therapeutic cancer vaccines have been unsuccessful in the past likely because T cell growing capacity in the tumor is suppressed by the tumor itself. Vigorous growth of vaccine-induced T cells in the tumour can be successfully achieved by the cytokine NKTR-214, overcoming the tumor inhibitory activity. NKTR-214 was shown to be safe and well tolerated in cancer patients and GAd vaccines have been safely used in many healthy volunteers, opening up the opportunity for a fast evaluation of a combo treatment in the clinic.
P435 Immunogenomics for the development of personalized T cell therapy for ovarian cancer
Muzamil Want, Sebastiano Battaglia, Takemasa Tsuji, Richard Koya
Roswell Park Cancer Institute, Buffalo, NY, USA
Correspondence: Sebastiano Battaglia (muzamil.want@roswellpark.org)
Background
Ovarian cancer (OC) is the fifth leading cause of cancer death in the United States with approximately 20,000 women being diagnosed every year. Since OC development is mainly asymptomatic, patients are often diagnosed at late stage and present local and distal metastases. This offers a clinical challenge, as roughly 70% of the patients develop chemoresistant disease. With the advent of cancer immunogenomics it is now possible to identify tumor specific mutations, or neoantigens/neoepitopes, that can be exploited for the development of personalized T cell therapies via DC vaccines or adoptive cell therapy (ACT). We therefore hypothesize that tumor cells isolated from patients with ovarian cancer offer a unique opportunity to identify clinically actionable tumor neoantigens.
Methods
Somatic mutations were identified by whole exome sequencing (WES) and RNASeq analysis in tumor samples, using PBMCs as germline control. In parallel ovarian cancer PDXs were established in NSG mice by injecting 2×106 patient tumor cells/mouse subcutaneously (SQ, n=20). Tumor growth was monitored weekly via caliper measurement. Mutational analysis and immunogenicity prediction for the mutated peptides was done using a combination of home-developed bioinformatic approaches and NetMHC. Immunogenicity predictions were validated in-vitro by pulsing wild type and mutated peptides into CD8+ depleted PBMCs and co-cultured with CD8+ T cells for up to 20 days. T cell activation was quantitated via ELISA measuring IFN-g release and via flow cytometry by staining for CD137 expression.Lastly, the therapeutic potential of ACT targeting neoantiges was tested in-vivo in ovarian cancer PDXs and tumor size is being monitored weekly.
Results
We have established patient-derived xenografts (PDX) mice and interrogated the tumor mutational landscape. Targeted mutations were confirmed in the following PDX passages by NGS and Sanger sequencing. Out of 168 mutated peptides predicted from the patient tumor, 13 were found to have high affinity for different HLAs compared to the wild type peptide. In vitro screening of high affinity peptides showed that few of the mutated peptides can activate the T cells from patient PBMCs and tumor infiltrating lymphocytes thereby confirming the presence of neoantigen reactive T cells.
Conclusions
In this study, we demonstrate that the patient’s own T lymphocytes can recognize tumor neoantigens and could be used as personalized therapy in ovarian cancer patients
P436 In vivo self-assembled albumin/vaccine nanocomplexes for lymph-node-targeted vaccine delivery in combination cancer immunotherapy
Guizhi Zhu, Xiaoyuan Chen
National Institutes of Health, Bethesda, MD, USA
Correspondence: Guizhi Zhu (zhu.julian21@gmail.com)
Background
Cancer immunotherapy aims to normalize the immune system in patients to harness the power of the immune system to systemically treat cancer. Molecular vaccines hold tremendous potential for tumor immunotherapy, but its clinic outcome has yet been optimal, largely due to inefficient co-delivery of heterogeneous peptide antigens and adjuvants to secondary lymphoid organs such as lymph nodes (LNs). Nanovaccines were attempted to improve vaccine delivery, however, clinical translation of most of them have been impeded by complicated large-scale manufacturing and safety concerns.
Methods
We developed an albumin-binding Evans blue derivative as LN-homing moieties and cell uptake mediators for cancer vaccine delivery. The safety of this derivative has been validated in 2 ongoing clinical trials involving more than 200 patients and healthy volunteers. By conjugating this derivative with molecular vaccines (CpG adjuvant, B16F10 melanoma-associated antigen Trp2, and MC38 colon adenocarcinoma-specific neoantigen Adpgk), we developed albumin-binding vaccine (AlbiVax) (Fig. 1A) to efficiently co-deliver molecular adjuvants and antigens to LNs. By PET imaging of radiolabeled AlbiVax in mice, we systematically optimized AlbiVax for LN-targeted delivery, and light-sheet fluorescence imaging and super-resolution confocal microscopy were employed to study vaccine behaviors in transparent LNs and single antigen-presenting cells (APCs). In three tumor models, we studied the antigen-specific immunogenicity and tumor therapy efficacy of AlbiVax, either alone or in combination with anti-PD-1 or Abraxane.
Results
Quantitative PET imaging revealed that AlbiVax was delivered to LNs more efficiently (20-fold for CpG, up to 91-fold for antigens) than unconjugated vaccines or vaccines emulsified in Incomplete Freud’s Adjuvant (IFA) (Fig. 1B, Fig. 1F). Light-sheet fluorescence imaging elucidated the distribution of AlbiVax in transparent draining LNs (Fig. 1, Fig. 1D); and super-resolution imaging revealed efficient intracellular co-delivery of AlbiVax via albumin into endolysosome of APCs (Fig. 1E). AlbiVax elicited >21-fold higher frequency of Antigen-specific CD8+ cytotoxic T lymphocytes (CTLs) than IFA-emulsified vaccines and induced immune memory for > 5 months (Fig. g). AlbiVax dramatically regressed or inhibited the progression of established primary or lung metastatic EG7.OVA (Fig. 1G), B16F10 (Fig. 1H), and MC38 tumors (Fig. 1I). Remarkably, combining AlbiVax with immune checkpoint inhibitor anti-PD-1 further improved the therapeutic efficacy, and led to the complete regression of 60% MC38 tumors for at least 5 months, and triple combination of AlbiVax, anti-PD-1, and Abraxane exhibited synergistic therapeutic efficacy (Fig. 1H, Fig. 1I). AlbiVax showed excellent safety profile throughout the study.
Conclusions
AlbiVax represents a promising safe, widely applicable, and robust T cell vaccine for combination cancer immunotherapy.
In vivo self-assembled albumin/vaccine nanocomplexes for lymph-node-targeted vaccine delivery in combination cancer immunotherapy. (a) Schematics of albumin-binding vaccine (AlbiVax) for LN-targeted vaccine delivery for cancer immunotherapy. (b, f) by PET imaging of radiolabeled AlbiVax in small animals, AlbiVax was systematically optimized for optimal LN-targeted delivery. (c, d) By light-sheet fluorescence imaging, we elucidated the distribution of AlbiVax in draining LNs that were cleared to be transparent. (e) By super-resolution imaging, we discovered efficient intracellular co-delivery of AlbiVax via mouse serum albumin (MSA) into endolysosome of bone marrow derived dendritic cells (BMDCs). (g) AlbiVax elicited 21-fold higher frequency of Antigen-specific CD8+ cytotoxic T lymphocytes (CTLs) than IFA-emulsified vaccine. (h-j) AlbiVax dramatically regressed or inhibited the progression of EG7.OVA (g), B16F10 (h), and MC38 tumors (i)
PHYSICIAN/NURSE/PHARM: Best Practices for Improving Cancer Immunotherapy
P437 Mapping the treatment pathway for metastatic uveal melanoma (mUM) patients in England: A qualitative pilot study
Elisabeth Adams1, Chih-Yuan Cheng1, Joseph Sacco2, Sarah Danson3, Pippa Corrie4, Paul Nathan5, Peter Szlosarek6, Joanne Upton2, Abolore Amuludun6, Toby Toward7
1Aquarius Population Health Limited, London, United Kingdom; 2The Clatterbridge Cancer Centre, Wirral, United Kingdom; 3Weston Park Hospital, Sheffield, United Kingdom; 4Addenbrooke's Hospital, Cambridge, United Kingdom; 5Mount Vernon Cancer Centre, London, United Kingdom; 6Barts Cancer Institute, London, United Kingdom; 7Immunocore Limited, Abingdon, United Kingdom
Correspondence: Toby Toward (chihyuan.cheng@aquariusph.com)
Background
mUM is a rare disease (annual UK incidence <150 patients), with a reported median overall survival of 7-12 months. In 2015, the UK published their first national uveal melanoma guidelines [1]. However, with few therapeutically effective options available for metastatic patients, it is unclear what patients actually receive as standard of care (SoC). This pilot study aimed to map the real-world pathway for recently diagnosed mUM patients, through qualitatively evaluating their clinical management and treatments received within SoC.
Methods
Based on treatment recommendations within national guidelines, an outline decision-tree for mUM patients was developed. On presenting the decision-tree to 5 specialist centres across England (January-June, 2017), 6 senior clinicians were interviewed about their typical: clinical management; medical resource use; treatment decision-making and patient flow(s) between local and regional facilities, when treating mUM patients. The interview data were analysed to populate centre-level decision-trees, and consolidated to inform a consensus SoC pathway.
Results
Interviews revealed considerable variation in first-line treatments offered at specialist centres:
hepatic resection for liver metastases [1-25%];
loco-regional therapies for liver metastases (including percutaneous hepatic perfusion and radiofrequency ablation) [0-20%];
new treatments in clinical trials (TRAP, SelPac or IMCgp100) [10-80%];
immunotherapies (Ipilimumab, Pembrolizumab or Nivolumab) [0-54%]; and
chemotherapies (Dacarbazine or Temozolomide) [0-1%].
There was, however, consensus on treatment priorities - if liver metastases were operable then surgical options were considered first. If non-operable, clinical trials were subsequently considered, before other therapeutic options. Whilst centres agreed that treatment should be initiated at a specialist (supra-regional) mUM centre, there were mixed attitudes on “if” and “how” patients could receive ongoing treatments at more local centres.
Conclusions
The main reason suggested for treatment pathway variations amongst centres, was the absence of effective treatments. Introducing an effective therapeutic option at a defined optimum point in the pathway would be considered a “catalyst to transform the management of care”. This in turn would lend itself to greater consistency of practice, harmonise care pathways and improve overall outcomes for mUM patients.
References
1. Nathan P, Cohen V, Coupland S, Curtis K, Damato B, Evans J, Fenwick S, Kirkpatrick L, Li O, Marshall E, McGuirk K, Ottensmeier C, Pearce N, Salvi S, Stedman B, Szlosarek P, Turnbull N. United Kingdom Uveal Melanoma Guideline Development Working Group, Uveal Melanoma UK National Guidelines. European Journal of Cancer. 2015; 51: 2404– 2412.
P438 Continued challenges and opportunities in oncologists’ COMPREHension of clinical immunology and its relationship with cancer immunotherapy
Tara Herrmann1, Charlotte Warren1, Haleh Kadkhoda2, Lianne Wiggins3
1Medscape Education Oncology, New York, NY, USA; 2Medscape Education, New York, NY, USA; 3SITC, Milwaukee, WI, USA
Correspondence: Tara Herrmann (therrmann@medscape.net)
Background
Cancer immunotherapy (CIT) is revolutionizing the treatment of cancer. As part of an ongoing educational curriculum, assessment of oncologists' knowledge, competence, and performance in the use of CIT identified multiple challenges and barriers. This study’s objective was to assess and compare practicing oncologists’ comprehension of, and confidence in, the interplay between the immune system and cancer tumor development and metastasis.
Methods
An expert panel was convened to identify knowledge, competence, and performance gaps in the area of CIT. Educational curricula consisting of 8 activities that focused on foundational education were developed in 2014-2016, and posted online. Intra-activity questions embedded in each activity allowed learners to self-report their confidence with CIT concepts, while matching participants’ responses to knowledge-based questions presented before and after education were collected to measure reinforced learning and gains in comprehension. Confidentiality of respondents was maintained. Responses were collected from 5/2014-10/2016.
Oncologists (N=4,684) who participated in the activities demonstrated on average a 20% improvement from initial baseline between years 1 and 2. Additional education in year 3 was provided to help prevent decay in oncologists’ comprehension of immune system functions. However, comparison of oncologists' knowledge of the role of immune checkpoints indicates that while gains observed in year 1 were maintained, a drop off was observed between years 2 and 3. This is likely a result of more data becoming available on alternative CITs while simultaneously less foundational education was available.
Results
Oncologists continued to report a lack of confidence with the concepts of clinical immunology. Only 2%-11% of oncologists were very confident in their comprehension of the relationship between the immune system and tumor development. In addition, fewer than 50% of participants post-activity could recognize common T cell co-receptors that could be targeted as part of CIT. One quarter did not realize that tumors interfere with signal 1 by blocking MHC antigen processing.
Conclusions
This study demonstrates the value of and continued need for educational interventions on CIT. Retention of CIT concepts and improvements in knowledge of clinical immunology should facilitate competence among oncologists, which has valuable implications for use of CIT and for patient outcomes.
P439 PDL-1 status and value for Extrapulmonary small cell carcinomas
Mohammed Salhab, Yiqin Xiong, Karen Dresser, Benjamin Chen, James Liebmann
University of Massachusetts, worcester, MA, USA
Correspondence: Mohammed Salhab (mohammed.salhab@umassmemorial.org)
Background
Extrapulmonary small cell carcinomas (ESCCs) are rare but aggressive tumors with an overall 5-year survival rate of less than 15%. Relapse is common despite rigorous chemotherapy and radiotherapy. Cancer immunotherapy targeting PD-1/PD-L1 pathway emerges as an increasingly appealing strategy [1, 2, 3].
Methods
We investigated PD-L1 expression by immunochemistry (IHC) in ESCCs diagnosed at our institution from 1999-2016. 34 cases with sufficient material were selected for PD-L1 IHC analysis using clone E1L3N. Sites of origin include bladder (9), cervix (3), prostate (2), ovary (2), gallbladder (2), duodenum (1), esophagus (1), biliary tract (1), vagina (1), palate (1), ureter (1), and unknown primary (10).
Results
Two cases showed diffuse PD-L1 expression in both tumor and stromal cells, one from ureter and one from a gallbladder in origin. 10 cases (29%) showed at least patchy PD-L1 expression in stromal cells. 22 cases (65%) were completely negative for PD-L1 expression. Patients of both groups who was treated, did received chemotherapy +/- surgery or radiation. The overall response rate for PDL positive group was 80% while it was 67% for PDL-1 negative group (Table 1). The median overall survival for PD-L1 positive staining group, regardless of stage was 11.5 months’ vs 7 months for PDL-1 negative group (Fig. 1). Patients with limited stage disease with positive PDL-1 showed an impressive 53 months’ survival compared to PDL-1 negative limited disease with 15 months.
Conclusions
This study suggests that PD-L1 staining in extrapulmonary small cell carcinoma may be of prognostic relevance and further studies are required to understand the implications of immune dysregulation in these aggressive tumors. PD-1 inhibitors should be investigated in this group of patients.
References
1. Annemiek ME, Walenkamp A, Sonke G, Sleijfer D. Clinical and therapeutic aspects of extrapulmonary small cell carcinoma. Cancer Treatment Reviews. 2009; 35 :228–236.
2. Wong YN, Jack R, Mak V, Henrik M, Davies E. The epidemiology and survival of extrapulmonary small cell carcinoma in South East England, 1970–2004. BMC Cancer. 2009; 9:209.
3. Brennan S, Gregory D, Stillie A, Herschtal A, Manus M, Ball D. Should Extrapulmonary Small Cell Cancer BeManaged Like Small Cell Lung Cancer? Cancer.2010.
See text for description
See text for description
PHYSICIAN/NURSE/PHARM: Case Studies demonstrating exceptional responses
P440 Durable complete response with PANVAC and Trastuzumab in metastatic triple positive breast cancer (mTPBC)
Margaret Gatti-Mays1, Mahsa Mohebtash2, Ravi Madan1, Julius Strauss1, Marijo Bilusic1, Caroline Jochems1, Myrna Rauckhorst1, Elizabeth Jones3, William Dahut1, Jeffrey Schlom1, James Gulley1
1National Cancer Institute, NIH, Bethesda, MD, USA; 2Medstar Union Memorial Hospital, Baltimore, MD, USA; 3National Institutes of Health, Bethesda, MD, USA
Correspondence: Margaret Gatti-Mays (margaret.gatti-mays@nih.gov)
Background
First line treatment of metastatic HER2+ breast cancer with Docetaxel+Trastuzumab(TH) carries a median PFS of 12months and OS of 40months.1 The addition of Trastuzumab+/-Pertuzumab to chemotherapy prolongs survival in the metastatic setting1 but no therapies are curative. Trastuzumab has immune-related effects including antibody-dependent cell-mediated cytotoxicity and cross-presentation of tumor antigens. PANVAC is a recombinant poxviral vaccine that contains transgenes for MUC-1, CEA and T-cell costimulatory molecules (B7.1, ICAM-1 and LFA-3). For optimal response, PANVAC employs a prime-boost regimen, using vaccinia (PANVAC-V) as the primer and fowlpox (PANVAC-F) as the boost. We report an exceptional response of mTPBC to PANVAC+Trastuzumab after progression on Letrozole+Trastuzumab.
Methods
A pilot study of PANVAC+Sargramostim(NCT00088413) demonstrated minimal side effects and suggested clinical benefit in low disease burden.2 On Day 1(D1), patients received PANVAC-V 2x10e8pfu SC followed by PANVAC-F 1x10e9pfu SC every 2weeks x 3, then monthly x 12 followed by maintenance every 3months until progression. Sargramostim 100μg SC was given on D1-D4 of each PANVAC injection. Scans were performed every 2-3months.
Results
A 32-year-old female, BRCA wildtype, with poorly differentiated, invasive ductal carcinoma, ER60%, PR70%, HER2+(IHC3+) had biopsy-confirmed metastasis to an axillary node and liver shortly after diagnosis. After bilateral oophorectomy, she received THx6cycles followed by a partial hepatectomy, which demonstrated a pathologic complete response. She received radiation to supraclavicular lymph nodes with a tumor bed boost. After 12months on Letrozole+Trastuzumab she recurred. While on Letrozole she completed 6injections for a HER2/neu peptide vaccine trial(NCT00194714). Due to progression in bilateral hilar and subcarinal(2.8cm) lymph nodes determined by PET, she enrolled on trial at NCI in January 2008. Per protocol above, she received PANVAC-V followed by PANVAC-F. Restaging at 10months showed a partial response(0.8cm) and at 18months showed a complete response(CR). She has remained on-study and receives PANVAC-F+Sargramostim every 90days and Trastuzumab every 3weeks. Restaging in May 2017 shows a continued CR at 113months.
Conclusions
This case report highlights the potential of immunotherapy and possibly multiple immunotherapies in a patient with breast cancer. Further study is required to understand the mechanism of response in this patient and if it can be applied on a broader scale.
Trial Registration
NCT00088413
References
1. Swain SM, Baselga J, Kim SB, et al. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N Engl J Med. 2015;372(8):724-734.
2. Mohebtash M, Tsang KY, Madan RA, et al. A pilot study of MUC-1/CEA/TRICOM poxviral-based vaccine in patients with metastatic breast and ovarian cancer. Clin Cancer Res. 2011;17(22):7164-7173.
P441 Activity of Nivolumab (Opdivo®) in relapsed progressive peripheral T-cell Lymphoma-NOS (PTCL-NOS)
Philip A Haddad1, David Sommerhalder2
1Overton Brooks VAMC, Shreveport, LA, USA; 2LSUHSC-S, Shreveport, LA, USA
Correspondence: Philip A Haddad (haddad8838@msn.com)
Background
Nivolumab is a member of the family of check-point inhibitors. It is a PD-L1 inhibitor that has been approved by the FDA as treatment for several solid tumors including non-small cell lung cancer. It’s also approved to treat Hodgkin’s Lymphoma after failure of several lines of therapy. PTCL is often aggressive and incurable. Compared to B-cell lymphomas, they tend to present at more advanced stages and respond less frequently and with shorter duration to the same chemotherapy regimens. Nevertheless, PCTL is still approached with suboptimal B-cell therapeutic strategies. As to the recently approved PTCL specific therapies, they are palliative in nature. An urgent need for better therapeutic agents in this desperate lymphoma continues to exist.
Methods
We report on the activity of Nivolumab as a 4th line treatment in a case of relapsed and rapidly progressive PTCL-NOS.
Results
Our patient was a 62 y.o. white man who was diagnosed with poor risk stage IVA PTCL and Stage IA lung squamous cell carcinoma in 2014. Due to his borderline functional performance status and decreased left ventricular ejection fraction, he was treated with Pralatrexate that is approved for PTCL and has activity in lung cancer. He received 2 cycles of Pralatrexate which stabilized both malignancies for 3 months after which they progressed. Subsequently, he was given Carboplatin and Gemcitabine combination that has activity in both malignancies. This combination seemed to stabilize his lung cancer and PTCL for 4 months. After the 5th cycle, the patient’s lymphoma progressed. At this time, the patient was started on Belinostat to address his PTCL. Initially, PTCL responded to Belinostat and then stabilized for 5 months. After the fifth cycle, his lung cancer started progressing requiring lung cancer directed therapy. He was started on Nivolumab which stabilized his lung cancer. After the 4th dose of Nivolumab further unexpected PTCL response was noted clinically and on follow up imaging. This response was maintained for a period of 15 months spanning 27 cycles of Nivolumab without any significant toxicity. Unfortunately, by the 27th cycles the patient’s lung cancer progressed and he decided to stop all treatment and enroll in hospice where he eventually expired due to his progressive lung cancer.
Conclusions
This case demonstrates for the first time the notable activity and clinical benefit of Nivolumab as a single agent in PTCL-NOS. Further evaluation of Nivolumab’s activity in PTCL as well its incorporation into PTCL treatment approaches need additional study and validation.
P442 Pseudoprogression manifesting as recurrent ascities with anti-PD-1 therapy
Randy Sweis, Yuanyuan Zha, Russell Szmulewitz, Lomax Pass, Tara Chongsuwat, Jason Luke, Thomas Gajewski
University of Chicago, Chicago, IL, USA
Correspondence: Randy Sweis (rsweis@uchicago.edu)
Background
Pseudoprogression occurs when patients treated with immunotherapy have an initial increase in apparent tumor size prior to regression. This phenomenon results from an influx of activated immune cells, occurring in up to 7% of patients [1]. Ascites is also a common manifestation of progressive malignancy from peritoneal metastases. However, ascites has not been previously reported as a manifestation of pseudoprogression. We describe a metastatic urothelial cancer patient with progressive ascites and concurrent reduction in tumor burden on anti-PD-1 therapy.
Methods
Pembrolizumab was given 200 mg IV every 3 weeks. Computed tomography scans were obtained and tumor burden assessed by RECIST v1.1. Peritoneal fluid was analyzed for cytology and fluorescence-activated cell sorting (FACS) using FlowJo10.1r5. Multiplex cytokine assay was performed on peritoneal fluid. Normal donor PBMCs stimulated with phorbol myristate acetate/ionomycin was used as a positive control.
Results
A 62-year-old female with urothelial bladder cancer previously treated with chemotherapy and radiation presented with recurrent metastatic disease including two peritoneal nodules and an enlarged para-aortic lymph node. Pembrolizumab was started and she had a significant partial response with a 49% reduction in target lesions. During therapy, she developed large-volume ascites. Initial paracentesis showed a serum-ascites albumin gradient >1.5, total protein of 1.2 g/dL, and leukocyte count of 231/μL including 45% lymphocytes. Cytology from four separate paracenteses over 8 weeks showed reactive cellular changes/inflammation, but no malignant cells were identified.
FACS analysis on peritoneal fluid showed CD8+ lymphocytes comprised 53% of CD45+CD3+ cells. The majority of CD8+ lymphocytes were PD-1+ (78%). Class II-MHC (HLA-DR) and CD40 ligand (CD154) were expressed on 43% and 20% of CD8+ lymphocytes, respectively. All three markers were increased relative to normal donor PBMC controls, suggesting a locally high number of antigen-experienced, activated T cells. FoxP3+CD25+ Tregs were also detected in 17% of the CD45+CD3+CD4+ compartment. Multiplex cytokine assay for 38 analytes was performed. The most significantly elevated analytes were IL-6, IL-15, G-GSF and FGF-2 (each >2-fold above control).
Conclusions
Pseudoprogression in cancer patients treated with immunotherapy can manifest as recurrent ascites due to a rapid influx of activated T lymphocytes into the peritoneum. Since ascites can also be caused by disease progression, it is important to clinically distinguish the two, so that treatment may be appropriately continued or stopped.
References
1.Blumenthal, et al. Treatment beyond progression with immune checkpoint inhibitors—known unknowns. JAMA Oncology. 2017.
P443 Malignant melanoma metastatic to the heart: exploring patient outcomes and cardiovascular complications in the era of immunotherapy
Laura Talamo, William Grosh
University of Virginia, Charlottesville, VA, USA
Correspondence: William Grosh (lt3j@virginia.edu)
Background
Cardiac metastasis (CM) from melanoma is being recognized more frequently, but often presents without signal symptoms. A rate of CM of 64% has been quoted in one autopsy series [1], though the clinical diagnosis rate is considerably lower [2]. We evaluated patients with known CM for cardiac complications and impact of therapy, and defined survival among these patients.
Methods
This retrospective review identified patients with CM seen in the University of Virginia Oncology clinic between 2010 and 2017. Eligible patients were at least 18 years old with metastatic melanoma (MM) and CM.
Our primary outcome metrics were 2 year survival from time of diagnosis of melanoma (M) and MM. We recorded cardiac complications, diagnostic procedures, patient outcomes, and treatment modality. Ten patients were identified with the demographics outlined below. Longest follow up was 21 months from diagnosis of CM (DCM).
Results
Two year survival from M diagnosis was 70%. Two year survival from DCM could not be calculated (two patients are still living at 9 and 21 months from DCM). Figure 1 shows Kaplan-Meier survival analysis. Of the eight patients who died, all died within one year of DCM. Responses to immunotherapy (IO) are shown in Table 2.
Conclusions
The rate of cardiac complications was low. Only one death was a direct result of CM (patient 10).
90% of patients received IO. At the time of data collection, two patients remain (2 and 3) alive nine and 21 months since DCM; both received >20 cycles of P. Patients 1, 2, 3, 8, 9, and 10 received IO after DCM, but only patient 3’s CM responded to IO (IN induction, P maintenance). Patient 1 did not respond to I; patient 2 was lost to follow up on IL-2; patient 8’s response cannot be determined due to death from septic shock; patient 9 progressed through P; and patient 10 did not respond after one cycle of I.
This study is limited by small patient numbers, but supports a poor prognosis for patients with CM. Responses can be seen with IO, specifically PD-L1 inhibitors, which may improve survival. Other treatments cannot be assessed. The only patient who survived one year after DCM has had an ongoing response to P.
References
1. Glancy DL, Roberts WC. The heart in malignant melanoma. A study of 70 autopsy cases. Am J Cardiol. 1968;21:555–71.
2. Villa et al. Cardiac metastases of melanoma as first manifestation of the disease. J Radiol Case Rep. 2014;1;8(4):8-15.
PHYSICIAN/NURSE/PHARM: irAE Management: Clinical Care and Best Practices
P444 Inflammatory arthritis induced by the use of checkpoint inhibitors for immunotherapy of cancer
Noha Abdel-Wahab1,2, Jean H. Tayar1, Adi Diab1, Sang T. Kim1, Huifang Lu1, Maria E. Suarez-Almazor1
1The University of Texas MD Anderson Cancer Center, Houston, TX, USA; 2Assiut University Hospitals, Faculty of Medicine, Assiut, Egypt
Correspondence: Maria E. Suarez-Almazor (nahassan@mdanderson.org)
Background
Checkpoint inhibitors (CPI) have revolutionized cancer treatment with remarkable survival benefits in multiple cancer patients. Yet, their use can be hampered by frequent immune-related adverse events (irAEs), affecting any organ, with signs and symptoms usually seen in inflammatory and autoimmune diseases. There is paucity of data on CPI induced arthritis, primarily from case reports or small series.
Methods
Patients developed arthritis while using CPI were identified from the Rheumatology Clinic at UT MD Anderson Cancer Center. We diagnosed arthritis if patients developed joint swelling after receiving any FDA approved CPI, after ruling out other potential underlying causes or pathologies. Here we report 13 pts. Cancer types included melanoma (n=5), non-small cell lung cancer (n=4), renal cell carcinoma (n=2), acute myeloid leukemia, and cancer colon (1 each). CPI included nivolumab (n=8), pembrolizumab (n=3), and ipilimumab and atezolizumab (1 each).
Results
Patients age ranged from 47-74 years and 62% were male. CPI induced arthritis was the sole irAE in eight pts (62%), and was associated with other irAEs including sicca symptoms (xerostomia, dry eyes), colitis, pancreatitis, or dermatitis in the remaining five (38%). Overall, median time to onset of arthritis after the initiation of CPI was 6 (range 1 to 15) months. When occurred with other irAEs, arthritis always manifested up to 4 months after the first occurring irAE despite sufficient immunosuppressant treatment (infliximab and/or high dose steroid) and symptoms control, with the exception of one patient with sicca symptoms. Initial symptoms varied widely between small and large joints of upper and lower extremities. Over time, symmetrical polyarthritis resembling rheumatoid arthritis, predominantly involving small joints of the hands and wrists, along with larger joints was reported in six patients. Two had oligoarthritis resembling seronegative spondyloarthropathy with axial pain and enthesopathy. Four had asymmetrical inflammatory polyarthritis, and one had oligoarthritis. Rheumatoid factor and/or anti-citrullinated peptide antibodies were positive in two patients, and antinuclear antibodies in two others. Eleven patients were treated with steroids, six were controlled, while five needed additional biological agents (4 tocilizumab, 1 infliximab) to achieve appropriate steroid taper (< 7.5 mg). One patient improved with intra-articular steroid injection, and one received tocilizumab as first line.
Conclusions
Our data suggest that arthritis is a late occurring irAE that may be due to a different immunopathology. A multi-institutional collaborative work including tissue analysis is needed to enhance our understanding of the pathogenesis and design optiomal therapy without dampening antitumor immunity.
P445 Clinical features of immune checkpoint inhibitor-induced inflammatory arthritis differ in those treated with combination versus single agent therapy
Laura Cappelli, Clifton Bingham, Julie Brahmer, Patrick Forde, Dung Le, Evan Lipson, Jarushka Naidoo, Lei Zhang, Ami Shah
Johns Hopkins, Baltimore, MD, USA
Correspondence: Laura Cappelli (lcappel1@jhmi.edu)
Background
Multi-system immune-related adverse events (iRAE) can occur as a result of immune checkpoint inhibitors (ICIs) targeting PD-1, PD-L1, and CTLA-4, with variations depending on tumor type and agent used. Inflammatory arthritis (IA) has been increasingly recognized as an iRAE that may occur as a result of anti-PD-1/PD-L1/CTLA-4 therapy used in the treatment of malignancy [1, 2]. We evaluated associations between ICI regimen and clinical presentation of IA in a single institution.
Methods
Patients referred to rheumatology for suspected ICI-induced IA, were identified January 2013- July 2017. Patients were included if they developed de novo IA during or after ICI therapy and had no history of pre-existing autoimmune disease. Patients were included for the single agent group if they were on a PD-1 or PD-L1 inhibitor without CTLA-4 inhibition. Those in the combination group were treated with PD-1 and CTLA-4 inhibition. Clinical, demographic and laboratory data were obtained from the medical records.
Results
Tumor types for included patients were melanoma, non-small cell lung cancer, small cell lung cancer, colon cancer, Hodgkin or cutaneous lymphoma, renal cell carcinoma, endometrial carcinoma, duodenal carcinoma, merkel cell carcinoma, basal cell carcinoma, or squamous cell carcinoma. The group treated with combination CTLA-4/PD-1 inhibition was significantly younger than the monotherapy group; those in the combination group were also more likely to be male and to have melanoma (Table 1). Combination therapy was associated with higher levels of C-reactive protein (4 mg/dl vs. 0.7 mg/dL) and a higher likelihood of having a large joint affected first in the arthritis course (table 1). All instances of reactive arthritis-like presentation (arthritis + sterile urethritis, conjunctivitis) were in the combination therapy group. Patients who received PD-1/PD-L1 monotherapy were more likely to have IA as their first IRAE. No patients had CCP antibodies; two patients were positive for rheumatoid factor.
Conclusions
This study illustrates the diversity in clinical phenotypes of patients with ICI-induced IA. Differences in presentation may be explained by the regimen used to treat a patient’s malignancy. Understanding the diversity of clinical presentations will help oncologists identify and treat this increasingly important iRAE.
References
1. Cappelli LC, Gutierrez AK, Baer AN, et al. Inflammatory arthritis and sicca syndrome induced by nivolumab and ipilimumab. Ann Rheum Dis 2017; 761):43-50.
2. Calabrese C, Kirchner E, Kontzias K, et al. Rheumatic immune-related adverse events of checkpoint therapy for cancer: case series of a new nosological entity. RMD open 2017;3(1):e000412.
See text for description
P446 Immunosuppression use and clinical outcomes in patients with immune checkpoint inhibitor -induced inflammatory arthritis
Laura Cappelli, Clifton Bingham, Julie Brahmer, Patrick Forde, Dung Le, Evan Lipson, Jarushka Naidoo, Lei Zheng, Ami Shah
Johns Hopkins, Baltimore, MD, USA
Correspondence: Laura Cappelli (lcappel1@jhmi.edu)
Background
Inflammatory arthritis (IA) resulting from immune checkpoint inhibitors (ICIs) is an increasingly recognized immune-related adverse event. The requirement for immunosuppression to treat IA, including TNF-inhibitors (TNF-I), has been documented in previous case series [1, 2]. How often immunosuppression is required to treat ICI-induced IA is unclear. The consequences of long-term immunosuppression on tumor response from ICIs is also uncertain.
Methods
Patients treated at Johns Hopkins Rheumatology for IA due to ICIs were included if they received anti-CTLA-4, anti-PD-1, and/or anti-PD-L1 therapy and developed IA during or after therapy. Patients with pre-existing autoimmune disease were excluded. Patients were managed by rheumatology and oncology based on preliminary recommendations for ICI-induced IA treatment [3].
Results
Included patients (n=32) had melanoma, non-small cell lung cancer (NSCLC), small cell lung cancer, colon cancer, Hodgkin or cutaneous lymphoma, renal cell carcinoma, endometrial carcinoma, duodenal carcinoma, merkel cell carcinoma, basal cell carcinoma, or squamous cell carcinoma. Of these patients, 20 (63%) had a partial or complete tumor response to ICIs clinically or by RECIST criteria. Twenty six (81.3%) required corticosteroids for management of their IA. Eleven patients (34.3%) required additional immunosuppression. TNF-Is were used in 7 patients (tumor types: melanoma n=6, RCC n=1). Methotrexate or leflunomide was used in 4 patients (tumor types: NSCLC, small cell lung cancer, duodenal carcinoma, endometrial carcinoma). Duration of TNF-I use ranged from 2-16 months. Four patients treated with TNF-I, all with melanoma, had complete response by RECIST criteria or no evidence of disease at ICI start. None of these patients had tumor progression while on TNF-I (duration: 3-16 months). Of the patients treated with methotrexate or leflunomide, 3 had a complete or partial response to ICIs, and none had tumor progression during 2-12 months of IA management.
Conclusions
In a cohort of patients with ICI-induced IA, the majority of patients required corticosteroids to control IA symptoms. In this series, TNF-inhibitors and other forms of non-corticosteroid immunosuppression did not affect tumor response in those who had an initial response to ICIs for malignancy.
References
1. Cappelli LC, Gutierrez AK, Baer AN, et al. Inflammatory arthritis and sicca syndrome induced by nivolumab and ipilimumab. Ann Rheum Dis 2017;76(1):43-50.
2. Calabrese C, Kirchner E, Kontzias K, et al. Rheumatic immune-related adverse events of checkpoint therapy for cancer: case series of a new nosological entity. RMD open 2017;3(1):e000412.
3. Naidoo J, Cappelli LC, Forde PM, et al. Inflammatory Arthritis: A Newly Recognized Adverse Event of Immune Checkpoint Blockade. Oncologist 2017;22(6):627-30.
P447 Infliximab associated with faster symptom resolution compared to corticosteroids alone for management of immune related enterocolitis
Daniel H Johnson1, Chrystia Zobniw2, Van A Trinh2, Roland Bassett2, Junsheng Ma2, Jamie Anderson2, Jennifer Davis2, Jocelyn Joseph2, Marc Uemura2, Cassian Yee2, Rodabe Amaria2, Sapna Patel2, Hussein Tawbi2, Isabella Glitza2, Michael A Davies2, Michael Wong2, Scott Woodman2, Patrick Hwu2, Wen-Jen Hwu2, Yinghong Wang2, Adi Diab2
1University of Texas MD Anderson Cancer Center, Houston, TX, USA; 2UT-MD Anderson Cancer Center, Houston, TX, USA
Correspondence: Daniel H Johnson (dhjohnson@mdanderson.org); Adi Diab
Background
Immune-related enterocolitis (irEC) is the most common serious complication from checkpoint inhibitors (CPIs) that can lead to bowel perforation, sepsis, and death. The current front-line treatment for irEC is high-dose corticosteroids (CS). However, prolonged high-dose CS therapy has significant toxicities and side effects, and prolonged therapy may reduce CPI anti-tumor activity. Early addition of TNF-alpha inhibitors such as infliximab may expedite symptom resolution and shorten CS duration. The only prospective trial evaluating infliximab with CS versus CS alone for irEC (NCT02763761) has recently been terminated. Thus we conducted a retrospective study to evaluate symptom resolution in patients with irEC treated with infliximab + CS versus CS alone.
Methods
The medical records of patients with irEC between January 1, 2012 and June 30, 2017 were reviewed under an institution review board-approved protocol. Patient demographics, CPI regimen (aCTLA-4, aPD-1/L-1, alone or in combination), colitis grade, colitis treatment, treatment-related toxicities, time to symptom resolution, and CS duration were collected. The primary-endpoint was time to symptom resolution for irEC (times to diarrhea resolution and to initiation of steroid titration) for patients managed with versus without infliximab. Duration of CS and rate of CS-associated toxicities were secondary-endpoints. Categorical and continuous variables were assessed between groups by Fisher's exact test and Kruskal-Wallis test (or two-sample t-test).
Results
Among 75 patients with irEC, 39 (52%) received CS alone and 36 (48%) received infliximab within a median of 8 days after CS initiation. Patient characteristics (age, comorbidities, cancer type, immunotherapy regimen, and initial steroid dose), were similar between the two arms. The incidence of grade 3+ colitis was higher in the infliximab + CS arm (86.1% vs. 34.2%, p<0.001). Despite this, times to diarrhea resolution (median 3 vs. 9 days, p<0.001) and to steroid titration (median 4 vs. 13 days, p<0.001) were shorter in patients treated with infliximab + CS versus CS alone. Rates of CS-associated toxicities (39% vs. 54%; p= 0.249) and total steroid duration (median 35 days vs. 51 days; p= 0.241) were numerically lower in the infliximab-treated patients (Table 1).
Conclusions
To our knowledge this is the first study to compare time to symptom resolution for irEC with infliximab + CS versus CS alone. Despite higher incidence of grade 3+ colitis, infliximab treatment was associated with a statistically significant shorter time to symptom resolution without additional toxicities. The data suggest that early introduction of infliximab should be considered for patients with irEC until definitive prospective clinical trials.
See text for description
P448 Incidence of thyroid test function abnormalities in patients receiving immune-checkpoint inhibitors for cancer treatment
Nisha Patel, Anais Oury, Gregory Daniels, Lyudmila Bazhenova, Sandip Patel
University of California San Diego, La Jolla, CA, USA
Correspondence: Nisha Patel (nip011@ucsd.edu)
Background
With the advent of immune-checkpoint inhibitor (ICI) therapy (anti-CTLA-4, anti-PD-1), immune related adverse events (irAE) such as thyroid function test abnormalities (TFTA) are common with a reported incidence range of 2-15% depending upon the ICI utilized [1,2]. The aim of this study was to describe the incidence of TFTAs retrospectively in patients who received ICI therapy.
Methods
A total of 285 patients were reviewed (178 male, 107 female; ages 16-94) of which 218 had no baseline TFTA, 61 had baseline TFTAs, and 6 had history of thyroidectomy and were excluded. Patients received at least one dose of ipilimumab and/or nivolumab or pembrolizumab. Post-ICI therapy TFTA was classified according to definitions of thyroid abnormalities when possible [3].
Results
A total of 35% (76/218) patients had new onset TFTAs on ICI therapy. Of note, 70.5% (43/61) had baseline TFTA that were exacerbated by ICI therapy. Median time to new onset or exacerbated baseline TFTA were 46 & 33 days respectively. Of note, 65% (20/31) of patients on both ipilimumab and nivolumab had new onset TFTA, compared to 31.3% (15/48) with ipilimumab, 31.5% (28/89) nivolumab, 26% (13/50) pembrolizumab (Table 1).
Conclusions
The incidence of TFTAs with ICI therapy was higher than previously reported. Patients with baseline TFTA and/or receiving ipilimumab and nivolumab combination therapy had a higher incidence of TFTA than one agent ICI therapy. In conclusion, we recommend more frequent evaluation of TFT in the first two months, especially in those with baseline TFTA.
References
1. Corsello S, Barnabei A, Marchetti P, et al. Endocrine Side Effects Induced by Immune Checkpoint Inhibitors.J Clin Endocrinol Metab. 2013;98(4):1361-1375.
2. Morganstein D.L, Lai Z, Spain L, et al. Thyroid abnormalities following the use of cytotoxic T-lymphocyte antigen-4 and programmed death receptor protein -1 inhibitors in the treatment of melanoma. Clinical Endocrinology. 2017;(0) 1-7.
3. Rossi E, Sgambato A, De Chiara G, et al. Thyroid-Induced Toxicity of Check-Point Inhibitors Immunotherapy in the Treatment of Advanced Non-Small Cell Lung Cancer. J Endocrinol Diabetes. 2016; 3 (1):1-10.
See text for description
P449 Neurotoxicity in patients with metastatic solid tumors treated with immune checkpoint inhibitors: a single institution retrospective analysis
Bianca Santomasso, Rachna Malani, Aya Haggiagi, Jenessa Holder, Yelena Shames, Samuel Briggs, Margaret Callahan
Memorial Sloan Kettering Cancer Center, New York, NY, USA
Correspondence: Bianca Santomasso (Santomab@mskcc.org); Margaret Callahan
Background
Immune checkpoint inhibitors (ICIs) have dramatically improved survival for patients with melanoma and are playing an increasingly important role in the management of other tumor types. Anti-CTLA4, anti-PD1, and anti-PDL1 antibodies have been approved for multiple tumor types based on their clinical benefit, however they can be associated with a unique set of toxicities termed immune related adverse events (irAEs). We aimed to identify the incidence and clinical manifestations of patients who developed neurologic irAEs with ICIs at our institution in order to inform investigation and management guidelines.
Methods
An IRB approved retrospective study involved review of charts and institutional databases at MSKCC. We identified patients who developed neurotoxicity while on at least one of the following ICIs: anti-CTLA4, anti-PD1 or anti-PD-L1 over a 6 year period (1/1/2010-11/16/2016).
Results
We identified a total of 3,804 patients who were treated with one or more ICI during the period of review. Neurotoxicity was observed in 81 patients (2.1%) and affected both central and peripheral nervous systems. 31 patients (38.2%) received more than one ICI. Median number of cycles prior to developing toxicity was 3 (1-29). Ten patients had more than one neurotoxicity. The various neurologic phenotypes observed in patients included: sensory neuropathy, encephalopathy, headache and aseptic meningitis, myasthenia gravis-like syndrome, myopathy, autonomic neuropathy, radiculopathy, brachial plexitis, mononeuritis multiplex, Guillain-Barre syndrome or chronic inflammatory demyelinating polyneuropathy, paraneoplastic neurologic disorder, and PRES. 43 patients required hospital admission and 2 required ICU level of care. A diagnosis of neurotoxicity was based upon the temporal association with ICIs and appropriate workup including but not limited to neurologic consultation, lumbar puncture, and neuroimaging. Patients were treated with drug holiday, observation, corticosteroids, plasma exchange and/or IVIG.
Conclusions
As ICIs are being used with increased frequency, oncologists and other health care providers should be aware of the varied manifestations of neurotoxicity in order to ensure appropriate diagnosis, work up and treatment. Prompt treatment may relay a benefit in regards to outcome.
PHYSICIAN/NURSE/PHARM: Patient Experience: Patient Education Regarding I
P450 Educating cancer patients about immunotherapy: gains from attending a national educational immunotherapy workshop
Claire Saxton, Heather Hollen, Marni Amsellem
Cancer Support Community, Washington, DC, USA
Correspondence: Claire Saxton (hhollen@cancersupportcommunity.org)
Background
Immunotherapy continues to offer promising treatments for many tumor types. Unfortunately, patients potentially eligible for immunotherapy are often not aware of options. According to a 2014 Cancer Support Community (CSC) online survey, only 34.8% of cancer survivors knew the term “immuno-oncology” and 64.9% had heard of “immunotherapy.” Despite low awareness, most (84%) wanted to know more about these topics. Recognizing this need, the CSC developed a national psychoeducational immunotherapy workshop. The current analyses investigate the program’s informational and empowerment outcomes among attendees with cancer.
Methods
From 2014-16, 706 program attendees of the CSC’s Frankly Speaking About Cancer: Your Immune System & Cancer Treatment workshop completed a post-program evaluation assessing knowledge (on 5-point scale) and program outcomes, including program satisfaction. Individuals with cancer (n=478) comprised 68.0% of participants in the workshop. These attendees were White (81.7%), female (79.6%), and averaged 62.6 years old [1]. Workshop outcomes were ascertained by descriptive analyses and ANOVAs.
Results
Over half of workshop attendees with cancer (53.0%) reported generally being very involved in their overall treatment decision-making, yet 47.2% had been unsure whether immunotherapy was a treatment option for them[m1]. [AKZ2] About one-third (36.8%) had spoken with their doctor about immunotherapy and 26.2% inquired about potential side effects of immunotherapy; 44.1% had searched for information about immunotherapy on their own before the workshop. The majority (74.6%) reported low knowledge about immunotherapy before the workshop.
After the workshop, most attendees with cancer reported a high level of knowledge of immunotherapy (57.8%), representing a significant gain from attending the program (F (4,416) = 39.2, p <.01). Post-workshop, most (86.5%) felt confident speaking about immunotherapy with their doctors. Furthermore, a majority felt “more confident” discussing potential side effects (86.3%), asking about treatment options generally (90.3%), and making decisions with their doctors (84.8%) post-workshop. Nearly all (92.5%) strongly or moderately recommend attending the workshop.
Conclusions
Results from the attendee evaluations demonstrate significant informational and confidence gains from attending a two-hour psychoeducational workshop. Attendees reported increased awareness of options, knowledge, and efficacy in discussing potential options for them. Results suggest that improving access to comprehensive information about immunotherapy and promoting communication between the patient and the healthcare team about treatment options and decision-making via an educational workshop is viable and can be implemented nationally to reach many.
References
1. Cancer Support Community. CSC Immuno-Oncology online survey. 2014.
Tumor Microenvironment (Mechanisms and Therapies)
P451 Alpha-TEA demonstrates antitumor activity against trastuzumab sensitive and resistant breast cancer
Emmanuel Akporiaye1, Mary Disis2, Ekram Gad2, Tobias Hahn1, Brook Cole3, Michael Gough1, Talicia Savage4, Hailing Lu5
1Providence Portland Medical Center, Portland, OR, USA; 2University of Washington School of Medicine, Seattle, WA, USA; 3University of Minnesota, Minneapolis, MN, USA; 4Albert Einstein College of Medicine, Bronx, NY, USA; 5Immune Design, Seattle, WA, USA
Correspondence: Emmanuel Akporiaye (etakporiaye@gmail.com)
Background
Alpha-tocopheryloxyacetic acid (α-TEA) is an orally administered, small molecule, multi-pathway cancer therapeutic agent that drives tumor cell apoptosis and autophagy to stimulate antigen cross-presentation. Anti-tumor activity of α-TEA in combination with trastuzumab was evaluated in vivo against established Her2/neu-expressing mouse tumors (MMC) and in vitro against HER2 positive, human trastuzumab-sensitive (BT-474) and trastuzumab-resistant (HR6) tumor cells.
Methods
In vivo studies: Alpha-TEA was formulated in mouse chow and administered to mice ad libitum. FVB/N-TgN (MMTVneu) on nutrient-matched control chow mice received s.c. implant of 1 million Her2/neu positive murine mammary cancer (MMC) cells. On day 25, after tumors were established, mice were transferred to α-TEA diet, and treatment with anti-Her2/neu monoclonal antibody (7.16.4) was initiated. Mice received 3 injections of 7.16.4 antibody 3 times a week for 1 month and tumor growth was monitored until day 60. Mice demonstrating complete tumor regression were challenged with viable tumor cells to assess the establishment of a memory response. Spleen cells were removed from treated mice and assessed for cytokine production by ELISPOT assay.
In vitro studies: Human trastuzumab-sensitive (BT-474) and a resistant sub-clone (HR-6) were treated continuously with α-TEA alone or in combination with trastuzumab, both at various doses and evaluated for clonogenic potential and activation of ERK and AKT signaling pathways.
Results
Treatment of mice harboring established MMC tumors with α-TEA plus trastuzumab caused complete tumor regression, and increased cytokine production by splenic lymphocytes. Individually, α-TEA and trastuzumab caused cell death and reduced colony formation of BT-474 tumor cells in vitro; however only α-TEA was able to kill and reduce the clonogenicity of the trastuzumab-resistant HR6 cell line. Furthermore, α-TEA-mediated cell death of HR6 tumor cells involved inhibition of AKT and ERK phosphorylation. Treatment with α-TEA did not affect the expression of HER2/neu by both cell lines.
Conclusions
Alpha-TEA synergizes with trastuzumab to cause regression of established Her2/neu-expressing tumors.
Anti tumor activity of α-TEA is due to apoptosis induction and suppression of AKT and ERK signaling pathways.
Alpha-TEA represents a potential opportunity for combination treatment with Herceptin in HER2 positive breast cancer.
P452 Defining IL-15 expressing myeloid cells in the tumor microenvironment
Figen Beceren-Braun, Rosa M. Santana Carrero, Sarai C. Rivas, Kimberly S. Schluns
University of Texas MD Anderson Cancer Center, Houston, TX, USA
Correspondence: Figen Beceren-Braun (fbeceren@mdanderson.org)
Background
Loss of IL-15 within colorectal tumors correlates with higher risk of relapse, decreased survival, lower T cell density and decreased T cell proliferation [1] suggesting IL-15 expression within the tumor is important for anti-tumor responses. Furthermore, we have found that soluble IL-15 (sIL-15) complexes are produced in early tumors of mice implanted with tumor cell lines. However, the cell types expressing IL-15 and the mechanisms regulating IL-15 responses in the tumor microenvironment are not known. The goal of this study was to identify myeloid cell populations expressing IL-15 in the tumor microenvironment and investigate the mechanisms of expression.
Methods
Both IL-15 transcriptional and translational EGFP (enhanced green fluorescent protein) reporter mice were injected subcutaneously with 3x105 B16 melanoma, MCA-205 fibrosarcoma or MC-38 murine adenocarcinoma cell line. After two weeks tumors were stained for myeloid cell surface markers and analyzed via flow cytometry. Distribution of myeloid cell populations in tumors were also monitored via confocal laser-scanning microscopy of IL-15 reporter mice. CCR2+ IL-15+ double reporter mice were used to further characterize myeloid cells. To investigate the cellular source of sIL-15 complexes, we used mice with a conditional deletion of IL-15Rα from DCs (CD11c-Cre x IL-15Rαfl/fl) or macrophages (LysM-Cre x IL-15Rαfl/fl); or mice were treated with anti-Ly6G depleting antibodies.
Results
In both IL-15 reporter lines, we found that the EGFP expressing cells in the tumor microenvironment are composed of three main populations: CD11b+Ly6ChiLy6G-, CD11b+Ly6C+Ly6G+ and CD11b+Ly6C-/loLy6G- cells with the CD11b+Ly6ChiCCR2+ cells being the most abundant. Similar composition of EGFP+ myeloid populations was also seen in MCA-205 and MC-38 tumors. Imaging studies of MCA-205 tumors showed that the majority of the F4/80+ or CCR2+ cells were also EGFP+. In MCA-205 tumors, EGFP expression was decreased in CD11b+Ly6C+Ly6G+ cells compared to the spleen. Analyses with conditional knockout mice or depleting antibodies showed that the monocytic cells and macrophages are major sources of sIL-15 complexes in early B16 tumors.
Conclusions
In the tumor microenvironment, IL-15 is primarily produced by inflammatory monocytes and macrophages in early tumors and is down regulated with tumor growth.
References
1. Mlecnik B, et al. Functional Network Pipeline Reveals Genetic Determinants Associated with in Situ Lymphocyte Proliferation and Survival of Cancer Patients. Sci Transl Med. 2014; 6228:228ra37.
P453 Targeted tumor stroma depletion in combination with a whole cell tumor vaccine enhances effector T cell infiltration in a mouse model of pancreatic cancer
Alex Blair1, Victoria Kim1, Stephen Muth1, Todd Armstrong1, Sanna Rosengren2, Lei Zheng1
1Johns Hopkins Medical Institution, Baltimore, MD, USA; 2Halozyme Therapeutics, San Diego, CA, USA
Correspondence: Lei Zheng (ablair9@jhmi.edu)
Background
Pancreatic ductal adenocarcinoma (PDAC) is an aggressive disease with dismal prognosis due to conventional therapy resistance. The tumor microenvironment (TME) of PDAC is characterized by a desmoplastic reaction that excludes effector immune cell infiltration, thus preventing optimal efficacy of immunotherapy. A human recombinant hyaluronidase (PEGPH20) has been clinically formulated to enzymatically deplete hyaluronan (HA) in the tumor extracellular matrix. We tested the hypothesis that combining the stromal targeting agent PEGPH20 with a GM-CSF secreting allogenic pancreatic tumor vaccine (GVAX) would increase immune cell infiltration and improve survival of PDAC bearing mice.
Methods
C57BL/6 mice were orthotopically transplanted with 2 x 106 murine KPC pancreatic tumor cells to form liver metastases by a hemisplenectomy technique on day 0. Following tumor transplantation, mice were treated subcutaneously with GVAX in combination with intravenous PEGPH20 or the appropriate control. PEGPH20 was given on day 6 and GVAX on post-operative day 7. An immune modulating dose of Cyclophosphamide (Cy) was given the day before GVAX in all groups. Tumor immune lymphocyte infiltration was assessed by immunohistochemistry and flow cytometry. RT-PCR was performed on tumor associated macrophages isolated with CD11b beads. Mice were sacrificed on day 16 for immune and gene expression analysis. Additional survival analysis followed mice until death.
Results
Combination therapy with PEGPH20 and GVAX yielded higher infiltration of effector CD8+ and CD4+ tumor infiltrating lymphocytes in the TME compared to untreated and monotherapy groups (all p<0.05) (Fig. 1). Murine survival was improved in the combination therapy group as compared to GVAX therapy alone (Hazard Ratio: 0.286 [0.10-0.82] p=0.02). Additionally, a larger percent of tumor-infiltrating CD8+ cells were noted to express the lymphocyte activation surface marker CD137 in combination therapy compared to monotherapy controls. A trend towards lower CXCR4 expression was noted in the combination therapy group, suggesting a possible mechanism to CD8+CD137+ infiltration beyond decreased vascular impedance.
Conclusions
Our preclinical murine model of PDAC demonstrates enhanced efficacy of GVAX in combination with the stromal targeting agent PEGPH20 with increased immune cell infiltration and a survival advantage. This study provides rationale for further study of this combination for PDAC treatment.
Total number of live (A) CD8+ and (B) CD4+ tumor infiltrating lymphocytes after KPC hemispleen surgery and indicated therapy. Data represent mean ± SEM from one representative experiment with three mice per group that was repeated twice. (C) Kaplan-Meier survival curves of mice that were implanted with PDAC cells and treated with Cy/GVAX with and without combination of PEGPH20. * = p.

Combination therapy of PEGPH20 with Cy/GVAX improves immune cell infiltration and survival in a PDAC mouse model
P454 Molecular targeted radiotherapy facilitates in situ vaccination in a syngeneic murine melanoma model
Peter M Carlson1, Clinton Heinze1, Joseph Grudzinski1, Reinier Hernandez1, Stephen D Gillies2, Hans Loibner3, Alexander L Rakhmilevich1, Mario Otto1, Bryan Bednarz1, Jamey Weichert1, Paul M Sondel1, Zachary S Morris1
1University of Wisconsin School of Medicine and Public Health, Madison, WI, USA; 2Provenance Biopharmaceuticals, Carlisle, MA, USA; 3Apeiron Biologics, Vienna, Austria
Correspondence: Zachary S Morris (pcarlson2@wisc.edu)
Background
We reported that 12Gy external beam radiation (EBRT) and intratumoral (IT) administration of the hu14.18-IL2 immunocytokine (IC) [an anti-disialoganglioside (GD2) mAb fused to IL2] five days later elicits an in situ vaccination effect in GD2+ murine tumors. An established distant, untreated secondary tumor exerts an antagonistic effect, “concomitant immune tolerance” (CIT), which drastically reduces the efficacy of in situ vaccination. We have shown that CIT is mediated, in part, by tumor-infiltrating regulatory T cells (Tregs) and that CIT can be overcome by delivering 12Gy EBRT to both tumors. Clinically, it is not feasible to deliver this dose to all sites of metastatic disease. Systemically delivered molecular targeted radiotherapy (MTRT) with the tumor-selective alkyl phosphocholine analog 131I-NM404 could overcome this challenge, and potentially deplete immunosuppressive cells responsible for CIT in practically all sites of metastasis. We evaluated doses of EBRT to distant tumor sites needed to eliminate CIT, performed dosimetry to determine the administered activity of 131I-NM404 that approximates this EBRT absorbed dose, and tested whether systemic administration of MTRT may overcome CIT in a syngeneic murine melanoma model.
Methods
We monitored tumor volume, survival, and infiltrating lymphocyte populations in primary and secondary tumors treated with EBRT+IT-IC+/-intravenous MTRT in C57BL/6 mice bearing one or two ~250mm3 syngeneic GD2+ B78 melanoma flank tumors.
Results
We demonstrate that CIT can be substantially attenuated using low-dose (2Gy) EBRT to the secondary tumor. This lower dose is not adequate to generate an in situ vaccine response but appeared comparable to 12Gy in its capacity to overcome CIT. Using longitudinal 124I-NM404 PET/CT imaging with Monte Carlo dosimetry, we estimated that 60 μCi of 131I-NM404, a dose which does not destroy B78 tumors, given IV provides approximately 2Gy to both tumors in animals bearing two B78 tumors. Following 60μCi of 131I-NM404 to mice with two B78 tumors, we observed depletion of tumor-infiltrating Tregs by IHC staining over a 4-week period, without bone marrow suppression (thrombocytopenia/leukopenia/anemia). We observed that pretreatment with systemic 131I-NM404 one week prior to 12Gy EBRT+IT-IC to a single tumor site may substantially attenuate CIT and rescue a systemic response to in situ tumor vaccination.
Conclusions
We demonstrate that 2Gy EBRT is sufficient to attenuate CIT, and that this dose can be approximated using systemic MTRT. This work suggests that MTRT may enable conditioning of the tumor immune microenvironment to overcome intrinsic immune evasion mechanisms and restore efficacy of in situ tumor vaccination.
P455 Investigating the specificity and response of a native leukemia-reactive CD8+ T cell clone
Xiufen Chen1, Brendan MacNabb1, Emily Curran1, Bruce Blazar2, Justin Kline1
1University of Chicago, Chicago, IL, USA; 2University of Minnesota, Minneapolis, MN, USA
Correspondence: Justin Kline (xchen82@bsd.uchicago.edu)
Background
We previously reported that high-affinity, antigen-specific CD8+ T cells were induced to undergo abortive proliferation and deletion in leukemia-bearing hosts, and have subsequently found that this deletional CD8+ T cell tolerance requires cross-presentation of the leukemia antigen by splenic Batf3-lineage dendritic cells.
Methods
Recently, we generated a T cell receptor (TCR) transgenic mouse strain (referred to as Tg101) that expresses the TCR-α and -β chains of a CD8+ T cell clone specific for a native leukemia antigen.
Results
Having confirmed that Tg101 CD8+ T cells specifically recognize C1498 acute myeloid leukemia (AML) cells in the context of the MHC class I molecule, H-2Db, we investigated the behavior and fate these cells in naïve and C1498 leukemia-bearing hosts. In naïve mice, Tg101 thymocytes predominantly developed along the CD8+ lineage, and existed as naïve CD8+ T cells in peripheral lymphoid organs, suggesting that the Tg101 antigen is likely leukemia-specific. In leukemia-bearing mice, antigen encounter by Tg101 T cells was delayed (day 10-12), and primarily occurred in the liver - a prominent site of leukemia progression. Subsequently, Tg101 cells expanded and upregulated expression of co-inhibitory receptors, including PD-1, LAG-3, TIM-3 and TIGIT, which correlated with a progressively dysfunctional phenotype. Surprisingly, antigen encounter by Tg101 T cells in leukemia-bearing mice occurred independently of Batf3-lineage DCs, suggesting that the Tg101 antigen was poorly cross-presented in vivo. Given this finding, we deleted H-2Db in C1498 cells to determine the extent to which direct antigen presentation by leukemia cells is responsible for the acquisition of dysfunctional properties in Tg101 T cells in vivo. In vitro expansion was 2-fold decreased in Tg101 T cells compared to high-affinity 2C CD8+ T cells (which recognize the SIY peptide in the context of H-2Kb) upon culture with SIY-expressing C1498 cells (C1498.SIY), indicative of a lower avidity of Tg101 vs the 2C T cells for antigen. In contrast to Tg101 T cells, 2C T cells encountered leukemia antigen rapidly (day 3-4) in the context of splenic CD8a+ DCs, proliferated briefly, and were effectively deleted from the host by day 12.
Conclusions
Collectively, our data reveal that CD8+ T cell tolerance is effectively generated in the context of AML. However, the nature of the leukemia antigen, as well as the context in which it is presented, are critical determinants in the underlying mechanism of tolerance that ensues.
P456 CDK4 & 6 inhibitor Abemaciclib exerts intrinsic immunomodulatory effects on human T cells in vitro
Darin Chin1, Carmine Carpenito1, Nelusha Amaladas1, Linda Chung2, Kirk Staschke2, David Schaer3, Richard Beckmann2, Ruslan Novosiadly1, Michael Kalos1
1Eli Lilly & Co, New York, NY, USA; 2Eli Lilly & Co, Indianapolis, IN, USA; 3Eli Lilly & Co, Brooklyn, NY, USA
Correspondence: Carmine Carpenito (chin_darin@lilly.com); Kirk Staschke, David Schaer, Richard Beckmann, Ruslan Novosiadly, Michael Kalos
Background
Abemaciclib is an inhibitor of cyclin-dependent kinases 4 and 6 (CDK4 & 6) that suppresses DNA synthesis and cell proliferation as result of cell cycle arrest in G1 phase. Significant clinical activity of abemaciclib has been observed in hormone receptor-positive breast cancer patients highlighting the therapeutic potential of this compound in oncology, including combinations with other therapeutic modalities. In preclinical studies, pretreatment with abemaciclib followed by PD-L1 checkpoint blockade has demonstrated synergistic immune activation and antitumor efficacy. This has generated interest in combination studies of abemaciclib with immunotherapeutic agents. However, the mechanisms underlying immunomodulatory activity of abemaciclib have not been fully elucidated. Consequently, the objective of this study was to understand the direct effects of abemaciclib on primary human T cells.
Methods
Jurkat and primary human T cells were stimulated in vitro with anti-CD3/CD28 and treated with increasing concentrations of abemaciclib ranging between 0 uM to 1 uM for up to ten days. Cells were harvested at the end of the experiments and nuclear factor of activated T cells (NFAT) activation, cell surface immune markers (costimulatory and coinhibitory receptors and ligands) and expression of immune-related genes were analyzed by signal reporter assay, flow cytometry and Quantigene Plex, respectively.
Results
Treatment of Jurkat T cells showed a dose dependent increase in NFAT reporter activation during abemaciclib treatment which was replicated in primary human T cells. Additionally, treatment of primary T cells with abemaciclib resulted in a distinct phenotype exemplified by up-regulation of CD137, GITR, TIM3, PD-1, PD-L1, and TIGIT on the surface of both CD4+ and CD8+ T cells correlating with gene expression changes associated with T cell activation phenotype (upregulated Granzyme B, CD137, OX40, TNF-alpha, IL2R, IP10, ICAM-1) with only a modest concentration-dependent decrease in T cell expansion. Furthermore, intermittent exposure of T cells to abemaciclib during various stages of expansion revealed that T cells were capable of recovering from abemaciclib-induced growth inhibition whereas expression of costimulatory/coinhibitory molecules was maintained once the drug was removed.
Conclusions
Taken together, these results demonstrate that abemaciclib can exert T cell-intrinsic effects at concentrations comparable to clinical exposure. This is exemplified by increased T cell activation phenotype with only a modest and reversible decrease in T cell expansion, providing further rationale for clinical exploration of combination studies with immunomodulatory agents such as PD-1 pathway inhibitors.
P457 Pharmacodynamic activity of MEDI9197, a TLR7/8 agonist, administered intratumorally in subjects with solid subcutaneous or cutaneous tumors
Zachary A. Cooper1, Christopher A. Morehouse1, Yuling Wu1, Keith Steele1, David Brucker2, Leo Tseng1, Maria Jure-Kunkel1, Nancy Mueller1, Patricia Ryan1, Juneko Grilley-Olson3, David Hong4, Aurélien Marabelle5, Pamela Munster6, Rahul Aggarwal6, Sandrine Aspeslagh5, Robert G. Dixon3, Manish Patel7, Vivek Subbiah4, Joshua Brody8, Shilpa Gupta7
1MedImmune, Gaithersburg, MD, USA; 2Definiens, Cambridge, MA, USA; 3University of North Carolina Lineberger Comprehensive Cancer Center, Chapel Hill, NC, USA; 4MD Anderson Cancer Center, Houston, TX, USA; 5Institut Gustave Roussy, 94800 Villejuif, France; 6UCSF Helen Diller Family Comprehensive Cancer Center, San Francisco, CA, USA; 7Masonic Cancer Center, Minneapolis, MN, USA; 8Mount Sinai (Hess Center for Science and Medicine), New York, NY, USA
Correspondence: Zachary A. Cooper (robert.mulvey@ashfieldhealthcare.com)
Background
MEDI9197 (formerly 3M-052), is a novel TLR7/8 dual agonist formulated for intratumoral (IT) injection and optimized for retention within the tumor after IT injection. IT administration aims to focus antigen presentation in the tumor by activation of both plasmacytoid (TLR7 expressing) and conventional dendritic (TLR8 expressing) cells, thus minimizing systemic inflammatory toxicities. We report the pharmacodynamic results of MEDI9197 from a phase I, first-in-human, dose-escalation study in patients with solid tumors (NCT02556463).
Methods
The dose-escalation trial of MEDI9197 (0.005 to 0.055 mg) has enrolled 24 patients with subcutaneous/cutaneous tumors. As reported at AACR 2017, the maximum tolerated dose is 0.037 mg and IT administration is feasible and safe. Here we present updated tumoral and peripheral pharmacodynamic effects in patients treated with MEDI9197 0.005, 0.012, and 0.037 mg.
Results
Local pharmacodynamic effects assessed by immunohistochemistry (IHC) in longitudinal biopsies demonstrated the majority of patients treated with 0.037 mg had an increase (≥2 fold) in CD8, CD40, CD56, or PD-L1 (tumor and immune cells) markers 3 weeks after a single dose of MEDI9197. RNAseq analysis of paired tumor biopsies showed an increase in TLR7/8-downstream regulated genes and innate and adaptive immune activation signatures such as Type 1 IFN, IFNγ and T effector signatures (≥1.5 fold) consistent with IHC.
Increases in peripheral levels of IFNγ, CXCL10, and CXCL11 were observed across all 3 doses (0.037, 0.012, and 0.005 mg). Within 24 hours of IT administration of MEDI9197 0.037 mg, median peak values of IFNγ, CXCL10, and CXCL11 observed were 236, 9286, and 558 pg/mL respectively. The fold change increase ranged from 4.5-43, 30-132, and 3.7-52 vs. baseline for IFNγ, CXCL10, and CXCL11 respectively. Peripheral levels of IFNγ showed a significant trend for a dose response with median peak values for those treated with MEDI9197 0.005 mg 4.1-fold less than those treated with 0.037 mg. Analysis of whole blood microarray data across all doses demonstrated increases (≥2 fold) in TH1 and Type 1 IFN gene expression signatures with a transient decrease (≥1.5 fold) in CD8A transcript and NK cell signature expression, suggesting trafficking of T and NK cells.
Conclusions
IT administration of MEDI9197 induces Type 1 and 2 IFN as well as TH1 responses, suggesting activation of both plasmacytoid and conventional dendritic cells. A combination trial of MEDI9197 with durvalumab (anti-PD-L1) and radiation in patients with advanced solid tumors is currently ongoing.
Trial Registration
NCT02556463.
P458 Nitric oxide synthase inhibition results in PDL-1 upregulation and ADAR1 inhibition, triggering immunogenic cell death: an in-vitro and in-vivo analysis
Daniel Davila-Gonzalez, Roberto Rosato, Wei Qian, Anthony Kozielski, Wen Chen, Dong Soon Choi, Patrick Tucker, Jenny Chang
Houston Methodist, Houston, TX, USA
Correspondence: Jenny Chang (ddavilagonzalez@houstonmethodist.org)
Background
Response failure in checkpoint inhibitors, such as death ligand 1 (PD-L1) and its receptor programmed death 1 (PD-1), is associated with cancer-related immunosuppression. Inducible nitric oxide synthase (iNOS) product, nitric oxide, has been implicated in immunosuppressive environments. In triple negative breast cancer (TNBC), iNOS expression is associated with poor survival and increased tumor aggressiveness. We’ve shown ADAR1 (adenosine deaminase acting on RNA 1) is upregulated by iNOS activation in TNBC cell lines. ADAR1 RNA editing suppresses inflammation, thereby damping the immune response. This study aims to evaluate whether NOS inhibition plus anti-PD-1 is a feasible therapeutic combination in TNBC.
Methods
TNBC cell lines BT-549, SUM-159, SUM-157, HCC-70, MDA-MB-231 and MDA-MD-468 were treated with NOS inhibition therapy (L-NMMA, 4mM) + amlodipine (5μM). PDL-1, HMGB1, and ADAR1 expression was assessed via western blot. BALBc mice growing orthotopically injected 4t1 cells were treated weekly with: 1) vehicle (saline, oral/Rat-IgG2 i.p.); 2) NOS inhibition [L-NMMA (200mg/kg oral) + amlodipine (10mg/kg, i.p.), days 1-5]; 3) anti-PD-1 antibody (10 mg/kg i.p.; Bio X Cell Clone: RMP1-14, days 1, 3, 5); or 4) anti-PD-1 + NOS inhibition. Humanized mice were developed by injecting hematopoietic stem cells (HSC, CD34+) into the tail vein of irradiated NOD-scid IL2Rγnull mice. Mice were sorted and TNBC patient-derived xenografts (PDXs), representing 12 different patients, were implanted into the mammary fat pad (3 mice/PDX). Mice were sorted and treated weekly with: 1) vehicle; 2) pembrolizumab (anti-PD-1, 200μg, day 1, IV); or 3) pembrolizumab + NOS inhibitor.
Results
NOS inhibition increased PD-L1 in most cell lines. 4T1 syngeneic model helped assess the potential benefit of combined therapy. There were no significant differences in tumor growth between monotherapy and control group. However, anti-PD1 + NOS-inhibition resulted in a significant reduction in tumor growth. In immune-humanized TNBC PDXs, 40% responded to anti-PD1 monotherapy, 60% had improved response following combination therapy, and 33% responded only to combination therapy. In total, 66% of PDXs analyzed benefited from combination therapy. Furthermore, both in-vitro and in-vivo samples showed downregulation of ADAR1 levels when treated with NOS inhibition, with a subsequent increase in HMGB1, a marker of immunogenic cell death.
Conclusions
NOS inhibition upregulates PDL-1 expression in TNBC cell lines. Furthermore, combining NOS inhibition with anti-PD1 therapy improved antibody response by activating the immunogenic cell death pathway. This combination treatment may benefit cancer patients who do not respond to anti-PD-1 monotherapy.
P459 Class IIa HDAC inhibition promotes an anti-tumor macrophage phenotype that induces breast tumor regression and inhibits metastasis
Jennifer Guerriero1, Alaba Sotayo1, Holly Ponichtera1, Jessica Castrillon1, Alexandra Pourzia1, Sara Schad1, Shawn Johnson1, Ruben Carrasco1, Suzan Lazo1, Roderick Bronson2, Scott Davis3, Mercedes Lobera3, Michael Nolan3, Anthony Letai1
1Dana-Farber Cancer Institute, Boston, MA, USA; 2Harvard Medical School, Boston, MA, USA; 3GlaxoSmithKline, Cambridge, MA, USA
Correspondence: Jennifer Guerriero (Jennifer_Guerriero@dfci.harvard.edu)
Background
The breast tumor environment is complex and includes both neoplastic and immune cells. Among the most prevalent immune cell population within breast tumors is tumor associated macrophages (TAMs). Macrophages have the ability to polarize into either M1 cells, which have potent anti-tumor capabilities, or M2 macrophages which promote tumor progression by stimulating tumor vasculature, invasion, metastasis, and can enhance tumor resistance to chemotherapy. Generally TAMs in breast tumors are considered M2 macrophages and a high tumor density of TAMs clinically correlates to both overall worse prognosis and increased metastasis. Several therapeutic strategies exist to modulate TAMs clinically, focusing on depleting or inhibiting TAMs. However, macrophage are required for optimal tumor regression in response to both chemotherapy and immunotherapy. Their embedded location and their untapped potential provide impetus to the discovery of strategies to turn them against tumors and to harness for cancer therapy. Therefore here, we describe a novel method to polarize pro-tumor macrophages to an anti-tumor phenotype.
Methods
We recently reported that a first in class selective class IIa HDAC inhibitor (TMP195) influenced human monocyte responses to colony stimulating factors CSF-1 and CSF-2 in vitro. Here, we utilize a macrophage-dependent autochthonous mouse model of breast cancer to demonstrate that in vivo TMP195 treatment alters the tumor microenvironment and reduces tumor burden and pulmonary metastases through macrophage modulation.
Results
Here, we demonstrate that a first in class, class IIa histone deacetylase (HDAC) inhibitor, TMP195, can activate tumor macrophages in vivo to induce tumor regression and inhibit pulmonary metastases in a mouse model of breast cancer. We find that TMP195 induces macrophage recruitment and differentiation of highly phagocytic cells within the tumor, which increases tumor cell death while decreasing angiogenesis. Strikingly, we find that TMP195 enhances chemotherapy and immunotherapy to induce durable tumor reduction.
Conclusions
Our findings reveal an immunostimulatory effect of class IIa HDAC inhibition that contrasts with strategies of depleting or inhibiting TAMs for cancer therapy. Class IIa HDAC inhibition leverages the effector functions of macrophages, opening the door to clinically-relevant cooperation of checkpoint blockade, agonistic aCD40 or inhibitory aCD47 therapy, peptide vaccine therapy or the antibody-dependent cellular phagocytosis (ADCP) associated with mAb therapy. Indeed, the innate immune system is the natural complement to the adaptive immune system that surveys and fights tumors, and these studies demonstrate a novel approach to harness innate immune cells to cooperate with agents that stimulate an adaptive anti-tumor immune response in an otherwise resistant cancer.
P460 Selenium, the element of the moon, improves immunotherapies on earth
Claudia Lennicke1, Shamim Ahmad1, Rajeev Shrimali1, Barbara Seliger2, Youcef Rustum3, John Janik1, Mikayel Mkrtichyan1, Seema Gupta1, Samir Khleif1
1Augusta University, Augsuta, GA, USA; 2Institute of Medical Immunology, Halle, Germany; 3University of Buffalo, Buffalo, NY, USA
Correspondence: Samir Khleif (segupta@augusta.edu)
Background
Recent advances in immunotherapy have shown amazing results in cancer patients, albeit in small fraction. Attempts in developing immunotherapy to generate effective response against tumors are still faced with immune suppressive environment. The abundance of tumor-infiltrated T regulatory cells (Tregs) highly is one of the main mechanism by which tumors overcome immune responsiveness. Tregs also correlates with poor patient prognosis.
Selenium (Se), an essential trace element named after the goddess of the moon, is described to exhibit anti-carcinogenic potentials [1]. However, the underlying mechanisms of Se action are insufficiently understood. Since Se is also known to exhibit immune-modulatory properties [2] we hypothesized that Se in combination with vaccine-based immunotherapy might enhance anti-tumor responses.
Methods
TC-1 tumor-bearing C57BL/6 mice were treated either with Vaccine (E7, 100 μg/mouse; PADRE, 20 μg/mouse; every 7th day), Methylselenocysteine, as source of Se (MSC, 100 μg/mouse; daily) or with a combination of both. In addition, an untreated group served as the control. Spleens and tumor tissues were analyzed for T cell subsets via flow cytometry. ELISpot assays were performed to determine IFNg production in E7/PADRE restimulated splenocytes. In vitro, CD4+ T cells isolated from spleens of C57BL/6 mice were cultured under iTreg conditions, treated with Se and analyzed via Flow Cytometry and Immunoblotting.
Results
We found that combining Selenium with specific antigen vaccine led to significant enhancement of immune response leading to delay in tumor growth thereby prolonging the survival rate of mice. Importantly, Se significantly reduced the infiltration of Tregs into the tumor environment thereby enhancing the CD8+/Treg ratio leading to an increased vaccine-induced immune response. In vitro analysis of iTregs revealed that Se reduces Foxp3 expression and destabilizes Tregs.
Conclusions
Here we show for the first time that Se an essential trace element improves vaccine-based immunotherapies thereby opening a new window for the development of novel strategies to fight against cancer.
References
1. Misra S, Boylan M, Selvam A. Redox-active selenium compounds--from toxicity and cell death to cancer treatment. Nutrients. 2015;7:3536–56.
2. Huang Z, Rose AH, Hoffmann PR. The role of selenium in inflammation and immunity: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal. 2012;16:705–43.
P461 Listeria monocytogenes-based immunotherapies alter the suppressive phenotype of intratumoral regulatory T cells
Rachelle E. Kosoff, Nithya Thambi, Lauren K. Pettit, Daniel O. Villarreal, Kimberly Ramos, Michael F. Princiotta, Robert G. Petit, Sandra M. Hayes
Advaxis, Inc., Princeton, NJ, USA
Correspondence: Sandra M. Hayes (hayes@advaxis.com)
Background
Advaxis’ Listeria monocytogenes (Lm)-based immunotherapies are live attenuated bacterial vectors that elicit tumor-reactive T cells through the delivery of tumor-associated antigens directly to dendritic cells. In addition, our Lm-based immunotherapies reduce the frequency and function of intratumoral regulatory T cells (Tregs), thereby counteracting the immunosuppressive tumor microenvironment (TME) and sustaining the newly generated antitumor immune response. The purpose of this study is to investigate further the mechanism by which Advaxis’ Lm-based immunotherapies alter Treg function.
Methods
C57BL/6 mice were implanted with TC-1 tumor cells, which express the human papillomavirus (HPV)16 E6 and E7 proteins. On day 8, when tumors were palpable, mice were treated with either one dose of ADXS-HPV-Quad, an Lm-based vector targeting the E6 and E7 proteins from both HPV16 and HPV18, or PBS. Five days post-treatment, tumor infiltrating lymphocytes were phenotyped by flow cytometry and intact tumors were analyzed by immunofluorescence microscopy.
Results
We reasoned that Lm-based immunotherapies, to be effective at tumor control, must reprogram the immunosuppressive TME early during the course of treatment in order to induce and sustain infiltration of tumor-specific T cell effectors. Accordingly, we analyzed the TME 5 days after a single dose of ADXS-HPV-Quad. By flow cytometry, we compared the proliferative status and viability of Tregs in tumors of mice treated with either PBS or ADXS-HPV-Quad. The percentage of proliferating (Ki-67+) Tregs was comparable between the two treatment groups, but the percentage of Tregs undergoing apoptosis was 2-fold higher in ADXS-HPV-Quad-treated mice than in PBS-treated mice. Further phenotypic analysis revealed that the percentage of Tregs expressing CCR8, a chemokine receptor whose expression is essential for Treg survival and function, was almost 2-fold lower in ADXS-HPV-Quad-treated mice than in PBS-treated mice. Expression levels of other Treg phenotypic markers, namely Foxp3 and CD39, were also reduced in the CCR8-negative Tregs. The phenotypic changes in intratumoral Tregs were associated with a loss in in vivo suppressive activity, as there was a dramatic increase in the infiltration of activated cytokine-producing T cell effectors into the tumor core of ADXS-HPV-Quad-treated mice but not into the tumor core of PBS-treated mice.
Conclusions
Within 5 days of administration, a single dose of ADXS-HPV-Quad alters the tumor microenvironment by impairing Treg survival and function and by promoting effector T cell recruitment and function. Changes in the suppressive Treg phenotype, specifically the downregulation of CCR8 expression, may be a key mechanism by which Advaxis’ Lm-based immunotherapies impair Treg function.
P462 Ubiquitin-specific protease 6 (USP6) oncogene promotes immune cell recruitment in sarcoma
Ian Henrich1, Laura Quick2, Rob Young2, Andre Oliveira3, Margaret Chou1
1University of Pennsylvania, Philadelphia, PA, USA; 2Children's Hospital of Pennsylvania, Philadelphia, PA, USA; 3Mayo Clinic, Rochester, MN, USA
Correspondence: Ian Henrich (ihenrich@mail.med.upenn.edu)
Background
The de-ubiquitylating enzyme USP6 is a target of chromosomal translocation and the key etiologic agent in several benign bone and soft tissue tumors (BSTTs) [1]. We have shown that USP6 drives tumorigenesis by directly de-ubiquitylating the Jak1 kinase, leading to its stabilization and activation of STAT transcription factors [2]. We recently found that USP6 is also upregulated in sarcomas, malignancies of mesenchymal origin. Activation of the Jak-STAT pathway plays a key role in modulating the tumor microenvironment, however it is not known if or how USP6 affects immune cell infiltration and/or composition. We sought to determine USP6 effects on sarcoma tumor growth and immune cell migration/infiltration.
Methods
USP6 was expressed in a doxycycline-inducible manner in the patient-derived sarcoma cell lines A673 and RD-ES. USP6 expression levels were confirmed to approximate those in primary patient tumor samples.
Results
Overexpression of USP6 results in the production of several immunomodulatory cytokines, most notably CXCL10, CCL5, and CCL20. USP6 expression led to the hallmarks of a heightened immune response, such as enhanced migration, polarization, and activation of several classes of immune cells, such as CD4+ and CD8+ T cells, monocytes, and macrophages. In murine xenografts, sarcoma cells expressing USP6 had attenuated tumor growth and enhanced immune cell infiltration compared to parental lines. Furthermore, analysis of sarcoma patient datasets revealed that elevated USP6 expression correlated with an immune cell gene signature, and immunohistochemistry of USP6-translocated tumors revealed a high degree of CD4+, CD8+, and CD163+ infiltration.
Conclusions
USP6 expression results in the production of several immunomodulatory cytokines that are known to promote the activation and migration of several classes of immune cells such as T cells, monocytes, and natural killer cells. High USP6 expression correlated with increased immune cell infiltration in mouse xenografts and patient samples. The ability of USP6 to provoke an enhanced immune response could potentially serve as a biomarker for susceptibility to immunotherapy.
References
1. Oliveira A, Chou M. The TRE17/USP6 oncogene: a riddle wrapped in a mystery inside an enigma. Front Biosci (Schol Ed).2012;4:321-340.2.
2. Quick L, Young R, Henrich I, Wang X, Asmann Y, Oliveira A, Chou M.Jak1-STAT3 signals are essential effectors of the USP6/TRE17 oncogene in tumorigenesis. Cancer Res. 2016;76(18):5337-47.
P463 TAM RTKs facilitate MDSC-mediated tumor immunosuppression
Alisha Holtzhausen1, William Harris1, Debra Hunter1, Eric Ubil1, Xiaodong Wang1, Douglas Graham2, Stephen Frye1, H. Shelton Earp1
1University of North Carolina at Chapel Hill, Chapel Hill, NC, USA; 2Emory University, Atlanta, GA, USA
Correspondence: H. Shelton Earp (alisha_holtzhausen@unc.edu)
Background
The innate immune-regulatory receptor tyrosine kinases Tyro3, Axl and MerTK (TAM RTKs) and their ligands, Gas 6 and Protein S, suppress the immune response through various mechanisms, including down regulating M1 cytokines, suppressing antigen presenting cell function and decreasing CD8+ tumor infiltration. Myeloid-derived suppressor cells (MDSCs) are potently immunosuppressive and inhibit T cell activity via various mechanisms, including increased expression of arginase, inducible nitric oxide synthase (iNOS), and the production of reactive oxygen species (ROS).
Methods
We studied the expression of TAM RTKs in MDSCs and their respective roles in MDSC-mediated immune suppression in the setting of cancer. Using syngeneic murine tumor models implanted into WT and TAM RTK-/- mice, we FACS isolated MDSCs and analyzed the TAM RTKs’ effect on MDSC suppressive mechanisms. To complement our genetic models, we employed a pan-TAM RTK inhibitor, UNC4241, and evaluated MDSC function and tumor growth.
Results
TAM RTKs and their ligands are significantly (M-MDSCs>20 fold, G-MDSCs>15 fold) upregulated in MDSCs from tumor-bearing mice compared to non-tumor bearing controls. MDSCs isolated from tumor bearing TAM RTK-/- mice have significantly diminished arginase, iNOS and ROS activity compared to WT counterparts. Consistent with these findings, M-MDSC functional suppression assays showed that loss of either mertk or tyro3 yielded tumor-elicited M-MDSCs exhibiting reduced suppressive capabilities in T cell proliferation and IFN-g ELISPOT assays. We performed tumor and MDSC co-implantation studies and demonstrated that WT and axl-/- MDSCs migrate readily to the TDLN while mertk-/- and tyro3-/- MDSCs exhibit a migration defect and remain within the tumor, suggesting that the TAM RTKs play several roles in MDSC-mediated inhibition of T cell priming. Adoptive transfer of TAM RTK-/- MDSCs results in diminished B16 melanoma development in syngeneic hosts relative to those receiving WT MDSCs. To evaluate whether inhibition of TAM RTKs has clinical potential, we employed a pan-TAM inhibitor, UNC4241, and found this agent to suppress MDSC arginase, iNOS and ROS activity, all while increasing T cell proliferation and diminishing the growth of a BRAFV600EPTEN-/- melanoma model. In vivo MDSC ablation reversed the effect of UNC4241 indicating that the mechanism of this agent is MDSC-dependent.
Conclusions
These results indicate that coordinate TAM RTK action mediates MDSC suppressive capabilities and that their pharmacologic inhibition reverses MDSC-dependent T cell suppression and diminishes tumor growth. These findings suggest that inhibition of TAM RTKs represents a novel approach for modulating MDSC function and augmenting the efficacy of checkpoint inhibitor therapy.
P464 Use of evofosfamide for targeting immune-suppressive hypoxia in head and neck squamous cell carcinoma
Francis Hunter1, Avik Shome1, Way Wong1, Pratha Budhani2, Neil Senzer3, E. Gabriela Chiorean4, Dan Li1, Peter Tsai1, Nooriyah Poonawala1, Purvi Kakadiya1, Maria Kondratyev5, Courtney Lynch1, Cho Rong Hong1, Tet-Woo Lee1, Dennis Kee6, Andrew Macann6, Nicholas McIvor6, Kevin Hicks1, Stefan Bohlander1, Cristin Print1, Stew Kroll7, Charles Hart7, Bradly Wouters5, William Wilson1, Michael Curran2, Stephen Jamieson1
1University of Auckland, Auckland, New Zealand; 2MD Anderson Cancer Center, Houston, TX, USA; 3Mary Crowley Cancer Research Center, Dallas, TX, USA; 4Indiana University Melvin and Bren Simon Cancer Center, Indianapolis, IN, USA; 5University Health Network, Toronto, ON, Canada; 6Auckland City Hospital, Auckland, New Zealand; 7Threshold Pharmaceuticals, San Francisco, CA, USA
Correspondence: Francis Hunter (f.hunter@auckland.ac.nz)
Background
Hypoxia is prevalent in head and neck squamous cell carcinoma (HNSCC), where it limits radiotherapy outcomes and may create an immune-suppressive microenvironment. The hypoxia-activated prodrug (HAP) evofosfamide (TH-302) targets hypoxia by undergoing oxygen-sensitive reductive activation (Figure 1). Evofosfamide is being clinically evaluated in combination with ipilimumab in solid tumours, including HNSCC (NCT03098160). The present study investigated the efficacy and sensitivity determinants of evofosfamide in HNSCC.
Methods
Case reports were retrieved from an unpublished phase 2 evofosfamide monotherapy expansion cohort in solid tumours (480 or 575 mg.m2 on days 1, 8 and 15 of 28-day cycles). A collection of 22 human papillomavirus-negative HNSCC cell lines derived from lesions of varying TNM stages was assessed for sensitivity to 3 HAPs – evofosfamide, PR-104A and SN30000 – in addition to cisplatin, 5-fluorouracil and the active metabolite of evofosfamide, bromo-iso-phosphoramide mustard (Br-IPM). Reductive activation of evofosfamide in cultured cells was measured by LC–MS/MS. The lines were molecularly characterised by RNAseq and whole-exome sequencing and their genomic and transcriptomic features compared to public domain HNSCC clinical samples. Molecular predictors of evofosfamide sensitivity were investigated using hierarchical clustering, differential expression and correlation analyses. The antitumour activity of evofosfamide (50 mg.kg-1 qdx5 for 2-3 cycles with/without 10 Gy radiation on day 5 of cycle 1) was evaluated in two HNSCC xenografts and two HNSCC PDX models derived in-house. Evofosfamide was evaluated in combination with CTLA-4 blockade in the murine SCC-VII model. Hypoxic fraction was assessed using pimonidazole.
Results
Of 5 metastatic or locally-advanced HNSCC patients who received evofosfamide after failing standard-of-care, two showed partial responses and three showed stable disease. Evofosfamide was highly selective for hypoxic HNSCC cells and more potent and selective than PR-104A or SN30000. Cell line sensitivity to evofosfamide was correlated with Br-IPM and cisplatin but not with PR-104A, SN30000 or 5-FU, indicating distinct sensitivity determinants. Evofosfamide sensitivity was associated with the expression of genes relating to proliferation. Accordingly, a proliferation metagene identified subtypes within the cell lines that were differentially sensitive to evofosfamide. Xenografts chosen on the basis of putative predictive biomarkers (tumour hypoxia, proliferation subtype) showed the expected patterns of response. Two PDX models were also highly responsive to evofosfamide. SCC-VII was refractory to evofosfamide monotherapy but showed increased growth delay when evofosfamide was combined with CTLA-4 inhibition.
Conclusions
This study provides a rationale for the clinical evaluation of evofosfamide with immunotherapy and/or radiotherapy in genetically defined subsets of HNSCC.
P465 Characterization of immune cells in the pre-metastatic niche in a murine model of rhabdomyosarcoma
Sabina Kaczanowska, Meera Murgai, Daniel Beury, Rosandra Kaplan
National Cancer Institute, Bethesda, MD, USA
Correspondence: Rosandra Kaplan (sabina.kaczanowska@nih.gov)
Background
Tumor metastasis is a critical step in the progression of cancer that is associated with patient mortality. Prior to the arrival of tumor cells at the metastatic site, hematopoietic stem and progenitor cells expand in the bone marrow and mobilize to the pre-metastatic niche, where they differentiate into immunosuppressive myeloid cells [1,2]. Furthermore, recent data has demonstrated that perivascular cells undergo phenotypic changes in the lung that are dependent on the transcription factor KLF4 and enhance metastasis in response to tumor-derived factors [3]. Targeting the pre-metastatic niche by either antibody-mediated depletion of myeloid cells or inhibition of perivascular cell phenotypic switching significantly decreases metastasis [2,3]. However, changes in other immune cell populations in the pre-metastatic niche are not well defined. We hypothesize that primary tumor growth will alter immune cell infiltration and/or function in the pre-metastatic niche.
Methods
Lungs and bone marrow from mice bearing M3-9-M, a metastatic embryonal rhabdomyosarcoma cell line, were harvested at various time points and analyzed by flow cytometry for lymphoid and myeloid populations.
Results
Primary tumor progression was correlated with infiltration of immune cells into the lung before detectable metastasis. There was a dramatic increase of CD11b+Ly6G+ and CD11b+Ly6C+ myeloid cells in the lung, and a shift in macrophage and dendritic cell populations and phenotypes in the lung and bone marrow. Furthermore, the proportion of both CD4+ and CD8+ T cells that express PD-1 is significantly enhanced in the lung, as is the number of leukocytes expressing PD-L1.
Conclusions
We demonstrate that the immune compartment in the bone marrow and in pre-metastatic lungs changes significantly in response to primary tumor growth. The differences in immune populations that were observed in the lung and bone marrow were distinct, indicating that the immune changes occurring at these sites in response to tumor progression are specific to their unique microenvironment. We postulate that disrupting the formation of the pre-metastatic niche by targeting stromal cell populations combined with immunotherapy could have anti-metastatic potential. These studies will provide insight into the immune signature of metastatic disease and identify new targets for therapeutic intervention with the potential to reduce metastasis.
References
1. Rosandra Natasha Kaplan et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005; 438(7069):820-7.
2. Amber Jin Giles et al: Activation of Hematopoietic Stem/Progenitor Cells Promotes Immunosuppression Within the Pre–metastatic Niche. Cancer. Res. 2016;76(6):1335-47.
3. Meera Murgai et al. KLF4-dependent perivascular plasticity mediates pre-metastatic niche formation and metastasis. Nat. Med. In Press.
P466 Rational design of immuno-oncology biologics with improved safety and efficacy
Margaret Karow1, Kurt Jenkins2, Miso Park2, Parker Johnson2, Asaul Gonzalez2, Veronica Flesch2, Tim Miles2, Ulrich Rodeck3, John Williams2
1Akriveia Therapeutics, Thousand Oaks, CA, USA; 2City of Hope, Duarte, CA, USA; 3Thomas Jefferson University, Philadelphia, PA, USA
Correspondence: Margaret Karow (karow@akriveiatx.com)
Background
The benefit of enhancing anti-tumor immune response has been demonstrated by the unprecedented clinical responses of immune checkpoint antagonists and cell-based therapies1. However, the challenge with these therapeutic approaches is that they necessarily involve a systemic activation of the immune system2. This leads to treatment-limiting and often debilitating immune related adverse events (irAEs). Attempts to control irAEs include reducing dosing frequency and dosing levels, co-dosing immune suppressants, and drug withdrawal; all of which may limit the utility of immune therapies3. Focusing the activity of immuno-oncology agents to the tumor microenvironment is expected to overcome the limitations posed by irAEs.
Methods
Using anti-mouse CTLA4 antibody 9D9 and anti-mouse PD1 antibody J43 as a proof of concept, we have applied a rational approach to design molecules that are activated in the tumor microenvironment by tumor-associated proteases. In vitro testing using antigen binding by Surface Plasmon Resonance (SPR), antigen-capture ELISA and kinetic assays provided important information to support the design process. Candidates were further characterized in vivo in C57Bl/6 mice implanted with syngeneic MC38 tumors using tumor regression as a measure of efficacy. The potential for improved safety resulting from inhibition of systemic activity was evaluated using a Fluorescent Activated Cell (FAC) analysis of the T cell population in the spleen for the CTLA4 targeting agents. Induction of diabetes in NOD mice was used to evaluate safety potential for the anti-PD1 agents.
Results
The tumor-activated antibodies exhibit significantly reduced affinity for their targets without activation, and full binding affinity is restored with proteolytic activation. In vivo testing of the tumor-activated antibodies led to tumor reduction similar to the parental antibody. Critically, the tumor-activated antibodies exhibit reduced levels of the systemic activity compared to their parental antibody.
Conclusions
We have developed and demonstrated a rational approach to efficiently design immunotherapeutics that are locally activated in the tumor microenvironment and exhibit reduced systemic activities. Our approach, which we have termed Aklusion™, has the potential to improve the safety and efficacy of immuno-oncology therapeutics.
References
1. Hoos, A. Development of immuno-oncology drugs — from CTLA4 to PD1 to the next generations. Nat Rev Drug Discov. 2016;4:235-47.
2. June CH, Warshauer, JT, Bluestone JA. Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat Med. 2017;23(5):540-547.
3. Abdel-Wahab N, Shah M, Suarez-Almazor ME. Adverse Events Associated with Immune Checkpoint Blockade in Patients with Cancer: A Systematic Review of Case Reports. PLoS ONE. 2016;11 (7): e0160221.
P467 CCR4 antagonists inhibit regulatory T cell (Treg) recruitment and increase effector T cell (Teff) numbers in the tumor microenvironment potentiating an anti-tumor response
Oezcan Talay1, Lisa Marshall1, Cesar Meleza1, Maureen Reilly1, Omar Robles1, Mikhail Zibinsky1, Abood Okal1, Jenny McKinnell1, Scott Jacobson1, Erin Riegler2, Sashie Marubayashi1, Emily Karbarz1, David Chian1, Angela Wadsworth1, David Wustrow1, Jordan Fridman2, Paul Kassner1
1FLX Bio Inc., South San Francisco, CA, USA; 2Formerly at FLX Bio Inc., South San Francisco, CA, USA
Correspondence: Paul Kassner (pkassner@flxbio.com)
Background
Regulatory T cells (Treg) are essential for immune tolerance and Treg-mediated suppression of effector T cells (Teff) is important to control inflammation and prevent autoimmune diseases. However, the presence of Treg in the tumor microenvironment (TME) has been shown to dampen anti-tumor immune responses. Human Treg express CCR4, the receptor for the chemokines CCL17 and CCL22. Preclinical and clinical data supports a role for CCR4-mediated recruitment and accumulation of Treg in the TME which can be associated with poor prognosis. Furthermore, patients receiving immunomodulatory agents demonstrate an influx of Treg in responding patients which may dampen optimal anti-tumor responses. Therefore, CCR4 is an ideal target to selectively block Treg recruitment into the TME.
Methods
We have developed a structurally unique series of small molecule antagonists of CCR4 with cellular potencies in multiple assays (e.g. chemotaxis of primary human Treg in 100% serum) in the low double-digit nM range. Moreover, these compounds have excellent in vitro and in vivo ADME properties, consistent with convenient oral dosing. These CCR4 antagonists were tested in murine syngeneic tumor models alone and in combination with immunomodulatory agents. During and following treatment, CCR4 ligand levels, tumor infiltrating lymphocytes, and tumor volumes were evaluated.
Results
Preclinically, these CCR4 antagonists block Treg migration and support expansion of activated Teff in the tumor. Our antagonists reduce Treg in the tumor, but not in peripheral tissues such as blood, spleen or skin; which presents a potential safety advantage to the non-selective approach of depleting anti-CCR4 antibodies. In preclinical efficacy studies, treatment with various checkpoint inhibitors and immune stimulators (e.g., anti-CTLA-4 or anti-CD137) induce the upregulation of CCR4 ligand expression. Combination therapy with CCR4 antagonist and immunomodulatory agents reduced intratumoral Treg number and increased number of activated and total Teff, resulting in an increase in the intratumoral ratios of both CD4+ and CD8+ Teff to Treg. The change in these Teff to Treg ratios is greater for our CCR4 antagonist in combination than with the immunomodulatory agent alone and correlates with enhanced tumor growth inhibition and increased tumor regression.
Conclusions
Combination therapy with CCR4 antagonist and immunomodulatory agents overcome Treg-mediated suppression in tumors and tips the balance toward tumor rejection.
P468 A novel 3D ex vivo platform of fresh patient tumor tissue with intact tumor microenvironment for immuno-oncology drug development and biomarker discovery
Melanie Mediavilla-Varela, Melba Marie Page, Jenny Kreahling, Scott Antonia, Soner Altiok
Nilogen Oncosystems, Tampa, FL, USA
Correspondence: Soner Altiok (jenny@nilogen.com)
Background
Cancer immunotherapy delivers treatment of high specificity, low toxicity and prolonged activity in subsets of patients. There is an unmet need for clinically relevant preclinical models and translational strategies that recapitulate the complexity of tumor immune microenvironment to test the therapeutic potential of immuno-oncology (IO) drugs, identify rational combination therapies and to develop novel predictive biomarkers of clinical response. Here we describe a novel 3D ex vivo drug assay utilizing fresh patient tumor samples with intact immune microenvironment to facilitate oncology and IO drug development and biomarker discovery.
Methods
3D ex vivo studies were performed with fresh tumor tissue obtained from consented patients with non-small cell lung, renal, urothelial, head & neck and breast carcinoma. 3D tumor microspheroids (150 μm in size) prepared according to a proprietary protocol were treated in their intact immune microenvironment with PD1 inhibitors pembrolizumab or nivolumab at 10μg/ml for 36 hours. Culture media was collected over the course of the experiments to simultaneously analyze the differential release of cyto-and chemokines. Treatment-mediated changes in T-cell activation and immune cell populations were monitored by flow cytometry while NanoString PanCancer Immune Profiling platform containing probes to quantitate 770 immune function genes was used to determine positive and negative associations between expression of immune function genes and TIL activation by ex vivo treatment. Immunohistochemical studies were performed to identify PD-L1 expression and immune cell composition in tumor samples.
Results
3D ex vivo samples treated with pembrolizumab or nivolumab demonstrated PD1 occupation in all tumors, while approximately 30% of tumors showed increased CD8 T-cell activation that closely correlated with proinflammatory cytokine release in the conditioned media. Immune gene expression profiling studies revealed increased expression of IFNg inducible genes in tumors showing activated CD8 population by flow cytometry. No significant correlation was found between tumor PD-L1 expression and ex vivo response to treatment. Furthermore, we showed pembrolizumab or nivolumab treatment ex vivo led to changes in tumor immune cell composition in specifically including changes in monocyte populations in subset of tumors used in the ex vivo experiments.
Conclusions
Our studies showed that this approach can be used to identify rationale drug combinations and to develop potential companion diagnostics to facilitate biomarker-driven drug development efforts and personalized medicine in immuno-oncology.
P469 Tumor oxygenator OMX reverses multiple modes of immunosuppression, activates anti-tumor immunity and cures as a single agent, and in combination with anti-PD-1 in resistant orthotopic tumors
Ana Krtolica, Natacha Le Moan, Philberta Leung, Sarah Ng, Tina Davis, Carol Liang, Jonathan Winger, Stephen P. L. Cary
Omniox, Inc., San Carlos, CA, USA
Correspondence: Ana Krtolica (akrtolica@omnioxinc.com)
Background
Hypoxia, a common feature of solid tumors that correlates with poor patient outcomes, is a central mediator of escape from innate and adaptive anti-tumor immune responses. Numerous classes of immunotherapy target specific pathways of hypoxia-driven immunosuppression. Reversing tumor hypoxia, however, would work upstream at one of the primary nodes of immunosuppression to stimulate effective responses. The hypoxic tumor microenvironment arises due to dysfunctional tumor vasculature, and oxygen diffusion is restricted to within ~80 mm of active capillaries. To reverse tumor hypoxia and create an immunopermissive microenvironment, we engineered OMX, a stable, non-vasoactive, high affinity oxygen carrier that preferentially accumulates in solid tumors and specifically re-oxygenates hypoxic microenvironments without affecting normoxic tissues. OMX improves anti-tumor immune responses.
Methods
We used immunohistochemical, flow cytometric, and multiplex cytokine analyses to evaluate OMX’s ability to weaken immunosuppression in the tumor microenvironment of orthotopic GL261 brain tumors and promote tumor cures.
Results
A single dose of OMX in GL261 tumor-bearing mice reduces tumor hypoxia, enhances T cell accumulation into the tumor, decreases Tregs and increases activation and proliferation of cytotoxic T lymphocytes (CTLs). Specifically, OMX increases the Teff/Treg ratio by ~3-fold, indicating a switch from an immunosuppressive to an immunopermissive microenvironment. In addition, OMX stimulates secretion of a major chemoattractant for Th1 cells, and increases the accumulation of CD8+ tumor-specific T cells in the tumor tissue. When combined with anti-PD-1, OMX increases CTL infiltration, proliferation and cytotoxic activity, and modulates IFNγ and IFNγ-inducible cytokines that may polarize T cells towards a Th1 phenotype. OMX alone resulted in ~50% tumor cures when treatment is initiated at day 7 in GL261 tumor-bearing mice. Furthermore, the combination of OMX with anti-PD-1 in late-stage GL261 tumor-bearing mice increases mouse survival by ~80% and in large PD-1 resistant GL261 tumors, OMX significantly increases the median survival by ~50%. The survival benefit observed with OMX could be predicted with an identified circulating chemokine signature (post-hoc test).
Conclusions
By delivering oxygen specifically to the hypoxic tumor microenvironment, OMX may restore anti-cancer immune responses in cancer patients through activation of inflammatory cytokine cascades and mobilization and activation of tumor-specific T cells. As a unique, pleiotropic immunotherapy, OMX may enhance immune control of tumors to improve patient outcomes.
P470 Tertiary lymphoid organs as a good prognostic indicator following neoadjuvant chemo (radio) therapy for pancreatic cancer
Shota Kuwabara, Takahiro Tsuchikawa, Yoshitsugu Nakanishi, Toshimichi Asano, Takehiro Noji, Yo Kurashima, Yuma Ebihara, Soichi Murakami, Toru Nakamura, Keisuke Okamura, Toshiaki Shichinohe, Satoshi Hirano
Hokkaido University Graduate School of Medicine, Sapporo Hokkaido, Japan
Correspondence: Takahiro Tsuchikawa (medicineman424@yahoo.co.jp)
Background
Pancreatic cancer (pancreatic ductal adenocarcinoma; PDAC) is a highly malignant tumor that frequently develops local recurrence and distant metastasis even after a curative resection. Prognosis of PDAC is very poor, and the overall 5-year survival rate for patients with PDAC treated by curative resection is 15-25%. Recently, neoadjuvant chemo (radio) therapy had been reported to improve treatment outcome. Therefore, it is an urgent need to explore surrogate markers predicting patients’ outcome. We herein focused on the tumor microenvironment as one of the candidates for monitoring local pathological and immunological reactions, and put a spotlight on tertiary lymphoid organs (TLO, alternatively, ectopic lymphoid tissue) as a biomarker reflecting the effect of preoperative neoadjuvant chemotherapy (NAC).
Methods
In this study, we retrospectively analyzed the area of TLO, dividing 140 patients diagnosed with PDAC from January 2009 to December 2015 into two groups, those who performed upfront surgery (SF group; n=93) and those who underwent NAC (NAC group; n=47). Then we measured the area (mm2) of TLO, the total area (mm2) of the tumor, and calculated the area ratio of total tumor to the total TLO (TLO/ Tumor ratio) by microscopic observation, and statistically analyzed the association between the level of TLO formation and prognosis in the tumor microenvironment. All the microscopic images were imported as digital photo files using a NanoZoomer Digital Pathology system (Hamamatsu Photonics, Hamamatsu, Japan), and imaging analysis were performed using Image J software (NIH, Bethesda, MD, USA).
Results
The TLO formation was recognized in 128 patients (91.4%; 128/140). There were no significant difference in terms of TLO formation nor TLO/ Tumor ratio between the two groups (94.6%; 88/93 v.s. 85.1%; 40/47, p=0.0575 and 0.66±1.22 v.s. 0.64±8.62, p=0.2342, respectively). The 5-year-overall survival rate of NAC group had been significantly better than SF group (41% v.s.16%, p=0.0203). On multivariate analysis, lymphnode metastaisis (HR 0.047, 95% CI: 0.005-0.265, p<0.0001) and high TLO/ Tumor ratio (HR 0.108, 95% CI: 0.010-0.593, p=0.008) were revealed as independent favorable prognostic factors.
Conclusions
Our present results indicate that the level of TLO serves as one of the valuable independent prognostic markers following neoadjuvant chemo (radio) therapy for PDAC.
P471 Patterns of immune cell infiltration in murine models of melanoma: roles of antigen and tissue site in creating inflamed tumors
Katie Leick1, Joel Pinczewski2, Donna Deacon1, Amber Woods1, Marcus Bosenberg3, Victor Engelhard1, Craig Slingluff1
1University of Virginia, Charlottesville, VA, USA; 2Dorevitch Pathology, North Melbourne, Australia; 3Yale University, New Haven, CT, USA
Correspondence: Victor Engelhard, Craig Slingluff (kml2h@virginia.edu)
Background
Immune cell infiltration is associated with improved survival in melanoma and other cancers. Melanoma metastases in humans may be grouped into three Immunotypes representing patterns of immune cell infiltration: A (sparse), B (perivascular cuffing), and C (diffuse)1. Prior work suggests that anatomical site and intratumoral vascularity can have dramatic effects on T cell responses and immune infiltration. Murine models provide opportunities to understand factors that control immune infiltrates, but Immunotypes have not been defined for common murine models. We hypothesize that patterns of immune cell infiltration can be defined in Braf/Pten and B16 murine models that are similar to Immunotypes observed in human melanoma metastases1, and that AAD and OVA transfectants and intraperitoneal (i.p.) location will facilitate greater immune infiltration in the B16 model.
Methods
We performed immunohistochemistry for S100, CD31, and CD45, with immune cell enumeration, Immunotyping, and scoring of vascular density in spontaneous melanoma models and in transplantable melanoma models (B16-F1, B16-OVA, and B16-AAD). The transplantable tumors were grown either in subcutaneous (s.c.) or i.p. locations.
Results
The Braf/Pten and Braf/Pten/β-catenin tumors had low immune cell counts in 6/6 tumors that were consistent with Immunotype A (Fig. 1A, Fig. 1B), which was also seen in 11/12 B16-F1 tumors (Fig. 1C). In comparison, 9/10 tumors in B16-OVA s.c. and i.p. and 5/6 B16-AAD s.c. tumors were characterized as Immunotype B. Only the B16-AAD i.p. tumors were Immunotype C in 4/6 tumors. The i.p. location was characterized by significantly higher immune cell counts in B16-OVA tumors (P = 0.0008, Fig. 1D) and higher tumor vascularity. Interestingly, mutated cancer antigens in mutant BRAF models and in B16 melanoma were insufficient to induce significant immune infiltrates, suggesting the existence of barriers to immune infiltration. Addition of model neoantigens (OVA or AAD) overcame existing barriers in a manner that was dependent in part on tumor site.
Conclusions
These murine models may be useful for preclinical studies of combination immune therapy, and suggest that both tumor antigen expression and tumor location contribute to the extent of immune cell infiltration and Immunotype in murine melanoma.
Spontaneous Braf/Pten and Braf/Pten/β-catenin dermal melanomas lacked significant intratumoral immune cell infiltrates characteristic of Immunotype A (A), and the mean CD45+ cell counts for 3 mice of each tumor type were all less than 10 per hpf in murine melanoma (B). Associations of B16 tumor cell type and tumor location with immune infiltrate. B16-F1 melanomas (B16-F1), or those transfected with OVA or AAD, were grown in from s.c. or i.p. locations and evaluated for Immunotype (C) and overall CD45+ cell counts (D). Significance is noted by asterisks: * < 0.05, ** < 0.001, **** <0.0001.
References
1. Erdag G, Schaefer JT, Smolkin ME, et al. Immunotype and immunohistologic characteristics of tumor-infiltrating immune cells are associated with clinical outcome in metastatic melanoma. Cancer Res. 2012;72(5):1070.

Immunotype and immune cell density of Braf/Pten and B16 murine melanomas
P472 DARPin®-based fibroblast activation protein-targeted agonists of 4-1BB and OX40 co-stimulate T-cells in a Fc-receptor-independent, tumor-restricted manner
Alexander Link, Julia Hepp, Ulrike Fiedler, Christian Reichen, Clara Metz, Alexan der Titz, Ivana Tosevski, Joanna Taylor, Zita Arany, Laurent Juglair, Patricia Schildknecht, Yvonne Kaufmann, Denis Villemagne, Simon Fontane, Ralph Bessey, Christof Zitt, Keith Dawson, Dan Snell, Daniel Steiner, Michael Stumpp, Victor Levitsky
Molecular Partners AG, Schlieren-Zürich, Switzerland
Correspondence: Victor Levitsky (victor.levitsky@molecularpartners.com)
Background
In addition to check point blockade, immunosuppressive tumor microenvironment can be counteracted by enhanced triggering of T-cell co-stimulatory receptors. In vivo agonistic activity of antibodies against such receptors usually requires Fc-dependent crosslinking and comes at a cost of various systemic, immune related toxicities. We developed a new class of biologics based on the DARPin® technology platform which allow tumor-restricted engagement of co-stimulatory molecules independently of Fc-receptors. Our preclinical in vitro and in vivo results indicate that DARPin®-based co-stimulatory receptor agonists are comparable to or exceed conventional antibodies in agonistic potency but restrict their activity to the tumor site effectively widening the therapeutic window for compounds exploiting this mode of action.
Methods
We generated multi-specific DARPin® therapeutics, which bind to human fibroblast activation protein (FAP) and either to 4-1BB or OX40 co-stimulatory receptor, preferentially expressed on CD8+ and CD4+ lymphocytes, respectively. The agonistic activity of both drug candidates was analyzed using NFkB-pathway activation reporter cells lines and several types of T-cell activation assays. The 4-1BB-agonist was tested in a xenotransplantation tumor model established in NOG mice humanized by PBMCs.
Results
Both 4-1BB and OX40-specific FAP-targeted DARPin® drug candidates efficiently induced activation of the NFkB pathway in 4-1BB or OX40 expressing reporter cells but only after crosslinking on FAP, either surface immobilized or expressed on cells. The capacity of the molecules to enhance lymphokine secretion and upregulate expression of activation markers on T-cells stimulated by polyclonal T-cell receptor crosslinking or specific antigen recognition also strictly required FAP-dependent oligomerization.
Urelumab, a clinically tested 4-1BB agonistic antibody associated with liver toxicity, led to signs of severe graft-versus-host disease (GVHD) and weight loss greater than 20% in the humanized NOG mice transplanted with HT-29 tumor cells at termination of the experiment. In contrast, mice treated with several doses of the 4-1BB-specific DARPin® drug candidate showed only minor signs of GVHD and marginal weight loss. Further analysis indicated that urelumab induced CD8+ T-cell expansion both in the peripheral blood and tumor tissue of animals while the 4-1BB-specific DARPin® drug candidate triggered a similar change only in the tumor sites.
Conclusions
Thus, tumor targeted DARPin® agonists of co-stimulatory molecules can enhance T-cell activity in a manner independent of Fc-crosslinking and limited to the tumor site, thereby avoiding systemic toxicity caused by antibodies against the same immunologic targets. This approach with DARPin® therapeutics can be broadly applied to a range of tumor targets and immune receptors.
P473 Immuno-oncological efficacy of RXDX-106, a novel TAM (TYRO3, AXL, MER) family small molecule kinase inhibitor
Erin Lew1, Yumi Yokoyama1, Colin Walsh1, Jack Lee1, Joanne Oh1, Elizabeth Tindall1, Robin Navarez1, Amy Dilberto1, Heather Ely1, Ruth Seelige2, Amanda Albert1, Jack Bui2, Gary Li1
1Ignyta, Inc, San Diego, CA, USA; 2University of California, San Diego, San Diego, CA, USA
Correspondence: Gary Li (elew@ignyta.com)
Background
The receptor tyrosine kinases (RTKs), TYRO3, AXL, and MER (TAM), are emerging targets for cancer therapy. Particularly, TAM RTKs play a key homeostatic role as negative regulators of immune responses. RXDX-106 is a potent and selective TAM family inhibitor in preclinical development. Here, we sought (1) to evaluate the efficacy of RXDX-106 as a single agent and in combination with immune checkpoint inhibitors; (2) to identify the immuno-modulatory mechanisms of action; and (3) to decipher the reciprocal regulation of TAM expression on cancer and immune cells, and how pharmacological inhibition of TAM signaling would be beneficial, given their complex interaction and regulation in the tumor microenvironment.
Methods
The anti-tumor efficacy of RXDX-106, as single agent or in combination with checkpoint inhibitors, was studied in tumors grown either in syngeneic models or in immunocompromised mice. Immuno-phenotyping was performed in tissue and tumor samples collected during or at the end of studies by flow cytometry, gene expression and protein analyses, and Luminex bead-based multiplex assays.
Results
Here, we demonstrated that tumor growth inhibition in an MC38 model was associated with increase in intra-tumoral M1 macrophages, CD169hi antigen presenting macrophages, and PD-L1 expression. Also observed were a higher ratio of CD8+/CD4+ T cells and an increased expression of CD69 and PD-1 on CD8+ T cells, indicative of activation of cytotoxic T cells. Additionally, an increase in Granzyme B and IFNγ with a concomitant decrease in VEGF in tumor cell lysates indicted T cell activation and M1 polarization of macrophages. Furthermore, in a CT26 syngeneic model, we demonstrated that RXDX-106 inhibited tumor growth as a single agent, and the effect was further potentiated by combination therapy with immune checkpoint inhibitors, as evidenced by upregulation of anti-tumor gene expression patterns, upregulation of anti-tumor cytokines in the tumor cell lysates, and an increase in T cell function. Finally, in an AXL-driven tumor model, we demonstrated that AXL expressing tumors induced a pro-tumorigenic immune environment, and treatment with RXDX-106 resulted in complete tumor regression and re-polarization of macrophages towards an M1, anti-tumor phenotype.
Conclusions
RXDX-106 has the potential to restore and enhance immune function in macrophages and T cells, resulting in repolarization of the immune response towards an anti-tumor microenvironment. The unique mechanism of activating both innate and adaptive immunity, plus regulating cross-talk between immune cells and tumor cells by RXDX-106 supports clinical development of RXDX-106 to potentially treat a wide variety of cancers.
P474 Gene expression in MHC II pathway may predict triple negative breast cancer prognosis
Amy Zhao1, Gavin Bond1, Katherine Varley2, Andres Forero-Torres3, Albert LoBuglio3, Dongquan Chen3
1University of Pennsylvania, Philadelphia, PA, USA; 2University of Utah, Salt Lake City, UT, USA; 3University of Alabama at Birmingham, Birmingham, AL, USA
Correspondence: Amy Zhao (yli@uabmc.edu)
Background
Triple negative breast cancer (TNBC) is a subtype with heterogeneous patient outcomes. It behaves aggressively, and patients are not a candidate for ER or HER2/Neu targeted therapy. Although there have been major research efforts directed at establishing genomic signatures to assess prognosis of breast cancer, prognostic gene expression signatures are not well developed for TNBC. In this study, we investigated twenty-four genes [1] related to the MHC II pathway in 47 TNBC patients and explored gene(s) that can be used to distinguish who will relapse. This work may lead to TNBC management and treatment and could also provide mechanism for the generation of the anti-tumor immune response.
Methods
Forty-seven snap frozen primary TNBC tumor specimens were analyzed using RNA-Seq. Individual gene expressions were transformed to fit a normal distribution. The Logistic regression analysis was used to identify genes that can predict no relapse. The optimal cutpoint was determined from the receiver operating characteristic analysis. This study has the approval of UAB’s Institutional Review Board.
Results
CIITA had significantly high expression in TNBC patients who had not relapsed (P<0.0001). With high CIITA expression, TNBC were 10 times more likely to have not relapsed than those with low expression (OR =10.8, 95% CI 2.8-41.9, p=0.006). The sensitivity and specificity are 75% and 78.3%, respectively. Using multivariable approach with all 24-gene panel, we identified three genes, CIITA, CTSH and KRT14, which together can predict no relapse with 87.5% sensitivity, and 78.3% specificity. Further, at the optimal cut point, we found CIITA alone can predict TNBC no relapse with 84% sensitivity and 77% specificity and accuracy of 81%. It predicted 17/ 22 relapse, and 21/25 no relapse correctly. These biomarkers were independent predictors of TNBC prognosis regardless of age, race, tumor grade and stage. The findings were confirmed in public microarray data from 199 patients with TNBC confirmed.
Conclusions
High expression of genes in the MHC II pathway, e.g. CIITA, may predict TNBC prognosis with high sensitivity and specificity regardless of age, race, and tumor grade and tumor stage. This work may lead TNBC management and treatment. Although we explored the optimal point to distinguish patients’ outcome, it is important to further establish the threshold in a larger TNBC population.
References
1. Forero A, et al. Expression of the MHC Class II Pathway in Triple-Negative Breast Cancer Tumor Cells is Associated with a Good Prognosis and Infiltrating Lymphocytes. Cancer Immunol Res. 2016; 4(5): 390–399.
P475 Profiling Immune Infiltration of Glioblastoma
Camilo Fadul, Craig Slingluff, Ileana Mauldin
University of Virginia, Charlottesville, VA, USA
Correspondence: Craig Slingluff (ileanasoto@virginia.edu)
Background
The median survival for patients with glioblastoma (GBM) treated with standard of care therapies is 15-20 months. The efficacy of immunotherapy for many types of cancer make this an attractive strategy to improve outcomes for patients with GBM; unfortunately, thus far these have not had demonstrated efficacy in GBM. In other cancers, the presence of immune cell infiltration, the pattern of infiltration, and the expression of immune markers have been associated with survival and response to some types of immunotherapy. However, the immune infiltrate in GBM has not been well-characterized.
Methods
Multispectral immunohistochemistry images were obtained from 6 GBM specimens stained with CD8, CD20, FoxP3, PNAD, CD83 and Ki67, and analyzed for tumor infiltrating lymphocytes using digital software.
Results
In preliminary studies, we have observed that GBM can have dense B cell infiltrates located near blood vessels; however, the functional role of B cells in GBM, and the prevalence of these cells has been unstudied. Few studies have been performed evaluating B cells in GBM and suggest contradictory roles for B cells in GBM. Candolfi et al. studied the role of B cells in a GBM murine model and concluded that B cells act as antigen presenting cells, lending a critical role in clonal expansion of tumor antigen specific T cells, and brain tumor regression in mice. Conversely, Saraswathula et al. showed that patients with GBM have Bregs in peripheral blood. Bregs downregulate effector T cell responses through the secretion of immunosuppressive cytokines such as IL-10 and through cell-cell contact. Thus, these studies present conflicting roles for B cells in the GBM setting. As a preliminary assessment, we examined the association between survival and overexpression of the MS4A1 gene which encodes for CD20, using RNA-seq gene expression data obtained from TCGA. The expression data showed that patients with overexpression of the gene encoding for CD20 had decreased overall survival (p = 0.0111). While these data provide interesting preliminary findings, they need to be further investigated, by evaluating markers of B cell effector functions in GBM and the impact of B cells on T cells infiltrating tumor. However, considering these gene expression data, we hypothesize that CD20+ B cells in GBM may negatively impact patient survival, and that these cells may constitute Bregs.
Conclusions
To optimize immunotherapy, there is a necessity to understand the prevalence and the functional role that B cells play in GBM and their impact on patient survival.
P476 Targeting the IDO/TDO pathway through degradation of the immunosuppressive metabolite kynurenine and inhibition of the downstream AHR pathway
Karen McGovern1, X. Michelle Zhang1, Everett Stone3, Todd Triplett3, Kendra Garrison3, Silvia Coma1, Luis Felipe Campesato2, Meghan Walsh1, Jeremy Tchaicha1, Candice Lamb3, Christos Karamitros3, John Blazek3, Taha Merghoub2, Jedd Wolchok2, George Georgiou3, Mark Manfredi1
1Kyn Therapeutics, Cambridge, MA, USA; 2Memorial Sloan Kettering Cancer Center, New York, NY, USA; 3University of Texas, Austin, TX, USA
Correspondence: Karen McGovern (karen@kyntherapeutics.com)
Background
Checkpoint inhibitors have become the cornerstone of immune-based oncology therapy. Several orthogonal immune pathways are currently being investigated to relieve suppression or boost activity of the innate and adaptive immune system, including the study of the immune-modulating role of metabolites. Both Indoleamine-pyrrole 2,3-dioxygenase 1 (IDO1) and tryptophan 2,3-dioxygenase 2 (TDO2) enzymes metabolize tryptophan forming kynurenine, which binds the aryl hydrocarbon receptor (AHR) in multiple innate and adaptive immune cell types, causing a net immunosuppressive environment. Both enzymes are upregulated across many tumor types, and the IDO1 enzyme has been clinically validated with small molecule inhibitors in combination with checkpoint inhibition, lending credence to the tumor microenvironment containing small molecule metabolites that induce immune cell tolerance, and leaving the opportunity for broader efficacy if both pathways can be targeted. Kynureninase (Kynase), an enzyme degrading kynurenine generated from IDO1 and TDO2 pathway, as well as AHR antagonism through small molecule inhibition, can potentially relieve the immunosuppression on multiple cells types and provides novel approaches to inhibiting this pathway in multiple tumor types.
Methods
For Kynase, a pegylated enzyme was used for in vitro and in vivo studies. Pharmacodynamic (kynurenine depletion) and efficacy studies were performed in syngeneic tumor models as single agent or in combination with checkpoint inhibitors (anti-PD1 or anti-CTLA4). Ex-vivo studies were done to analyze the immune cell composition of tumors post-treatment and the effect on AHR signaling. For AHR antagonists, in vitro and in vivo studies were performed to assess kynurenine-dependent signaling through AHR.
Results
Kynase depleted kynurenine produced by IDO1+, TDO2+ and dual positive human cancer cells whereas, the IDO1 inhibitor epacadostat or TDO2 inhibitor 680C91 selectively inhibited Kyn production in IDO1+ or TDO2+ cells, respectively. Kynase ameliorated the suppressive effects of kynurenine on CD4+ and CD8+ T-cells. In B16F10 tumor-bearing mice, a single subcutaneous dose of Kynase depleted kynurenine in the plasma and tumors, leading to an increase in tumor infiltrating CD8+ and CD4+ lymphocytes. Kynurenine activation of AHR in cells, measured by gene expression, is inhibited with AHR antagonists. Kynase demonstrated significant tumor growth inhibition and survival benefit either as a single agent or in combination with checkpoint inhibitors in B16F10, CT26 and 4T1 models. Interestingly, Kynase combined with anti-PD1, showed greater efficacy than epacadostat / anti-PD1 combination.
Conclusions
Our data support clinical development of human Kynase for the treatment of cancers where both IDO1/TDO2 pathways play a significant immunosuppressive role through kynurenine production.
P477 Novel effector phenotype of Tim3+ regulatory T cells leads to enhanced suppressive function in head and neck cancer patients
Zhuqing Liu1, Elizabeth McMichael2, Gulidanna Shayan3, Jing Li3, Rghvendra Srivastava2, Lawrence Kane2, Robert Ferris2
1Tongji University, Shanghai, China; 2University of Pittsburgh, Pittsburgh, PA, USA; 3Tsinghua University, Beijing, China
Correspondence: Robert Ferris (cmichaele@upmc.edu)
Background
Regulatory T (Treg) cells are important suppressive cells among tumor infiltrating lymphocytes. Treg express the well-known immune checkpoint receptor PD-1, which is reported to mark “exhausted” Treg with lower suppressive function. T cell immuglobulin mucin (Tim)-3, a negative regulator of Th1 immunity, is expressed by a sizable fraction of TIL Tregs, but the functional status of Tim-3+ Tregs remains unclear.
Methods
CD4+CTLA-4-CD25high Treg were sorted from freshly excised head and neck squamous cell carcinoma (HNSCC) TIL based on Tim-3 expression. Functional and phenotypic features of these Tim-3+ and Tim-3- TIL Tregs were tested by in vitro suppression assays and multi-color flow cytometry. Gene expression profiling and NanoString analysis of Tim-3+ TIL Treg was performed. A murine HNSCC model was used to test the effect of anti-PD-1 immunotherapy o nTim-3+ Treg.
Results
Despite high PD-1 expression, Tim-3+ TIL Treg displayed a greater capacity to inhibit naïve T cell proliferatin than Tim-3- Treg. Tim-3+ Treg from human HNSCC TIL also displayed an effector-like phenotype, with more robust expression of CTLA-4, PD-1, CD39, and IFN-γ receptor. Exogenous IFN-γ treatment could partially reverse the suppressive function of Tim-3+ TIL Treg. Anti-PD-1 immunotherapy downregulated Tim-3 expression on Tregs isolated from murine HNSCC tumors, and this treatment reversed the suppressive function of HNSCC TIL Tregs.
Conclusions
Tim-3+ Treg are functionally and phenotypically distinct in HNSCC TIL, and are highly effective at inhibiting T cell proliferation despite high PD-1 expression. IFN-γ induced by anti-PD-1 immunotherapy may be beneficial by reversing Tim-3+ Treg suppression.
P478 Formation and functional associations of CD49a-, CD49b- and CD103-expressing CD8 T cell populations in human metastatic melanoma
Marit Melssen1, Walter Olson1, Nolan Wages1, Ciara Hutchison1, Ileana Mauldin1, Cornelis Melief2, Timothy Bullock1, Victor Engelhard1, Craig Slingluff, Jr1
1University of Virginia, Charlottesville, VA, USA; 2ISA Pharmaceuticals, Leiden, Netherlands
Correspondence: Marit Melssen (mm2xz@virginia.edu)
Background
Integrins α1β1 (CD49a), α2β1 (CD49b) and αEβ7 (CD103) mediate retention of lymphocytes in peripheral tissues, and their expression is upregulated on CD8+ tumor infiltrating lymphocytes (TIL) compared to circulating lymphocytes. Little is known about what induces expression of these retention integrins (RI). We hypothesized that RI expression marks functionally distinct T cell subsets in the tumor microenvironment (TME) and that these subsets can be induced by selected cytokines and T cell receptor (TCR) stimulation.
Methods
CD8+ TIL from 19 human melanoma metastatic lesions were stained for CD49a, CD49b, CD103, and cytokines either directly or after PMA/Ionomycin stimulation. For in vitro studies, circulating CD8+ T cells from normal donors (n=4-7) were cultured with cytokines and/or CD3/CD28 stimulation and evaluated by flow cytometry.
Results
Among CD8+ TIL from melanoma metastases, RIneg, CD49a+CD49bnegCD103neg and CD49a+CD49b+CD103neg subpopulations were found in virtually all patients. However, only a subset of patients had CD103+ subpopulations (CD49a+CD49bnegCD103+ and CD49a+CD49b+CD103+). Each subset was assessed for cytokine production. A large fraction of CD49a+CD49bnegCD103neg TIL was multifunctional, producing IFNγ, TNFα and IL-2. In contrast, CD49a+CD49bnegCD103+ TIL only expressed IFNγ, and both CD49a+CD49b+CD103neg and CD49a+CD49b+CD103+ subsets were incapable of inducing IFNγ, TNFα or IL-2 at comparable levels (Fig.1). Next, we evaluated RI expression of naïve T cells in response to different stimuli in vitro. TCR or IL-2 stimulation alone induced two RI+ cell populations: one that co-expressed CD49a and CD49b and another that expressed CD49a alone. Adding TNFα to TCR stimulation further induced these populations, whereas TGFβ + TCR stimulation generated two additional populations; CD49a+CD49bnegCD103+ and CD49a+CD49b+CD103+. Each of these subsets was found among melanoma TIL, suggesting that TCR stimulation, IL-2, TNFα and/or TGFβ may be responsible for the generation of RI+ subsets in the TME.
Conclusions
These observations suggest that CD49b and/or CD103 expression is upregulated as effector TIL lose the capability of producing cytokines and become more exhausted. Generation of CD103+ subsets in the TME may be driven by TCR stimulation and TGFβ and given their absence in a fraction of tumors, may only be present in tumors producing sufficient TGFβ. The other RI+ subsets can be induced by IL-2, TCR engagement and/or TNFα, which we hypothesize will be available in all tumors. Together, our findings identify opportunities to modulate RI expression in the TME by cytokine therapies and to augment subsets with specific RI expression in the interest of improving immune therapies for cancer or other immune-related diseases.
P479 MPL-5821, an ESM™-p38 MAPK Inhibitor, modulates macrophage plasticity leading to enhanced IL-12p70 and IFN gamma, reduced IL-10 and the reversal of macrophage induced T-cell suppression
Martin Perry1, David Moffat1, Justyna Rzepecka2, Lucia Janicova2, Anastasia Nika2, Darryl Turner2, Clare Doris2, Stephen Anderton2
1Macrophage Pharma Limited, Windsor, United Kingdom; 2Aquila BioMedical Limited, Edinburgh, United Kingdom
Correspondence: David Moffat (david@macrophagepharma.com)
Background
Myelomonocytic cells orchestrate both innate and adaptive immune response to tumours but frequently adopt an immunosuppressive, tumor supportive phenotype (M2-like). However, their plasticity offers the opportunity for therapeutic repolarization to one that is non-immunosuppressive and supportive of an anti-tumor immune response (M1-like). A challenge in identifying such pharmaceuticals is their counter-active functionality in other important immune cell types such as lymphocytes. Macrophage Pharma Limited’s Esterase Sensitive Motif (ESM™) technology predominantly targets myelomonocytic cells [1]. We describe the preliminary in vitro characterisation of MPL-5821, an ESM-p38 MAPK inhibitor, contrasting it with conventional inhibitors, with reference to its cell type specificity, sparing of myelomonocytic – lymphocyte communication and ability to reverse macrophage driven lymphocyte immunosuppression.
Methods
Human Macrophage Assays: M2 polarised macrophages (M-CSF, +/- IL-4, IL-10, TGFβ) were stimulated with LPS/IFNγ +/- compounds overnight. Cytokine production and macrophage markers were measured by ELISA and flow cytometry respectively.
Human PBMC Assay: PBMCs were stimulated with either LPS or anti-CD3 +/- compounds for 72 hours and cytokine production measured by ELISA. CD4 and CD8 T-cell proliferation was measured by flow cytometry.
Macrophage suppression of Human T-cells: M2-polarised macrophages were incubated with autologous PBMCs +/- compound. T cell stimulation was provided by anti-CD3/anti-CD28. Cytokine production was analysed by ELISA and expression of CD4, CD8 and Ki67 by flow cytometry.
Results
Human Macrophage Assays: MPL-5821 and reference p38 MAPK inhibitors exhibited a similar profile in inhibiting IL-10 production and enhancing IL-12p70 production.
Human PBMC Assay: In comparison with reference p38 MAPK inhibitors, MPL-5821 was several orders of magnitude more potent in enhancing IL-12p70 production and was the only inhibitor to enhance IFNγ production under LPS stimulation. Similarly, MPL-5821 was the only inhibitor to enhance IFNγ production in response to anti-CD3 stimulation.
Macrophage suppression of Human T-cells: MPL-5821 reversed the suppressive effects of the M2 macrophages as evidenced by a return of CD4 and CD8 proliferation to control levels accompanied by a concomitant increase in IFNγ and reduction in IL-10.
Conclusions
MPL-5821 modulates the macrophage phenotype to IL-12p70Hi / IL-10Lo and reverses the M2 macrophage immune suppression of T-cell functionality. The ESM™ cell selectivity differentiates MPL-5821 from other p38 MAPK inhibitors by its sparing of other immune cells such as lymphocytes. Thus MPL-5821 enables the maintenance of the myelomonocytic/lymphocyte IL-12p70/IFNγ axis, key to an effective anti-tumour immune response.
References
1. Needham LA et al, J Pharmacol Exp Ther. (2011), 339 :132-42.
P480 Tumor infiltrating T regulatory cells in head and neck cancer patients treated with cetuximab demonstrate increased inhibitory receptors and survival
Jessica Moskovitz, Tullia Bruno, Robert Ferris, Dario Vignali
University of Pittsburgh, Pittsburgh, PA, USA
Correspondence: Jessica Moskovitz (jessiemoskovitz@gmail.com)
Background
Patients with locally advanced head and neck squamous cell carcinoma (HNSCC) incur significant treatment morbidity and have a poor 5 year overall [1]. The idea of treating HNSCC by targeting epidermal growth factor receptor (EGFR) developed from the demonstration of EGFR overexpression correlating with advanced tumor size and decreased survival [2]. Clinical anti-tumor activity of cetuximab, a monoclonal antibody that prevents ligand binding of EGFR, did not correlate with expected response rates despite high HNSCC tumor cell EGFR expression. Further studies to clarify the mechanism of action of cetuximab in HNSCC revealed an increase in CTLA4+ T regulatory cells (Tregs) that suppressed NK cell mediated cytotoxicity, which correlated with poor response to neoadjuvant cetuximab treatment [3].
Methods
We aimed to further measure inhibitory receptor [e.g. PD1, TIM3, TIGIT, CTLA4, neuropilin-1(NRP1)] expression on T cells in tumor and peripheral blood lymphocytes (TIL/PBL) of cetuximab treated patients. Patients with locally advance HNSCC were treated with neoadjuvant cetuximab for four weeks prior to definitive surgery with CT scans performed prior to each intervention. Nine samples (TIL and PBL) pre and post cetuximab were analyzed using flow cytometry, and tumor area change was measured using delta CT.
Results
Tumor infiltrating CTLA4+ Tregs had increased expression of NRP1 and TIGIT after cetuximab therapy, specifically in patients that had a poor response to cetuximab. Further, tumor infiltrating CTLA4+TIGIT+ Tregs correlated with patients having larger baseline tumors and minimal change in tumor area post cetuximab. Finally, Tregs from these patients displayed an increase in mean flouresecence intensity of pro-survival markers such as Bcl2.
Conclusions
Increased inhibitory receptors such as TIGIT and NRP1 on CTLA4+ Tregs combined with increased pro-survival markers in patients that had a minimal response to cetuximab suggests that these Tregs maintain suppressive function and survival despite cetuximab treatment. Further characterization of the immune resistance mechanism to cetuximab will provide the basis for designing knowledgeable combinatorial trials with other immunotherapy agents i.e. CTLA-4.
Trial Registration
clinical trials.gov:NCT01218048,NCT0193592.
References
1. Vokes EE, Weichselbaum RR, et al. Head and neck cancer. N Engl J Med 1993;328:184-194.
2. Ang KK, Berkey BA,et al. Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Res. 2002;62(24):7350-6.
3. Jie HB, Ferris RL, et al. CTLA4+ Regulatory T cells are increased in head and neck cancer patients, supress NK cell cytotoxicity and correlate with poor prognosis. Cancer Res. 2015;75 (11):2200-2210.
P481 Non-oncogenic acute viral infection modulates the innate immune response and reduces tumor growth in hosts with established cancer
Jenna Newman, Charles Brent Chesson, Andrew Zloza
Rutgers Cancer Institute of New Jersey, New Brunswick, NJ, USA
Correspondence: Jenna Newman jhn33@gsbs.rutgers.edu
Background
The impact of infection on the development and progression of cancer has been a source of debate throughout the past century. Several pathogens have been linked to the development of cancer, while conversely, pathogen components (including Coley’s toxins) and oncolytic viruses have demonstrated tumor regression. Since infection and cancer are two often-concomitant challenges to the immune system, we sought to understand the impact of acute, non-oncogenic, non-oncolytic infections – which constitute the majority of infections affecting humans – on tumor growth and host immunity.
Methods
C57BL/6 (B6) or NOD-scid-gamma (NSG) mice were challenged with 1.2 x 105 cells B16-F10 melanoma intradermally (right flank) on day 0 and influenza A/PR8/H1N1 (10,000 PFU) administered intranasally (prior to tumor challenge, after tumor challenge before clinical appearance of the tumor, or once the tumor was established). Tumor growth and influenza infection were monitored via caliper measurement and host weight, respectively, every 2-3 days. Immune cell populations within tumors, lungs, and spleens were analyzed by flow cytometry at day 20. Statistical comparisons between groups were determined using the student’s t test with p<0.05 considered statistically significant.
Results
Mice infected with influenza prior to or during the subclinical phase of melanoma development displayed accelerated tumor growth relative to influenza-naïve mice (p<0.01). Contrarily, mice with established (~3 x 3 mm) tumors displayed slower tumor growth after infection compared to uninfected controls (p<0.01). For the latter, infected mice were found to have a 9-fold reduction of systemic MHC-II- Ly6-G+ Ly6-C+ myeloid-derived suppressor cells (MDSCs) in the spleen, relative to that observed in influenza-naïve counterparts. Furthermore, the result of curtailed tumor growth in concomitantly challenged mice was observed in immunocompromised NSG mice as well.
Conclusions
Previously we reported that influenza infection in the lung accelerates tumor growth of subclinical melanoma in the flank. However, our recent data demonstrate that the reverse effect is observed if influenza is administered to hosts with an established tumor in the flank. The latter effect is observed also in NSG mice, which lack adaptive immunity and maintain limited innate immunity. These findings, in conjunction with a decrease in MDSCs observed in immunocompetent infected B6 mice exhibiting reduced tumor growth, suggest that the innate immune system may promote anti-tumor immunity in the context of infection. Further research into the mechanisms by which influenza alters tumor growth are being investigated in preparation for clinical trials that will focus on harnessing microbial immunity for the treatment of cancer.
P482 Somatic TP53 mutations alter the immune micro-environment after chemotherapy in breast cancer
Mellissa Nixon1, M. Valeria Estrada2, Susan Opalenik1, Donna Hicks3, Michael Korrer1, Mark Pilkinton1, Melinda Sanders1, Roberto Salgado4, Simon Mallal1, Young Kim1, Rebecca Cook3, Carlos Arteaga1, Justin Balko1
1Vanderbilt University Medical Center, Nashville, TN, USA; 2UC San Diego, La Jolla, CA, USA; 3Vanderbilt University, Nashville, TN, USA; 4Institut Jules Bordet, Brussels, Belgium
Correspondence: Justin Balko (mellissa.nixon@vanderbilt.edu)
Background
Neoadjuvant chemotherapy (NAC), followed by surgery, is the standard of care for triple negative breast cancer (TNBC) patients. Unfortunately, only 30% achieve a pathological complete response (pathCR). In patients who do not achieve a pathCR, tumor infiltrating lymphocytes (TILS) after NAC correlate with improved survival. This suggests there is an immune response after NAC which may be augmented by immune checkpoint blockade (ICB). Clinical trials are underway to examine the efficacy of ICB, therefore it is vital to identify biomarkers of response to identify patients who may benefit from these modalities. As more than 80% of TNBC patients harbor somatic TP53 mutations, we investigated the role of TP53 mutations on the immune microenvironment of breast cancer after NAC.
Methods
We examined matched pre and post NAC primary breast cancer. TILs were scored by a pathologist. RNA and DNA were extracted and analyzed using Nanostring Pan-Cancer Immune panel (>700 genes) and ImmunoSeq T-cell Receptor (TCR) sequencing respectively. CRISPR technology was applied to mouse tumor cell lines to develop Trp53 mutations commonly found in TNBC. Cells were treated with chemotherapy in vitro to determine tumor specific changes in cytokines and PD-L1 expression. To determine alterations in the immune microenvironment, cell lines were injected into syngeneic mice or genetically engineered mouse models (GEMM) were utilized Expression of antigen presentation by the tumor as well as immune phenotyping of TIL after NAC was performed.
Results
TILs in residual disease of patients correlated with improved survival. A third of patients had increased immune gene signatures after NAC which correlated with increased TCR clonality and increased overall survival. CRISPR induced Trp53 mutant cells were used to elucidate the role of p53. Cells in vitro had higher basal levels of immune-recruiting chemokines which were further induced with chemotherapy treatment. Trp53 mutant cells also exhibited increased antigen presentation (MHC-I/II) and PD-L1 expression after NAC. In a GEMM of breast cancer that had recurred after chemotherapy, TILs had increased expression of CXCR3, LAG3 and PD-1 by flow cytometry.
Conclusions
Our work suggests that NAC induces immune gene signatures in a subset of TNBC which is correlated with a better prognosis. TP53 mutations in the tumor may contribute to an increased immune infiltrate through upregulating chemokines, antigen presentation, and increased cytokine receptor CXCR3+ T cells that display increased exhaustion marks PD-1 and LAG3. Ongoing studies with ICB will indicate if p53-mutant tumors have a better response to ICB after NAC.
P483 A novel non-competitive and non-brain penetrant adenosine A2A receptor antagonist designed to reverse adenosine-mediated suppression of anti-tumor immunity
Erica Houthuys, Reece Marillier, Théo Deregnaucourt, Margreet Brouwer, Romain Pirson, Joao Marchante, Paola Basilico, Shruthi Prasad, Julie Preillon, Anne-Catherine Michaux, Kim Frederix, Annelise Hermant, Florence Nyawouame, Charlotte Moulin, Vanesa Bol, Bruno Gomes, Florence Lambolez, Véronique Bodo, Jakub Swiercz, Gregory Driessens, Noémie Wald, Chiara Martinoli, Michel Detheux, Christophe Quéva, Stefano Crosignani
iTeos Therapeutics, Gosselies, Belgium
Correspondence: Christophe Quéva (christophe.queva@iteostherapeutics.com)
Background
High levels of extracellular adenosine in the tumor microenvironment promote tumor immune evasion by suppressing Th1 cytokine production and the activation and cytolytic activity of T cells and NK cells.
Methods
We measured extracellular adenosine concentration using intratumoral microdialysis of 11 patient-derived xenografts from various cancer indications. In parallel, we defined the expression of the four adenosine receptors in primary human immune cells using nanostring.
Results
Intratumoral adenosine concentrations reached a range of 1 to 37 μM (mean, 11 μM), significantly higher than the 0.1-0.2 μM reported in normal brain tissue.
A2A was the main adenosine receptor expressed by CD4 and CD8 T lymphocytes and monocytes, and the only one in mature monocyte-derived dendritic cells and NK cells. A2B mRNA were not abundant in T cells and monocytes, while A1 and A3 mRNA were not detected in any of the cell types evaluated. We further studied A2A functions in primary human T lymphocytes and monocytes and demonstrated that selective A2A agonists such as CGS-21680 suppressed cytokine production by activated T lymphocytes and monocytes, highlighting the role of A2A as the main receptor mediating adenosine signaling in these cells.
We showed that A2A antagonists initially designed for Parkinson’s disease but recently repurposed for immuno-oncology dramatically lost potency in a high adenosine environment. We therefore developped a novel non-brain penetrant and non-competitive inhibitor of A2A with sub-nanomolar Ki and selectivity versus A1, A2B, and A3 receptors. Our compound potently inhibited A2A signaling in human T lymphocytes independently of adenosine concentrations (IC50 in T cell cAMP assay = 2.8 and 5.5 nM in 1 and 100 μM adenosine, respectively), and rescued cytokine production in the presence of high concentrations of A2A agonists. iTeos A2A antagonist potently rescued TH1 cytokine production in human whole blood treated by A2A agonists, and increased CD8 T cell cytotoxicity in a co-culture assay of effector CD8 T cells and target cancer cells.
Conclusions
iTeos Therapeutics non-competitive A2A receptor antagonist is uniquely designed to address the challenge of counteracting elevated adenosine concentrations in tumors in order to restore antitumor immunity.
P484 A novel metabolic therapeutic that harnesses the anti-tumor immune response
Dayana Rivadeneira, Nicole Scharping, Greg Delgoffe
University of Pittsburgh, Pittsburgh, PA, USA
Correspondence: Greg Delgoffe (rivadeneirad@upmc.edu)
Background
In the past decade immunotherapy has shown impressive clinical responses, quickly becoming the first line treatment for cancers such as melanoma. Despite these promising results, all patients do not have the same level of response, and recent efforts have focused on delineating avenues to understand and avoid therapy resistance. Our lab and others have recently proposed that tumor cells may compromise T cell function by generating a metabolically inhospitable microenvironment. Most importantly immune or tumor metabolism can be modified to modulate the T cell response to immunotherapy.
Methods
We hypothesize that leptin, a hormone that affects a wide range of metabolic processes, might be utilized as a means to remodel the metabolic state of the tumor microenvironment, consequently modulating tumor growth and immunotherapy response. Previous studies have shown that leptin can enhance T cell proliferation and activation, but the effects of leptin on tumor-infiltrating T cells have not been thoroughly investigated. In order to assess the effects of leptin in the tumor microenvironment, we obtained tumor infiltrating T cells (TIL) from tumor-bearing mice treated with recombinant leptin. Additionally, we generated PTEN/BRAF melanoma cell lines stably overexpressing leptin.
Results
As we have previously shown, glucose uptake in TIL is decreased compared to ndLN. When mice were treated with recombinant leptin, we observed an increase in TIL glucose uptake, suggesting metabolic remodeling. Furthermore, PTEN/BRAF melanoma cells which overexpress leptin have significantly slower growth kinetics compared to control tumors and an increase in overall survival. Leptin overexpressing tumors have increased T cell infiltration compared to control tumors, and these TIL are metabolically and functionally superior.
Conclusions
Taken together, these data suggest leptin can have a direct effect on tumor infiltrating T cell function and tumor growth. Our goal is to further characterize how leptin can metabolically remodel the tumor microenvironment and modulate immunotherapeutic response.
P485 Use of the NanoString gene expression profiling platform to capture the immunological status of the leukemia microenvironment
Jayakumar Vadakekolathu1, Tasleema Patel2, Stephen Reeder1, Heidi Schaarschmidt3, Marc Schmitz4, Martin Bornhäuser3, Sarah Warren5, Tressa Hood5, Patrick Danaher5, Alessandra Cesano5, Joseph M. Beechem5, A. Graham Pockley1, Sarah Tasian2, Sergio Rutella1
1Nottingham Trent University, Nottingham, United Kingdom; 2Children's Hospital of Philadelphia, Philadelphia, PA, USA; 3University Hospital Carl Gustav Carus, Dresden, Germany; 4Medical Faculty Carl Gustav Carus, Dresden, Germany; 5NanoString Technologies, Inc., Seattle, WA, USA
Correspondence: Sergio Rutella (sergio.rutella@ntu.ac.uk)
Background
Acute myeloid leukemia (AML) is characterized by clonal expansion of poorly differentiated myeloid precursors and results in impaired hematopoiesis and often bone marrow failure. Immune responses are defective in patients with AML due to powerful immune suppressive circuits that are activated by soluble factors and immune checkpoint molecules, including PD-L1 and IDO1. The development and delivery of new therapeutic strategies for high-risk AML, including immunotherapy, therefore remains a priority.
Methods
This multi-institutional study was undertaken to collect comprehensive immune profiles across the biological heterogeneity of childhood and adult AML, with the aim to implement new molecularly targeted agents for patients with specific immunologic subtypes of AML. We employed the nCounter™ system (NanoString Technologies, Seattle, USA) to characterize bone marrow (BM) specimens from 70 patients with non-promyelocytic AML (42 children and 28 adults). Ninety BM samples (63 de novo AMLs, 18 AMLs in complete remission [CR] and 9 relapsed AMLs) were analyzed on the nCounter® FLEX platform, using the RNA Pan-Cancer Immune Profiling Panel. Transcriptomic data were normalized using the nSolver™ software package.
Results
Hierarchical clustering identified patient subgroups with heightened expression of CD8 T-cell, Th1-cell, B-cell and cytotoxicity-related genes, including genes related to NK-cell function, in the leukemia microenvironment (“immune enriched” patients). Interestingly, a select group of patients over-expressed genes associated with monocyte/macrophage, neutrophil and mast cell functions, with low expression of T-cell and B-cell genes. AML patients with ‘immune enriched’ gene profiles also expressed CD8A, IFNG, FOXP3, and inhibitory molecules including IDO1 and the immune checkpoints LAG3, CTLA4 and PD-L1.
Gene set analyses identified differences in immune gene expression levels between childhood and adult AMLs, as well as patients with newly diagnosed AML, relapsed AML or AML in CR. In particular, genes highly expressed in adult AMLs compared with childhood disease included chemokine genes, dendritic cell and macrophage genes and NF-kB inhibitor-a. In contrast, immune genes down-regulated in adult compared with childhood AMLs included integrin family members, cytokine receptors, TCR complex components and lipocalin-2, a neutrophil gelatinase-associated molecule implicated in suppression of tumor invasiveness.
Conclusions
Our analysis has captured the immunological status of the leukemia tumor microenvironment in children and adults with AML at different disease stages. From a clinical standpoint, ‘immune enriched’ AMLs might be amenable to immunotherapy approaches tailored to the BM microenvironment.
P486 Utilization of dual IHC and quantitative image analysis techniques to evaluate LAG-3-positive T cells in the tumor microenvironment of NSCLC tissue
Malik Khenkhar1, Philipp Uhlig1, Nickels Winkler1, Hartmut Juhl1, Alison Bigley2, Lorcan Sherry2
1Indivumed GmbH, Hamburg, Germany; 2OracleBio Limited, Glasgow, United Kingdom
Correspondence: Lorcan Sherry lorcan.sherry@oraclebio.com
Background
Non-small cell lung cancer (NSCLC) patients have impaired immune responses, where the expression of inhibitory checkpoint molecules on T cells may lead to reduced immune cell activity. The presence of lymphocyte activation gene-3(LAG-3)-positive T cells in the tumor microenvironment and their potential impact on prognosis have been investigated over many years. One implication has been that the engagement of LAG-3 inhibits T cell proliferation resulting in T cell suppression and poor prognosis [1]. We aimed to quantify CD3 and LAG-3 immune cell relationships in terms of cell infiltrations and proportions within the tumor microenvironment of NSCLC tissue.
Methods
Ten individual NSCLC clinical tissue samples were immunohistochemically (IHC) stained by Indivumed using the Ventana DISCOVERY XT automated staining platform. The first section was dual-labeled for LAG-3 and CD3 and the second for pan-Cytokeratin (CK). Image analysis was performed by OracleBio using Indica Labs Halo software. Tumor and stroma regions of interest (ROI) were classified using the CK section. ROI classifications were automatically transferred, during analysis, from the sample-associated CK section to the co-registered dual IHC section. Cellular analysis was performed on the ROI using threshold established to identify and count CD3, LAG-3 and dual-labeled cells.
Results
Evaluation of tumor samples demonstrated a mean number of LAG-3/CD3-dual positive cells within the stroma and tumor of 132 ± 59 and 54 ± 20 per mm2 ROI area, respectively. Interestingly however, when the proportion of LAG-3/CD3-dual positive cells was normalized to the total population of CD3-positive cells within these regions, an opposite relationship was observed where the mean % LAG-3/CD3-positive cells relative to the total CD3 immune cell population was 5 ± 2% within the stroma and 12 ± 3% within the tumor.
Conclusions
These example data highlight the benefits of utilizing dual IHC staining with whole slide image analysis to characterize immune cell relationships within the tumor microenvironment. These techniques can assist in the assessment of changing relationships between proportions of immune cells between tissue compartments and evaluation of LAG-3 as a potential target for modulating T cell responses in NSCLC.
References
1. He Y, Yu H, Rozeboom L. LAG-3 Protein Expression in Non-Small Cell Lung Cancer and Its Relationship with PD-1/PD-L1 and Tumor-Infiltrating Lymphocytes. Journal of Thoracic Oncology. 2017;12(5):814-823.
P487 Multiplex immunofluorescence evaluation of an immune cell marker panel in the tumor microenvironment of NSCLC tissue using tailored analysis of multi-spectral image component data
Richard Bystry1, Edward Durley1, Lee Dawson1, Rebecca Houliston1, Alison Bigley2, Lorcan Sherry2
1Asterand Bioscience, Royston, United Kingdom; 2OracleBio Limited, Glasgow, United Kingdom
Correspondence: Lorcan Sherry lorcan.sherry@oraclebio.com
Background
Multiplex fluorescence immunohistochemistry (MIF), multispectral imaging and image analysis of immune cells within the tumor microenvironment has been increasingly applied to profile and correlate cell interactions in relation to cancer treatment and predicting patient outcome. Tumor-infiltrating T lymphocytes are known to play an important role in anti-cancer mechanisms where coexisting infiltrations of CD8 and CD4 T cells affords a good prognosis in non-small-cell lung carcinoma (NSCLC) [1]. Here we use CD8/CD4 evaluation to exemplify the use of MIF combined with multispectral imaging and tailored spectral and spatial quantification to examine relationships between immune cell populations within tumor and stroma.
Methods
An exemplar NSCLC lung tumor section was MIF labelled by Asterand for 5 immune cell markers, including CD4 and CD8, a pan cytokeratin tumor marker and DAPI nuclear marker. The stained slide was digitised using the Vectra Polaris multispectral scanner (Perkin Elmer) and exemplar region of interest (ROI) images exported in component data format. The component data images were analysed by OracleBio using a 3-tiered sequence of tailored applications developed in Visiopharm Oncotopix Software. The first tier enabled the classification of tumor and stroma ROI, the second tier facilitated cell detection, classification and analysis and the third tier refined cell relationships and measured immune cell distances to the nearest tumor cell or between defined immune cells.
Results
Selected ROI displayed a range of tumor and stroma content with varying populations of CD4 and CD8 positive immune cell infiltrations. The mean number of CD4 and CD8 positive cells in tumor was 30 and 250 per mm2 respectively and in stroma was 474 and 915 per mm2 respectively. Evaluation of tumor-immune cell spatial relationships within the stroma, at a 20μm distance to the tumor border, highlighted that the percentage of CD4 and CD8 positive cells within this stromal border was 33% and 54% respectively and the average distance between the 2 immune cell types was 13μm.
Conclusions
The exemplar data generated provides information relating to the potential influences of immune cell recruitment and activation within the tumor microenvironment, correlation of spatial distributions and the inter-dependencies between immune cells and cancer cells, which may be indicative of response to treatment or predicting patient outcome.
References
1. Hiraoka K, Miyamoto M, Cho Y: British Journal of Cancer. 2006;94:275–280.
P488 Single-cell RNA sequencing of CD8+ T cells to prioritize immunotherapy combinations
Yashaswi Shrestha, Michael Kuziora, Lydia Greenlees, Brandon Higgs, Katie Streicher, Rajiv Raja, Koustubh Ranade
MedImmune, Gaithersburg, MD, USA
Correspondence: Yashaswi Shrestha (robert.mulvey@ashfieldhealthcare.com)
Background
Immunotherapy has transformed cancer treatment. To address remaining unmet needs, combinations of checkpoint inhibitors and agonists are under investigation, although prioritization strategies are unclear. We hypothesized that molecular understanding of T-cell phenotypes under chronic stimulation that mimics the tumor microenvironment could aid in optimizing combinations. Additionally, target co-expression is important for designing bispecific monoclonal antibodies (mAbs). Although aggregate gene expression analysis of CD8+ T cells is valuable, it does not show target co-expression at the single-cell level. Hence, we explored single-cell RNAseq to identify targetable CD8+ T-cell subpopulations at various dysfunctional states.
Methods
CD8+ T cells were negatively selected from PBMCs and treated with IL2 (NonStim) or IL2+anti-CD3,antiCD28 antibodies (Stim) for 13 days, including two cycles of treatment separated by a two-day rest in IL2 [1].
Over 1500 Stim and NonStim cells were profiled on 10X Genomics platform and analyzed with corresponding pipeline. Gene expression corresponded to 2 unique molecular indices. Published consensus gene-sets for functional states of CD8+ T cells in cancer [2] were used for Gene-Set Variant Analysis (GSVA) to identify populations with significant differences in the four states – dysfunctional, senescent, terminal-effector, and stem-like memory – between Stim and NonStim.
Results
GSVA scores indicated significant upregulation of a dysfunction signature and downregulation of terminal-effector function in Stim versus NonStim cells (P<0.001). PD-1 was most differentially expressed between top and bottom tertiles of Stim cells with dysfunctional signature. Similarly, cells with high expression of TIM3, another dysfunction marker, showed downregulation of cytotoxicity and antigen-presentation associated genes, compared to those with low TIM3 expression.
The percentage of cells expressing dysfunction markers increased under chronic stimulation, while those producing effector cytokines decreased. Cells expressing PD-1, CTLA-4 and GATA3 increased from 5%, 3% and 16% to 17%, 80% and 21%, respectively, whereas cells expressing IFNγ decreased from 16% to 7%. Of the Stim cells, 11%, 15% and 71% co-expressed PD-1 and TIM3, PD-1 and CTLA-4, and CTLA-4 and LAG3, respectively. CD137 and TIM3 were co-expressed in >30% of Stim cells, but only 1% of NonStim.
Conclusions
Our results provide new insights into the heterogeneity of activated/exhausted/dysfunctional CD8+ T cells at the single-cell level, which could aid in prioritizing immunotherapy combinations and designing new bispecific mAbs.
References
1. McKinney E, et al. T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature. 2015;523:612–616.
2. Apetoh L, et al.Consensus nomenclature for CD8+ T cell phenotypes in cancer. Oncoimmunology. 2015;4:e998538.
P489 Studying the cellular heterogeneity and spatial arrangement of immune cells in FFPE sections of Hodgkin Lymphoma and normal lymph node using a highly multiplexed Imaging Mass Cytometry
Mohan Singh, Parvesh Chaudhry, Erik Gerdtsson, Lisa Harton, James Hicks, Peter Kuhn, Wendy Cozen, Imran Siddiqi, Akil Merchant
University of Southern California, Los Angeles, Los Angeles, CA, USA
Correspondence: Akil Merchant (mohansin@usc.edu)
Background
Clinical successes with immuno-oncology agents have demonstrated the potency of the immune system in controlling cancers, most strikingly in Hodgkin lymphoma (HL), where response rates to PD1/PDL1 inhibitors approach 90%. The majority of tumor bulk in HL is made up of non-malignant immune cells. High parameter methods such as gene expression profiling or flow cytometry have been applied to the study of the tumor immune microenvironment (TME) in Hodgkin disease, however due to the inherent limitations of these techniques, several questions remain unanswered. Data on cellular heterogeneity and rare cells is lost with gene expression studies while the spatial relationship between tumor and immune cells is lost with flow cytometry. The Fluidigm Hyperion imaging mass cytometry (IMC) system combines a CyTOF mass cytometer with a laser ablation system allowing for 40+ parameter simultaneous immunophenotyping on a single slide of FFPE tissue, with sub-cellular resolution. Here we describe our efforts to develop and optimize a 26 parameter antibody panel for the characterization of the lymphoma TME on the IMC platform.
Methods
Literature was reviewed to identify markers relevant to HL TME. Antigen retrieval, blocking, and antibody staining concentrations were optimized to identify universal staining conditions. Slides were imaged using the Fluidigm Hyperion IMC system and image analysis was performed with MCD viewer (Fluidigm) or CellProfiler/miCAT (https://doi.org/10.1101/109207).
Results
We successfully performed IMC imaging of Hodgkin lymphoma, NK lymphoma and normal lymph nodes (LN) using a 26 parameter panel. We were able to clearly identify tissue architecture (Fig. 1, normal LN, germinal center(GC), mantle zone(MZ), cortex/paracortex(PC), Fig. 2A) and characterize specific cellular populations such as CD8+Tcells, Th1/Th2 subsets, FoxP3+Treg, tissue associated macrophages, CD56+NK cells and tumor vasculature. Important immuno-oncology targets such as LAG3, PDL1/PDL2 and PD1 are included. Cellular resolution is similar to IHC, allowing for imaging of tumor, effector and regulatory cell interactions at a single cell level (Fig. 2B, Reed Sternberg(RS)cells (red). CD8+(green) adjacent to RS cell flanked by TREG (FoxP3-teal) and Th2(CCR4-yellow) CD4+(not shown) cells. Abundant CD68+(magenta) cells which co-express PDL1/PDL2 (not shown) in the background.)
Conclusions
IMC allows for successful multiplex imagining of the TME in FFPE tissues. The method does not require special processing and can be applied to archival specimens and tumor microarrays. Methods developed here should be applicable for the study of the TME in other tumor types and could be used to identify additional biomarkers of response to immuno-oncology agents.

FFPE section of Normal lymph node

FFPE section of Hodgkin lymphoma
P490 PSMA-specific CARTyrin T-stem cell memory therapy eliminates solid tumor in subcutaneous prostate cancer model
Jenessa B. Smith, Rebecca Codde, Yening Tan, Burton E. Barnett, David Hermanson, Srinivas Rengarajan, Eric M. Ostertag, Devon J. Shedlock
Poseida Therapeutics, San Diego, CA, USA
Correspondence: Devon J. Shedlock (jbsmith0711@gmail.com)
Background
Prostate-specific membrane antigen (PSMA) is a transmembrane glycoprotein overexpressed in >80% of metastatic prostate cancer. Although PSMA is a promising target for peptide and antibody drug conjugate therapies, early chimeric antigen receptor (CAR) T-cell therapies lacked clinical efficacy. Here we developed a novel CAR T-cell product (P-PSMA-101) via piggyBac™ transposition of a tri-cistronic transgene encoding a safety switch, a PSMA-specific Centyrin-based CAR (CARTyrin), and a selection gene, features that may improve safety and efficacy compared with previous anti-PSMA CAR T-cell therapies.
Methods
We developed and identified a lead anti-PSMA CARTyrin from over 250 available Centryin binders. We also tested the previously clinically-tested anti-PSMA J591 scFv-based CAR for comparability. Initial assessment utilized mRNA delivery of candidate CARTyrins to confirm CAR surface expression and specific T-cell degranulation against PSMA+ prostate tumor cells or PSMA-engineered cells. We then used piggyBac to deliver our tri-cistronic vector system encoding the lead PSMA CARTyrin (P-PSMA-101), J591 scFv CAR, or a BCMA-specific CARTyrin to T-cells, resulting in >95% CAR+ T-cells after selection and expansion. Importantly, our unique production methodology leads to >60% T-stem cell memory (Tscm) cells, an early memory population that correlates with complete responses in CD19 CAR T-cell clinical trials. In vitro, P-PSMA-101 and J591 CAR T-cells specifically proliferated, lysed, and secreted IFN-γ against PSMA+ LNCaP or PSMA-engineered K562s. No evidence of tonic signaling or exhaustion was detected.
Results
P-PSMA-101 demonstrated significantly enhanced anti-tumor efficacy and survival in comparison to J591 CAR T-cell-treated mice in a low “stress test” dose of T-cells against established subcutaneous LNCaP(fLuc+) solid tumors in NSG mice. The >60% Tscm P-PSMA-101 expanded in vivo and gave rise to differentiated effector CAR+ T-cells that were detected in the peripheral blood concomitant with a decrease in tumor burden below detectable caliper and bioluminescent imaging limits. P-PSMA-101 then contracted, yet persisted in the peripheral blood with >70% of T-cells exhibiting a Tscm phenotype. On the contrary, J591 CAR T-cells did not significantly control tumor burden. In a dose titration study, P-PSMA-101 eliminated established LNCaP tumor in 100% of animals for the duration of the studies (42 days post-treatment), while 2/3 low-dose animals remained tumor-free.
Conclusions
P-PSMA-101 is a first-in-class Centyrin-based CAR T-cell therapeutic that exhibits a persistently high frequency of Tscm and mediates durable anti-solid tumor efficacy that surpasses previously established anti-PSMA CAR T-cell therapy in our in vivo model. Future efforts will continue towards clinical application of P-PSMA-101 in patients with metastatic castrate resistance prostate cancer.
P491 Alerting the immune system by removing epigenetic silencing of TH1 chemokines
Heather Sonnemann, Amber Giles, Jinkyu Jung, Caitlin Reid, Marsha-Kay Hutchinson, Deric Park, Mark Gilbert
NIH, NCI, Bethesda, MD, USA
Correspondence: Amber Giles (heather.sonnemann@nih.gov)
Background
BACKGROUND: Solid tumors employ multiple mechanisms to evade immunologic detection and destruction. However, the potential to enhance the immune response to cancer has been proven in several malignancies and is under investigation in many others, including primary CNS tumors. Brain tumors, in particular, lack robust T-cell infiltration, suggesting poor immune surveillance. Recent studies have found that tumor cells express T-cell attracting chemokines, CXCL9 and CXCL10, in response to interferon gamma (IFNγ). Chemokine expression can be further amplified by drugs that prevent epigenetic silencing, such as histone methyltransferase inhibitors or histone deacetylase (HDAC) inhibitors. This approach increases T-cell trafficking to tumors in vivo and enhances the anti-tumor efficacy of checkpoint blockade. We hypothesized that T-cell trafficking to brain tumors could be increased by epigenetically enhancing CXCL9 and CXCL10 expression.
Methods
Assays were performed on human glioma brain tumor cell lines. CXCL9 and CXCL10 expression were measured by real-time PCR and Western blot. GSK126, a commercially available methyltransferase inhibitor, was utilized to demethylate histone H3 (K9 and K27). LB201 (Lixte Biotechnology, Setatuket, NY) and SAHA were used to inhibit class 1 and class 2 HDACs and Entinostat was used to inhibit class I and class III HDACs. Histone methylation and acetylation status were examined using Western blot analysis. T-cell migration was measured using transwell migration assays. RESULTS: Expression of CXCL9 and CXCL10 was induced by IFNγ in brain tumor lines. Chemokine expression was further increased when cells were treated with GSK126 and HDAC inhibitors compared to IFNγ alone. Migration assays confirmed that tumor cells treated with GSK126 and HDAC inhibitors released chemotactic factors that increased T-cell migration.
Results
Expression of CXCL9 and CXCL10 was induced by IFNγ in brain tumor lines. Chemokine expression was further increased when cells were treated with GSK126 and HDAC inhibitors compared to IFNγ alone. Migration assays confirmed that tumor cells treated with GSK126 and HDAC inhibitors released T-cell chemotactic factors that increased T-cell migration.
Conclusions
These studies demonstrate that brain tumors express T-cell attracting chemokines CXCL9 and 10 in response to IFNγ. Further, single agent and the combination of GSK126 and HDAC inhibitors enhanced expression of CXCL9 and CXCL10, resulting in increased T-cell migration toward tumor cells. Together, these data provide a potential means to enhance the efficacy of immune therapies for brain tumors by promoting T-cell trafficking toward tumor cells.
P492 Galectin-3 inhibition with GR-MD-02 synergizes with agonist anti-OX40 mAb therapy leading to reduced immune suppression and improved overall survival
Elizabeth Sturgill, Stephanie Linch, Courtney Mick, Melissa Kasiewicz, William Redmond
Providence Portland Medical Center, Portland, OR, USA
Correspondence: William Redmond (elizabeth.sturgill@providence.org)
Background
Cancer immunotherapy aims to harness the immune system to destroy tumor cells as well as reverse tumor-induced immune suppression. One such molecule reported to contribute to immune suppression is galectin-3 (Gal3). Indeed, Gal3 is upregulated on a wide variety of tumors and is associated with poor patient prognosis. In addition, increased amounts of Gal3 are found in patients with metastatic disease. Gal3 has been shown to recruit myeloid derived suppressor cells (MDSCs) into the tumor microenvironment, thus aiding in tumor escape. Therefore, we hypothesized that the addition of a galectin-3 inhibitor (GR-MD-02) in conjunction with an agonist anti-OX40 antibody (aOX40) would synergize to promote tumor regression and increased survival via a reduction in tumor-induced immune suppression.
Methods
MCA-205 sarcoma cells or TRAMP-C1 prostate adenocarcinoma cells were implanted into the flank of C57Bl/6 mice; 4T-1 mammary carcinoma cells were implanted orthotopically into the mammary fat pad. GR-MD-02 (2.4 mg/dose) and aOX40 (250 μg/dose) were administered to mice intraperitoneally (ip) either 3x/week for 2 weeks or 2x/week for 1 week, respectively. Tumor growth (area) was assessed 2x/week and mice were sacrificed when tumors exceeded 150 mm2. Tumors were either digested and immune cells were analyzed by flow cytometry or homogenized into tumor lysate was assayed by cytokine bead array.
Results
We observed that combined GR-MD-02/aOX40 therapy significantly improved survival of MCA-205, 4T-1, and TRAMP-C1 tumor-bearing mice, and additionally reduced lung metastases in the 4T-1 model. Further analysis revealed that GR-MD-02/aOX40 therapy significantly reduced the amount of both monocytic (Mo-) and granulocytic (Gr-) MDSCs within the tumor compared to aOX40 alone (p<0.05). In addition, we observed a significant decrease in PD-L1, iNOS, arginase 1 (p<0.05), and CD206 (p<0.001) expression in both Mo- and Gr-MSDCs. Moreover, GR-MD-02/aOX40 therapy led to increased CD8+ T cell proliferation in the lymph node and increased intratumoral concentrations of IL-17a and IL-15 (p<0.05), suggesting a potential role in modulating immune infiltrate and/or prolonged T cell survival.
Conclusions
In summary, our data suggests that Gal3 inhibition plus agonist aOX40 therapy may reduce myeloid recruitment to the tumor microenvironment, thus reducing immune suppression and subsequently mediating tumor regression and increased survival.
P493 LTX-315: A first-in-class oncolytic peptide that reshapes the tumor microenvironment
Baldur Sveinbjørnsson1, Ketil Camilio2, Meng-Yui Wang2, Janne Nestvold3, Gunhild Mælandsmo2, Gunnar Kvalheim2, Øystein Rekdal1
1Lytix Biopharma, Oslo, Norway; 2Radiumhospitalet, Oslo, Norway; 3Rikshospitalet, Oslo, Norway
Correspondence: Øystein Rekdal (baldur.sveinbjornsson@lytixbiopharma.com)
Background
The oncolytic peptide LTX-315, which has been de novo designed based on structure-activity relationship studies of host-defense peptides, has the ability to kill human cancer cells and induce long-lasting anticancer immune response when injected locally into tumors established in immunocompetent murine models. The oncolytic effect of LTX-315 involves perturbation of the plasma membrane and the mitochondria with subsequent release of danger-associated molecular pattern molecules (DAMPs) such as ATP, Cytochrome C and HMGB1. Furthermore, LTX-315 effectively disintegrates the cellular compartments with subsequent release of tumor antigens as demonstrated by a greater T-cell infiltration (TILs), TILs clonality and the number of clones with greater abundance in the tumor microenvironment. In experimental tumor models, LTX-315 exerts abscopal effects and reshapes the tumor microenvironment by decreasing the local abundance of immunosuppressive cells and by increasing the frequency of effector T-cells. LTX-315’s ability to convert immunogenically “cold” tumors to “hot” makes it ideal combination partner with other immunotherapies or immunochemotherapy. Indeed, in preclinical tumor models, a combination of LTX-315 and immune checkpoint inhibitor (anti-CTLA4) demonstrates significant synergy.
Methods
In the present study the antitumor efficacy and potential synergy of LTX-315 in combination with low-dose chemotherapy was investigated in experimental mouse models. Subcutaneously established murine A20 lymphoma was treated with LTX-315 alone, cyclophosphamide alone or LTX-315 in combination with cyclophosphamide. Similarily, orthotopically established 4T1 murine mammary carcinoma was treated with either LTX-315 alone, liposomal doxorubicin (CAELYX) alone or in combination.
Results
LTX-315 showed significantly enhanced anticancer efficacy against A20 lymphomas and 4T1 breast carcinomas when combined with cyclophosphamide and doxorubicin, respectively.
Conclusions
The LTX-315 unique “release and reshape” properties make it a promising candidate for combination with several types of anticancer therapies. Phase 1b study combining LTX-315 with ipilimumab (anti-CTLA-4) in malignant melanoma patients, as well as LTX-315 with Pembrolizumab (anti-PD1) in metastatic breast cancer is ongoing.
P494 Reversal of adenosine-mediated immune suppression by AB421, a potent and selective small-molecule CD73 inhibitor
Joanne BL Tan, Annette Becker, Daniel DiRenzo, Kristen Zhang, Ada Chen, Rhiannon Thomas-Tran, Joel Beatty, Debashis Mandal, Guifen Xu, Steve Young, Matthew Walters, Ulrike Schindler, Jay Powers
Arcus Biosciences, Hayward, CA, USA
Correspondence: Joanne BL Tan jtan@arcusbio.com
Background
Extracellular adenosine-triphosphate (ATP) is efficiently hydrolyzed to adenosine by ecto-nucleotidases CD39 and CD73, which converts adenosine-monophosphate (AMP) into adenosine (ADO). ADO suppresses immune responses including those of T cells, natural killer (NK) cells, and dendritic cells (DCs) through activation of A2aR and A2bR receptors. CD73 inhibition is a promising therapeutic approach for preventing ADO-mediated immunosuppression in the tumor microenvironment.
Methods
Human CD8+ T cells, CD4+ T cells, and CD14+ monocytes were isolated from buffy coats using various RosetteSep and EasySep Enrichment cocktails. The ability of AB421 to rescue AMP-mediated inhibition of T cell activation was evaluated using CD3/CD28/CD2 stimulation. Mixed lymphocyte reactions (MLRs) were established by mixing GM-CSF and IL-4 differentiated monocytic DCs with allogeneic CD4+ T cells. AMP and ADO levels in C57BL/6J mice were measured by mass spectrometry in plasma samples isolated at various time points after administration of AB421.
Results
We have designed a highly potent and selective small-molecule CD73 inhibitor, AB421, which inhibits endogenous CD73 activity in human CD8+ and CD4+ T cells with IC50 values of 4.5 pM and 6.2 pM, respectively. AB421 prevents AMP-mediated inhibition of T cell activation in CD3/CD28/CD2 activated human CD4+ and CD8+ T cells, as well as allogeneic CD4+ T cell activation in MLRs. Addition of exogenous AMP abrogated the enhanced allogeneic CD4+ T cell activation and IFN-γ production mediated by α-PD-L1 monoclonal antibody (mAb), an effect that was blocked by AB421. Mechanistically, addition of AMP repressed expression of activation markers (CD25) and immune checkpoint proteins (CTLA-4, PD-1, TIM-3, LAG-3, ICOS, CD28) in the MLR assays. This suggests that activation of the adenosinergic pathway is dominant and may limit the utility of most antibodies targeting immune checkpoint proteins by curtailing their expression and/or upregulation (as was seen with an α-PD-L1 mAb and AMP co-culture systems). Analysis of TCGA databases and tumor microarrays showed differential expression of CD73 across tumor types, with high expression detected in non-small cell lung carcinoma (adenocarcinoma), colorectal, head and neck, esophageal, and stomach cancers. Analysis of dissociated tumor cells by flow cytometry showed CD73 expression in both hematopoietic and non-hematopoietic cells. Finally, we show that AB421 can elevate AMP-to-ADO ratios in vivo in a dose-dependent manner, reflecting systemic inhibition of CD73.
Conclusions
AB421 represents a class of potent, reversible and selective CD73 inhibitors that exhibit picomolar potency in primary human immune cells and is currently undergoing preclinical evaluation as a candidate for clinical evaluation.
P495 Separate molecular pathways mediate anti-tumor versus tumor-promoting Aspects of dsRNA signaling in cancer microenvironments
Marie-Nicole Theodoraki1, Saumendra Sarkar1, Ravi Muthuswamy1, Jamie Voyten1, Robert Edwards2, David Bartlett1, Kalinski Pawel1
1University of Pittsburgh, Pittsburgh, PA, USA; 2Magee-Womens Hospital of UPMC, Pittsburgh, PA, USA
Correspondence: Marie-Nicole Theodoraki (marie.nicole.theodorakis@gmail.com)
Background
Infiltration of tumors with cytotoxic T cells (CTLs) predicts improved prognosis and has been shown critical for antitumor effectiveness of checkpoint blockers. In contrast, tumor infiltration with regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs) typically predicts rapid progression and poor outcomes. Poly-I:C, a frequently used adjuvant which induces not only the CTL-attracting chemokines but also Treg attractants. Here we evaluated the molecular pathways, which lead to the induction of chemokines by dsRNA in TME and different types of tumor-associated cells, in order to develop improved adjuvants which selectively attract the desirable effector cells rather than suppressive cells.
Methods
Isolated cells or human cancer biopsies were cultured in the absence or presence of one of two synthetic TLR3 ligands Poly-I:C (non-selective activator of TLR3 and helicases) or rintatolimod (selective TLR3 ligand) and in the absence or presence of a COX-1/2 inhibitor, NF-kB- or TNFa inhibitors. mRNA assays. ELISA, chemotaxis assays and molecular biology assays were used to analyze the chemokine production and tumor-associated suppressive factors. Confocal microscopy of macrophage cultures treated with the TLR3 ligands was performed to evaluate the activation of the different signaling pathways by the TLR3 receptor and cytoplasmic helicases.
Results
We observed that poly-I:C induced activation of NF-kB- and COX-2 pathways leading to induction of COX2-dependent suppressive factors and Treg- and MDSC-attracting chemokines. These undesirable effects were blocked with inhibitors of both NF-kB- or COX-2 pathways. In contrast rintatolimod selectively induced the desirable chemokines, which were associated with lack of direct activation of NF-kB- or COX-2 pathways, and strongly suppressed attraction of Tregs and MDSCs, with elevated CTL attraction in ex vivo migration assays. Looking at the upstream signaling pathways, both TLR3 ligands induced IRF3 (Type-1 interferon pathway). However, only Poly-I:C induced activation of TRAF3 and RIP-1 (TLR3 dependent NF-kB pathway) as well as MAVS (cytoplasmic helicases).
Conclusions
Our data implicate an important new role of helicases by the induction of tumor-promoting factors by dsRNA and points out to new targets to enhance the immunogenic and antitumor activities of adjuvants.
P496 Degradation of hyaluronan (HA) by PEGPH20 promotes anti-tumor immunity and enhances the effect of checkpoint blockade in an HA-accumulating mouse syngeneic tumor model
Benjamin J Thompson, Renee Clift, Trevor B Kimbler, Jesse D Bahn, Chunmei Zhao, Barbara Blouw, Curtis B Thompson, Sanna Rosengren
Halozyme, San Diego, CA, USA
Correspondence: Benjamin J Thompson (bthompson@halozyme.com)
Background
The glycosaminoglycan hyaluronan (HA) is abundant in many solid tumors, and its accumulation is often associated with poor patient outcomes. Its degradation by intravenously administered PEGylated recombinant human hyaluronidase PH20 (PEGPH20) remodels the tumor stroma, reduces intratumoral pressure, decompresses tumor blood vessels, and facilitates drug delivery. Recently, PEGPH20 was shown to enhance tumor growth inhibition induced by the immune checkpoint inhibitor anti-PD-L1 in HA-accumulating pancreatic and orthotopic breast syngeneic tumor models, and to enhance tumor T cell infiltration. Here, the HA content of commonly used syngeneic models was systematically studied, and MC38 (Charles River Laboratories) was identified to further evaluate the combinatorial effect of PEGPH20 and anti-PD-L1 in HA-accumulating tumors.
Methods
Eight syngeneic tumor models were screened for HA content by ELISA, and immune activity by qPCR for a panel of transcripts that included surface markers, cytokines, chemokines, immune checkpoints, and immunomodulatory enzymes. For tumor growth and immunophenotyping studies, MC38 tumors were implanted subcutaneously, and PEGPH20 (37.5 μg/kg) was administered intravenously 24 h prior to anti-PD-L1 (10F.9G2, 5 mg/kg) using a biweekly dosing regimen. Tumor HA was visualized by immunohistochemistry followed by positive pixel count. Tumor-infiltrating immune cells were evaluated by flow cytometry following tumor dissociation.
Results
Of the eight models tested, MC38 tumors had the highest HA content, and displayed evidence of robust immune activity in the qPCR screen. PEGPH20 reduced MC38 tumor HA by nearly 90% 24 h after either a single dose, or the last of three biweekly doses. In two out of three blinded studies, PEGPH20 significantly enhanced inhibition of MC38 tumor growth induced by anti-PD-L1, and combination treatment also significantly prolonged the survival of tumor-bearing mice compared to either treatment alone. PEGPH20-treated tumors contained significantly higher numbers of CD8+ and CD4+ (both Th and Tregs) T cells, as well as natural killer cells, compared to controls. Among CD45+ cells, tumor-associated macrophages (TAMs) were the most abundant cell type; PEGPH20 significantly reduced the frequency of TAMs, while increasing the frequency of dendritic cells.
Conclusions
Collectively, these results suggest that degradation of tumor HA by PEGPH20 can facilitate an anti-tumor immune response by promoting effector cell infiltration and skewing the immune microenvironment toward a more anti-tumor composition, thereby enhancing the effect of anti-PD-L1. These findings support the ongoing clinical evaluation of PEGPH20 in combination with checkpoint inhibitors in HA-accumulating solid tumors, and provide rationale for investigating PEGPH20 in combination with additional immune modulating cancer therapies.
P497 Tumor microenvironment immune gene signature asscoiated with axicabtagene ciloleucel (axi-cel, KTE-C19), an anti-CD19 chimeric antigen receptor (CAR) T cell, in a multicenter trial (ZUMA-1)
John Rossi1, Jérôme Galon2, Sarah Turcan2, Corinne Danan2, Frederick Locke3, Sattva Neelapu4, David Miklos5, Nancy Bartlett6, Caron Jacobson7, Ira Braunschweig8, Olalekan Oluwole9, Tanya Siddiqi10, Yi Lin11, John Timmerman12, Patrick Reagan13, Lazaros Lekakis14, Sherryonne Unabia1, William Go1, Jeff Wiezorek1, Adrian Bot1
1Kite Pharma, Santa Monica, CA, USA; 2HalioDx, Marseille, France; 3Moffitt Cancer Center, Tampa, FL, USA; 4MD Anderson Cancer Center, Houston, TX, USA; 5Stanford University School of Medicine, Stanford, CA, USA; 6Washington University Medical School, Saint Louis, MO, USA; 7Dana-Farber Cacner Institute, Boston, MA, USA; 8Montefiore Medical Center, Albert Einstein College of Medicine, Bronx, NY, USA; 9Vanderbilt-Ingram Cancer Center, Nashville, TN, USA; 10City of Hope National Medical Center, Duarte, CA, USA; 11Mayo Clinic, Rochester, MN, USA; 12UCLA David Geffen School of Medicine, Santa Monica, CA, USA; 13University of Rochester Medical Center, Rochester, NY, USA; 14University of Miami Health System, Sylvester Comprehensive Care Center, Miami, FL, USA
Correspondence: Adrian Bot (kate.trueblood@nexusggmed.com)
Background
The efficacy of axicabtagene ciloleucel (axi-cel), an autologous anti-CD19 CAR T cell therapy, was evaluated in the multicenter, pivotal ZUMA-1 study in patients with refractory, aggressive non-Hodgkin lymphoma (NHL). In a pre-specified interim analysis, ZUMA-1 met its primary endpoint with an objective response rate of 82% and a complete response rate of 54% [1]. Based on an analysis of patients enrolled in ZUMA-1, we describe for the first time a tumor microenvironment immune gene signature associated with CAR T cell treatment of patients with aggressive NHL.
Methods
In this post-hoc analysis, paired biopsies were taken before and within 3 weeks of axi-cel treatment from 14 patients enrolled in ZUMA-1. These biopsies were analyzed by digital gene expression (NanostringTM) and a pre-specified bioinformatics algorithm was then applied to IGES15 and IGES21 genes, which are hypothesized to be involved in immune-mediated tumor regression (Immunosign®) [2]. The Immunosign® profiles evaluated a pre-defined set of genes for effector T cells, Th1 cells, chemokines, and cytokines. Expression analysis and hierarchical clustering were used to define an axi-cel–related tumor immune gene signature. Wilcoxon signed rank test with multiple test correction by false discovery rate (FDR; Benjamini-Hochberg) was used.
Results
A comparison of gene expression profiles of biopsies from 14 patients on ZUMA-1 taken before and after axi-cel treatment showed profound changes in gene expression within the tumor environment after infusion. The most upregulated genes post–axi-cel treatment were CCL5 (RANTES), CTLA4, and GZMA (log2 fold change >2, P<0.05, FDR <0.050). Immune checkpoints PD-L1 and LAG3 were also upregulated post–axi-cel treatment (log2 fold change >1.6, P<0.05, FDR <0.055). Other genes associated with T cell proliferation, homing, and effector function that were upregulated included IL-15, GZMK, CXC3CL1 (Fractalkine), CD8A, and STAT4 (log2 fold change >1.6; P< 0.05, FDR <0.074). Additional baseline tumor characteristics and associative analysis will be presented.
Conclusions
We define a mechanistic tumor immune gene signature in NHL patients associated with axi-cel treatment. This signature comprises upregulation of T cell activation, effector, chemokine, and immune checkpoint genes. These data will potentially lead to rational optimization of T cell interventions in cancer.
Trial Registration
NCT02348216.
References
1. Locke FL, Neelapu SS, Bartlett, NL, et al. AACR 2017. 9986.
2. Galon J, Angell HK, Bedognetti D, Farincola FM. Immunity. 2013;39: 11-26.
P498 AB928, a dual antagonist of the A2aR and A2bR adenosine receptors for the treatment of cancer
Matthew Walters, Daniel DiRenzo, Joanne Tan, Ulrike Schindler, Dana Piovesan, Devika Ashok, Tim Park, Nell NarasappaFangfang Yin, Wan Hsin Lim, Stefan Garrido-Shaqfeh, Jenna Jeffrey, Laurent Debien, Brandon Rosen, Ehesan Sharif, Dillon Miles, Manmohan Leleti, Juan Jaen, Jay Powers
Arcus Biosciences, Hayward, CA, USA
Correspondence: Matthew Walters (mwalters@arcusbio.com)
Background
In the tumor micro-environment (TME), extracellular ATP is sequentially hydrolyzed to adenosine by the ecto-nucleotidases CD39 (ATP→AMP) and CD73 (AMP→adenosine), where it acts to suppress immune cell function. A2aR is expressed by a variety of lymphocytes, while myeloid cells express both A2aR & A2bR. A2aR activation results in decreased T cell activation, while binding of adenosine to A2bR on myeloid cells is thought to promote a tolerogenic phenotype. To alleviate immune suppression within the TME, we have designed AB928, a potent and selective dual antagonist of A2aR and A2bR
Methods
Human immune cells were isolated from healthy donor PBMC and cultured as appropriate for the cell type. pCREB assays were performed using freshly collected whole mouse blood.
Results
AB928 inhibits both A2aR and A2bR with similar potencies (KB: 1.4 nM and 2 nM, respectively). In PBMC sorted from healthy donors, similar levels of A2aR mRNA were observed in naïve and central memory CD4 and CD8 T cells, as well as NK and B cells; A2aR was also expressed in both dendritic cells and monocytes. In contrast to this, A2bR was absent from lymphocytes but was present in both dendritic cell and monocyte populations. Adenosine-mediated suppression of CD8 T cell activation was reversed by AB928, which restored IFN-g and granzyme B production from these cells. Adenosine-mediated suppression of NK lytic activity was inhibited by AB928, resulting in a significant increase in cell killing. Monocyte-derived dendritic cells (moDC) express high levels of A2bR and those cells, when cultured in the presence of adenosine, had lower levels of costimulatory molecules upon activation and a reduced capacity to stimulate CD4 T cells in a mixed-lymphocyte reaction (MLR). Inhibition of adenosine-mediated signaling by AB928 resulted in a significant increase in the frequency of activated T cells in the MLR assay. Adenosine receptor activation leads to phosphorylation of CREB, which can be detected in circulating leukocytes, allowing for the establishment of PK/PD relationships in mice. AB928 exhibited a dose dependent inhibition of pCREB in whole blood.
Conclusions
In conclusion, AB928 is a potent dual inhibitor of A2aR and A2bR receptors with the potential to block all immunosuppressive effects of extracellular adenosine in the TME. This molecule is anticipated to enter clinical development later in 2017.
P499 Expression pattern and prognostic value of IL-36g in colorectal cancer
Aliyah M Weinstein1, Nicolas A Giraldo2, Catherine Julie3, Laetitia Lacroix2, Florent Petitprez2, Jean-François Emile3, Wolf H Fridman2, Walter J Storkus1, Catherine Sautès-Fridman2
1University of Pittsburgh, Pittsburgh, PA, USA; 2UMR_S1138, Centre de Recherche des Cordeliers, Paris, France; 3Hopital Ambroise Paré, Boulogne, France
Correspondence: Walter J Storkus (alw151@pitt.edu); Catherine Sautès-Fridman
Background
IL-36g/IL-1F9 is a member of the IL-1 cytokine superfamily and a mediator of Type 1 immunity. It was recently reported to be a necessary mediator of tertiary lymphoid organ (TLO) formation in a murine model of colon carcinoma [1]. Specifically, when IL-36g was introduced therapeutically into the tumor microenvironment of MC38-bearing mice, tumors exhibited delayed progression in correlation with TLO formation; this was not observed in control-treated mice. Co-treatment with the antagonist to IL-36g, IL-1F5, blocked the efficacy of the therapy. CD11c+ dendritic cells (DC) were observed to be a major source of IL-36g in the tumor microenvironment (TME), and signaling through IL-36R+ host cells (including those of the vasculature) was necessary for the efficacy of IL-36g-based treatment. In the present study, we sought to identify a role for IL-36g in the de novo formation of TLO.
Methods
In a cohort of 33 primary colorectal cancer patients, the histologic localization and magnitude of expression of IL-36g, IL-1F5/IL-36RA, CD20, CD138, CD8, CD31, PNAd, DC-LAMP, CD68, and Tbet were determined using immunohistochemical and immunofluorescent imaging. Images were analyzed using CaloPix and Visiopharm software packages.
Results
Both IL-36g and IL-1F5 can be expressed by vasculature-associated smooth muscle cells/myofibroblasts as well as immune cells. Expression of IL-36g by the tumor vasculature positively correlates with CD20+ B cell density in TLO, but not the number of TLO observed in the tumor; neither observation holds for IL-1F5. Some IL-36g+ vessels appear to be high endothelial venules, specialized vessels that aid in recruiting lymphocytes to TLO. Interestingly, while B cells are the primary cellular component of TLO in this model, they do not produce IL-36g. Within the immune compartment, CD8+ T cells (including those expressing Tbet) and CD68+ macrophages are major sources of IL-36g in the TME, while DC-LAMP+ DC are less so.
Conclusions
These results support that the pro-inflammatory conditions of colorectal cancer can promote a Type 1-polarized immune microenvironment; that IL-36g is secreted by diverse cell types within the TME; and that IL-36g expression may play a role in the maintenance of TLO and in B cell recruitment to these structures, within the immune contexture of colorectal cancer.
References
1. Weinstein AM, Chen L, Brzana EA, Patil PR, Taylor JL, Fabian KL, Wallace CT, Jones SD, Watkins SC, Lu B, Stroncek DF, Denning TL, Fu YX, Cohen PA, Storkus WJ. Tbet and IL-36g cooperate in therapeutic DC-mediated promotion of ectopic lymphoid organogenesis in the tumor microenvironment. OncoImmunology. 2017; 6(6).
P500 CD73 blockade restores the abscopal response to radiation therapy and anti-CTLA-4 in cGAS-deficient tumors
Erik Wennerberg, Claire Vanpouille-Box, Silvia Chiara Formenti, Sandra Demaria
Weill Cornell Medicine, New York, NY, USA
Correspondence: Erik Wennerberg (erik.ag.wennerberg@gmail.com)
Background
We have recently shown that in poorly immunogenic tumors resistant to immune checkpoint blockers (ICB), radiation therapy (RT) used at 8GyX3 induces cancer cell-intrinsic interferon type I (IFN-I) through cGAS-activation. This process is required for recruitment of BATF3-dependent CD103+ dendritic cells (DCs) and for priming of tumor-specific CD8+ T cells that mediated abscopal responses in the presence of ICBs [1]. Importantly, knockdown of cGAS in the cancer cells at the irradiated tumor site abrogates the induction of abscopal responses by RT+ICB. Extracellular ATP, which is released from irradiated cancer cells, provides an alternative signal for recruitment and activation of DCs. However, rapid conversion of ATP into immunosuppressive adenosine effectively abrogates this process. Therefore, we hypothesized that antibody blockade of the adenosine-converting ectoenzyme CD73 could restore abscopal responses by RT+ICB in cGAS-deficient tumors by augmenting ATP-mediated DC recruitment.
Methods
Mice bearing subcutaneously inoculated flank tumors (cGAS-silenced TSAshcGAS or mock-silenced TSAshNS as primary tumors and TSAshNS as abscopal tumors) were treated with (1) control antibody; (2) RT; (3) RT+anti-CD73; (4) RT+anti-CTLA-4 or (5) RT+anti-CD73+anti-CTLA-4. Anti-CD73 was administered on day 11, 14, 17 and 20 and anti-CTLA-4 on day 14, 17 and 20. RT was administered to the primary tumor in doses of 8 Gy on day 12, 13 and 14. Tumors were analyzed for DC infiltration on day 18.
Results
In mice bearing primary TSAshNS tumors, RT+anti-CTLA-4 elicited abscopal responses. In mice bearing primary TSAshcGAS tumors, RT+anti-CTLA-4 did not induce abscopal responses, while RT+anti-CTLA-4+anti-CD73 did (Tumor size day 37: 873±533 mm3 in control; 827±732 mm3 in RT+anti-CTLA-4; 131±169 mm3 in RT+anti-CTLA-4+anti-CD73, p<0.01). Treatment of mice bearing TSAshcGAS tumors with RT+anti-CD73 resulted in significantly increased tumor infiltration of CD103+ DCs among intratumoral leukocytes compared to mice receiving RT alone (1.2±0.8% in RT v. 2.5±0.7% in RT+anti-CD73, p<0.05).
Conclusions
Our results demonstrate that blocking CD73 recovers the ability of RT to induce recruitment of CD103+ DCs to cGAS-deficient tumors and restores the abscopal anti-tumor responses elicited by RT+ICB.
References
1. Vanpouille-Box C, et al. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun. 2017;8:15618.
P501 Involvement of local renin-angiotensin system in immunosuppression of tumor microenvironment and its modulation for augmentation of cancer immunotherapy
Tomonori Yaguchi1, Kenta Nakamura2, Kenji Morii1, Yutaka Kawakami1
1Keio University School of Medicine, Shinjuku-ku, Tokyo, Japan; 2Shinshu University School of medicine, Matsumoto, Nagano, Japan
Correspondence: Yutaka Kawakami beatless@rr.iij4u.or.jp
Background
To improve the current cancer immunotherapies, strategies to modulate various immunosuppressive stromal cells need to be developed. Local renin–angiotensin system (RAS) in cancer tissues has been reported to be involved in cellular migration, proliferation, inflammation, and angiogenesis in the tumor and supporting stromal cells, few studies have evaluated the effect of local RAS on anti-tumor immune responses. In this study we have evaluated the role of local RAS in the tumor immune-microenvironment.
Methods
We have evaluated the effect of administration of angiotensin receptor blockers (ARBs) on phenotypes of CD11+ myeloid cells and cancer associated fibroblasts (CAF) in cancer microenvironments, induction of tumor antigen specific T cells, and therapeutic effects of PD-1/PD-L1 immune checkpoint blockade therapies, using murine models bearing tumor cell lines in which RAS was not involved in their proliferation and angiogenetic ability.
Results
Administration of ARBs to tumor-bearing mice resulted in significant enhancement of tumor antigen specific T cells. The ARB administration did not change the numbers of CD11b+ myeloid cells in tumors, but significantly reduced their T-cell inhibitory ability along with decreased production of various immunosuppressive factors including IL-6, IL-10, VEGF, and arginase by CD11b+ cells in tumors. ARB also decreased expression of immunosuppressive factors such as chemokine ligand 12 and nitric oxide synthase 2 in CAFs. Lastly, combination of ARB and anti-programmed death-ligand 1 (PD-L1) antibodies resulted in significant augmentation of anti-tumor effects in a CD8-positive T cell-dependent manner.
Conclusions
These results demonstrated that RAS is involved in generation of immunosuppressive tumor microenvironments caused by myeloid cells and fibroblasts, other than the previously shown proliferative and angiogenetic properties of cancer cells and macrophages, and that ARB can transform the immunosuppressive properties of MDSCs and CAFs and could be used in combination with PD-1/PD-L1 immune checkpoint blockade therapies.
P502 IDO1, an immune checkpoint with distinct mechanisms and prognostic significance in glioblastoma
Lijie Zhai1, Derek Wainwright1, Erik Ladomersky1, Carlos Dostal2, Kristen Lauing1, Robert McCusker2, David Binder3, Meijing Wu1, Matthew Genet1, Galina Gritsina1, Balaz Gyorffy4, Priscilla Brastinanos5, Jeffrey Sosman1, Craig Horbinski1, David James1, Roger Stupp1, Francis Giles1
1Northwestern University Feinberg School of Medicine, Chicago, IL, USA; 2University of Illinois at Urbana-Champaign, Urbana, IL, USA; 3University of Colorado School of Medicine, Aurora, CO, USA; 4Semmelweis University, Budapest, Hungary; 5Massachusetts General Hospital, Boston, MA, USA
Correspondence: Derek Wainwright (zhailijie12@gmail.com)
Background
It is commonly accepted that indoleamine 2, 3 dioxygenase 1 (IDO1), a pivotal immune checkpoint, mediates potent immunosuppression in cancers through its tryptophan (Trp)-to-kynurenine (Kyn) catabolizing activity. However, this enzymatic-dependent mechanism of IDO1 has not been established in glioblastoma (GBM), the most common malignant brain tumor in adults. Additionally, the basis for elevated IDO1 expression in GBM is also poorly understood. The major objective of this study is to address this gap in our understanding of how IDO1 contributes to the biology of GBM and immunotherapy of this deadly malignance, as well as whether its level of expression is a determinant of GBM patients’ outcome.
Methods
Freshly resected human GBM, human T cell:GBM co-cultures, HPLC analysis, CTLA-4 and PD-L1 immune checkpoint blockade, IDO1-/- and IDO1+/+ C57BL/6 mice, spontaneous GBM [GFAP(ERT2)→Cre+/-;pTENfl/fl;Rbfl/fl;p53fl/fl;(+/-IDO1fl/fl);(+/-IDO1-/-)] mice, nu/nu mice, NOD-scid and humanized (NSG-SGM3-BLT) mice engrafted human GBM as well as the cancer genome atlas form the basis of our investigation.
Results
(1) Depletion of GBM-derived IDO1 did not affect brain tumor Trp catabolism, while systemic knockout of IDO1 resulted in decreased Trp->Kyn conversion in both naïve and tumor-bearing mice; (2)CTLA-4 and PD-L1-based dual immunotherapy did not alter Trp/Kyn level in IDO1-/- and IDO1+/+ mice, but only reached to maximal treatment effect in IDO1+/+ mice; (3) In situ hybridization for IDO1 revealed consistent transcript expression in all human GBM samples (n=172), whereas immunohistochemical IDO1 expression was highly variable; (4) Multivariate analysis revealed that higher levels of IDO1 transcript predicted/correlated with shorter survival in the TCGA dataset (P=0.0076) and IDO1 mRNA levels positively correlate with increased gene expression for markers of cytolytic and regulatory T cells, but no correlation with IFNs gene expression; (5) Humanized mice intracranially-engrafted with human GBM revealed an IFNγ-dependent and T cell-mediated increase of intratumoral IDO1 expression.
Conclusions
Our data indicate that tumor cell-derived IDO1 mediates its immunosuppression in GBM via a non-enzymatic mechanism, while the non-tumor IDO1 dominantly contributes to its Trp catabolic function and is essential for the immune checkpoint blockade therapy. TCGA analysis also revealed high intratumoral IDO1 mRNA levels correlate with inferior GBM patient outcome and infiltrating T cells. Future immunotherapeutic efforts aiming to increase T cell-mediated immune response should consider combinatorial approaches that inhibit potential T cell-mediated IDO1 upregulation during therapy.
P503 Comprehensive characterization of immune microenvironment of cholangiocarcinoma
Tao Xia1, Dwayne Thomas1, Linda Chen1, Qingfeng Zhu1, Jin He1, Robert Anders1, Shafat Quadri2, Elaine Pinherio2, Adrian Murphy1, Lei Zheng1
1Johns Hopkins University School of Medicine, Baltimore, MD, USA; 2Merck, Merck, NJ, USA
Correspondence: Lei Zheng lzheng6@jhmi.edu
Background
Background: Biliary tract cancers (BTC) are groups of tumors that include both intrahepatic cholangiocarcinoma (ICC), extrahepatic cholangiocarcinoma (ECC) and gallbladder carcinoma (GC). BTC comprise an uncommon cancer type that is currently on the rise with a high mortality rate. New modalities of treatment for BTC are highly demanded. Immunotherapy has emerged as a promising treatment for many malignant diseases. Although many risk factors of BTC are associated with chronic inflammatory conditions, the immune microenvironment of BTC has been examined to a very limited extent. Therefore, we perfored a comprehensive analysis of tumor microenvironment of BTCs to identify immune mechanistic targets for immunotherapy and identify optimal strategies of combinational immunotherapy.
Methods
Materials and methods: Surgically resected tumors from 31 cases of ECC and 24 cases of ICC were stained with multiplex immunohistochemistry of 14 immune cell markers including CD3, CD4, CD8, CD20, CD45, CD68, Cd163, CSF1R, DC-LAMP, DC-Sign, Foxp3, Ki-67, PD-1, PD-L1 on single slide section and analyzed by the Halo software. Nanostring of immune panels and cytokine arrays PCR were also performed by RNA purified from FFPE slides.
Results
Results: Higher density of CD8 T cells is significantly associated with longer overall survival (p=0.024). Higher densities of PD-1+ T cells and PD-L1+ myeloid cells are significantly associated with longer overall survival in ECC (PD-L1, p=0.019; PD-1, p=0.018) (Fig. 1, Fig. 2), but higher density of PD-L1+ cells are significantly associated with shorter overall survival in ICC (p=0.003) (Fig. 3, Fig. 4). CD68 is not prognostic. However, higher density of CD163+CD68+ M2 macrophages is associated with poorer survival in ECC (Fig. 5). Nanostring and cytokine array PCR results are being analyzed. More cases are being stained by multiplex immunothistochemistry.
Conclusions
Conclusion: Our study represents so far the most comprehensive characterization of immune cell subtypes in the tumor microenvironment of cholangiocarcinoma. The results suggest that PD-1/PD-L1 is not a good target in ECC, but a potentially good target in ICC. An ideal macrophage-targeting agent would be the one that depletes the M2 macrophages more specifically.

See text for description

See text for description

See text for description

See text for description

{kind=link}
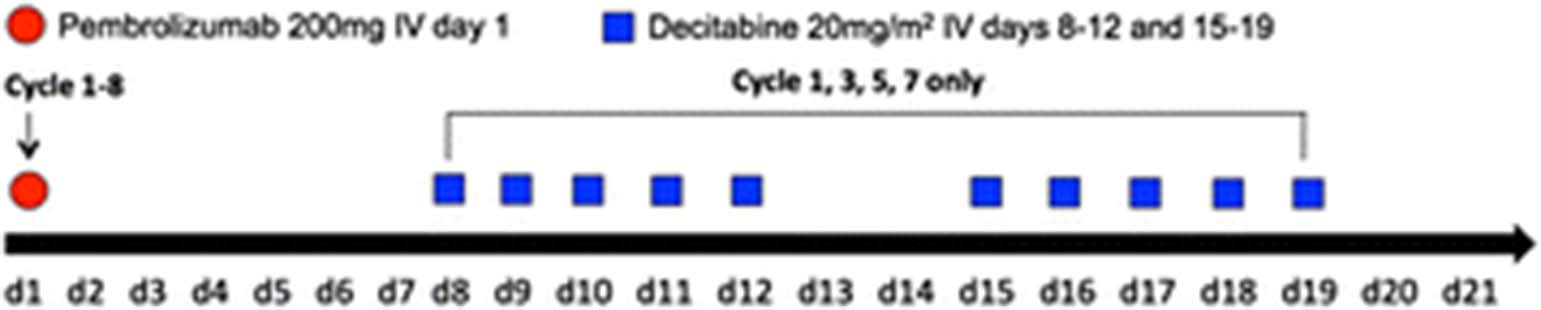{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
See text for description
P504 Enhancement of CAR-T cell tumor tropism through expression of CXCR2 chemokine receptors
Ismahene Benzaid1, Daniel Santiago1, Cecilia Ramello1, Mibel Pabon1, Wendy Kandell1, Theresa Boyle1, Anders Berglund1, Scott Antonia1, Daniel Abate-Daga1
1Moffitt Cancer Center, Tampa, FL, USA
Correspondence: Daniel Santiago Daniel.AbateDaga@moffitt.org
Background
Chimeric Antigen Receptor (CAR)-T cell-based immunotherapies are novel approaches to cancer therapy. With the expected FDA approval of an anti-CD19 CAR for B-cell malignancies, adoptive immunotherapies will become the standard of care for a large number of patients. However, the clinical success of CAR-T cells beyond hematological malignancies has been scarce. We hypothesize that this is in part due to suboptimal bioavailability of CAR-T cells in solid tumors, and we propose to manipulate the expression of chemokine receptors to enhance the CAR-T cells homing and increase their therapeutic efficacy against the tumor.
Methods
We first analyzed the expression of chemokines by human lung tumors, using publicly available databases of RNA expression (TCGA). In addition, we measured their expression in the supernatants of short-term cultures of human lung tumors. Retroviral vectors were generated for the expression of chemokine receptors, in combination with an anti-mesothelin (MSLN) second generation CAR. T cell migration and function were assessed using migration transwell and cytotoxic assays. Anti-tumor efficacy was assessed using a xenograft model of lung cancer.
Results
IL-8 (CXCL8) was consistently expressed by lung tumors, and secreted by short-term cultures of surgically-removed lesions. Co-expression of an anti-MSLN CAR and CXCR2 or CXCR1 increased the migration of human T cells towards medium containing recombinant IL-8, or conditioned media from lung cancer cell cultures, in transwell assays. Importantly, co-expression of chemokine receptors did not compromise CAR expression or function. Furthermore, expression of CXCR2 (but not CXCR1) in CAR T cells increased the number of intratumoral T cells, and significantly enhanced antitumor efficacy in a xenograft tumor model based on H2110 cells.
Conclusions
Tumor homing and therapeutic efficacy of CAR-T cells can be improved through co-expression of chemokine receptors. This approach may apply to other types of T cell-based immunotherapies, for the treatment of lung cancer, and other solid tumors.
P505 First in human, single ascending dose study in healthy volunteers of SNDX-6352, a humanized IgG4 monoclonal antibody targeting colony stimulating factor-1 receptor (CSF-1R)
Renger Tiessen1, Anniek Visser2, Henko Tadema2, Caryn Peterson2, Helen Pentikis3, Lei Wang3, Michael L. Meyers4, Peter Ordentlich3
1PRA Health Sciences, Groningen, Netherlands; 2PRA Bioanalytical Laboratory, Assen, Netherlands; 3Syndax Pharmaceuticals, Inc., Waltham, Massachussets, USA; 4Syndax Pharmaceuticals, Inc., New York, New York, USA
Correspondence: Renger Tiessen tiessenrenger@prahs.com
Background
CSF-1R is expressed on immunosuppressive tumor associated macrophages (TAMs). High numbers of TAMs in tumors are associated with poor prognosis. SNDX-6352 is a humanized IgG4 monoclonal antibody with high affinity against CSF-1R and has demonstrated antitumor activity in preclinical tumor models by enhancing the immune response.
Methods
Study SNDX-6352-0001 is a first-in-human, double-blind, randomized, placebo-controlled, single ascending dose, phase I trial in healthy volunteers. The planned dose levels were 0.15, 1.0, 3.0, 6.0, and 10.0 mg/kg. All treated subjects were observed for 72 hours post-infusion and underwent follow-up evaluations for 12 weeks. Safety assessments included adverse event (AE) monitoring, clinical laboratories, vital signs, 12-lead electrocardiogram (ECG), eye examination, and physical examination. Blood samples were collected to characterize the pharmacokinetics (PK) and pharmacodynamics (PD; change in plasma colony stimulating factor-1 [CSF-1] and interleukin-34 [IL-34]), as well as to evaluate changes in circulating classical and non-classical CD16+ monocytes in blood and change in CSF-1 receptor occupancy (RO).
Results
The first three placebo-controlled cohorts were completed with 14 subjects receiving SNDX-6352 (2 at 0.15mg/kg, 6 at 1mg/kg and 6 at 3mg/kg) and 6 subjects receiving placebo (2 in each cohort). Dosing up to 1.0 mg/kg was tolerated well with the most common complaint being mild to moderate itching (lasting 2 or 3 days). At the 3 mg/kg dose, similar itching complaints were observed. At this dose, mild to moderate eyelid swelling was observed in all subjects (median duration of 40 days), with no impact on vision. Dose escalation was then terminated. In blood, a transient reduction in non-classical CD16+ monocytes was observed, and an upward trend in LDH and ASAT. Cmax increases corresponded with dose, while AUC increases were greater than dose proportional, indicating non-linear PK. Incr.
Acknowledgments
Notes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
About this supplement These abstracts have been published as part of Journal for ImmunoTherapy of Cancer Volume 5 Supplement 2, 2017. The full contents of the supplement are available online at https://jitc.biomedcentral.com/articles/supplements/volume-5-supplement-2. Please note that this is part 2 of 2