
Abstract
Members of the PI3K/Akt/mTor signaling cascade are among the most frequently altered proteins in cancer, yet the therapeutic application of pharmacological inhibitors of this signaling network, either as monotherapy or in combination therapy (CT) has so far not been particularly successful. In this review we will focus on the role of PI3K/Akt/mTOR in two distinct tumors, Glioblastoma multiforme (GBM), an adult brain tumor which frequently exhibits PTEN inactivation, and Neuroblastoma (NB), a childhood malignancy that affects the central nervous system and does not harbor any classic alterations in PI3K/Akt signaling. We will argue that inhibitors of PI3K/Akt signaling can be components for potentially promising new CTs in both tumor entities, but further understanding of the signal cascade’s complexity is essential for successful implementation of these CTs. Importantly, failure to do this might lead to severe adverse effects, such as treatment failure and enhanced therapy resistance.
Similar content being viewed by others

Introduction
Modern cancer therapy aims at eradicating tumors by predominately inducing apoptosis, a form of programmed cell suicide, via DNA damage and is thus limited by emerging resistance of cancerous cells and toxic side effects on healthy tissue. In order to circumvent these limitations, combination therapy (CT) that combines cytotoxic agents, such as chemo- or radiotherapy at metronomic doses with at least one molecular-targeted agent, i.e. a pharmacological inhibitor of signaling cascades, has been proposed to break apoptosis resistance, while concurrently lowering adverse side effects [1]. The PI3K/Akt/mTOR survival signaling cascade is frequently considered a promising target in modern CT, particularly as PTEN, its negative regulator, is among the most frequent mutated proteins in cancer [2]. Small molecule compounds that inhibit members of the PI3K signaling network, either pan-specific, on the level of individual protein classes or even isoform-specific, are being developed and clinically evaluated by several companies [3, 4]. However, despite promising preclinical data, clinical successes so far have been limited. There is mounting evidence that the discrepancy between preclinical and clinical data is due to the intrinsic complexity of the PI3K/Akt/mTOR signaling cascade, so that the reductionist view of PI3K signaling as a mere linear 'survival pathway' seems no longer appropriate:
-
1)
Signaling cascades, such as the PI3K/Akt/mTOR pathway, are often depicted as a linear cascade, their activation akin to a row of dominoes falling, with individual signaling arms running parallel (at the same speed) with occasional feedback loops. However, the reality is more in line with a Rube Goldberg Machine. These pathways have evolved out of more primitive cascades, often with different functions, have recruited proteins and whole side arms and thus created overlapping or even diametrically opposite functions. A signaling cascade is not maintained in a population – be it mammal, human or tumor – due to its efficacy or elegance, but remains active in a population if it is the best current option that a) does what it needs to do and b) is available to said population. This, in turn, means that there is no simple linear relationship between degree of inhibition and tumor cell death. In other words our data, published and unpublished, as well as others clearly indicate maximal PI3K inhibition does not equal maximal sensitization for cell death [5, 6].
-
2)
Taking the evolutionary origins of cellular signaling pathways into account, the notion of a survival pathway that can be inhibited by taking out a central component appears greatly simplistic. The notion of distinct signaling cascades that overlap is a human construct, probably not dissimilar to the Linnæan classification system, useful most of the time, but not reflecting the whole biological truth. For example, there is mounting evidence that the PI3K/Akt/mTOR pathway and the Ras/MEK/Erk signaling cascade are – at least in some cellular systems – so entwined that one could really view them as a single, even more complex survival machinery (with a canopy of additional functions) [7]. Therefore, blocking an individual component is not comparable to figuratively blowing up a railroad bridge and thus stopping the cancer train, but more like blocking the highway forcing cancer to take the byroads and local diversions, slowing down progression, but with the minor risk of the discovery of a novel shortcut.
Taking these two caveats into account our answer to the question Is the PI3K signaling network still a promising target for cancer therapy? is Yes, but it is not the magic bullet hoped for.
Review
Glioblastoma and PI3K signaling
Glioblastoma multiforme (GBM) is the most common primary tumor of the central nervous system in adults [8] and with a mean patient survival of 15 months after treatment initiation, it is considered to be among the most lethal cancers [9]. The current standard of care consists of tumor resection followed by radiotherapy and a course of Temozolamide [10]. As PTEN expression is reduced by mutation or loss of heterozygocity in 5-40% and 60-80%, respectively [11], and PI3K/Akt/mTOR signaling is elevated in ~88% of all glioblastoma [12, 13], the addition of pharmacological inhibitors to current treatment schedules should lead to clear therapeutic benefits. Indeed several clinical trials have been initiated to study this hypothesis [14], but most have led to no significant therapeutic improvement, or – in the case of bevacizumab – have yielded rather controversial results [15]. So far, aside from two mTOR (complex 1) inhibitors, Everolimus and Temsirolimus, no drug targeting the PI3K/Akt/mTOR pathway specifically has been approved [14].
The first interesting point of note is that inhibition of PI3K signaling does not lead to apoptosis induction, although this has been reported for other tumor entities, such as Hodgkin lymphoma [16]. Furthermore, as soon as patients are treated with Temozolamide the presence or absence of PTEN within the tumor is irrelevant for any future prognostic development [11], the same phenomenon holds true for activated Akt [17]. In contrast, activity of the mTOR arm of the signaling cascade is associated with poor patient survival [17]. However, due to its complex role in several cellular processes [18], in particular autophagy, which may lead to increased or reduced cell survival [19] and the upregulation of ERK signaling upon its inhibition [20], mTOR is generally considered a difficult target in glioblastoma [21]. Furthermore, frequently the promising in vitro data regarding mTOR inhibition in Glioblastoma cells does not translate well into an in vivo setting (for example [22]).
Our own initial data analyzing a panel of Glioblastoma cell lines responding to CT combining inhibition of PI3K signaling and various chemotherapeutic agents, as well as death receptor ligands also yielded several interesting points [23, 24]: PI3K signaling contributes to therapy resistance in GBM cell lines, independently of PTEN status. However, this contribution seems stronger when cells are additionally stressed, i.e. serum-starved. Importantly, the observed effects often do not appear particularly strong, making it hard to argue that the PI3K/Akt/mTOR signaling cascade is the main mediator of apoptosis resistance in GBM. The above described clinical data seem to support these inferences, which leads to the question: Why then is this pathway so frequently mutated in GBM? We suggest three possible reasons that might explain this apparent contradiction. They are by no means mutually exclusive and are all supported by the literature.
-
1)
Activation of PI3K signaling functions as a driver mutation for the cell of origin to re-acquire stem cell characteristics/re-enter the cell cycle. While the cell of origin is still strongly debated in GBM [25], primary GBM, the more common form making up 91-95% of all GBM [26], apparently arise de novo within 3–6 months [26], suggesting rapid proliferation in a tissue which generally exhibits rather little cell division. This is of particular interest if GBM does not arise from neural stem cells or oligodendrocyte precursor cells [27], but de-differentiated astrocytes [25], and would fit with the observation that inhibition of PI3K signaling GBM primary affects proliferation [28, 29]. The most compelling data for this hypothesis comes from work in T cell acute lymphoblastic leukemia where it could be shown that activated AKT signaling enhanced the frequency of leukemia propagating cells, which can be considered in this context as tumor stem cells [30].
-
2)
PI3K/Akt facilitate the invasive phenotype which is characteristic for GBM, both in terms of motility and survival under stress. An association with FAK and Src has long been established for PI3K (for example [31, 32], thus linking it to signaling complexes associated with adhesion, motility and invasion [33]. Recent data seem to link Akt/mTOR activity directly to GBM motility [34], which is of particular interest as this would suggest a connection between two almost ubiquitous features of GBM, high activity of the PI3K signaling cascade and tumor dissemination throughout the whole brain [35, 36].
-
3)
The central role of PI3K/Akt/mTOR in GBM biology has led us grossly to underestimate its importance. As shown above PI3K has clearly other roles besides survival in GBM, it contributes to motility (point 2) and proliferation (point 1). This would suggest that PI3K signaling has several contradictory functions in GBM. For example, high proliferation is usually associated with therapy sensitivity [37], yet inhibition of PI3K signaling can lead to both reduced proliferation [28, 29] and chemosensitization [23, 24]. This notion does not negate the use of pharmacological PI3K inhibitors in CT, but rather indicates the design of CT needs to be carefully considered.
Interestingly, the importance of the last point is emphasized by our work mainly conducted on a different tumor system, Neuroblastoma (NB).
Neuroblastoma and PI3K signaling
NB is a common childhood neoplasia of the sympathetic nervous system that is generally characterized as a highly heterogeneous disease and categorized into four stages, of which stage 1 and 2 have a good prognosis. However, prolonged survival for patients with stage 3 and 4 is only 18-30% [38, 39]. Unfortunately 45% of patients exhibit high-risk tumors, most of which have already metastasized at clinical presentation [40]. Interestingly, NB lacks the mutations in PI3K signaling that characterize GBM and other tumors and a role for PI3K in NB was first considered due to its close association with the mycN oncogene [41] and its prominent role downstream of growth factor-initiated signaling, such as IGF-1 and IGF-2 [42]. More recently, the role of the ALK receptor tyrosine kinase has also been highlighted in NB. It is expressed in over 90% of all NB [43] and found to be activated by mutation in 6.9% [44]. One of the downstream survival cascades activated by ALK is PI3K/Akt signaling [42]. Our own group previously showed that in NB phosphorylated Akt correlates with poor patients' prognosis [45], while others subsequently demonstrated a link between PI3K signaling and growth/survival [46], as well as resistance to chemotherapy [47].
However, more recently following some previous hints concerning the underlying molecular mechanisms of how PI3K signaling affects chemosensitivity in NB [48], we were surprised to discover that the relationship between PI3K signaling and chemosensitivity is not as simple as we had assumed [5]. Maximal, prolonged inhibition of PI3K signaling did not sensitize NB cell lines for Doxorubicin-induced apoptosis. Indeed under certain conditions even a desensitization could be observed [5]. This might be due to a combination of the potent anti-proliferative effect of PI3K/mTOR inhibition in NB cells and the induction of autophagy [5], although it should be pointed out that autophagy in NB is controversially discussed, as in most tumor entities, and can be a survival mechanism as well as a cell death enhancer (for example [49, 50]). While concurrent treatment with a chemotherapeutic agent and a pharmacological inhibitor of PI3K signaling led mostly to good apoptosis sensitization, it was the application of the inhibitor several hours after the cell death inducer that yielded the best results [5]. We traced this particular effect to the mitochondria, i.e. the modulation of VDAC1 via Akt-mediated phosphorylation of GSK3β [5].
Importantly, as we found a similar temporal effect in GBM cells, this led us to propose a model which de-emphasizes the maximal inhibition of a target signaling cascade (clinically: high plasma levels of a pharmacological inhibitor prior to chemotherapy) and concentrates on the temporal relationship of the two treatment components, i.e. sequential dosing [5].
Pathway complexity: when and where?
Next, one needs to consider at which point in the signaling cascade to intervene. While in the past a preferred target seems to have been the dual inhibition of PI3K and mTOR [3], thus taking out a major section of the signaling cascade and ablating a potent feedback mechanism, currently there seems to be a shift towards inhibitors of Akt [4]. However, as emerging data suggest that Akt is not the central mediator of PI3K, i.e. inhibition of Akt signaling is not identical with inhibition of PI3K signaling [7], this target appears less promising. In particular, inhibition of PI3K can also block Ras/ERK signaling, while inhibition of Akt does not and is thus less efficient [7].
To consider the optimal target of PI3K signaling inhibition one has first to understand its role. Our data [5] provides strong evidence for the third proposition we put forward, indicating that we have previously underestimated the complexity of PI3K signaling. Importantly, this does not necessarily invalidate the other two options. In a model we and others [5, 6] suggest the PI3K signaling cascade regulates two or three aspects of cellular behavior relevant for the success of CT: On the one hand proliferation and metabolism (the inhibition of which can have adverse effects on chemo- and radiotherapy [5, 6], e.g. hyperactivation of Akt in this context has even been proposed as a therapeutic option [51]), on the other hand survival, particularly under stress. Importantly, these competing effects of the PI3K signaling network are mediated by different arms of the cascade at different speeds, giving us two therapeutic strategies to employ.
The approach we – and also, apparently, the Djuzenova group – favor is the use of existing, clinically evaluated pharmacological inhibitors of the PI3K signaling, while optimizing the treatment schedule so as concurrently to maximize the effect on survival and minimize the effect on proliferation/metabolism [5, 6]. Alternatively, a strategy could be envisioned whereby not the main components of the PI3K signaling cascade are targeted, but either proteins that are further upstream, apical to several, potentially compensatory networks, such as the MEK/Erk signaling cascade [7], or further downstream at the level of molecules that clearly only contribute to survival in the context of CT, such as the Bcl-2-family [48].
The epidermal growth factor receptor (HER1/EGFR) is one of the most frequently dysregulated oncogenes in glioblastoma [52]. About half of GBM show amplification of HER1/EGFR and of these about another half coexpress EGFRvIII, the continuously activated mutant form of the receptor [53]. In preclinical studies, treatment with HER1/EGFR-targeted agents such as monoclonal antibodies or small-molecule inhibitors, e.g. erlotinib (Tarceva; Genentech Inc.) were shown to exert promising antineoplastic activity in various in vitro and in vivo models in the setting of GBM [52, 54, 55]. These effects were at least in part related to inhibition of PI3K/Akt and MAPK signaling. However, when taken into the clinics HER1/EGFR inhibitors did not hold up to the expectations derived from the promising preclinical results [56–58]. Treatment with erlotinib did not result in a survival benefit either as a single agent therapy [58] or when combined with temozolomide and radiation [57]. Thus, at this point, upstream targeting of PI3K by inhibition of HER1/EGFR did not fulfill hopes in improving the fate of patients with GBM. One potential explanation for this finding is that PI3K signaling may be uncoupled from upstream control by inactivation of PTEN, the previously mentioned negative regulator of PI3K signaling (Figure 1).

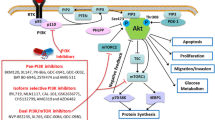
The PI3K/Akt/mTOR signaling cascade. Simplified schemata of the PI3K/Akt/mTOR signaling cascade. While indisputable involved at several levels, directly as well as indirectly with cell survival, other important functions of this signaling network are also highlighted here. Key molecules which have been implicated in cancer or have been selected as potential therapeutic targets are: 1. Receptor tyrosine kinases, such as EGFR, IGFR and ALK, often overexpressed or activated by mutations in many different cancers, including glioblastoma and neuroblastoma. These transmembrane proteins are apical of several interconnected signaling cascades. 2. PI3K, phosphatidylinositol 3'-OH kinase, a lipid kinase, predominately consisting of a p110 catalytic and a p85 regulatory subunit, of which the former has been found to be mutationally activated in certain cancers. 3. PTEN, phosphatidylinositol (3,4,5)-triphosphate [PtdIns(3,4,5)P 3 ] phosphatase and tensin homologue, is a phosphatase that counters PI3K-mediated phosphorylation and as such functions as a negative regulator of the PI3K/Akt/mTOR signaling cascade. It is among the most frequently inactivated, either by mutation or promoter methylation, proteins in cancer. 4. The serine/threonine kinase Akt, v-akt murine thymoma viral oncogene homologue (Protein Kinase B), is often considered the central downstream mediator of PI3K signaling, as it phosphorylates a diverse array of targets that are involved in all key functions of this pathway. 5. mTOR, mammalian target of Rapamycin, depending on its complex partners, can either be up- or downstream of Akt. It is frequently found to be associated with the process of autophagy, which depending on the context, can be either a survival strategy or a form of cell death. (modified from [33]).
In vitro, Fan and coworkers showed that PTEN mutant U87 cells were much less responsive to a treatment with erlotinib alone when compared to PTEN wild type LN229 cells [59]. Others reported similar findings [60]. In both studies, additional inhibition of either mTOR by rapamycin or PI3K/mTOR by PI-103 resulted in a sensitizing effect on PTEN mutated glioma cells towards HER1/EGFR inhibition by erlotinib. However, in a clinical setting, the results of a phase I/II pilot study of 22 patients with recurrent GBM treated with the mTOR inhibitor everolimus and the HER1/EGFR inhibitor gefitinib were rather mixed [61].
While identification of potential upstream targets seems less advanced in NB than in GBM, ALK presents itself as a promising candidate, but – although pharmacological inhibitors such as crizotinib are available – preclinical and early clinical trials do not appear promising. It seems that an almost complete inhibition of ALK is needed for a clinical response, while resistance to the inhibitor can develop relative rapidly, as is also the case with HER1/EGFR [62]. Similar to the combination of an HER1/EGFR inhibitor with blocking mTOR signaling in GBM, the combination of mTOR and ALK inhibition in NB is also of therapeutic benefit [63].
These findings in GBM and NB clearly indicate that CT with several inhibitors (of a single signaling network) of which some are upstream of PI3K can lead to enhanced therapeutic responses.
To avoid the inhibition of additional side arms of the signaling cascades or activation of compensatory mechanisms rather specific individual targets should be considered, but therefore the precise executor molecule for survival needs to be identified. We have identified DNA-PK, i.e. the DNA repair axis of PI3K signaling, as the crucial inhibitor of survival when GBM cells are challenged with Doxorubicin [24]. However when NB are treated with the same drug GSK-3 and VDAC, i.e. the mitochondrial arm of PI3K signaling, are crucial [5] (Figure 2). Further experiments are needed to find out whether these differences are indeed due to the specific tumor entity, the interplay between treatment and genetic make-up, or different role of the PI3K signaling cascade in different cells. As long as these questions are unanswered downstream molecules, despite the presumed lower side effects, remain undesirable, particularly, as a single end-point target increases the risk of mutational escape, as seen, for example, with Gleevec [64].

Potential tumor-specific differences in therapeutic end points. Although inhibition of PI3K/Akt/mTOR signaling is a feasible approach in both glioblastoma and neuroblastoma, the molecular background of those two malignancies is very different. While glioblastoma exhibits amplification of HER1/EGFR with coexpression of the activated EGFRvIII variant [53], their main feature with regards to the PI3K signaling network is the frequent inactivation of PTEN [11]. In contrast, neuroblastoma does not exhibit any frequent mutation within the PI3K/Akt/mTOR signaling cascade, but often displays overexpression of several receptor tyrosine kinases [42, 43]. Interestingly, our own data suggest that the downstream effectors that need to be inhibited to chemosensitize these tumors to chemotherapy are different. Glioblastoma is sensitized for doxorubicin-induced apoptosis via blocking DNA-PK activation, i.e. DNA repair [24], while in neuroblastoma this seems to be mediated at the level of mitochondria via VDAC1 [5]. It remains to be seen whether this is an isolated phenomenon, due to low numbers of cell lines investigated, or whether different arms of the PI3K network have a different weighting dependent on the malignancy.
Importantly, our data suggest that one does not have to pick a single or – in case of the dual PI3K/mTOR inhibitors - a double target, but that multiple, carefully timed inhibitions of what is generally considered a single signaling cascade can cooperate and thus produce a greater sensitizing effect [5]. Therefore it is feasible, from a purely biological perspective, to imagine a therapeutic approach that uses a stepwise inhibition of PI3K signaling both to extent the therapeutic window and enhance cell death (Figure 3). How such an approach could be translated into a clinical setting and whether cumulative inhibition of signaling might lead to unforeseen side effects are the key remaining challenges that need to be overcome.

Possible future treatment schedule. A possible complex combination therapy that consists of a cell death inducer and several sensitizer that all target individual components of the PI3K signaling cascade. 1. Take out the cancer cells' ability to move, thus preventing invasion and metastasis, extending the therapeutic window. Continue blocking this arm throughout therapy. 2. Block the DNA repair mechanisms prior to treatment. Importantly do not affect cell cycle progression, thus making cancer cells amenable to most standard treatments. 3. Administer chemo- or radiotherapy. 4. Block the survival pathways mediated by PI3K signaling in the cancer cells stressed by treatment. If, as outlined in Figure 2, details of key mediators are know, e.g. DNA-PK in glioblastoma (4.1), or VDAC1 in neuroblastoma (4.2), target these, otherwise Akt seems the most promising target. 5. Finally, block growth factor receptors to maximize cell cycle arrest, thus preventing a cancer repopulation by the cells that escaped treatment-induced apoptosis. Point 5 might appear superfluous, as 3 and 4 should already block proliferation, however, this inhibition should also target cells that have activated additional signaling cascades or have (epi)genetically escaped sensitivity to treatment.
Conclusions
Crosstalk between proneoplastic signaling pathways (or, depending on the perspective: underestimated complexity of a single signaling network) is abundant in both GBM [65] and NB [66, 67] and is regarded as one of the major culprits responsible for the failure of targeted therapies. In addition, it has also become apparent in the last decade or so that the number of mutations found in cancers is considerably higher than estimated: While only a handful are needed to initiate tumor progression, an excess of 10,000 mutations is found in many malignancies [68]. Although many of these will not contribute to the malignant phenotype and might even be present in benign neoplasms [68], other mutations will affect the cancer cells' behavior. With such a complexity, it is not surprising that blocking a single signaling cascade, either alone or as part of a CT approach, has not been successful. Indeed, viewed in this context it is actually surprising what a potent tool inhibition of PI3K signaling is!
As a consequence, multi-targeting has been developed as a therapeutic strategy to overcome this potential mechanism of resistance by combining two or more agents targeting different oncogenic signaling pathways, or targeting a single oncogenic signaling pathway differently in the sense of a combined forces alliance driving the cancer cells to undergo cell death [5, 69–72]. Frequently, the inhibition of PI3K signaling is considered a promising contributor to CTs.
In single therapy inhibition of PI3K signaling often leads to a cytostatic effect, not only in the aforementioned Hodgkin lymphoma [16], but also in both GBM [28, 29] and NB [41, 73]. We have argued elsewhere [33] that an approach that aims to chronify a malignancy rather than an attempt to cure a patient can be the preferable strategy, while others have also pointed out that prolonged cytostasis can ultimately lead to a cytotoxic event, if the tumor cells fail to escape this proliferation block [74]. For CT an antiproliferative effect of the sensitizer is often considered a hindrance, as it has often been suggested that rapid proliferation coincides with increased treatment sensitivity [37]. This is by no means an universal truth [75]. In addition, there are several inducers of cell death which are of therapeutic interest that exert their effects in a proliferation-independent manner, such as death receptor triggering [76, 77], or treatment with amiloride derivatives [78]. Most crucial, as outlined above, there is an emerging line of argument that the carefully timed application of sensitizer and inducer in CT can drastically affect the outcome [5, 6], suggesting that a carefully timed sequential application of inhibitor and sensitizer, together with carefully selected targets for inhibition, quite possibly several targets may lie within the same cascade, can greatly enhance the anti-tumorigenic effect of CT. In such a setting, pharmacological inhibition of PI3K/Akt/mTOR may prove to be an invariable tool in our therapeutic arsenal.
While this review focuses on glioblastoma and neuroblastoma, the inhibition of PI3K-mediated signaling is currently also being clinically evaluated in several additional tumor entities, such as lymphocytic leukemias, colorectal cancer, head and neck cancer, several forms of lymphoma, non-small cell lung cancer and renal cancer [79]. Indeed, it is well-worth remembering that PTEN, the negative regulator of this signaling cascade has often been cited as the second most frequently mutated gene in cancer and even as 'a new guardian of the genome' [80]. Since neuroblastoma and glioblastoma are, with respect to PI3K/Akt/mTOR signaling, two very different entities, there is no a priori reason to assume that other tumors will not be subject to the principles discussed here. For example, sequential dosing, has already been demonstrated to also be important in other malignancies, such as breast cancer [81]. However, further research will be needed to fully clarify this point.
With the current rapid advancement of sequencing techniques [82, 83], profiling [84, 85] and molecular histology [8], our understanding of which targets in which signaling cascade might represent promising targets will become much clearer within the next few years. However, other challenges remain, such as how successfully to translate the preclinical data into a clinical setting. For example, our own cell culture work suggested that in a complex CT approach to treat childhood GBM the inhibition of PI3K was preferable to targeting mTOR, however, applying these two combinations to an orthotopic xenotransplant mouse model indicated that the CT that blocked PI3K had no beneficial effect on mouse survival, while the therapy that targeted mTOR significantly prolonged survival [72]. Importantly, on analysing the tumours, we found indications that suggested treatment failure of the first combination was due to an additional effect on tumour vasculature that prevented the chemotherapeutic agent from reaching the malignancy [72]. This effect would have been undetectable in both cell culture and under local instead of systemic treatment of the mice. In addition, while a cell culture model and – to a certain extent – a preclinical animal model enable the precisely sequential-timed application of clearly defined concentrations allowing us fully to understand which amount of a substance reaches the target within what time frame, this luxury is not given in the clinic. A systemic application of treatment is here the norm and thus the precise understanding and control of the complex pharmacokinetics will be essential.
References
Li F, Zhao C, Wang L: Molecular-targeted agents combination therapy for cancer: developments and potentials. Int J Cancer. 2014, 134: 1257-1269.
Shi Y, Paluch BE, Wang X, Jiang X: PTEN at a glance. J Cell Sci. 2012, 125: 4687-4692.
Brana I, Siu LL: Clinical development of phosphatidylinositol 3-kinase inhibitors for cancer treatment. BMC Med. 2012, 10: 161.
Porta C, Paglino C, Mosca A: Targeting PI3K/Akt/mTOR Signaling in Cancer. Front Oncol. 2014, 4: 64.
Westhoff MA, Faham N, Marx D, Nonnenmacher L, Jennewein C, Enzenmuller S, Gonzalez P, Fulda S, Debatin KM: Sequential Dosing in Chemosensitization: Targeting the PI3K/Akt/mTOR Pathway in Neuroblastoma. PLoS One. 2013, 8: e83128.
Kuger S, Graus D, Brendtke R, Gunther N, Katzer A, Lutyj P, Polat B, Chatterjee M, Sukhorukov VL, Flentje M, Djuzenova CS: Radiosensitization of Glioblastoma Cell Lines by the Dual PI3K and mTOR Inhibitor NVP-BEZ235 Depends on Drug-Irradiation Schedule. Transl Oncol. 2013, 6: 169-179.
Will M, Qin AC, Toy W, Yao Z, Rodrik-Outmezguine V, Schneider C, Huang X, Monian P, Jiang X, de Stanchina E, Baselga J, Liu N, Chandarlapaty S, Rosen N: Rapid induction of apoptosis by PI3K inhibitors is dependent upon their transient inhibition of RAS-ERK signaling. Cancer Discov. 2014, 4: 334-347.
Huse JT, Holland EC: Targeting brain cancer: advances in the molecular pathology of malignant glioma and medulloblastoma. Nat Rev Cancer. 2010, 10: 319-331.
Wen PY, Kesari S: Malignant gliomas in adults. N Engl J Med. 2008, 359: 492-507.
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO: Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005, 352: 987-996.
Carico C, Nuno M, Mukherjee D, Elramsisy A, Dantis J, Hu J, Rudnick J, Yu JS, Black KL, Bannykh SI, Patil CG: Loss of PTEN is not associated with poor survival in newly diagnosed glioblastoma patients of the temozolomide era. PLoS One. 2012, 7: e33684.
Network CGAR: Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008, 455: 1061-1068.
Fan QW, Weiss WA: Targeting the RTK-PI3K-mTOR axis in malignant glioma: overcoming resistance. Curr Top Microbiol Immunol. 2010, 347: 279-296.
Wen PY, Lee EQ, Reardon DA, Ligon KL, Alfred Yung WK: Current clinical development of PI3K pathway inhibitors in glioblastoma. Neuro Oncol. 2012, 14: 819-829.
de Groot JF, Fuller G, Kumar AJ, Piao Y, Eterovic K, Ji Y, Conrad CA: Tumor invasion after treatment of glioblastoma with bevacizumab: radiographic and pathologic correlation in humans and mice. Neuro Oncol. 2010, 12: 233-242.
Georgakis GV, Li Y, Rassidakis GZ, Medeiros LJ, Mills GB, Younes A: Inhibition of the phosphatidylinositol-3 kinase/Akt promotes G1 cell cycle arrest and apoptosis in Hodgkin lymphoma. Br J Haematol. 2006, 132: 503-511.
Pelloski CE, Lin E, Zhang L, Yung WK, Colman H, Liu JL, Woo SY, Heimberger AB, Suki D, Prados M, Chang S, Barker FG, Fuller GN, Aldape KD: Prognostic associations of activated mitogen-activated protein kinase and Akt pathways in glioblastoma. Clin Cancer Res. 2006, 12: 3935-3941.
Laplante M, Sabatini DM: mTOR signaling in growth control and disease. Cell. 2012, 149: 274-293.
Das G, Shravage BV, Baehrecke EH: Regulation and function of autophagy during cell survival and cell death. Cold Spring Harb Perspect Biol. 2012, 4:
Albert L, Karsy M, Murali R, Jhanwar-Uniyal M: Inhibition of mTOR Activates the MAPK Pathway in Glioblastoma Multiforme. Cancer Genomics Proteomics. 2009, 6: 255-261.
Akhavan D, Cloughesy TF, Mischel PS: mTOR signaling in glioblastoma: lessons learned from bench to bedside. Neuro Oncol. 2010, 12: 882-889.
Mendiburu-Elicabe M, Gil-Ranedo J, Izquierdo M: Efficacy of rapamycin against glioblastoma cancer stem cells. Clin Transl Oncol. 2014, 16: 495-502.
Opel D, Westhoff MA, Bender A, Braun V, Debatin KM, Fulda S: Phosphatidylinositol 3-kinase inhibition broadly sensitizes glioblastoma cells to death receptor- and drug-induced apoptosis. Cancer Res. 2008, 68: 6271-6280.
Westhoff MA, Kandenwein JA, Karl S, Vellanki SH, Braun V, Eramo A, Antoniadis G, Debatin KM, Fulda S: The pyridinylfuranopyrimidine inhibitor, PI-103, chemosensitizes glioblastoma cells for apoptosis by inhibiting DNA repair. Oncogene. 2009, 28: 3586-3596.
Friedmann-Morvinski D, Bushong EA, Ke E, Soda Y, Marumoto T, Singer O, Ellisman MH, Verma IM: Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science. 2012, 338: 1080-1084.
Ohgaki H, Kleihues P: The definition of primary and secondary glioblastoma. Clin Cancer Res. 2013, 19: 764-772.
Alderton GK: Tumorigenesis: the origins of glioma. Nat Rev Cancer. 2011, 11: 627.
Bagci-Onder T, Wakimoto H, Anderegg M, Cameron C, Shah K: A dual PI3K/mTOR inhibitor, PI-103, cooperates with stem cell-delivered TRAIL in experimental glioma models. Cancer Res. 2010, 71: 154-163.
Zhu Y, Shah K: Multiple lesions in receptor tyrosine kinase pathway determine glioblastoma response to pan-ERBB inhibitor PF-00299804 and PI3K/mTOR dual inhibitor PF-05212384. Cancer Biol Ther. 2014, 15: 815-822.
Blackburn JS, Liu S, Wilder JL, Dobrinski KP, Lobbardi R, Moore FE, Martinez SA, Chen EY, Lee C, Langenau DM: Clonal evolution enhances leukemia-propagating cell frequency in T cell acute lymphoblastic leukemia through Akt/mTORC1 pathway activation. Cancer Cell. 2014, 25: 366-378.
Chen HC, Appeddu PA, Isoda H, Guan JL: Phosphorylation of tyrosine 397 in focal adhesion kinase is required for binding phosphatidylinositol 3-kinase. J Biol Chem. 1996, 271: 26329-26334.
Johnson D, Agochiya M, Samejima K, Earnshaw W, Frame M, Wyke J: Regulation of both apoptosis and cell survival by the v-Src oncoprotein. Cell Death Differ. 2000, 7: 685-696.
Westhoff MA, Bruhl O, Nonnenmacher L, Karpel-Massler G, Debatin KM: Killing me softly-future challenges in apoptosis research. Int J Mol Sci. 2014, 15: 3746-3767.
Gulati N, Karsy M, Albert L, Murali R, Jhanwar-Uniyal M: Involvement of mTORC1 and mTORC2 in regulation of glioblastoma multiforme growth and motility. Int J Oncol. 2009, 35: 731-740.
Giese A: Glioma invasion--pattern of dissemination by mechanisms of invasion and surgical intervention, pattern of gene expression and its regulatory control by tumorsuppressor p53 and proto-oncogene ETS-1. Acta Neurochir Suppl. 2003, 88: 153-162.
Ene CI, Fine HA: Many tumors in one: a daunting therapeutic prospect. Cancer Cell. 2011, 20: 695-697.
Cotterill SJ, Pearson AD, Pritchard J, Kohler JA, Foot AB: Late relapse and prognosis for neuroblastoma patients surviving 5 years or more: a report from the European Neuroblastoma Study Group "Survey". Med Pediatr Oncol. 2001, 36: 235-238.
Maris JM, Hogarty MD, Bagatell R, Cohn SL: Neuroblastoma. Lancet. 2007, 369: 2106-2120.
Chabner BA, Amrein PC, Druker BJ, Michaelson MD, Mitsiades CS, Goss PE, Ryan DP, Ramachandra S, Richardson PG, Supko JG, Wilson WH: Chemotherapy of Neoplastic Diseases. Goodman & Gilman's The Pharmacological Basis of Therapeutics. Edited by: Brunton LL, Lazo JS, Parker KL. 2006, New York: The Graw-Hill Companies, Inc., 315-1403. 11
Shimada H, Stram DO, Chatten J, Joshi VV, Hachitanda Y, Brodeur GM, Lukens JN, Matthay KK, Seeger RC: Identification of subsets of neuroblastomas by combined histopathologic and N-myc analysis. J Natl Cancer Inst. 1995, 87: 1470-1476.
Chesler L, Schlieve C, Goldenberg DD, Kenney A, Kim G, McMillan A, Matthay KK, Rowitch D, Weiss WA: Inhibition of phosphatidylinositol 3-kinase destabilizes Mycn protein and blocks malignant progression in neuroblastoma. Cancer Res. 2006, 66: 8139-8146.
Megison ML, Gillory LA, Beierle EA: Cell survival signaling in neuroblastoma. Anticancer Agents Med Chem. 2013, 13: 563-575.
Lamant L, Pulford K, Bischof D, Morris SW, Mason DY, Delsol G, Mariame B: Expression of the ALK tyrosine kinase gene in neuroblastoma. Am J Pathol. 2000, 156: 1711-1721.
De Brouwer S, De Preter K, Kumps C, Zabrocki P, Porcu M, Westerhout EM, Lakeman A, Vandesompele J, Hoebeeck J, Van Maerken T, De Paepe A, Laureys G, Schulte JH, Schramm A, Van Den Broecke C, Vermeulen J, Van Roy N, Beiske K, Renard M, Noguera R, Delattre O, Janoueix-Lerosey I, Kogner P, Martinsson T, Nakagawara A, Ohira M, Caron H, Eggert A, Cools J, Versteeg R: Meta-analysis of neuroblastomas reveals a skewed ALK mutation spectrum in tumors with MYCN amplification. Clin Cancer Res. 2010, 16: 4353-4362.
Opel D, Poremba C, Simon T, Debatin KM, Fulda S: Activation of Akt predicts poor outcome in neuroblastoma. Cancer Res. 2007, 67: 735-745.
Boller D, Schramm A, Doepfner KT, Shalaby T, von Bueren AO, Eggert A, Grotzer MA, Arcaro A: Targeting the phosphoinositide 3-kinase isoform p110delta impairs growth and survival in neuroblastoma cells. Clin Cancer Res. 2008, 14: 1172-1181.
Li Z, Thiele CJ: Targeting Akt to increase the sensitivity of neuroblastoma to chemotherapy: lessons learned from the brain-derived neurotrophic factor/TrkB signal transduction pathway. Expert Opin Ther Targets. 2007, 11: 1611-1621.
Bender A, Opel D, Naumann I, Kappler R, Friedman L, von Schweinitz D, Debatin KM, Fulda S: PI3K inhibitors prime neuroblastoma cells for chemotherapy by shifting the balance towards pro-apoptotic Bcl-2 proteins and enhanced mitochondrial apoptosis. Oncogene. 2010, 30: 494-503.
Castino R, Bellio N, Follo C, Murphy D, Isidoro C: Inhibition of PI3k class III-dependent autophagy prevents apoptosis and necrosis by oxidative stress in dopaminergic neuroblastoma cells. Toxicol Sci. 2010, 117: 152-162.
Saiki S, Sasazawa Y, Imamichi Y, Kawajiri S, Fujimaki T, Tanida I, Kobayashi H, Sato F, Sato S, Ishikawa K, Imoto M, Hattori N: Caffeine induces apoptosis by enhancement of autophagy via PI3K/Akt/mTOR/p70S6K inhibition. Autophagy. 2011, 7: 176-187.
Nogueira V, Park Y, Chen CC, Xu PZ, Chen ML, Tonic I, Unterman T, Hay N: Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell. 2008, 14: 458-470.
Karpel-Massler G, Schmidt U, Unterberg A, Halatsch ME: Therapeutic inhibition of the epidermal growth factor receptor in high-grade gliomas: where do we stand?. Mol Cancer Res. 2009, 7: 1000-1012.
Weller M, Kaulich K, Hentschel B, Felsberg J, Gramatzki D, Pietsch T, Simon M, Westphal M, Schackert G, Tonn JC, von Deimling A, Davis T, Weiss WA, Loeffler M, Reifenberger G: Assessment and prognostic significance of the epidermal growth factor receptor vIII mutation in glioblastoma patients treated with concurrent and adjuvant temozolomide radiochemotherapy. Int J Cancer. 2014, 134: 2437-2447.
Halatsch ME, Gehrke EE, Vougioukas VI, Botefur IC, A-Borhani F, Efferth T, Gebhart E, Domhof S, Schmidt U, Buchfelder M: Inverse correlation of epidermal growth factor receptor messenger RNA induction and suppression of anchorage-independent growth by OSI-774, an epidermal growth factor receptor tyrosine kinase inhibitor, in glioblastoma multiforme cell lines. J Neurosurg. 2004, 100: 523-533.
Lund-Johansen M, Bjerkvig R, Humphrey PA, Bigner SH, Bigner DD, Laerum OD: Effect of epidermal growth factor on glioma cell growth, migration, and invasion in vitro. Cancer Res. 1990, 50: 6039-6044.
Karpel-Massler G, Westhoff MA, Kast RE, Wirtz CR, Halatsch ME: Erlotinib in glioblastoma: lost in translation?. Anticancer Agents Med Chem. 2011, 11: 748-755.
Peereboom DM, Shepard DR, Ahluwalia MS, Brewer CJ, Agarwal N, Stevens GH, Suh JH, Toms SA, Vogelbaum MA, Weil RJ, Elson P, Barnett GH: Phase II trial of erlotinib with temozolomide and radiation in patients with newly diagnosed glioblastoma multiforme. J Neurooncol. 2010, 98: 93-99.
van den Bent MJ, Brandes AA, Rampling R, Kouwenhoven MC, Kros JM, Carpentier AF, Clement PM, Frenay M, Campone M, Baurain JF, Armand JP, Taphoorn MJ, Tosoni A, Kletzl H, Klughammer B, Lacombe D, Gorlia T: Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. J Clin Oncol. 2009, 27: 1268-1274.
Fan QW, Cheng CK, Nicolaides TP, Hackett CS, Knight ZA, Shokat KM, Weiss WA: A dual phosphoinositide-3-kinase alpha/mTOR inhibitor cooperates with blockade of epidermal growth factor receptor in PTEN-mutant glioma. Cancer Res. 2007, 67: 7960-7965.
Wang MY, Lu KV, Zhu S, Dia EQ, Vivanco I, Shackleford GM, Cavenee WK, Mellinghoff IK, Cloughesy TF, Sawyers CL, Mischel PS: Mammalian target of rapamycin inhibition promotes response to epidermal growth factor receptor kinase inhibitors in PTEN-deficient and PTEN-intact glioblastoma cells. Cancer Res. 2006, 66: 7864-7869.
Kreisl TN, Lassman AB, Mischel PS, Rosen N, Scher HI, Teruya-Feldstein J, Shaffer D, Lis E, Abrey LE: A pilot study of everolimus and gefitinib in the treatment of recurrent glioblastoma (GBM). J Neurooncol. 2009, 92: 99-105.
Carpenter EL, Mosse YP: Targeting ALK in neuroblastoma–preclinical and clinical advancements. Nat Rev Clin Oncol. 2012, 9: 391-399.
Berry T, Luther W, Bhatnagar N, Jamin Y, Poon E, Sanda T, Pei D, Sharma B, Vetharoy WR, Hallsworth A, Ahmad Z, Barker K, Moreau L, Webber H, Wang W, Liu Q, Perez-Atayde A, Rodig S, Cheung NK, Raynaud F, Hallberg B, Robinson SP, Gray NS, Pearson AD, Eccles SA, Chesler L, George RE: The ALK(F1174L) mutation potentiates the oncogenic activity of MYCN in neuroblastoma. Cancer Cell. 2012, 22: 117-130.
von Bubnoff N, Schneller F, Peschel C, Duyster J: BCR-ABL gene mutations in relation to clinical resistance of Philadelphia-chromosome-positive leukaemia to STI571: a prospective study. Lancet. 2002, 359: 487-491.
Sumazin P, Yang X, Chiu HS, Chung WJ, Iyer A, Llobet-Navas D, Rajbhandari P, Bansal M, Guarnieri P, Silva J, Califano A: An extensive microRNA-mediated network of RNA-RNA interactions regulates established oncogenic pathways in glioblastoma. Cell. 2011, 147: 370-381.
Yan X, Kennedy CR, Tilkens SB, Wiedemeier O, Guan H, Park JI, Chan AM: Cooperative Cross-Talk between Neuroblastoma Subtypes Confers Resistance to Anaplastic Lymphoma Kinase Inhibition. Genes Cancer. 2011, 2: 538-549.
He J, Gu L, Zhang H, Zhou M: Crosstalk between MYCN and MDM2-p53 signal pathways regulates tumor cell growth and apoptosis in neuroblastoma. Cell Cycle. 2011, 10: 2994-3002.
Tomlinson I, Sasieni P, Bodmer W: How many mutations in a cancer?. Am J Pathol. 2002, 160: 755-758.
Karamouzis MV, Konstantinopoulos PA, Papavassiliou AG: Targeting MET as a strategy to overcome crosstalk-related resistance to EGFR inhibitors. Lancet Oncol. 2009, 10: 709-717.
Karpel-Massler G, Westhoff MA, Zhou S, Nonnenmacher L, Dwucet A, Kast RE, Bachem MG, Wirtz CR, Debatin KM, Halatsch ME: Combined inhibition of HER1/EGFR and RAC1 results in a synergistic antiproliferative effect on established and primary cultured human glioblastoma cells. Mol Cancer Ther. 2013, 12: 1783-1795.
Kast RE, Boockvar JA, Bruning A, Cappello F, Chang WW, Cvek B, Dou QP, Duenas-Gonzalez A, Efferth T, Focosi D, Ghaffari SH, Karpel-Massler G, Ketola K, Khoshnevisan A, Keizman D, Magne N, Marosi C, McDonald K, Munoz M, Paranjpe A, Pourgholami MH, Sardi I, Sella A, Srivenugopal KS, Tuccori M, Wang W, Wirtz CR, Halatsch ME: A conceptually new treatment approach for relapsed glioblastoma: coordinated undermining of survival paths with nine repurposed drugs (CUSP9) by the International Initiative for Accelerated Improvement of Glioblastoma Care. Oncotarget. 2013, 4: 502-530.
Nonnenmacher L, Westhoff MA, Fulda S, Karpel-Massler G, Halatsch ME, Engelke J, Simmet T, Gorbacioglu S, Debatin KM: RIST: A potent new combination therapy for glioblastoma. Int J Cancer. doi:10.1002/ijc.29138
Ho R, Minturn JE, Hishiki T, Zhao H, Wang Q, Cnaan A, Maris J, Evans AE, Brodeur GM: Proliferation of human neuroblastomas mediated by the epidermal growth factor receptor. Cancer Res. 2005, 65: 9868-9875.
Rixe O, Fojo T: Is cell death a critical end point for anticancer therapies or is cytostasis sufficient?. Clin Cancer Res. 2007, 13: 7280-7287.
Mitchison TJ: The proliferation rate paradox in antimitotic chemotherapy. Mol Biol Cell. 2012, 23: 1-6.
Roth W, Wagenknecht B, Dichgans J, Weller M: Interferon-alpha enhances CD95L-induced apoptosis of human malignant glioma cells. J Neuroimmunol. 1998, 87: 121-129.
Fulda S, Debatin KM: Sensitization for tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by the chemopreventive agent resveratrol. Cancer Res. 2004, 64: 337-346.
Pasupuleti N, Leon L, Carraway KL, Gorin F: 5-Benzylglycinyl-amiloride kills proliferating and nonproliferating malignant glioma cells through caspase-independent necroptosis mediated by apoptosis-inducing factor. J Pharmacol Exp Ther. 2013, 344: 600-615.
Akinleye A, Avvaru P, Furqan M, Song Y, Liu D: Phosphatidylinositol 3-kinase (PI3K) inhibitors as cancer therapeutics. J Hematol Oncol. 2013, 6: 88.
Yin Y, Shen WH: PTEN: a new guardian of the genome. Oncogene. 2008, 27: 5443-5453.
Lee MJ, Ye AS, Gardino AK, Heijink AM, Sorger PK, MacBeath G, Yaffe MB: Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell. 2012, 149: 780-794.
Kim M, Lee KH, Yoon SW, Kim BS, Chun J, Yi H: Analytical tools and databases for metagenomics in the next-generation sequencing era. Genomics Inform. 2013, 11: 102-113.
Lee SH, Sim SH, Kim JY, Cha S, Song A: Application of Cancer Genomics to Solve Unmet Clinical Needs. Genomics Inform. 2013, 11: 174-179.
van Oostrum J, Calonder C, Rechsteiner D, Ehrat M, Mestan J, Fabbro D, Voshol H: Tracing pathway activities with kinase inhibitors and reverse phase protein arrays. Proteomics Clin Appl. 2009, 3: 412-422.
Carragher NO, Unciti-Broceta A, Cameron DA: Advancing cancer drug discovery towards more agile development of targeted combination therapies. Future Med Chem. 2012, 4: 87-105.
Acknowledgments
The authors wish to thank Bianca Welz, Angelika Vollmer and Sara Barry for their critical reading of early versions of the manuscript and Nicolas Marschall for his help in preparing its final version.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MAW and KMD conceived the review, MAW and GKM wrote the final version, OB and KLFB conceived and designed the illustrations, LN, SE and MDS helped drafting the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Westhoff, MA., Karpel-Massler, G., Brühl, O. et al. A critical evaluation of PI3K inhibition in Glioblastoma and Neuroblastoma therapy. Mol and Cell Ther 2, 32 (2014). https://doi.org/10.1186/2052-8426-2-32
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2052-8426-2-32