

Abstract
Background/Aims: Non-small cell lung cancer (NSCLC) is the leading cause of death worldwide. Although aquaporin-3 (AQP3) is widely distributed in mammalian tissues and over-expressed in NSCLC cells, there are limited studies on the effects of AQP3 knockdown on NSCLC cells under hypoxic conditions. Methods: The CCK-8 assay was used to calculate cell viability. Scratch-wound healing and transwell assays were used to detect cell migration and invasion. Apoptotic cells were measured by the TUNEL assay. mRNA expression levels were calculated via quantitative RT-PCR. Relative protein levels were determined by immunoblot assays. Results: AQP3 knockdown substantially reduced proliferation, migration, and invasion of A549 and NCI-H460 cells under hypoxic conditions. Moreover, AQP3 knockdown clearly induced cell apoptosis. Further analysis identified levels of HIF-1α, VEGF, Raf, phosphor-MEK, and phosphor-ERK, whose activities were significantly attenuated in the AQP3 knockdown group. Conclusions: These findings indicate that AQP3 knockdown retards the growth of NSCLC cells partially through inhibiting HIF-1α/VEGF and Raf/MEK/ERK signalling pathways.
Introduction
Lung cancer is a common and lethal malignancy, and it accounts for 1.59 million deaths annually. With the development of antitumour drugs, one-year all-stage survival has been increasing. However, five year survival has remained unchanged. The incidence of non-small cell lung cancer (NSCLC) accounts for approximately 85% of all lung cancers [1]. Therefore, clarifying the pathogenesis and finding new therapeutic targets is crucial.
Aquaporins (AQPs) are small, intrinsic membrane proteins that facilitate the transport of water across cell plasma membranes and are involved in numerous physiological as well as pathophysiological processes [2]. Currently, 13 isoforms of mammalian AQPs have been identified in various tissues [3,4,5]. AQPs are notably abundant in aggressive tumours [6,7,8] and their expression is closely correlated with the severity of histological tumours. Recent evidence has shown that AQPs play an essential role in tumour cell proliferation, migration, and even tumour angiogenesis [9,10]. The AQP AQP3 is widely distributed in mammalian tissues, especially in human neoplastic tissues, and is markedly dysregulated in breast cancer [11], colorectal carcinoma [12] and lung cancer [13]. Recently, several studies have shown that AQP3 plays a critical role in tumour growth, cell proliferation, cell invasion [9,14,15], and other cellular processes in various cancers. Shen et al. [16] have documented that AQP3 regulates proliferation, invasion and even migration in gastric carcinoma cells by disrupting E-cadherin activation. A recent study has demonstrated that an AQP3 knockdown suppresses tumour growth and angiogenesis in experimental NSCLC cells under normoxic conditions [17].
Hypoxia is a common phenomenon that occurs in most human tumours and has been shown to be associated with proliferation, invasion, and migration [18,19,20]. However, the effects of AQP3 on NSCLC cells under hypoxic conditions have not been studied. Therefore, this study sought to explore the effects of AQP3 and determine the underlying mechanisms in the effects of AQP3 knockdown on NSCLC cells under hypoxic conditions.
Materials and Methods
Cell Culture
The NSCLC A549 (adenocarcinoma) and NCI-H460 (large cell carcinoma) cell lines were from the American Type Culture Collection (ATCC) and were cultured in RPMI 1640 supplemented with 10% (v/v) foetal bovine serum. Briefly, exponentially growing cells were maintained at 37°C in a 5% CO2/95% air incubator. For hypoxic exposure, cells were cultured in a Concept 400 anaerobic incubator (Ruskinn Technology Ltd., Bridgend, UK, E.U.) under 1% O2/5% CO2/balanced N2 and incubated at 37°C.
RNA interference
To knock down the AQP3 gene, cells were starved for 12 h to normalize any group differences before treating the cells with small interfering RNA (siRNA) for 24 h. Then, A549 and NCI-H460 cells were transfected with the siRNA, which was designed and synthesized by Invitrogen. Non-targeted control siRNA (siRNA-NC) was used as a negative control. The siRNAs duplexes targeted AQP3 (siRNA-1, siRNA-2, and siRNA-3) and the non-targeted control siRNA were as follows: AQP3 siRNA-1: (sense) 5'-CCU CUG GAC ACU UGGAUAUTT-3', (antisense) 5'-AUAUCC AAG UGU CCAG AGGTT-3'; siRNA-2: (sense) 5'-CUGUAU UAU GAU UGG AUAU TT-3', (antisense) 5'-AGC CUAAUACUU CCA GAG GTT-3'; siRNA-3: (sense) 5'-GGC UGU AUU AUG AUG CAA UTT-3', (antisense) 5'-AUU GCA UCA UAA UAC AGC CTT-3'; non-targeted siRNA: (sense) 5'-UUC UCC GAA CGU GUC ACGU-3', (antisense) 5'-ACG UGA CAC GUU CGG AGAA-3'. Briefly, cells grew to 80% confluence, and then 1 µg of siRNA and 10 µl of the X-tremeGene siRNA Transfection Reagent was mixed with serum-free Opti-MEM medium. The mixture was incubated at room temperature for 20 min and then was added directly onto the cells; this was followed by a 24 h incubation at 37°C.
Cell Counting Kit-8
The cell viability was calculated using the Cell Counting Kit-8 (CCK-8) (Beyotime Biotechnology, Jiangsu, China) according to the manufacturer's protocol. Briefly, the A549 and NCI-H460 cells were seeded on 96-well plates in RPMI 1640 with 10% foetal bovine serum for 24 h. Then, the cells were transfected at 80% confluence using siRNAs against AQP3 for 24 h. Finally, the viable cells were detected using the Cell Counting Kit-8, where the absorbance for each sample was calculated at 450 nm using a microplate reader (TECAN, Salzburg, Austria).
Scratch-Wound assay
The confluent A549 and NCI-H460 cells cultured in 6-well plates were scratched by pipette tips, which led to a 1-mm-wide lane per well, and the remaining cells were washed with PBS. Then, the cells were treated with 10% FBS in RPMI 1640 containing siRNA. Wounded areas were measured at time point zero and after the 24 h treatment; photographs were taken of the same areas recorded at time point zero.
Cell migration and invasion analysis
For the migration assay, cells were transfected with siRNA and incubated for 24 h, and then resuspended cells were placed in the top layer of the transwell chamber with 8 µm pores (Millipore). The lower layer of the chamber contained 10% FBS as a chemoattractant. The chambers were maintained at 37°C in 5% CO2 for 24 h. Non-migrating cells on the top of the membrane were removed with cotton swabs. Cells that had migrated to the bottom were fixed with 95% ethanol, stained with 0.2% crystal violet (Sigma), photographed, and counted. Each experiment was performed in triplicate (magnification, ×200). Matrigel-coated transwell inserts (Millipore) were used in the cell invasion assay. After a 24 h treatment, cells that invaded the lower layer of the transwell membrane were fixed, stained, and counted (magnification, × 200).
TUNEL assay
The DNA fragmentation in A549 and NCI-H460 cells was measured by using a commercially available kit (Cell Death Detection Kit, Roche Biochemicals, Mannheim, Germany) as described previously [21]. Briefly, the cells were fixed with 4% paraformaldehyde at room temperature and cleaned with phosphate-buffered saline (PBS), and then permeabilized with 1% Triton X-100 at 4°C. Then, each slide was treated with the TdT-labelled nucleotide mix and maintained at 37°C for 1 h in the dark. Finally, slides were rinsed and then counterstained with 10 mg/ml 4,6-diamidino-2-phenylindole (DAPI) at 37°C. The fluorescence staining was viewed with laser scanning confocal microscopy (FV300, Olympus, Japan). TUNEL positive cells were counted in five fields (200 × magnification).
Immunoblot Analysis
Total protein extracts were isolated from A549 and NCI-H460 cells. The protein extracts were separated with 10% SDS-PAGE and then blotted onto a nitrocellulose membrane, then blocked with 5% non-fat milk at room temperature for 1 h. Then, the membranes were probed with primary antibodies for HIF-1α, Raf, total-MEK and total-ERK, phosphor-MEK and phosphor-ERK (Cell Signalling Technology, Danvers, MA, USA), AQP3, tubulin, VEGF (Abcam Inc., Cambridge, USA) and GAPDH (Kangcheng Bio-tech Inc., Shanghai, China), and incubated overnight at 4°C. Membranes were rinsed three times for 10 min with PBS containing 0.5% Tween-20, and incubated with a secondary antibody (Alexa Fluor, Molecular Probes, Eugene, OR, USA) for 1 h at room temperature. Western blot bands were scanned by Imaging System (LI-COR Biosciences, Lincoln, NE, USA) and detected by Odyssey v3.0 software in each group and normalized to GAPDH as an internal control.
RNA isolation and quantitative reverse transcription-PCR (qRT-PCR)
qRT-PCR was performed to measure the mRNA expression of HIF-1α and VEGF in A549 and NCI-H460 cells. Briefly, total RNA was isolated from cell lines using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. Total RNA (1 µg) was reverse-transcribed in a 20 µl volume at 37°C for 15 min and 98°C for 5 min using ReverTra Ace qPCR RT Kit (TOYOBO CO., LTD, Osaka, Japan). The PCR reactions were performed with real-time PCR Master Mix. The cDNA sample quality was assessed using an ABI PRISM 7500HT unit (Applied Biosystems, CA, USA). The cycle threshold (CT) fluorescence values were calculated using the SDS 2.4 software (Applied Biosystems). The relative expression of VEGF and HIF-1α were normalized to GAPDH using the 2-ΔΔCt method. All primers (Invitrogen) were as follows: VEGF sense primer: 5'- TAC CTC CAC CAT GCC AAGTG-3', antisense primer: 5'-TGATGATTC DTG CCC TCC TCC-3'; HIF-1α sense primer: 5'-CTG AGG TTG GTT ACT GTT GGT ATC-3', antisense primer: 5'-AGT GTA CCC TAC TAG CCG AGGA-3'; GAPDH sense primer: 5'-AAG AAG GTG GTG AAG CAGGC-3', antisense primer: 5'-TCC ACC ACC CAG TTG CTGTA-3'.
Statistical analysis
The values are expressed as the mean ± SEM. One-way ANOVA was used for statistical analyses followed by Bonferroni's test using a two-tailed p < 0.05 as statistically significant.
Results
Aquaporin 3 (AQP3) knockdown reduced A549 and NCI-H460 cell proliferation
To determine whether AQP3 was involved in the proliferation of NSCLC cells, A549 and NCI-H460 cells were transfected with specific siRNA targeting AQP3. The protein expression of endogenous AQP3 was markedly reduced by siRNA-3 compared with siRNA-NC in A549 cells (Fig. 1A). Similar effects were also identified in NCI-H460 cells (Fig. 1B). Based on the above results, siRNA-3 was selected for the following experiment. As shown in Fig. 2, siRNA-3 against AQP3 substantially reduced cell proliferation under hypoxic conditions in both cell lines (Fig. 2). This result demonstrated that AQP3 plays a role in proliferation under hypoxic conditions.
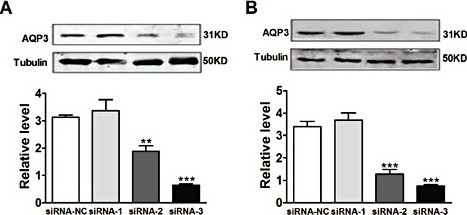
Effect of siRNA knockdown of Aquaporin 3 (AQP3) on protein expression of AQP3. The A549 and NCI-H460 cells were cultured and transfected with AQP3 siRNA-1 siRNA-2 or siRNA-3. Twenty-four hours after transfection, the protein expression of AQP3 was detected in A549 cells (A) and NCI-H460 cells (B). All of the values are denoted as the mean±SEM; **p < 0.01 or ***p < 0.001 vs. siRNA-NC, n = 3 independent experiments.
Effect of siRNA knockdown of Aquaporin 3 (AQP3) on protein expression of AQP3. The A549 and NCI-H460 cells were cultured and transfected with AQP3 siRNA-1 siRNA-2 or siRNA-3. Twenty-four hours after transfection, the protein expression of AQP3 was detected in A549 cells (A) and NCI-H460 cells (B). All of the values are denoted as the mean±SEM; **p < 0.01 or ***p < 0.001 vs. siRNA-NC, n = 3 independent experiments.

Effect of AQP3 on hypoxia-induced proliferation in A549 and NCI-H460 cells. The A549 and NCI-H460 cells were cultured and transfected with AQP3 siRNA or non-targeted control siRNA. Twenty-four hours later, cell proliferation was determined by CCK-8 in A549 cells (A) and NCI-H460 cells (B). All of the values are denoted as the mean±SEM; **p < 0.01 or ***p < 0.001 vs. siRNA-NC of normoxia, ##p < 0.01 or ###p < 0.001 vs. siRNA-NC of hypoxia, n = 6 independent experiments.
Effect of AQP3 on hypoxia-induced proliferation in A549 and NCI-H460 cells. The A549 and NCI-H460 cells were cultured and transfected with AQP3 siRNA or non-targeted control siRNA. Twenty-four hours later, cell proliferation was determined by CCK-8 in A549 cells (A) and NCI-H460 cells (B). All of the values are denoted as the mean±SEM; **p < 0.01 or ***p < 0.001 vs. siRNA-NC of normoxia, ##p < 0.01 or ###p < 0.001 vs. siRNA-NC of hypoxia, n = 6 independent experiments.
AQP3 downregulation retarded A549 and NCI-H460 cell migration
To further determine whether cell migration was regulated by AQP3, a scratch-wound assay and a transwell migration assay were conducted. The A549 cells transfected with AQP3 siRNA exhibited a significant decrease in cell migration after a 24 h incubation (Fig. 3B and F). The representative images are shown in Fig. 3A and E. Similarly, in the NCI-H460 cells treated with AQP3 siRNA, cellular migration was reversed nearly to normal levels (Fig. 3D and H). The photographs are shown in Fig. 3C and G. These results indicated that a significant down regulation of cellular migration was correlated with the reduced expression of AQP3 in A549 and NCI-H460 cells exposed to hypoxia.

Effect of AQP3 on hypoxia-mediated migration in A549 and NCI-H460 cells. Images showing migration changes after treatment with the AQP3 siRNA in scratch-wound and transwell migration assays for A549 (A, E) and NCI-H460 cells (C, G). Migration promotion of endogenous AQP3 was dominantly blocked by siRNA in A549 (B, F) and NCI-H460 cells (D, H). All values are denoted as the mean ± SEM; ** p < 0.01 or *** p < 0.001 vs. siRNA-NC of normoxia, ## p < 0.01 or ### p < 0.001 vs. siRNA-NC of hypoxia, n = 6 independent experiments.
Effect of AQP3 on hypoxia-mediated migration in A549 and NCI-H460 cells. Images showing migration changes after treatment with the AQP3 siRNA in scratch-wound and transwell migration assays for A549 (A, E) and NCI-H460 cells (C, G). Migration promotion of endogenous AQP3 was dominantly blocked by siRNA in A549 (B, F) and NCI-H460 cells (D, H). All values are denoted as the mean ± SEM; ** p < 0.01 or *** p < 0.001 vs. siRNA-NC of normoxia, ## p < 0.01 or ### p < 0.001 vs. siRNA-NC of hypoxia, n = 6 independent experiments.
Endogenous AQP3 knockdown retarded cell invasion and facilitated apoptosis
The transwell assay verified that AQP3 knockdown inhibited cell invasion. Representative images are shown in Fig. 4A and B, respectively. The proportions of invasive A549 cells were dramatically decreased (59.92%) after treatment with the AQP3 siRNA (Fig. 4C). Similarly, the invasive proportions of the NCI-H460 cells were reduced to 38.32% after AQP3 knockdown (Fig. 4D). The apoptotic cells were measured with the TUNEL assay. The A549 and NCI-H460 cells were cultured and transfected with AQP3 siRNA or non-targeted control siRNA. Twenty-four hours after transfection, high TUNEL positive cells were present in AQP3 siRNA treated A549 and NCI-H460 cells. The representative images are shown in Fig. 4E and F and the quantified results of cell apoptosis are shown in Fig. 4G and H.
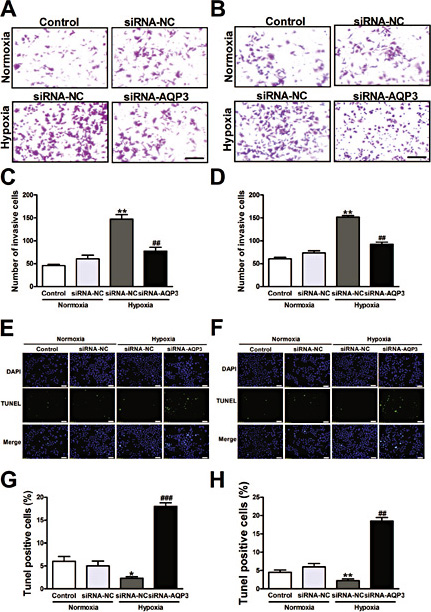
Effect of AQP3 knockdown on invasion and apoptosis in A549 and NCI-H460 cells. Photographs represent the cells passing through the Matrigel in an invasion assay in A549 (A) and NCI-H460 cells (B). The histogram shows that AQP3 knockdown substantially reduced cell invasion after transfection with the AQP3 siRNA for 24 h in A549 (C) and NCI-H460 cells (D). The TUNEL assay studies show that AQP3 knock-down induced cells apoptosis. The representative images are shown for A549 (E) and NCI-H460 cells (F). Quantitative analysis was performed to document that AQP3 knockdown significantly enhanced the number of apoptotic nuclei in A549 (G) and NCI-H460 cells (H). All of the values are denoted as the mean±SEM; * p < 0.05 or ** p < 0.01 vs. siRNA-NC of normoxia, ## p < 0.01 or ### p < 0.001 vs. siRNA-NC of hypoxia, experiments were repeated six times.
Effect of AQP3 knockdown on invasion and apoptosis in A549 and NCI-H460 cells. Photographs represent the cells passing through the Matrigel in an invasion assay in A549 (A) and NCI-H460 cells (B). The histogram shows that AQP3 knockdown substantially reduced cell invasion after transfection with the AQP3 siRNA for 24 h in A549 (C) and NCI-H460 cells (D). The TUNEL assay studies show that AQP3 knock-down induced cells apoptosis. The representative images are shown for A549 (E) and NCI-H460 cells (F). Quantitative analysis was performed to document that AQP3 knockdown significantly enhanced the number of apoptotic nuclei in A549 (G) and NCI-H460 cells (H). All of the values are denoted as the mean±SEM; * p < 0.05 or ** p < 0.01 vs. siRNA-NC of normoxia, ## p < 0.01 or ### p < 0.001 vs. siRNA-NC of hypoxia, experiments were repeated six times.
AQP3 knockdown inhibited the mRNA and protein levels of HIF-1α and VEGF
To validate the roles of HIF-1α and VEGF in AQP3-mediated antiproliferation, western blotting and qRT-PCR were performed. The mRNA and levels of HIF-1α and VEGF were measured in A549 and NCI-H460 cells transfected with AQP3 siRNA. The results showed that the mRNA level of HIF-1α was substantially decreased by AQP3 knockdown (Fig. 5A and C). Similarly, AQP3 knockdown clearly reduced hypoxic conditions and mediated the protein upregulation of HIF-1α in A549 and NCI-H460 cells (Fig. 5B and D). To further investigate whether VEGF is involved in AQP3-mediated cellular effects, we detected mRNA and protein expression of VEGF after AQP3 siRNA transfection. The results showed that AQP3 knockdown substantially decreased the mRNA and protein expression of VEGF in A549 and NCI-H460 cells, respectively (Fig. 5E, F and G, H).

Effect of AQP3 knockdown on mRNA and levels of HIF-1α and VEGF. Quantitative real-time PCR was performed to verify that AQP3 knockdown substantially reduced the mRNA levels of HIF-1α and VEGF in A549 (A, E) and NCI-H460 cells (C, G). Moreover, the corresponding levels of HIF-1α and VEGF was detected by western blot. Protein levels of HIF-1α and VEGF were significantly decreased in A549 (B, F) and NCI-H460 cells (D, H). All of the values are denoted as the mean±SEM; ** p < 0.01 vs. siRNA-NC of normoxia, ## p < 0.01 vs. siRNA-NC of hypoxia, experiments were repeated six times.
Effect of AQP3 knockdown on mRNA and levels of HIF-1α and VEGF. Quantitative real-time PCR was performed to verify that AQP3 knockdown substantially reduced the mRNA levels of HIF-1α and VEGF in A549 (A, E) and NCI-H460 cells (C, G). Moreover, the corresponding levels of HIF-1α and VEGF was detected by western blot. Protein levels of HIF-1α and VEGF were significantly decreased in A549 (B, F) and NCI-H460 cells (D, H). All of the values are denoted as the mean±SEM; ** p < 0.01 vs. siRNA-NC of normoxia, ## p < 0.01 vs. siRNA-NC of hypoxia, experiments were repeated six times.
Downregulation of AQP3 suppressed levels of Raf and phospho-MEK (p-MEK) and phospho-ERK (p-ERK)
To confirm the regulatory effects of AQP3 on Raf, p-MEK, and p-ERK in A549 and NCI-H460 cells, changes in Raf, p-MEK, and p-ERK protein levels following transfection with AQP3 were measured. After transfection with the AQP3 siRNA, the protein levels of Raf were decreased in the A549 cells (Fig. 6A). Similar results were also identified in the NCI-H460 cells (Fig. 6B). In addition, the levels of both p-MEK and p-ERK were reduced after transfection with the AQP3 siRNA in the A549 (Fig. 6D and H) and NCI-H460 cells (Fig. 6F and J). However, the total levels of MEK and ERK did not change between different groups in the A549 (Fig. 6C and G) and NCI-H460 cells (Fig. 6E and I). These results suggested that the effects of AQP3 knockdown are at least partly the result of the regulation of the Raf/MEK/ERK pathway.

Effect of AQP3 knockdown on levels of Raf, phospho-MEK (p-MEK) and phopho-ERK (p-ERK). The levels of Raf, Total-MEK (T-MEK), p-MEK, Total-ERK (T-ERK), and p-ERK were measured in western blotting assays. The results showed that AQP3 knockdown significantly inhibited levels of Raf, p-MEK and p-ERK in A549 cells (A, D and H) and NCI-H460 cells (B, F and J), respectively In addition, figures C, G (A549 cells) and E, 1 (NCI-H460 cells) show that protein levels of T-MEK, T-ERK had no noticeable changes.. All of the values are denoted as the mean±SEM; ** p < 0.01 vs. siRNA-NC of normoxia, ## p < 0.01 vs. siRNA-NC of hypoxia; experiments were repeated three times.
Effect of AQP3 knockdown on levels of Raf, phospho-MEK (p-MEK) and phopho-ERK (p-ERK). The levels of Raf, Total-MEK (T-MEK), p-MEK, Total-ERK (T-ERK), and p-ERK were measured in western blotting assays. The results showed that AQP3 knockdown significantly inhibited levels of Raf, p-MEK and p-ERK in A549 cells (A, D and H) and NCI-H460 cells (B, F and J), respectively In addition, figures C, G (A549 cells) and E, 1 (NCI-H460 cells) show that protein levels of T-MEK, T-ERK had no noticeable changes.. All of the values are denoted as the mean±SEM; ** p < 0.01 vs. siRNA-NC of normoxia, ## p < 0.01 vs. siRNA-NC of hypoxia; experiments were repeated three times.
Discussion
Lung cancer is one of the most prevalent malignancies worldwide. One class of lung cancer, NSCLCs, is relatively insensitive to chemotherapy and radiation compared with small cell carcinoma [22,23]. Thus, clarifying the carcinogenic mechanisms and the potential treatment targets are critical. Hypoxia is a common phenomenon occurring in most human tumours, including NSCLCs. It has been proven to play a pivotal role in tumour progression and chemo- or radio-resistance [24]. However, the regulatory mechanisms involved in hypoxia still remain largely unknown.
The aquaporins (AQPs), a large family of ‘‘water channels'', are conserved throughout animals, lower organisms, and plants. Recently, AQPs have been recognized as potential modulators in therapy for some types of refractory edema, brain swelling, neuroinflammation, pain, obesity, and even cancer [25]. Recent evidence has suggested that AQPs are involved in the regulation of cancer cell proliferation, migration and invasion [11,17,26,27,28].
AQP3 belongs to the family of ‘‘water channels'', and it often participates in the regulation of various cellular processes in cancer. Recently, in urothelial bladder cancer patients, Denzinger et al. [29] have indicated that 59% of patients exhibit AQP3-positive tumours. In hepatocellular carcinoma (HCC) patients, Gao et al. [30] have found that HCC patients expressing high levels of AQP3 protein have poorer 5-year disease-free survival and 5-year overall survival. Although multiple studies support the role of AQP3 in cell proliferation, several studies have indicated contrasting effects. Choudhary et al. [31] have indicated that re-expression of AQP3 has no effect on the expression of the proliferation makers keratin 5 and cyclin D1. Matsuo et al. [32] have suggested that decreased AQP3 expression is related to a more aggressive tumour behaviour. Recently, Kim et al. [33] have described that reduced AQP3 inhibits expression of phosphorylated PI3K, E-cadherin, β-catenin and decreases cell survival. Thus, the mechanisms of AQP3 remain to be uncovered. Recently, Katoh et al. [13] have identified AQP3 expression in 100% of their tested samples from pituitary adenomas, thymomas, thymic carcinomas, and pulmonary adenocarcinomas. In pulmonary adenocarcinomas, AQP3 has been detected in all the tumours within the bronchioloalveolar subtype. In contrast, AQP3 is scarcely detected in small cell carcinoma, pleomorphic carcinoma, or metastatic colon adenocarcinoma [34]. This study, for the first time, investigated the role of AQP3 in proliferation, migration, and invasion, and determined potential mechanisms of the AQP3 in A549 and NCl-H460 cell lines under hypoxic conditions. The results showed that AQP3 knockdown substantially reduced proliferation, migration and invasion in both the A549 and NCI-H460 cells (Fig. 2, 3 and 4). The mechanisms underlying the regulation of A549 and NCI-H460 cell proliferation, migration and invasion by AQP3 are often attributed to targeting important factors or key pathways. HIF-1α is considered to be the master transcriptional regulator of cellular, developmental and pathological responses to hypoxia [35]. In most situations, HIF-1α is rapidly degraded under normoxic conditions, but its level accumulates under hypoxic conditions. Recently, Wu et al. [36] manifested that high HIF-1α expressions had a significantly higher chance to be non-responder to chemotherapy in NSCLC patients. Therefore, we speculated that HIF-1α might be involved in AQP3 mediated cellular effects. In this study, we identified that AQP3 knockdown markedly reduced the expression of HIF-1α (Fig. 5). Notably, HIF-1α binds to the VEGF promoter and enhances its transcription [37], which is closely associated with proliferation, migration, invasion, angiogenesis and poor prognosis in cancer [38,39,40,41]. As expected, the results showed that AQP3 knockdown inhibited VEGF protein levels (Fig. 5F and H). Aberrantly expressed HIF-1α often participates in the regulation of lung cancer cells survival/death [42]. A recent report has shown that during hypoxic conditions, the Raf-MEK-ERK signalling pathway is activated. The activation of this pathway leads to angiogenesis and uncontrolled growth of cells [43]. Thus, it is conceivable that increased levels of Raf-MEK-ERK may be induced by the loss of AQP3 expression in tumour cells. In the present study, after introducing an AQP3 knockdown, the level of Raf was clearly inhibited. In addition, p-MEK and p-ERK were both downregulated with no significant changes in total-MEK and total-ERK (Fig. 6).
Though AQP3 downregulation exerts anticancer properties, more in vivo studies and related molecular mechanisms are need to further investigate and determine the molecular mechanisms of AQP3 in NSCLCs and other types of cancers. Moreover, the effect of AQP3 on migration closely parallels on proliferation, while the current data do not allow ruling out interdependence between the increase in the number of cells and in migration. Therefore, it is still need to further define the effect of AQP3 on migration and proliferation in NSCLCs in future. As mentioned above, AQP3 knockdown inhibited the growth of NSCLC cells under hypoxic conditions, which was associated with the HIF-1α/VEGF and Raf-MEK-ERK pathways. Our findings provide insight about new regulatory mechanisms involved in hypoxia and evidence for the participation of AQP3 in NSCLCs and as a therapeutic target for NSCLCs.
Abbreviations
Non-small-cell lung cancer (NSCLC); Aquaporin-3 (AQP3); Hypoxia-inducible Factor 1α (HIF-1α); Vascular Endothelial Growth Factor (VEGF).
Acknowledgements
This work was supported by the Heilongjiang Postdoctoral Financial Assistance (LBH-Z12170) and the Health and Family Planning Commission of Heilongjiang Province (2011-119).
Disclosure Statement
None.
References
S.-Y. Hou and Y.-P. Li contributed equally to this work.

