Abstract
We investigated the performance of several carbon-supported RuxPty electrocatalysts for their alkaline hydrogen oxidation and oxygen reduction performance in the presence of carbonate and compared their performance with monometallic, carbon-supported Pt. Our results indicate a strong dependence of HOR upon pH for the monometallic Pt catalysts (22 mV/pH) and a weak dependence upon pH for the Ru-containing electrocatalysts (3.7, 2.5, and 4.7 mV/pH on Ru0.2Pt0.8, Ru0.4Pt0.6, and Ru0.8Pt0.2, respectively). These results are consistent with our previous findings that illustrate a change in rds from electron transfer (on monometallic Pt) to dissociative hydrogen adsorption (on RuxPty catalysts). Analysis of the kinetic currents to determine the rate-determining step via Tafel slope analysis provides additional data supporting this conclusion. There is no difference in the performance at comparable pH values in the presence or absence of carbonate on monometallic Pt indicating that water/hydroxide is the primary proton acceptor for alkaline HOR in 0.1 M KOH aqueous electrolyte. Finally, we observe no pH or carbonate dependence for the ORR on monometallic Pt.
Export citation and abstract BibTeX RIS

This is an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial No Derivatives 4.0 License (CC BY-NC-ND, http://creativecommons.org/licenses/by-nc-nd/4.0/), which permits non-commercial reuse, distribution, and reproduction in any medium, provided the original work is not changed in any way and is properly cited. For permission for commercial reuse, please email: oa@electrochem.org.
Ruthenium-platinum nanomaterials are currently the best performing alkaline hydrogen oxidation reaction (HOR) catalysts in fuel cells.1 Their improved activity vs. platinum has been realized through the understanding and application of ligand and bi-functional effects to reduce the metal-hydrogen, M-H, binding energy of Pt and to improve Hads reactivity with adsorbed hydroxyl groups, OHads.1–4 Hydrogen oxidation on Pt electrocatalyst is sensitive to pH because the rate-determining step is concerted electron/proton transfer (Volmer/Heyrovsky step).3,5 However, we have demonstrated on Ru-Pt alloy catalysts that the rds changes to the pH-insensitive hydrogen dissociative adsorption (Tafel step). The impact of pH and carbonate on Ru-Pt alloy catalysts, consequently, remains unresolved for HOR in an alkaline environment.
It is well documented that carbon dioxide in air partitions into the aqueous alkaline electrolyte at the cathode as carbonate and is purged at the anode during fuel cell operation.6–8 Carbonate has not been demonstrated to poison the anode catalyst; however, alkaline carbonate has been proposed as an active proton acceptor,9 according to Eq. 1, competitive with hydroxide, Eq. 2.
![Equation ([1])](https://content.cld.iop.org/journals/1945-7111/163/3/F291/revision1/d0001.gif)
![Equation ([2])](https://content.cld.iop.org/journals/1945-7111/163/3/F291/revision1/d0002.gif)
It has been suggested by some authors that because of water's ability to rapidly self-associate/dissociate that it is an active participant in the oxidation of Hads during alkaline hydrogen oxidation.3 Water is the major component of the electrolytic environment in both traditional, three-electrode electrochemical cells and in solid polymer electrolyte fuel cells. Consequently, if water is an active participant in the HOR, concentration effects should mask the contribution of carbonate. Resolving this question has important implications for modeling the alkaline fuel cell environment, as well as developing improved catalysts.
Using a facile chemical vapor deposition (CVD) technique for the synthesis of high-surface-area catalysts on carbon supports,10–14 we here investigate the nature of the HOR/HER and ORR on state-of-the-art ruthenium-platinum alloys in the presence of alkaline carbonate. The surface composition of these catalysts has been previously demonstrated to reflect the bulk phase composition of the RuxPty alloys using extended X-ray absorption fine structure (EXAFS) and energy-dispersive spectroscopy–scanning transmission electron microscopy (EDS-STEM).4 A change in the rate-determining step was illustrated for hydrogen oxidation from electron transfer (Heyrovsky/Volmer step) to hydrogen dissociative adsorption (Tafel step) with the formation of the RuxPty alloy. These results are here again confirmed and are used to explain the insensitivity of the alloy catalysts to changing pH for alkaline HOR in contrast with the stark sensitivity to pH change for monometallic Pt catalysts.
Experimental
Nanoparticle synthesis
RuxPty and Pt electrocatalysts were synthesized via a single-step, chemical-vapor-phase deposition onto as-received Vulcan carbon XC-72R supports (∼46% wt platinum group metal, PGM) using metal-acetylacetonate precursors and similar methods to those previously published.11,12,15 The solid precursors (Sigma-Aldrich) were mixed with as-received Vulcan XC-72R (Cabot) and thermally treated for 15 h in a water-N2 atmosphere at 240°C. During the thermal treatment, the liquid water vaporized, the organometallic precursors sublimed then decomposed, and metallic nanoparticles were deposited onto the support. As-synthesized C-PGM powders (5 mg) were dispersed in 25% isopropanol solutions (4 mL) with 5 mg of Nafion from a 5% solution (Ion Power) and further diluted to approximately 0.25 μg/μL to make electrodes for electrochemical analysis.
Spectroscopy/microscopy
High-Resolution Transmission Electron Microscopy was conducted on a Hitachi HF-3300 TEM/STEM at 300 kV.
Electrochemical characterization
Electrocatalysts were tested at room temperature in a standard three-electrode electrochemical cell (Pine Instruments) with a double-junction Ag/AgCl reference electrode (Pine Instruments) and Pt-wire reference electrode in H2-saturated potassium hydroxide (semi-conductor grade, Sigma-Aldrich) and potassium carbonate (semi-conductor grade, Sigma-Aldrich) electrolyte. See Table I for electrolyte compositions. Electrodes were made by depositing well-dispersed catalyst inks onto glassy-carbon electrodes (A = 0.196 cm2) with rotation while drying.16 The target loading of electrocatalyst was 6 μg Pt-group metal (PGM) per cm2. Polarization curves were obtained over the range −0.1–1.025 V vs. RHE using a Bio-Logic VMP3 research-grade multi-channel potentiostat with EC-Lab software. Potentiostatic electrochemical impedance spectroscopy (EIS) spectra were recorded from 200 kHz to 1 Hz at 0.6 V vs. RHE with a 10 mV sine perturbation amplitude under mass transport limited conditions and 1 atm H2. The high-frequency intercept of the EIS spectrum was used to eliminate the ohmic resistance of the electrochemical cell from the polarization curves. Electrochemical active surface areas (ECSAs) were obtained using Cu-stripping.17 The background-corrected charging currents were integrated, and the ECSAs were calculated using a charge density of 420 μC/cm2metal.17
Table I. Electrolyte composition for HOR and ORR measurements.
| [KOH] | [K2CO3] | pH | pH | [K2CO3] |
|---|---|---|---|---|
| (mol/L) | (mol/L) | (HOR) | (ORR) | [OH−] |
| 0.10 | 0 | 12.95 | 12.93 | 0 |
| 0.10 | 0.1 | 12.95 | 12.94 | 0.1 |
| 0.10 | 0.1 | 13.03 | 12.91 | 1 |
| 0.10 | 1.0 | 12.48 | 12.75 | 35 |
| 0.010 | 1.0 | 12.37 | 12.00 | 45 |
| 0.0010 | 1.0 | 12.10 | 11.87 | 80 |
| 0.00010 | 1.0 | 12.08 | 11.87 | 85 |
The solution pH was determined using the HOR/HER reversible potential measured under hydrodynamic conditions vs. Ag/AgCl reference electrode and calculated using the Nernst equation according to Eq. 3.
![Equation ([3])](https://content.cld.iop.org/journals/1945-7111/163/3/F291/revision1/d0003.gif)
R is the ideal gas constant, T is the electrolyte temperature, F is Faraday's constant, and EAg/AgCl is the reference electrode potential at standard conditions.
The narrow pH range, but wide range of K2CO3/KOH was chosen to approximate the pH and carbonate concentrations that the anode may experience during fuel cell operation. The upper anion concentration is determined by the polyelectrolyte binder's ion exchange capacity,6 which is typically ∼0.5–2.5 M.18 The high buffer capacity used for the electrolyte here, i.e., > ∼0.2 M, serves to mitigate surface pH swings during hydrogen oxidation measurements.5
Results and Discussion
The impact of carbonate on alkaline HOR and ORR was investigated using a series of carbon-supported RuxPty nanoparticle catalysts whose TEM micrographs are given in Fig. 1. The representative micrographs show that the nanoparticles are reasonably uniform in size, dispersed evenly over their respective carbon supports, and possess the anticipated nanosize typical to the vapor-grown catalysts synthesized by this technique.11,12,14

Figure 1. Representative TEM images of the as-synthesized electrocatalysts: a-b) Ru0.2Pt0.8 and c-d) Ru0.8Pt0.2.
Extensive characterization has been conducted on these catalysts, including X-ray diffraction (XRD), EXAFS, X-ray absorption near edge spectroscopy (XANES), EDS-STEM, and high-resolution transmission electron microscopy (HR-TEM). These data have been presented previously for similar electrocatalysts.4 The data confirm that the as-synthesized catalyst surface matches the bulk phase composition.
Electrochemical removal of adsorbed Cu, used to determine the electrochemically active surface areas (ECSAs), produced charging currents in accord with surface compositions that are similar to their respective bulk compositions. Such data demonstrate the in situ surface composition of a nanoparticle alloy surface.
Ru increases the oxophilicity of the electrocatalyst's surface by shifting its d-band center closer to the Fermi level vs. Pt.2,19,20 This phenomenon increases the electron density in the catalysts' conduction band and decreases its interaction with Hads. We have demonstrated experimentally that this behavior enhances HOR activity in alkaline electrolyte.4 In addition, the electrochemical charging data obtained in N2-saturated, 0.1 M KOH, illustrated in Fig. 3, show a systematic cathodic (leftward) shift of the surface oxide reduction peak vs. monometallic Pt with increasing Ru content (demarcated on Fig. 3). Cathodic shifts of the surface oxide reduction peak indicate that surface oxides are more difficult to reduce and are a consequence of the changed oxophilicity of the surface because of contributions from both Ru and Pt to the charging current.
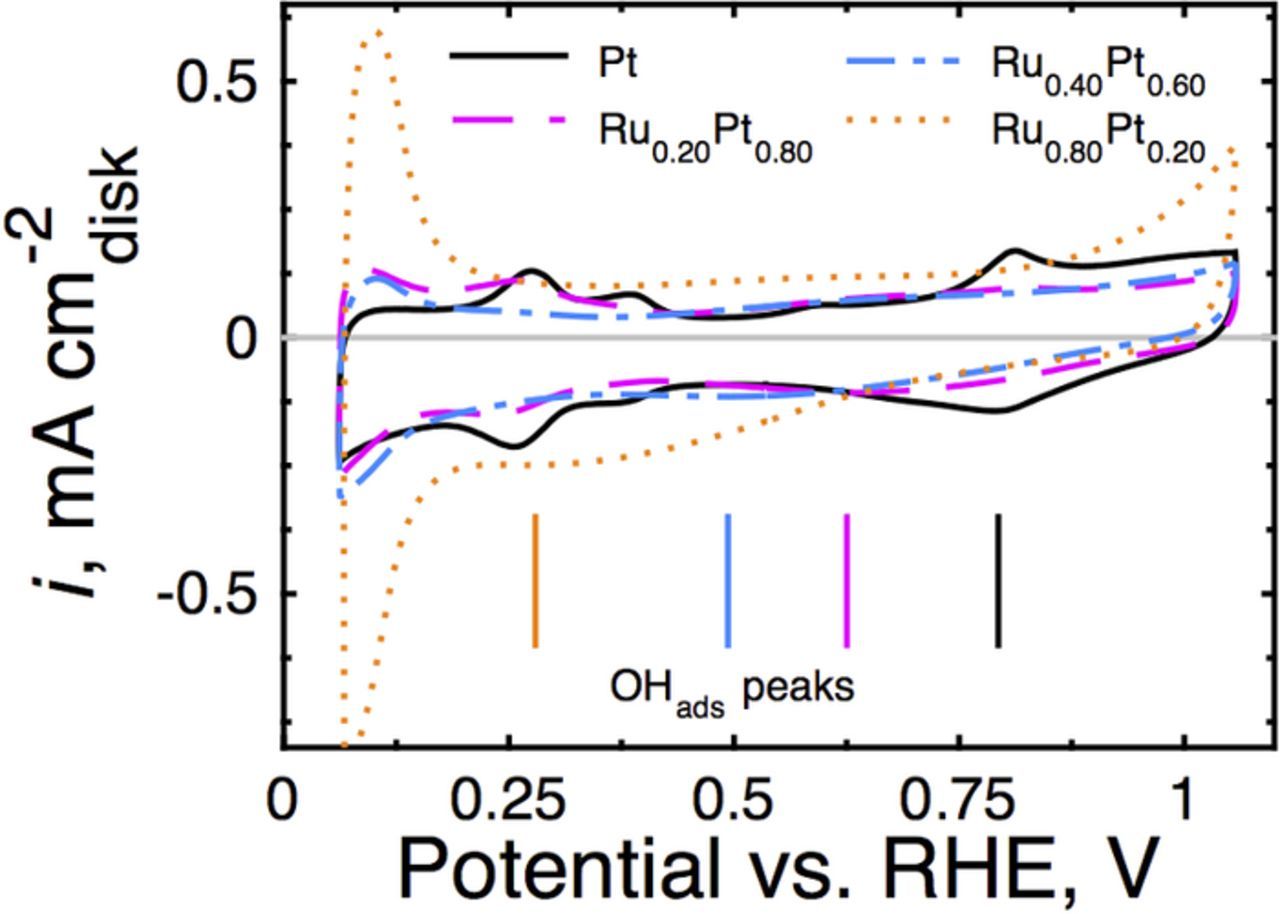
Figure 3. Electrochemical charging currents collected in static, N2-saturated, 0.1M KOH at 50 mV/s for the indicated electrocatalysts. A compositionally dependent, cathodic shift (leftward) in the surface oxide reduction peak is observed illustrating increased oxophilicity of the nanoparticle surface with Ru addition.
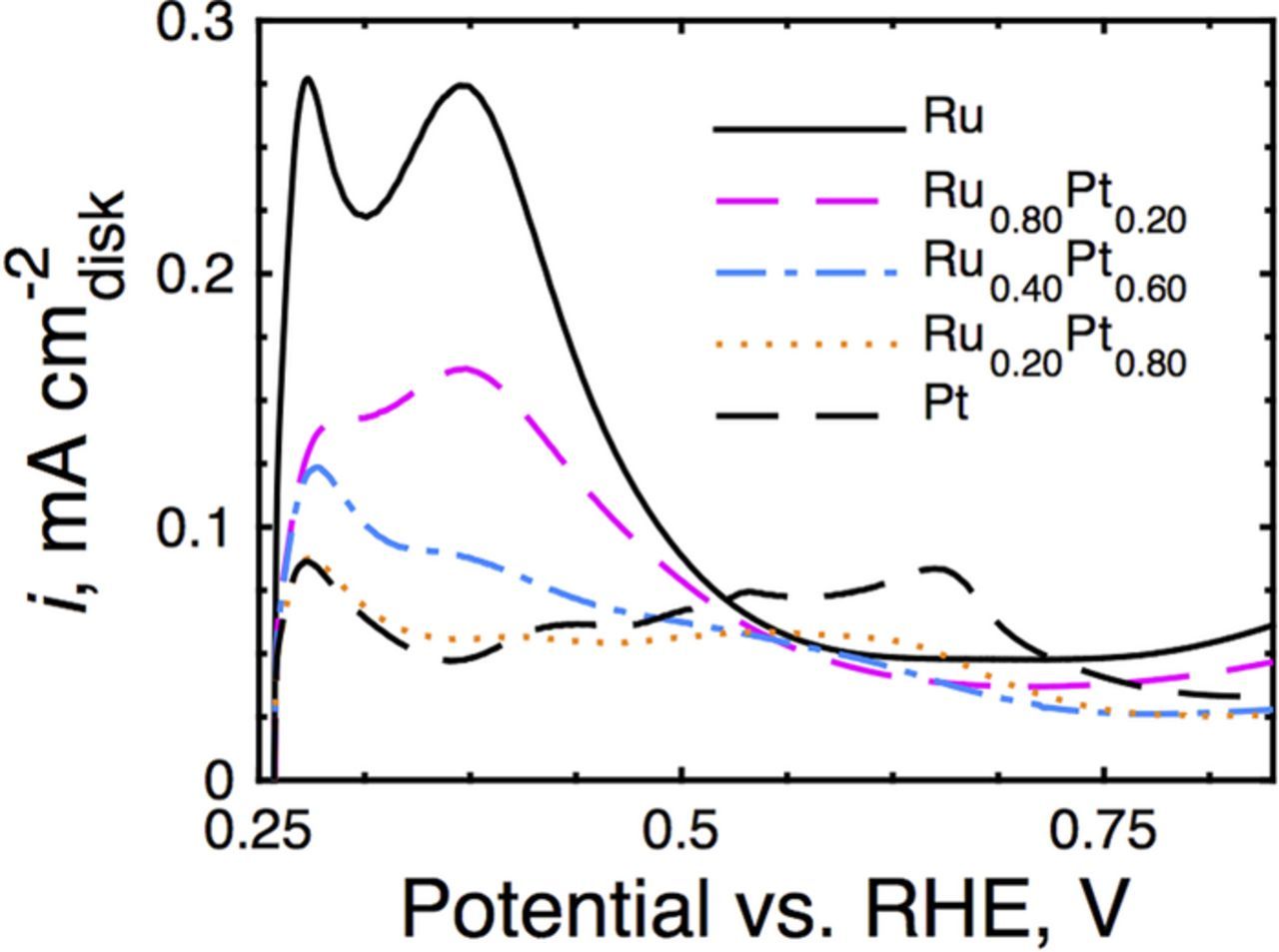
Figure 2. Electrochemical data in static, N2-saturated, 0.1M H2SO4 with 2.5 mM CuSO4 collected at 10 mV/s for positive-going scans. The charging currents for the alloy nanoparticles are intermediate to those obtained for their constituent monometallic nanoparticles and suggest surface compositions consistent with bulk compositions. Monometallic Ru has been included for comparison but was not tested for HOR here.
Hydrogen oxidation data were collected under rotation of the electrode at 1600 rpm with a 10 mV/s sweep rate in H2-saturated electrolyte. The results, illustrated in Fig. 4, are normalized to their respective mass-transport-limited currents calculated using the Levich equation (Eq. 4).21
![Equation ([4])](https://content.cld.iop.org/journals/1945-7111/163/3/F291/revision1/d0004.gif)
The mass-transport-limited current varied with electrolyte composition22 because of differences in bulk H2 concentration, C*H2, diffusivity, DH2, and kinematic viscosity, ν, where n is the number of electrons transferred, ω is the rotation speed, and A is the projected area of the electrode. Normalizing to the mass-transport-limited current provides a clearer visualization of the changes in the polarization response compared to the uncorrected data.
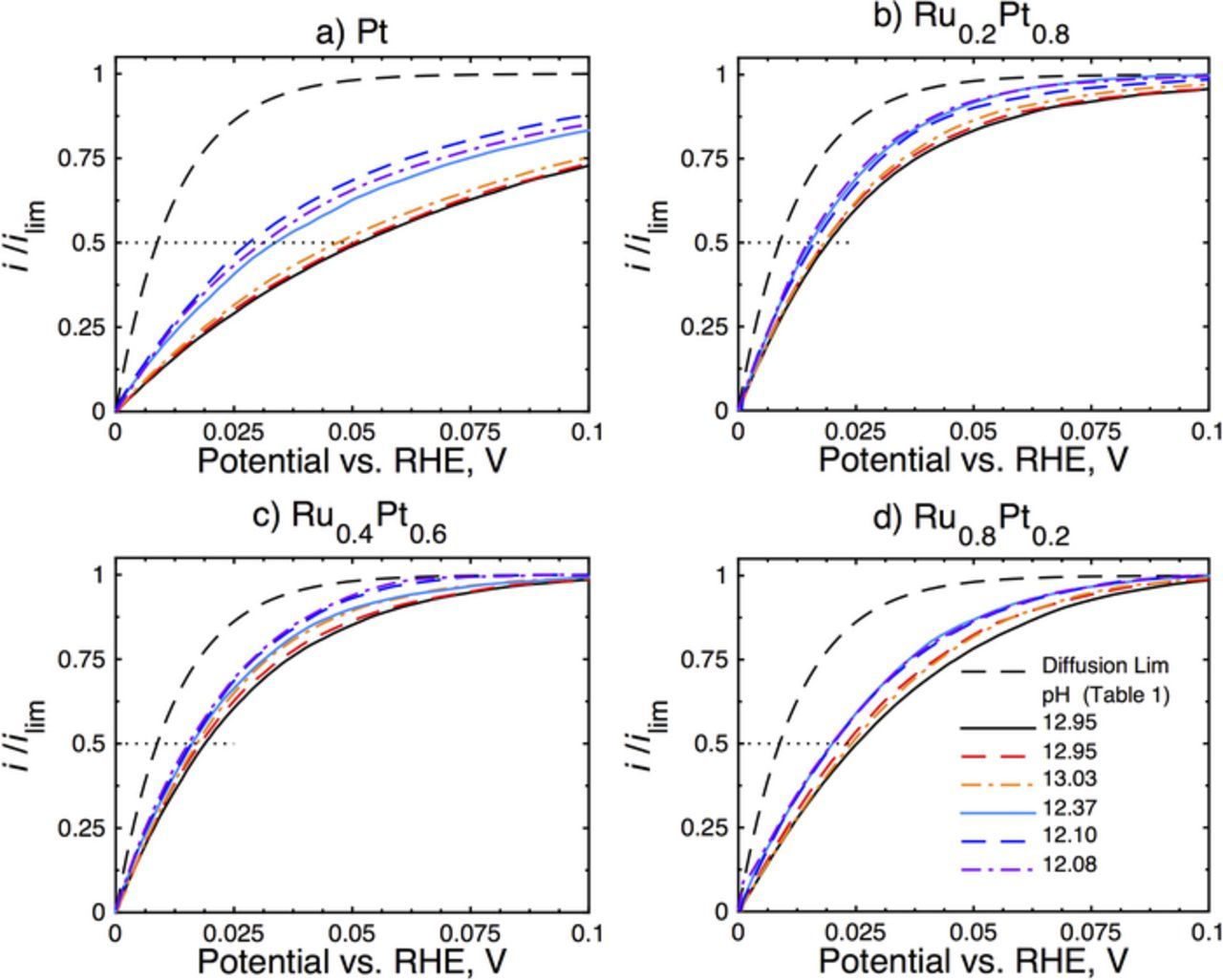
Figure 4. Electrochemical hydrogen oxidation data collected in H2-saturated electrolyte 1600 rpm and 10 mV/s for a) Pt; b) Ru0.2Pt0.8; c) Ru0.4Pt0.6; and d) Ru0.8Pt0.2 electrocatalysts at several different pH values. The compositions of the various electrolytes investigated are given in Table I. Hydrogen oxidation currents have been normalized to their mass-transport-limited currents. The oxidation half wave has been illustrated (...) to guide the eye, as well as the mass-transport-limited current density (– –).
The illustrated polarization data in Fig. 4 show that as the pH of the supporting electrolyte is reduced the hydrogen oxidation current moves toward the mass-transport-limited current calculated using Eq. 5.23
![Equation ([5])](https://content.cld.iop.org/journals/1945-7111/163/3/F291/revision1/d0005.gif)
where η is the overpotential.
It has been demonstrated5 on monometallic Pt that the polarization response for HOR is not differentiated from the mass-transport-limited current with infinitely fast kinetics at a pH = ∼6, while monotonically increasing half-wave potential shifts are observed at higher pH. The data here show a similar monotonically increasing half-wave potential for small pH variations over the range ∼12–13. The HOR current shifts are most noticeable for the Pt electrocatalyst (22 mV/pH) while the overall pH sensitivity of the HOR is significantly decreased on the Ru-containing catalysts: 3.7, 2.5, and 4.7 mV/pH on the Ru0.2Pt0.8, Ru0.4Pt0.6, and Ru0.8Pt0.2 catalysts, respectively. We believe the lack of sensitivity to pH for the Ru-containing catalysts can be explained by a change in the rate-determining step (rds) from an electron transfer (Heyrovsky/Volmer step) to a dissociative hydrogen adsorption (Tafel step). A consequence of this change would be an apparent insensitivity to pH of HOR for the Ru-containing catalysts.
Tafel Reaction:
![Equation ([6])](https://content.cld.iop.org/journals/1945-7111/163/3/F291/revision1/d0006.gif)
Heyrovsky Reaction:
![Equation ([7])](https://content.cld.iop.org/journals/1945-7111/163/3/F291/revision1/d0007.gif)
Volmer Reaction:
![Equation ([8])](https://content.cld.iop.org/journals/1945-7111/163/3/F291/revision1/d0008.gif)
Further evidence of this change in the rate-determining step comes from a Tafel analysis of the kinetic currents determined by applying Eq. 9 to HOR data collected at 1600 RPM for a reaction order of 1/2.
![Equation ([9])](https://content.cld.iop.org/journals/1945-7111/163/3/F291/revision1/d0009.gif)
where ik is the kinetic current. Tafel slopes of ∼120 mV/dec. are consistent with an electron transfer rds, while Tafel slopes of ∼30 mV/dec. are consistent with a hydrogen dissociative adsorption rds.23,24 The average Tafel slope for the monometallic Pt catalyst at all of the pH values measured was 127 ± 18 mV/dec. while the average Tafel slopes for the Ru-containing catalysts were 31 ± 3, 30 ± 7, and 30 ± 4 mV/dec. for the Ru0.2Pt0.8, Ru0.4Pt0.6, and Ru0.8Pt0.2 electrocatalysts, respectively.
To better illustrate the HOR half-wave shifts, the overpotential at half of the mass-transport-limited current has been plotted vs. electrolyte pH in Fig. 5. Some authors have suggested that changes in activity in the presence of carbonate are related to the direct participation of carbonate in the oxidation of adsorbed hydrogen, Hads, on the electrocatalyst surface.9 In order to investigate this hypothesis, we measured the HOR polarization response in 1.0 M potassium phosphate supporting electrolyte for the monometallic Pt catalyst (filled square). The HOR half-wave shifts in phosphate electrolyte follow an identical trend as that observed in the carbonate electrolyte on the monometallic Pt. To ensure that we were observing the electrolyte pH and not an artifact of its determination using Eq. 1 in the presence of carbonate, we also measured the pH using a bulb-meter. The pH values measured using both methods were identical.

Figure 5. The HOR half-wave overpotential is (a) plotted vs. electrolyte pH and is (b) compared to the half-wave overpotential shifts determined by Sheng et al. (cone area).5 The half-wave shifts of the Ru0.2Pt0.8, Ru0.4Pt0.6, Ru0.8Pt0.2 catalysts are less sensitive to pH than the Pt-only electrocatalyst and are significantly shifted toward the mass-transport-limited overpotential. The half-wave shift in carbonate (open symbols) and in phosphate (filled symbols) alkaline electrolyte is considered for monometallic Pt only.
The results, we believe, do not discount the involvement of carbonate in the HOR so much as they suggest that water is as active a participant as hydroxide in 0.1M KOH aqueous electrolyte. Because water can rapidly dissociate and is a fast proton acceptor, it is a proton shuttle to bulk hydroxide during alkaline HOR. These data and this conclusion are similar with the discussion of alkaline HOR provided by Durst et al.3 The authors were not able to provide experimental evidence that water and its ability to rapidly self-associate/dissociate impacted the HOR. However, we believe water's rapid self-dissociation is the only explanation for why carbonate, a good proton acceptor, is not observed to impact the HOR when present in two-fold excess to hydroxide (yet still at a concentration more than 50-times less than that of water).
An emerging hypothesis suggests that on fast catalysts for the HOR/HER, like Pt, the most significant barrier to reaction is the configurational entropy penalty paid to move protons through the double layer25 where the double layer composition itself is the source of the pH sensitivity. This is consistent with our results here where the HOR activity is indifferent to the proton acceptor in this narrow pH range on Pt. It is also consistent with results that we've published previously4 and those obtained by other authors26 for Pt and Pt-shell catalysts. Within this framework, catalysts with a demonstrated bi-functional effect would have a more muted pH response because the surface reaction of adsorbed hydroxyl/hydrogen species obviates the need for proton transport through the double layer. Again, this result is consistent with the behavior of the PtRu alloy catalysts here and those we have presented previously.4 If this emerging hypothesis were reinforced with more experimental data, it would reify the importance of understanding the impact of double layer composition on electrocatalysis in addition to understanding the impact of reactant/product binding energies.
To highlight the compositional dependence of HOR activity, we have plotted in Fig. 6 shifts in HOR half-wave potential vs. Pt content for the catalysts investigated here in carbonate as well as for additional catalyst compositions in 0.1 M KOH without carbonate. The illustrated results indicate that there is a minimum shift for compositions containing ∼60–80% Pt. This optimum in the compositionally dependent response suggests a path toward highly active HOR catalysts in alkaline electrolyte.
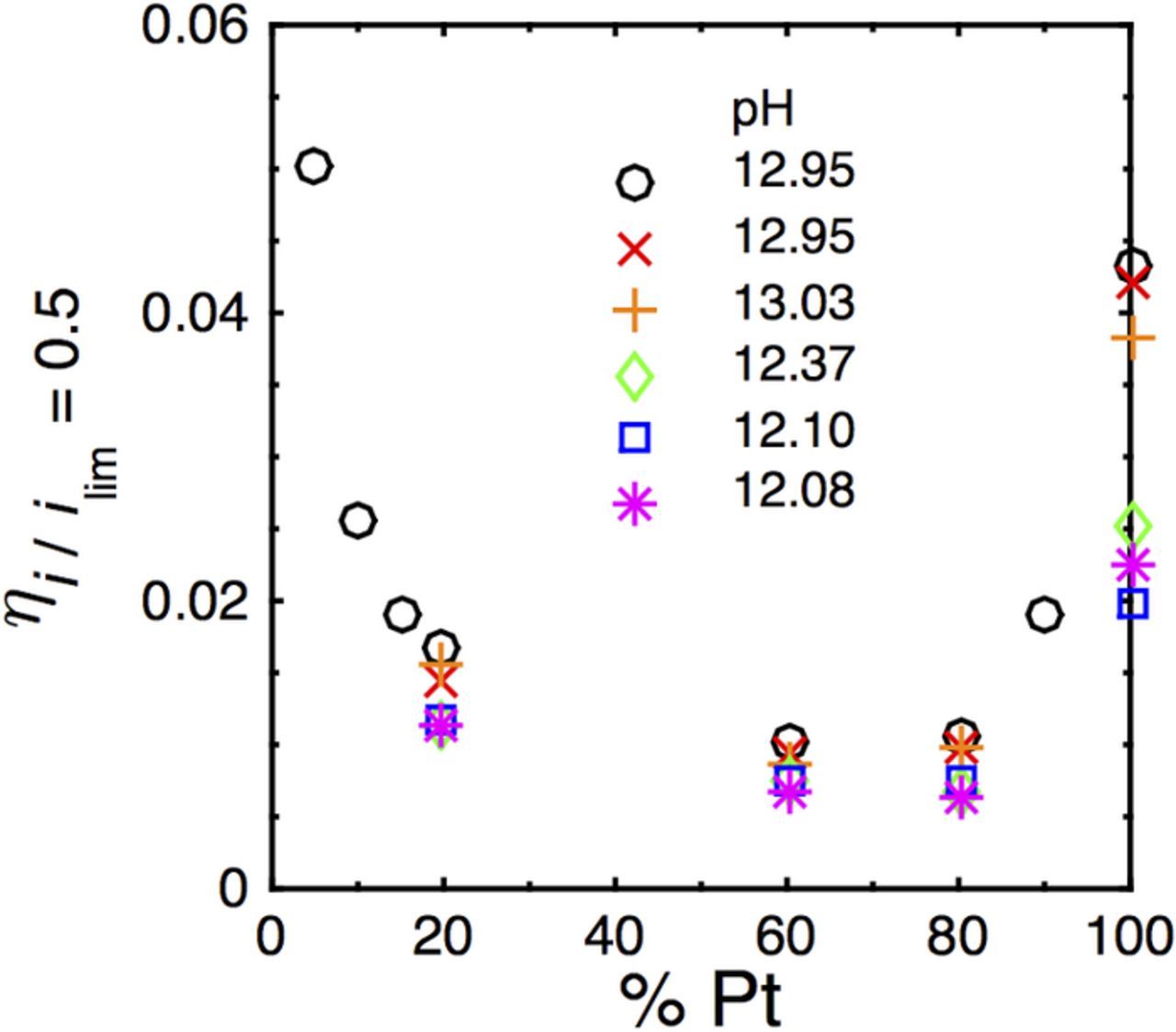
Figure 6. HOR half-wave overpotential vs. Pt content in the nanoparticle alloys indicate that the Ru-containing alloys are less sensitive to changes in pH than monometallic Pt catalysts. A minimum in the half-wave shift is observed for Pt content ∼60–80%. Data for carbonate-free electrolyte (◯) have been included for additional catalyst compositions.
In operating fuel cells at low current density and high gas flow rates (cell start-up), it has been demonstrated that carbonate is present at both the anode and cathode.6,8 Additionally, carbon dioxide would continuously enter the fuel cell system at the cathode via partitioning from air.6–8 Consequently, we are as interested in any impact of carbonate on the ORR as well. Oxygen reduction data were collected under rotation of the electrode at 1600 rpm with a 10 mV/s sweep rate in O2-saturated electrolyte on the monometallic Pt catalyst. The results, illustrated in Fig. 7, are normalized to their mass-transport-limited current calculated using Eq. 227–29 and compared to data collected in non-adsorbing perchloric acid.
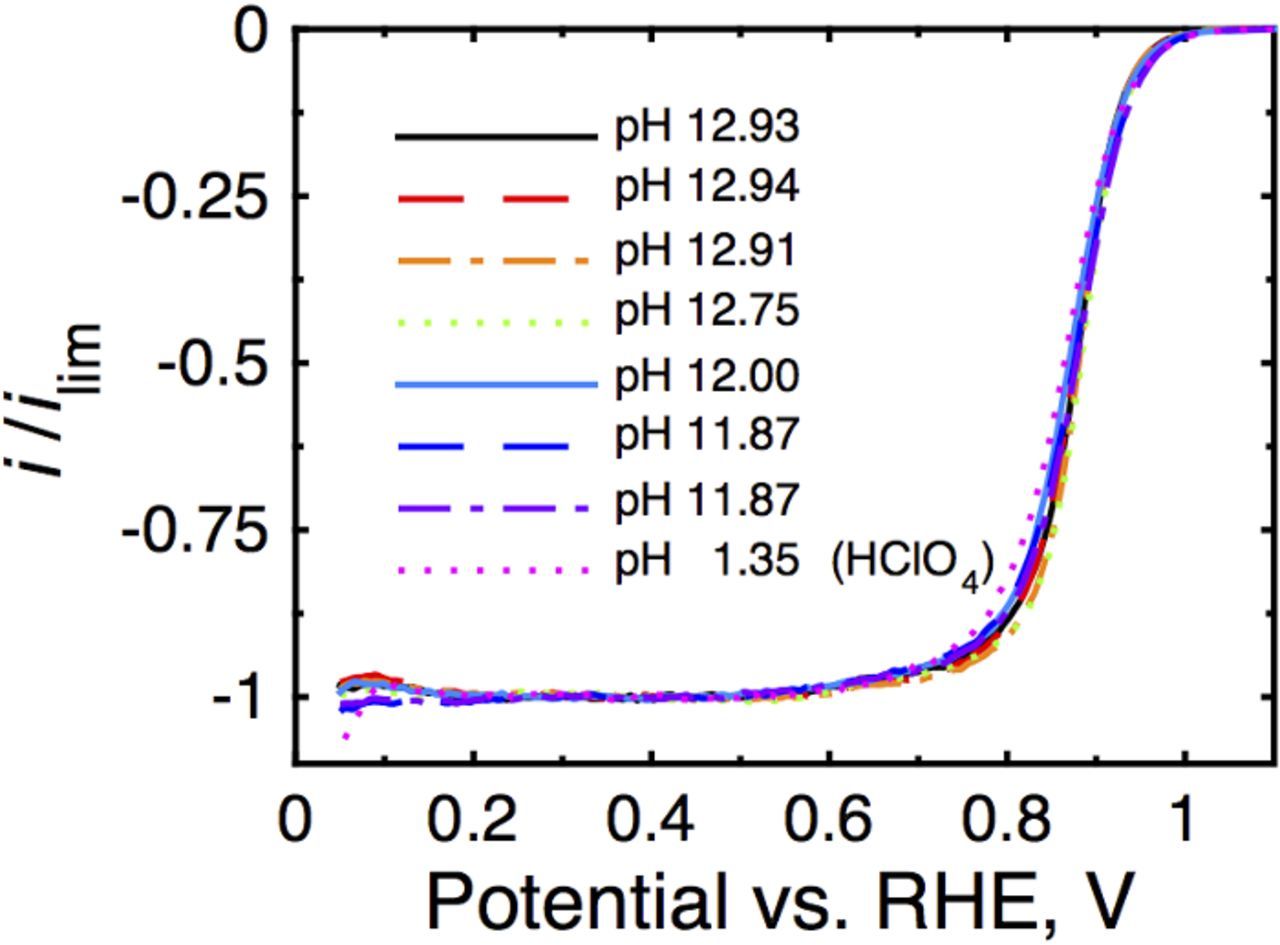
Figure 7. Electrochemical oxygen reduction data collected in O2-saturated electrolyte 1600 rpm and 10 mV/s for carbon-supported Pt electrocatalyst at several different pH values. Oxygen reduction currents have been normalized to their transport limited currents. A comparison is made to the value in 0.1M HClO4 (pH 1.35) instead of to the mass-transport-limited current as in the HOR example. No change in ORR activity with respect to pH or carbonate content is observed for the ORR on Pt nanoparticles.
No change in the half-wave potential is observed with respect to pH or carbonate content. These results are similar to those presented on polycrystalline Pt surfaces in a variety of alkaline and acidic electrolytes.30 This is a well known result for polycrystalline (stepped) Pt surfaces.31,32
Conclusions
The polarization response of monometallic Pt and RuxPty alloy catalysts was investigated for aqueous alkaline hydrogen oxidation and oxygen reduction performance in the presence of carbonate. The data were used to illustrate a strong dependence of HOR upon pH for the monometallic Pt catalysts (22 mV/pH) and a weak dependence upon pH for the Ru-containing electrocatalysts (3.7, 2.5, and 4.7 mV/pH on Ru0.2Pt0.8, Ru0.4Pt0.6, and Ru0.8Pt0.2, respectively). These results corroborate our previous findings that showed a change in rds from electron transfer (on monometallic Pt) to dissociative hydrogen adsorption (on RuxPty catalysts). Tafel slope analysis of the kinetic currents revealed an average slope for the monometallic Pt catalyst at all of the pH values as 127 ± 18 mV/dec., while the average Tafel slopes for the Ru-containing catalysts were 31 ± 3, 30 ± 7, and 30 ± 4 mV/dec. for the Ru0.2Pt0.8, Ru0.4Pt0.6, and Ru0.8Pt0.2 electrocatalysts, respectively. There is no difference in the performance at comparable pH values in the presence or absence of carbonate on monometallic Pt indicating that water/hydroxide is the primary proton acceptor for alkaline HOR in 0.1M KOH. Finally, we observe no pH or carbonate dependence for the ORR on monometallic Pt.
Acknowledgments
This work was supported by the Office of Naval Research, N00014-12-1-0887 and the NSF-funded, TN-SCORE program, EPS-1004083, under Thrust 2. Electron microscopy was conducted at the Center for Nanophase Materials Sciences, which is sponsored at Oak Ridge National Laboratory by the Scientific User Facilities Division, Office of Basic Energy Sciences, U.S. Department of Energy. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.