
Abstract
Formic acid presents several advantages for use as a fuel in fuel cells. We present a first-principles based analysis of electrooxidation trends for formic acid on the close-packed facets of eight fcc metals: Au, Ag, Cu, Pt, Pd, Ni, Ir, and Rh, and four hcp metals: Co, Os, Ru, and Re. To explore the structure sensitivity of this reaction on the fcc metals, we also studied the open (100) facet of these eight metals. We find that the open facets of Au, Ag, Cu, Pt, and Pd are more energy-efficient (i.e. require less overpotential) for formic acid electrooxidation when compared to their close-packed facets. The opposite is true for the stronger-binding metals: Ni, Ir, and Rh. Using the free energy of adsorbed CO* and OH* as reactivity descriptors, we cast the thermochemistry of the reaction network into phase diagrams showing regions of rate-determining steps, together with their calculated free energies. This allows the identification of bimetallic alloys, potentially possessing improved electrocatalysis for formic acid electrooxidation, compared to Pt or Pd. We discuss the need for anode catalysts for direct formic acid fuel cells (DFAFCs) to diminish CO* poisoning, by promoting the formation of formate instead of carboxyl intermediate.
Export citation and abstract BibTeX RIS

This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 License (CC BY, http://creativecommons.org/licenses/by/4.0/), which permits unrestricted reuse of the work in any medium, provided the original work is properly cited.
Rising standards of living and growing global populations have exacerbated the dependence of humanity on fossil fuels, giving rise to unprecedented challenges regarding excessive greenhouse gas (e.g. CO2) emissions. Unlike stationary emission sources, where CO2 can be captured on site, mobile sources pose the difficult challenge of capturing CO2 from ambient atmosphere, where CO2 is 300 times more dilute than it is in flue gases.1 This necessitates the economical production and utilization of carbon-free (e.g. H2, NH3) or carbon-neutral (e.g. CH3OH, HCOOH, if produced from CO2 or biomass) fuels in the transportation sector and in mobile applications at large. Low-temperature polymer electrolyte membrane (PEM) fuel cells are efficient candidates for taking advantage of such fuels, being of higher thermodynamic efficiency than internal combustion engines which are limited by Carnot efficiency.2 Moreover, electrochemical power generation enables convenient storage, management, and transportation of energy in the form of chemical fuels.2,3
Among these fuels, hydrogen is the cleanest, producing only water upon its oxidation at the anode of fuel cells. However, hydrogen transportation and storage is problematic,4 and other liquid fuels can be more convenient for fuel cells. Methanol has been extensively studied for this purpose; however, its crossover to the cathode limits its efficient utilization.5,6 Dimethyl ether has also been considered as a possible fuel for low-temperature fuel cells, showing high power densities and milder crossover than methanol.7 In this work, we focus on utilizing formic acid (FA) as a fuel for direct formic acid fuel cells (DFAFCs). FA has the potential of being a renewable fuel, since it is a by-product of levulinic acid production; a biorefinery platform chemical,8 or can be produced by direct reduction of CO2.9 FA is a safe and non-toxic10 liquid compared to methanol,11 less flammable than dimethyl ether,11 and its crossover flux through Nafion membranes is considerably less than that of methanol.12 Particularly, FA electro-oxidation (FAO) exhibits a strong electromotive force, as evidenced by an equilibrium cell voltage of 1.40 V, which is higher than that for hydrogen (1.23 V), methanol (1.21 V), and dimethyl ether (1.20 V) fuel cells, among others.11 Unlike several other liquid fuels for fuel cells – including methanol and dimethyl ether – a direct oxidation mechanism of FA is not limited by the need for supplying oxygenated species (e.g. hydroxyl), as evidenced by an oxidation onset potential that is cathodic to that of water activation on Pt.13 Furthermore, FAO has been described as a model reaction for studying electrochemical oxidation of small organic molecules,14 and FA and its oxidation intermediates appear in the electrochemical oxidation networks of other fuels like methanol15 and formaldehyde.16 As a result, FAO in DFAFCs has attracted considerable attention, with FA being a suitable liquid fuel for fuel cells that can surpass the activity of methanol.17
FAO is generally considered to proceed through a dual path mechanism (Scheme 1), in which FA can be oxidized to CO2 directly via a reactive intermediate, or indirectly via an adsorbed CO* species.18 The nature of the reactive intermediate in the direct mechanism has been debated in the literature. Osawa and coworkers have advocated for formate (either as a bidentate adsorbed species or as an ion) as the reactive intermediate on Pt13,19–24 (see Schwarz et al.25 for an interesting theoretical study of formate ion electrooxidation on Pt). In fact, formate was suggested as a common intermediate in both direct and indirect mechanisms on Pt and Au.19,21 Spectroscopically, bridge-bonded formate was indeed detected on Pt electrodes during FAO, along with linear and bridge-bonded CO*,13,20,22,23,26 and weakly adsorbed water.20,22 On Pd, formate was hypothesized to be a short-lived reaction intermediate,27 and was recently detected28 at potentials above 0.3 VRHE using in situ electrochemical attenuated-total-reflection surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS).
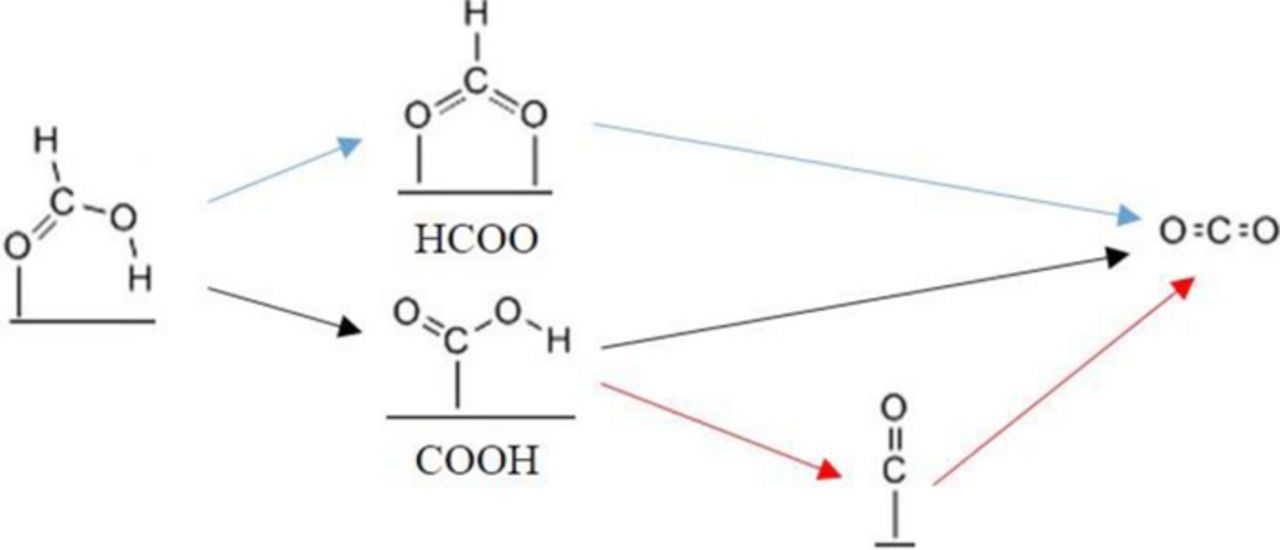
Scheme 1. A general FAO mechanism on anodes of DFAFCs. HCOOH can decompose directly to CO2(g) via a reactive intermediate, the nature of which is still elusive. Alternatively, an indirect route to CO2(g) involves the formation of adsorbed CO*. The overall reaction mechanism consists of three paths: direct via formate (blue), direct via carboxyl (black), and indirect via carboxyl (red). Formate does not generate CO* easily on the metal surfaces studied here.
Several synchronous features29 between the oxidation current and the formate band intensity have been suggested as evidence that formate is the reactive intermediate. Such evidence includes the simultaneous rise in the current and formate band intensity (which is proportional to formate coverage when obtained via ATR-SEIRAS) upon rapid oxidative removal of CO*.23 Moreover, formate deprotonation to CO2 has been suggested as rate-determining, based on potential-step experiments,22,23 isotopic solution exchange experiments,22,26,30 and the non-linear relationship between current (i.e. reaction rate) and formate coverage.20,22,23 However, in other reports, formate was rejected as the reactive intermediate, and was even described as an inhibiting species. In a recent report by Okamoto et al.,31 asynchronous features were detected during electrochemical measurements between current peaks and bidentate formate band intensities, thus challenging the idea that formate could be the reactive intermediate on Pt. Behm and coworkers'14 chronoamperometric transient data on a Pt film electrode suggested that the formate path cannot account but for a minority of the observed anodic current, and suggested the direct oxidation of weakly adsorbed HCOOH to CO2 as the dominant pathway. In a later report,32 they investigated the kinetic isotope effect for HCOOH and DCOOH solutions on a Pt film electrode, and showed that both show similar formate coverages, indicating that formate adsorption/desorption equilibrium (that does not involve breaking the C-H/C-D bond) is far faster than formate decomposition; thus concluding that formate is rather a spectator or inhibiting species. Neurock et al.33 have also arrived at a similar conclusion, citing unfavorable energy barriers for formate oxidation to CO2 or recombination to FA. A recent literature review on the FAO reaction mechanism is very comprehensive.34
Pt and Pd are the most widely studied monometallic catalysts for FAO. However, they come with their problems of easy CO* poisoning, instability and easy dissolution in acidic environments typical of DFAFCs, as well as their high cost.34 Platinum group metal (PGM) costs remain the major contributor to inadaptability of fuel cells to mass implementation in transportation, which necessitates the reduction of their content in fuel cells below 6 gPGM/vehicle.35 Alloying Pt or Pd with less precious metals is a viable way to attend to this goal. In this respect, theory can play an important role in elucidating reaction mechanisms, and together with experiments, it plays a synergistic role toward the efficient screening of a broad array of materials for improved electrocatalysis.36,37 Specifically, density functional theory (DFT) has increasingly enabled accurate and efficient modeling of catalytic systems on the atomic scale.38–40 While the vapor-phase counterpart of FAO, FA vapor-phase decomposition, has been studied from a trends analysis viewpoint,41,42 to the best of our knowledge there has been no trends analysis for the electrochemical oxidation of FA. As recently done for other electrochemical systems,43–52 we provide here a systematic reactivity trends analysis and a structure sensitivity study for FAO on model transition metal surfaces.
Our contribution is organized as follows: firstly, we describe the computational setup, model systems, and proposed reaction network. Secondly, we discuss in detail the FAO mechanisms in terms of the onset potentials on the (111) and (100) facets of Pt and Pd, the most-widely studied monometallic systems. We then expand our discussion to the remainder of the surfaces studied. Subsequently, we cast our findings into phase diagrams allowing the visual representation and rationalization of alloying strategies for improved catalysis for FAO on bimetallic surfaces. Finally, we discuss recent examples of improved binary alloy catalysts for FAO, reflecting on the challenges facing anode catalysts for DFAFCs.
Methods
We perform periodic GGA-PW91 DFT calculations, as implemented in VASP,53,54 to calculate the free energies of HCOO*, COOH*, CO*, and OH*, whereby * denotes an adsorbed species. Calculations are performed on the close-packed facets: (111) of Au, Ag, Cu, Pt, Pd, Ni, Ir, and Rh; (0001) of Co, Os, Ru, and Re; as well as the open (100) facets of the fcc metals Au, Ag, Cu, Pt, Pd, Ni, Ir, and Rh. The detailed computational setup is provided in the Supplemental Material. Since we do not explicitly include the electrochemical environment at the anode/electrolyte interface, we employ the computational hydrogen electrode; a framework that has been successfully used for studying a variety of electrochemical systems, including: methanol electrooxidation,48–50 dimethyl ether electrooxidation,51,52,55 formic acid electrooxidation,56 ammonia electrooxidation,43 ethanol and ethylene glycol electrooxidation,57 ammonia electrosynthesis,44–46,58 CO2 electroreduction,59,60 and the oxygen reduction reaction.61–66 Here, we offer a brief overview of the method, but more details can be found in the original work of Nørskov and coworkers.61 According to Nørskov's computational hydrogen electrode,61 hydrogen gas is in equilibrium with protons and electrons under standard conditions of temperature (298 K) and pressure (1 bar of H2) and at all pH, at a defined potential of 0.00 V referenced to the reversible hydrogen electrode (RHE), according to the following reaction:
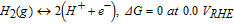
All potentials hereafter are relative to RHE. Applied bias (U) is taken into account by shifting the free energy of an electron by -|e|U, where e is the absolute charge of an electron. A pH of 0 is assumed throughout. The free energy (G) is calculated from the ZPE-corrected total energies as:
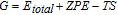
where, Etotal is the total energy calculated from DFT, ZPE is the calculated zero-point energy, S is the calculated entropy, and T is the standard temperature of 298 K. For compatibility with previously published work on methanol electrooxidation,48–50 all species have their G referenced to those of gas-phase CO2, H2, and H2O. For example, the G of adsorbed CO* (GCO*) is calculated as:
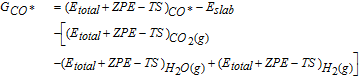
We study FA electrooxidation in the context of the following elementary steps, and in accordance with Scheme 1:
![Equation ([1])](https://content.cld.iop.org/journals/1945-7111/165/15/J3109/revision1/d0004.gif)
![Equation ([2])](https://content.cld.iop.org/journals/1945-7111/165/15/J3109/revision1/d0005.gif)
![Equation ([3])](https://content.cld.iop.org/journals/1945-7111/165/15/J3109/revision1/d0006.gif)
![Equation ([4])](https://content.cld.iop.org/journals/1945-7111/165/15/J3109/revision1/d0007.gif)
![Equation ([5])](https://content.cld.iop.org/journals/1945-7111/165/15/J3109/revision1/d0008.gif)
![Equation ([6])](https://content.cld.iop.org/journals/1945-7111/165/15/J3109/revision1/d0009.gif)
![Equation ([7])](https://content.cld.iop.org/journals/1945-7111/165/15/J3109/revision1/d0010.gif)
![Equation ([8])](https://content.cld.iop.org/journals/1945-7111/165/15/J3109/revision1/d0011.gif)
This reaction network consists of three main paths. First, direct oxidation via formate (HCOO*), which consists of steps (1) and (2). It is worth noting that monodentate formate was always found to be significantly less stable (by more than ∼0.4 eV) than bidentate formate on all surfaces studied, and therefore, all energetics for this path are given for the bidentate configuration of formate. Second, direct oxidation via carboxyl (COOH*), which consists of steps (3) and (4). Finally, we consider an indirect oxidation mechanism via carboxyl, which consists of steps (3) and (5)-(8). Importantly, we do not consider an indirect oxidation via the formate mechanism, which would entail the possibility of forming CO* from formate; an energetically less favorable process than forming CO* from carboxyl, based on previous DFT work.67,68 Relevant DFT studies by Yoo et al.42 and Herron et al.41 have also omitted the possibility of CO* formation from formate on similar grounds. The C-OH bond scission in COOH* can result in CO* and OH* in step (5), which can – in turn – react off to CO2(g) in step (6). Alternatively, OH* in the presence of protons can spontaneously form water. We explore this possibility via step (7); in such case, it becomes necessary to activate water: via the reverse of step (7) to form COOH*, or via step (8) to form OH* necessary for oxidizing CO* in step (6). We do not consider the possibility of directly converting CO*+H2O(g) to CO2(g), since this is a multiple electron-transfer step. We note that step (7) – as written – could be thought of as a dehydration step, since water is formed via a single electron-transfer step. Therefore, we have three main paths; two direct: the formate mechanism, and the carboxyl mechanism; and the indirect mechanism which includes the dehydration step.
Notice that step (5) – through which the CO-OH bond is broken – is the only non-faradaic step in the proposed reaction network. Hence, it is the only step that is not affected by any applied electrochemical potential. The thermochemistry and kinetics of an elementary step are empirically correlated via Brønsted-Evans-Polanyi correlations, in which the more endothermic (or less exothermic) an elementary step is, the higher its activation energy is.69,70 We do not calculate activation energy barriers for this step, and rely on its thermochemistry alone as a first approximation to estimating its kinetics difficulty. All remaining steps in the proposed scheme above are proton-electron transfer steps, and are directly affected by applied bias as explained earlier. As established previously, the kinetic barriers of these steps have been approximated by their free-energy change (ΔG) if ΔG is positive, or zero if ΔG is negative.71 This effectively sets the barrier of these steps to their theoretical minimum. The overall anode reaction is HCOOH(g)→CO2(g) + 2(H++e−), with a calculated equilibrium potential of −0.13 V (half the free-energy change for a two-electron transfer reaction), in reasonable agreement with the reported formal potential of −0.25 V.72
The activity of a chosen path is given by:
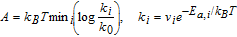
where, kB is Boltzmann's constant, T is absolute temperature (taken as 298 K), ki is the forward rate constant for reaction i, with a prefactor νi and a barrier of Ea, i, and k0 is a normalization factor for non-activated proton-electron transfer steps ( Ea= 0) with a prefactor ν0 = νi. Therefore, and following the policy for activation barriers mentioned above, the expression for activity is reduced to  , with more active paths having less positive or more negative ΔGi. To relate calculated energetics to measurable rates, a difference in activity of 0.06 eV at room temperature corresponds to an order-of-magnitude difference in rate constants of corresponding elementary steps.
, with more active paths having less positive or more negative ΔGi. To relate calculated energetics to measurable rates, a difference in activity of 0.06 eV at room temperature corresponds to an order-of-magnitude difference in rate constants of corresponding elementary steps.
Onset potential for a specific reaction path, which can be compared to that reported in the experimental literature, is defined as the minimum potential that must be applied for all proton-electron transfer elementary steps within this path to take place spontaneously (ΔG ≤ 0). This definition excludes non-faradaic steps, which, for the purpose of our analysis, are potential independent. Hence, the onset potential is dictated by the most endergonic (or least exergonic) faradaic step within a given path. Such a step is referred to as the potential-determining step (PDS). Therefore, for each of the three paths considered in this study, we report an onset potential corresponding to the respective PDS.
In addition to any previously stated assumptions, we discuss here the following limitations to our model. Importantly, we ignore solvation effects on the binding energy or free energy of adsorbed species. Previous DFT work has indicated that solvation can selectively stabilize adsorbed species e.g.: hydroxyl (OH*) up to ∼0.50 eV on Pt(111),61,73 but only up to ∼0.10 eV on Pt(100).74 It is reasonable to assume that other adsorbates, especially those that can form hydrogen bonding with solvent molecules like OH* does, will be stabilized by a similar extent, regardless of the surface they are adsorbed on. Since neither we nor others in the literature75,76 have systematically studied the effect of solvation on all adsorbates across varying surfaces, we omit this effect in our analysis. Further, van der Waals interactions are not included in this study, assuming that these interactions will likely be similar across surfaces, and hence will not affect the overall conclusions of our study. Finally, our study is conducted at the low coverage limit (1/9 ML), and does not take into account possible effects arising from higher coverage of spectator species. Generally, higher coverage might tend to destabilize various adsorbates on transition metal surfaces.77,78
Results
Because Pt and Pd are the most widely studied monometallic catalysts for FAO, before discussing reactivity trends across all surfaces studied, we start by describing our results on the (111) and (100) facets of these two metals.
Structure sensitivity of FAO on Pt
Figure 1 shows the free energy diagram at 0.00 V for the three mechanisms on Pt(111) and Pt(100). Similar free energy diagrams are provided at potentials corresponding to the positive onset potentials of the three mechanisms in Supplemental Material (Figure S1 and Figure S2 for Pt(111) and Pt(100), respectively).
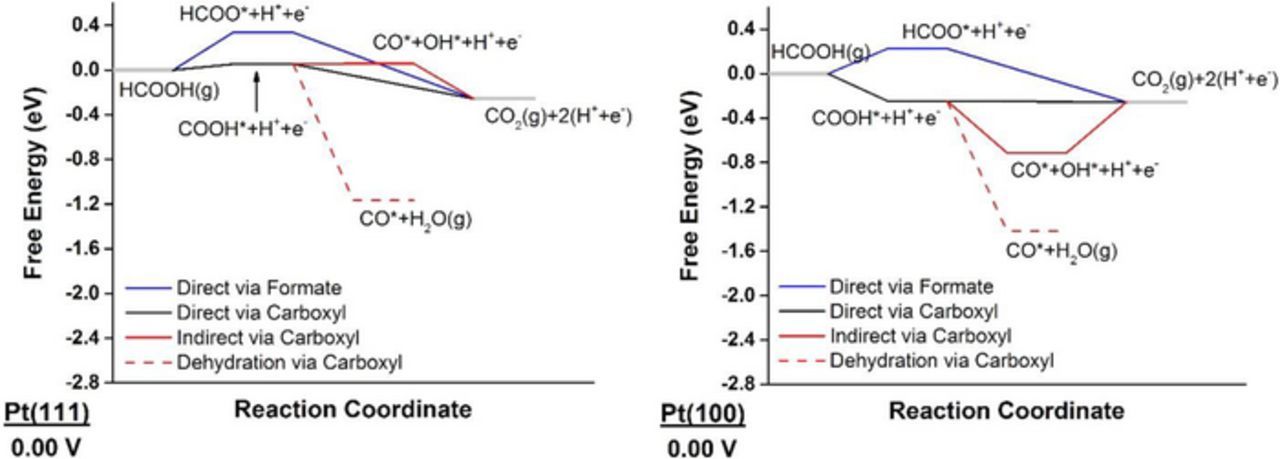
Figure 1. Free-energy diagrams of FAO on Pt(111) and Pt(100) at 0.00 V showing the direct oxidation via formate mechanism in blue, the direct oxidation via carboxyl mechanism in black, and the indirect mechanism via carboxyl in red; the dehydration step is dashed.
We first focus on Pt(111). Starting from HCOOH(g), the first deprotonation step could either result in formate (HCOO*) or carboxyl (COOH*). The carboxyl pathway, with an onset potential of just 0.05 V, is less endergonic than the formate pathway by 0.29 eV. We therefore predict that the reaction flux will initially proceed through forming carboxyl at low potentials. After forming carboxyl, we can deprotonate COOH* once more to produce CO2(g), or can produce CO*+H2O(g) or CO*+OH*. Apart from the last, both first and second options are exergonic, with CO*+H2O(g) being more exergonic than CO2(g) production by 0.91 eV. We note here that a difference of 0.06 eV in calculated energetics corresponds to an order-of-magnitude difference in reaction rates. Hence, Pt will be easily poisoned by CO* at low potentials, as is well-demonstrated by electrochemical and spectroscopic measurements,79–81 and rationalized theoretically.82,83 Our analysis is thermodynamic in nature, and overlooks the existence of kinetic barriers. In a recent study of FA vapor-phase decomposition on Pt(111),68 Scaranto and Mavrikakis calculated the decomposition energetics of HCOOH on Pt(111) to COOH*+H* or HCOO*+H*. They found that carboxyl is more stable than formate by ∼0.30 eV, which agrees with our finding in this work that carboxyl is produced less endergonically than formate. They also found that the activation energy barrier for carboxyl formation is higher than that for formate formation, and yet carboxyl decomposes to CO2(g) easier than formate does.68 Further, previous theoretical studies reported high kinetic barriers for CO* formation from HCOOH on Pt(111); nevertheless, CO* formation was exergonic with and without solvation effects84,85 which is in agreement with our calculated energetics. These calculated high barriers for CO* formation have motivated the proposition of a concerted path on Pt(111) to simultaneously form CO* and CO2(g) from FA dimers.82
To gain a deeper mechanistic insight of FAO on Pt(111), we analyze the three paths shown in Figure 1 for onset potentials and associated PDS. The direct oxidation via formate has the formation of formate as the only endergonic step, with a calculated ΔG of 0.34 eV. Since formate appears in our reaction network in conjunction with a single proton-electron pair, its formation becomes more exergonic by 1 eV for each 1 V of applied potential. We therefore calculate an onset potential of 0.34 V for the formate path on Pt(111), which is close to the value of 0.305 V at which a small band for HCOO* was detected on time-dependent ATR-SEIRA spectra on Pt.19 Similarly, the onset potential for the direct oxidation via carboxyl is calculated to be 0.05 V, and is reflecting carboxyl formation. Once carboxyl is formed, CO* formation is downhill in energy, which results in poisoning the surface until water is activated. Water can be activated to form OH* at a potential as high as 1.23 V, or to form COOH* at a slightly lower potential of 1.22 V, a value corresponding to the onset potential of the indirect mechanism. Experimentally, cyclic voltammograms31,86 show two peaks for FAO on Pt, around 0.6 V and 0.9 V, which were attributed to the direct and indirect paths, respectively. Within the limitations of our analysis (see methods section), these two potentials align reasonably well with our predicted onset for the direct path and the indirect path, respectively; yet other interpretations for these peaks attribute them both to the direct path.16,23,26 Since the carboxyl path becomes exergonic at very low potentials, direct evidence for it can be compromised by the hydrogen underpotential deposition (H-UPD) region, as well as the formation of CO* poison.
We now turn our attention to Pt(100). The onset potential for the formate and the indirect mechanism is calculated to be 0.23 V and 0.70 V (corresponding to water activation to OH*), respectively. The carboxyl path is readily exergonic at 0.00 V, where carboxyl is preferred to its formate isomer by 0.47 eV. Compared to Pt(111), Figure 1 shows that the free-energy surface for FAO on Pt(100) is shifted to more negative (or less positive) free energy, which implies stabilization of the various intermediates on the open facet. Therefore, Pt(100) is predicted to be more readily poisoned at low potentials than Pt(111), which explains the lower current densities obtained experimentally in the forward sweep on Pt(100).87 Nevertheless, Pt(100) is more capable of removing CO*, as evidenced by its lower onset potential for the indirect mechanism. The fundamental reason behind this is that water is more easily activated on Pt(100) than on Pt(111), which is in agreement with results from Koper and coworkers.88 We predict that, because of its increased ability to remove CO*, and due to its stronger adsorption of COOH* (the more stable product of the first deprotonation step), Pt(100) should be more active for FAO than Pt(111). This structure sensitivity is clearly manifested in higher current density in the backward scan on Pt(100), when compared to that on Pt(111).87
Structure sensitivity of FAO on Pd
Figure 2 shows the free energy diagram at 0.00 V for the three FAO mechanisms on Pd(111) and Pd(100). Similar free energy diagrams are provided at potentials corresponding to the positive onset potentials of the three mechanisms in Supplemental Material (Figure S3 and Figure S4 for Pd(111) and Pd(100), respectively).
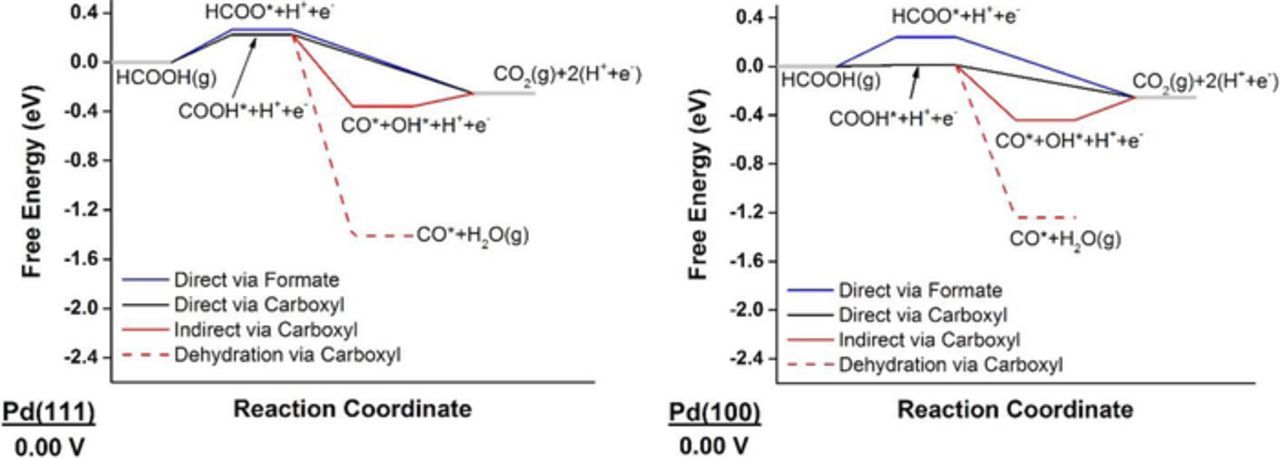
Figure 2. Free-energy diagrams of FAO on Pd(111) and Pd(100) at 0.00 V showing the direct oxidation via formate mechanism in blue, the direct oxidation via carboxyl mechanism in black, and the indirect mechanism via carboxyl in red; the dehydration step is dashed.
On Pd(111), the direct mechanisms via formate and carboxyl have onset potentials of 0.26 V and 0.22 V, respectively. Compared to Pt(111), on Pd(111) carboxyl is destabilized whereas formate is stabilized, thereby closing the gap between the onset potentials of the two direct mechanisms. Since this gap is only 0.04 V wide, one would expect the reaction flux to proceed through both paths, which is in agreement with recent theoretical findings.67,89 This is an interesting result because the difference in these onset potentials should influence the selectivity toward full oxidation to CO2(g) against forming the CO* poison, which can only be formed from COOH*. As shown in Figure 2, CO*+H2O(g) is the most thermodynamically favored state, and therefore Pd(111) should get poisoned by CO*, despite the close onset potentials of both direct mechanisms. CO* remains on the surface until water is activated to OH* at 1.05 V, a voltage marking the onset potential of the indirect mechanism. Pd anodes have generally offered lower tendency to get poisoned by CO* than Pt anodes.34,90,91 In fact, CO* on Pd was not readily detected using spectroscopic techniques;92 it has not been until recently that concrete spectroscopic evidence of CO* formation over Pd has been obtained by combining surface-enhanced infrared spectroscopy with attenuated total reflection and thin-layer flow cell configurations.93 We attribute less CO* poisoning of Pd(111) compared to Pt(111) to: 1) the higher contribution of HCOO* path to the overall activity, and 2) lower onset potential (by 0.18 V) of the indirect mechanism, which would contribute toward cleaning the surface from CO*.
To explore FAO's structure sensitivity on Pd, we next discuss our results on Pd(100). Onset potentials for formate, carboxyl, and indirect mechanisms (corresponding to water activation to OH*) are determined to be 0.24 V, 0.01 V, and 0.80 V, respectively. Notice that all these onset potentials are more positive than their counterparts on Pt(100), indicating less energy efficiency (i.e. higher overpotential) for Pd(100). The gap between carboxyl and formate onset potentials is 0.23 V, far larger than it is on Pd(111), thus significantly limiting the contribution of the HCOO* path toward the reactive flux. Compared to Pd(111), water activation is easier on Pd(100) by 0.25 eV. Accordingly, we predict that Pd(100) would show higher activity than Pd(111), because it is better at reactively removing CO*. Additionally, CO* is destabilized by 0.17 eV on the open facet (see free energies in Figures 2), which explains the higher collected current density during the forward scan of Pd(100), as compared to Pd(111).94
Before discussing the other metal surfaces, we will comment on some debated aspects of the FAO reaction mechanism on Pt and Pd surfaces.34 There have been contradicting reports about the role of formate, with some suggesting that it is the reactive intermediate,13,19–23,27 and others that it is a spectator species.32,33 Furthermore, carboxyl has been elusive to detect spectroscopically, making it difficult to be identified as an active intermediate.34 Although our computational model is simple with no detailed kinetic barriers in the electrochemical environment, we still can draw some insights from the thermochemistry alone. First, COOH* is elusive to detect because CO* is the more thermodynamically-preferred product. As discussed earlier, we consider CO* poisoning as direct evidence that COOH* is formed. The high CO* coverage should destabilize HCOO* formation and further COOH* formation, pushing the onset potentials for both direct mechanisms higher than they appear in Figures 1 and 2. Upon further increase of potential to the onset potential of the indirect path, the surface is freed of CO* and the reaction proceeds downhill to CO2(g). Since carboxyl is calculated to be more stable than formate on both Pt and Pd (based on calculated free energies), we predict that at high potentials, it would be carboxyl that carries the majority of the reaction flux, while at low potentials CO* poisons the surface. More informed assessments as to the contribution of various intermediates to the FAO reaction flux should come from detailed microkinetic modeling under relevant electrochemical conditions.
Having discussed the two most experimentally studied monometallic systems, we next discuss trends among the remainder of transition metals studied. To facilitate this discussion, we first investigate – for each fcc metal – the direct oxidation mechanisms, in order to determine the preference of each metal to either carboxyl or formate mediation, which gives insights into the propensity of each surface to accumulate CO* poison. Following this, we discuss the indirect oxidation mechanism for each metal, in order to understand how different metals would likely oxidize CO* to CO2(g). In parallel, we compare close-packed and open facets for each metal to elucidate the extent of FAO structure sensitivity. Since we do not study FAO structure sensitivity on hcp metals, we discuss them all at the end. Metals are arranged according to periodic table group in descending order and, within each group, by decreasing atomic number; an order that generally reflects increasing adsorption strength. Table I lists the free energies of key reaction intermediates relative to HCOOH(g) on all surfaces studied. Figure 3, Figure 4, and Figure 5 show the onset potentials for the direct oxidation via formate, the direct oxidation via carboxyl, and the indirect oxidation mechanism, respectively, on all surfaces studied. Supplemental Material contains the free energy diagrams on all surfaces studied at their calculated positive onset potentials.
Table I. Free energies of key intermediates relative to HCOOH(g) at 0.00 V on close-packed and open facets of transition metals. For CO*+OH* and CO*+H2O(g), energies are given at infinite separation of involved species. For COOH*, CO*+OH*, and CO*+H2O(g), stoichiometry is balanced with protons and electrons, which are not written explicitly here. For each metal facet, among all isomeric intermediates, the most stable isomer is indicated in bold. The close-packed facet for the hcp metals Co, Os, Ru, and Re is (0001); no open facet was studied for these metals (entry –). Metals are arranged according to periodic table group in descending order and, within each group, by decreasing atomic number. The final state of CO2(g)+2(H++e−) is surface-independent, and is calculated to be –0.26 eV relative to HCOOH(g) at 0.00 V.
| Free energy (eV) relative to HCOOH(g) at 0.00 V | ||||||||
|---|---|---|---|---|---|---|---|---|
| Metal | COOH* | HCOO* | CO*+OH* | CO*+H2O(g) | ||||
| (111) | (100) | (111) | (100) | (111) | (100) | (111) | (100) | |
| Au | 1.07 | 1.01 | 0.91 | 0.80 | 1.86 | 1.50 | 0.27 | 0.16 |
| Ag | 1.29 | 1.14 | 0.37 | 0.29 | 1.38 | 1.28 | 0.43 | 0.45 |
| Cu | 0.79 | 0.63 | −0.04 | −0.20 | 0.14 | 0.23 | −0.28 | −0.15 |
| Pt | 0.05 | −0.25 | 0.34 | 0.23 | 0.06 | −0.71 | −1.17 | −1.42 |
| Pd | 0.22 | 0.01 | 0.26 | 0.24 | −0.36 | −0.44 | −1.41 | −1.24 |
| Ni | 0.26 | −0.03 | −0.12 | −0.39 | −0.90 | −1.13 | −1.26 | −1.24 |
| Ir | −0.21 | −0.43 | −0.15 | −0.28 | −0.59 | −1.11 | −1.44 | −1.43 |
| Rh | −0.06 | −0.19 | −0.11 | −0.22 | −0.66 | −0.90 | −1.34 | −1.28 |
| Co | 0.23 | − | −0.28 | − | −0.93 | − | −1.08 | − |
| Os | −0.31 | − | −0.63 | − | −1.08 | − | −1.46 | − |
| Ru | −0.16 | − | −0.56 | − | −1.07 | − | −1.28 | − |
| Re | −0.09 | − | −0.67 | − | −1.21 | − | −1.11 | − |
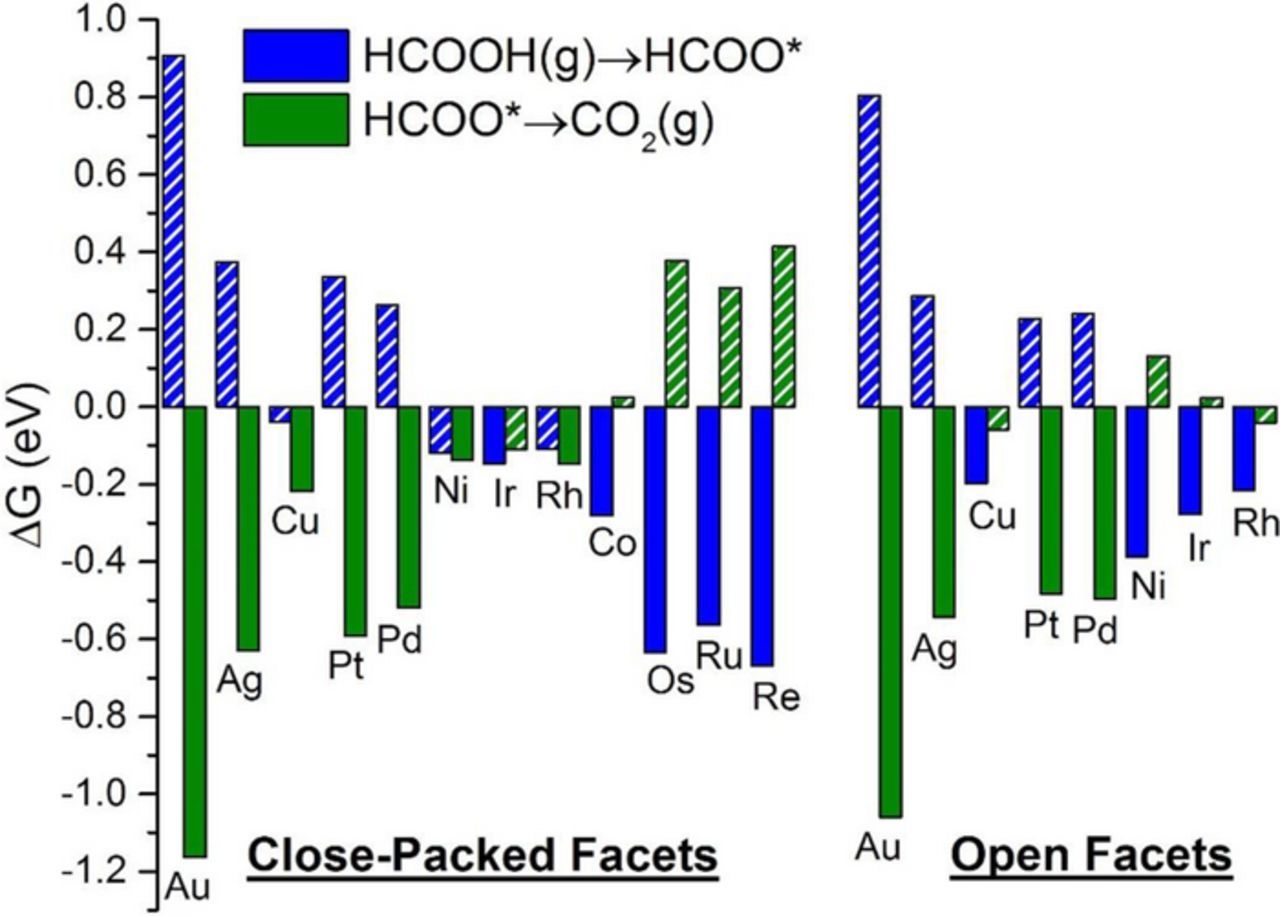
Figure 3. Free energy change for the elementary steps of direct oxidation via formate on the close-packed and open facets of metals studied. For each surface, blue bars correspond to formate formation from FA; green bars correspond to deprotonation of formate to CO2(g). The PDS for each surface is the more endergonic or less exergonic step, and is represented by the hatched bar. For each elementary step listed in the legend, stoichiometry is implicitly balanced with protons and electrons.
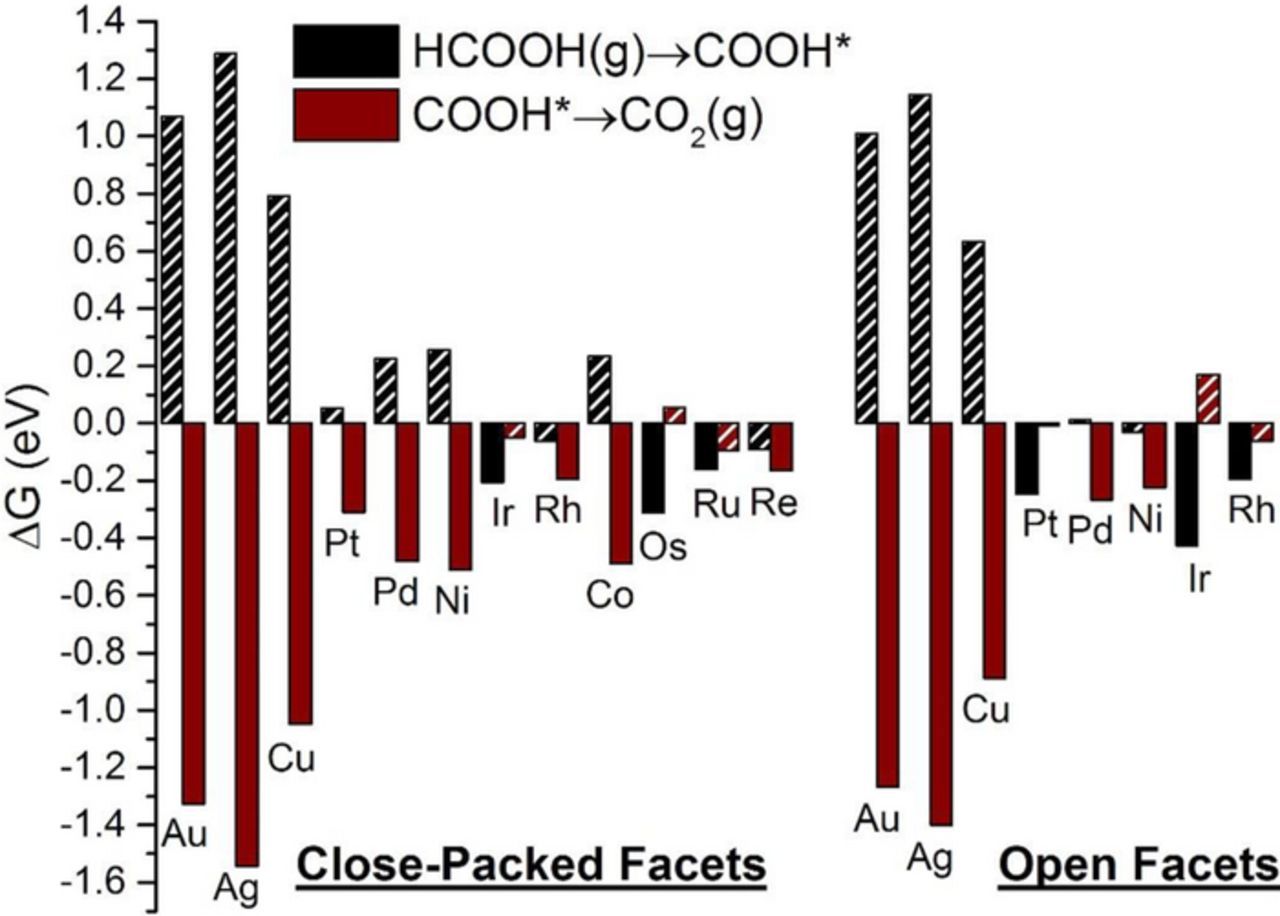
Figure 4. Free energy change for the elementary steps of direct oxidation via carboxyl on the close-packed and open facets of metals studied. For each surface, black bars correspond to carboxyl formation from FA; brown bars correspond to deprotonation of carboxyl to CO2(g). The PDS for each surface is the more endergonic or less exergonic step, and is represented by the hatched bar. For each elementary step listed in the legend, stoichiometry is implicitly balanced with protons and electrons.
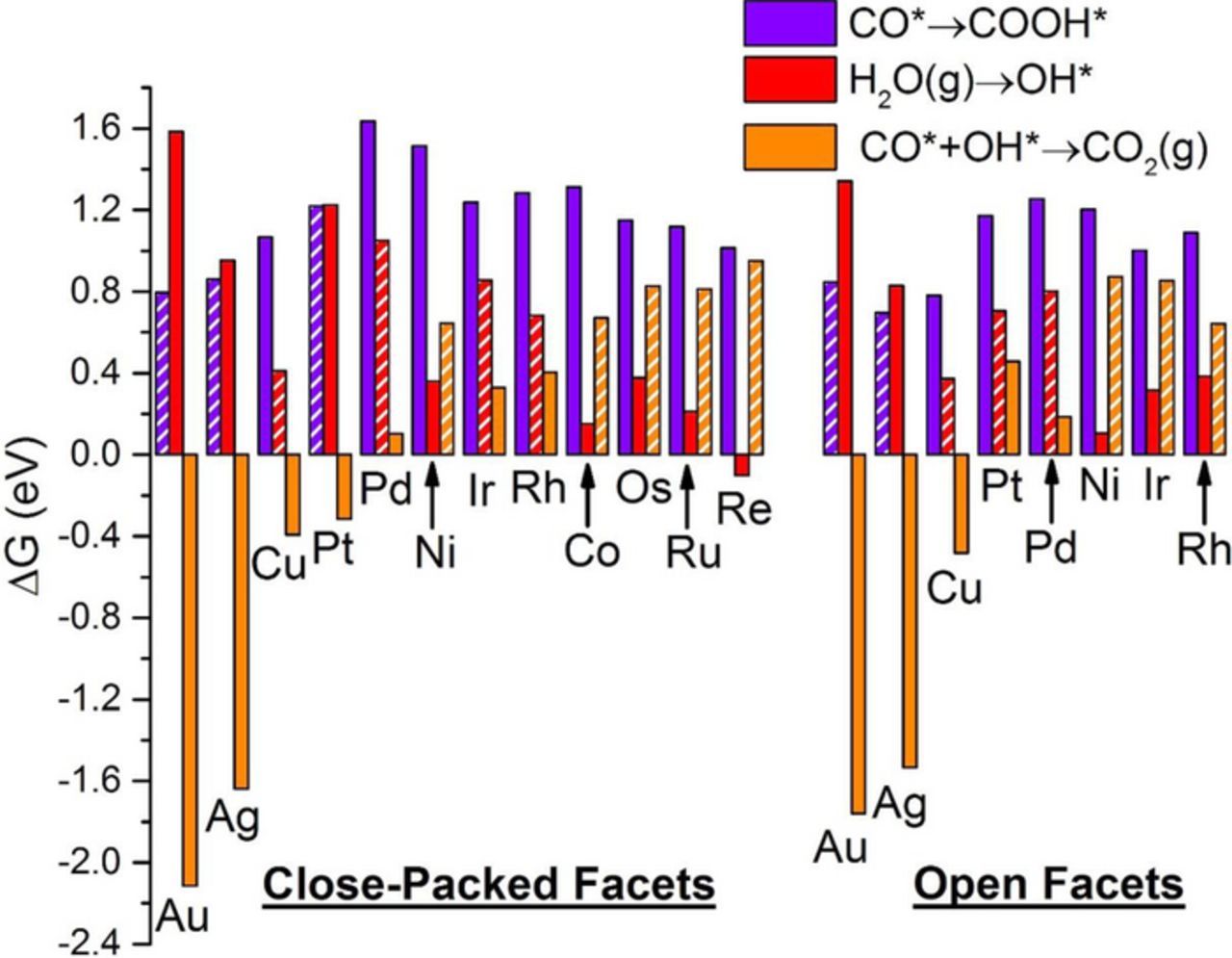
Figure 5. Free energy change of elementary steps involved in CO* removal. Violet corresponds to formation of COOH* from CO*, red corresponds to water activation to form OH*, and orange corresponds to CO* oxidation to CO2(g) via OH*. For each surface, the hatched bar represents the PDS for the indirect mechanism. For each elementary step listed in the legend, stoichiometry is implicitly balanced with water, protons and electrons.
Structure sensitivity of FAO on Au
On Au(111), the onset potentials (Figure 3 and Figure 4) for the formate and carboxyl paths are 0.91 V and 1.07 V, respectively; see Figure S5 for free energy diagrams at the onset potentials. The high onset potentials for either mechanism on Au(111) indicate its poor activity toward FAO. This is in agreement with Capon and Parsons90 who observed that FAO on Au anodes only takes place at high overpotentials at which an oxide phase of Au is formed. In turn, this can generally be attributed to the weak interaction of Au surfaces, specifically Au nanoparticles larger than 5 nm,95 with adsorbates. As depicted in Figure 3 and Figure 4, for Au(100), we calculate onset potentials of 0.80 V and 1.01 V for the formate and carboxyl paths, respectively; see Figure S6 for free energy diagrams at the onset potentials. Therefore, Au(100) stays largely inactive toward FAO compared to either facet of Pt or Pd. We observe that the formate path is dominant on either Au facet. This comes in agreement with a recent ATR-SEIRAS study of FAO on Au, in which Cuesta et al.29 determined formate as the reactive intermediate. They, however, argue based on electro-kinetic data, that formate oxidation to CO2 is the rate determining step, similarly to what we suggested previously for FA vapor-phase decomposition on Au.96 A more rigorous scheme for calculating barriers of electrochemical steps, as well as microkinetic modeling, will be needed to extract definitive conclusions on the mechanism on a given surface. Our calculations of the thermochemistry suggest that formate deprotonation is exergonic, and that the main challenge for the weak-adsorbing Au catalysts is to form formate in the first place. It is due to this challenge that the (100) facet shows lower onset potentials – for either direct mechanism – than the (111) facet. This is the case because the PDS for both mechanisms on either Au facets results in the formation of an adsorbed species, a step that is less endergonic on the open facet. Accordingly, FAO shows structure sensitivity on Au, as the dominant path (formate) is calculated to have a lower onset potential on the (100) facet than the (111) facet by ∼0.10 V. Yet, both Au facets would appear largely inactive compared to Pt or Pd.
We do not consider the indirect mechanism to be relevant on either facet of Au for the following reasons: 1) HCOO* is more easily formed than COOH*, the CO* precursor, and 2) the formation of CO* from COOH* is endergonic at the onset potential of the carboxyl path; see Table I, Figure S5, and Figure S6. We conclude that both facets of Au will be free of CO* poisoning. In this respect, it is worth mentioning that Cuesta et al.29 could not detect any CO* on an Au anode, as expected from our theoretical results.
Structure sensitivity of FAO on Ag
Compared to Au, the onset potential for the formate path imparts higher activity to Ag, especially for the open facet. The onset potential for the formate path is 0.37 V on Ag(111), and 0.29 V on Ag(100); see Figure 3, Figure S7, and Figure S8 for free energy diagrams at the onset potential. Compared to the corresponding Au facets, both facets of Ag: 1) destabilize COOH*, 2) stabilize HCOO*, and, thus, 3) have much stronger preference to HCOO* than to COOH*. This stronger preference to formate on Ag, relative to Au, reflects the stronger binding of adsorbates binding through O (HCOO*, OH*, and O*), compared to those binding through C (COOH*, CO*, and C*) on Ag.41,48,49,51 This trend suggests that the carboxyl path may not contribute to any observed current on Ag anodes, as indicated by the large gap in calculated onset potentials for the formate and carboxyl paths. Similar to what we found on Au, the Ag(100) facet shows lower onset potentials toward the formation of adsorbed species, indicating strong structure sensitivity for FAO on Ag. Owing to the high onset potentials for the carboxyl mechanism (1.29 V on Ag(111) and 1.14 V on Ag(100); see Figure 4, Figure S7, and Figure S8), we predict that Ag surfaces, like Au surfaces, would be free of CO* poison, especially because forming CO* from COOH* is endergonic at the onset potential of the carboxyl mechanism; see Table I, Figure S7, and Figure S8. Consequently, the indirect mechanism on Ag surfaces appears to be less relevant and is not discussed further. Despite being CO* free while retaining reasonable onset potentials for the formate mechanism, Ag is likely to experience dissolution and/or oxidation in acidic media,97 which would tend to diminish Ag's relevance for practical application in FAO.
Structure sensitivity of FAO on Cu
While the formate mechanism proceeds downhill on both facets of Cu at 0.00 V (Figure 3), the carboxyl mechanism is characterized by high onset potentials: 0.79 V and 0.63 V on the (111) and (100) facets, respectively. The high onset potentials originate from the difficulty of activating FA to COOH* (Figure 4), hence the lower onset potential on Cu(100), which stabilizes COOH* with respect to Cu(111). At the high onset potentials of the carboxyl mechanism on either facet, CO2(g), not CO*, becomes the more thermodynamically-favorable product of the COOH* decomposition; see Figure S9 and Figure S10. As a result, CO* is not predicted to poison Cu catalysts. Like Ag, the tolerance toward CO* poisoning of Cu does not originate from its weak binding and inability to activate FA, but rather from its strong preference to forming HCOO*. This comes in agreement with a recent study of FA vapor-phase decomposition on Cu catalysts, where HCOO* was found to be the dominant reactive intermediate.98 Owing to the stronger stabilization of HCOO* on the (100) facet, Cu(100) should show higher activity than Cu(111). However, Cu, like Ag, exhibits poor oxidation resistance99 and will likely dissolve in electrochemical environments, thus limiting its practical use in monometallic anodes of DFAFCs.
Structure sensitivity of FAO on Ni
We first consider the formate path. Table I indicates higher stability of HCOO* relative to COOH*, on both facets of Ni, with HCOO* being more stable on Ni(100) compared to Ni(111). Interestingly, this higher stability of formate on the open facet does not correspond to lower onset potential for the formate path (see Figure 3, Figure S11, and Figure S12); while the formate path proceeds downhill on Ni(111) at 0.00 V, it is not activated on Ni(100) until 0.13 V. The fundamental reason behind this result is the different PDS for the formate path on the two facets. While Ni(111) adsorbs and dissociates HCOO* spontaneously, Ni(100) adsorbs HCOO* strong enough that its dissociation to CO2(g) is the most endergonic step of the formate path. On the other hand, the carboxyl path has the formation of COOH* as the PDS for both Ni facets (see Figure 4, Figure S11, and Figure S12), therefore showing a higher onset potential (0.26 V) for Ni(111), while proceeding spontaneously on Ni(100). Hence, while we predict that formate is the active intermediate of FAO over Ni(111), we predict that it will result in HCOO* spectators on Ni(100), poisoning the surface at low potentials. Given that COOH* is not spontaneously formed on Ni(111) at 0.00 V, we predict a narrow operating window via the formate path at potentials below 0.26 V, beyond which COOH* is formed and, in turn, CO* poisons the surface.
Ni also shows interesting features for the indirect mechanism; see Figure 5, Figure S11, and Figure S12. Water activation (H2O(g)→OH*) is endergonic on both facets of Ni, although less so on the open facet which stabilizes OH*. However, water activation is not the PDS on either facet; rather CO*+OH*→CO2(g) is. While water activation needs 0.36 V on Ni(111), the PDS needs up to 0.64 V to be activated, thus setting the onset potential of the indirect mechanism. Similarly, the onset potential of the indirect mechanism on Ni(100) is 0.87 V, while water activation to produce OH* needs only 0.11 V. The higher onset potential of the indirect mechanism on Ni(100) again points out to the stronger stabilization of both CO* and OH* on the open facet relative to the close-packed facet.
The discussion above reveals that FAO is structure sensitive on Ni catalysts. Not only do onset potentials change, but also the PDS of the formate mechanism depends on the Ni facet. We predict that Ni(111) will be active predominantly via the formate path at low potentials, where carboxyl formation is uphill. At moderate potentials, carboxyl would be formed, resulting in CO* which can be removed at 0.64 V. On the other hand, Ni(100) would have to free the surface from accumulating HCOO* and CO*, with the latter not leaving the surface until 0.87 V. Therefore, Ni(111) is predicted to be less-poisoned and more active than Ni(100).
Structure sensitivity of FAO on Ir
As shown in Figure 3, Figure 4, and Figure S13, both direct mechanisms proceed spontaneously to CO2(g) at 0.00 V on Ir(111). This is not the case on Ir(100); see Figure 3, Figure 4, and Figure S14, where the deprotonation of HCOO* or COOH* to CO2(g) requires 0.02 V and 0.17 V, thereby setting the onset potentials of the formate and carboxyl paths, respectively. For the indirect mechanism on Ir(111), FAO needs to overcome an onset potential of 0.86 V to activate water to OH* in order to oxidize the readily-formed CO*. Ir(100), however, activates water at a much milder potential of 0.32 V (see Figure 5, Figure S13, and Figure S14), but needs 0.85 V to oxidize CO* with the formed OH*. We conclude that FAO is structure-sensitive on Ir, showing different PDS (for the indirect mechanism) and different onset potentials on each facet. Ir(100), which stabilizes all reaction intermediates relative to Ir(111), is predicted to be more poisoned, as corroborated by the lower current in the forward potential scan on Ir(100).100
Structure sensitivity of FAO on Rh
The FAO reaction mechanism on Rh(111) is very similar to that on Ir(111); both direct mechanisms proceed spontaneously at 0.00 V. Unlike Ir(100), which stabilizes HCOO* and COOH* enough to show positive onset potentials for their activation, Rh(100) retains their spontaneous dissociation to CO2(g) despite stabilizing them relative to Rh(111), as shown in Figure 3, Figure 4, Figure S15, and Figure S16. For the indirect mechanism, the PDS is different on the two facets; see Figure 5, Figure S15, and Figure S16. While Rh(111) requires 0.68 V to activate water to OH* (followed by the spontaneous removal of CO* with the formed OH*), OH* formation proceeds much easier on Rh(100) at 0.39 V. However, the strongly bound CO* and OH* are not removed from the Rh(100) surface until 0.64 V. Accordingly, both surfaces will suffer from CO* poisoning, which agrees with cyclic voltammograms on both surfaces at low potentials,101 as well as spectroscopic evidence for CO* on Rh electrodes.102
Before discussing our results on the hcp metals, we conclude this section on the fcc metals by comparing Ni, Ir, and Rh to Pt and Pd. For the more stable close-packed facet, Ni, Ir, and Rh all show lower onset potentials for the indirect mechanism than Pt or Pd. However, Ni is known to leach in acidic media typical of DFAFCs.103 Hence, the advantage of high activity of Ni(111) is offset by its low stability. Additionally, Ir and Rh are shown to be poor FAO catalysts due to a limited double layer region,90 the effect of which is largely neglected in our model. Moreover, Ir and Rh are known for their strong binding of adsorbates, as well as their propensity to form oxides at low potentials.100 Our results confirm that OH* and HCOO* bind more strongly on either facet of these two metals relative to Pt and Pd. Additionally, CO* is adsorbed strongly on either facet of Ir and Rh. In fact, because Ir and Rh do show some activity toward FAO while being extensively covered by CO*, it has been suggested104 that CO* should not be thought of as a poison, but rather a reaction intermediate that tangibly contributes to collected currents. This is inferred from slow 13CO/12CO isotopic exchange at low potentials, but fast 13CO/12CO isotopic exchange at potentials beyond that of FAO onset.104 Further, the open facets of Pt or Pd are more energy-efficient than the open facets of Ni and Ir, which tend to get more easily poisoned by reaction intermediates than Pt or Pd do. This leaves Pt and Pd as the only monometallic catalysts among those studied here so far, which strike a compromise between activity and stability.34
FAO on hcp metals: Co, Os, Ru, and Re
We studied the (0001) facet of these four hcp metals. Co(0001) prefers HCOO* to COOH* by 0.51 eV, rendering the formate path as the dominant path at low applied bias; the onset potential for the formate path (corresponding to decomposing HCOO* to CO2(g)) is only 0.02 V (see Figure 3 and Figure S17). At 0.23 V, the carboxyl path is activated, spontaneously poisoning the surface with CO* (see Figure 4). Hence, similar to Ni(111), we predict a narrow operating window for Co(0001) between 0.02 V and 0.23 V. Water is easily activated at 0.15 V, leaving CO* oxidation with OH* to be the PDS of the indirect mechanism with an onset potential of 0.67 V (see Figure 5 and Figure S17).
As shown in Figure 3, all remaining three hcp(0001) surfaces are characterized by too strong binding of HCOO* such that CO2(g) formation is potential-determining for the formate mechanism. The onset potential for the formate mechanism is 0.38 V for Os (Figure S18), 0.31 V for Ru (Figure S19), and 0.41 V for Re (Figure S20). By contrast, COOH* is less stable than HCOO* on all these surfaces, yielding a spontaneous deprotonation of COOH* to CO2(g) on Ru and Re (see Figure 4, Figure S19, and Figure S20), and a slightly endergonic COOH* deprotonation to CO2(g) (by 0.05 V) for Os (see Figure 4 and Figure S18). As we move from Os, to Ru, to Re, CO* binding weakens, while OH* binding gets stronger.41,48,51 The net effect is increased poisoning by oxygenated species – most likely atomic O*41,105–107 – as we move from Os, to Ru, to Re, again indicating the propensity of these surfaces to form oxides. The weakening trend of CO* binding is also reflected in weaker COOH* binding, and thus the observed declining onset potential for the COOH* path (corresponding to COOH* deprotonation to CO2(g)) as we move from Os (0.05 V), to Ru and Re (spontaneous). Nevertheless, it is still exergonic to form COOH* followed by CO*, which is likely to poison the surface, together with O*/OH*. The PDS of the indirect mechanism on all three surfaces, as shown in Figure 5, is CO*+OH*→CO2(g), with onset potentials of 0.83 V on Os (Figure S18), 0.81 V on Ru (Figure S19), and 0.95 V on Re (Figure S20).
To conclude the discussion of FAO on hcp metals, we note that Os, Ru, and Re tend to get more poisoned by reaction intermediates due to the strong stabilization of various adsorbates on their surfaces. Although Co shows moderate binding characteristics (it binds the weakest among the hcp metals), much like Ni, it too suffers from poor corrosion resistance in acidic media.108
We conclude our discussion of onset potentials by recognizing the Sabatier principle, which states that an optimal catalyst would bind adsorbates not too strongly and not too weakly. In a descending order of periodic table grouping, Au and Ag bind adsorbates too weakly, rendering them poor catalysts for FAO because of their inability to activate HCOOH(g). On the other hand, surfaces that bind adsorbates too strongly are likely to get poisoned by reaction intermediates, or develop oxide phases, also rendering them largely inactive toward FAO. With Ni and Co being largely unstable under acidic media, Pt and Pd are the only monometallic catalysts that strike the delicate balance between activity and stability.
Figure 6 summarizes the onset potentials (Figures 3–5) of all three mechanisms on all surfaces studied. An interesting general trend is observed; the open facets of the noble metals (Au, Ag, Cu), and of Pt and Pd have lower overpotentials, thereby offering higher energy-efficiency, than their close-packed facets, while the opposite is true for the more reactive metals: Ni, Ir, and Rh (see Figure 6). Further, Au, Ag, and Pt(111) require unpractically high overpotentials to activate water for the indirect mechanism. These trends are attributed to the fact that the close-packed facets of the less reactive metals (Au, Ag, Cu, Pt, and Pd) bind adsorbates too weakly, while the open facets of the more reactive metals (Ni, Ir, and Rh) bind adsorbates too strongly. Accordingly, the PDS on close-packed surfaces of the less reactive metals have their PDS yielding adsorbed species, whereas open facets of the more reactive metals have their PDS yielding gas-phase species from adsorbed intermediates.
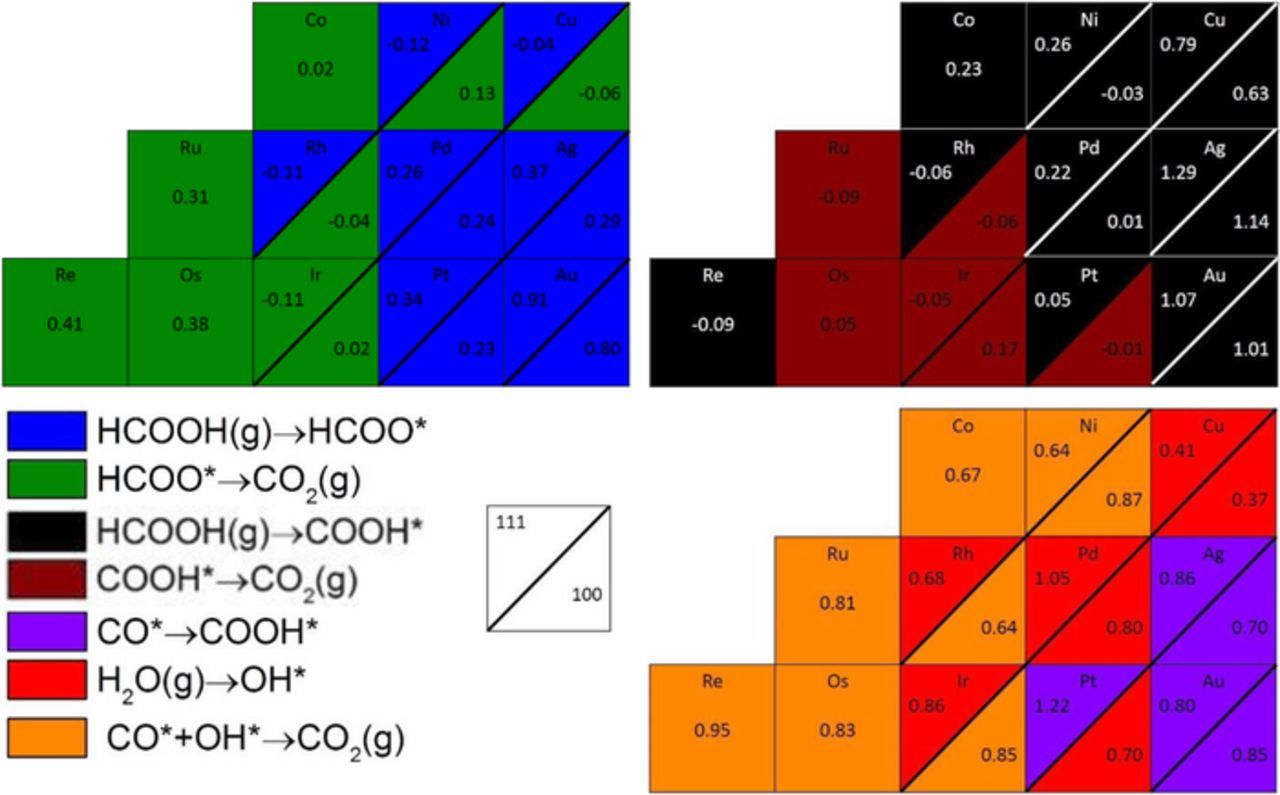
Figure 6. A summary of Figures 3–5; onset potentials for: direct oxidation via formate mechanism (top-left), direct oxidation via carboxyl mechanism (top-right), and indirect oxidation mechanism (bottom-right). Within each split box, the top left corner displays the onset potential of the respective mechanism for the close-packed facet, while bottom right corner displays the onset potential of the respective mechanism for the open facet. Negative onset potentials indicate spontaneous paths at 0.00 V. All boxes are color-coded according to their PDS as described in the legend. Water, protons, and electrons balance the elementary steps in the legend, and are not written explicitly.
Phase diagrams
In this section, we formulate the insights gained above into phase diagrams (or volcanoes) at 0.0 V and 0.6 V (a value chosen high enough to show the effect of applied bias, but low enough to avoid the potential region of oxide formation (∼0.80 V) on Pt13,16 and Pd),27 see Figure 7, showing the activity and rate determining steps on a continuous spectrum, which may enable strategies for improved catalyst design. We start by developing linear scaling relations (see Supplemental Material, Figures S21 and S22) between the free energies (relative to CO2(g), H2O(g), and H2(g)) of COOH* and HCOO* and those of CO* and OH*, respectively. This allows us to map out the complete energetics of the reaction network using only values for the free energies of CO* and OH*. Such scaling relations have been shown to exist109 among carbon-containing species and among oxygen-containing species, and have been used extensively in the literature.42,48–52 Consequently, the free energy change of all elementary steps could be readily calculated at a given pair of the free energies of CO* and OH*, rendering these two as reactivity descriptors for FAO. The overall phase space is divided into regions defined by rate determining steps, as estimated from scaling relations. We note that the free energies of CO* and OH* correlate poorly to each other, leading to the need for these two distinct descriptors. It is relevant to note that these two descriptors have also been found to dictate the energetics of the electrooxidation of methanol,48–50 dimethyl ether,51,52,55 and ethanol and ethylene glycol,57 thus establishing the free energies of CO* and OH* as near-universal descriptors for the activities of metallic surfaces toward the electrooxidation of small oxygenate molecules.
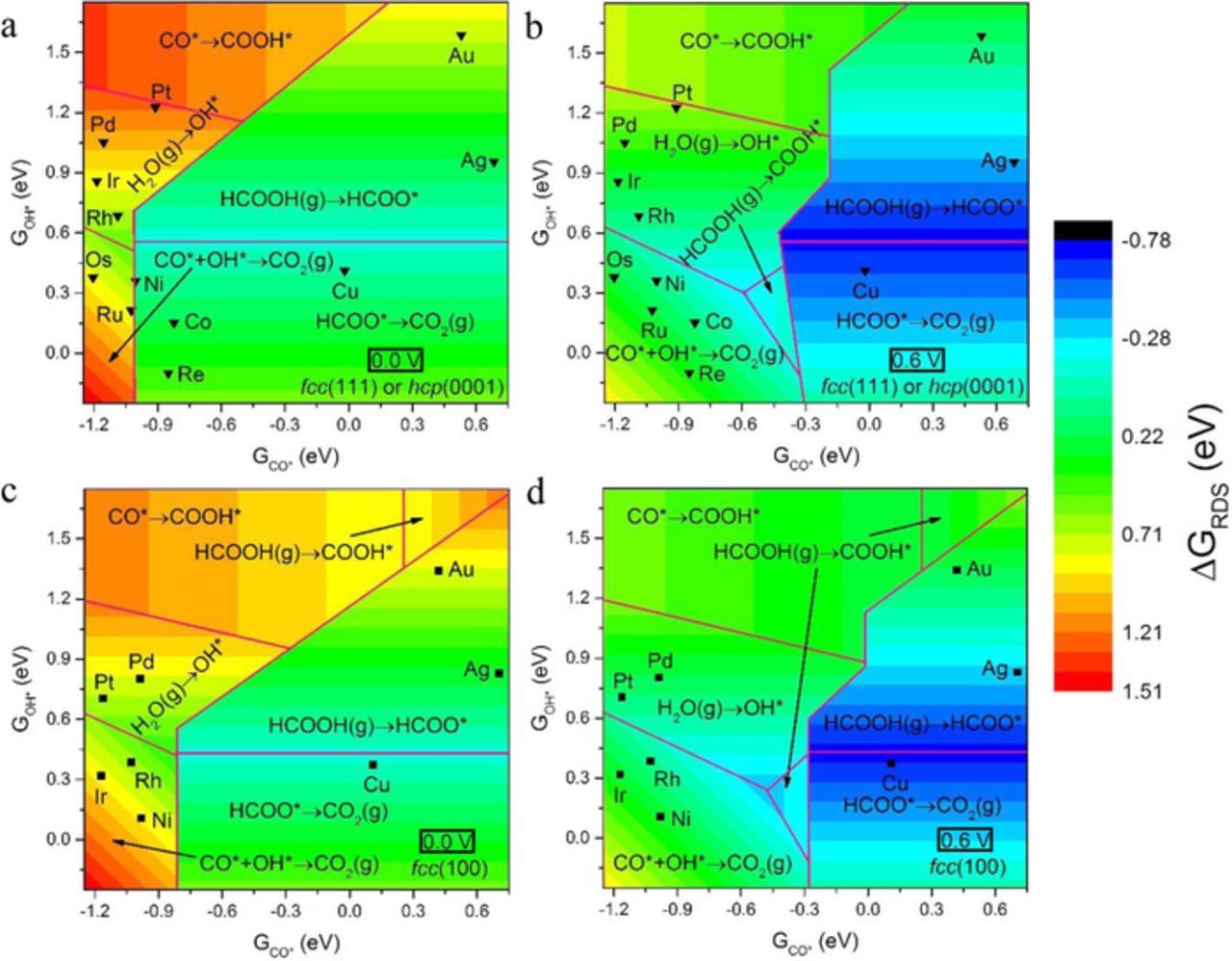
Figure 7. Calculated phase diagrams for FAO on close-packed facets a) at 0.0 V and b) at 0.6 V, and open facets c) at 0.0 V and d) at 0.6 V of transition metals. Linear scaling relations between reactivity descriptors (GCO* and GOH*) and (GCOOH* and GHCOO*) allow the description of reactivity across the whole phase space. The free energy of the most endergonic step is signified by the color code explained in the right hand side bar, as a function of the two reactivity descriptors. The pink lines divide the phase space into regions by the nature of the most endergonic step. Metals are scattered on the phase space (inverted triangles for close-packed facets, and squares for open facets) according to their values of the reactivity descriptors, as calculated with DFT. Because of the approximate nature of the underlying scaling relations, the rigorously calculated ΔGRDS for specific surfaces could differ from what is shown here.
In what follows, we analyze the various regions in these phase diagrams, starting with the low applied voltage (0.0V) cases first (Figures 7a and 7c). The top right corners of the phase diagrams represent regions where the binding of both CO* and OH* is weak, while bottom left corners represent strong binding of both species. In the top right side of the volcano, the high values of the descriptors (GCO* and GOH*) indicate weak enough binding of COOH* and HCOO*, so that CO* formation is not feasible, leaving the direct oxidation via carboxyl or formate paths to dominate reactivity. Following their weak binding, COOH* or HCOO* are readily oxidized to CO2(g), leaving the first deprotonation step of FA as rate determining. It is important to note that HCOO* is more stable than COOH* for the close-packed facets (Figure 7a) in the top right corner of the phase space, while the opposite is true for the open facets, giving rise to the region HCOOH(g)→COOH* in Figure 7c. As we descend vertically from this region along stronger binding of OH* (Figures 7c), the binding of HCOO* gets stronger until it becomes more stable than COOH*. Accordingly, the mechanism changes into the direct oxidation via formate. At intermediate binding of OH* (see Figures 7a and 7c), HCOO* is bound weakly enough to readily decompose to CO2(g), rendering HCOO* formation the rate determining step. As the binding of OH* moves to less positive values, the binding of HCOO* gets stronger such that its deprotonation to CO2(g) – rather than its formation from FA – becomes rate determining.
A similar path can be taken, starting from the top right corners and moving left along stronger binding of CO* (or interchangeably COOH*). As we move left, COOH* formation no longer becomes unfavorable (as in Figure 7a) or limiting (as in Figures 7c). At such values of GCO*, it is downhill to form CO*, thus signaling a change to the indirect mechanism of FAO. In the top left corner, OH* binding is much weaker than that of CO*, leaving CO* removal to depend on COOH*, which is stable enough following its linear correlation to CO*. As a result, the rate determining step in the top left corner is the back-formation of COOH* from CO*.
Moving along a vertical line from the top left corner to stronger binding of OH* makes water activation (to OH*) more feasible than the formation of COOH* from CO*. Therefore, water activation to OH* becomes rate determining at intermediate binding of OH*. As we transition to the left bottom corner, the binding of OH* becomes even stronger, such that its formation from water is no longer difficult. Together with the strongly-bound CO*, the rate determining step becomes the removal of these two strongly bound species.
Before we describe the higher applied voltage cases, it is instructive to compare the low bias phase diagrams (0.0 V) for close-packed and open facets (Figures 7a and 7c). Starting with similarities, a shallow peak exists at the boundaries of the two rate determining steps of the direct oxidation via formate mechanism (the light cyan region), at an intermediate binding of OH* (or interchangeably, HCOO*). Once CO* binding becomes strong enough to make COOH* favorable to HCOO*, the mechanism shifts into the indirect oxidation pathway, which is inactive because a catalytic surface would be poisoned by CO* at low potentials, since all steps that might remove CO* are more endergonic or less exergonic than those involved in the direct oxidation via formate mechanism. This is manifested in the apparent discontinuity in the phase diagrams, where the peak in the formate mechanism abruptly ends when the indirect mechanism is invoked. Although both facets are qualitatively similar, there are some subtle differences, brought about mainly by the quantitatively distinct linear scaling relations used for the two types of facets. Firstly, the border between CO*→COOH* and H2O(g)→OH* regions is shifted downward for the open facets, again due to the stabilization of COOH* on the open facets, which allows the region of CO*→COOH* to expand at the expense of that of H2O(g)→OH*. Generally, open facets stabilize COOH* more than HCOO*, when compared to close-packed facets. In fact, our linear scaling relations predict that HCOO* would rather be marginally destabilized on the open facets relative to the close-packed facets, in the free energy sense. We remind here of the approximate nature of the linear scaling relations, especially that the scaling relations of the close-packed facets show mean absolute errors greater than 0.10 eV (Figure S21). As a consequence, the formate mechanism retreats on the open facets (Figure 7c), and gives way to larger regions for the indirect mechanism.
We now turn our attention to the phase diagrams developed for higher applied voltage: 0.6 V (Figures 7b and 7d). As the potential is increased to higher positive bias, all faradaic steps become more exergonic, and the more negative region (blue color) of ΔGRDS becomes accessible. The formate regions retreat for both facets compared to low bias (e.g.: for 0.0 V), since at high bias COOH* is formed at moderate GCO*, giving rise to the carboxyl and indirect mechanisms. The high bias cases show similar regions as do the low bias cases, with the notable exception of the region HCOOH(g)→COOH*, which emerges at the stronger binding end of GOH* at high bias. In this region, CO* is not stabilized enough to make CO*+OH*→CO2(g) rate-determining, nor is OH* destabilized enough to make its formation rate-determining. The emergence of this new region at high bias gives rise to a shallow peak, which is less active than the horizontal peak at the interface between the two steps of the direct formate path.
It is instructive to characterize the regions of higher activity, or the tip of the volcano. At low potentials, we notice a definite active region at the interface between the two regions of the formate path. However, the carboxyl region does not show such a peak, since any surfaces that form carboxyl at the low potential would likely be inactive due to CO* poisoning. When the problem of CO* poisoning is alleviated at high potentials however, one notices two active peaks; one in each of the formate or carboxyl regions, with the peak in the latter region being less active. We note that the region of maximum activity, or interchangeably, more negative ΔGRDS, takes place at intermediate binding of CO* or OH*. This is in accord with the Sabatier principle, stating that an active catalyst should bind adsorbates neither too weakly nor too strongly. Relative to the optimal values of the two descriptors, more positive free energies of GCO* or GOH* indicate weaker binding to the surface, and thus less activity in initiating the reaction. On the other hand, more negative values of the descriptors indicate much stronger binding of reaction intermediates, which poisons the catalyst and inhibits the catalytic cycle. We also note that the less active peak takes place at stronger binding of CO* as compared to the more active peak, indicating the higher activity of surfaces that bind CO* less strongly.
Before attempting to utilize the volcano plots of Figure 7 for designing improved FAO catalysts, we wish to emphasize two important aspects of the catalyst design problem, which are not accounted for in these phase diagrams: (i) catalyst stability and metal dissolution potential, which could be determined in suitable durability tests; and (ii) metal oxide formation. Both are challenges that many metals are faced with under typical DFAFCs operational conditions.110
Strategies for catalyst design
We start by noting that all close-packed and open facets of the metals studied have been placed in the four panels of Figure 7, based on their DFT-calculated GCO* and GOH*. The linear scaling relations used for preparing the volcano plots in Figure 7 introduce some error due to their approximate nature (Figure S21 and Figure S22). The scatter in these linear scaling relations is partially attributed to the different preferred adsorption sites of the correlated species on the different surfaces.109 We provide specific geometric information on preferred adsorption configurations in Figures S23–S27, and in Tables S1-S4. As a result, true areas of higher activity and true regions defining the rate-determining steps (RDS) may differ from what is shown in Figure 7. Nevertheless, Figure 7 is still useful in informing strategies for catalyst design.
Pt(111) is the non-group-11 metal with the weakest CO* binding among the close-packed facets, while this is the case for Pd(100) among the open facets. Although these two surfaces bind CO* relatively weakly, they also bind OH* weakly, resulting in their inefficiency at activating water. The latter is reflected in the high onset potential calculated for that step (Figure 5). This points to a key challenge for DFAFC anode catalysts allowing the carboxyl pathway: for them to be better than Pt or Pd in FAO, they should be able to activate water easier than Pt or Pd, a condition analogous to what has been suggested for methanol electrooxidation.48,49 At the same time, they should be able to efficiently remove OH*. Our results suggest that the hcp metals, as well as Ni(100), Ir(100), and Rh(100) all bind OH* and CO* too strongly, as demonstrated by their PDS for the indirect mechanism; see Figure 5. We also note that improved catalysts allowing the COOH* path should also exhibit weaker binding of CO* (together with stronger binding of OH* to promote water activation), more so for the open facets which have their optimal GCO* less negative (i.e. at weaker binding) than their close-packed counterparts.
Figure 7 suggests that the activity of Pt or Pd could be improved by alloying with metals characterized by stronger binding of OH*, e.g.: Ir, Ni, Co, Ru. Following a simple interpolation logic,111 one might expect such alloys to exhibit GOH* intermediate between those of the two alloying elements. Alternatively, two alloyed elements could infer synergistic effects on each other, rendering the descriptors' values for the alloy beyond the range limited by the individual's values for the same descriptors. Cai and coworkers34 have recently reviewed improved Pd and Pt alloy catalysts for FAO. Figure 7 suggests that both facets of Cu and Ag lie in a region of low FAO overpotential, but both metals suffer from low stability in the acidic environment of FAO. One possible strategy for identifying improved FAO electrocatalysts would be to alloy these metals with other more stable metals (e.g.: Pd or Pt), so that their stability is enhanced while maintaining local ensembles of active sites rich in Cu/Ag. Indeed, higher activities and better stabilities were reported for PtCu,112 PdCu,113 PtAg,114 and PdAg115 alloys, compared to the corresponding monometallic Pt or Pd catalysts.
Enhanced activity has also been reported for PdCo,116,117 PdIr,118 PdNi,119,120 and PtPd.121,122 In addition, three-dimensional PtCo nanowire assemblies123 have shown improved performance for methanol electrooxidation. Since we have identified similar requirements for improved catalysis for methanol electrooxidation and FAO (i.e. the requirement to oxidize accumulating CO*), we expect catalysts showing improved behavior for one to also improve the other reaction. This has also been suggested for the bifunctional PtRu alloy,124 as well as PdRu.125 Enhanced activity has largely been attributed to improved CO* tolerance and/or easier water activation. The improved CO* tolerance can also be a result of ensemble effects, which discourage the dehydration pathway due to the lack of favorable geometrical arrangements of Pt atoms toward CO* formation. This has been reported for PtPd122,126 and PtAu127–131 alloys. More successful examples of Pd-based alloys have also been recently reviewed,132,133 while Demirci reviews the theoretical tools for screening bimetallic anode catalysts for electrochemical oxidation of liquid fuels, including FA.134 Skillful inorganic synthesis plays a major role in realizing the predicted improved active sites by tuning their morphology and composition.135,136 Accordingly, there have been some excellent recent reviews of this research area.135,137,138
On the other hand, not all alloying attempts have been successful in improving activity. For instance, Pd90Ir10 and Pd90Pt10 alloys139 were found to be more poisoned by CO* than Pd. A later study identified Pd79Ir21 as more active and CO* tolerant than Pd,118 whereas a previous study has identified a Pd90Pt10 alloy as most active among a series of PdxPt1-x compared to Pd.122 These studies speak to the importance of tuning atomic ratios as well as shapes of bimetallic alloys toward improving electrocatalyst activity and stability.
In addition to bulk alloys, near-surface alloys (NSAs) have gained popularity due to their unique properties and controllable syntheses. Defined as alloys in which the solute element exists near the surface at concentrations different from those in the bulk,140 the solute could form an overlayer structure, or shell in nanoparticles, on top of the host/core metal. Reactivity of the overlayer/shell metal is modulated by the core metal through strain and ligand effects.141,142 In the context of FAO, Kibler et al.143 have pseudomorphically deposited a monolayer of Pd atop a number of (111) single crystals of transition metals, as well as a PtRu bulk alloy. While some systems exhibited inferior activities compared to pristine Pd, the monolayers on Pt and Au exhibited higher current density than that of pure Pd(111). The Pd monolayer on the PtRu alloy far exceeded the activity of all systems studied.
In addition to electronic and geometrical modification of the overlayer metal, its deposition only in the near surface increases its dispersion and improves its utilization efficiency.35 The synthesis of these overlayer structures with a controlled number of layers has been well-established,144,145 and have been found to be more stable than pristine metals in electrochemical environments.146 Accordingly, Pt and Pd overlayers on transition metals could be a promising class of catalysts for FAO. We are currently exploring reactivity trends for FAO on Pt and Pd monolayers on transition metals.
Finally, we briefly discuss the implications of the FAO mechanism on the challenges facing the design of DFAFCs anode electrocatalysts. The reaction mechanism recognizes COOH* as the only precursor to CO* poison formation. On all surfaces studied, forming CO* is guaranteed if COOH* is more stable than HCOO*, or if COOH* formation is downhill in free energy. This establishes an important strategy of CO* tolerant anode electrocatalysts for DFAFCs: they need to stabilize HCOO*, and destabilize COOH*. On a related topic, Yoo et al.60 have recently identified HCOO*, not COOH*, to be the desired intermediate for selective CO2 electroreduction to HCOOH; this reaction is the reverse of FAO. This leaves HCOO* as a common desirable intermediate for HCOOH electrochemistry. To explore the feasibility of attaining such a constraint, we plot in Figure 8 the free energy of COOH* versus that of HCOO*, for all surfaces studied. A poor correlation between these two quantities is found, indicating the possibility of designing transition metal catalysts that selectively stabilize HCOO*.
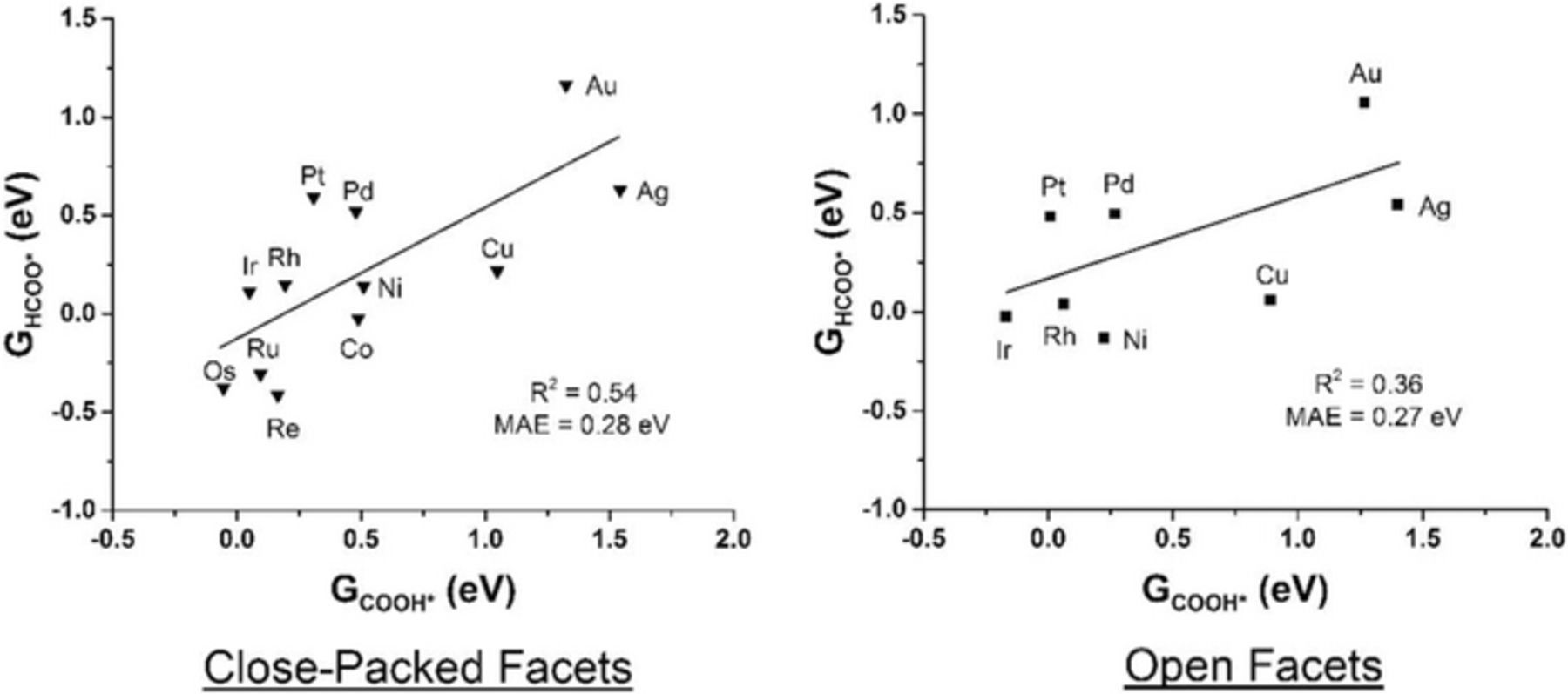
Figure 8. Attempts to linearly correlate the free energy of adsorbed COOH* (GCOOH*) and adsorbed HCOO* (GHCOO*). All energies are relative to CO2(g), H2O(g), and H2(g). Close-packed facets are plotted as inverted triangles, and open facets as squares. Marked R2 indicate a poor correlation between these two quantities.
Conclusions
We presented reactivity trends and structure sensitivity of FAO on model monometallic transition metal surfaces. We conclude that Pt and Pd offer a compromise between activity and stability, with weaker binding metals being inactive, and stronger binding metals easily poisoned by reaction intermediates. In terms of reaction structure sensitivity, FAO is structure-sensitive on most metals studied. Importantly, we conclude that weaker-binding metals have their open facets more energy-efficient (i.e. show less overpotential) than their close-packed facets, while the opposite is true for stronger-binding metals. After developing suitable scaling relations, we described FAO reactivity as a function of the free energy of CO* and OH*, and developed volcano plots identifying rate determining steps and estimated onset potentials, as a function of these two descriptors. The developed volcanos for both close-packed and open facets are capable of rationalizing recent literature reports for improved binary alloys of Pt or Pd. Our analysis suggests that improved FAO catalysts should selectively promote the formate path, and hinder the formation of carboxyl intermediate, which usually leads to forming the CO* poison on monometallic surfaces.
Acknowledgments
This manuscript is dedicated to Dr. Radoslav Adzic with the fondest memories of a very productive 15-year collaboration. His work has been most inspiring to generations of scientists with the desire of understanding electrocatalysis at the most fundamental level. This work has been supported by DOE-BES, Division of Chemical Sciences, grant DE-FG02-05ER15731. We thank Benjamin Chen and Lang Xu for their thoughtful comments on the manuscript. This work was performed in part using supercomputing resources from the following institutions: EMSL, a National scientific user facility at Pacific Northwest National Laboratory (PNNL) sponsored by the Department of Energy's Office of Biological and Environmental Research; the Center for Nanoscale Materials at Argonne National Laboratory (ANL) supported by the U.S. Department of Energy, Office of Science, under contract DE-AC02-06CH11357; the National Energy Research Scientific Computing Center (NERSC), supported by the U.S. Department of Energy, Office of Science, under contract DE-AC02-05CH11231; and UW-Madison Center for High Throughput Computing (CHTC), supported by UW-Madison, the Advanced Computing Initiative, the Wisconsin Alumni Research Foundation, the Wisconsin Institutes for Discovery, and the National Science Foundation, and is an active member of the Open Science Grid, which is supported by the National Science Foundation and the U.S. Department of Energy's Office of Science.
ORCID
Ahmed O. Elnabawy 0000-0002-8911-1916
Manos Mavrikakis 0000-0002-5293-5356