
Abstract
A one-dimensional (1D) core/shell hybrid electrocatalyst for alkaline direct ethanol fuel cells (ADEFC) has been fabricated using metal@oxide (Ni@NiO) nanowire arrays (NWA) as a novel support for porous Pt nanoparticles (Pt NP). The Ni@NiO NWA has been fabricated by direct electrodeposition inside a polycarbonate template and the subsequent conversion of the outer shell of Ni into an oxide performed by oxygen plasma annealing after template removal. The Pt NP have been deposited onto the Ni@NiO NWA surface by hydrothermal reduction using a borohydride technique. Transmission electron microscopy (TEM) has shown that the PtNP contain a homogeneous distribution of fine pores. The electro-catalytic activity for the electro-oxidation of ethanol in alkaline media for Ni@NiO NWA/Pt has been found to be 126 mA/cm2. Chronoamperometry has been used to show that the Ni@NiO NWA/Pt electrocatalyst exhibits favorable stability and activity as well as tolerance to CO-like poisoning during ethanol electro-oxidation at temperatures of 21°C and 50°C. The data described indicates that the Ni@NiO NWA/Pt electrocatalyst is a potential candidate to replace the commercial Pt/C NP anode electrocatalysts currently used in ADEFC.
Export citation and abstract BibTeX RIS
Alkaline direct ethanol fuel cells (ADEFC) have attracted increasing interest over the past decade as an ideal power source.1–5 The primary attraction of ADEFC systems is the relatively high energy densities that can be achieved (8.1 kW/kg versus 6.1 kW/kg for methanol)5 as well as the fact that the kinetics of ethanol oxidation6 and oxygen reduction7,8 reactions are faster in alkaline than in acid media. Pt is currently considered to be the optimum electrocatalyst for low temperature fuel cells.9 Both Pt and Pt-based nano-electrocatalysts have been extensively studied for the anodic electro-oxidation of ethanol4,5,10–13 and the cathodic reduction of oxygen7,8,14–17 in fuel cell applications. However, Pt-based electrocatalysts are far from ideal and are known to exhibit slow electron transfer kinetics for cathodic oxygen reduction reactions and inefficient electro-oxidation of ethanol to CO2 due to catalytic poisoning as well as being of high cost and limited availability.13,16,18–20 There appear to be few, if any, alternative electrocatalysts that can break the C–C bond of alcohols efficiently to the required end-products of CO2 and water. For this reason, recent research efforts have centered on the poisoning issue (by the adsorption of CO-like species) and it is this work that has led to the evolution of Pt based systems modified with foreign metals such as Ru, Ni, Co, Mo and Sn.19–21 At present, Pt decorated Ru bimetallic nanostructures are considered to be the most effective anode electro-catalytic systems for the electro-oxidation of alcohols,22 and which have also been shown to improve the CO tolerance of the electrocatalyst.23,24 Nevertheless, a high loading of the precious metals is generally required to achieve reasonable fuel cell performance whereas factors such as commercialization and maximizing the activity of the electrocatalyst necessitates a reduction of this loading into the electrode. High surface area, inexpensive supporting nanostructures potentially afford one means of optimizing the efficiency of the electrocatalysts without sacrificing their performance. The design of core/shell nanostructures that form layers of Pt onto inexpensive materials has emerged as an effective way of increasing electro-catalytic activity.1,10,25–28 One-dimensional (1D) nanostructures, particularly carbon nanotubes (CNT), have been shown to be promising support materials for Pt nanoparticles (NP) due to their high electrochemical surface area (ECSA) and unique physical properties.29,30 However, agglomeration of CNT can significantly reduce the surface area of the electrocatalystic support and increase contact resistance31 whilst there are a number of challenges associated with the synthesis of these systems that need to be overcome if Pt NP are to be reliably deposited onto the walls of such support structures. In contrast, 1D metal nanowire arrays are potentially more attractive support media for fuel cell applications, particularly where the precious metal can be coated or deposited on the nanowire surface of inexpensive metals.32–34
Metal oxide promoted Pt electrocatalysts are known to be efficient for direct ethanol electro-oxidation both in acid and alkaline media. Pt-based electrocatalysts that are promoted by metal oxides such as Pt/SnOx,35 Pt-MgO/C,36 Pt-CeO2/C,37,38 Pt-ZrO2/C,39 CNT/SnO2/Pt thin films,40 Pt-Mo/CNT@TiO2,41 and Pt–ATO(platinum–antimony tin oxide)/MWCNT42 have been shown to improve electrocatalytic activity and stability for ethanol electro-oxidation by comparison with pure Pt or Pt/C electrocatalysts. NiO is a very attractive material for various applications such as Li-ion battery anode,43 supercapacitors44,45 and fuel cell electrodes.46,47 Herein, we report a novel hybrid electrode fabricated using highly ordered conductive metal@oxide (Ni@NiO) nanowire arrays (NWA) as a support for porous Pt NP. We have developed a facile method for the fabrication of the electrode by direct electrodeposition of Ni nanowires using a template and controlled conversion of the deposited Ni nanowires into a core/shell Ni@NiO nanostructure by low power oxygen plasma annealing after chemically removing the template. The Pt NP are reduced onto the Ni@NiO NWA surface by chemical reduction using a borohydride technique thereby forming a 1D Ni@NiO NWA/Pt electrocatalyst. The synthesized material has been evaluated and the results suggest that Ni@NiO NWA/Pt could be used as an efficient nano-electrocatalyst for ethanol electro-oxidation in ADEFC.
Experimental
Synthesis of core/shell Ni@NiO NWA
A polycarbonate template (Isopore Membrane Filters of 47 mm diameter, nanopore diameter of ∼0.22 μm, thickness of ∼25–30 μm and porosity of ∼13.8%) was purchased from Millipore Ireland B.V. A 400 nm Ni seed layer was thermally evaporated (Temescal FC-2000) and a copper wire connected to the seed layer (back side of the template) using silver paste (Radionics Ltd. Ireland). A low stress nickel sulfamate bath was prepared using nickel sulfamate (120 g/L), boric acid (40 g/L), nickel bromide (4 g/L) and a wetting agent, ANKOR (R) F (10 mL/L). The pH of the solution was adjusted to 3.8 using 1 M sulfamic acid. A two electrodes cell was used with Ni pellets in a Ti-basket as an anode and the Ni/porous polycarbonate template as a working electrode (cathode). The electrodeposition was conducted at a constant current density (50 mA/cm2 as the ratio of the total template area) at 50°C with slow stirring of the solution. The deposition rate of the Ni nanowires was found to be 0.45 μm per minute. A thin film of Ni (3–4 μm) was also deposited onto the back side of the template which prevented the nanowires from collapsing after removing the template by immersion in dichloromethane. The Ni NWA was washed in deionized water, dried in air and attached to a silicon substrate by Kapton adhesive tape (DuPont, USA) so that only the surface regions were converted to an oxide during the plasma treatment. The Ni NWA was plasma annealed (March Plasmod GCM 200) for 2 minutes at an input power of 50 W and at a constant oxygen flow (30 cc/min). A NiO thin electrode was also prepared by conversion of the surface of electrodeposited Ni film to oxide using the plasma annealing technique described above.
Synthesis of porous core/shell Ni@NiO NWA/Pt electrocatalyst
The core/shell Ni@NiO NWA supported porous Pt NP were prepared by a hydrothermal reduction method from a solution containing a metal precursor salt (5 mM Na2PtCl6 × 6H2O) and a reducing agent (30 mM NaBH4). A piece of the Ni@NiO NWA sample was immersed in a 5 mL glass tube containing 500 μL aliquots of the Pt metal precursor salt at 80°C and stirred for 20 minutes. An excess quantity of freshly prepared NaBH4 solution was subsequently added drop wise to the mixture and stirred slowly until its color turned from yellowish to colorless (typically within 10 minutes). After completion of the reaction, the resulting hybrid electrocatalyst was removed from the mixture and washed repeatedly with deionized water for the complete removal of residual Cl− ions (the filtrate water was tested by 1 M silver nitrate solution and no white colored precipitate (AgCl) was obtained). For a comparison of electro-catalytic activities, Pt NP were also deposited onto the metallic Ni NWA, thin Ni and NiO surfaces using the NaBH4-reduction method described above. The synthesized electrocatalysts were dried in an oven at 100°C for 1.5 hours. The amount of Pt reduced onto the surface of the Ni NWA, Ni@NiO NWA, thin Ni and NiO electrodes was determined using a high precision microbalance (Mettler Toledo, XS 105 Dual Range, repeatability (SD), d = 0.01 mg) from the weight difference before and after the reduction of Pt.
Characterization
The morphology of the nanostructured electrocatalyst was examined by scanning electron microscopy (SEM, QUANTA FEG 650) coupled with energy dispersive X-ray spectroscopy (EDS Oxford Instruments INCA energy system) and transmission electron microscopy (TEM, JEOL 2100 operating at an accelerating voltage of 200 kV). Samples for TEM examination were encapsulated in a low melting point wax and thinned to electron transparency using the standard focused ion beam (FIB) thinning procedures described elsewhere.48 X-ray diffraction (XRD) analysis was carried out with a Philips X'pert PW3710-MPD diffractometer using filtered Cu-Kα (λ = 1.54 Å) radiation operating at 40 kV and 40 mA. Cyclic voltammetry (CV) and chronoamperometry (CA) were performed with a test cell filled with 1 M ethanol (EtOH) and 0.5 M KOH electrolyte at a temperatures of 21 ± 1°C and 50 ± 1°C using a platinized titanium mesh and a Hg/HgO (1 M KOH solution) as the counter and reference electrodes respectively. Electrodes (0.5 × 0.5 cm2) comprised of 13 μm long 1D Ni NWA/Pt and Ni@NiO NWA/Pt were attached onto the Ni coated polymer substrate by conductive glue (silver paste, Radionics Ltd. Ireland), and the edges were covered by nail varnish to avoid electrolyte leakages to the back side. CV measurements were carried out in the potential range of 0.4 to −1.0 V at different scan rates of 2 to 100 mV/s and conditioned at the open circuit potential (−0.65 V, OCP) for 20 s. CA measurements were carried out at a fixed potential of −0.2 V.
Results and Discussion
Figure 1 shows SEM images of the as deposited and plasma annealed 1D Ni NWA dispersed onto the surface of an Au/Si wafer. Comparison of Fig. 1a and 1b demonstrates that the surface roughness of the Ni NWA increases after the oxygen plasma treatment and the formation of nickel oxide (NiO) and a core/shell Ni@NiO NWA nanostructure, as reported in our recent work.45 The 1D Ni@NiO NWA was used to coat Pt NP to form a novel Ni@NiO NWA/Pt hybrid nano-electrocatalyst via a simple hydrothermal NaBH4-reduction method. Figure 2a–2b shows SEM images of the Ni@NiO NWA after reaction with the metal precursor salt solution, which clearly reveal that Pt NP are adsorbed on the surface of the NiO. Further insight into the microstructure of the NiO and Pt NP formed at the surface of the Ni@NiO nanowires was obtained by TEM. The bright field (BF) TEM images seen in Fig. 3a–3c show different regions of the Pt NP supported Ni@NiO nanowires at increasing magnification. The Pt NP can clearly be seen at the surface of the core/shell Ni@NiO nanowires and the grain size of the Pt NP typically varies from ∼3 to 15 nm although some NPs (often at the core of the 75 nm agglomerates) are somewhat larger at ∼30 nm. The diffraction pattern shown in Fig. 3d was obtained from a typical region of the Pt NP and demonstrates the presence of face centered cubic (fcc) Pt for which the measured (111), (200), (220) and (311) spacing are 0.2256, 0.1957, 0.1385 and 0.1208 nm, respectively. The Pt NP has also been shown to contain a reasonably homogeneous distribution of fine pores. Figure 4 shows a higher magnification through focal series of BF images where the reverse Fresnel effects that can be seen as a function of the defocus conditions are consistent with a distribution of fine pores. The diameter of the pores is typically in the region of 2–3 nm. The NiO shell has been found to have a characteristically 'ribbon-like' morphology with a typical thickness of ∼15 nm and to be made up of irregular grains that vary in diameter from ∼3 to 15 nm. The measured d-spacing for the (111), (200), (220) and (311) fcc oxide are 0.2399, 0.2077, 0.1468 and 0.1253 nm respectively.

Figure 1. SEM images of (a) as deposited Ni NWA and (b) plasma annealed Ni NWA. SEM images clearly reveal the rough morphology around the circumference of the Ni nanowires after the plasma treatment.
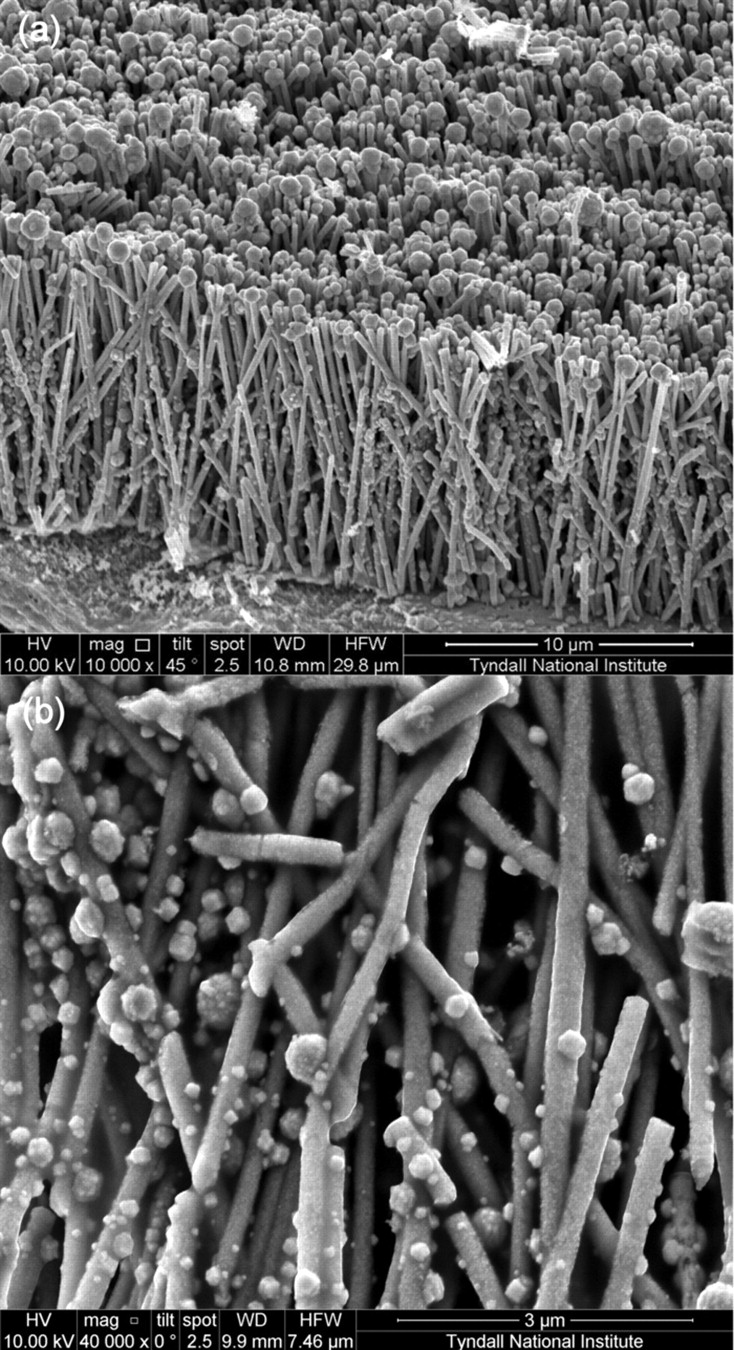
Figure 2. SEM images of 1D Ni@NiO NWA supported Pt NP cross-sectional (a) low and (b) higher magnified view.

Figure 3. BF TEM images of (a-b) Pt supported Ni@NiO NWA where Pt NP can be seen at both the sidewalls and tip of the elongated core/shell Ni@NiO nanowires. (c) A higher magnification BF image of a sidewall Pt NP at a Ni@NiO nanowire surface where the irregular NiO layer formed at the surface of the Ni can be distinguished. (d) A diffraction pattern confirming the presence of Ni, NiO and Pt for which the lattice parameters of the NiO and Pt are found to be 0.4120 nm and 0.3936 nm respectively.
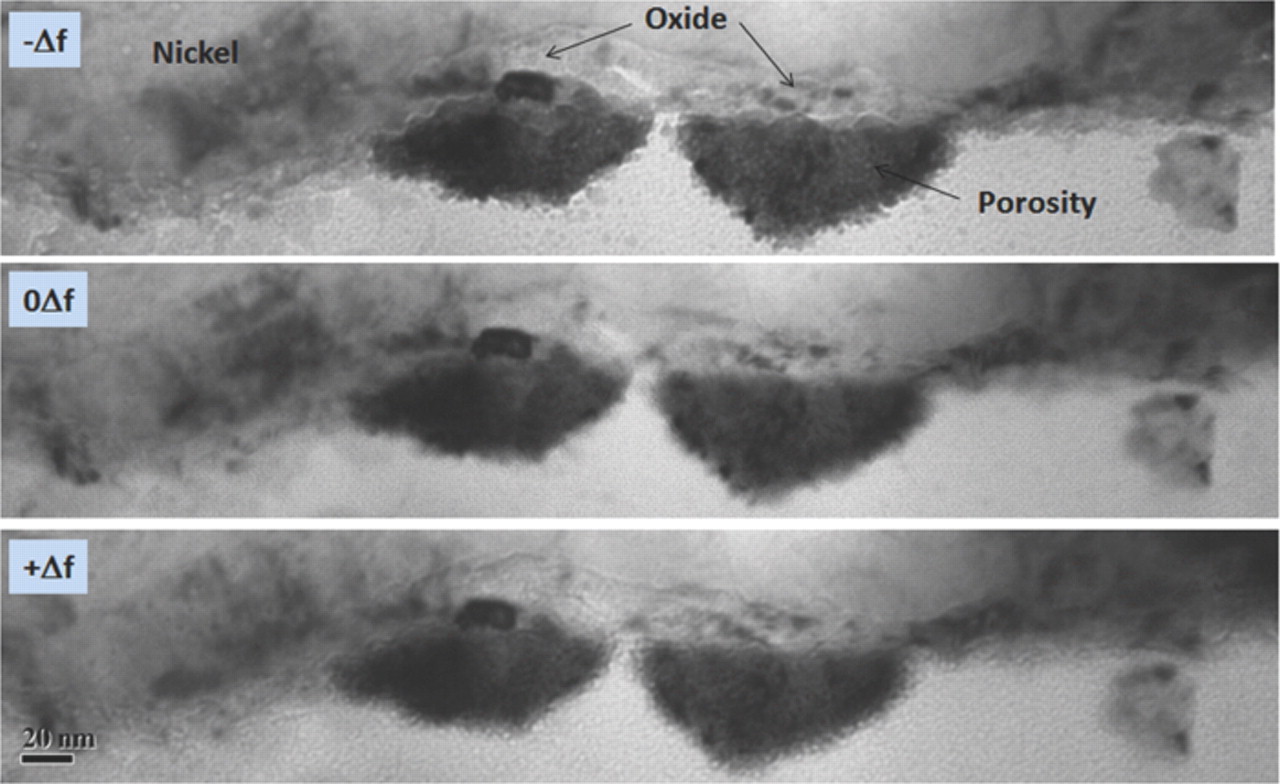
Figure 4. Higher magnification BF images of a sidewall Pt NP agglomerate taken in an (a) under-focus (b) in-focus and (c) over-focus condition showing a distribution of 2–3 nm pores. Note the reverse Fresnel effects as a function of the defocus conditions.
The XRD pattern of the as synthesized Ni@NiO NWA/Pt nano-electrocatalyst is shown in Fig. 5. Sharp and well defined peaks are observed for Pt at 2θ values of 39.8, 46.2, and 67.6° corresponding to the (111), (200), and (220) reflections respectively of the fcc metal. The average crystal size of the Pt NP was determined from the full-width at half maximum (FWHM) of the (111) plane using the Debye-Scherrer equation (B2θ = 0.94λ/rcosθ, where B2θ is FWHM, λ incident wavelength, r is the crystallite diameter, and θ is the diffraction angle). The average Pt crystallite size was found to be 12.6 nm. The XRD pattern is consistent with both the diffraction pattern shown in Fig. 3d, which demonstrates the polycrystalline nature of the Pt NP, and the TEM images shown in Fig. 3a–3c where the crystallite sizes were found to vary from 3 to 15 nm. The XRD pattern of Ni@NiO NWA/Pt similarly show three well-resolved characteristic peaks for NiO that can be readily indexed to (111, 2θ = 37.2°), (200, 2θ = 43.2°), and (220, 2θ = 62.8°) for the fcc oxide. The peaks marked with asterisks are associated with the metallic Ni phase and show (111), (200) and (220) reflections at 2θ values of 44.38°, 51.8° and 76.37° respectively for the fcc metal.
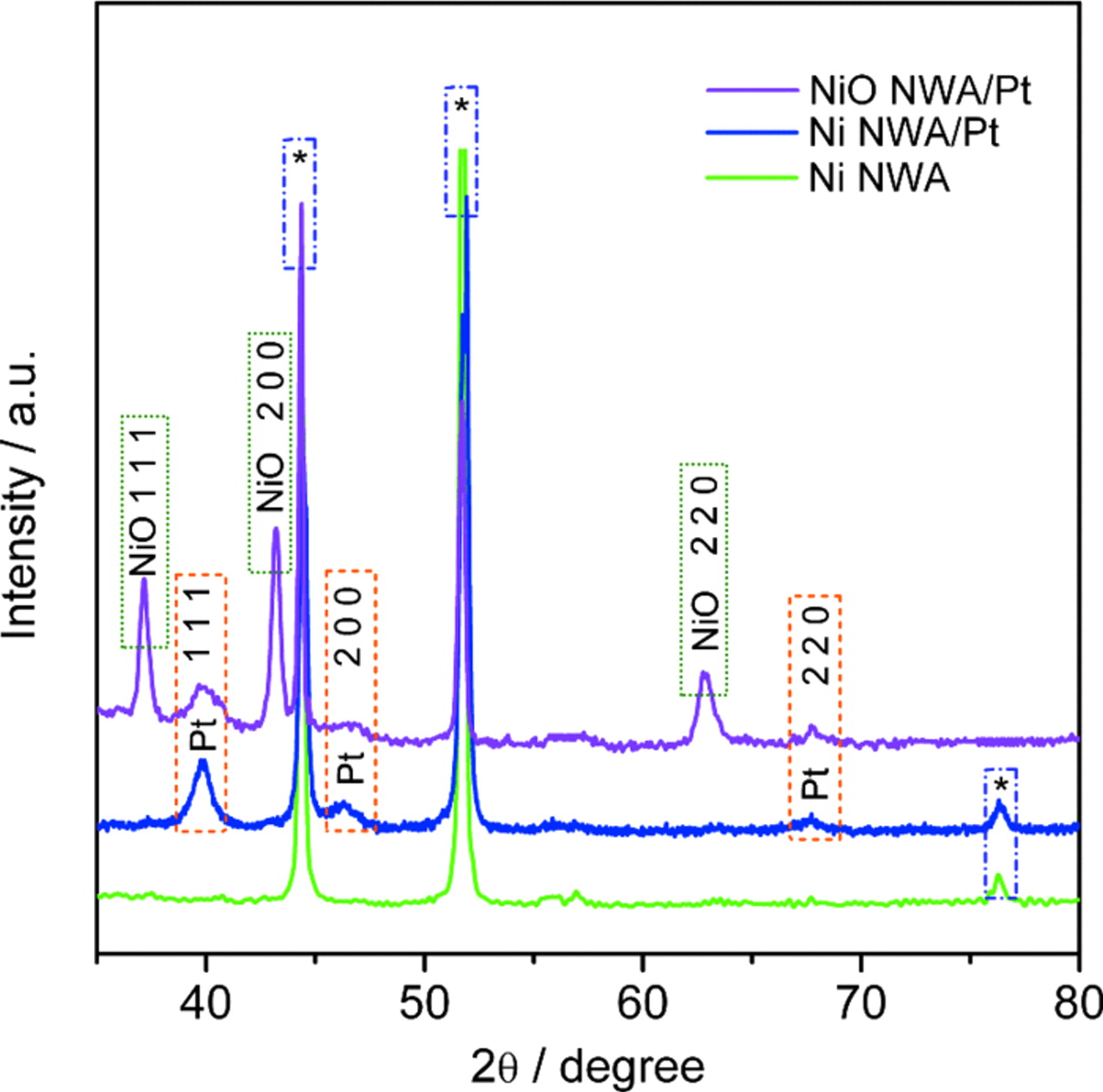
Figure 5. XRD patterns of the 1D Ni NWA, Ni NWA/Pt, and Ni@NiO NWA/Pt. The core/shell Ni@NiO NWA was prepared by oxygen plasma annealing at 50 W for 120 s. The peaks marked by asterisks correspond to the metallic Ni phase.
The cyclic voltammograms recorded in 0.5 M KOH after 20 scans are shown in Fig. 6. The characteristic features of hydrogen adsorption/desorption and oxide formation/reduction are observed for both the 1D Ni NWA and the Ni@NiO NWA supported porous Pt NP electrocatalysts. No electro-catalytic activities, however, are observed in the specified potential window for the bare Ni NWA and Ni@NiO NWA support structures. The real electrochemical surface area (ECSA) can be calculated from the expression, ECSA = (QH/QM WPt),49 where QM is the charge due to the monolayer adsorption of hydrogen (0.21 mC/cm2 for Pt), QH is the charge (mC/cm2) due to hydrogen absorption onto the electrode, and WPt is the Pt loading in the electrocatalyst. The ECSA values are estimated using the hydrogen desorption peak in the potential region of −0.9 V to −0.4 V in the anodic scan of the cyclic voltammograms (see Fig. 6) and are summarized in Table I. The ECSA of Pt supported Ni@NiO NWA (120 m2/gPt) can be seen to be 1.3 times higher than that of the Ni NWA (92 m2/gPt) structure. It should also be noted that the ECSA for the Ni@NiO NWA/Pt electrocatalyst is significantly higher than the reported values for Pt nanorods (30 m2/gPt) synthesized by galvanic displacement reaction34 as well as a commercially available carbon supported Pt NP electrocatalyst (∼53 m2/gPt)19 indicating higher utilization efficiency of the Pt NP on the conductive core/shell metal@oxide NWA. The dramatic increase in the measured ECSA is due to the 1D nanostructure which is free from any surfactants/ancillaries and that is enhanced by the homogenous fine porous microstructure of the Pt NP at the surface of the conductive robust core. The favorable properties are likely to be further improved by the interspacing of the nanowires for the fast ion adsorption/desorption, which would enhance the electrochemically active sites.
Table I. Cyclic voltammetry analysis of the Ni@NiO NWA supported Pt electrocatalysts. The specific and mass activities are measured at the respective peak potential (Ep).
| ECSA | Specific activity (mA/cm2) | Mass activity (mA/mgPt) | |||
|---|---|---|---|---|---|
| Electrocatalyst | (m2/gPt) | Fw. scan, Ep/V | Rev. scan, Ep/V | Fw. scan, | Rev. scan |
| Ni NWA/Pt | 92 | 84, 0.068 | 75, −0.14 | 350 | 312 |
| Ni@NiO NWA/Pt | 120 | 126, 0.062 | 103, −0.16 | 525 | 429 |
2* Pt loading in both electrodes is 0.24 mg/cm.
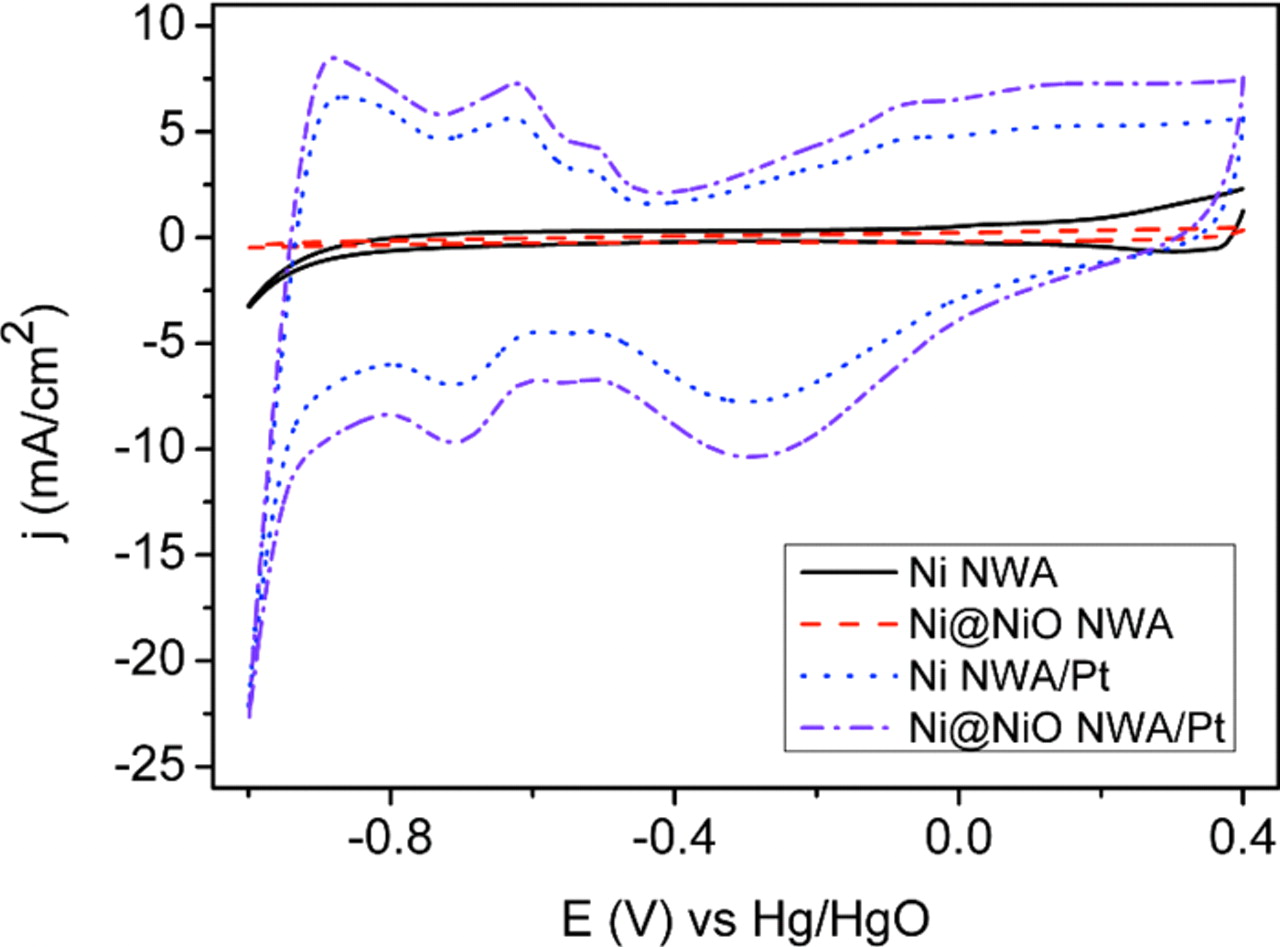
Figure 6. Comparison of cyclic voltammograms of 1D Ni NWA, Ni@NiO NWA, Ni NWA/Pt and Ni@NiO NWA/Pt in 0.5 M KOH solution at 21°C, scan rate: 50 mV/s.
The electro-catalytic activity for ethanol electro-oxidation using porous Pt NP supported Ni@NiO NWA was investigated by cyclic voltammetry and chronoamperometry. Figure 7a shows the typical behavior for ethanol electro-oxidation on Pt based electrocatalysts with the forward oxidation peak (i.e. under anodic condition) and the backward oxidation peak (i.e. under cathodic condition). The oxidation current peak in the forward scan is associated with the electro-oxidation of the freshly chemisorbed ethanol species onto the Pt surface, and the peak in the backward scan is attributed to the reduction of the oxidized Pt oxide and the removal of the incompletely oxidized CO-like carbonaceous species formed in the forward scan.18 The electro-oxidation of ethanol on the Ni@NiO NWA/Pt electrocatalyst surface commences at a more negative potential than that of Ni NWA/Pt, as shown Fig. 7b–7c, indicating that the NiO nano-shell around the highly conductive Ni NWA influences the faster electrode kinetics. The electro-oxidation current of Ni@NiO NWA/Pt for both the forward and backward peaks increases significantly (see Fig. 7a) and is almost 1.5 times higher than that of Ni NWA/Pt (see Table I). The oxide promotion effect for the electro-oxidation of ethanol is also realized in the thin film electrode version of Ni/NiO/Pt, as shown Fig. 7d. The specific peak current density of the Ni/NiO/Pt electrocatalyst is 5 mA/cm2 (based on the forward anodic peak), which is ∼1.5 times higher than that of the Ni/Pt (3 mA/cm2) electrode. It is clear that the addition of oxide to the Ni/Pt system significantly promotes the electro-catalytic activity for the electro-oxidation of ethanol at the same Pt loading. The specific peak current density (based on the forward anodic peak, If) of 1D Ni NWA/Pt and Ni@NiO NWA/Pt electrocatalysts are 84 and 126 mA/cm2 and their mass peak current density are 350 and 525 mA/mgPt respectively. The higher specific and mass activity of Ni@NiO NWA/Pt electrocatalyst could be attributed to the active role played by the NiO nano-shell around the conductive Ni core. It is interesting to note that the specific activity of the 1D Ni@NiO NWA/Pt electrocatalyst is much higher than the corresponding activities reported for 1D Pt/Co nanowires (∼6 mA/cm2),33 Te/Pt hybrid nanowires (∼1 mA/cm2),32 commercial Pt/C NP (∼1 mA/cm2) and Pt-Co alloy NP (∼2 mA/cm2).19 The specific activity of the oxide promoted 1D core/shell Ni@NiO NWA/Pt electrocatalyst is approximately 20 and 100 times higher than those reported for 1D Pt/Co nanowires and the commercial Pt/C NP electrocatalysts respectively. These results imply that even with a reduced Pt loading within the Ni@NiO NWA/Pt electrode system, it could be possible to replace the commercial Pt/C NP anode electrocatalysts currently used in ADEFC. The enhanced electro-catalytic activity due to the oxide support can be attributed to the synergistic interaction between the finely porous Pt NP and the NiO nano-shell formed at the outer surface of the conductive 1D metallic Ni core.42,47
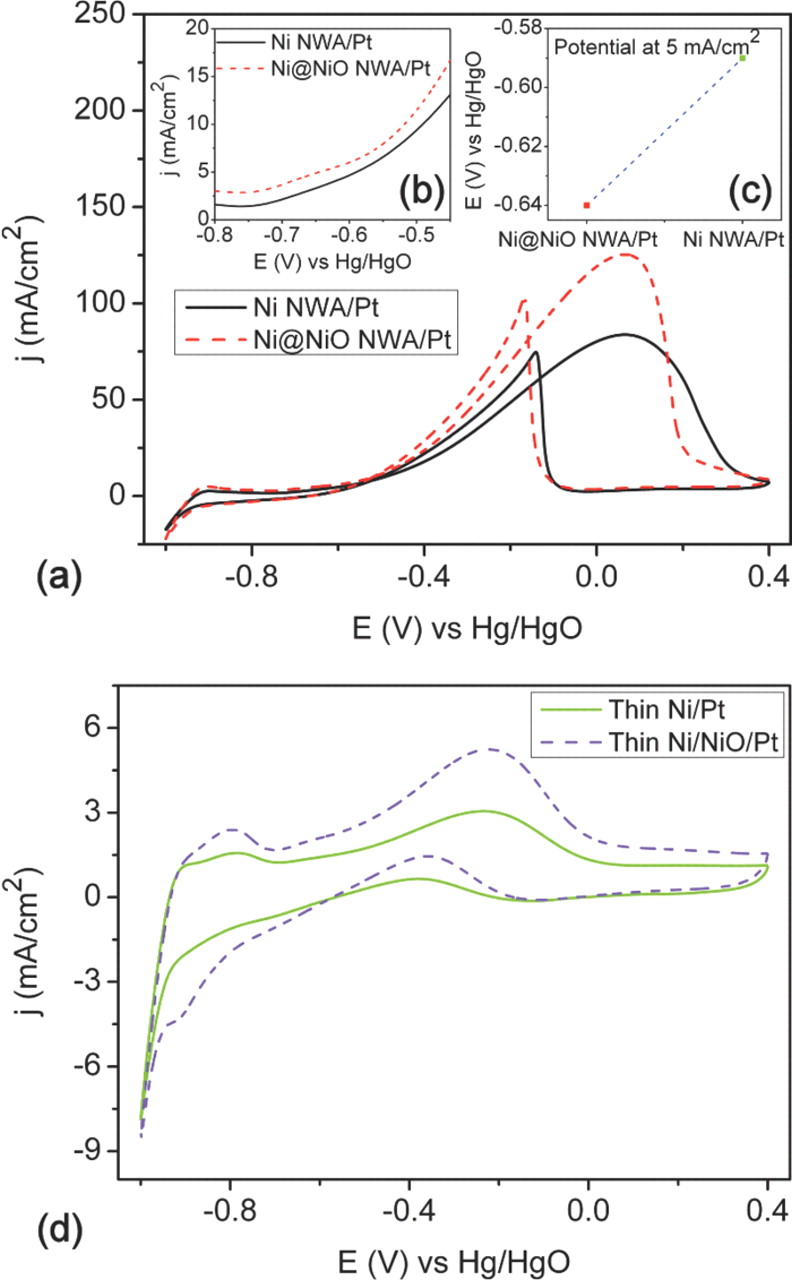
Figure 7. (a) Cyclic voltammograms of 1D Ni NWA/Pt and Ni@NiO NWA/Pt electrocatalysts in 0.5 M KOH/1 M ethanol solution at 21°C, scan rate: 20 mV/s (Pt loading, 0.24 mg/cm2). (b) Magnified view of onset potential region of cyclic voltammograms. (c) Electro-oxidation potential of the respective electrocatalyst at 5 mA/cm2. (d) Cyclic voltammograms of thin Ni/Pt and NiO/Pt electrocatalysts in 0.5 M KOH/1 M ethanol solution at 21°C, scan rate: 20 mV/s (Pt loading, 0.1 mg/cm2).
The ratio of the forward anodic peak current density (If) to the backward anodic peak current density (Ib) is an important index for the electrocatalyst through the provision of a measurement of the poisoning tolerance to CO-like carbonaceous oxidative intermediates formed during the electro-oxidation of alcohols (in the forward scan). The If/Ib value of the Ni@NiO NWA/Pt electrocatalyst was found to be 1.2, and this value is higher than those reported for 1D Pt/Co nanowires (0.88) and commercial Pt/C NP (0.76) electrocatalysts.19,33 The higher ratio of If/Ib indicates a superior poisoning tolerance, and this may be attributed to the dual effects of the NiO nano-shell and the finely porous Pt NP at the surface of the core/shell 1D nanostructure.
The nature of the mass transport behavior of the Ni@NiO NWA/Pt electrocatalyst has been investigated. Figure 8a shows a series of cyclic voltammograms at different scan rates. The correlation between the peak current density (in the forward scan) and the square root of the scan rate of the Ni@NiO NWA/Pt electrode is shown in Fig. 8b where the onset of a linear relationship can be seen at 20 mV/s. This observation indicates that the electrocatalytic process for the electro-oxidation of ethanol on the Ni@NiO NWA/Pt structure is not controlled by the mass transport or the concentration polarization until the scan rate is higher than 20 mV/s. However, at lower scan rates, the electro-oxidation of ethanol on the Ni@NiO NWA/Pt is dominated generally by the activation polarization. The electrode kinetic parameters for ethanol electro-oxidation are determined from the Tafel plots, as shown in Fig. 9. A linear region of the Tafel plots on both Ni NWA/Pt and Ni@NiO NWA/Pt electrocatalysts are found and the measured Tafel slopes are 111 mV/decade (dec) and 87 mV/dec respectively: the similarity in the Tafel slopes indicate that the same reaction mechanism occurs for both electrocatalysts. The lower Tafel slope obtained for the Ni@NiO NWA/Pt structure demonstrates a higher charge transfer rate during the electro-oxidation of ethanol by comparison with the Ni NWA/Pt electrocatalyst.
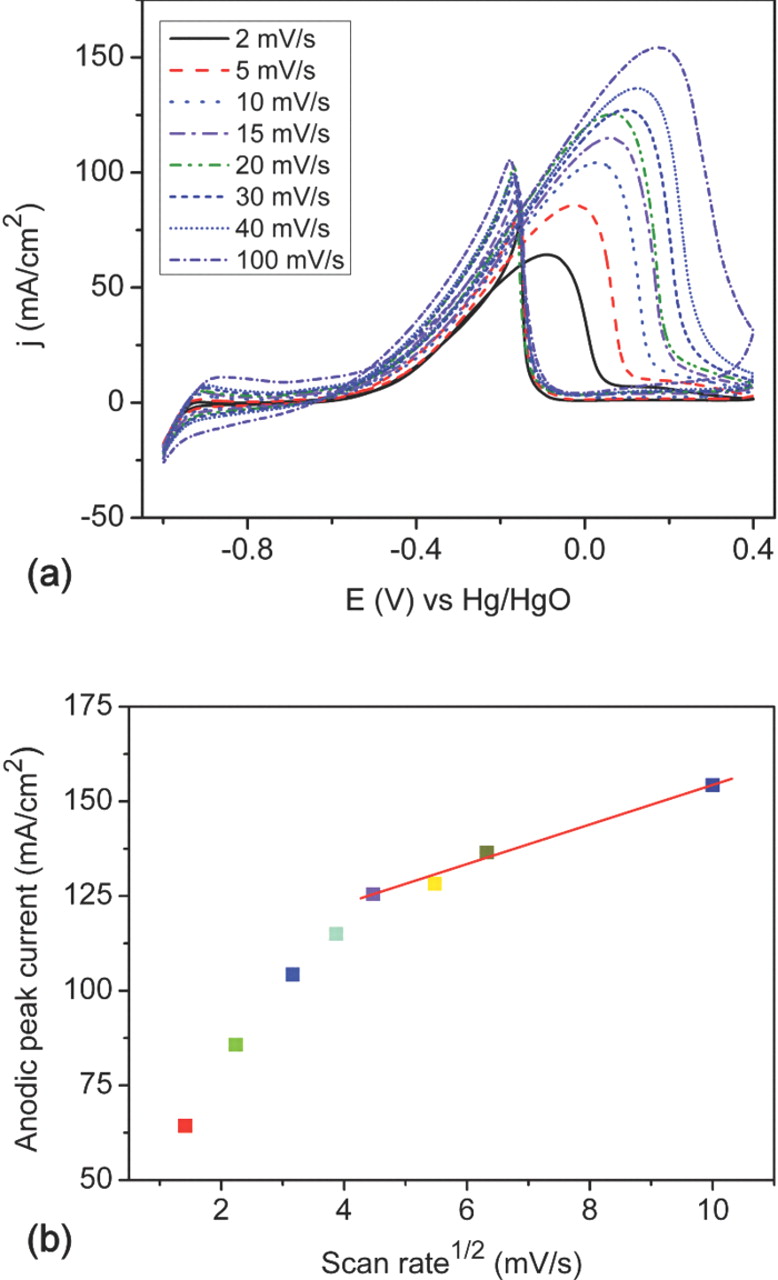
Figure 8. (a) Cyclic voltammograms of 1D Ni@NiO NWA/Pt electrocatalysts at different scan rates in 0.5 M KOH/1 M ethanol solution at 21°C, (b) Plot of forward anodic peak current density and the square root of the scan rate.
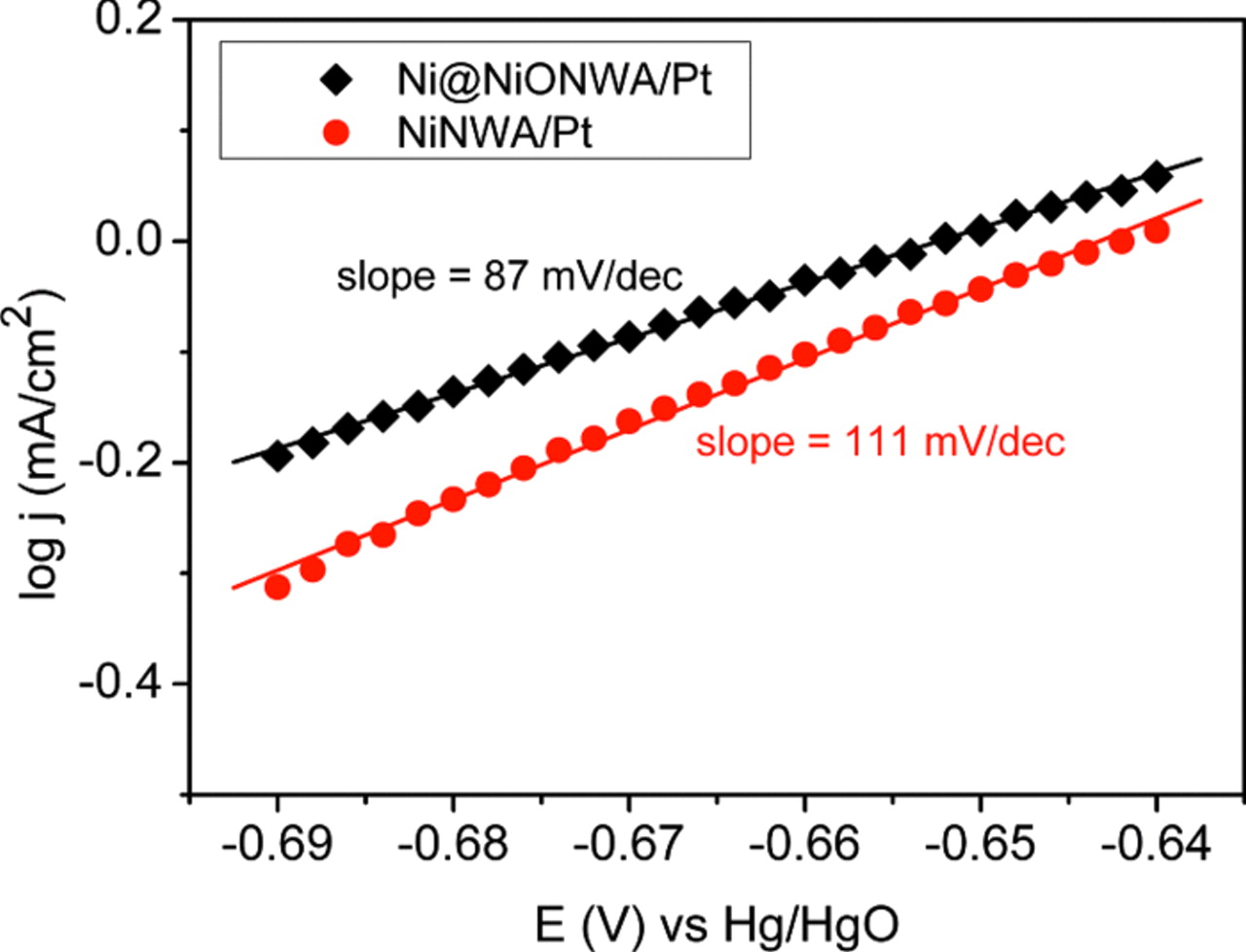
Figure 9. Tafel plots for ethanol electro-oxidation of 1D Ni NWA/Pt and Ni@NiO NWA/Pt electrocatalysts measured in 0.5 M KOH/1 M ethanol solution at 21°C, scan rate 2 mV/s.
Figure 10 shows chronoamperometric data obtained at different temperatures in order to investigate the stability of the electrocatalysts. The potential of the working electrodes was held at −0.2 V (Vs. Hg/HgO) and the changes in ethanol electro-oxidation current with time were plotted. A high current density is observed in the initial stages for both electrocatalysts which is due to double layer charging. The electro-oxidation current of ethanol was found to decay continuously for both electrocatalysts, most likely due to electrode poisoning by chemisorbed CO-like carbonaceous oxidative intermediates. In general, chemisorbed CO starts to accumulate on the electrocatalyst surface if the kinetics of their removal becomes significantly slower than those of the electro-oxidation of ethanol. In this way, a more steady decay rate of the electro-oxidation current with time implies a higher stability of the electrocatalyst and a greater tolerance to poisoning. However, the Ni NWA/Pt electrocatalyst shows a rapid current decay at low temperature (21°C) by comparison with the decay characteristics seen at 50°C. The rate of electro-oxidation current decay with time for the Ni@NiO NWA/Pt system was found to be much lower at both temperatures. The electro-oxidation current densities at the end of 3600 s test at 21°C are 3 and 8 mA/cm2 for the Ni NWA/Pt and Ni@NiO NWA/Pt electrocatalysts respectively whereas at 50°C, these current densities rise to 6 and 11 mA/cm2 respectively. The Ni@NiO NWA/Pt electrocatalyst using core/shell metal@oxide NWA as the support exhibits the higher electrocatalytic stability toward ethanol electro-oxidation both at the two operating temperatures investigated.
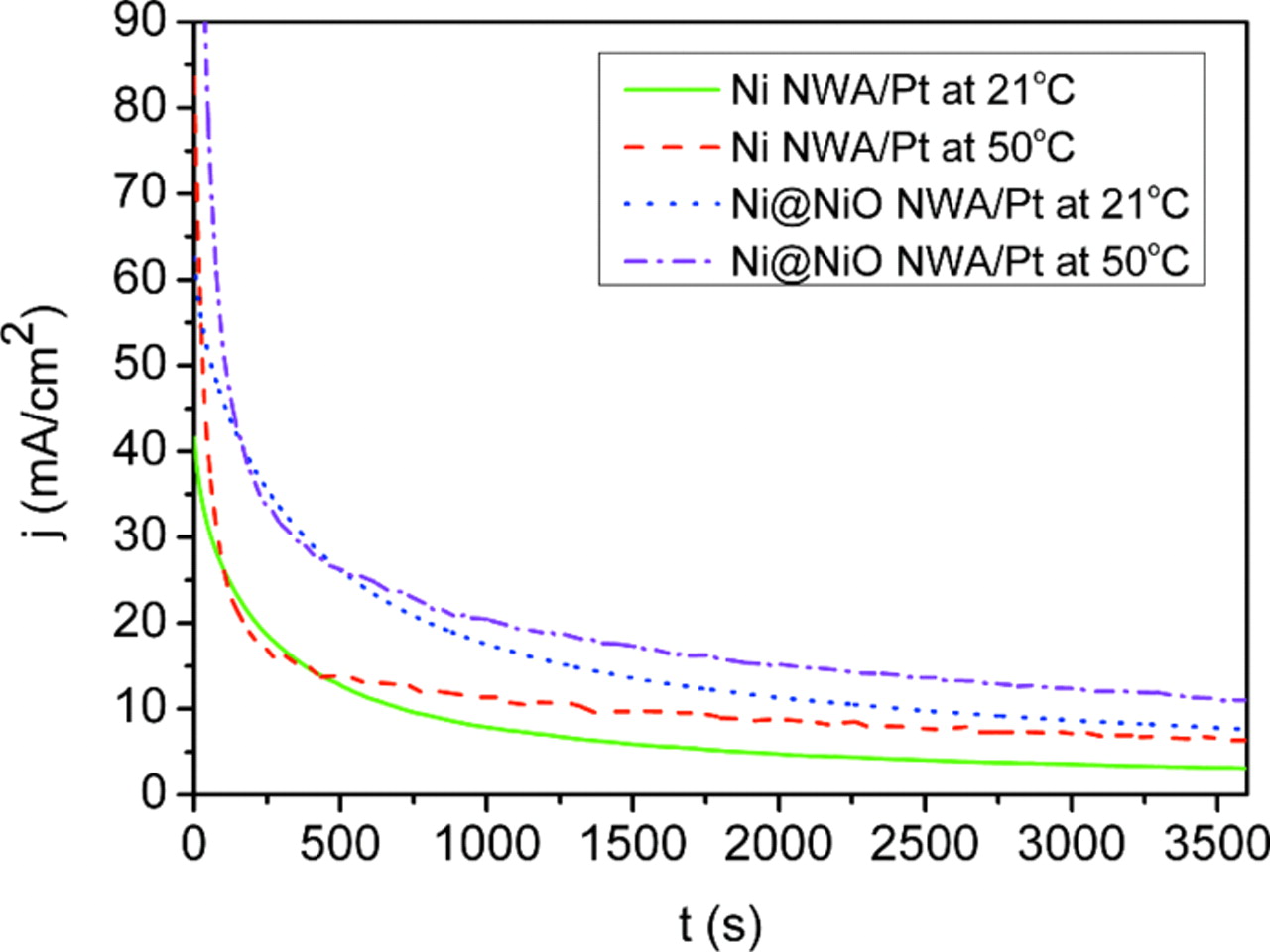
Figure 10. Chronoamperograms for the electro-oxidation of ethanol on 1D Ni NWA/Pt and Ni@NiO NWA/Pt at −0.2 V (vs. Hg/HgO) in 0.5 M KOH/1 M ethanol solution at 21°C and 50°C.
Conclusions
A novel Ni@NiO NWA/Pt electrocatalyst with porous Pt NP supported on 1D Ni@NiO NWA has been synthesized by a simple fabrication route. The hybrid nano-electrocatalyst has been shown to exhibit excellent electro-catalytic activity and stability for the electro-oxidation of ethanol in alkaline media. The observed enhancement in electro-catalytic activity performance by comparison with commercial Pt/C NP anode electrocatalysts can be attributed to (i) the 1D uniform nanostructure which is free from any surfactants/ancillaries (ii) the Pt NP containing a distribution of fine pores at the NiO surface that is supported by a conducting robust core of metallic Ni, and (iii) the interspacing of the nanowires ensuring that the access of alcohol for fast ion adsorption/desorption is easy. The specific and mass activity for the electro-oxidation of ethanol on the Ni@NiO NWA/Pt structure is approximately 1.5 times higher than that on Ni NWA/Pt at the same Pt loading. The Ni@NiO NWA/Pt electrocatalyst exhibits higher stability and tolerance to CO-like poisoning during ethanol electro-oxidation at 21°C and at 50°C. The enhancement in electro-catalytic activity due to the oxide can be attributed to the synergistic interaction between the Pt NP and the NiO nano-shell formed at the outer surface of conductive 1D metallic Ni core. The work described here has demonstrated a new method for the fabrication of a highly active and stable 1D Ni@NiO NWA/Pt electrocatalyst, which could be developed as a potential candidate system to replace the commercial Pt/C NP anode electrocatalysts currently used in ADEFC. The porous 1D Ni@NiO NWA/Pt nanostructure similarly has potential in sensor applications for gaseous or aqueous species such as NO2, CO, CO2 or C2H5OH.
Acknowledgments
This work is financially supported by Enterprise Ireland (EI) under the commercialization fund CFTD/2008/322.