Abstract
An equimolar mixture of lithium bis(trifluoromethylsulfonyl)amide (Li[TFSA]) and triglyme (G3) or tetraglyme (G4) yields the stable molten complexes, [Li(G3)][TFSA] or [Li(G4)][TFSA], respectively, classified into solvate ionic liquids (SILs). The Li-conducting SIL electrolytes have favorable thermal and electrochemical properties, but their intrinsic high viscosities and low ionic conductivities impede widespread application. In this study, SILs were diluted with organic solvents, such as toluene, hydrofluoroether (HFE) and propylene carbonate (PC), to enhance their ionic conductivity. Subsequently, the performance of a battery consisting of diluted SILs, LiCoO2, and graphite electrodes was evaluated. The electrochemical stability and charge/discharge behavior of the LiCoO2 cathode and graphite anode were greatly influenced by the stability of the complex cations, [Li(G3)]+ or [Li(G4)]+, in the diluted SILs. Unfavorable ligand exchange between the glyme and PC occurred in PC-diluted SILs. Oxidative decomposition of the uncoordinated glyme and pitting corrosion of Al current collector deteriorated the battery performance of LiCoO2 half-cell with PC-diluted SILs. We demonstrate that toluene- and HFE-diluted SILs, which do not contain chemicals such as carbonate solvent and LiPF6 used in commercialized Li-ion batteries, allow both LiCoO2 cathode and graphite anode to operate stably.
Export citation and abstract BibTeX RIS

This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 License (CC BY, http://creativecommons.org/licenses/by/4.0/), which permits unrestricted reuse of the work in any medium, provided the original work is properly cited.
Because of their high specific energy density, Li-based secondary batteries have been the most attractive candidates among a variety of energy storage systems. Research aimed at the development of Li-ion battery technologies has focused mainly on positive and negative electrode material,1,2 while Li-conducting electrolytes have played a "supporting" role. Indeed, the major components of the electrolyte in commercialized Li-ion batteries, i.e., a mixture of polar ethylene carbonate (EC) and low-viscous linear carbonates such as diethyl carbonate (DEC) with ∼1 mol dm−3 of lithium hexafluorophosphate (LiPF6),3 have been in use since these batteries were first developed for practical application in 1991.
The standard electrolyte (1 mol dm−3 LiPF6 in EC/DEC) has been selected for the mature Li-ion battery technology, for several reasons,3–7 and the battery reaction relies on each electrolyte component. The polar EC not only assists the supporting Li-salt to dissociate to a higher degree, but also forms a good solid electrolyte interphase (SEI) on the graphite anode. The SEI is formed via sacrificial initial decomposition and it allows reversible intercalation/deintercalation reactions of Li ions during charge/discharge. Less viscous linear carbonates such as DEC are mixed with the more viscous EC to improve the ionic conductivity of the electrolyte without compromising the high degree of salt dissociation and formation of the SEI. Despite its poor chemical stability, LiPF6 is used as the supporting salt because of its high degree of dissociation. Moreover, LiPF6 plays an important role in suppressing the corrosion of the aluminum current collector at the cathode.8
Carbonate-based standard electrolytes suffer from flammability issues in large, high-capacity Li-ion batteries.9 Additionally, recent studies show that replacement of carbonate-based electrolytes by others can help in avoiding unfavorable side reactions between the carbonate solvents and the intermediates of the discharge products in ultra-high energy density batteries, so-called beyond Li-ion batteries, e.g., Li-sulfur and Li-air batteries.10,11 Accordingly, innovative design of Li-battery electrolytes is key to developing high energy density batteries.12 The alternatives studied thus far range from organic electrolytes using fluorinated solvents,13,14 phosphorus-based solvents,15,16 and sulfones17,18 to ionic liquid (IL)-based electrolytes.19
Highly concentrated (or superconcentrated) electrolytes are becoming the focus of attention for Li-conducting electrolytes.20 It was found that simply increasing the salt concentration to near saturation resulted in various anomalies on bulk and interfacial properties, e.g., higher thermal stability, improved electrochemical stability, and fast electrode reaction. These characteristics are favorable for the electrolyte in advanced Li-ion and beyond Li-ion batteries.21–24 Equimolar mixtures of lithium bis(trifluoromethanesulfonyl)amide (Li[TFSA]) and triethylene glycol dimethylether (triglyme, G3) or tetraethylene glycol dimethylether (tetraglyme, G4) yield a room-temperature molten complex, abbreviated as [Li(G3)][TFSA] or [Li(G4)][TFSA].25 These complexes are not only deemed representative of highly concentrated electrolytes, but also considered a new subset of ILs, i.e., solvate ILs (SILs)26 because of their IL-like characteristics. Therefore, SILs are being investigated as an alternative electrolyte for advanced Li-ion and next generation batteries.27–30
A shortcoming of highly concentrated electrolytes, including SILs, is the poor ionic conductivity resulting from their high viscosity. In previous work,31 SILs [Li(G3)][TFSA] and [Li(G4)][TFSA] were diluted with molecular solvents to reduce their viscosity, and they were found to be soluble in various solvents. The SILs were fully miscible with less polar solvents such as hydrofluoroether (HFE, HCF2−CF2−O−CH2−CF2−CF2H) and DEC as well as with highly polar solvents such as water, propylene carbonate (PC), acetonitrile, and acetone. These SILs were also partially miscible with the apolar toluene, in a SIL/toluene molar ratio of 1/4, corresponding to salt concentrations of 1.33 and 1.28 mol dm−3 at 30°C for [Li(G3)][TFSA] and [Li(G4)][TFSA], respectively. The electrolytes, the mixtures of SIL/organic solvent, can also be regarded as the solutions in which Li[TFSA] is dissolved in the mixed solvent of glyme and another solvent. Li[TFSA] can be dissolved easily in glyme and PC owing to their strong solvating power. In contrast, Li[TFSA] cannot be dissolved in the low polar solvents, such as HFE and toluene, without the co-solvent. Hence, the high miscibility of SILs opens up various options for solvent selection in designing novel Li-conducting liquid electrolytes, where the SIL serves as a highly soluble supporting salt. The SIL electrolytes diluted with HFE can also be used as the battery electrolyte. The reversible charge/discharge of the graphite anode in this electrolyte and the battery performance of Li-ion and Li-sulfur batteries have been reported in our previous papers.28,32,33
In this study, organic solvents, PC, HFE, and toluene, were selected for their compatibility with metallic Li and lithiated graphite. Previous work reveals that glyme molecules of the SILs in toluene or HFE remain coordinated to Li+ ions in the form of a crown-ether like complex cations, [Li(G3)]+ or [Li(G4)]+, whereas ligand exchange between the glyme and solvent takes place in the presence of PC.31 Here, we report the electrochemical stability and charge/discharge behavior of the LiCoO2 cathode and graphite anode in the diluted SILs, which are dependent on the co-solvent species, i.e., toluene, HFE or PC. The long-lived complex cations [Li(G3)]+ and [Li(G4)]+ play a substantial role in the successful charge/discharge of Li-ion batteries in diluted SILs. With toluene- and HFE-diluted SIL electrolytes, we demonstrate that both LiCoO2 cathode and graphite anode can be operated stably even in carbonate- and LiPF6-free electrolytes.
Experimental
Materials
Triglyme (G3, water content <50 ppm) and tetra-glyme (G4, water content <50 ppm) were kindly supplied by Nippon Nyukazai. Li[TFSA] (battery-grade, water content <50 ppm) was kindly supplied by Solvay Japan. Super-dehydrated toluene was purchased from Wako. Battery-grade propylene carbonate (PC) and lithium bis(pentafluoroethanesulfonyl)amide (Li[BETA]) were obtained from Kishida Chemical. A hydrofluoroether, 1,1,2,2-tetrafluoroethy-l 2,2,3,3-tetrafluoropropyl ether, which has a chemical structure of HCF2−CF2−O−CH2−CF2−CF2H (hereafter, abbreviated as HFE), was purchased from Kanto Kagaku and dried with molecular sieves. All these electrolyte components were used as received. The electrolyte solutions were prepared and stored in an Ar-filled glove box (VAC, [H2O] < 1 ppm).
For the LiCoO2 positive electrode, LiCoO2 powder (kindly supplied by AGC Seimi Chemical), acetylene black (AB, Denki Kagaku Kogyo), and poly(vinylidene fluoride) (PVDF, Kureha Chemical) were used as an active material, a conduction supporting agent, and a binder, respectively. The mass ratio of LiCoO2/AB/PVDF was 85/9/6. A slurry with N-methylpyrrolidone (NMP, Kanto Chemical) was spread on an Al foil current collector and dried at 80°C. The prepared LiCoO2 electrode sheet was cut into a circular shape (16 mm diameter) and compressed at 300 kgf cm−2 using a hydraulic press. The thickness and active material loading were ca. 17 μm and ca. 3 mg cm−2, respectively. For the graphite negative electrode, artificial graphite powder (kindly supplied by Hitachi Chemicals), AB, and PVDF were mixed with the mass ratio of graphite/AB/PVDF at 90/3/7. The NMP slurry was spread on a Cu foil current collector and dried at 80°C. This graphite sheet was cut into a circular shape (16 mm diameter) and compressed at 300 kgf cm−2 using a hydraulic press. The typical thickness of the graphite anode was ca. 40 μm, which corresponds to a loading of graphite of ca. 4 mg cm−2. The above electrodes were used for half-cell tests. Commercially available electrode sheets of LiCoO2 (1.5 mAh cm−2) and graphite (1.6 mAh cm−2) (both obtained from Piotrek), were also employed for full-cell tests.
Measurements
The oxidative electrochemical stabilities of the electrolytes were studied using linear sweep voltammetry (LSV) with an electrochemical measurement system (VMP3, Bio-Logic). LSV was performed at 30°C at a scan rate of 1 mV s−1 in a three-electrode cell with Li metal foil as the reference and counter electrodes and a platinum or aluminum disk as the working electrode.
For half-cell tests, a 2032-type coin cell was fabricated with the composite electrode (LiCoO2 or graphite), a glass filter as a separator (GA-55, Advantec), a Li metal disk (16 mm in diameter, Honjo Metal), and electrolyte in an Ar-filled glove box. Galvanostatic charge/discharge measurements were performed on the coin cells using an automatic charge/discharge instrument (HJ1001SD8, Hokuto Denko) at 30°C. The cutoff potentials were set to 3.0 and 4.2 V for the LiCoO2 half-cell and at 0.001 and 2.0 V for the graphite half-cell, respectively. The specific capacity of the cell was calculated on the basis of the active material loading. The gravimetric current density was 137 mA g−1 and 372 mA g−1 (C/1 rate) for the LiCoO2 and graphite half-cell, respectively. For full-cell tests, the commercial LiCoO2 and graphite electrodes (10 and 12 mm in diameter, respectively) and the glass filter separator were used for fabricating a 2032-type coin cell.
Results and Discussion
Transport properties of diluted solvate IL electrolytes
Table I summarizes salt concentration (c); transport properties such as conductivity (σ), viscosity (η), and molar conductivity ratio (Λimp/ΛNMR); and the ratio of self-diffusion coefficients for glyme and lithium (DG/DLi) for neat SILs and SILs diluted by toluene, PC or HFE as reported in our previous work.31,34 Except for [Li(G3)][TFSA]/toluene, ionic conductivities were enhanced to a level comparable with that of the standard electrolyte by dilution with less viscous solvents. However, increases in ionic conductivities were not as high as expected given that dilution lowered viscosity values by two orders of magnitude. This phenomenon can be attributed to a decrease in the number of charge carriers resulting from dilution and ionic association. The molar conductivity ratio (so-called ionicity, Λimp/ΛNMR) has been used to estimate the apparent degree of dissociation or correlative motion of ions in various types of electrolytes.35,36 Λimp is the molar conductivity, and ΛNMR is determined from the ionic self-diffusion coefficients DLi and DTFSA using the Nernst-Einstein equation: ΛNMR = F2(DLi + DTFSA)/RT, where F is the Faraday constant, R is the gas constant, and T is the absolute temperature. As can be seen in Λimp/ΛNMR values (Table I), the addition of toluene decreased Λimp/ΛNMR for both SILs and ionic association is more pronounced in [Li(G3)][TFSA]/toluene. The lower ionicity and increased ionic association caused a minor increase in conductivity for [Li(G3)][TFSA]/toluene.
Table I. Properties of SIL-based electrolyte measured at 30°C; salt concentration c, ionic conductivity σ, viscosity η, molar conductivity ratio (ionicity) Λimp/ΛNMR, and diffusivity ratio DG/DLi.
| Electrolyte | c (mol dm−3) | σ (mS cm−1) | η (mPa s) | Λimp/ΛNMR (-) | DG/DLi (-) |
|---|---|---|---|---|---|
| [Li(G3)][TFSA]a | 3.06 | 1.1 | 188 | 0.74 | 1.00 |
| [Li(G3)][TFSA]/tolueneb | 1.33 | 2.8 | 3.0 | 0.26 | 1.00 |
| [Li(G3)][TFSA]/PCb | 1.00 | 7.6 | 5.4 | 0.65 | 1.09 |
| [Li(G4)][TFSA]a | 2.75 | 1.6 | 106 | 0.64 | 1.00 |
| [Li(G4)][TFSA]/tolueneb | 1.28 | 7.6 | 3.1 | 0.41 | 0.97 |
| [Li(G4)][TFSA]/PCb | 1.00 | 7.3 | 5.7 | 0.66 | 1.06 |
| [Li(G4)][TFSA]/HFEb | 1.04 | 5.2 | 5.2 | 0.40 | 0.95 |
The DG/DLi ratio is a useful tool for evaluating the stability of complex cations [Li(G3)]+ and [Li(G4)]+ in the diluted SILs (Table I). DG/DLi is close to unity for neat and toluene- and HFE-diluted SILs, suggesting the formation of glyme-Li complex cations. However, DG/DLi is > 1 in PC-diluted SILs, indicating the glyme diffused faster than Li+. Likely, ligand exchange between the glyme and PC was responsible for the DG/DLi > 1. Obviously, the stability of glyme-Li complex cations is greatly influenced by solvent species and uncoordinated glymes were present in the PC-diluted SILs.
Electrochemical oxidative stability
Figure 1 shows LSV curves at a Pt electrode in the neat and diluted SILs. Previous studies have shown that glyme solvents are oxidized around 4 V (vs Li/Li+) in the uncoordinated form, but that anodic stability is enhanced to 5 V by complex formation with Li+ because strong glyme-Li interactions lower the highest occupied molecular orbital (HOMO) energy levels of the glymes.37,38 The diluted electrolytes of [Li(G3)][TFSA] and [Li(G4)][TFSA] showed similar voltammograms regardless of their cationic structure, [Li(G3)]+ or [Li(G4)]+. The onset potentials of current flow in toluene-, PC- or HFE-diluted SILs were lower than those in neat SILs. Modest anodic current in the range of 4−4.5 V can be measured in the PC-diluted SILs. Because the co-solvent PC is electrochemically stable in this potential range, the anodic current is attributed to the oxidative decomposition of the uncoordinated glyme. Discernible current flow followed by peak current was observed at potentials higher than 4.5 V in the toluene-diluted SILs, likely due to anodic decomposition of the co-solvent toluene. Although the anodic stability of the toluene- and HFE-diluted SILs was lower than those of the neat SILs, they have a potential to withstand the operation of 4 V-class Li-ion batteries.
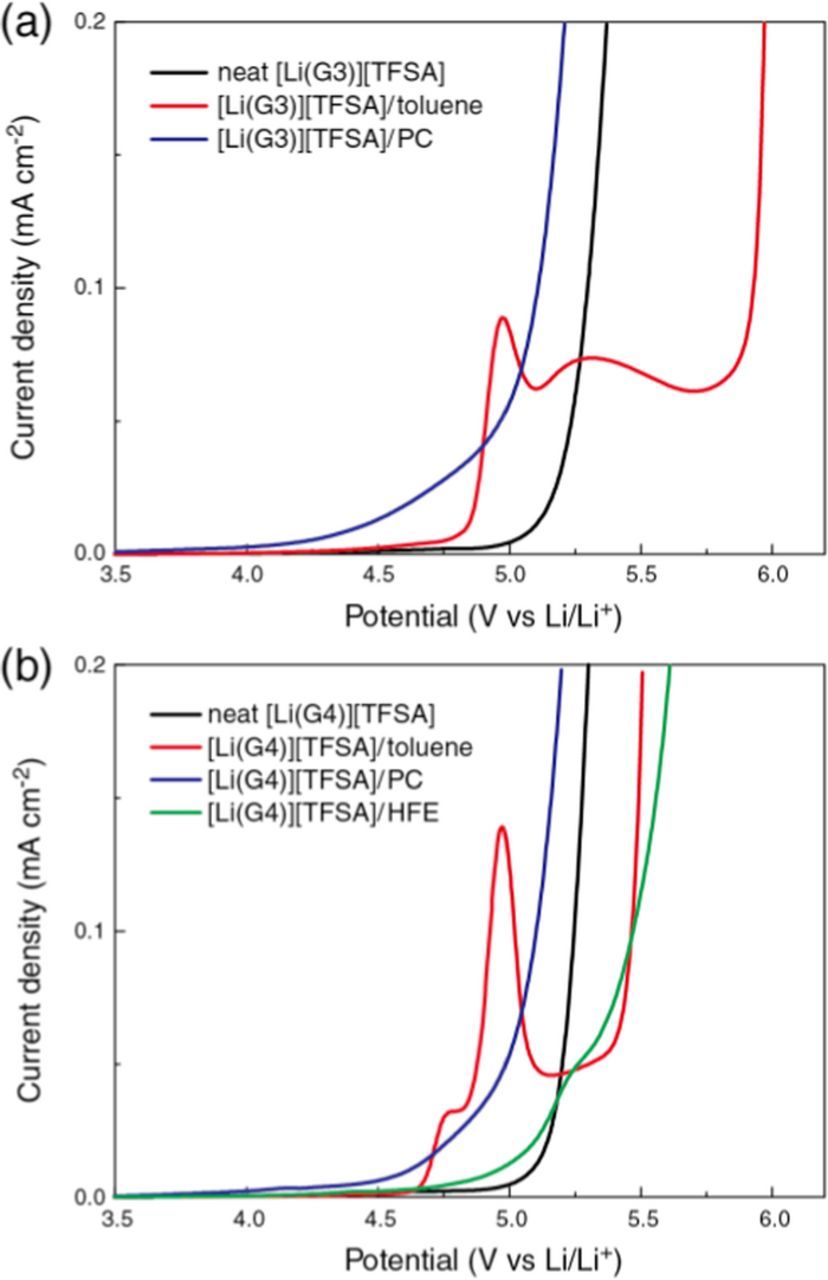
Figure 1. Linear sweep voltammograms (LSVs) on a Pt electrode in neat SILs and SILs diluted with toluene, PC or HFE; (a) [Li(G3)][TFSA] and (b) [Li(G4)][TFSA] at scan rate of 1 mV s−1 at 30°C.
Charge/discharge of LiCoO2 half-cell
To examine the applicability of the diluted SIL electrolytes to 4 V-class Li-ion batteries as noted in the former section, we investigated the charge/discharge behavior of Li-LiCoO2 half-cell. The theoretical capacity of LiCoO2 in this voltage range is 137 mA h g−1 based on the electrochemical reaction: LiCoO2 ⇄ Li1/2CoO2 + 1/2Li+ + 1/2e−. Figures 2 and 3 show the galvanostatic charge/discharge curves and cycle characteristics of [Li metal anode | diluted SIL electrolyte | LiCoO2 cathode] cells measured in the cell voltage range 3.0−4.2 V at a gravimetric current density of 17 mA g−1 (C/8 rate), corresponding to a geometric current density of 50 μA cm−2.
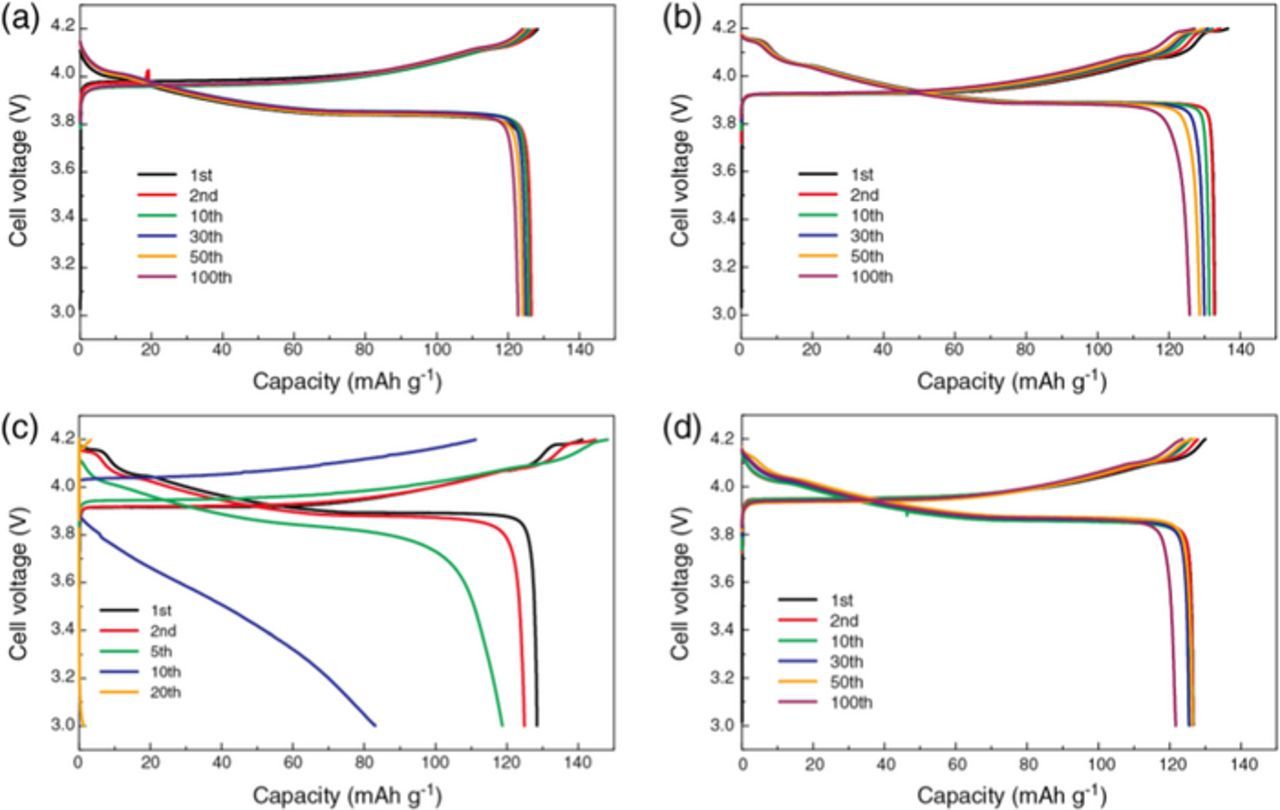
Figure 2. Galvanostatic charge/discharge curves of [Li metal anode | diluted SIL electrolyte | LiCoO2 cathode] cells with a constant current density of 50 μA cm−2 (C/8 rate) at 30°C with (a) [Li(G3)][TFSA]/toluene, (b) [Li(G4)][TFSA]/toluene, (c) [Li(G3)][TFSA]/PC, and (d) [Li(G4)][TFSA]/HFE.
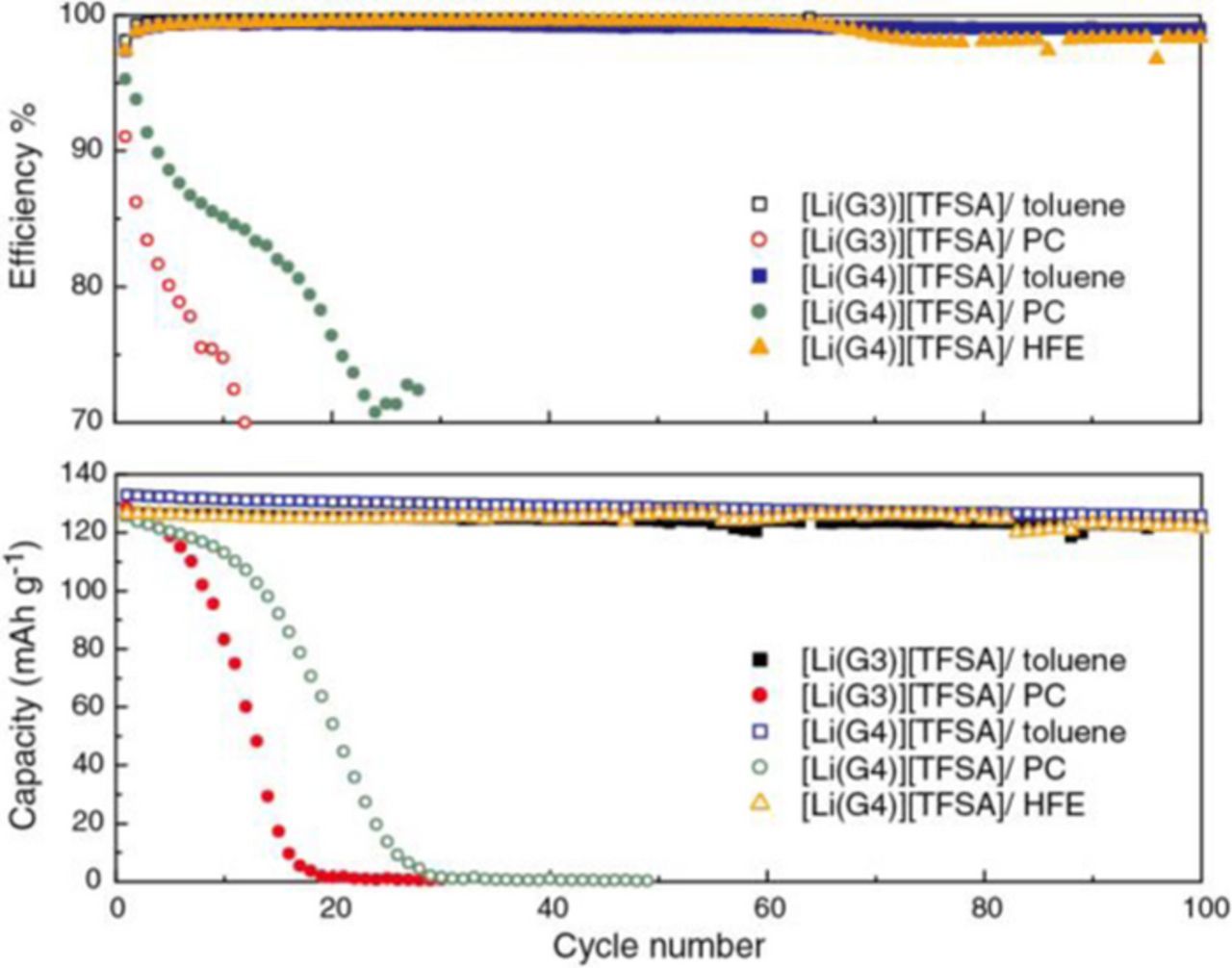
Figure 3. Coulombic efficiency (top) and discharge capacity (bottom) as a function of cycle number for [Li metal anode | diluted SIL electrolyte | LiCoO2 cathode] cells with a constant current density of 50 μA cm−2 (C/8 rate) at 30°C.
Due to the enhanced anodic stability, the toluene-diluted SILs allowed reversible and stable charge/discharge behaviors with a high Coulombic efficiency (99%) of the LiCoO2 half-cells. Cells retained 97% and 95% discharge capacities even after 100 cycles for [Li(G3)][TFSA]/toluene and [Li(G4)][TFSA]/toluene, respectively. The cell with [Li(G3)][TFSA]/toluene showed a lower initial capacity (126 mAh g−1) compared to the cell with [Li(G4)][TFSA]/toluene that delivered the initial charge and discharge capacity (132 mAh g−1) close to the theoretical value. As shown in Figures 2a and 2b, the charge and discharge voltage plateaus of the cell with [Li(G3)][TFSA]/toluene are slightly higher and lower, respectively, than those of the cell with [Li(G4)][TFSA]/toluene. This is indicative of higher internal resistance in the cell with [Li(G3)][TFSA]/toluene. The lower ionic conductivity of [Li(G3)][TFSA]/toluene is probably responsible for the observed lower capacities and larger overvoltage of the LiCoO2 half-cell. For comparison, the charge-discharge curves and the cycling performance of the LiCoO2 half-cell with [Li(G4)][TFSA]/HFE is also shown in Figures 2d and 3. HFE is not a common organic solvent but can provide non-flammability39 and high fluidity to the SILs.31 Similar to the cell with the toluene-diluted SILs, [Li(G4)][TFSA]/HFE showed reversible and stable charge/discharge behaviors with a high Coulombic efficiency (99%) because the glyme-Li complex cations also remained stable in HFE.31
In contrast to the cells using the toluene-diluted SILs, the charge/discharge behavior of the cells using the PC-diluted SILs was very unstable, as shown in Figures 2c and 3. At the beginning of the cycles (1st−5th), the charge capacities exceeded the theoretical capacity of 137 mAh g−1 suggesting unfavorable side reactions occurred in the cell, e.g., the oxidative decomposition of the glyme as discussed in the former section. The overvoltage in the charge/discharge curves increased with repeating charge/discharge cycles and both capacity and efficiency rapidly faded within 20 or 30 cycles. The rapid failure of the cell is possibly due to the electrochemical instability of the PC-diluted SIL electrolyte, i.e., the uncoordinated glymes, at high charging potential (∼4.2 V) in the LiCoO2 half-cell.
The Al electrode is known to undergo pitting corrosion around 3.8 V in electrolytes containing Li[TFSA] salt.40–42 Therefore, both corrosion of the Al current collector and electrolyte decomposition may occur in diluted SILs. This corrosion may be another factor behind the deteriorating charge/discharge performance of the cells with PC-diluted SILs. Previous reports proposed that Al corrosion in Li[TFSA]-based electrolytes takes place through decomposition of the oxide layer, followed by the anodic dissolution of Al metal into the electrolyte in form of a complex with [TFSA]− such as Al[TFSA]3.43,44 Recent studies have also shown that Al corrosion can be suppressed in concentrated electrolytes45 and neat SILs30 as well as in aprotic IL-based electrolytes, in which no effective solvents that are capable of dissolving the Al-[TFSA] complexes are present.44
To study the Al corrosion behavior in the diluted SILs, cyclic voltammetry was performed on the coin cell with Al foil as a working electrode and metallic Li-foil as counter and reference electrodes. Figure 4 shows cyclic voltammograms of an Al electrode in diluted SIL electrolytes. As expected, there is no detectable current in the voltammograms for [Li(G3)][TFSA]/toluene at a potential less than 5 V. The Al corrosion was evidently suppressed in [Li(G3)][TFSA]/toluene because Al-[TFSA] complexes would be insoluble in both toluene and [Li(G3)][TFSA]. Similarly, the Al corrosion is suppressed in the [Li(G3)][TFSA]/HFE electrolyte because Al-[TFSA] complexes is insoluble in HFE.32 Therefore, the LiCoO2 half-cell with the HFE-diluted SIL can be cycled for more than 100 cycles (Figure 2d). However, a drastic increase in the anodic current can be seen in [Li(G3)][TFSA]/PC. The surface Al2O3 layer was decomposed at the first anodic scan. Then, oxidation of the fresh Al surface and the anodic dissolution of Al-[TFSA] complexes occurred simultaneously during the reverse scan. SEM image of the Al electrode after measurement (Figure 4 inset) confirmed the pitting corrosion of the Al electrode in [Li(G3)][TFSA]/PC. Both polar co-solvent PC and the uncoordinated glyme can solubilize the Al-[TFSA] complexes, leading to the observed pitting corrosion. The same Al corrosion tests were also performed in [Li(G4)][TFSA]/toluene, [Li(G4)][TFSA]/PC, and [Li(G4)][BETA]/PC, but the results (data not shown) were very similar to those in the diluted SILs based on [Li(G3)][TFSA] or [Li(G3)][BETA] as shown in Figure 4.
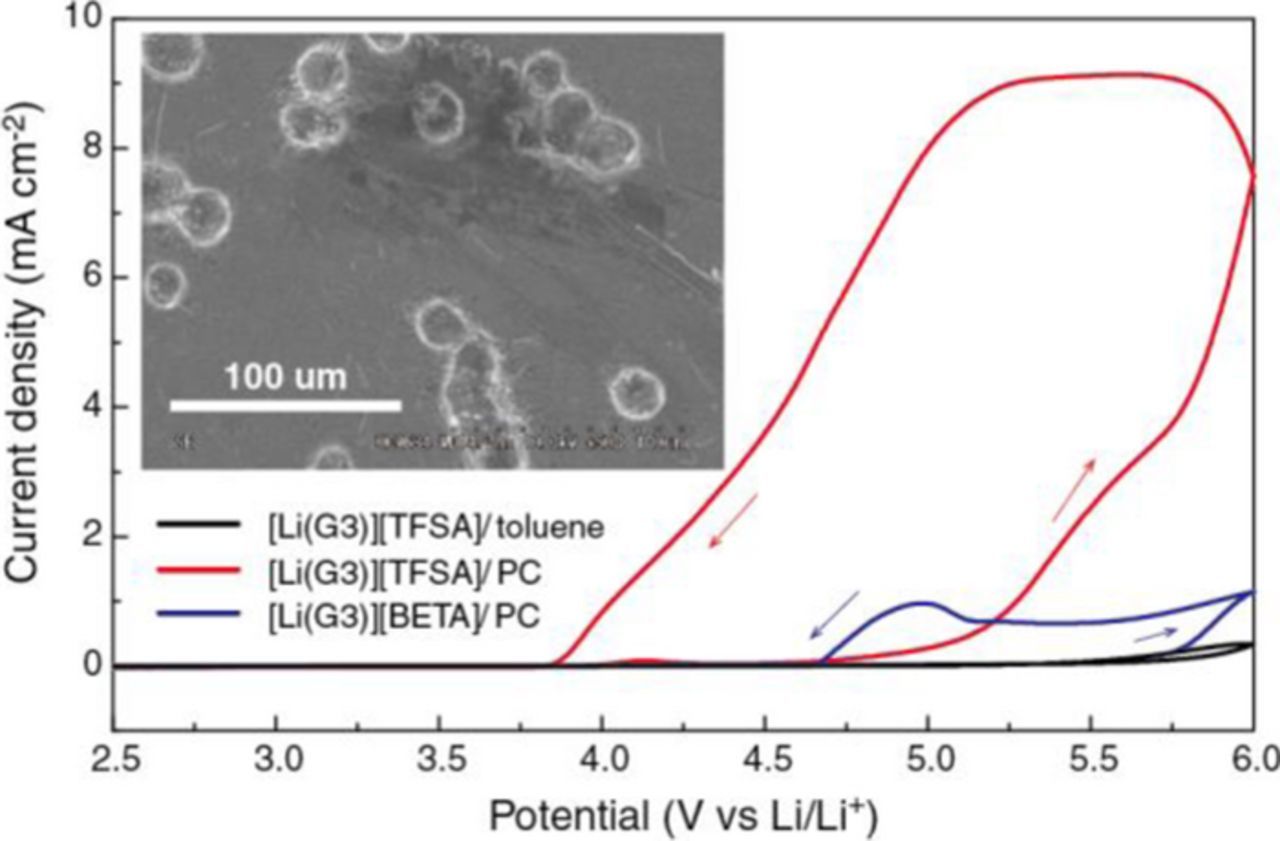
Figure 4. Cyclic voltammograms (1st cycle) on an Al working electrode in diluted SIL electrolytes at a scan rate of 5 mV s−1 at 30°C. Inset shows SEM image of Al foil after measurement in [Li(G3)][TFSA]/PC. Scale bar is 100 μm.
Unlike Li[TFSA]-based electrolytes, organic electrolytes containing Li[BETA] salt were reported to mitigate the pitting corrosion of Al current collector.43 Therefore, we also examined the Al corrosion behavior in [Li(G3)][BETA]/PC. The anodic dissolution of the Al electrode (and/or electrolyte decomposition) was suppressed up to 4.7 V in [Li(G3)][BETA]/PC. Likely, insoluble Al-[BETA] complexes could be formed on the electrode surface during the anodic scan and further Al corrosion did not take place in this electrolyte. Nevertheless, the capacities of the LiCoO2 half-cells with [Li(G3)][BETA]/PC faded upon cycling, as shown in Figure 5. This result indicates the anodic decomposition of the uncoordinated glymes (as suggested by DG/DLi = 1.09 for [Li(G3)][BETA]/PC) also contributed to capacity fading. The decline was not as drastic as for the LiCoO2 half-cells with [Li(G3)][TFSA]/PC and these cells retained 53% discharge capacity after 100 cycles. Coulombic efficiency (98−99%) was greater for the cells with [Li(G3)][BETA]/PC than that of the cell with [Li(G3)][TFSA]/PC. These results suggest that the rapid capacity fading and low efficiency of the cell with [Li(G3)][TFSA]/PC (Figure 3) were predominantly attributed to Al corrosion.
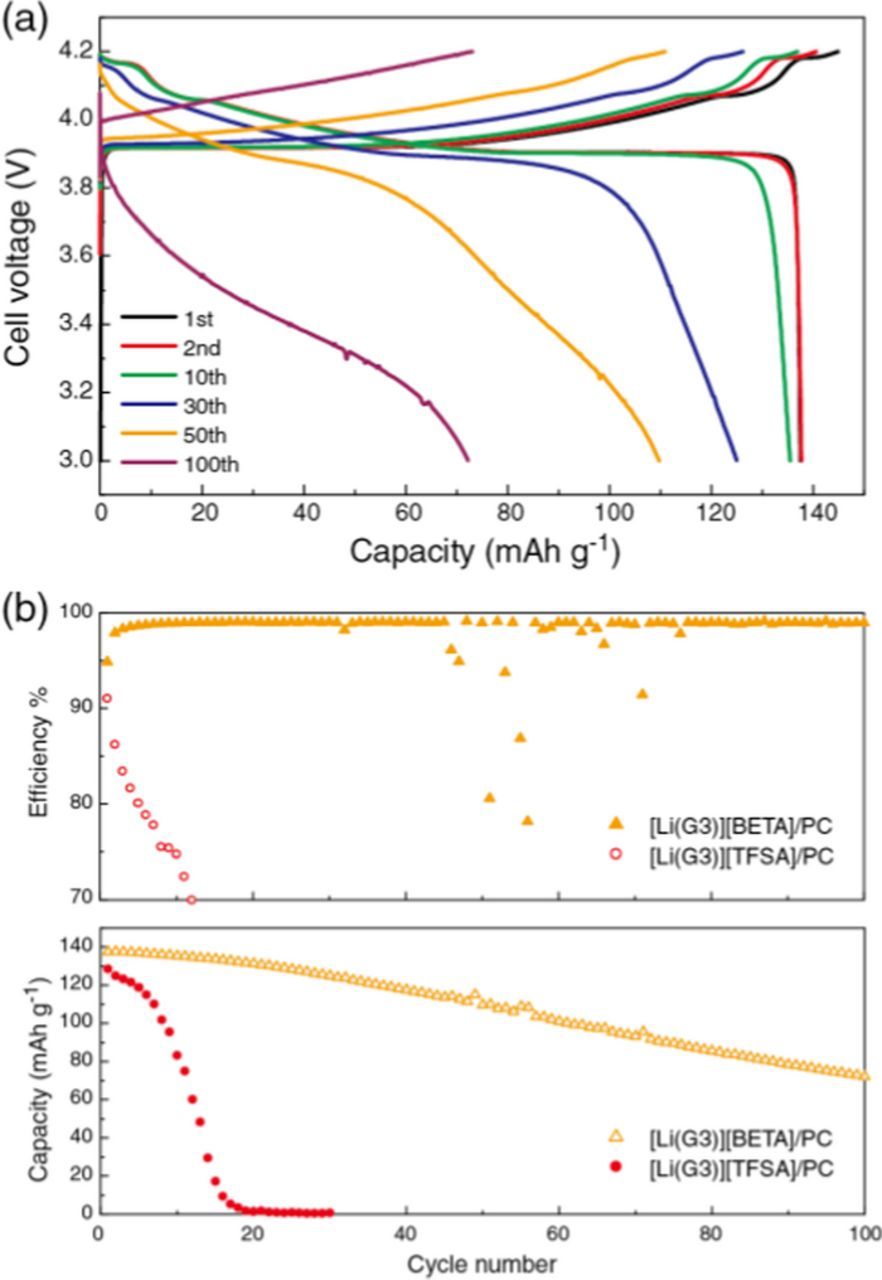
Figure 5. Charge/discharge characteristics of Li metal anode | [Li(G3)][BETA]/PC | LiCoO2 cathode cells with a constant current density of 50 μA cm−2 (C/8 rate) at 30°C; (a) charge/discharge curves, and (b) Coulombic efficiency and discharge capacity vs cycle number. Data of Coulombic efficiency and discharge capacity for [Li(G3)][TFSA]/PC are also shown for comparison.
Consequently, electrolyte components in diluted SILs greatly influenced the electrochemical stability and charge/discharge performance of the LiCoO2 half-cells. The SILs diluted with apolar solvents, such as toluene and HFE, allowed stable and reversible cycling of the LiCoO2 half-cells, owing to their higher anodic stability and suppression of the Al corrosion.
Charge/discharge of graphite half-cell
Reversible charge-discharge of graphite anode depends on the formation of an effective SEI layer on the graphite surface to yield a graphite intercalation compound accompanied with desolvation of Li+. This is one of the reasons that the standard electrolyte has been thus far irreplaceable. Many electrolytes were excluded from candidacy for Li-ion batteries using a graphite electrode due to their incompatibility with the graphite anode. For instance, the intercalation of the organic cations hinders the successful intercalation of Li+ in aprotic IL-based electrolyte,46 except for in special cases such as bis(fluorosulfonyl)amide (FSA) anion based ILs.47 The reversible intercalation/deintercalation of the cationic species of the ILs from/into graphite was confirmed instead of the Li+ intercalation/deintercalation. Sulfone-based organic electrolytes generally reduced at a potential higher than the intercalation/deintercalation potential of Li+, but did not form the effective SEI. Even organic electrolyte with PC as a main component does not achieve successful charge/discharge of the graphite anode due to the co-intercalation of the Li+ solvated with PC solvents into the graphite layer and continuous reductive decomposition of PC on the graphite. Likewise, ether-based organic electrolytes including glyme also caused the solvent intercalation and the associated graphite exfoliation.48 SEI-forming additives such as ethylene sulfite, vinylene carbonate, and fluoroethylene carbonate have often been added to electrolytes for the reversible intercalation/deintercalation.49 Recently, Song et al. reported that the co-intercalation can be suppressed in a PC-based electrolytes by the addition of small amount of diglyme.50 In this case, the diglyme-solvated Li+ ion preferentially intercalated and decomposed within the graphite and form a surface film (SEI), and the SEI effectively suppresses the co-intercalation of PC-solvated Li+, i.e. the diglyme acts as SEI-forming additive in PC-based electrolytes.
Recent works have shown that highly concentrated electrolytes of various popular solvents such as PC, ethyl acetate, dimethoxyethane, tetrahydrofuran, acetonitrile, sulfolane, and dimethyl sulfoxide allow reversible charge/discharge of graphite anode.51–54 The same thing was also true for the SILs [Li(G3)][TFSA] and [Li(G4)][TFSA].32,33,48 A possible explanation by Yamada et al. for this general phenomenon in the highly concentrated electrolytes is that an effective SEI can be formed by sacrificial decomposition of counter anions of a Li salt such as [TFSA]−.55 Dokko et al. reported that the desolvation of Li+ occurred at the graphite interface when the activities of Li+ and uncoordinated solvent in the electrolyte are significantly high and low respectively, whereas the co-intercalation of Li+ and solvent takes place in the electrolyte containing the uncoordinated solvent (i.e., high activity of the uncoordinated solvent).48 The activities of Li+ and the uncoordinated solvent were responsible for the reversible intercalation/deintercalation.32,48
Apolar solvents such as HFE and toluene do not behave as a solvent for Li+ solvation so are likely not responsible for the reversible intercalation/deintercalation. Our previous study revealed that the HFE-diluted SILs allowed the reversible Li+ intercalation into graphite with a relatively high capacity of 300 mAh g−1.32,33
Figure 6 shows the galvanostatic charge/discharge curves of [Li metal anode | diluted SIL electrolyte | graphite cathode] cells measured in the cell voltage range from 0.001−2.0 V at a current density of 18.6 mA g−1 at 30°C (C/20 rate). The Li+ intercalation process is defined as "charging" for the graphite electrodes. In Figure 6a, irreversible capacity was observed in the range 0.25−0.8 V at 1st cycle during the charging for the cell with [Li(G3)][TFSA]/toluene. This phenomenon can be attributed to co-intercalation of Li+ and G3 into the graphite, leading to initial Coulombic efficiency of 70%. However, in 2nd and 3rd cycles, the irreversible charge capacity became significantly smaller and a large charge capacity appeared in the potential range <0.2 V due to the expected Li+ intercalation for the formation of graphite intercalation compound with Li+. The discharge capacity of the cell reached approximately 270 mA h g−1, and the Coulombic efficiency of 2nd and 3rd cycles were ca. 94−96%. This suggests that desolvation of Li+ occurred successfully at the graphite/electrolyte interface and the co-intercalation of G3 was suppressed in [Li(G3)][TFSA]/toluene. The capacity being lower than the theoretical value may be ascribed to the lower ionic conductivity of [Li(G3)][TFSA]/toluene. In Figure 6b, irreversible capacity for the cell with [Li(G4)][TFSA]/toluene was larger than that for the cell with [Li(G3)][TFSA]/toluene, and was still discernible at 2nd and 3rd cycles. Discharge capacities (approximately 200 mA h g−1) and Coulombic efficiency (64−82%) were also smaller for the cell with [Li(G4)][TFSA]/toluene. These results suggest that the unfavorable co-intercalation of the solvent was more pronounced in the cell with [Li(G4)][TFSA]/toluene. Highly reversible charge/discharge of the graphite half-cell with [Li(G3)][TFSA]/toluene is probably due to higher activity of Li+ in [Li(G3)][TFSA] than in [Li(G4)][TFSA] as reported in previous measurements of Li/Li+ electrode potential.56
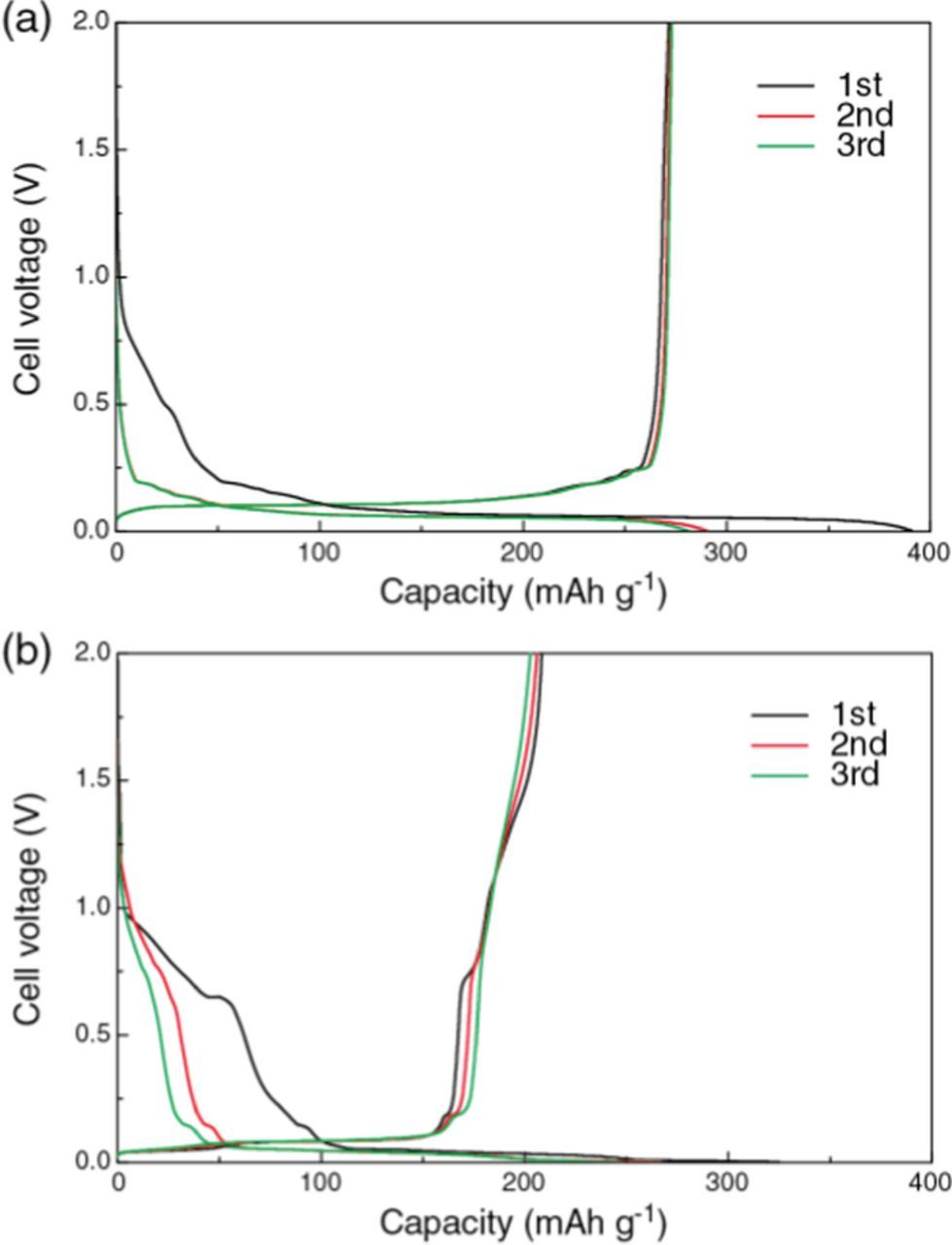
Figure 6. Galvanostatic charge/discharge curves of Li metal anode | diluted SIL electrolyte | graphite cathode cells with a constant current density of 74 μA cm−2 (C/20 rate) at 30°C with (a) [Li(G3)][TFSA]/toluene, and (b) [Li(G4)][TFSA]/toluene.
Charge/discharge of LiCoO2-graphite full-cell
The rate capability of the [LiCoO2 cathode | diluted SIL electrolyte | graphite anode] cell is shown in Figure 7, where [Li(G3)][TFSA]/toluene was chosen as the electrolyte because of its better compatibility with the LiCoO2 and graphite electrodes. The initial charge capacity exceeded the theoretical capacity of the cell (1.5 mAh cm−2), but 1st discharge capacity was ca. 1.2 mAh cm−2, corresponding to a Coulombic efficiency of 66%. These capacities are likely due to the irreversible reactions at the graphite/electrolyte interface as mentioned in the former section. After the 1st cycle, the efficiencies greatly improved to 95−99% at subsequent cycles depending on the charge/discharge rate, suggesting more reversible charge/discharge reactions at both LiCoO2 positive and the graphite negative electrodes. The capacities decreased with an increase in charge/discharge rate, but the full-cell still delivered charge/discharge capacities of ca. 0.4 mAh cm−2 (C/1 rate) with the efficiency of 99% and discharge capacity retention of 75% after 30 cycles at a C/20 rate. As can be seen in Figure 7b, the overpotential on charge/discharge increased at a higher rate, due to the limited Li+ transport in [Li(G3)][TFSA]/toluene as well as Li+ diffusion inside the active materials and Li+ transfer across the electrode/electrolyte interfaces. Therefore, the increased overpotential (internal resistance of the cell) is responsible for the lower capacity at higher charge/discharge rates. Although the performance of the full-cell with [Li(G3)][TFSA]/toluene was not as excellent as that of commercialized Li-ion cells, further optimizing diluted SILs with electrolyte additives holds potential to improve full-cell performance. These results clearly show that diluted SILs can be used as the electrolyte in rocking chair type Li-ion batteries without any of the components of the well-designed standard electrolyte, such as carbonate solvents and LiPF6.
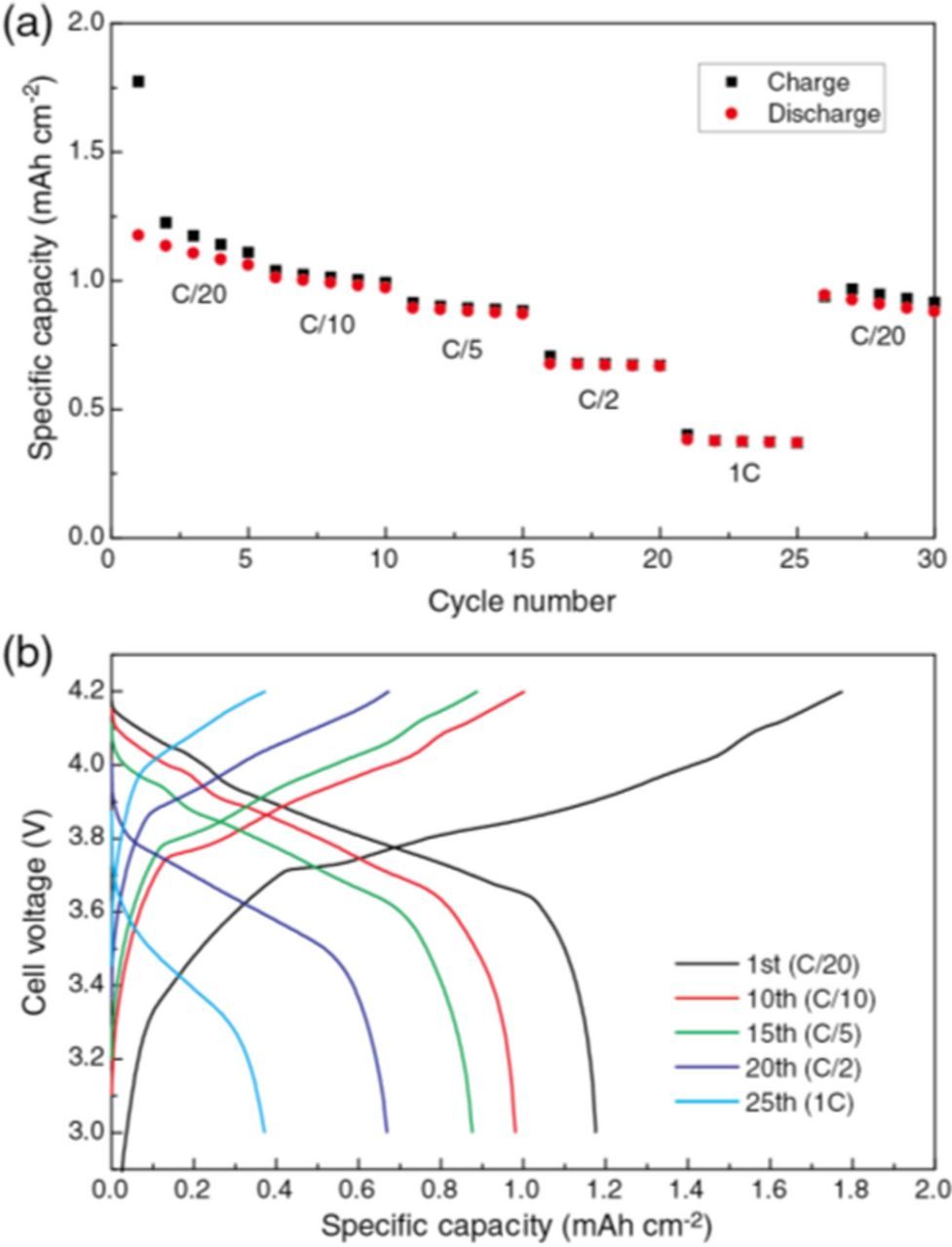
Figure 7. Galvanostatic charge/discharge curves of [graphite anode | diluted SIL electrolyte | LiCoO2 cathode] cells with difference charge/discharge current density in the range from 75 μA cm−2 (C/20 rate) to 1.5 mA cm−2 (C/1 rate) at 30°C with [Li(G3)][TFSA]/toluene: (a) cycling performance, and (b) charge/discharge curves.
Conclusions
In this study, we investigated the Li-ion battery performance using SILs diluted with toluene, HFE or PC. The PC-diluted SILs oxidatively decomposed around 4 V due to the presence of the uncoordinated glyme. Competitive solvation and ligand exchange between the glyme and PC occurred in the PC-diluted SILs and thus, the glyme-Li complex cations were unstable. By contrast, the anodic stability of the toluene- and HFE-diluted SILs were enhanced to ∼4.5 V, where all the glyme remained coordinated tightly to Li+. The choice of the diluting solvents also affected the Al pitting corrosion for the current collector. Remarkable Al corrosion occurred in the PC-diluted SILs because the uncoordinated glyme and the co-solvent PC can dissolve Al-[TFSA] complexes. However, the Al corrosion was suppressed in the toluene- and HFE-diluted SILs due to the insolubility of the Al-[TFSA] complexes in the electrolytes. As a result of the enhanced oxidative stability of the electrolyte and suppressed Al corrosion, LiCoO2 half-cells using the toluene- and HFE-diluted SILs could realize long-life charge/discharge cycling. Successful charge and discharge of graphite half-cells were also achieved in the toluene- and HFE-diluted SILs. The graphite half-cell with [Li(G3)][TFSA]/toluene exhibited higher capacity and efficiency than the graphite half-cell with [Li(G4)][TFSA]/toluene, likely due to the higher activity of Li+ in [Li(G3)][TFSA] than in [Li(G4)][TFSA]. We showed that the [Li(G3)][TFSA]/toluene enabled successful operation of the graphite/LiCoO2 full-cell without any components of the standard electrolyte consisting of carbonate solvents and LiPF6. Battery performance was not sufficient for the practical use, but could be further improved by optimizing the SIL-based electrolyte. The present study using the diluted SILs gives insight into a novel design approach to alternative electrolytes for advanced Li-ion and beyond Li-ion batteries.
Acknowledgments
This study was supported in part by the Advanced Low Carbon Technology Research and Development Program (ALCA) of the Japan Science and Technology Agency (JST), and by the Japan Society for the Promotion of Science (JSPS) via JSPS KAKENHI (No. 15H03874 for KD, No. 15H06439 and No. 16H06053 for KU).